
Structure of the Noncompetitive Antagonist-binding Site of the ...
11
THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1992 by The American Society for Biochemistry and Molecular Biology, Inc. Vol. 267, No. 15, Issue of May 25, pp. 10489-10499,1992 Printed in U.S.A. Structure of the Noncompetitive Antagonist-binding Site of the Torpedo Nicotinic Acetylcholine Receptor [3H]MEPROADIFEN MUSTARD REACTS SELECTIVELY WITH a-SUBUNIT Glu-262* (Received for publication, January 22, 1992) Steen E. PedersenS, Shannan D. Sharp, Wu-Schyong Liu, and Jonathan B. Cohens From the Department of Anatomy and Neurobiology, Washington University School of Medicine, St. Louis, Missouri 63110 [‘HIMeproadifen mustard, an affinity label for the noncompetitive antagonist site of the nicotinic acetyl- choline receptor (AChR), specifically alkylates the AChR a-subunit when the acetylcholine-binding sites are occupied byagonist(Dreyer, E. B., Hasan, F., Cohen, S. G., and Cohen, J. B. (1986) J. Biol. Chem. 261,13727-13734). In this report, we identify the site of alkylation within the a-subunit as Glu-262. AChR- rich membranes from Torpedo californica electric or- gan were reacted with r3H]meproadifen mustard in the presence of carbamylcholine and in the absence or presence of nonradioactive meproadifen to define spe- cific alkylation of the noncompetitive antagonist site. Alkylated a-subunits were isolated and subjected to chemical or enzymatic cleavage. When digests with CNBr in 70% trifluoroacetic acid or 70% formic acid were fractionated by gel filtration high performance liquid chromatography (HPLC), specifically labeled material was recovered in thevoid volume fractions. Based upon NH2-terminal sequence analysis, for both digests, the void volume fractions contained a fragment beginning at Gln-208 before the M1 hydrophobic se- quence, whereas the sample from the digest in trifluo- roacetic acid also contained as a primary sequence a fragment beginning at Thr-244 and extending through the M2 hydrophobic sequence. Sequence analysis re- vealed no release of 3H for the sample from digestion in formic acid, whereas for the trifluoroacetic acid digest, there was specific release of ‘H in cycle 19, which would correspond to Glu-262. This site of al- kylation was confirmed by isolation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and re- versed-phase HPLC of a specifically labeled fragment from an endoproteinase Lys-C digest of the alkylated a-subunit. NH2-terminal amino acid sequencing re- vealed release of 3H at cycle 20 from a fragment begin- ning at Met-243 and extending into the M3 hydropho- bic sequence. Because r3H]meproadifen mustard con- tains, as its reactive group, a positively charged quaternary aziridinium ion, Glu-262 of the a-subunit is identified as a contributor to the cation-binding do- * This work was supported in part by United States Public Health Service Grants NS 19522 and NS 22828 (Senator Jacob Javits Center for Excellence in Neuroscience). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accord- ance with 18 U.S.C. Section 1734 solely to indicate this fact. $ Present address: Dept. of Molecular Physiology and Biophysics, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030. I To whom correspondence should be addressed Dept. of Anatomy and Neurobiology, Washington University School of Medicine, 660 S. Euclid Ave., St. Louis, MO 63110. Tel.: 314-362-3539; Fax: 314- 362-3446. main of the noncompetitive antagonist-binding site and thus of the ion channel. The nicotinic acetylcholine receptor (AChR)’ from Torpedo electric organ is a ligand-gated cation channel composed of five transmembranesubunitsina stoichiometry of az/3y6 (Reynolds and Karlin, 1978; Raftery et al., 1980).The homol- ogous subunits (Noda et al., 1983) are arranged with circular pseudosymmetry about a central axis presumed to correspond to thecation channel (Toyoshima and Unwin, 1990; Mitra et al., 1989). The AChR contains two binding sites for agonists and competitive antagonists (Neubig and Cohen, 1979,1980). These sites have been shown to be located at the a-y and a-6 subunit interfaces (Blount and Merlie, 1989; Pedersen and Cohen, 1990), with the a-subunits containing the primary sites of labeling by agonist and antagonist affinity reagents (Kao et al., 1984; Dennis et al., 1988; Abramson et al., 1989; Galzi et al., 1990; Middleton and Cohen, 1991; Cohen et al., 1991). Other functionally important domains of the AChR have been identified by determining the sites of action of noncom- petitive antagonists, compounds that block the permeability response without preventing the binding of acetylcholine. This class of compounds includes aromatic amines, alcohols, fatty acids, detergents, and general anesthetics (for reviews, see Peper et al. (1982),Colquohoun (1986),Cohen et al. (1986), and Forman and Miller (1989)). Whereas these compounds all can act as nonspecific perturbants of membrane structure, electrophysiological studies provide evidence that some of them may block ion permeation by binding to andphysically occluding the open channel (Neher and Steinbach, 1978; Ogden et al., 1981; Neher, 1983). Direct measurement of the binding of radiolabeled amine noncompetitive antagonists to Torpedo postsynaptic mem- branes shows that these compounds bind with high affinity (Keg - pM) to a site distinct from, but coupled allosterically to, the acetylcholine sites (reviewed by Cohen et al. (1985)). At equilibrium, many, but not all, of these compounds bind with highest affinity to the AChR when acetylcholine sites are occupied by agonist, i.e. they bind preferentially to the desensitized state of the AChR. In addition to binding with high affinity to the AChR, noncompetitive antagonists bind with low affinity to sites present at substantially higher con- centration than the concentration of AChRs, perhaps as a result of interactions at the protein-lipid interface (Heidmann The abbreviations used are: AChR, nicotinic acetylcholine recep- tor; HPLC, high performance liquid chromatography; PTH, phenyl- thiohydantoin; SDS, sodium dodecyl sulfate; PAGE, polyacrylamide gel electrophoresis; Tricine, N-[2-hydroxy-l,l-bis(hydroxymethyl)- ethyllglycine. 10489
Transcript of Structure of the Noncompetitive Antagonist-binding Site of the ...

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1992 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 267, No. 15, Issue of May 25, pp. 10489-10499,1992 Printed in U.S.A.
Structure of the Noncompetitive Antagonist-binding Site of the Torpedo Nicotinic Acetylcholine Receptor [3H]MEPROADIFEN MUSTARD REACTS SELECTIVELY WITH a-SUBUNIT Glu-262*
(Received for publication, January 22, 1992)
Steen E. PedersenS, Shannan D. Sharp, Wu-Schyong Liu, and Jonathan B. Cohens From the Department of Anatomy and Neurobiology, Washington University School of Medicine, St. Louis, Missouri 63110
[‘HIMeproadifen mustard, an affinity label for the noncompetitive antagonist site of the nicotinic acetyl- choline receptor (AChR), specifically alkylates the AChR a-subunit when the acetylcholine-binding sites are occupied by agonist (Dreyer, E. B., Hasan, F., Cohen, S . G., and Cohen, J. B. (1986) J. Biol. Chem. 261,13727-13734). In this report, we identify the site of alkylation within the a-subunit as Glu-262. AChR- rich membranes from Torpedo californica electric or- gan were reacted with r3H]meproadifen mustard in the presence of carbamylcholine and in the absence or presence of nonradioactive meproadifen to define spe- cific alkylation of the noncompetitive antagonist site. Alkylated a-subunits were isolated and subjected to chemical or enzymatic cleavage. When digests with CNBr in 70% trifluoroacetic acid or 70% formic acid were fractionated by gel filtration high performance liquid chromatography (HPLC), specifically labeled material was recovered in the void volume fractions. Based upon NH2-terminal sequence analysis, for both digests, the void volume fractions contained a fragment beginning at Gln-208 before the M1 hydrophobic se- quence, whereas the sample from the digest in trifluo- roacetic acid also contained as a primary sequence a fragment beginning at Thr-244 and extending through the M2 hydrophobic sequence. Sequence analysis re- vealed no release of 3H for the sample from digestion in formic acid, whereas for the trifluoroacetic acid digest, there was specific release of ‘H in cycle 19, which would correspond to Glu-262. This site of al- kylation was confirmed by isolation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and re- versed-phase HPLC of a specifically labeled fragment from an endoproteinase Lys-C digest of the alkylated a-subunit. NH2-terminal amino acid sequencing re- vealed release of 3H at cycle 20 from a fragment begin- ning at Met-243 and extending into the M3 hydropho- bic sequence. Because r3H]meproadifen mustard con- tains, as its reactive group, a positively charged quaternary aziridinium ion, Glu-262 of the a-subunit is identified as a contributor to the cation-binding do-
* This work was supported in part by United States Public Health Service Grants NS 19522 and NS 22828 (Senator Jacob Javits Center for Excellence in Neuroscience). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accord- ance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ Present address: Dept. of Molecular Physiology and Biophysics, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030.
I To whom correspondence should be addressed Dept. of Anatomy and Neurobiology, Washington University School of Medicine, 660 S. Euclid Ave., St. Louis, MO 63110. Tel.: 314-362-3539; Fax: 314- 362-3446.
main of the noncompetitive antagonist-binding site and thus of the ion channel.
The nicotinic acetylcholine receptor (AChR)’ from Torpedo electric organ is a ligand-gated cation channel composed of five transmembrane subunits in a stoichiometry of az/3y6 (Reynolds and Karlin, 1978; Raftery et al., 1980). The homol- ogous subunits (Noda et al., 1983) are arranged with circular pseudosymmetry about a central axis presumed to correspond to the cation channel (Toyoshima and Unwin, 1990; Mitra et al., 1989). The AChR contains two binding sites for agonists and competitive antagonists (Neubig and Cohen, 1979,1980). These sites have been shown to be located at the a-y and a-6 subunit interfaces (Blount and Merlie, 1989; Pedersen and Cohen, 1990), with the a-subunits containing the primary sites of labeling by agonist and antagonist affinity reagents (Kao et al., 1984; Dennis et al., 1988; Abramson et al., 1989; Galzi et al., 1990; Middleton and Cohen, 1991; Cohen et al., 1991).
Other functionally important domains of the AChR have been identified by determining the sites of action of noncom- petitive antagonists, compounds that block the permeability response without preventing the binding of acetylcholine. This class of compounds includes aromatic amines, alcohols, fatty acids, detergents, and general anesthetics (for reviews, see Peper et al. (1982), Colquohoun (1986), Cohen et al. (1986), and Forman and Miller (1989)). Whereas these compounds all can act as nonspecific perturbants of membrane structure, electrophysiological studies provide evidence that some of them may block ion permeation by binding to and physically occluding the open channel (Neher and Steinbach, 1978; Ogden et al., 1981; Neher, 1983).
Direct measurement of the binding of radiolabeled amine noncompetitive antagonists to Torpedo postsynaptic mem- branes shows that these compounds bind with high affinity (Keg - pM) to a site distinct from, but coupled allosterically to, the acetylcholine sites (reviewed by Cohen et al. (1985)). At equilibrium, many, but not all, of these compounds bind with highest affinity to the AChR when acetylcholine sites are occupied by agonist, i.e. they bind preferentially to the desensitized state of the AChR. In addition to binding with high affinity to the AChR, noncompetitive antagonists bind with low affinity to sites present at substantially higher con- centration than the concentration of AChRs, perhaps as a result of interactions at the protein-lipid interface (Heidmann
The abbreviations used are: AChR, nicotinic acetylcholine recep- tor; HPLC, high performance liquid chromatography; PTH, phenyl- thiohydantoin; SDS, sodium dodecyl sulfate; PAGE, polyacrylamide gel electrophoresis; Tricine, N-[2-hydroxy-l,l-bis(hydroxymethyl)- ethyllglycine.
10489

10490 Noncompetitive Antagonist Site of the Acetylcholine Receptor
et al., 1983). As a consequence, it has been difficult to quantify the concentration of high affinity noncompetitive antagonist sites. Where high affinity binding can be quantitated, how- ever, there is one site/AChR monomer both for [3H]phency- clidine, which binds with highest affinity to the desensitized state of the AChR (Heidmann et al., 1983; Herz et al., 1987)) and for [3H]tetracaine, which binds with 30-fold higher affin- ity in the absence of agonist than in its presence (Cohen et al., 1985; Strnad, 1988).
Regions of the AChR contributing to the noncompetitive antagonist site have been identified by use of photoaffinity reagents. In desensitized AChRs, [3H]chlorpromazine can be photoincorporated with similar efficiency into all AChR sub- units, and sites of incorporation have been mapped in the 6- subunit to Ser-254 within the M2 hydrophobic segment and to homologous residues in the other subunits (Giraudat et al., 1987, 1989; Revah et al., 1990). The same residues have been associated with the binding site for another desensitizing noncompetitive antagonist, [3H]triphenylmethylphosphon- ium (Hucho et al., 1986). These results suggest a structural model of the desensitized AChR with M2 segments from each subunit associating around the central axis of the AChR. In contrast, when [3H]quinacrine azide is photoincorporated into AChRs after exposure to agonist for short times, the primary sites of labeling are unidentified residues within the M1 hydrophobic segment of the a-subunit (DiPaola et al., 1990).
Results of site-directed mutagenesis also implicate the M2 region as being important for ion permeation. Charged resi- dues near the ends of the M2 sequence of each subunit affect channel conductance and especially the sensitivity to M2' (Imoto et al., 1988). Whether these residues also contribute to the site for cationic noncompetitive antagonists was not determined, but mutation of the residues within M2 labeled by [3H]chlorpromazine alters the potency of compounds act- ing as open channel blockers (Leonard et al., 1988; Charnet et al., 1990).
To identify regions of the AChR interacting with the posi- tive charge of noncompetitive antagonists, [3H]meproadifen mustard (2-(chloroethylmethylamino)ethyl-2,2-diphenylpen- tenoate HCl) was introduced (Dreyer et al., 1986) as an alkylating analog of the desensitizing noncompetitive antag- onists proadifen and meproadifen. In its active form, the positive charge is present as a reactive quaternary aziridinium ion that is well suited to react with nucleophiles that might be present within the noncompetitive antagonist site (Dermer and Ham, 1969). [3H]Meproadifen mustard alkylated the noncompetitive antagonist site only in the presence of high concentrations of agonist. The predominant site of incorpo- ration was within the AChR a-subunit, localized to a 20-kDa fragment beginning at Ser-162/Ser-173 and including the three putative transmembrane segments M1, M2, and M3 (Pedersen et al., 1986). In this report, we demonstrate that the site of alkylation by [3H]meproadifen mustard is con- tained within the M2 region of the AChR. [3H]Meproadifen mustard does not react with Ser or Thr within M2, but with Glu-262 located at the predicted extracellular end of M2 and implicated by site-directed mutagenesis (Imoto et al., 1988) in the control of channel conductance.
EXPERIMENTAL PROCEDURES
Materials-AChR-rich membranes from Torpedo californica elec- tric organ were prepared by the method of Sobel et al. (1977) as described by Pedersen et al. (1986). [3H]Meproadifen mustard (35 Ci/ mmol) was prepared by reductive tritiation of the 2,2-diphenyl-4- pentenoate precursor as described by Dreyer et al. (1986). [3H]Me- proadifen mustard was stored at -80 "C in ethanol and was diluted isotopically to 1 Ci/mmol with nonradioactive material for all exper-
iments. Meproadifen iodide was synthesized as described (Krodel et al., 1979). CNBr was from Fluka. Endoproteinase Lys-C, acrylamide, bisacrylamide, and dithiothreitol were from Boehringer Mannheim. Lubrol-PX, guanidine, sodium dodecyl sulfate (SDS), and trifluoro- acetic acid were from Pierce Chemical Co. Formic acid was from J. T. Baker Inc. and proteinase K and diisopropylphosphorofluoridate were from Sigma. Prestained low molecular weight gel standards were from GIBCO-Bethesda Research Laboratories.
Alkylution and Isolation of PHIMeproadifen Mustard-lubeled a- Subunit-Membranes were reacted with [3H]meproadifen mustard essentially as described by Dreyer et al. (1986). Briefly, AChR-rich membranes (1 mg/ml) were incubated in Torpedo physiological saline (250 mM NaC1, 5 mM KCI, 3 mM CaC12, 2 mM MgCI,, 5 mM sodium phosphate, pH 7.0) with 3 mM carbamylcholine and 10 p~ [3H] meproadifen mustard that had been precyclized with base for 30 min. A parallel incubation that contained 200 p~ meproadifen in addition to the other reagents was used to define nonspecific labeling. After a 30-min reaction at ambient temperature, the reaction was quenched with 10 mM sodium thiosulfate, and membranes were then incubated for 1 h at 4 "C in 10 mM lithium diiodosalicylate, 2 mM EDTA, 10 mM Tris, pH 8.1, to release peripheral proteins (Porter and Froehner, 1983). The [3H]meproadifen mustard-labeled a-subunit was isolated by preparative gel electrophoresis on 8% acrylamide slab gels (1.5 mm thick) as described (Pedersen et al., 1990) using passive elution from diced gel slices according to Hager and Burgess (1980). After acetone precipitation, subunits were redissolved in 0.1% SDS, 1 mM dithiothreitol, 1 mM EDTA, 10 mM Napi, pH 7.0, and stored at -80 'C. The labeled a-subunit was usually isolated at 50-60% overall yield, with 500-600 pg of a-subunit recovered from 10 mg of mem- branes loaded on two gels. In four experiments using different isotope dilutions of [3H]meproadifen mustard, total incorporation into the a- subunit varied from 1200 to 3500 cpm/pg when the alkylation reaction was carried out in the absence of meproadifen, and in each case, 50- 65% of the labeling was specific, i.e. inhibited by 0.2 mM meproadifen. Because of the reactivity and instability of [3H]meproadifen mustard, its radiochemical purity was not assessed routinely. Based upon the nominal specific activity, the level of specific incorporation was equivalent to alkylation of 6-9% of a-subunits.
Digestions of a-Subunit-For digestions with CNBr and some enzymatic digestions, the [3H]meproadifen mustard-labeled a-sub- unit was reduced with dithiothreitol and alkylated with iodoacetamide in the presence of 6 M guanidine, 0.1% Lubrol-PX as described (Pedersen et al., 1990). Prior to cleavage with CNBr, reagents were removed by gel filtration chromatography on a Sephadex G-50 column that was pre-equilibrated and eluted with 70% formic acid. For CNBr digestion in 70% formic acid, the Sephadex G-50 column eluate was added to 0.25 volume of freshly prepared 1 M CNBr in 70% formic acid. For CNBr digestion in 70% trifluoroacetic acid, the Sephadex G-50 column eluate was immediately dried by vacuum centrifugation (Savant SpeedVac) and then redissolved in 0.2 M CNBr in 70% trifluoroacetic acid. The reaction mixtures were incubated at ambient temperature for 16 h under argon, and in the dark. The reagents were then removed by vacuum centrifugation, and the dried samples were redissolved in 50-100 p1 of 70% formic acid for chromatography.
Prior to digestion with proteinase K, the [3H]meproadifen mustard- labeled a-subunit was reduced with dithiothreitol and alkylated with iodoacetamide, with the reagents removed on a Sephadex (2-50 col- umn pre-equilibrated with 0.1 M ammonium bicarbonate, 0.1% Lu- brol-PX. The labeled a-subunit (70 pg in 0.6 ml of 0.1 M ammonium bicarbonate, 0.5% SDS, 0.1% Lubrol-PX) was digested with protein- ase K (20 pg) for 16 h at ambient temperature. After digestion, the sample was stored at -20 "C. For TSK 2000SW chromatography, the samples were dried by vacuum centrifugation, redissolved in 70% formic acid, applied to the column, and eluted with 70% formic acid.
Digestion of the labeled a-subunit with endoproteinase Lys-C was carried out without prior reduction or alkylation. The a-subunit (0.2 mg/ml) was incubated for 2-4 days at room temperature with enzyme (4 units/ml) in 1 mM sodium phosphate, 15 mM Tris, pH 8.1, 0.1% SDS. Reactions were terminated by addition of diisopropylphospho-
were digested, with half the sample analyzed by SDS-PAGE and half rofluoridate to 1 mM. For analytical scale experiments, 20-pg aliquots
by reversed-phase HPLC. For preparative scale digests, 100-pg ali- quots were digested and then fractionated first by SDS-PAGE and then by reversed-phase HPLC. Detailed digestion conditions are provided in the figure legends.
Chromatography-Peptides generated by CNBr or proteinase K cleavage were fractionated by high performance gel filtration chro- matography using a TSK 2000SW column (75 X 300 mm) eluted with

Noncompetitive Antagonist Site of the Acetylcholine Receptor 10491
70% formic acid at 0.5 ml/min. The chromatography system was composed of two Waters Model 510 pumps, a Model 680 gradient controller, and a U6K injector. Elution of peptides was detected at 280 nm by a Waters 4402 absorbance detector. Formic acid was chosen as a suitable solvent because of its strongly denaturing effect on proteins and because it was not expected to dehydrate the column matrix. Previous work demonstrated the feasibility of isolating hy- drophobic peptides from bacteriorhodopsin (Gerber et al., 1979) and myelin proteolipid protein (Stoffel et al., 1984) by gel filtration chromatography in formic acid. The TSK 2000SW column allowed good separation of peptides in the size range of 2-10 kDa, as shown by the elution times of standard proteins and peptides in Fig. 1.
Peptides generated by endoproteinase Lys-C were fractionated by reversed-phase HPLC using a Brownlee Aquapore C, column (100 X 2.1 mm, with a 30-mm guard column). Solvent A was 0.05% trifluo- roacetic acid in 60% CH3CN, 40% 2-propanol; and solvent B was 0.08% trifluoroacetic acid in water. Elution was at 0.2 ml/min using a linear gradient increasing from 25 to 100% solvent A in 80 min, followed by 5 min at 100% solvent A and a return to 25% solvent A in 5 min. Fractions were collected at 1-min intervals in microcentri- fuge tubes, and the elution of peptides was monitored by absorbance at 210 nm using a Kratos 757 detector.
lyzed by SDS-PAGE using the buffer system of Laemmli (1970) with Gel Electrophoresis-In preliminary experiments, digests were ana-
modifications as described (Pedersen et al., 1986). For analysis and isolation of peptides from endoproteinase Lys-C digests, the Tricine gel system of Schagger and von Jagow (1987) was used. Gels were composed of an 11-cm-long small-pore separating gel (16.5% T, 6% C), a 2-cm spacer gel (10% T, 3% C), and a 2-cm stacking gel (4% T, 3% C). Analytical and preparative scale digests were fractionated on gels of 0.75- and 1.5-mm thickness, respectively. Analytical gels were stained with Coomassie Blue, and then the wet gels were cut into 3- mm slices for determination of 3H distribution (Middleton and Cohen, 1991). The molecular weights of labeled bands were estimated using prestained standards including ovalbumin (43,000), carbonic anhy- drase (29,000), @-lactoglobulin (18,400), lysozyme (14,300), bovine trypsin inhibitor (6200), and the B (3400) and A (2300) chains of insulin. Based upon the results of the analytical digests, specifically labeled fragments migrated in bands between 6 and 16 kDa. For the preparative scale digests, four strips (each 7 mm wide) were excised from that region of the gel that was identified by the migration of the prestained standards. Proteolytic fragments were eluted passively from diced gel slices, and the eluates were concentrated in Centricon 3 concentrators (Amicon Corp.). To maximize elution efficiency, gel slices were eluted two times, initially for 2 days and then for 3 more days. Eluates after concentration were combined to a final volume of ~0.2-0.3 ml, and samples were then centrifuged to remove particu- lates before HPLC.
Amino Acid Sequence Analysis-NHz-terminal sequence analysis of the cleavage fragments of the AChR a-subunit was performed on an Applied Biosystems Model 470A Protein Sequencer. All chemicals used for sequence analysis were from Applied Biosystems, Inc. Ap-
t 00000 . , . , . , . , . , . , r l
5 6 7 8 9 1 0 1 1 Elution Volume (mls)
FIG. 1. TSK 2000SW chromatography of molecular weight standards in 70% formic acid. The following compounds were dissolved in 70% formic acid, applied to a TSK 2000SW column, and eluted at 0.5 ml/min: bovine serum albumin, M , = 66,000; carbonic anhydrase, M, = 29,000; cytochrome c, M, = 12,100; a-bungarotoxin, M, = 8000; insulin, M, = 6000; insulin B chain, M, = 3496; insulin A chain, M, = 2530; d-tubocurarine, M , = 693; and ethylene glycol, M, = 62.1. The elution volume of each compound as detected by absorb- ance at 280 nm is plotted uers'sus the molecular weight.
proximately 40% of the released PTH-derivative was separated by an on-line Model 120A PTH-derivative analyzer, and 40% was used for determination of radioactivity. Release of amino acids was quanti- tated by comparison of integrated peak areas with a standard run performed before each sequence run. Initial yields were estimated by back extrapolation to the zeroth cycle from the yields of the quanti- fiable amino acids (i.e. excluding Ser, Thr, Trp, Arg, Cys, and His). For radioactive samples, the amount of 3H loaded was compared to the amount retained on the filter and filter support at the end of the run and to the amount released with the PTH-derivatives. About 50% of the radioactivity loaded could be accounted for in long sequencing runs.
In initial experiments, peptides were loaded onto trifluoroacetic acid-treated glass-fiber filters coated with 1.5 mg of Polybrene. Iden- tification of the sites of labeling by [3H]meproadifen mustard was hindered by the fact that the labeled peptides isolated from CNBr digests were sequenced at very low repetitive yield. This appeared to result in part because the labeled peptide was rich in serines and threonines and susceptible to 0- to N-acyl transfer and NH2-terminal blockage during sequence analysis (Allen, 1989) and also because the hydrophobic peptides were eluted from the filter by the solvents used during normal sequence operation. To address the latter problem, we utilized a hydrophobic test peptide to evaluate alternative solid sup- ports and solvent wash conditions. Repetitive yield was improved when samples were immobilized on chemically modified glass-fiber discs (part 191101) from Porton Instruments (Tarzana, CA) without Polybrene coating. As recommended by the manufacturer, a modified degradation cycle was developed for use with the Porton sample support disc. Two major modifications from the standard Applied Biosystems O3CPTH cycle were as follows: 1) the solvent wash steps after phenyl isothiocynate coupling were shortened substantially; and 2) ethyl acetate was used to transfer the anilinothiazolinone-deriva- tives from the reaction cartridge to the conversion flask.
Protein Determination-Protein was assayed using the method of Lowry et al. (1951) or of Schaffner and Weissman (1973).
RESULTS
In previous work, it was shown that the primary site of specific alkylation by the noncompetitive antagonist [3H] meproadifen mustard is between Ser-173 and Glu-338 of the AChR a-subunit (Pedersen et al., 1986; White and Cohen, 1988). Preliminary experiments established that 3H is incor- porated into the a-subunit via bonds that are stable under the pH conditions necessary for gas-phase protein microse- quencing and for chemical cleavage with CNBr. This was a concern because the reactive aziridinium of meproadifen mus- tard was separated from 3H by an ester bond, and a second potentially labile ester bond would result if the aziridinium reacted with a side-chain carboxylate. When the [3H]me- proadifen mustard-labeled a-subunit (14,000 cpm) was sub- jected to two cycles of Edman degradation with all solvent washes recovered manually, <2% of loaded 3H was recovered in the washes. Acid stability of the 3H-labeled a-subunit was assessed by size-exclusion chromatography on a TSK 2000SW column. After 12 h in 70% trifluoroacetic acid, >85% of 3H was recovered with the a-subunit, with only 5% recovered in the column-included volume (Mr < 700). In contrast, after 18 h in 0.25 M acetic acid at 105 "C, <lo% of the counts/minute were recovered with the a-subunit, and -50% were recovered in the included volume.
Digestion with CNBr in Formic Acid-As a first approach to identify the sites of labeling by [3H]meproadifen mustard, we utilized CNBr to cleave at methionines. Cleavage of the AChR a-subunit at methionines could produce 16 fragments ranging in size from 0.5 to 12.2 kDa (Table I), with potential cleavage sites preceding the hydrophobic sequences M1 (Met- 207) and M2 (Met-243) as well as two sites within M3 (Met- 278 and Met-282) and a site after M3 (Met-308). AChR a- subunits labeled in the absence and presence of meproadifen were reduced with dithiothreitol, alkylated with iodoaceta- mide, and then digested with CNBr in 70% formic acid.

10492 Noncompetitive Antagonist Site of the Acetylcholine Receptor
TABLE I CNBr fragments of the T. californica AChR a-subunit
based upon the sequence of Noda et al. (1982)
M, Fragment Starting residue
I Ser-1 12,257 I1 Thr-107 1397 111 Tv-118 3218 IV Lys-145 3047 V (2111-172 838 VI Lys-179 3718 VI1 Gln-208 4138 VI11 Thr-244 3720 IX Leu-279 511 X Ile-283 2904 XI Pro-309 1958 XI1 Phe-325 632 XI11 Lys-330 6378 XIV LYS-387 2074 xv Val-405 1303 XVI Leu-416 2322
Attempts to isolate the labeled fragments by reversed-phase HPLC were largely unsuccessful. Labeled fragments were not eluted by acetonitrile from C4 columns (25 cm X 4.5 mm), but could be eluted from wide-pore microbore C4 columns (10 cm X 2 mm) with reasonable recovery (>50%) of labeled material. Elution of labeled material could be made nearly quantitative using mixtures of isobutyl alcohol and acetonitrile as the eluant; but the resolution of the hydrophobic material was poor, and the peak shapes were frequently amorphous. This led to attempts at gel filtration HPLC for the purification of the labeled peptide.
Separation of CNBr cleavage fragments was achieved by gel filtration HPLC carried out in 70% formic acid. The elution pattern of the a-subunit cleavage fragments as de- tected by absorbance at 280 nm is shown in Fig. !2A, with the corresponding elution of [3H]meproadifen mustard-labeled fragments shown in Fig. 2B. The specifically labeled frag- ments eluted in the void volume corresponding to a sharp peak of absorbance (peak A). A number of other absorption peaks (peaks B-G) that did not contain specific label were also observed. For a CNBr digest of the a-subunit labeled with 4-(N-maleimido)benzyltri[3H]methylamm~nium iodide, (10% of loaded 3H eluted in the void volume, and -60% eluted in peak D (data not shown). Since 4-(N-maleim- ido)benzyltri[3H]methylammonium iodide is known to alky- late Cys-192/Cys-193 (Kao et d., 1984), which reside in a CNBr fragment beginning at Lys-179 (Table I, fragment VI), it appeared that the [3H]meproadifen mustard-labeled site lay beyond Met-207, the COOH terminus of fragment VI.
NH2-terminal amino acid sequencing of the void volume peak (peak A), which included material specifically labeled with [3H]meproadifen mustard, revealed a predominant se- quence beginning at Gln-208 (fragment VII, initial yield of 74 pmol) as well as other sequences beginning at Val-405 (frag- ment XV, 33 pmol), Lys-179 (fragment VI, 30 pmol), and Pro- 309 (fragment XI, 8 pmol). PTH-derivatives corresponding to the primary sequence (Gln-208) were readily quantified for 20 cycles (Fig. 3) and were sequenced at a repetitive yield of 92%. The repetitive yields of the secondary sequences were lower, and by cycle 10, no other PTH-derivatives were re- leased at a level >5-10% of that of fragment VII. No specific release of 3H was observed during NH,-terminal amino acid sequencing of this material for up to 39 cycles (Fig. 3) or in several other similar runs. These results suggested that the labeled site was not within the M1 putative transmembrane
0.08, . , . , . , , , , , . , I
Elution Volume (mls)
FIG. 2. Fractionation of CNBr digest of [3H]meproadifen mustard-labeled a-subunit by gel filtration chromatography. The [3H]meproadifen mustard-labeled a-subunit (1 nmol) was re- duced, alkylated, and incubated with CNBr in 70% formic acid as described under "Experimental Procedures." The dried samples were redissolved in 50 p1 of 70% formic acid and applied to a TSK 2000SW column. Elution of peptide fragments was detected by absorption at 280 nm ( A ) . Elution of [3H]meproadifen mustard-labeled fragments was monitored by scintillation counting of 20 pl of each 250-pl fraction ( B ) . Parallel experiments with the a-subunit that had been labeled in the absence ( 0 1540 cpm/pg) or presence (0,580 cpm/pg) of 200 p~ meproadifen are shown to define elution of specifically labeled fragments. Based upon recovery of radioactivity, 80% of the starting material was recovered in the void volume pool after Sepha- dex G-50 chromatography. After digestion with CNBr, recovery from the TSK 2000SW column was >95% for the a-subunit labeled in the absence of meproadifen (ie. containing specific label), and 65% of the counts were recovered in the void volume fractions; for the nonspecifically labeled a-subunit, 34% of the counts were recovered in these fractions.
. I80
0 5 10 15 20 25 30 35 Sequencing Cycle
FIG. 3. NHz-terminal amino acid sequencing of ['Hlme- proadifen mustard-labeled fragment from CNBr digest in 70% formic acid. The void volume fractions from the experiment shown in Fig. 2 (5.25-5.75 ml) were combined and dried for storage at -20 "C. The sample was redissolved in 88% formic acid (21,400 cpm) and applied to a Porton glass-fiber filter for sequence analysis. The sample contained multiple peptides (see text), with the picomoles released for the primary sequence beginning at Gln-208 shown for each cycle (V). Missing data points indicate that the amino acid was not integrated. The counts/minute released in each cycle are also shown (0).
region (Leu-212 to Leu-235), but lay on the COOH-terminal side of this region.
To assess the extent of cleavage by CNBr, a digest was fractionated by gel filtration, and fractions corresponding to the peaks were characterized by sequence analysis (Table 11). As expected, the peptide beginning at Gln-208 was the pri- mary sequence identified in peak A (the void volume), and most of the other possible fragments were detected in the other peaks, indicating fairly complete cleavage at Met resi- dues. A notable exception was fragment VI11 (Thr-244 et seq.), which was not detected. Cleavage at Met-243 may have been prevented by the presence of a Thr residue in the following position (Schroeder et al., 1969; Fontana and Gross,

Noncompetitive Antagonist Site of the Acetylcholine Receptor 10493
TABLE I1 NH2-termiml amino acid sequencing of CNBr cleavage fragments
isolated by gel filtration chromatography The [3H]meproadifen mustard-labeled a-subunit was cleaved with
CNBr in either 70% trifluoroacetic acid (TFA) (1 nmol) or 70% formic acid (4 nmol) as described under “Experimental Procedures.” Digests were separated by gel filtration chromatography, and frac- tions were pooled for NH2-terminal amino acid sequencing according to the peaks observed in the chromatograph (peaks A-G as in Figs. 2 and 4). The fragment number scheme from Table I is used to represent the seauences observed in the seauencine runs.
HCOOH fragments Poak
TFA fragments - _”. Major Minor Major Minor
A VI1 VI. xv VIII. VI1 VI. x. xv B I 11, XI11 I, xi11 VI; 11’ C x111 IV. xv. XI XIII. IV XIV. VI. xv D X , i I VI: v. XVI VI, x , IV, 111 xm: XIV E XVI, XI XIV ’ XIV, XVI, IV 111 F 11. xv. XVI XI G IX, XI1 v; IX, ’XI1
1986). These observations suggested that the inability to detect release of label upon sequencing the void volume frac- tion was because the label was within a large peptide begin- ning at Gln-208 and extending through a possible cleavage site at Met-243. It seemed likely that such a fragment would end at the next possible cleavage site at Met-278 since the next sequential fragment (fragment IX) was detected in high yield in peak G, indicating good cleavage of Met-278. Thus, the results suggested that the site of [3H]meproadifen mustard labeling was between the end of the M1 sequence (about Leu- 235) and Met-278.
Digestion with CNBr in Triflluoroacetic Acid-To improve the yield of CNBr cleavage at Met-243, the [3H]meproadifen mustard-labeled a-subunit was cleaved using 70% trifluoroa- cetic acid as the solvent (Tarr, 1986; DiPaola et al., 1990). Fractionation of this digest by gel filtration chromatography in 70% formic acid (Fig. 4) yielded an absorption profile similar to the digest in 70% formic acid, with the elution of [3H]meproadifen mustard-labeled material also coinciding with the void volume peak (Fig. 4B).
Based upon NH2-terminal sequence analysis, each of the peaks from the CNBr digest in 70% trifluoroacetic acid con- tained essentially the same fragments as the corresponding peaks from a digestion in 70% formic acid (Table 11). The most notable difference was the fact that digestion in triflu- oroacetic acid resulted in cleavage at Met-243 as evidenced by the presence of fragment VI11 in peak A. The void volume fraction peak A contained, as its major sequence, the peptide beginning at Thr-244 (fragment VIII, initial yield of 300 pmol) as well as secondary sequences beginning at Gln-208 (frag- ment VII, 60 pmol), Lys-179 (fragment VI, 60 pmol), Ile-283 (fragment X, 40 pmol), and Val-405 (fragment XV, 50 pmol). Despite the presence of the secondary sequences, PTH-deriv- atives associated with fragment VI11 could be quantified for 20 cycles, and this peptide was sequenced at a repetitive yield of 75% (Fig. 5 ) . As was true for the digestion in formic acid, the peptide beginning at Gln-208 sequenced at much higher repetitive yield (-93%) (data not shown), so that by cycle 10, the sequence was present at twice the level of fragment VIII (and by cycle 20, at six times the level).
The pattern of 3H release during NH2-terminal sequencing of the void volume fraction (peak A) revealed a clear release of 3H in cycle 19 (Fig. 5 ) , a release associated with the site of specific alkylation of the a-subunit since it was not detected during sequence analysis of peak A from a parallel digest of the a-subunit labeled nonspecifically with [3H]mepr~adifen
Elution Volume (mls)
FIG. 4. Fractionation of CNBr digest in 70% trifluoroacetic acid of [3H]meproadifen mustard-labeled a-subunit by gel fil- tration chromatography. The [3H]meproadifen mustard-labeled a-subunit (2 nmol) was reduced, alkylated, and incubated with CNBr in 70% trifluoroacetic acid as described under “Experimental Proce- dures.” The dried samples were redissolved in 50 gl of 70% formic acid and applied to a TSK 2000SW column. Elution of peptide fragments was detected by absorption at 280 nm ( A ) . Elution of [3H] meproadifen mustard-labeled fragments was monitored by scintilla- tion counting of 10 pl of each 25O-wl fraction ( B ) . Parallel experiments with the a-subunit that had been labeled in the absence ( 0 1220 cpm/gg) or presence (0, 474 cpm/pg) of 200 pM meproadifen are shown to define elution of specifically labeled fragments. Based upon recovery of radioactivity, 80% of the starting material was recovered in the void volume pool after Sephadex G-50 chromatography. After digestion with CNBr, recovery from the TSK 2000SW column was >95% for the a-subunit labeled in the absence of meproadifen (i.e. containing specific label), with 45% of the counts recovered in the void volume fractions; for the nonspecifically labeled a-subunit, 19% of the counts were recovered in these fractions.
h : v T) 600 m v) 0
400 LL:
I 5 200
n
300
0
200 2 3
;D E
100 E m a
4 0 -
0 5 10 15 20 25 Sequencing Cycle
FIG. 5. NHz-terminal amino acid sequencing of [‘Hlme- proadifen mustard-labeled fragment from CNBr digest in 70% trifluoroacetic acid. The void volume fractions from the experiment shown in Fig. 4 (5.25-5.75 ml) were combined and dried for storage at -20 “C. The samples were redissolved in 88% formic acid and applied to Porton glass-fiber filters for sequence analysis. The picomoles released from the sequence beginning at Thr-244 (fragment VIII) are shown for each cycle (V). Missing data points indicate that the amino acid was not integrated; the low values in cycles 1, 3, and 5 reflect poor integration of Thr and Ser residues. The counts/minute released in each cycle are shown from parallel sequencing runs for a-subunits labeled in the absence (0) or presence (0) of 200 pM meproadifen to define nonspecific labeling. For the a- subunit labeled in the absence of meproadifen, 24,700 cpm were applied to the filter; after 30 cycles, 3250 cpm remained on the filter. For the nonspecifically labeled a-subunit, 5020 cpm were loaded, with 880 cpm remaining after 25 cycles.
mustard. Comparison of the sequences detected in the void volume fraction (peak A) of the trifluoroacetic acid digest with those of the formic acid digest suggests that the specific release of 3H in cycle 19 was likely to result from the reaction of [3H]meproadifen mustard with Glu-262 of the primary sequence. The void volume peaks (peak A) from both digests contained the specifically labeled fragment (Figs. 2B and 4B)

10494 Noncompetitive Antagonist Site of the Acetylcholine Receptor
and contained similar sets of CNBr cleavage fragments (Table 11), except for fragment VIII, which appeared only in the trifluoroacetic acid digest. Therefore, the release of 3H was most likely due to sequencing of fragment VI11 beginning at Thr-244. In addition, the other sequences present in peak A were each present at higher levels in other fractions where no release of 3H was detected fragment VI1 beginning at Gln- 208 was present at high levels in peak A of the formic acid digest; fragments VI and X, both longer than 20 amino acids, were present at substantially higher levels in peak D than in peak A. The sequence beginning at Val-405 (fragment XV) in peak A actually extended beyond Met-415; and its 19th resi- due would be Val-424, which was also contained within frag- ment XVI, present at a higher level in peak F. In addition, the Val-405 sequence is outside the previously defined bounds of the labeled site (Pedersen et al., 1986).
We attempted, unsuccessfully, to utilize reversed-phase HPLC to isolate fragment VI11 from CNBr/trifluoroacetic acid digests of the labeled a-subunit. When digests were fractionated on a microbore C4 column (Brownlee BU-300) using an isobutyl alcohol, acetonitrile, 0.1% trifluoroacetic acid gradient, specifically labeled material eluted at -50% recovery in a broad peak that contained the same mixture of hydrophobic sequences detected in peak A of the gel filtration column. The chromatographic profile was not substantially improved when samples were pretreated with aminoethanol, a treatment that in some cases decreases peak broadening caused by esterification of serine and threonine residues (Tarr and Crabb, 1983). Because of these results, we did not attempt to use reversed-phase HPLC to purify fragment VI11 from the other peptides present in peak A; instead we utilized enzy- matic digestion to confirm that Glu-262 was the primary site of labeling by ['Hlmeproadifen mustard.
Enzymatic Fragmentation-Both specific and nonspecific proteases were assayed for their ability to produce cleavages within the a-subunit hydrophobic domains. Digestions were carried out on labeled subunits that were then fractionated by gel filtration or reversed-phase HPLC or examined by SDS-polyacrylamide gel electrophoresis. After digestion with trypsin (20%, w/w; 24 h), specifically incorporated ['Hlme- proadifen mustard was recovered only within a large fragment produced by cleavage at Arg-209 before M1. No specific re- lease of radioactivity was detected upon sequencing through M1, and there was no evidence of efficient cleavage at Lys- 242 before M2. Digestion with chymotrypsin (20%, w/w) or Staphylococcus aurezu protease V8 (20%, w/w) also failed to produce evidence of efficient cleavage at sites within the hydrophobic core. The nonspecific endoproteinases Pronase (5% (w/w) in 50 mM Tris, pH 6.8,0.5% SDS) and thermolysin (10% (w/w) in 0.1 M NH4HC03, 0.1% Lubrol) also produced only large fragments that contained the sites of specific al- kylation.
In contrast, proteinase K (10% (w/w) in 0.1 M NH4HC03, 0.5% SDS) generated 3H-labeled fragments of low molecular weight. In analytical experiments (-10 pg of a-subunit), the 3H elution profiles observed after proteinase K digestion were not readily reproducible. In some instances, both specifically and nonspecifically labeled materials were recovered between 8.5 and 10.5 ml (MI 5000 to 500), whereas in other experi- ments, the specifically labeled material was resolved from the nonspecifically labeled material. The source of this variability was likely to result from variations in sample handling as digests were frozen and thawed or dried down and resuspended for HPLC.
In Figure 6 are shown the TSK 2000SW column elution profiles of parallel proteinase K digests of a-suuunits isolated
0.02 f
1000
0 0 2 4 6 8 1 0 1 2
Elution Volume (mls) FIG. 6. Fractionation of proteinase K digest of [%]me-
proadifen mustard-labeled a-subunit by TSK 2000SW chro- matography. The [3H]meproadifen mustard-labeled a-subunit (1.4 nmol) was digested with proteinase K as described under "Experi- mental Procedures," dried, redissolved in 75 pl of 70% formic acid, and applied to a TSK 2000SW column that was eluted with 70% formic acid. Elution of peptide fragments was detected by absorption at 280 nm ( A ) . Elution of [3H]meproadifen mustard-labeled frag- menta was monitored by scintillation counting of 25 pl of each 250- pl fraction ( B ) . Parallel digests of a-subunit that had been labeled in the absence ( 0 187,000 cpm loaded, 4250 cpm/pg) or presence ( 0 ; 43,900 cpm loaded, 2000 cpm/pg) of 200 p~ meproadifen are shown to define elution of specifically labeled fragments. Based upon recov- ery of radioactivity, recovery from the sizing column was >95%. For the a-subunit labeled in the absence of meproadifen, 40% of the counts were recovered between 11.5 and 12.25 ml; for the nonspecif- ically labeled a-subunit, 22% of the counts were recovered in these fractions.
from AChRs labeled in the absence and presence of meproad- ifen. As judged by the absorption profile (Fig. a), proteinase K cleaved the a-subunit into small fragments, with only a minimal amount of material eluting with an apparent M, above 1000. Less than 5% of specifically incorporated [3H] meproadifen mustard eluted in the void volume fraction, and 50% eluted at 12 ml (Fig. 6E), well after the included volume (10.7 ml; see Fig. 1). This retardation of the specifically labeled material suggested that it interacts with the column matrix rather than eluting according to molecular weight.
Fractions containing specifically labeled material (11.75- 12.25 ml) (Fig. 6) were pooled and sequenced. The major sequence detected corresponded to Thr-254 et seq. of the a- subunit, which could be quantified for 13 cycles (Fig. 7). In addition to this peptide within M2, secondary sequences were present, one beginning at Trp-86 and extending at least to Asn-95 and a second beginning at Ser-252. This latter se- quence begins only 2 amino acids prior to the primary se- quence. Several additional amino acids within each of the first five cycles were observed that were not readily reconciled with the a-subunit sequence. Release of 3H from the sequenc- ing run (Fig. 7) showed a peak of release at cycle 9, corre- sponding to Glu-262 of the major sequence as well as a shoulder at cycle 11, corresponding to Glu-262 of the second- ary sequence beginning at Ser-252.
Digestion with Endoproteinuse Lys-C-A limitation of the use of size-exclusion chromatography to fractionate a-subunit digests was that it was impossible to purify from the digests a single radiolabeled peptide free of contaminating sequences. Similarly, when reversed-phase HPLC has been used to frac- tionate trypsin or CNBr digests of the a-subunit, the fractions that contain peptides beginning at Met-243 or Thr-244 before M2 also contain additional sequences (see Hucho et al. (1986) and Giraudat et al. (1989); but also see DiPaola et al. (1990)). An alternative approach is to first fractionate by SDS-PAGE

Noncompetitive Antagonist Site of the Acetylcholine Receptor 10495
1250 v 13 $ 1000 0 4 750
x 500 n 0
250
0
LIC
40 0
30 0 3
XI
20 $
10%
0 UI
v 4
0 0 5 10 15
Sequencing Cycle
FIG. 7. NHa-terminal amino acid sequencing of [‘Hlme- proadifen mustard-labeled fragment from proteinase K di- gest. The specifically labeled fractions from the experiments shown in Fig. 6 (11.75-12.25 ml) were combined and dried for storage at -20 “C. The sample was redissolved in 88% formic acid and loaded onto a polybrene-treated glass-fiber filter for sequence analysis. The picomoles released from the sequence beginning at Thr-254 are shown for each cycle (V). Missing data points indicate that the amino acid was not integrated. The counts/minute released in each cycle are shown (0). Of the 34,940 cpm loaded onto the filter, 2570 cpm remained on the filter after 18 cycles.
and then to repurify labeled fragments by reversed-phase HPLC. This approach has been used to identify sites of specific incorporation of an uncharged noncompetitive antag- onist, 3-trifluoromethyl-3-(m-[’26I]iodophenyl)diazirine, in the M2 region of AChR B- and d-subunits (White, 1991; White et al., 1991; White and Cohen, 1991) as well as sites of nonspecific incorporation of 3-trifluoromethyl-3-(rn-[’251]io- dopheny1)diazirine and 1-azidopyrene in the M1, M3, and M4 regions of the a-subunit (Blanton and Cohen, 1992).
We examined the use of endoproteinase Lys-C, which cleaves at the COOH-terminal side of lysine residues and is active in 0.1% SDS (Steffens et al., 1982), in such a two-step purification strategy. If cleavage occurs at all in lysines of the a-subunit, the M1 hydrophobic sequence would be contained in a long peptide (a186-242, ~ 6 . 8 kDa), M2 in a243-276 (3.6 kDa), and M3 in a277-314 (3.0 kDa). In analytical experi- ments, the labeled a-subunit (20 pg) was digested with a range of concentrations of enzyme for 1-3 days, and the digests were analyzed separately by reversed-phase HPLC and SDS- PAGE in a Tricine-based gel system. Although digestion conditions that produced only a single peak of 3H were not identified, conditions that resulted in two well-defined peaks of 3H on reversed-phase HPLC were identified (Fig. &4, fractions 53-55 (67% solvent A) and fractions 58-60 (72% solvent A)). When analyzed by SDS-PAGE (Fig. 8B), sites of specific labeling migrated at =8 kDa, with additional material migrating between 12 and 16 kD, and there was no evidence for significant aggregation of labeled peptides.
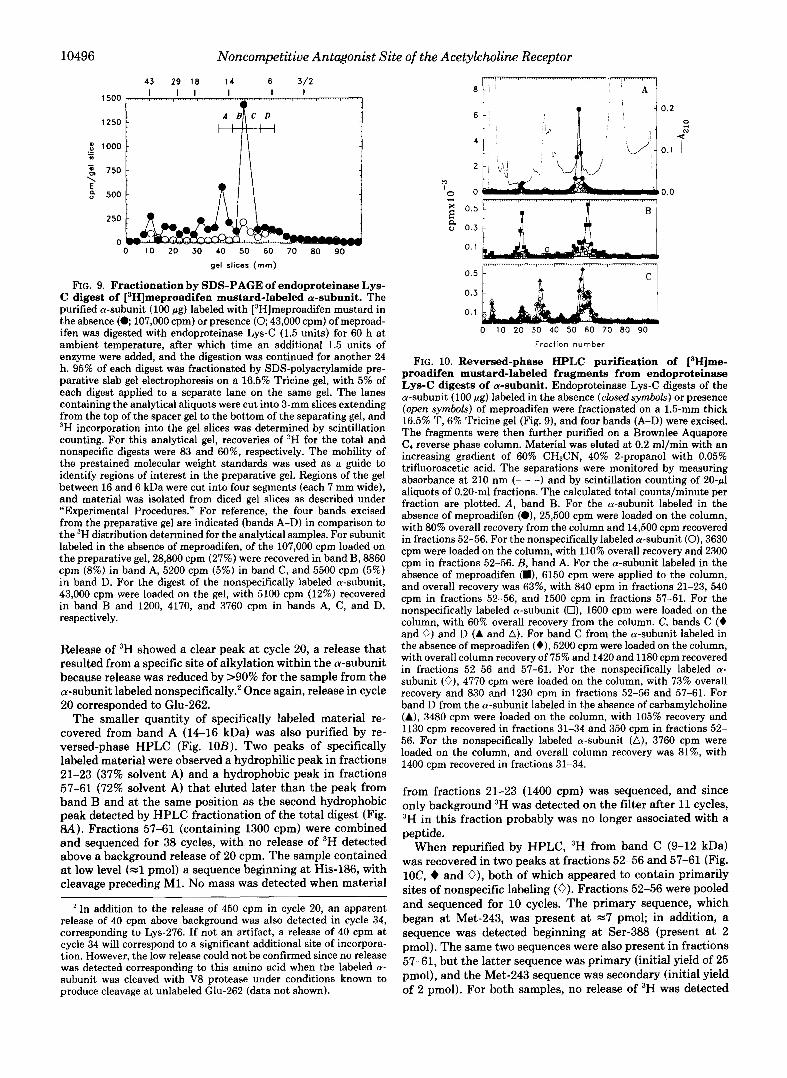
Larger quantities (100 pg) of the a-subunit labeled in the absence or presence of meproadifen were then digested with endoproteinase Lys-C and fractionated by preparative SDS- PAGE, with aliquots (5 pg) run on the same gel in separate lanes for analysis of the distribution of 3H (Fig. 9). Sites of specific labeling migrated at =lo kDa, with additional mate- rial migrating at 12-16 kDa. Material was eluted from four strips of the preparative gel (referred to as bands A-D) that together spanned the region of the gel from 16 to 6 kDa. For the digest of the a-subunit labeled in the absence of meproad- ifen, 107,000 cpm were loaded on the gel, and 28,800 cpm (27%) were recovered in band B, with smaller amounts re- covered in bands A (8860 cprn), C (5200 cpm), and D (5500 cpm). For the digest of the a-subunit labeled nonspecifically, 43,000 cpm were loaded on the gel, with 5100 cpm recovered in band B and 1200,4170, and 3760 cpm in bands A, C, and
1 00
50 %A
N- 3 1 0
X -
2
n 1 v X
n 0 10 20 30 40 50 60 70 80 90 100
Fraction number
43 29 18 14 6 3/2
0 20 40 60 80 100 120
mm gel
FIG. 8. Fractionation by reversed-phase HPLC and SDS- PAGE of endoproteinase Lys-C digest of [‘Hlmeproadifen mustard-labeled a-subunit. The purified a-subunit (20 pg) labeled with [3H]meproadifen mustard in the absence (V; 21,200 cpm) or presence (V; 8,300 cpm) of meproadifen was digested with endopro- teinase Lys-C (0.3 unit) for 42 h a t ambient temperature. The reaction was terminated by addition of diisopropylphosphorofluoridate to 1 mM, and half of each digest was fractionated either by reversed-phase HPLC ( A ) or SDS-PAGE (E). A , the digest was applied to a Brownlee BU-300 column and eluted at 0.20 ml/min with an increasing gradient of 60% CHaCN, 40% 2-propanol with 0.05% trifluoroacetic acid. Fractions of 0.2 ml were collected, and aliquots of 50 (V) or 100 (V) rl were assayed by scintillation counting. The calculated total counts/ minute per fraction are plotted. Recovery from the column was -40% for the a-subunit labeled in the absence of meproadifen and -65% for the nonspecifically labeled a-subunit. E, aliquots (10 pg) of digests of total (V) and nonspecifically labeled (V) a-subunits as well as 2- pg aliquots of total (0) and nonspecifically labeled (0) a-subunits were fractionated by SDS-PAGE on a 16.5% T, 5% Tricine gel as described under “Experimental Procedures.” The gel was stained with Coomassie Blue, and the lanes were cut into 3-mm gel slices from the top of the spacer gel to the bottom of the separating gel. 3H incorpo- ration into gel slices was quantified by scintillation counting. For undigested a-subunits, recoveries of total and nonspecific labels were 100 and 80%, respectively, whereas for the endoproteinase Lys-C digests, recoveries were 70 and 85%, respectively.
D, respectively, Thus, specifically labeled fragments were isolated from bands A and B, whereas material from bands C and D was labeled nonspecifically.
Labeled fragments from band B were further purified by reversed-phase HPLC (Fig. 1OA). About 60% (14,500 cpm) of loaded 3H was recovered in a single sharp peak (fractions 52- 56, 67% solvent A) associated with a peak in the absorption profile, and the peak of 3H was reduced by 85% for the digest of the a-subunit labeled nonspecifically. Material in fractions 52-56 was pooled and sequenced (Fig. 11). For both specifi- cally and nonspecifically labeled a-subunits, a single sequence was detected beginning at Met-243, with any other released PTH-derivatives present at <lo% of that level. The samples were subjected to 38 cycles of Edman degradation, and PTH- derivatives corresponding to a243-280 were identified. Con- sistent with its mobility on the polyacrylamide gel, the peptide extended beyond the next possible cleavage site (Lys-276 between M2 and M3). In both samples, the peptide was present at similar levels (initial yields of 11 and 14 pmol).
10496 Noncompetitive Antagonist Site of the Acetylcholine Receptor
43 29 18 14 6 3/2
1500 I
250
0 0 10 20 30 40 50 60 70 80 90
gel slices (rnm)
FIG. 9. Fractionation by SDS-PAGE of endoproteinase Lys- C digest of [‘Hlmeproadifen mustard-labeled a-subunit. The purified a-subunit (100 pg) labeled with [3H]meproadifen mustard in the absence ( 0 107,000 cpm) or presence (0; 43,000 cpm) of meproad- ifen was digested with endoproteinase Lys-C (1.5 units) for 60 h at ambient temperature, after which time an additional 1.5 units of enzyme were added, and the digestion was continued for another 24 h. 95% of each digest was fractionated by SDS-polyacrylamide pre- parative slab gel electrophoresis on a 16.5% Tricine gel, with 5% of each digest applied to a separate lane on the same gel. The lanes containing the analytical aliquots were cut into 3-mm slices extending from the top of the spacer gel to the bottom of the separating gel, and “H incorporation into the gel slices was determined by scintillation counting. For this analytical gel, recoveries of 3H for the total and nonspecific digests were 83 and 60%, respectively. The mobility of the prestained molecular weight standards was used as a guide to identify regions of interest in the preparative gel. Regions of the gel between 16 and 6 kDa were cut into four segments (each 7 mm wide), and material was isolated from diced gel slices as described under “Experimental Procedures.” For reference, the four bands excised from the preparative gel are indicated (bands A-D) in comparison to the 3H distribution determined for the analytical samples. For subunit labeled in the absence of meproadifen, of the 107,000 cpm loaded on the preparative gel, 28,800 cpm (27%) were recovered in band B, 8860 cpm (8%) in band A, 5200 cpm (5%) in band C, and 5500 cpm (5%) in band D. For the digest of the nonspecifically labeled a-subunit, 43,000 cpm were loaded on the gel, with 5100 cpm (12%) recovered in band B and 1200, 4170, and 3760 cpm in bands A, C, and D, respectively.
Release of 3H showed a clear peak at cycle 20, a release that resulted from a specific site of alkylation within the a-subunit because release was reduced by >90% for the sample from the a-subunit labeled nonspecifically.’ Once again, release in cycle 20 corresponded to Glu-262.
The smaller quantity of specifically labeled material re- covered from band A (14-16 kDa) was also purified by re- versed-phase HPLC (Fig. 10B). Two peaks of specifically labeled material were observed a hydrophilic peak in fractions 21-23 (37% solvent A) and a hydrophobic peak in fractions 57-61 (72% solvent A) that eluted later than the peak from band B and at the same position as the second hydrophobic peak detected by HPLC fractionation of the total digest (Fig. a). Fractions 57-61 (containing 1300 cpm) were combined and sequenced for 38 cycles, with no release of 3H detected above a background release of 20 cpm. The sample contained at low level (=l pmol) a sequence beginning at His-186, with cleavage preceding MI. No mass was detected when material
In addition to the release of 450 cpm in cycle 20, an apparent release of 40 cpm above background was also detected in cycle 34, corresponding to Lys-276. If not an artifact, a release of 40 cpm at cycle 34 will correspond to a significant additional site of incorpora- tion. However, the low release could not be confirmed since no release was detected corresponding to this amino acid when the labeled a- subunit was cleaved with V8 protease under conditions known to produce cleavage at unlabeled Glu-262 (data not shown).
t 0 : i 0.2
0.1
0.5
0.3
0.1
0 10 20 30 40 50 60 70 80 90
Fraction number
FIG. 10. Reversed-phase HPLC purification of [‘Hlme- proadifen mustard-labeled fragments from endoproteinase Lys-C digests of a-subunit. Endoproteinase Lys-C digests of the a-subunit (100 pg) labeled in the absence (closed symbols) or presence (open symbols) of meproadifen were fractionated on a 1.5-mm thick 16.5% T, 6% Tricine gel (Fig. 9), and four bands (A-D) were excised. The fragments were then further purified on a Brownlee Aquapore
increasing gradient of 60% CRCN, 40% 2-propanol with 0.05% Ca reverse phase column. Material was eluted at 0.2 ml/min with an
trifluoroacetic acid. The separations were monitored by measuring absorbance at 210 nm (- - -) and by scintillation counting of 2 0 4 aliquots of 0.20-ml fractions. The calculated total counts/minute per fraction are plotted. A, band B. For the a-subunit labeled in the absence of meproadifen (01, 25,500 cpm were loaded on the column, with 80% overall recovery from the column and 14,500 cpm recovered in fractions 52-56. For the nonspecifically labeled a-subunit (0), 3630 cpm were loaded on the column, with 110% overall recovery and 2300 cpm in fractions 52-56. B, band A. For the a-subunit labeled in the absence of meproadifen (m), 6150 cpm were applied to the column, and overall recovery was 63%, with 840 cpm in fractions 21-23, 540 cpm in fractions 52-56, and 1500 cpm in fractions 57-61. For the nonspecifically labeled a-subunit (O), 1600 cpm were loaded on the column, with 60% overall recovery from the column. C, bands C (+ and 0) and D (A and A). For band C from the a-subunit labeled in the absence of meproadifen (e), 5200 cpm were loaded on the column, with overall column recovery of 75% and 1420 and 1180 cpm recovered in fractions 52-56 and 57-61. For the nonspecifically labeled a- subunit (O), 4770 cpm were loaded on the column, with 73% overall recovery and 830 and 1230 cpm in fractions 52-56 and 57-61. For band D from the a-subunit labeled in the absence of carbamylcholine (A), 3480 cpm were loaded on the column, with 105% recovery and 1130 cpm recovered in fractions 31-34 and 350 cpm in fractions 52- 56. For the nonspecifically labeled a-subunit (A), 3760 cpm were loaded on the column, and overall column recovery was 81%, with 1400 cpm recovered in fractions 31-34.
from fractions 21-23 (1400 cpm) was sequenced, and since only background 3H was detected on the filter after 11 cycles, 3H in this fraction probably was no longer associated with a peptide.
When repurified by HPLC, 3H from band C (9-12 kDa) was recovered in two peaks at fractions 52-56 and 57-61 (Fig. 1oC, + and 0), both of which appeared to contain primarily sites of nonspecific labeling (0). Fractions 52-56 were pooled and sequenced for 10 cycles. The primary sequence, which began at Met-243, was present at =7 pmol; in addition, a sequence was detected beginning at Ser-388 (present at 2 pmol). The same two sequences were also present in fractions 57-61, but the latter sequence was primary (initial yield of 25 pmol), and the Met-243 sequence was secondary (initial yield of 2 pmol). For both samples, no release of 3H was detected

Noncompetitive Antagonist Site of the Acetylcholine Receptor 10497
a243-YTLSISVLLSLTVFLLVIVELIPSTSSAVPLIGKYMLF Detected Amino Acid Sequence
, . . 600
IO v - v t 500 0
m
s i
i e: - a
0.1 0 5 10 15 20 25 30 35 40
Sequencing Cycle
FIG. 11. NHz-terminal sequence analysis of [3H]meproadi- fen mustard-labeled fragment from endoproteinase Lys-C di- gest of a-subunit. HPLC fractions 52-56, containing the peak of specifically labeled material purified from band B (Fig. lOA), were pooled and loaded into Porton glass filters for sequence analysis. For the material isolated from membranes labeled in the absence of meproadifen (V and O), 14,000 cpm were pooled for loading on the filter, and 1560 cpm remained after 38 cycles. For the nonspecifically labeled a-subunit (O), 1100 cpm were loaded on the filter, and 230 cpm remained after 38 cycles. For both samples, 40% of each cycle of Edman degradation was analyzed for released PTH-derivatives (V, total digest), and the remainder for 3H (0 and 0). For both samples, a single peptide was detected corresponding to an a-subunit fragment beginning at Met-243 and extending at least to Phe-280. . . . , fit of the data based upon nonlinear least-squares regression of the ob- served release ( M ) for each cycle (n): M = lan, where lo is the initial yield and R is the repetitive yield. For the sample containing specific label, I , = 11.4 & 1.4 pmol and R = 91 & 1%. For the sample isolated from the a-subunit labeled nonspecifically, the peptide was sequenced with lo = 14 & 1 pmol and R = 93 & 1% (data not shown).
above a background of 20-30 cpm. 3H-containing material from band D (6-9 kDa) was also recovered in fractions 52- 61, with similar levels of 3H recovered from digests of the a- subunit labeled in the absence (A) or presence (A) of carba- mylcholine. Fractions 52-56 and 57-61 were each pooled and sequenced separately, and in this case, both samples contained peptides beginning at Ser 388 (5 pmol) and Tyr-401 (2 pmol).
DISCUSSION
The noncompetitive antagonist [3H]mepr~adifen mustard reacts with Glu-262 of the AChR a-subunit. This was first indicated by analysis of 3H release during sequencing of peaks containing sites of specific labeling recovered by gel filtration HPLC of CNBr digests and of a proteinase K digest of the labeled a-subunit. In these experiments, sites of specific label- ing were recovered in fractions containing multiple sequences, and confirmation of the identification of a-subunit Glu-262 as the site of labeling was achieved by sequencing material isolated by SDS-PAGE and reversed-phase HPLC from an endoproteinase Lys-C digest of the labeled a-subunit.
In the analysis of the CNBr digests, Glu-262 was identified as the most likely site of labeling based upon a comparison of the results obtained for digests carried out in 70% formic acid or 70% trifluoroacetic acid. For both digests, specifically in- corporated 3H was recovered in the void volume fractions, which, based upon the elution profile of standard proteins, would indicate an M , >20,000. Since essentially all predicted CNBr fragments were identified in appropriate fractions of the column eluate (Table 11), elution of the specific label in the void volume probably occurred because the label was contained in smaller fragments that aggregated under the conditions used. For the void volume fraction of the formic acid digest, no specific release of 3H was detected during 35 cycles of repetitive Edman degradation (Fig. 3), whereas for
the corresponding sample from the trifluoroacetic acid digest, there was clear release of 3H in cycle 19 (Fig. 5). Comparison of the a-subunit fragments present in the two samples as well as the distribution of peptides in the other column fractions led us to conclude that the observed release of 3H in cycle 19 of the trifluoroacetic acid digest void volume fraction was most likely to result from alkylation of Glu-262 contained within a peptide beginning at Thr-244. This peptide was the primary sequence detected in this fraction, and it was the only sequence not detected in the formic acid digest. The other sequences detected in the void volume fraction of the trifluoroacetic acid digest were also present at higher levels either in other fractions of the column eluate or in the void volume fraction of the formic acid digest.
After CNBr digestion in formic acid, the sequence begin- ning at Thr-244 (fragment VIII) was not detected in any HPLC fraction, and it was likely that specifically incorporated [3H]meproadifen mustard was present in a peptide beginning at Gln-208 and extending though Met-243 to Met-278. How- ever, we cannot rule out that some part of 3H was contained in a peptide beginning at Thr-244 either refractory to Edman degradation or present at a substantially lower level than in the trifluoroacetic acid digest. The clear presence of fragment VI11 in the trifluoroacetic acid digest and its absence in the formic acid digest are consistent with the results of DiPaola et al. (1990) in their mapping of the site of incorporation of [3H]quinacrine azide in the AChR a-subunit and with the reported improved efficiency of cleavage at Met-Thr by CNBr in trifluoroacetic acid rather than in formic acid (Fontana and Gross, 1986). In addition, it is noteworthy that the peptide beginning at Gln-208 sequenced at >90% repetitive yield when isolated from either digest, whereas the peptide begin- ning at Thr-244 sequenced at only 75% repetitive yield. Frag- ment VI11 beginning at Thr-244 is particularly rich in serine and threonine, and it is known that formyl esters of serine and threonine are susceptible to NH2-terminal blockade dur- ing Edman degradation as a result of 0- to N-acyl transfer (Allen, 1989).
Gel filtration HPLC fractionation of a proteinase K cleav- age of the [3H]mepr~adifen mustard-labeled a-subunit also provided results consistent with the identification of Glu-262 as a site of specific labeling. In this instance, specifically labeled material apparently was retarded on the gel filtration column (Fig. 6), and sequence analysis of the peak of 3H revealed the presence of multiple sequences, including a frag- ment beginning at Thr-254, with release of 3H at cycle 9, that would correspond to Glu-262 (Fig. 7).
A limitation of the gel filtration fractionation of the CNBr digests and of the proteinase K digest was the fact that labeled peptides were always recovered in fractions containing more than one sequence. Consistent with results reported by others (Giraudat et al., 1989; Hucho et al., 1986), we were unable to use reversed-phase HPLC to purify a unique radiolabeled peptide from CNBr digests of the a-subunit. An alternative strategy to generate and isolate peptides from the hydrophobic core of AChR subunits is to fractionate digests first by SDS- PAGE and then by reversed-phase HPLC (White, 1991; White and Cohen, 1991). To identify sites of labeling by [3H] meproadifen mustard in the a-subunit, such a fractionation of an endoproteinase Lys-C digest yielded unambiguous evi- dence that a-subunit Glu-262 is in fact the principal site of labeling. When the labeled subunit was digested by endopro- teinase Lys-C and fractionated by SDS-PAGE, sites of spe- cific labeling were recovered in fragments of M , 10,000-16,000, with little evidence of high molecular weight aggregates. About 25% of specifically labeled material was recovered in

10498 Noncompetitive Antagonist Site of the Acetylcholine Receptor
band B (12-14 kDa), which, when repurified by reversed- phase HPLC, eluted as a single peak (fractions 52-56, 67% solvent A). Based upon NH2-terminal sequence analysis, that peak contained only a single sequence beginning at Met-243 before M2 and extending through Lys-276 into M3. No other sequence was identifiable, and other amino acids when de- tected were present at 4 0 % of the level of those of the major sequence. Release of 3H occurred only at cycle 20, correspond- ing to Glu-262. There was no release of 3H detected upon sequencing through Ser-248, the residue in the a-subunit identified as a primary site of photolabeling by [3H]chlor- promazine (Giraudat et al., 1989), or at any of the nucleophilic side chains in M2 before Glu-262.
Analysis of the 3H-~~ntaining material from other regions of the polyacrylamide gel establishes the importance of the two-stage purification procedure. For example, fragments con- taining the M4 region eluted at the same position on the HPLC gradient (fractions 52-56) as the fragment beginning at Met-243, but they were recovered from bands C (9-12 kDa) and D (6-9 kDa), regions of the gel containing primarily sites of nonspecific labeling. In addition, specifically labeled ma- terial was recovered in band A (14-16 kDa). This material eluted as a sharp peak at 72% solvent A, but the amount of 3H was only 10% of that recovered at 67% solvent A from band B. A fragment beginning at His-186 was detected at low levels (1 pmol), and it is thus likely that the site of specific labeling was contained within a peptide beginning at His-186 and extending at least through the M2 hydrophobic sequence.
Digestion with endoproteinase Lys-C provides an excellent means to identify sites of labeling within the M2 hydrophobic segment, but it is not useful to identify sites of labeling in M1 because Lys-185 is positioned 35 residues before M1 (a209- 236). However, the lack of release of 3H during sequence analysis of the void volume fractions of the CNBr digest in formic acid, which contained the peptide beginning at Glu- 208 at high levels, indicates that there is unlikely to be significant labeling within M1. In the analysis of the endo- proteinase Lys-C digest, there was no evidence of efficient cleavage at Lys-277, a cleavage necessary to reveal sites of labeling within the M3 hydrophobic segment. The labeled material isolated from band B clearly extended through that cleavage site, and no fragment beginning at Tyr-278 was identified in any of the HPLC fractions analyzed. Analysis of the CNBr digests provides evidence against specific labeling of the M3 segment since no specific label was associated with peak D of the elution profiles of CNBr digests that contained CNBr fragment X beginning at Ile-283 within M3. Thus, both the observed release of 3H corresponding to Glu-262 and the lack of release of 3H at other cycles of the M2 segment or of sequencing fractions containing the M1 or M3 hydrophobic segments led us to conclude that Glu-262 is the primary, if not the only, site of reaction within the a-subunit.
The positive charge of meproadifen mustard is contained within the reactive quaternary aziridinium ion that would react with nucleophilic side chains within the noncompetitive antagonist-binding site. Such side chains presumably stabilize the positive charge of the noncompetitive antagonists. There- fore, a-subunit Glu-262 contributes to the cation-binding domain of the noncompetitive antagonist site. It is noteworthy that meproadifen mustard aziridinium provides a direct iden- tification of a carboxylate involved in the noncompetitive antagonist-binding site, whereas acetylcholine mustard aziri- dinium reacts with a tyrosyl side chain and not a carboxylate in the cation-binding domain of the ACh-binding site (Cohen et al., 1991).
Since meproadifen mustard only alkylates the noncompet-
itive antagonist site when the ACh sites are occupied by high concentrations of agonists (Dreyer et al., 1986), alkylation occurs while the bulk of the AChRs are in the desensitized conformation. However, the concentrations of agonists asso- ciated with half-maximal alkylation (C60 = 20 WLM for acetyl- choline and 160 WM for carbamylcholine) (Dreyer et al., 1986) are 100-fold higher than those required for agonist binding to desensitized AChRs and are closer to the agonist concentra- tions required for channel activation. This suggests that label- ing may occur preferentially while the AChR adopts nonde- sensitized conformations, possibly an open channel confor- mation.
Glu-262 of the a-subunit, which lies at the COOH-terminal end of the M2 hydrophobic region, as well as the homologous residues in other subunits have been shown to contribute to the permeability of the open channel in a manner consistent with their presence at the extracellular vestibule of the ion channel (Imoto et al., 1988). Meproadifen mustard does not alkylate a-subunit Ser-248 within "2 or homologous residues in other subunits that have been identified as the primary sites of photoincorporation in the desensitized AChR of the noncompetitive antagonists [3H]chlorpromazine (Giraudat et al., 1989) and [3H]triphenylmethylphosphonium (Hucho et al., 1986). This is not surprising since meproadifen mustard identifies regions interacting with the positive charge of the noncompetitive antagonists, whereas reactive sites for pho- toincorporation are likely to be contained within the hydro- phobic aromatic domain of the noncompetitive antagonists. When the M2 domain is modeled as an a-helix, both Ser-248 and Glu-262 lie on a common face of the helix, consistent with the proposed association of M2 helices from each subunit at the central axis of the AChR to form the noncompetitive antagonist site and the ion channel (Giraudat et al., 1987). However, Ser-248 and Glu-262 are 21 A apart along the helix face, a distance considerably greater than the dimensions of chlorpromazine or meproadifen mustard in their maximally extended configurations (14 A). This indicates that it is not possible for either bound ligand to be in contact simultane- ously with both residues and suggests that the two antagonists bind to different regions within the central M2 domain of the AChR. Although we have not demonstrated that the AChR specifically alkylated by meproadifen mustard loses the ability to bind chlorpromazine, this is undoubtedly true since alkyl- ation by meproadifen mustard does prevent the binding of [3H]histrionicotoxin (Dreyer et al., 1986).
If the noncompetitive antagonist-binding site is at a cen- trosymmetric location in the AChR, it would be expected that homologous residues of the @-, y-, and &subunits (Asp, Gln, and Gln, respectively) should also contribute to this binding site. Nonetheless, the a-subunit was the predominant site of labeling, with the labeling in the @-subunit only 5% of that of the a-subunit and no detectable labeling in the y- and 8- subunits (Dreyer et al., 1986). This may be explained by the poor nucleophilicity of the Gln residues contributed by the y- and &subunits and by the fact that, in each subunit other than the a-subunit, the homologous residue is followed by basic amino acids (lysine in the @- and y-subunits and arginine in the &subunit). Thus, the positive charge of the noncom- petitive antagonists might be stabilized preferentially by in- teractions with residues from the a-subunit and not by equiv- alent interactions with residues from the other subunits. Analysis of the functional interaction of meproadifen and other reversible noncompetitive antagonists with mutant AChRs, differing at those residues, will provide a means to test that hypothesis.
Acknowledgments-We thank Benjamin H. White and Michael P.

Noncompetitive Antagonist Si
Blanton for many valuable discussions during the course of this work, and we thank David Chiara both for encouraging us to test endopro- teinase Lys-C and for assistance with protein sequence analysis.
REFERENCES Abramson, S. N., Li, Y., Culver, P., and Taylor, P. (1989) J. Biol.
Allen, G. (1989) Sequencing of Proteins and Peptides, Elsevier/North-
Blanton, M. P., and Cohen, J. B. (1992) Biochemistry, in press Blount, P., and Merlie, J. P. (1989) Neuron 3, 5349-5357 Charnet, P., Labarca, C., Leonard, R. J., Vogelaar, N. J., Czyzyk, L.,
Gavin. A., Davidson, N., and Lester, H. A. (1990) Neuron 2.87-95 Cohen, J . B., Medynski, D. C., and Strnad, N. P. (1985) in Effects in
Anesthesia (Covino, B. G., Fozzard, H. A,, Rehder, K., and Stri- chartz, G., eds) pp. 53-64, American Physiological Society, Be- thesda, MD
Cohen, J. B., Correll, L. A., Dreyer, E. B., Kuisk, I. R., Medynski, D. C., and Strnad, N. R. (1986) in Molecular and Cellular Mechanisms of Anesthetics (Roth, S., and Miller, K., eds) pp. 111-124, Plenum
Chem. 264,12666-12672
Holland Biomedical Press, Amsterdam
Press, New York . . ~~
Cohen, J. B., Sharp, S. D., and Liu, W. S. (1991) J. Biol. Chert. 2 6 6 , 23354-23364
Colquohoun, D. (1986) Hundb. Exp. Pharmacol. 79,59-113 Dennis, M., Giraudat, J., Kotzyba-Hibert, F., Goeldner, M., Hirth,
C., Chang, J.-Y., Lazure, C., Chretien, M., and Changeux, J.-P. (1988) Biochemistry 2 7 , 2346-2357
Dermer, 0. C., and Ham, G. E. (1969) Ethylenimine and Other Azirdines pp. 205-248,270-273, Academic Press, New York
DiPaola, M., Kao, P. N. and Karlin, A. (1990) J. Biol. Chem. 2 6 5 ,
Dreyer, E. B., Hasan, F., Cohen, S. G., and Cohen, J. B. (1986) J. Biol. Chem. 2 6 1 , 13727-13734
Fontana, A., and Gross, E. (1986) in Practical Protein Chemistry: A Handbook (Darbre, A., ed) pp. 67-120, John Wiley & Sons, New York
Forman, S. A., and Miller, K. W. (1989) Trends Pharmacol. Sci. 10 ,
Galzi, J. L., Revah, F., Black, D., Goeldner, M., Hirth, C., and Changeux, J. P. (1990) J. Biol. Chem. 2 6 5 , 10430-10437
Gerber, G. E., Anderegg, R. J., Herlihy, W. G., Gray, C. P., Biemann, K., and Khorana, H. G. (1979) Proc. Natl. Acad. Sci. U. S. A. 76, 227-231
Giraudat, J., Dennis, M., Heidmann, T., Haumont, P.-Y., Lederer, F., and Changeux, J.-P. (1987) Biochemistry 26,2410-2418
Giraudat, J., Galzi, J.-L., Revah, F., Changeux, J.-P., Haumont, P.- Y., and Lederer, F. (1989) FEBS Lett. 2 5 3 , 195-198
Hager, D. A., and Burgess, R. R. (1980) Anal. Biochem. 109, 76-86 Heidmann, T., Oswald, R. E., and Changeux, J.-P. (1983) Biochem-
Herz, J. M., Johnson, D. A., and Taylor, P. (1987) J. Biol. Chem.
Hucho, F., Oberthur, W., and Lottspeich, F. (1986) FEBS Lett. 205,
Imoto, K., Busch, C., Sakmann, B., Mishina, M., Konno, T., Nakai, J., Buju, H., Mori, Y., Fukuda, K., and Numa, S. (1988) Nature
Kao, P. N., Dwork, A. J., Kaldany, R.-R. J., Silver, M. L., Wideman, J., Stein, S., and Karlin, A. (1984) J. Biol. Chem. 2 5 9 , 11662- 11665
Krodel, E. K., Beckman, R. A., and Cohen, J. B. (1979) Mol. Phar- macol. 15, 294-312
11017-11029
447-452
istry 22,3112-3127
262,7238-7247
137-142
335,645-648
'te of the Acetylcholine Receptor 10499
Laemmli, U. K. (1970) Nature 227,680-685 Leonard, R. J., Labarca, C. G., Charnet, P., Davidson, N., and Lester,
Lowry, 0. H., Rosebrough, N. J., Farr, A. L., and Randall, R. J. (1951)
Middleton, R. E., and Cohen, J. B. (1991) Biochemistry 30 , 6987-
Mitra, A. K., McCarthy, M., and Stroud, R. M. (1989) J. Cell Biol.
Neher, E. (1983) J. Physiol. (Lond.) 335,123-137 Neher, E., and Steinbach, J. H. (1978) J. Physiol. (Lond.) 277 , 153-
Neubig, R. R., and Cohen, J . B. (1979) Biochemistry 18,5464-5475 Neubig, R. R., and Cohen, J. B. (1980) Biochemistry 19, 2770-2779 Noda, M., Takahashi, H., Tanabe, T., Toyosato, M., Furutani, Y.,
Nature 2 2 9 , 793-797 Hirose, T., Asai, M., Inayama, S., Miyata, T., and Numa, S. (1982)
Noda, M., Takahashi, H., Tanabe, T., Toyosato, M., Kikyotani, S., Furutani, Y., Hirose, T., Takashima, H., Inayama, S., Miyata, T., and Numa, S. (1983) Nature 302 , 528-532
Ogden, D. C., Siegelbaum, S. A., and Colquohoun, D. (1981) Nature
Pedersen, S. E., and Cohen, J . B. (1990) Proc. Natl. Acad. Sci. U. S.
Pedersen, S. E., Dreyer, E. B., and Cohen, J. B. (1986) J. Biol. Chem.
Pedersen, S. E., Bridgman, P. C., Sharp, S. D., and Cohen, J. B.
Peper, K., Bradley, R. J., and Dreyer, F. (1982) Physiol. Reu. 62,
Porter, S., and Froehner, S. C. (1983) J. Biol. Chem. 2 5 8 , 10034- 10040
Raftery, M. A,, Hunkapiller, M. W., Strader, C. D., and Hood, L. E. (1980) Science 208, 1454-1457
Revah, F., Galzi, J.-L., Giraudat, J., Haumont, P.-Y., Lederer, F., and Changeux, J.-P. (1990) Proc. Natl. Acad. Sci. U. S. A. 8 7 , 4675- 4679
H. A. (1988) Science 242,1578-1581
J. Biol. Chem. 193,265-275
6997
109,753-774
176
289,596-598
A. 87,2785-2789
261,13735-13743
(1990) J. Biol. Chem. 265,569-581
1271-1340
Reynolds, J. A., and Karlin, A. (1978) BiochemistTy 17, 2035-2038 Schaffner, W., and Weissman, C. (1973) Anal. Biochem. 56,502-514 Schagger, H., and von Jagow, G. (1987) Anal. Biochem. 166, 368-
Schroeder, W. A., Shelton, J. B., and Shelton, J. R. (1969) Arch.
Sobel, A., Weber, M., and Changeux, J.-P. (1977) Eur. J. Biochem.
Steffens, G. J., Giinzler, W. A., Otting, F., Frankus, E., and FlohB, L. (1982) Hoppe-Seyler's Z. Physiol. Chem. 363 , 1043-1058
Stoffel, W., Hillen, H., and Giersiefen, H. (1984) Proc. Natl. Acad. Sci. U. S. A. 81,5012-5016
Strnad, N. P. (1988) Binding of Agonist and Noncompetitive Antago- nists to the Nicotinic Acetylcholine Receptor. Ph.D. dissertation, Washington University
Tarr, G. E. (1986) in Methods of Protein Microcharacterization (Shively, J. E., ed) pp. 155-194, Humana Press, Clifton, NJ
Tarr, G. E., and Crabb, J. W. (1983) Anal. Biochern. 13, 99-107 Toyoshima, C., and Unwin, P. N. T. (1990) J. Cell Biol. 111 , 2623-
2635 White, B. H. (1991) Structural Characterization of the Nicotinic
Acetylcholine Receptor. Ph.D. dissertation, Washington University School of Medicine
379
Biochem. Biophys. 130,551-556
80,215-224
White, B. H., and Cohen, J. B. (1988) Biochemistry 27,8741-8751 White, B. H., and Cohen, J. B. (1991) Neurosci. Abstr. 17, 22A White, B. H., Howard, S., Cohen, S. G., and Cohen, J. B. (1991) J.
Biol. Chem. 266,21595-21607


















