
Astrophysically Relevant Large Carbonaceous Molecules and ...

Structure determination of small and large
molecules using single crystal X-ray
crystallography
A thesis submitted to The University of Manchester for the degree
of Master of Science by Research in the Faculty of Engineering
and Physical Sciences
2010
Richard Prendergast
School of Chemistry

1
Structure determination of small and large molecules using single crystal X-ray crystallography
List of figures 4
List of tables 9
Abstract 10
Declaration 11
Copyright statement 12
Acknowledgements 13
Part A - Small molecule X-ray crystallography Chapter 1 - A review of the single crystal method Page 1.1 Basic Principles of X-ray crystallography 15
1.2 Diffraction of X-rays by crystals 16
1.3.1 Crystal structure and symmetry 17
1.3.2 The Bragg equation 22
1.3.3 Miller Indices 23
1.4.1 Nature, production and generation of X-rays 24
1.4.2 X-ray tube source 24
1.4.3 Synchrotron source 26
1.4.4 Detection of X-rays 27
1.5 Crystal growth 28
1.6.1 Structure determination procedure 30
1.6.2.1 The measurement of intensities 30
1.6.2.2 Preparation and mounting of the crystal 30
1.6.2.3 The collection of the X-ray intensities 32
1.6.2.4 The diffraction images data reduction process 32
1.6.3.1 The phase problem and possible solutions 33
1.6.3.2 The Patterson synthesis 34
1.6.3.3 Direct methods 35
1.6.4 Refining the structure 36
Chapter 2 - Structure determination of a small molecule – C26H36N8018Cl2Co

2
2.1 Introduction to C26H36N8018Cl2Co 38
2.2 X-ray diffraction data collection and processing procedure 38
2.3 Crystal structure analysis 40
2.4 Crystal structure implications 43
Chapter 3 - Structure determination of a small molecule – C26H36N8010F12P2Co 3.1 Introduction to C26H36N8010F12P2Co 44
3.2 X-ray diffraction data collection and processing procedure 44
3.3 Crystal structure analysis 46
3.4 Crystal structure implications 48
3.5 Comparison of the crystal structures 48
Chapter 4 - Structure determination of two small molecules – C30H24N04Sn & C30H20Sn 4.1 Introduction to C30H24N04Sn & C30H20Sn 52
4.2 X-ray diffraction data collection and processing procedure for C30H20Sn 53
4.3 X-ray diffraction data collection and processing procedure for C30H24N04Sn 55
4.4 Crystal structure analysis for C30H20Sn 56
4.5 Crystal structure analysis for C30H24N04Sn 59
4.6 Crystal structure implications of C30H20Sn 63
4.7 Crystal structure implications of C30H24N04Sn 64
Part B - Macromolecular X-ray crystallography Chapter 5 - Macromolecular X-ray crystallography 5.1 Introduction 67
5.2.1 Crystallisation techniques 68
5.2.2 The batch method 69
5.2.3 Dialysis 69
5.2.4 Vapour diffusion methods 69
5.2.5 Hot box technique 70
5.3.1 Solving the phase problem in macromolecular X-ray crystallography 70
5.3.2 Isomorphous replacement 70
5.3.3 Anomalous scattering 74

3
5.3.4 Molecular replacement 76
5.4 Rigid body and restrained refinement 77
5.5 The R free factor
78
Chapter 6 - Crystal structure determination and model refinement of a co-crystallisation of HEWL and TA6Br12
6.1 Introduction 79
6.1.2 Introduction to lysozyme 79
6.1.3 Introduction to Ta6Br12 80
6.2 Co-crystallisation procedure of HEWL and Ta6Br12 81
6.3 X-ray diffraction data collection procedure 82
6.4 The model refinement procedure 84
6.5 Refinement of the occupancies of the Ta6Br12 binding sites using SHELX 88
6.6 Analysis of the three dimensional structure 91
6.7 Implications of the three dimensional structure 98
Chapter 7 - Crystal structure determination and model refinement of a co-crystallisation of HEWL and Carboplatin 7.1.1 Introduction to carboplatin 99
7.1.2 Previous work by Casini et al 101
7.1.3 Previous work by the Helliwell group 101
7.2 Co-crystallisation procedure and optimisation of the conditions 102
7.3 X-ray diffraction data collection procedure 106
7.4 The model refinement procedure 108
7.5.1 Analysis of the three dimensional structure 111
7.5.2 Comparison with HEWL and cisplatin crystal structure 114
7.5.3 Comparison with previous HEWL and carboplatin crystal structure 116
7.5.4 Comparison with HEWL and NAG trisaccaride crystal structure 116
7.6 Implications of the three dimensional structure 117
Chapter 8 - Conclusions and future work 120 References 121 Bibliography 126
Word count = 26,292

4
List of figures
Chapter 1 Page
1.1 An example of a diffraction pattern. 16
1.2 An example of the lattice of a crystal. 18
1.3 An example of a unit cell with the constituent axes and angles
labelled. 18
1.4 The fourteen Bravais lattices. 20
1.5 A 21 screw axis. 22
1.6 A pictorial representation of the Bragg equation. 23
1.7 The 111 Miller plane. 24
1.8 The 010 Miller plane. 24
1.9 A schematic representation of an X-ray tube. 26
1.10 The vapour diffusion method for small molecule crystallisation. 29
1.11 The vapour diffusion method for macromolecular crystallisation. 30
1.12 A crystal mounted within a loop. 32
Chapter 2 Page
2.1 An ORTEP diagram of C26H36N8018Cl2Co with 50% ellipsoid
probability. 41
2.2 Figure to show hydrogen bonding arrangement between cobalt
malonate molecules to form one dimensional chains. 42
2.3 A figure to show the crystal packing arrangement of
C26H36N8018Cl2Co. 43

5
Chapter 3 Page
3.1 An ORTEP diagram of C26H36N8O10F12P2Co with 50% ellipsoid
probability. 46
3.2 A figure to show the crystal packing arrangement of
C26H36N8O10F12P2Co. 47
3.3 A figure illustrating the common hydrogen bonding motif which is
present in both structures C26H36N8O18Cl2Co and
C26H36N8O10F12P2Co.
49
3.4 A figure to illustrate the difference in the hydrogen bonding
arrangements around the PF6 and perchlorate counter ions. 50
Chapter 4 Page
4.1 The expected chemical structure of the molecule in the crystal
MHB7. 52
4.2 The expected chemical structure of the molecule in the crystal
MHB8. 52
4.3 An ORTEP diagram of C24H20Sn with 50% ellipsoid probability 57
4.4 A figure to show the location of eight weak H…C-H interactions that
each C24H20Sn molecule forms. 58
4.5 A figure to show the stacking of the C24H20Sn molecule within the
crystal.. 58
4.6 A figure to show the stacking of layers of C24H20Sn molecules
stabilised by weak van der Waals interactions. 59
4.7 An ORTEP diagram of C30H24NO4Sn with 50% ellipsoid probability. 60
4.8 A figure to show the arrangement of the polymeric chains in
C30H24NO4Sn with weak van der Waals interactions shown as blue 61

6
lines. Hydrogen atoms are omitted for clarity.
4.9 A figure to show the distance between aromatic phenyl rings in
C30H24NO4Sn. Hydrogen atoms are omitted for clarity. 61
4.10 A figure to show the intramolecular hydrogen bond present within
the monomeric units. 62
4.11 A figure to show how SHELX views the molecules as discrete units
and not as a polymeric structure. 63
4.12 The crystallographically determined structure has this chemical
diagram with the highlighted area corresponding to the deviation
from the expected chemical structure.
65
Chapter 5 Page
5.1 The crystal growth phases. 68
5.2 A vectorial representation of the isomorphous replacement method. 72
5.3 A Harker construction for a native protein and a single heavy atom
derivative. 73
5.4 A Harker construction for a native protein and a second heavy atom
derivative. 74
Chapter 6 Page
6.1 The structure of the Ta6Br12 cluster. 80
6.2 A picture of the Ta6Br12 and HEWL crystals. 82
6.3 An X-ray diffraction pattern image from the Ta6Br12 & HEWL data
collection. 83
6.4 A figure to illustrate the gradual reduction of the conventional R
factor with each step of refinement performed.
88

7
6.5 A figure to show the evolution of the occupancies of the four binding
sites versus each step of refinement 91
6.6 A figure showing the electron density around the first Ta6Br12 to
lysozyme binding site. 92
6.7 A figure to show the distances between the tantalum atoms in the first
Ta6Br12 to lysozyme binding site. 92
6.8 A figure showing the electron density around the second Ta6Br12 to
lysozyme binding site. 94
6.9 A figure to show the distances between the tantalum atoms in the
second Ta6Br12 to lysozyme binding site. 94
6.10 A figure showing the electron density around the third Ta6Br12 to
lysozyme binding site. 95
6.11 A figure to show the distances between the tantalum atoms in the
third Ta6Br12 to lysozyme binding site. 96
6.12 A figure showing the electron density around the fourth Ta6Br12 to
lysozyme binding site. 97
6.13 A figure to show the distances between the tantalum atoms in the
fourth Ta6Br12 to lysozyme binding site. 98
Chapter 7 Page
7.1 The chemical structure of cisplatin. 99
7.2 The chemical structure of carboplatin. 99
7.3 A picture of the carboplatin and HEWL crystals. 105
7.4 A picture of an aggregate of carboplatin and HEWL crystals. 106
7.5 A picture of a carboplatin and HEWL crystal mounted onto a loop. 106
7.6 An X-ray diffraction pattern image from the carboplatin and HEWL
data collection. 108

8
7.7 A figure to illustrate the gradual reduction of the conventional R
factor with each step of refinement performed. 111
7.8 A figure to show the distances from the platinum atom to the two
ammonia groups in both carboplatin to lysozyme binding sites. 112
7.9 A figure to show the distances from the platinum atom to the nearest
nitrogen atom on the histidine 15 residue for both binding sites 113
7.10 A figure to show the electron density around the binding site present
on the left hand side of histidine 15. 113
7.11 A figure to show the electron density around the binding site present
on the right hand side of histidine 15. 114
7.12 A figure showing the superimposition of the histidine 15 residue in a
crystal of cisplatin and HEWL and a crystal of carboplatin and
HEWL.
115
7.13 A figure displaying the location of the DMSO molecule present
within the lysozyme active site for both the cisplatin and carboplatin
models.
116
7.14 Figure 7.13 – A figure showing the location of a NAG trisaccharide
and the DMSO molecules present in the cisplatin and carboplatin
models.
117

9
List of tables
Table Page
1 The essential symmetry and unit cell restrictions of the seven crystal
systems. 19
2 A summary of the X-ray diffraction and crystal data for
C26H36N8018Cl2Co. 40
3 The hydrogen bonding details for structure C26H36N8O18Cl2Co. 41
4 A summary of the X-ray diffraction and crystal data for
C26H36N8O10F12P2Co. 45
5 The hydrogen bonding details for structure C26H36N8O10F12P2Co. 46
6 A summary of the X-ray diffraction and crystal data for C24H20Sn. 53
7 A summary of the X-ray diffraction and crystal data for
C30H24NO4Sn. 55
8 A comparison of the details of the original 1970 C24H20Sn structure
and the structure reported in this thesis. 64
9 A summary of the X-ray diffraction data collection of a Ta6Br12 and
HEWL crystal. 84
10 A summary of the conditions attempted in the crystallisation of
HEWL in the presence of DMSO. 103
11 A summary of the X-ray diffraction data collection of a carboplatin
and HEWL crystal. 107

10
The University of Manchester
Richard Prendergast
Msc. in Chemistry by Research - Structure determination of small and large
molecules using single crystal X-ray crystallography
06/09/2010
Abstract
Single crystal X-ray crystallography can be applied to the entire spectrum of molecular size. If performed correctly the result is an unambiguous, three dimensional image of all the atoms located within a molecule. This applies to small chemical structures all the way through to biological macromolecules. In this thesis the method is used to solve the crystal structures of four small molecules and in addition to two macromolecular adducts. The first two molecules studied were believed to be closely isomorphous cobalt containing structures. The first small molecule was found to be C26H36N8018Cl2Co and crystallised in the triclinic space group P 1 . The structure was solved with an R factor of 0.0309. The second small molecule was found to be C26H36N8O10F12P2Co and crystallised in the triclinic space group P 1 . The structure was solved with an R factor of 0.0313. The remaining two small molecules were believed to be closely isomorphous tin containing structures. The third small molecule was found to be Ph4Sn and crystallised in the tetragonal space group P 4 21/c. The structure was solved with an R factor of 0.0353. The fourth small molecule was found to be C30H24NO4Sn and crystallised in the triclinic space group P 1 . The structure was solved with an R factor of 0.0245. In addition the crystal packing of all four small molecules were analysed. The implications of the determined crystal structures are discussed in terms of the relevant literature in each case. The method was also used to determine the structure of two macromolecular adducts. The first was a co-crystallisation of hen egg white lysozyme and Ta6Br12. The model refinement and a description of the Ta6Br12 binding sites are included. The second was a co-crystallisation of hen egg white lysozyme and carboplatin with the solubility of the carboplatin optimised using DMSO, whilst still obtaining crystals. The model refinement and a description of the carboplatin binding sites are included. Finally conclusions and possible routes for future work are offered.

11
Declaration
I declare that no portion of the work referred to in this thesis has been submitted in
support of an application for another degree or qualification at this or any other university
or other institute of learning.

12
Copyright
i. The author of this thesis (including any appendices and/or schedules to this
thesis) owns certain copyright or related rights in it (the “Copyright”) and s/he
has given The University of Manchester certain rights to use such Copyright,
including for administrative purposes.
ii. Copies of this thesis, either in full or in extracts and whether in hard or
electronic copy, may be made only in accordance with the Copyright, Designs
and Patents Act 1988 (as amended) and regulations issued under it or, where
appropriate, in accordance with licensing agreements which the University has
from time to time. This page must form part of any such copies made.
iii. The ownership of certain Copyright, patents, designs, trade marks and other
intellectual property (the “Intellectual Property”) and any reproductions of
copyright works in the thesis, for example graphs and tables
(“Reproductions”), which may be described in this thesis, may not be owned
by the author and may be owned by third parties. Such Intellectual Property
and Reproductions cannot and must not be made available for use without the
prior written permission of the owner(s) of the relevant Intellectual Property
and/or Reproductions.
iv. Further information on the conditions under which disclosure, publication and
commercialisation of this thesis, the Copyright and any Intellectual Property
and/or Reproductions described in it may take place is available in the
University IP Policy (see
http://www.campus.manchester.ac.uk/medialibrary/policies/intellectual-
property.pdf), in any relevant Thesis restriction declarations deposited in the
University Library, The University Library’s regulations (see
http://www.manchester.ac.uk/library/aboutus/regulations
)

13
Acknowledgements
I would like to begin by thanking Professor John R. Helliwell for his supervision and
for allowing me this opportunity.
I am extremely grateful to Dr Madeline Helliwell and Dr George Habash for their
help and patience regarding small molecule and protein X-ray crystallography
respectively.
I would also like to thank Dr Jim Raftery for his advice and for stimulating
discussions regarding subjects ranging from crystallography to politics.
Finally, I would like to thank my parents. Their loving support has made this possible.
RJP
Manchester
2010

14
Part A – Small molecule X-ray crystallography

15
Chapter 1
A review of the single crystal method 1.1 – Basic principles of X-ray crystallography A simple analogy to help visualise the basic principles that underpin X-ray
crystallography is that of a simple optical microscope. In both microscopy and
crystallography it is useful to view radiation in terms of a travelling wave of energy as
opposed to a particle.
In the case of the optical microscope a light source provides visible light waves which
pass through the sample under study and are subsequently diffracted. Each of these
diffracted waves has a characteristic intensity and phase associated with it. These
intensities and phases are then recombined by a lens in order to form an image.
As the name suggests X-ray crystallography utilises X-rays as opposed to visible light.
They are used as they are easily accessible and possess wavelengths comparable to bond
lengths allowing for visualisation down to the atomic level.
However the use of X-rays poses a problem – there is no known method capable of
recombining the scattered X-rays and thus forming an image. The intensity of the
diffracted waves can easily be determined by using an X-ray sensitive detector or
photographic plate. Unfortunately the phase information of the waves has been lost. This
is the physical basis of the phase problem that is inherently present within
crystallography.
Instead a branch of mathematics known as Fourier series are used in place of a lens to
recombine the scattered# X-rays.
# Technically the terms “scattered” and “diffracted” describe different wave-obstacle
phenomenon. Scattering is results in the wave changing direction with no form of
interference produced.

16
In comparison diffraction wavelength results in a change of direction of the wave as well
as the production of constructive and destructive interference. Therefore technically it is
diffraction and not scattering that produces the patterns observed in crystallography.
1.2 – Diffraction of X-rays by crystals
In theory a single molecule could be irradiated in order to produce a diffraction pattern.
However in practice this would lead to an immeasurably weak pattern and rapid
degradation of the molecule by the X-rays. Crystals are highly ordered structures which
are composed of a regular arrangement of units (these units could be atoms, molecules or
ions) that is repeated infinitely in three dimensions. Therefore instead of having one unit
in a particular orientation there are now in effect an infinite number – this leads to
“reinforcement” of the diffraction pattern and hence an averaged data set. In addition due
to the huge amount of identical units radiation damage is usually negligible.
Figure 1.1 – An example of a diffraction pattern. The particular position and symmetry
of the spots is illustrated in addition to the varying intensities of the spots.
To create a diffraction pattern a crystal is bathed in a beam of X-rays. The regular
arrangement of the atoms present in the crystal acts as a three dimensional diffraction

17
grating .The incident X-rays interact with the electrons of the crystal via inelastic
collisions which causes diffraction. The result is a pattern consisting of spots which
possesses three important properties directly related to the crystal under study (Figure
1.1).
The position, symmetry and intensity of the spots all hold information that must be
extracted.
However, one diffraction pattern is not sufficient to allow for structure determination.
This is because that only a small number of reflections will be excited at the particular
angle of the stationary crystal. As a result the crystal must be slowly rotated (through
small increments) whilst still fully immersed within the X-ray beam. In modern day
diffractometers this a a fully automated, computer controlled process which results in the
maximum number of reflections being recorded
The X-rays most commonly used in “home” laboratory based experiments are
monochromated MoKα (λ = 0.71Ǻ) and CuKα (λ = 1.54 Ǻ). These particular wavelengths
are favoured as they are comparable with the distances under study. (E.g. C-C = 1.54 Ǻ).
This helps to ensure appreciable diffraction occurs.
1.3.1 - Crystal structure and symmetry
As a consequence of their highly ordered structure, crystals also display a high degree of
symmetry. This symmetry is described by a number of different concepts which are
subsequently defined.
As previously mentioned crystals are composed of a regular, repeating arrangement of
units. If each of the constituent units was represented by a single point then the resulting
array would be representative of the repeating nature of the crystal. This array of points
(related to each other by translational symmetry) is known as the lattice of the crystal
(Figure 1.2).

18
Figure 1.2 – A demonstration of the crystal lattice, created by representing the
constituent units with points
An extension upon the theme of lattice points is the unit cell. A unit cell is a
parallelogram consisting of four lattice points. Crystals are defined by their unit cells –
they describe the simplest “building block” that is repeated in three dimensions to
produce the bulk crystal. A unit cell is characterised by three vectors a, b and c which lie
along the x, y and z directions respectively. Also of importance are the angles between
these vectors – alpha, beta and gamma. Convention dictates that alpha is the angle
between vectors b and c, beta is the angle between vectors a and c whilst gamma is the
angle between a and b (Figure 1.3).
Figure 1.3 – An example of a unit cell with the axes and angles labelled.
These vectors and the angles between them give rise to the seven crystal systems which
are used to describe the geometry of the unit cell. Rotational and reflection symmetry
place restrictions of the allowed vector lengths and angles. These restrictions allow for

19
classification into seven groups – triclinic, monoclinic, orthorhombic, tetragonal, trigonal,
hexagonal and cubic. The aforementioned restrictions are given in Table 1.
Crystal system Unit cell restrictions Essential symmetry of crystal
Triclinic None None
Monoclinic One diad axis (2 fold rotation)
or mirror plane (inverse diad
axis)
a ≠ b ≠ c
β ≠ α = γ = 90°
Orthorhombic Three orthogonal diad axes or
inverse diad axes
a ≠ b ≠ c
α = β = γ = 90°
Tetragonal One tetra axis (four fold
rotation) or inverse tetrad axis
a = b = c
α = β = γ = 90°
Trigonal One triad (three fold rotation)
axis or inverse triad axis
a = b = c
α = β = γ ≠ 90°
Hexagonal One hexad (five fold rotation)
axis or inverse hexad axis
a = b ≠ c
α = β = 90°, γ = 120°
Cubic Four triad axes or inverse triad
axes
a = b = c
α = β = γ = 90°
Table 1 – The essential crystal symmetry and unit cell restrictions of the seven crystal
systems.
Introducing translational symmetry into the seven crystal systems (which only include
rotational and reflection symmetry) forms the Bravais (or space) lattices. There are 14
possible Bravais lattices which involve four different ways of centring the lattice points
(Figure 1.4). The possible lattice centrings are –
- Primitive (P) – Lattice points are located at the corners of the unit cell.
- Body Centred (I) – All primitive points included plus an additional point at the
centre of the unit cell.
- Face Centred (F) – All primitive points included plus additional points at the
centre of each face of the unit cell.

20
- Centred (C) – All primitive points included plus an additional point at the
centre of one face of the unit cell.
Figure 1.4 – The fourteen Bravais lattices with the lattice points displayed1.
A point group is a mathematical descriptor for a group of symmetry operations that pass
through a central point. These symmetry operations must leave at least one point
unchanged and the appearance of the object unaltered.
There are four symmetry operations associated with point groups -
- n-fold rotation axes – a rotation through (360°/n) which leaves the object
unaltered (where n is an integer).
- Mirror planes – involves a reflection which takes place with respect to a
mirror plane.
- Inversions – involves moving every point x,y,z to –x,-y,-z.
- Improper rotations - a rotation followed by an inversion.
Compared to the point groups for an isolated object (such as a single molecule) there are
230 possible crystallographic space groups. This is because there are 32 possible ways to
combine the point group symmetry operations with the translational symmetry inherently
present within crystals (crystallographic restriction theorem). For point groups the
Schoenflies notation is most commonly used. For example the molecule SF6 belongs to

21
the octahedral point group (in Schoenflies notation represented by Oh) which contains 31
associated symmetry elements2.
The specific method of describing the symmetry present in a crystal is that of space
groups. Space groups describe the symmetry operations present in an infinitely repeating
three dimensional pattern (crystals are as an approximation to infinite repeating
structures). Therefore each space group is a combination of the point group symmetry
operations with translational (or space) symmetry operations.
There are 230 space groups which completely describe all possible combinations of the
aforementioned symmetry operations.
Typically the internationally recognised Hermann and Magiun notation is used.
A typical example is P 21/m –Where P is the type of Bravais lattice – Primitive in this
example.
The letters following the P represent the symmetry operations which lie along a special
direction in the crystal. In this example 21 represents a 21 screw axis in the direction of
the unique axis of the monoclinic crystal system. The ‘/m’ represents an ordinary
reflection plane which is perpendicular to the unique 21 axis.
The space groups and their associated symmetry operations are systematically detailed in
the International Tables for Crystallography3.
In addition to the symmetry operations possessed by point groups there are two space
symmetry operations which may be contained within space groups. These operations are
termed glide planes and screw axes.
A screw axis is a combination of a rotation of (360/n) followed by an appropriate
translation parallel to the axis of rotation to preserve the translational repetition (where n
is an integer). For example a 21 screw axis consists of a twofold rotation axis (360°/2)
followed by a translation along half of the lattice axis that is parallel to the rotation
(Figure 1.5).

22
Figure 1.5 – The effect a 21 screw axis has upon a particular point4.
A glide plane is a combination of a reflection in a mirror plane followed by a translation.
There are five possible glide planes – denoted a, b, c, n and d. For example a c glide plane
consists of a reflection in the xy plane followed by a translation along half of the c axis.
Screw axes and glide planes can cause the systematic absence of certain reflections in a
diffraction pattern. These systematic absences can help in the assignment of space groups
as the absences are well known and are listed in the International Tables for
Crystallography3 (although space group ambiguities do exist).
1.3.2 - The Bragg equation
In 1913 W. L. Bragg derived his now eponymous equation5 following on from work
conducted by Freidrich, Knipping and Laue. This work proved a proposal by Laue which
was stimulated by Ewald that crystals were capable of diffracting X-rays. The Bragg
equation is still used today to mathematically explain the diffraction geometry of X-rays
by crystals.
The equation treats crystals as being composed of a series of parallel planes of atoms
separated by a small distance. The planes are assumed to be capable of reflecting the X-
rays in a manner which results in the angle of incidence equalling the angle of reflection
(Figure 1.6).
The contributions from successive planes will be in phase (i.e. the difference in path
length between successive waves must be an integer number of wavelengths) only for
certain angles.
As a result constructive interference and the production of diffraction maxima can only
occur if the Bragg equation (Equation 1) is satisfied.

23
Equation 1 – The Bragg equation
If the waves are out of phase destructive inference will occur. Indeed since most of a
diffraction pattern consists of empty space this the common situation. The absence of
diffraction spots can provide as much information as their presence. For example space
groups can be assigned on the basis of systematically absent reflections .
Laue also derived a set of three equations that describe the same effect but these are less
widely used6.
Figure 1.6 – A pictorial depiction of the relationships that constitute the Bragg
equation7.
1.3.3 - Miller Indices
Named after W. H. Miller these indices are an unambiguous way of defining crystal
planes. They consist of three numbers (hkl) which correspond to the inverse of the ratio
Where –
n = An integer
λ = Wavelength of the radiation (m)
d = Interplanar spacing (m)
sin θ = Angle of incidence of radiation

24
of the intercepts on the a, b and c axes of the unit cell (for examples see Figures 1.7 &
1.8).
Figure 1.7 – A pictorial representation of the 111 Miller plane.
Figure 1.8 – A pictorial representation of the 010 Miller plane.
1.4.1 - Nature, production and detection of X-rays
X-rays are a form of electromagnetic radiation which possess wavelengths within the
range of 0.01nm (0.1 Ǻ) to 10nm (100 Ǻ) with wavelengths in the range 0.2 – 3 Ǻ being
useful in crystallography. As such they consist of an electric field and magnetic field
vector which are perpendicular to each other. These vectors oscillate in a sinusoidal
manner perpendicular to the direction of propagation.
X-rays are the favoured form of radiation in crystallography as they possess wavelengths
comparable to bond lengths and can also be easily generated in a “home” laboratory
setting. A more recent method of generating much more intense and finely tuneable X-
rays using a synchrotron source is also now widely used.
1.4.2 - X-ray tubes – For X-ray generation in the “home” laboratory
X
Y
Z
Y
X
Z

25
The X-ray tubes used in modern day crystallography are known as filament (or Coolidge)
tubes and date back to 1913. They consist of an evacuated glass enclosure which contains
a tungsten filament and a disk of a target metal (Figure 1.9). The target metal is
responsible for the production of the characteristic wavelength of the X-rays. The most
commonly used target metals are Molybdenum (wavelength = 0.71 Ǻ), Copper (1.54 Ǻ)
and Silver (0.56 Ǻ).
To initiate the production of X-rays the tungsten filament is heated by passing an electric
current through it. This results in the production of electrons which are accelerated by a
potential difference and directed towards the target metal. If the potential difference is
sufficiently high (typically 40kV) the electrons will possess enough energy to cause
ionisation of inner core electron`s of the target metal. To compensate an electron in a
higher atomic energy level for the metal will drop in energy to take the place of the
ejected electron. This results in the emission of a photon with a characteristic wavelength.
The characteristic wavelength produced is dependent upon the metal atom energy levels
from which each electron is ejected. For example MoKα emission corresponds to
electrons moving between the L and M shells (λ = 0.71Ǻ) of Molybdenum whilst MoKβ
emission corresponds to a movement between the L and K shells (λ = 0.63λ).
This characteristic wavelength created is defined by Equation 2.
Equation 2 – The equation for the characteristic wavelength generated from a
particular target material.
The spectra purity of the X-ray beam onto the crystal is created by using filters to remove
background and other unwanted wavelengths whilst a beryllium window allows the X-
rays to leave the tube head with minimum absorption.
Where –
h = Plancks constant (6.6261 x 10-34 J s)
c = Speed of light (2.9989 x 108 m/s)
E1 = Lower energy level of target atom
E2 = Higher energy level of target atom

26
This method of X-ray production can be considered quite inefficient as the vast majority
of the energy carried by the electrons is converted into heat rather than X-rays (literature
sources mention 1% X-ray conversion8). The heating of the target is largely compensated
by a water cooling system which prevents melting of the target material up to certain
current limits. An additional disadvantage of this method is that the X-rays generated are
quite divergent which may pose a problem if small crystals are under study.
Figure 1.9 – A schematic diagram of an X-ray tube9.
An improved method of generating X-rays in the home laboratory is known as a rotating
anode. In this apparatus the target metal is cylindrical and is spun about its axis. This
allows the energy of the X-rays to be spread out over a larger overall area thereby
reducing the heating problem. As a result much higher electrical currents can be
introduced which creates a much higher flux density. This method of X-ray generation is
important especially for molecules which possess large unit cells such as proteins which
are often in large complexes.
1.4.3 - Synchrotron source X-ray generation
Synchrotrons were initially developed as a tool in particle physics to accelerate beta
particles (electrons and positrons). It is observed that when such particles are accelerated
through magnetic fields at relativistic speeds they lose energy in the form of
electromagnetic radiation (this radiation covers the entire EM spectrum not just X-rays).

27
When the beta particles pass through the magnetic fields they change direction. This
causes the tangential emission of radiation. Although emission of radiation occurs at non
relativistic speeds, a feature of relativity known as the Lorentz transformation means that
the radiation is emitted in a highly collimated fashion at speeds approaching that of the
speed of light. This emission of radiation was first observed in 1946 at a 70MeV
synchrotron in Schenectady by F. R. Elder et al10. Today many synchrotron sources are
now operational as nationally and internationally shared facilities.
Synchrotrons consist of a linear accelerator (LINAC) which creates high energy electrons
(around 10MeV). These electrons are subsequently injected into a small accelerator
(known as a booster synchrotron) which increases the energy of the electrons to around
500MeV. Once this point has been reached the electrons are injected into the main
synchrotron ring where the energy is further increased via multiple passes through radio
frequency cavities. This produces X-rays which extends to the necessary short
wavelengths and are much more intense and well collimated than laboratory based
sources. This allows for extremely fast data collection times and smaller crystals to be
studied. The continuous spectrum allows for the fine tuning of the selected wavelengths
using monochromators. Alternatively the whole ‘white’ X-ray spectrum may be used in
Laue diffraction experiments.
1.4.4 - Detection of X-rays
In the beginnings of X-ray crystallography intensities were often measured by using
photographic films coated in silver halide. Exposure to X-rays causes silver halide to
darken. The darkness of the spots is related to the intensity of the absorbed radiation in a
given reflection (spot).
A vast improvement was the appearance of computer controlled diffractometers in the
1960`s. These routinely utilised an X-ray sensitive electronic device known as a
scintillation counter. A scintillation counter consists of a crystal mounted onto a
photomultiplier tube. A commonly used crystal is sodium iodide doped with a small
amount of Thallium (around 1%)11. These crystals produce light when irradiated by X-
rays. This light can then enter the photomultiplier which results in the ejection of
electrons and thus the generation of an electric current. This process results in the

28
production of an electric pulse for each individual X-ray allowing for the measurement of
intensities. There are disadvantages associated with scintillation counters. Foremost is
that the diffracted beams are measured one at a time which often translates to long data
collection times.
A more recently developed method of X-ray detection is known as a charge coupled
device (CCD). These detectors have the advantage of being able to record a number of
diffracted beams at the simultaneously, thereby reducing data collection times. A CCD
detector employs a semiconductor in which the incident X-rays induce the production of
free electrons and electron holes12. The electrons produced are trapped in potential wells,
and in addition to the electron holes, are read out as a current. The magnitude of this
current is proportional to the intensity of the diffracted beams .The various designs of
CCD detectors can be roughly divided into two groups depending on how the intensity of
the radiation is detected. This may be done by either measuring the intensity of the X-
rays directly or by conversion of the X-rays to visible light using a phosphor conversion
mechanism13.
Diffractometers that utilise a CCD detector are often known as three circle
diffractometers. This is because they possess three rotation axes (one in relation to the
detector and two in relation to the crystal). Scintillation counter based diffractometers
possess four rotation axes as the detector is smaller and can only record reflections which
occur in the horizontal plane. As a result an additional crystal rotation axes is required.
1.5 - Crystal growth
Crystals are formed as a result of chemical systems seeking to minimise the Gibbs free
energy. On the one hand the formation of crystals results in an unfavourable loss of
entropy. This arises because the individual molecules which constitute the crystal are in
effect “locked” in place. As a result they lose rotational and translational degrees of
freedom. Conversely this is coupled with a favourable increase in the enthalpy of a
system. This increase arises because the crystallisation process involves the formation of
many new, stable non covalent chemical bonds. This increase in enthalpy more than

29
counterbalances the unfavourable decrease in entropy and overall favourably decreases
the Gibbs free energy.
Small molecule and macromolecule crystal growth utilise different apparatus, although
common pricincples. The initial aim is to tailor the experimental conditions so that the
solution is just saturated. At this moment the saturation point should be very slowly
lowered whilst the rate of nucleation is limited. In theory this should yield well formed
and decently sized crystals possessing a high degree of regularity.
In practice however obtaining crystals of a suitable size and quality is often a major rate
limiting step in the structure determination process. The crystallisation process in a given
case is often poorly understood whilst the high number of variables involved (e.g.
temperature, pH, concentrations) further complicates the process. However there are
exceptions, for example the crystal growth of silicon is exceedingly well understood.
Techniques for inducing the crystallisation of small molecules are often much simpler
than those used in macromolecular crystallisation. Commonly used techniques to induce
small molecule crystallisation are the slow evaporation of a solution, slow precipitation
by vapour diffusion and sublimation.
Macromolecular crystallisation techniques are discussed in Chapter 5 although both areas
often use vapour diffusion (although the apparatus differs slightly as illustrated by
Figures 1.10 & 1.11).
Figure 1.10 – The vapour diffusion method for small molecule crystallisation.

30
Figure 1.11 – The hanging drop vapour diffusion method for macromolecular
crystallisation.
1.6.1 - Structure determination procedure
Once crystals of a suitable size have been grown the crystal structure determination
procedure can begin. This procedure can be thought of as being divided into three main
stages –
- The first stage involves the measurement of the intensities of the Bragg reflections
and the application of corrections to take into account various geometrical and
physical phenomena.
- The second stage involves using mathematical and computer program methods to
imitate the behaviour of a microscope lens to solve the phase problem.
- The final stage involves refining the initial structure so that there is an optimum
agreement between the observed and calculated structure factors.
The steps involved in each of these stages are further explained below.
1.6.2.1 - Stage 1 – Measurement of X-ray intensities
1.6.2.2 - Step 1 - The first step towards measuring X-ray intensities is the selection
and preparation of a suitable single crystal.
For use in home laboratory experiments single crystals in the order of 0.2-0.4mm are
routinely required. This is because the X-ray beam generated is relatively weak in

31
intensity (compared to a synchrotron source) and the diameter is less than 1mm; using
such a small size ensures that the crystal is fully immersed in the X-ray beam. It is
important to inspect crystals beforehand using a microscope to ensure that no visible
defects such as cracks or twinning are apparent. In addition crossed polarisers can be used
to ensure that the crystals extinguish. This can help to reveal defects within the crystal
that were previously not apparent. However should not be considered a conclusive test as
some crystals (depending upon their symmetry) do not extinguish.
Once a crystal of a suitable size and quality has been selected it can be mounted onto a
small loop or mesh (Figure 1.12). The crystal is held in place by using a small amount of
amorphous glue. If the data is to be collected at a cryogenic temperature then a viscous
oil can be used. The oil will freeze in the cryogenic stream thereby again fixing the
crystal in place. Data collection at cryogenic temperatures is advantageous as it reduces
the rate of radiation damage caused by the incident X-rays. In addition cryogenic
temperatures reduce atomic mobility which in turn enhances the diffraction spot
intensities (as this minimises disorder). In special cases such as if the sample is air
sensitive the crystal can be contained within a thin walled glass capillary. The glass has
an amorphous structure and hence does not appreciably contribute to the diffraction
pattern.
Finally, the loop, mesh or capillary containing the crystal is mounted onto a goniometer
head and placed onto the diffractometer. The goniometer head is a device that allows the
crystal to be easily centred in the X-ray beam. Additionally in modern day
diffractometers a high magnification video camera is used to ensure that the crystal is in
the correct position and to record a digital picture of the crystal for size determination.

32
Figure 1.12 – A crystal mounted within a loop held in place with a viscous oil. Pictured
via a high magnification video camera present on the diffractometer.
1.6.2.3 - Step 2 – The collection of the X-ray intensities
Once the crystal has been correctly centred with respective to the X-ray beam irradiation
can begin. This irradiation will produce a diffraction pattern that is commonly recorded
by a CCD detector. The CCD diffraction images collected then need to be integrated to
produce a list of reflections i.e. spots (hkl values) each with an associated intensity.
It is possible to determine the unit cell dimensions from the first few images. Other
factors such as the quality of the crystal (the mosaic spread and/or splitting) are also
obvious from the first few images obtained.
1.6.2.4 - Step 3 – The diffraction images data reduction process
This step includes the application of corrections to the measured intensities which take
into account various geometrical and physical phenomena.
A common geometrical correction applied is known as the Lorentz-polarisation factor.
The Lorentz factor is related to the amount of time the reflection is in a diffraction
position and is instrument dependent. The polarisation factor is required because the
reflected X-rays are partially polarised.
A commonly applied physical correction concerns the absorption of X-rays by crystals
(this is particularly true for inorganic crystals). Absorption corrections are needed for
crystals that are not approximately spherical and are calculated by analysing systematic

33
variations in the intensities of symmetry related reflections. This is because the amount of
absorption is dependent upon the path length the X-rays travel through the crystal.
Absorption of X-rays also increases the larger the crystal is; using a crystal as small as
possible helps to minimise this error. Finally absorption varies with elemental
composition; often heavy atoms strongly absorb X-rays.
The data reduction process also involves the merging of symmetry related reflections and
the calculation and application of scale factors to the measured reflections. The result is a
unique, scaled data set. The data reduction process is a ‘black box’ method that is
performed by computers.
1.6.3.1 - Stage 2 – The crystallographic phase problem and possible
solutions The phase problem is intrinsic to X-ray crystallography. Each of the diffracted X-rays
will have a particular phase and amplitude associated with it. X-ray sensitive detection
methods such as photographic film or CCD detectors are able to measure intensities from
which the amplitudes are easily obtained (intensities = ampltiude2). However the relative
phases of the waves are lost during the experiment. This is a problem because in order to
elucidate the crystal structure both the intensities and relative phases are required.
Therefore a method of obtaining approximate phases and hence solving the phase
problem is required. In small molecule crystallography two methods are almost
exclusively used which both utilise a branch of mathematics known as Fourier series.
Fourier series arise from Fourier’s theorem which states that any periodic function can be
represented by a summation of sine and cosine terms. The diffraction pattern and the
electron density of a crystal are related by a Fourier series. In addition a diffraction
pattern consists of well defined individual spots. Therefore a summation must be used as
opposed to integration which would be performed if the pattern was diffuse.
Crystals can be described by a Fourier series as the structure of a crystal is a periodically
repeating, (effectively) infinite array.

34
The structure factor equation is used to describe how the incident X-rays are diffracted by
the constituent atoms of a crystal. This equation takes into account the scattering power
of each atom (which is described by fj which is the scattering factor for the jth atom) and
is dependent upon electron density. This is described by Equation 3.
Equation 3 – The structure factor equation.
The electron density calculation must be performed in three dimensions in order for a
three dimensional structure to be produced. The unit cell volume (V) must also be taken
into account. The equation used to calculate the electron density at a particular point
(xyz) is given by Equation 4.
Equation 4 – The equation used to calculate the electron density at point ρ(xyz).
The two commonly used methods used to solve the phase problem in small molecule
crystallography are described below.
1.6.3.2 – The Patterson Synthesis
This method is commonly employed when there is one or a small number of heavy atoms
present in the structure.
In 1934 A. L. Patterson presented a synthesis (or Patterson map) that is obtained by
performing a Fourier series on the square of the amplitudes with all waves taken in
Where –
N = The number of atoms
within the structure
fj = Atomic scattering factor
for the jth atom

35
phase14. If there are an N number of atoms in a unit cell then there is a N2 number of
vectors running between these atoms. Therefore a Patterson map shows where atoms are
located relative to each other but not where they are located with respective to the unit
cell origin. The result is a map that has an appearance similar to that of an electron
density map in that it contains peaks of positive density located in particular positions.
However this is not a map of electron density, instead it is a map of the vectors between
pairs of atoms in the structure. The Patterson synthesis is described mathematically by
Equation 5.
Equation 5 – The mathematical representation of the Patterson synthesis.
1.6.3.3 – Direct Methods
This method was developed for equal atom structures i.e. those that contain no heavy
atoms.
Direct methods also use the measured intensities but takes advantage of the fact that
electron density within a crystal can not be negative. This places restrictions on the
possible phase angles between reflections. The process is almost a trial and error
approach – the reflections which contribute most to the Fourier transform are selected as
are approximations that appear promising (assessed by a numerical factor). The Fourier
series are calculated using the measured intensities and these approximate phases.
Sensible looking chemical fragments can be used to assess the different trial structures.
The resulting trial structure is only an initial approximation of the true structure and must
undergo further refinement.
Direct methods are often described as black box as the process is automated and
performed by computers.
Where –
V = The unit cell volume
(in Ȧ3).

36
Methods used to solve the phase problem in macromolecular crystallography differ and
are detailed in Chapter 5.
1.6.4 - Stage 3 – Refining the structure The final stage involves refining the initial structure so that there is a optimum agreement
between the observed structure factor amplitudes and the structure factor amplitudes
calculated for the current structure.
The measure by which these factors agree is described by the conventional residual factor
(commonly known as the R factor). The R factor is defined by Equation 6.
Equation 6 – The conventional residual factor.
As illustrated by Equation 6 the lower the R factor the better the agreement and the more
correct the structure is. That the earlier stages of the structure determination procedure
were performed correctly is essential if a low R factor is to be obtained.
Refinement uses a mathematical technique known as least squares analysis which adjusts
parameters such as atom positions and atomic displacement parameters in order to
produce the maximum agreement between two sets of data (in this case the observed and
calculated amplitudes). The refinement on F2 was used in this thesis and is defined by
Equation 7.
Where –
FO = Observed structure factor amplitude
FC= Calculated structure factor amplitude from model
Where –
yO = Observed structure factor amplitude
yC= Calculated structure factor amplitude from model

37
Equation 7 – The least squares refinement of the square of the structure factor
amplitudes.
Several cycles of refinement are required as the data used is calculated using Fourier
series which are non linear equations. Consequently cycles of refinement are required
until the adjustments of the parameters are insignificant (a process known as
convergence).
The refinement parameters can be split into two groups based on the mathematical detail
used to describe the atoms. Isotropic refinement uses three positional coordinates (x,y,z)
as well as a single vibrational parameter to approximate vibrating atoms as spheres.
Anisotropic refinement uses the same three positional coordinates as well as six
vibrational parameters to describe atoms in terms of ellipsoids. Although a perfect
ellipsoid can be described by three vibrational parameters atoms typically possess
distorted ellipsoids which are described by require six vibrational parameters .This results
in a significantly more accurate and realistic model structure. In addition, once
anisotropic refinement has been carried out small peaks can often be observed,
corresponding to hydrogen atom positions.
The process of refinement is complete when convergence is achieved and the electron
density map contains no undefined peaks or holes.
For small molecule X-ray crystallography a final R factor in the range 0.02 – 0.07 is an
indicator of a good quality structure.

38
Chapter 2
Structure determination of a small molecule – C26H36N8018Cl2Co
2.1 - Introduction to C26H36N8O18Cl2Co
This complex was synthesised by the group of Professor Subrata Mukhopadhyay of the
University of Jadavpur, India for study into the field of crystal engineering. The field of
crystal engineering seeks to utilise intermolecular interactions to aid molecular
recognition through the identification and control of recognition motifs. The field is still
relatively new and its full potential has yet to be realised. This is because there are
problems present which are poorly understood, for example weak interactions can prove
especially unpredictable and therefore difficult to control. However it is hoped that
molecular recognition may prove useful in the design and synthesis of future functional
materials.
In order for molecular recognition to be successfully implemented a full appreciation of
the intermolecular interactions present is required. This can be achieved using single
crystal X-ray crystallography to provide a complete unambiguous three dimensional
structure.
The probable composition of the complex was determined by the synthetic chemists as
[Co(mal)2(H2O)2](ClO4)2(LH)4 where mal indicates malonate and LH4 indicates
protonated 2-amino pyridine.
2.2 – X-ray diffraction data collection and processing procedure
The crystals provided were approximately 1-2 mm in length and pink in colour. The
crystals appeared to be of good quality and extinguished well under crossed polarisers.
As a result a single crystal was cut using a razor blade to around 0.5mm in length. The
crystal was then immersed in a viscous oil and fitted onto a loop, which was placed onto
a goniometer head and fitted onto the diffractometer. The data collection was performed

39
at a cryogenic temperature (100K), which caused freezing of the viscous oil and thus
fixation of the crystal. This is in addition to being beneficial by reducing atomic
displacement parameters. The crystal was centred using rotating screws on the
goniometer head and a video camera as a visual aide. Firstly the X-axis was adjusted at
phi 0˚ to ensure the crystal was centred. Once complete the screw was rotated to phi 180˚
and the crystal centred again. This process was repeated in the same manner for the Y-
axis but with phi angles 90˚ and 270˚. Once the crystal was completely centred it was
irradiated with monochromated MoKα radiation. The X-ray generator settings were 40kV
and 40 mA.
The computer program SAINT15 was used to collect and integrate the CCD frame images
in order to produce integrated intensities. This produced files with filename extensions
.p4p and .RAW. These files were then introduced into SHELX16 XPREP for
determination of the crystal system and space group. SHELX SADABS was used to
produce an X-ray absorption corrected data set which took into account absorption of X-
rays by the crystal, although this was a symmetrically sized crystal (crystal dimensions
listed in Table 2).
It was found that SHELX XS was unable to satisfactorily solve the structure using both
direct methods or a Patterson synthesis. This sort of unexplained failure of SHELX can
occur. As a result the direct methods program SIR 200417 was used to solve the structure.
Further refinement was carried out within the SHELX XSHELL program.
All non hydrogen atom positions were refined anisotropically. The hydrogen atom
positions were clearly visible using difference Fourier methods and were refined
isotropically.
Empirical formula C26H36N8018Cl2Co
Chemical formula weight 878.46 g mol-1
Crystal system Triclinic
Space group P 1

40
Unit cell dimensions a = 7.1122(7) Ǻ α = 86.908(2)°
b = 11.2696(10) Ǻ β = 84.168(2)°
c = 11.7951(11) Ǻ γ = 72.440(2)°
Unit cell volume 896.41(15) Ǻ3
Z 1
Data collection temp 100 K
Radiation MoKα, graphite monochromator
Diffractometer Bruker AXS Apex (3 circle)
Detector CCD area detector
Crystal size 0.5 x 0.4 x 0.4 mm
Tmin & Tmax 0.1756 & 0.8698
F (000) 453
Theta range 2.54 to 26.38°
Total reflections measured 5183
Independent reflections 3548 (Rint = 0.0242)
R indices [F2 > 2σ(F2)] 0.0309
R indices (F2) 0.0363
Largest diff. peak and hole 0.423 and -0.374 e.Å-3
Number of refined parameters 322
Table 2 – A summary of the X-ray diffraction and crystal data for C26H36N8O18Cl2Co.
2.3 - Crystal structure analysis
It was found that the small molecule C26H36N8O18Cl2Co crystallises in the triclinic space
group P 1 . The asymmetric unit contains half of the cobalt malonate anion
[Co(C3H2O4)2(H2O)2]- in addition to two protonated 2-amino pyridine cations (C5H7N2+)
and a perchlorate counter ion. The cobalt atom is located on an inversion centre bonded
to two oxygens on each of the equatorial malonates, as well as to a single oxygen on each
of the trans, axial, waters (Figure 2.1).
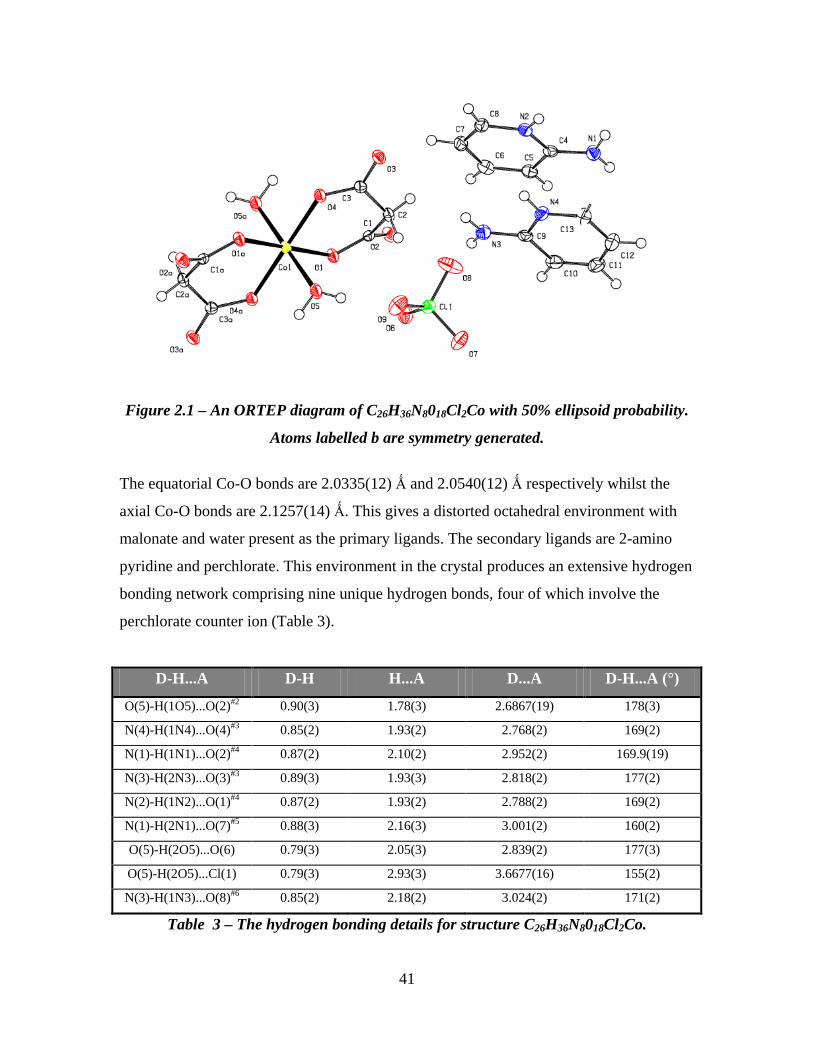
41
Figure 2.1 – An ORTEP diagram of C26H36N8018Cl2Co with 50% ellipsoid probability.
Atoms labelled b are symmetry generated.
The equatorial Co-O bonds are 2.0335(12) Ǻ and 2.0540(12) Ǻ respectively whilst the
axial Co-O bonds are 2.1257(14) Ǻ. This gives a distorted octahedral environment with
malonate and water present as the primary ligands. The secondary ligands are 2-amino
pyridine and perchlorate. This environment in the crystal produces an extensive hydrogen
bonding network comprising nine unique hydrogen bonds, four of which involve the
perchlorate counter ion (Table 3).
D-H...A D-H H...A D...A D-H...A (°)
O(5)-H(1O5)...O(2)#2 0.90(3) 1.78(3) 2.6867(19) 178(3) N(4)-H(1N4)...O(4)#3 0.85(2) 1.93(2) 2.768(2) 169(2)
N(1)-H(1N1)...O(2)#4 0.87(2) 2.10(2) 2.952(2) 169.9(19)
N(3)-H(2N3)...O(3)#3 0.89(3) 1.93(3) 2.818(2) 177(2)
N(2)-H(1N2)...O(1)#4 0.87(2) 1.93(2) 2.788(2) 169(2)
N(1)-H(2N1)...O(7)#5 0.88(3) 2.16(3) 3.001(2) 160(2)
O(5)-H(2O5)...O(6) 0.79(3) 2.05(3) 2.839(2) 177(3)
O(5)-H(2O5)...Cl(1) 0.79(3) 2.93(3) 3.6677(16) 155(2)
N(3)-H(1N3)...O(8)#6 0.85(2) 2.18(2) 3.024(2) 171(2)
Table 3 – The hydrogen bonding details for structure C26H36N8018Cl2Co.

42
Symmetry transformations used to generate equivalent atoms:
#1 -x+2,-y+1,-z+2 #2 x+1,y,z #3 x-1,y,z #4 -x+1,-y+1,-z+2
#5 x,y-1,z #6 -x+1,-y+1,-z+1
The cobalt malonate anions are linked by a hydrogen bond of length 1.78(3) Ȧ between a
axial water molecule and an equatorial carboxylate oxygen (Figure 2.2). As a result the
cobalt malonate molecules form one dimensional chains which run parallel to each other.
Figure 2.2 – A figure to show the hydrogen bonding arrangement linking two cobalt
malonate units to form a one dimensional chain.
The 2-amino pyridine and perchlorate ions form alternating layers between the chains of
cobalt malonate anions (Figure 2.3). This arrangement is further stabilised by π-π
interactions between 2-amino pyridine cations. Each pair of 2-amino pyridines that is
joined to a particular cobalt malonate anion can interact but pairs belonging to different
cobalt malonate anions are unable to interact due to ring slippage (different pairs do not
lie directly above each other and are therefore unable to interact).

43
Figure 2.3 – A figure to show the crystal packing arrangement of C26H36N8018Cl2Co .
The 2-amino pyridine and perchlorate molecules form alternating layers between the
parallel cobalt malonate chains.
2.4 - Crystal structure implications
The structure was solved with a low R factor of 0.031 (as described by Equation 6) and
was consistent with the expected chemical composition.
The presence of the electron accepting oxygen and chlorine atoms of the perchlorate
anion help in the formation of an extensive hydrogen bonding network in the crystal.
Further stabilisation is provided via π-π interactions between aromatic rings of the 2-
amino pyridine cations. These intermolecular interactions help to minimise the free
energy by decreasing the enthalpy and therefore help promote crystallisation.

44
Chapter 3
Structure determination of a small molecule – C26H36N8O10F12P2Co
3.1 Introduction to C26H36N8O10F12P2Co
Presented in this chapter is the data collection procedure and structural analysis of a
cobalt containing complex. This complex was again synthesis by the group of Professor
Subrata Mukhopadhyay of the University of Jadavpur, India, for study into the field of
crystal engineering. The complex and the previous example were expected to be closely
related with both expected to contain 2-amino pyridine and malonate ions.
The probable composition of the complex was determined by the synthetic chemists as
[Co(mal)2(H2O)2](PF6)2(LH)4 where mal indicates malonate and LH4 indicates protonated
2-amino pyridine.
3.2 – X-ray diffraction data collection and processing procedure
The crystals provided were pink in colour and had an average size of approximately 5mm
to 1cm in length. The crystals appeared to be of good quality and extinguished well under
crossed polarisers. As a result a single crystal was cut using a razor blade to around
0.3mm in length. The crystal was then immersed in a viscous oil then fitted and mounted
as described in chapter 2. Once centred the crystal was irradiated with monochromated
MoKα radiation. The X-ray generator settings were 40 kV and 40 mA.
The computer program SAINT15 was used to collect and integrate the CCD frame images
in order to produce integrated intensities. This produced files with filename extensions
.p4p and .RAW. These files were then introduced into SHELX16 XPREP for
determination of the crystal system and space group. SHELX SADABS was used to
produce an absorption corrected data set which took into account absorption of X-rays by
the crystal, although this was a symmetrically sized crystal (crystal dimensions listed in

45
Table 4). The direct methods program SHELX XS was used to solve the structure with
further refinement being performed in the SHELX XSHELL program
All non hydrogen atom positions were refined anisotropically. The hydrogen atom
positions were clearly visible using difference Fourier methods and were refined
isotropically.
Empirical formula C26H36N8O10F12P2Co
Chemical formula weight 969.50 g mol-1
Crystal system Triclinic
Space group P 1
Unit cell dimensions a = 7.1433(5) Ǻ α = 84.3130(10)°
b = 11.7421(9) Ǻ β = 84.2630(10)°
c = 11.8894(9) Ǻ γ = 72.3190(10)°
Unit cell volume 9421.90(12) Ǻ3
Z 1
Data collection temp 100K
Radiation MoKα, graphite monochromator
Diffractometer Bruker AXS Apex (3 circle)
Detector CCD area detector
Crystal size 0.50 x 0.50 x 0.20 mm
Tmin & Tmax 0.7328 & 0.8788
F (000) 493
Theta range 2.42 to 28.31°
Total reflections measured 8224
Independent reflections 4299
R indices [F2 > 2σ(F2)] 0.0313
R indices (F2) 0.0325
Largest diff. peak and hole 0.502 and -0.276 e.Å-3
Number of refined parameters 340
Table 4– A summary of the X-ray diffraction and crystal data for C26H36N8O10F12P2Co.

46
3.3 - Crystal structure analysis
It was found that the small molecule C26H36N8O10F12P2Co crystallises in triclinic space
group P 1 . The asymmetric unit contains half of the cobalt malonate anion
[Co(C3H2O4)2(H2O)2]- in addition to two protonated 2-amino pyridine cations (C5H7N2+)
and a PF6 counter ion (Figure 3.1). Like the previous structure the cobalt atom is located
on an inversion centre bonded to two oxygens on each of the equatorial malonates as well
as to a single oxygen on each of the trans axial waters.
Figure 3.1– An ORTEP diagram of C26H36N8O10F12P2Co with 50% ellipsoid
probability. Atoms labelled b are symmetry generated.
The equatorial Co-O bonds are 2.03275(10) Ǻ and 2.0554(10) Ǻ respectively whilst the
axial Co-O bonds are 2.1232(12) Ǻ . This gives a distorted octahedral environment with
malonate and water present as the primary ligands. The secondary ligands are 2-amino
pyridine and PF6. Again this environment in the crystal gave rise to an extensive
hydrogen bonding network comprising of nine unique hydrogen bonds, four of which
involve the PF6 counter ion (Table 5).
D-H…A D-H (Ǻ) H…A (Ǻ) D…A (Ǻ) D-H…A (º)
N(4)-H(1)…O(1) 0.83(2) 2.01(2) 2.8286(16) 167.1(18)

47
N(3)-H(2)...O(2) 0.85(2) 2.10(2) 2.9409(17) 175(2)
N(3)-H(1)...F(4) 0.89(2) 2.12(2) 2.9828(16) 162.6(18)
N(2)-H(1)...O(4) 0.84(2) 1.93(2) 2.7737(17) 176(2)
N(1)-H(2)...F(1) 0.83(2) 2.13(2) 2.9238(18) 161(2)
N(1)-H(1)...O(3) 0.86(2) 1.96(2) 2.8155(19) 171(2)
O(5)-H(2)...O(2) 0.80(3) 1.88(3) 2.6737(16) 172(2)
O(5)-H(1)...F(2) 0.78(2) 2.36(2) 3.0681(19) 152(2)
O(5)-H(1)...F3 0.78(2) 2.24(2) 2.9381(17) 149(2)
Table 5 – The hydrogen bonding details for structure C26H36N8O10F12P2Co.
Like the previous crystal structure the cobalt malonate anions are linked by a hydrogen
bond of length 1.88(3) Ǻ between a axial water and a equatorial carboxylate oxygen
(Figure 2.2). The cobalt malonate anions form one dimensional chains which run parallel
to each other. The 2-amino pyridine and PF6 ions form alternating layers between the
chains (Figure 3.2).
This arrangement is further stabilised by π-π interactions between 2-amino pyridine
cations. Each pair of 2-amino pyridines that is joined to a particular cobalt malonate
anion can interact but pairs belonging to different cobalt malonate anions are unable to
interact due to ring slippage (different pairs do not lie directly above each other and are
therefore unable to interact).
Figure 3.2– A figure to show the crystal packing arrangement of C26H36N8O10F12P2Co
. The 2-amino pyridine and PF6 molecules form alternating layers between the parallel
cobalt malonate chains.

48
3.4 - Crystal structure implications
The structure was solved with a low R factor of 0.031 (as described by Equation 6) and
was consistent with the expected composition.
The presence of the electron accepting fluorine atoms of the PF6 anion help in the
formation of an extensive hydrogen bonding network. Further stabilisation is provided
via π-π interactions between aromatic rings of the 2-amino pyridine cations. These
interactions help to minimise the free energy by decreasing the enthalpy and therefore
allowing crystallisation to occur.
3.5 - Comparison of the crystal structures
X-ray analysis revealed that the crystal structures are closely isomorphous. It was found
that both crystallised in triclinic P 1 space group with almost identical atomic
arrangements and unit cell dimensions.
Due to the similar atomic composition and arrangement both structures contained a
common hydrogen bonding motif composed of 2-amino pyridine and cobalt malonate
molecules (Figure 3.3) which accounted for five of the nine bonds present within the two
structures.

49
Figure 3.3 – A figure illustrating the common hydrogen bonding motif which is present
in both structures C26H36N8O18Cl2Co and C26H36N8O10F12P2Co .
Differences arise when the hydrogen bonding arrangements around the respective counter
ions are examined (Figure 3.4). In the crystal structure of C26H36N8O18Cl2Co the central
chorine of the perchlorate molecule is involved in hydrogen bonding. However in the
crystal structure of C26H36N8O10F12P2Co the central phosphorous of the counter ion is not
involved in the hydrogen bonding network. This may be expected due to the difference in
electro negativity between the two atoms (phosphorus has a value of 2.19 whilst chlorine
has a value of 3.16 on the Pauling scale of electro negativity).
In addition the respective counter ions form hydrogen bonds with different ions. As
illustrated the perchlorate counter ion forms hydrogen bonds with two 2-amino pyridine
cations and two hydrogen bonds with a malonate anion (the chlorine to malonate
interaction is not pictured).
In comparison the PF6 counter ion forms hydrogen bonds with three 2-amino pyridine
cations and only one hydrogen bond with the cobalt malonate anion.
The lengths of the hydrogen bonds between the two structures are comparable except for
the case involving the chlorine atom of the perchlorate. This bond is anomalously long in
comparison to the others at 2.93(3) Ǻ with the remaining hydrogen bonds in the two

50
complexes all with hydrogen to acceptor distances in the range of 1.78 Ǻ – 2.36 Ǻ. It is
unlikely that the electronegativity fully accounts for this anomaly as the fluorine atoms
form shorter hydrogen bonds (in the range of 2.12 – 2.36 Ǻ) even though they possess a
greater electronegativity.
Figure 3.4 – A figure to illustrate the differences in the hydrogen bonding
arrangements around the perchlorate and PF6 counter ions. A fourth interaction not
shown is present between the chlorine of the perchlorate counter ion and an oxygen of
the axial water.
The equatorial Co-O bond lengths of the two complexes reported here (2.0335(12) Ǻ,
2.03275(12) Ǻ, 2.03275(10) Ǻ, 2.0554(10) Ǻ) are consistent with Co-O bond lengths
observed in previously reported cobalt malonate complexes18 (2.034(1) Ǻ, 2.063(1) Ǻ)
synthesised by the group of Professor Subrata Mukhapadhyay.
In addition the type of intermolecular interactions seen here are known as previously
reported examples of transition metal, malonate complexes with 2-amino pyridine have
displayed the same hydrogen bonding motif as described here (Figure 12). This molecular
recognition phenomenon appears to be responsible for driving the crystal packing

51
arrangement in the solid state. Formation of this motif apparently requires the presence of
2-amino pyridine. Previous work by the by the group of Professor Subrata
Mukhapadhyay found no such motif was formed when 4-amino pyridine was used in
place of 2-amino pyridine18.
The hydrogen bond lengths reported here are consistent with those previously reported
cobalt malonate complexes. However differences arise due to the presence of the
perchlorate and PF6 counter ions. It appears that introducing varying counter ions into the
complex can subtly alter the hydrogen bonding arrangement and therefore the crystal
packing arrangement. This may hold useful implications for crystal engineering where
the tight control of intermolecular interactions is essential.

52
Chapter 4
Structure Determination of two Small Molecules – C30H24NO4Sn &
C24H20Sn (SnPh4)
4.1 - Introduction to C30H24NO4Sn & C24H20Sn (SnPh4)
A set of two tin containing, crystalline compounds were submitted to the university by a
Pakistani research group for structure determination. The two compounds were expected
to be closely related. The expected chemical structures were determined by synthetic
chemists with the predicted structures shown in Figures 4.1 & 4.2.
Sn OO
NH
CF3
Figure 4.1 – The expected chemical structure of the molecule in the crystal MHB7.
Sn OO
NH
OCH3
Figure 4.2 – The expected chemical structure of the molecule in the crystal MHB8.
Sample MHB7 consisted of colourless, needle shaped crystals which were extremely thin
(around 0.08mm across). In contrast sample MHB8 consisted of colourless, block shaped
crystals with fairly symmetric dimensions. Both samples appeared to be of good quality
when viewed under a microscope and both extinguished well under crossed polarisers.

53
4.2 – X-ray diffraction data collection and processing procedure for
C24H20Sn (SnPh4)
Sample MHB7 was the first to be analysed. A single needle shaped crystal measuring
approximately 0.30 x 0.08 x 0.08mm was selected and immersed in a viscous oil. The
crystal was fitted on a loop, which was then placed onto a goniometer head and fitted
onto the diffractometer. The data collection was performed at a cryogenic temperature
(100K), which caused freezing of the viscous oil and thus fixation of the crystal as well as
being beneficial to reduce atomic displacement parameters. The crystal was centred using
rotating screws on the goniometer head and a video camera as a visual aide. Firstly the X-
axis was adjusted at phi 0˚ to ensure the crystal was centred. Once complete the screw
was rotated to phi 180˚ and the crystal centred again. This process was repeated in the
same manner for the Y-axis but with phi angles 90˚ and 270˚. Once the crystal was
completely centred it was irradiated with monochromated MoKα radiation. The X-ray
generator settings were 40kV and 40 mA.
After a sufficient number of frames had been collected using the SAINT15 computer
program the unit cell dimensions were determined (using the SMART15 computer
program) whilst data collection was still continuing. It was found that the dimensions of
the unit cell were 12.0079(14) x 12.0079(14) x 6.3934(16)Ǻ with angles 90 x 90 x 90˚.
These dimensions correspond to a tetragonal crystal system, which is fairly unusual for
small molecules (most small molecules crystallise in monoclinic or triclinic). A search of
the unit cell dimensions and space group of the Cambridge Crystallographic Data Centre
(CCDC) 19 using CONQUEST20 software revealed the dimensions to correspond to
SnPh4. This compound was used as a starting material in the synthesis and typically
forms needle shaped crystals.
Empirical formula C24H20Sn
Chemical formula weight 427.09 g mol-1
Crystal system Tetragonal

54
Space group P 4 21/c
Unit cell dimensions a = 12.0079(14) Ǻ α = 90.00 ° b = 12.0079(14) Ȧ β = 90.00 ° c = 6.3934(16) Ȧ γ = 90.00 °
Unit cell volume 921.9(3) Ǻ 3
Z 2
Data collection temp 100 K
Radiation MoKα, graphite monochromator
Diffractometer Bruker AXS Apex (3 circle)
Detector CCD area detector
Crystal size 0.30 x 0.08 x 0.08 mm
Tmin & Tmax 0.6808 & 0.8971
F (000) 428
Theta range 2.40 to 26.35°
Total reflections measured 4976
Independent reflections 684 (Rint = 0.0205)
R indices [F2 > 2σ(F2)] 0.0353
R indices (F2) 0.0386
Largest diff. peak and hole 0.564 and -0.365 e.Ǻ-3
Number of refined parameters 77
Table 6 – Summary of the X-ray diffraction data and refinement for C24H20Sn. The computer program SAINT was used to collect and integrate the CCD frame images
in order to produce integrated intensities. This produced files with filename extensions
.p4p and .RAW. These files were then introduced into SHELX16 XPREP for
determination of the crystal system and space group. As the crystal contained a heavy tin
atom X-ray absorption corrections were applied using SHELX SADABS. These
corrections were also required because the crystal had quite unsymmetrical dimensions
(crystal dimensions listed in Table 6). The direct methods program SHELX XS was used
to solve the structure with further refinement being performed in the SHELX XSHELL
program.

55
All non hydrogen atom positions were refined anisotropically. The hydrogen atom
positions were clearly visible using difference Fourier methods and were refined
isotropically.
4.3 - X-ray diffraction data collection and processing procedure for
C30H24NO4Sn
Sample MHB8 was subsequently analysed. A single colourless, block shaped crystal
measuring approximately 0.40 x 0.30 x 0.30mm was selected and mounted as described
for the previous crystal (Chapter 4.2). Once the crystal was completely centred it was
irradiated with monochromated MoKα radiation. The X-ray generator settings were 40kV
and 40 mA.
The computer program SAINT15 was used to collect and integrate the CCD frame images
in order to produce integrated intensities. This produced files with filename extensions
.p4p and .RAW. These files were then introduced into SHELX16XPREP for
determination of the crystal system and space group. Although this was a symmetrical
sized crystal (crystal dimensions listed in Table 7) the presence of a heavy tin atom
required the application of X-ray absorption corrections to the data set, which was done
using SHELX SADABS
The direct methods program SHELX XS was used to solve the structure with further
refinement being performed in the SHELX XSHELL program.
All non hydrogen atom positions were refined anisotropically. The hydrogen atom
positions were clearly visible using difference Fourier methods and were refined
isotropically.
Empirical formula C30H24NO4Sn
Chemical formula weight 581.19 g mol-1
Crystal system Triclinic
Space group P 1
Unit cell dimensions a = 9.7556(5) Ǻ α = 73.1870(10) °

56
b = 11.3298(6) Ǻ β = 87.0820(10) ° c =12.0571(6) Ǻ γ = 79.8410(10) °
Unit cell volume 1255.69(11) Ǻ 3
Z 2
Data collection temp 100K
Radiation MoKα, graphite monochromator
Diffractometer Bruker AXS Apex (3 circle)
Detector CCD area detector
Crystal size 0.40 x 0.30 x 0.30 mm
Tmin & Tmax 0.6916 & 0.9023
F (000) 586
Theta range 1.91 to 26.35°
Total reflections measured 10025
Independent reflections 5034 (Rint = 0.0236)
R indices [F2 > 2σ(F2)] 0.0245
R indices (F2) 0.0250
Largest diff. peak and hole 0.938 and -0.516 e.Ǻ-3
Number of refined parameters 421
Table 7 – Summary of the X-ray diffraction data and refinement for C30H24NO4Sn.
4.4 - Crystal structure analysis of C24H20Sn (SnPh4) It was found that the small molecule SnPh4 crystallises in the tetragonal space group P 4
21/c. The asymmetric unit consists of the central tin atom bonded to a single phenyl
(C6H6) ring. The central tin atom is present in a tetrahedral coordination environment
with four Sn-C bonds of length 2.148(5) Ǻ (Figure 4.3).

57
Figure 4.3 – An ORTEP diagram of C24H20Sn with 50% ellipsoid probability. Atoms
labelled a,b or c are symmetry generated.
Using both the PLATON21 and SHELX computer programs it was found that no classical
hydrogen bonds were present within the crystal structure. This is due to the complete
absence of both suitable hydrogen bond donors and acceptors. There is however the
presence of weak van der Waals interactions (between carbon and hydrogen atoms) with
each SnPh4 molecule forming eight weak interactions (Figure 4.4) with eight other
neighbouring SnPh4 molecules.

58
Figure 4.4 – A figure to show the location of eight weak H…C-H interactions that each
C24H20Sn molecule forms.
It is found that the aromatic phenyl rings do not lie above each other in the crystal
packing arrangement. The adopted arrangement prevents the aromatic phenyl rings
interacting with each other via π-π stacking (Figure 4.5).
Figure 4.5 – A figure to show the stacking of the C24H20Sn molecule within the crystal.
Such an arrangement prevents the formation of π-π interactions.

59
Instead the molecules appear to form layers that are stabilised by the weak H…C-H
interactions running between them (Figure 4.6).
Figure 4.6 – A figure to show the stacking of layers of C24H20Sn molecules stabilised by
weak van der Waals interactions.
4.5 - Crystal structure analysis of C30H24NO4Sn
It was found that the small molecule C30H24NO4Sn crystallises in the triclinic space group
P 1 . The asymmetric unit contains two molecules of C30H24NO4Sn, which are joined to
each other via a O-Sn bond.

60
Figure 4.7 – An ORTEP diagram of C30H24NO4Sn with 50% ellipsoid probability.
Atoms labelled a or b are symmetry generated.
Using PLATON21 to analyse the crystal structure the molecules are shown to adopt a
polymeric structure (Figure 4.7) with the monomeric units linked by an unusually long O-
Sn bond of length 2.6534(15) Ǻ. In this arrangement the central tin atom is involved in a
trigonal bipyrimidal coordination environment with the remaining four bonds (to the
three phenyl groups and to an oxygen) possessing more reasonable lengths of 2.113(2),
2.117(2), 2.119(2) and 2.1152(15) Ǻ (with the final value corresponding to the Sn-O
bond).
The polymeric chains form layers which run parallel to each other. This allows the bulky
triphenyl groups to be arranged so as to minimise the steric clashes between the group
present on adjacent molecules (Figure 4.8).

61
Figure 4.8 – A figure to show the arrangement of the polymeric chains in
C30H24NO4Sn with weak van der Waals interactions shown as blue lines. Hydrogen
atoms are omitted for clarity.
It is found that the aromatic phenyl groups are arranged in a fashion which prevented the
formation of π-π stacking. Although Figure 4.8 appears to show aromatic phenyl rings
stacked above each other the distances involved (around 20Ǻ) are obviously far too great
for any significant interaction to occur (Figure 4.9).
Figure 4.9 – A figure to show the distance between aromatic phenyl rings in
C30H24NO4Sn. Hydrogen atoms are omitted for clarity.

62
Instead stabilisation of the crystal packing appears to be entirely dependent upon weak
van der Waals type interactions with each monomeric unit able to form twelve of these
weak interactions.
Using both the SHLEX and PLATON computer programs it was found that no hydrogen
bonds were present in the crystal packing. This is due to the absence of suitable hydrogen
bond donors although suitable hydrogen bond acceptors are present in the form of
carboxylate groups. However, the presence of an intermolecular hydrogen bond within
the monomeric units is observed (Figure 4.10). This interaction is around 2.700 Ǻ long
and is between a carboxylate group and the nitrogen atom.
Figure 4.10 – A figure to show the intramolecular hydrogen bond present within the
monomeric units.

63
An interesting observation is that SHELX does not detect the presence of the unusually
long O-Sn bond. As a result molecules are displayed as discrete units with the central tin
atom present in a tetrahedral environment (Figure 4.11). This difference arises because
the SHELX and PLATON computer programs use different values for the atomic radius
of the tin atom, which affects the bonds displayed.
Figure 4.11 – A figure to show how SHELX views the molecules as discrete units and
not as a polymeric structure.
4.6 - Crystal structure implications of C24H20Sn (SnPh4)
The structure was solved with a low R factor of 0.0353 (as described by Equation 6)
which indicates a good quality structure.
The complete absence of hydrogen bond donors and acceptors precluded the formation of
hydrogen bonds whilst the arrangement of the aromatic phenyl rings was not favourable
to allow for the formation of π-π interactions. The presence of weak van der Waals type
interactions was detected (of distance 3.062 Ǻ) and appears to be the major force in the
stabilisation of the crystal packing.
As the crystals provided consisted of starting material the obvious major implication is
that the attempted chemical synthesis was unsuccessful. As a result the synthesis must be
modified and improved upon if the desired product is to be obtained. This information
has been fed back to the synthetic chemist concerned.

64
A search of the CCDC revealed that C24H20Sn was a previously determined structure
which was first reported in 1970 by Chieh and Trotter22. This original structure was
obtained using a CuKα source and the measurement of 366 independent reflections. The
structure was solved using a Patterson synthesis. A comparison of the results between the
original structure and the structure presented in this thesis reveal a favourable agreement.
The same space group (P 4 21/c) was assigned in both cases as well as almost identical
unit cell dimensions (listed in Table 8).
Original 1970 structure
(Chieh & Trotter22)
Structure reported in this
thesis
Unit cell dimensions a = 12.058(1) Ǻ α = 90.00°
b = 12.058(1) Ǻ β = 90.00°
c = 6.568(1) Ǻ γ = 90.00°
a = 12.0079(14) Ǻ α = 90.00°
b = 12.0079(14) Ǻ β = 90.00°
c =6.3934(16) Ǻ γ = 90.00°
R indices [F2 > 2σ(F2)] 0.078 0.0309
Independent reflections 366 3548
Length of Sn-C bond 2.14 Ǻ 2.148(5) Ǻ
Table 8 – The differences between the original 1970 SnPh4 structure and the structure
determined in this thesis.
The R factor of the two structures differs with the structure reported in this thesis
possessing a significantly lower R factor. This improvement is likely to be a consequence
of improvements in instrumentation as well as the substantially increased number of
reflections measured for the structure reported in this thesis.
4.7 - Crystal structure implications of C30H24NO4Sn
The presence of the heavy tin atom and the relatively high number of reflections allowed
the structure to be solved with a low R factor of 0.0245 (as described by Equation 6).

65
No classical hydrogen bonds were present in the crystal packing arrangement. It is likely
that the presence of an intramolecular hydrogen bond is not relevant to the crystal
packing arrangement.
Although the expected chemical structure and crystallographically three dimensional
structure are similar there are clear differences (Figure 4.12). The crystallographically
determined structure contains an extra two C-H units in addition to an extra carboxylate
unit. These differences imply that the attempted chemical synthesis was unsuccessful and
therefore requires modification if the desired product is to be obtained, and which has
been relayed to the synthetic chemist concerned.
Sn OO
H H ON
OCH3
Figure 4.12– The crystallographically determined structure has this chemical diagram
with the highlighted area corresponding to the deviation from the expected chemical
structure.
The monomer units are linked by a O-Sn bond measuring 2.6534(15) Ǻ. This is an
unusually long bond with regular tin to oxygen bonds expected to be around 2.10 Ǻ. A
search of the CCDC19 using CONQUEST20 software revealed 198 previously reported
structures contained a O-Sn bond of at least 2.65 Ǻ with bond lengths of up to 2.9 Ǻ
being reported. However none of these reported crystal structures possessed a significant
structural similarity to the structure of C30H24NO4Sn.

66
Part B – Macromolecular X-ray crystallography

67
Chapter 5
Macromolecular X-ray Crystallography
5.1 - Introduction
The principles of small molecule X-ray crystallography (as detailed in chapter one) are
equally applicable to the study of macromolecules such as proteins and viruses. However,
there are differences in procedure for the crystal structure determination and refinement.
The field of macromolecular X-ray crystallography is still relatively new with the first
protein crystal structures (those of myoglobin and haemoglobin) being solved in 1958 by
Kendrew and Perutz respectively. However thanks to advances in instrumentation as well
as technique development larger and more complex structures are now routinely studied.
Foremost amongst these developments is the advent of synchrotron radiation which
allows for the production of much more intense, well collimated and finely tuneable X-
rays. These properties are important as proteins consists of light, poorly scattering
elements primarily carbon, nitrogen and hydrogen as well as containing a high solvent
content. In addition methods of solving the phase problem specifically for use in
macromolecular crystallography have been developed.
Recently the 2009 Nobel Prize in chemistry was awarded to Ramakrishnan, Steitz and
Yonath for “Studies of the structure and function of the ribosome”23. This involved the
determination of a bacterial 70S ribosome consisting of two subunits with molecular
weights 800,000 and 1,500,000 using synchrotron X-ray crystallography. Obtaining high
resolution three dimensional structures of such macromolecules is important as it helps to
elucidate the mechanisms by which they operate.
A dedicated repository for the three dimensional structure of biological macromolecules
now exists24 and is free to access (available at www.pdb.org). As of 13/07/10 there were
66,324 structures deposited within the Protein Data Bank (PDB). The vast majority of
these being solved via X-ray crystallography (approx 57,000)

68
Although the number of structures solved by X-ray crystallography is growing annually
at an exponential rate problems are still prevalent. Foremost is the issue of growing
suitably sized diffraction grade single crystals. As the measured resolution is dependent
upon the crystal quality, well ordered crystals must be obtained if accurate structures are
to be obtained.
5.2.1 - Macromolecular crystallisation techniques
Small molecule crystallisation is a relatively simple process in comparison to
macromolecular crystallisation. This is because macromolecular crystallisation involves a
larger number of complicated interactions and is still poorly understood. Obtaining
diffraction grade, single crystals is a notorious ‘bottleneck’ and is often the rate limiting
step in the crystal structure determination. For example the pH at which crystallisation is
attempted will affect the net charge of a protein (via the protonation states of titratable
side chains). The resulting net charge will affect the solubility of the protein and therefore
the crystallisation process. A wide range of experimental conditions must be considered
and tailored, which may involve many attempts to perfect. The techniques used to induce
macromolecular crystallisation can be divided into four broad categories25. All four
methods involve different steps that crystal growth passes through26(Figure 5.1) such as
super saturation (which involves the formation of nuclei) and nucleation (which leads to
the formation of larger crystals.
Figure 5.1 – A phase diagram for crystal growth26.

69
5.2.2 – The batch method
This is the simplest method of crystallisation and is most useful when the conditions of
crystallisation have been narrowed down to a small range. A number of small glass vials
containing a protein and a precipitant (which is present at a level slightly less than at
which the protein precipitates) are prepared. The level of the protein and the precipitant is
varied between the vials therefore allowing the effect that the different concentrations
have on the crystallisation process to be observed. Usually, only microlitre volumes are
required. For example in the research behind this thesis crystallisations of 1ml and 2ml
were set up using this method.
5.2.3 - Dialysis
Dialysis uses a semi permeable membrane the pore sizes of which permit the passage of
solvent and small molecules. However as macromolecules are significantly larger they
are unable to pass through the pores. The macromolecule is slowly brought towards
supersaturation and its precipitation point by dialysis against a concentration of a
precipitating agent. It is also possible to induce crystallisation by altering the pH
(achieved by altering the concentration of the buffer). Like the batch method dialysis can
be performed on a bulk or a microlitre scale.
5.2.4 - Vapor diffusion methods
There are a number of variations of this method. Examples include the sitting drop,
hanging drop (Figure 1.11) and sandwich drop methods.
The basic idea behind them is that a small amount of the macromolecule is mixed with a
small amount of a precipitating agent. This drop is then allowed to equilibrate with a
reservoir of the precipitating agent contained within a closed system. An equilibrium will
form which will result in the water present in the sample diffusing out. Conversely the
concentration of the protein will increase, eventually reaching the supersaturation and
precipitation points.

70
5.2.5 - Hot box technique
This technique uses a temperature gradient to induce crystallisation. In this technique the
protein is dissolved at a low ionic strength in a test tube. The test tube is then suspended
in a thermos at high temperature (around 60°)27. The high temperature seeks to render the
protein more soluble and, as it slowly cools, a supersaturation point should be reached.
5.3.1 - Solving the phase problem in macromolecular crystallography
Owing to the increased complexity of proteins with respect to small molecules different
methods have been developed to solve the phase problem in macromolecular X-ray
crystallography.
A conventional Patterson synthesis cannot be performed as proteins often contain no
heavy atoms. In addition even small proteins contain a large number of atoms which
would lead to an uninterpretable Patterson map. For example lysozyme contains 2303
(non hydrogen) atoms, which would correspond to 5,303,809 vectors between the atoms!.
This results in far too many peaks for meaningful information to be extracted from the
Patterson map.
Direct methods are not applied to macromolecules as they require a relatively low
number of reflections for the computations to be effective. The large number of
reflections from a protein crystal would require an unrealistic amount of calculation to
retrieve the phases, for the computations to be effective.
As a result phase retrieval methods specific to macromolecular crystallography have been
developed, which are described below.
5.3.2 - Isomorphous replacement method
The isomorphous replacement method is based on the variation in intensities of the
diffraction spots belonging to two or more isomorphous crystals. Crystals can be
described as isomorphous if they have the same space group and almost identical unit cell
dimensions and atomic arrangements.

71
One of the crystals must be the native protein with one or more derivatives containing at
least one heavy atom. The heavy atom can be introduced by soaking a pre formed crystal
in a solution containing the heavy atom. Alternatively the heavy atom may be introduced
via a co-crystallisation.
An essential condition is that the binding of the heavy atom must not result in a
significant conformational change of the protein. Usually this is not a problem as the
heavy atom will bind at specific sites within a protein. As a result only the local area
where the metal binds will be disturbed. As proteins are such large structures the overall
conformation of the native protein and its derivatives is largely identical allowing for
effective computational comparisons to be made.
The light atoms, which constituent a protein, scatter with different phases and essentially
cancel. In contrast a heavy atom contains a large number of electrons concentrated within
a small sphere (the atomic radius). As a result these electrons scatter in phase relative to
each other.
As the diffraction pattern is composed of contributions from all atoms in the unit cell the
addition of even a single heavy atom results therefore in a change in the intensities of the
spots.
Under isomorphous conditions the difference in intensities between the native and its
heavy atom derivatives can be attributed solely to the contribution of the heavy atom
present within the derivative, expressed as vector structure factor amplitudes (Equation 9
& Figure 5.2).
FPH = FP + FH
Equation 9 – The basic principle of the isomorphous replacement method.
Where –
FPH = Vector representing the structure factor
amplitude of the heavy atom derivative
FP = Vector representing the structure factor
amplitude of the native protein
FH= Vector representing the structure factor
amplitude of the heavy atom

72
Figure 5.2 – A vectorial representation of the isomorphous replacement method
The differences in structure factor amplitudes can be used to calculate a difference
Patterson map which will consist of just the vectors between the heavy atoms. Combining
the difference map with the crystal symmetry should allow the heavy atom positions to be
determined. Once the heavy atom positions are known their contributions to the structure
factors can be determined.
The phase angles may be determined graphically by considering structure factors as
vectors. The vectors possess a length equal to the amplitude of the structure factor and a
direction corresponding to the estimated phase.
If only one derivative is considered and taking the assumption that FPH = FP + FH and that
FH can be calculated then there are two possible values that the phase may possess
If a second derivative is prepared there are again two possible values for the phase.
However only one value will be consistent with the first case thereby providing one
possible solution for the phase angle
A graphical representation of the phase angle calculation is known as a Harker
construction. The case when only one derivative is available will be considered first. In
this method a circle with a radius corresponding to the amplitude of the native protein
structure factor (FP) is drawn centred on the origin. A line corresponding to the position
and phase angle of the heavy atom (calculated from the difference Patterson map and
crystal symmetry) is then drawn. A second circle corresponding to this heavy atom
derivative is drawn (FPH) with the origin cantered on the end of the heavy atom line. The
FP
FPH
FH αH

73
two circles will intersect at two points giving two possible values for the phase angle
(Figure 5.3).
Figure 5.3 – A Harker construction for a native protein and a heavy atom derivative.
The two possible phase angles are labelled as α (P1) and α (P2).
If a second heavy atom derivative is available the above process is repeated and extended
to include the second derivative. This will result in three circles with only one point
where all three circles intersect. This point of intersection corresponds to the value of the
phase angle (Figure 5.4).
α P(2)
α P(1) Circle FP with origin as centre and radius corresponding to amplitude.
Circle FPH with FH as origin and radius corresponding to amplitude.
FH
90º
0º

74
Figure 5.4 - A Harker construction for a native protein and two heavy atom derivatives.
The one possible phase angle is labelled as α (P1).
An inherent disadvantage of the method is the basically unavoidable introduction of some
level of errors through non-isomorphism. This is because crystals will never be
completely isomorphous. Therefore performing and comparing measurements of multiple
crystals will lead to the introduction of some level of errors. The effect of some errors
will progressively affect the higher resolution X-ray diffraction data and electron density
map details will become blurred.
5.3.3 - Anomalous scattering
Anomalous scattering is also dependent upon the presence of a heavy atom within a
protein. Conventional X-ray diffraction is a result of coherent scattering whereby the
incident X-rays cause electrons to vibrate. This effect generates radiation of a frequency
equal to the frequency of the incident radiation. However in the case of anomalous
scattering this is no longer true.
To enhance this effect the wavelength of the incident X-rays is tuned to correspond to an
absorption edge of a heavy atom present within the protein. An absorption edge involves
a small energy range and corresponds to an atomic transition, and which promotes the
α P(1) Circle (blue) FP with origin as centre and radius corresponding to amplitude.
Circle (red) FPH with FH as centre and radius corresponding to amplitude.
FH
90º
0º Circle (black) FPH2 with FH2 as centre and radius corresponding to amplitude.
FH2

75
heavy atom to an electronically excited state. This effect may involve the simple
promotion of a core electron to an unoccupied higher energy level or the complete
ejection of the electron from the atom (ionisation). This promotion or ejection of an
electron will alter the phase of the scattered radiation with respect to the scattering from
the light atoms. The effect of this phase change is equivalent to altering the path length of
the scattered radiation. This results in a change in intensities of the diffraction spots. The
increase in the anomalous scattering is coupled with a decrease in the coherent scattering.
This is because a proportion of the energy of the incident X-rays is used to create
transitions within the heavy atom.
An important use of anomalous scattering is in the determination of absolute
configurations. This can be achieved because anomalous scattering leads to a violation of
a condition known as Friedel`s law (which is assumed in what might be called
conventional X-ray crystallography). Friedel`s law states that a pair of symmetry related
reflections will have the same intensity and phases of equal magnitude but opposite in
sign (i.e. one will be positive the other negative). However at wavelengths close to an
absorption edge this condition is violated. This is because the heavy atoms will behave in
a different manner to the light atoms (in terms of how the phases are effected by the
scattering). This can be observed in the diffraction pattern as the intensities of the two
symmetry related spots being different e.g. F(hkl) will no longer equal F(-h,-k,-l).
The phases can be calculated graphically in the same way as in isomorphous replacement
using either a Harker construction or a vectorial representation (shown by Figures 5.2,
5.3, 5.4).
The expected structure factors for a pair of enantiomers can then be calculated and
compared with the observed structure factors which should then allow for assignment of a
specific enantiomer.
Anomalous scattering is now often the method of choice for solving the phase problem in
macromolecular X-ray crystallography. It is considered a more accurate method than
isomorphous replacement as it involves performing measurements on only one crystal as
opposed to two or three. This means that there are no errors introduced through non-
isomorphism. Isomorphous replacement can also be used in conjunction with anomalous
scattering resulting in an overall powerful method of phase determination.

76
5.3.4 - Molecular replacement28
The previous two methods of phase retrieval are required when the structure under study
is completely unknown. In contrast molecular replacement can be used when a suitably
related structure has been previously reported (known as the model). For example if a
model of oxyhaemoglobin is available it would assist in elucidating the structure of
deoxyhaemoglobin. As a rough guide a model can be considered suitable if the amino
acid sequence is greater than 30% identical to that of the structure under study. In the
case of oxyhaemoglobin and deoxyhaemoglobin this is obviously true (being 100%) but
for other examples it may be unclear as the success of the method is not guaranteed.
If this is the case the sequence identities may have to be determined.
If a suitable model is available then the phases from the model are used as initial
approximations for the phases of the structure under study. For this to be done the model
must firstly be correctly orientated and positioned in the unit cell of the structure under
study. This is done by systematically comparing predicted and observed structure factors
and thereby finding the orientation and position where there is an optimum correlation.
The orientation and positioning involves six positional parameters (three rotational angles
and three translational parameters). Performing calculations using all six parameters at
once presents an extremely large problem. This is because for N atoms in the asymmetric
unit there will be 6N parameters required to describe the solution. However Patterson
functions can be used, which allows the rotational and translational parameters to be
separated thereby simplifying the calculation convergence. Likelihood based methods
may also be used and are increasingly used in place of Patterson functions. These use
statistical methods in reciprocal space and can be divided into rotational and translational
functions in the same manner as for the Patterson methods.
As a result many molecular replacement programs choose a relatively small number of
good quality solutions provided by the rotational parameters and test these using the
translational parameters to finally provide a solution.
The rotational Patterson function involves calculating the Patterson map of the model and
rotating it over the observed Patterson map. The most probable orientation is found when
there is a close agreement between the two maps.

77
The translational Patterson function involves placing the centre of the model at all
positions in the unit cell of the structure under study. For each position attempted the
Patterson map can be calculated and compared to the observed Patterson map of the
structure under study. Where the two will agree yields the most likely position.
Once the correct orientation and position has been identified an electron density map of
the structure under study can be calculated. This electron density map is calculated using
the measured X-ray diffraction structure factor amplitudes from the structure under study,
and the estimated phases obtained form the model (that is correctly positioned and
orientated in the unit cell).
The difference map can be calculated, which will include areas of negative density
(corresponding to areas which are present in the model but do not fit the real density) and
areas of positive density (corresponding to areas which are not included in the model but
are present in the structure under study).
The molecular replacement method is increasingly popular as it is relatively quick to
perform. A high degree of automation is also involved with the rotation and translation
function computer programs now available. An example of a popular molecular
replacement computer program is PHASER.
5.4 – Rigid body and restrained refinement
Rigid body refinement is used as a first step in the refinement process for the
macromolecular adducts studied in this thesis. In rigid body refinement the distances
between the constituent atoms of a protein are fixed. In the simplest possible case this
means that the entire protein is treated as one large, rigid molecule. Alternatively the
protein may be divided into a small number of subunits (e.g. beta sheets or alpha helices).
The rigid blocks of the structure are then placed to match the experimentally determined
electron density.
In restrained refinement the bond lengths and angles of the protein are allowed a certain
degree of freedom. This means that the bond lengths and angles are allowed to vary
within a small range but not but a large amount i.e. they are restrained to within a certain
range. Although bond lengths and angles are perhaps the most important restraint used, a

78
large number of other restraints are possible (such as forcing peptide bonds to adopt a
planar conformation).
The aim is the same as small molecule refinement refinement, which is the optimal
correlation between the observed and calculated structure factor amplitudes.
5.5 - The R free factor
In addition to the conventional R factor detailed in Chapter one macromolecular
crystallography uses another criterion to describe the correctness of a structure. The
additional criterion is known as the R free factor and is calculated using the same
equation as the conventional R factor (Equation 6). However the R free factor is instead
calculated exclusively from a small percentage of reflections (typically around 5%) which
are excluded from the refinement process. This avoids using the same data to perform
refinement as well as measuring the correctness of the structure.
The R free factor is normally higher than the conventional R factor although both should
possess values relatively close together. A difference of up to 6% is usually tolerated.
In addition to monitoring the progress of the model refinement the R free factor is used to
validate that the conventional R factor is not being artificially lowered by the addition of
an increased number of parameters.

79
Chapter 6
Crystal structure determination and model refinement of a co-
crystallisation of HEWL and TA6Br12
6.1.1 - Introduction
This chapter details the data collection procedure and subsequent model refinement of a
co-crystallisation of hen egg white lysozyme (HEWL) and Ta6Br12. The motivation for
conducting this work lies in facilitating technique development. It is hoped that the
crystal structure determination will lead to a well resolved structure which is solved to a
satisfactory resolution. This model structure can then be compared to models obtained
using data gathered from newly developed methods. Ideally this will allow for the
accuracy of methods to be accessed and weaknesses identified and improved upon. The
advent of the free electron laser has catalysed technique development in a number of
areas. These include protein powder diffraction and the possibility of using nanoclusters
or even single molecules as opposed to crystals. Developments such as these which
remove the need for crystals (which are often difficult or sometimes impossible to obtain)
could yield new possibilities.
6.1.2 - Introduction to lysozyme
Lysozyme is an enzyme that catalyses the cleaving of polysaccharide chains present in
the cell walls of bacteria29, 30. This has the effect of causing the cell wall to rupture.
Without the rigidity supplied by the cell wall the bacteria burst as a result of intolerable
osmotic pressure. As a result of this antibacterial function lysozyme is often termed as a
natural antibiotic. It is commonly found in tears, saliva and in hen egg white (the form
used in this thesis).
Lysozyme is a commonly used test enzyme within crystallography for a variety of
reasons. It is cheap and easy to obtain. Its structure has been previously well studied

80
(which allows for molecular replacement) and it is relatively small in size (129 amino
acids). In addition it crystallises easily in a wide range of experimental conditions.
It is hoped that if a molecule binds in a particular site in lysozyme then this may act as a
model for a more complicated enzyme.
6.1.3 - Introduction to Ta6Br12
Ta6Br12 is a cluster used in the heavy atom derivatisation of macromolecules. Examples
present in the literature detail how the cluster has been used for phase determination
involving macromolecular structures31, 32. For example a paper by Szczepanowski et al33
published in 2005 details how crystals of mouse ubiquitin activating enzyme were soaked
in a solution containing the Ta6Br12 cluster. This produced promising heavy atom
derivatives that were used in a multiple anomalous scattering experiment using
synchrotron X-ray radiation.
The cluster is used because it contains two anomalous scatters with the tantalum L-ІІІ
edge at 1.2548 Ǻ and the bromine K edge at 0.9202 Ǻ34. In addition the cluster can also
be used to produce derivatives for use in the isomorphous replacement method.
The cluster is highly symmetrical consisting of six tantalum atoms in an octahedral
environment with twelve bridging bromine atoms located along the twelve edges of the
tantalum octahedron (Figure 6.1). Each tantalum is bonded to four other tantalum atoms
with a Ta-Ta bond length of 2.898 Ǻ. In addition each tantalum is bonded to four
bromine atoms with a Ta-Br bond length of 2.604 Ǻ.
Figure 6.1 – A figure of the crystallographically determined structure of the Ta6Br12
cluster with bromines in yellow and tantalums in purple. .

81
The Ta6Br12 was supplied by Jena Bioscience in the form of a fine green powder.
The cluster has a +2 charge which is countered by two bromine ions in the preparation
supplied. It is a possibility that the positive charge on the cluster will cause it to bind to
side chains in the protein which possess negative charges (a Coulombic interaction).
The binding of Ta6Br12 to lysozyme was first investigated using single crystal X-ray
crystallography by Corey et al in 196235. However due to the technological restrictions of
the time no three dimensional structure was produced. It is hoped that the subsequently
vast improvements in instrumentation and technique development will enable the three
dimensional structure to be determined.
6.2 – Co-crystallisation procedure of HEWL and Ta6Br12
The crystals of hen egg white lysozyme and Ta6Br12 were grown using a batch method
co-crystallisation (adopted from Blundell and Johnson25).
A sodium acetate buffer was used to regulate the pH. This was prepared by dissolving
0.54g of sodium acetate trihydrate (CH3COONa.3H2O) in 50ml of distilled water in a
volumetric flask. Once all solids had dissolved 229µl of acetic acid (CH3COOH) was
added to the solution which was then stirred for five minutes. The volume of the solution
was then accurately increased to 100ml. The resulting solution was pH 4.7 with an
acetate concentration of 0.04M.
The precipitating agent used was a 10% salt solution. This was prepared by adding 10g
of salt (NaCl) to a volumetric flask. Distilled water was then added to accurately bring
the volume to 100ml.
For the co-crystallisation 0.04M acetate buffer (1ml) was added to lysozyme (50mg) in a
small glass vial. The solution was stirred for 5 minutes to ensure the lysozyme powder
had fully dissolved. At this point one aliquot (1mg) of Ta6Br12 was added. Stirring the
solution for five minutes with the end of a Finn pipette was essential to ensure the Ta6Br12
had fully dissolved. The solution was now pale green in colour. Finally, 10% salt solution
(1ml ) was added over a five minute period to help induce crystallisation. The final

82
solution was stirred for five minutes. The vial was then left in an undisturbed position at
room temperature.
After three days it was found that a large number of single crystals were present on the
bottom of the vial. The crystals appeared to be of good quality with no visible defects. In
addition the crystals extinguished well under crossed polarisers. The crystals were green
in colour, like Ta6Br12 (Figure 6.2).
Figure 6.2 – A picture of the Ta6Br12 & HEWL crystals as viewed under a microscope
after 3 days. Crystals were approximately 0.1mm in length at this point in time.
6.3 – X-ray diffraction data collection procedure
Glycerol (4µl) was used as a cryoprotectant and was added to mother liquor (12µl) which
contained the crystals. A low level of glycerol (25%) was required as it appeared to
slowly interact with the crystals. From this a single, green crystal measuring 0.2mm
across was selected. The crystal was fitted onto a fibre mesh and then mounted onto an R-
Axis imaging plate diffractometer with a rotating copper anode source.
The detector to crystal distance was carefully considered. This is because moving the
detector further away from the crystal will reduce the amount of incoherent scattering
from the crystal and thus improve the accuracy of the data. However moving the detector
further away will also result in a smaller range of θ angles being recorded which will

83
cause a reduction in the measured resolution. In this case the crystal to detector distance
was set at 120mm and the data collection temperature at 100K.
A full 360º of data were collected with an exposure time of seven minutes per degree.
Figure 6.3 is one of the X-ray diffraction images obtained. A summary of the data
collection statics is listed in Table 9.
Figure 6.3 – An X-ray diffraction pattern image from the Ta6Br12 & HEWL data
collection.
The resulting data was processed, merged and scaled using the d*trek program36 (part of
the Rigaku suite of programs). It was decided to remove the images in the ranges 1-74º
and 342 - 360º from the processing as removing these images improved the value of
Rmerge. An initial model structure was obtained using the model replacement method.
This was done using the PHASER computer program which is part of the CCP4i suite37.
The resolution of the model was solved to 1.95Ȧ.

84
Crystal system Tetragonal
Space group P 43 21 2
Unit cell dimensions a = 78.9964 Ǻ α = 90.00°
b = 78.9964 Ǻ β = 90.00°
c = 36.8507 Ǻ γ = 90.00°
Unit cell volume 229964 Ǻ3
Data collection temperature 100 K
Radiation CuKα rotating anode
Diffractometer R-Axis
Detector Image plate
Crystal size 0.20 x 0.20mm
Crystal mosaicity 1.434°
Total reflections measured 177143
Independent reflections 16885
Data completeness 100% (100%)
<I σI> 13.2 (3.8)
Average redundancy 10.49 (10.44)
Rmerge 0.085 (0.433)
Resolution range 55.86 - 1.95 (2.02 - 1.95)
Table 9 – The summary of the X-ray diffraction data collection of HEWL and Ta6Br12
crystal. Values in parentheses indicate the last resolution shell
6.4 – Model refinement procedure The following steps were performed to move from an initial model to a final structure.
All the refinement steps were performed in the refmac5 program which is part of the
CCP4i suite. Map inspection and model building was performed in the COOT38 program.
Step 1
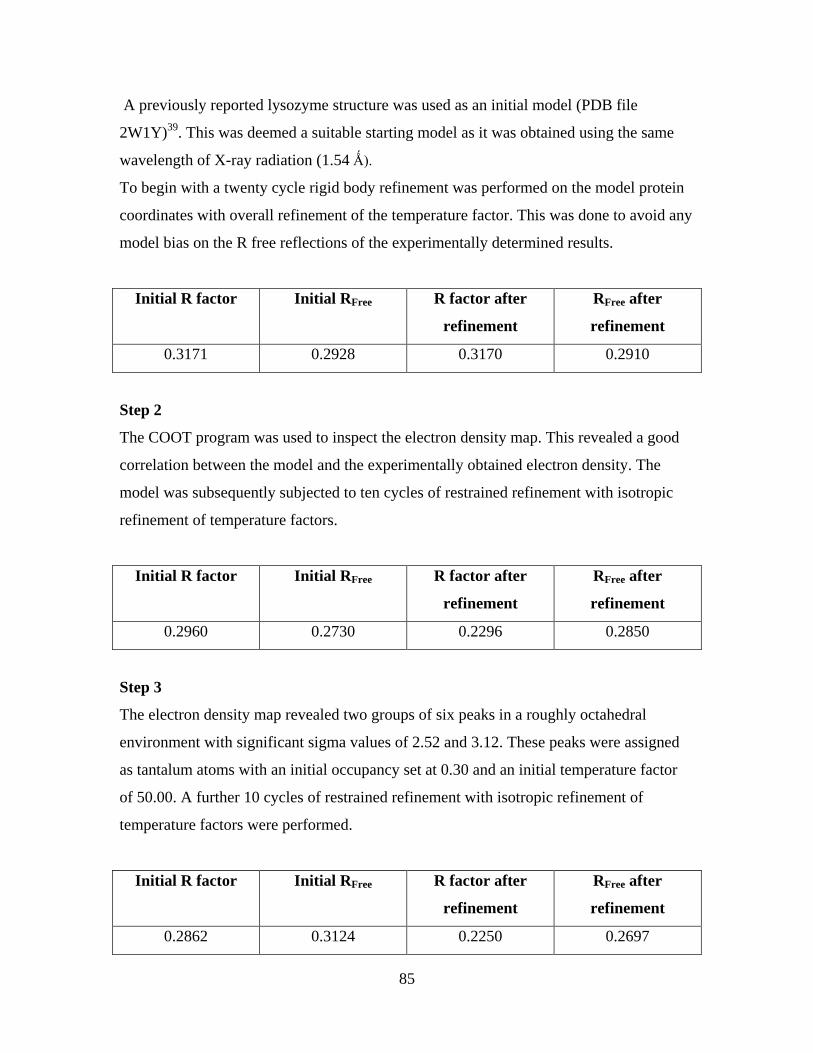
85
A previously reported lysozyme structure was used as an initial model (PDB file
2W1Y)39. This was deemed a suitable starting model as it was obtained using the same
wavelength of X-ray radiation (1.54 Ǻ).
To begin with a twenty cycle rigid body refinement was performed on the model protein
coordinates with overall refinement of the temperature factor. This was done to avoid any
model bias on the R free reflections of the experimentally determined results.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.3171 0.2928 0.3170 0.2910
Step 2
The COOT program was used to inspect the electron density map. This revealed a good
correlation between the model and the experimentally obtained electron density. The
model was subsequently subjected to ten cycles of restrained refinement with isotropic
refinement of temperature factors.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2960 0.2730 0.2296 0.2850
Step 3
The electron density map revealed two groups of six peaks in a roughly octahedral
environment with significant sigma values of 2.52 and 3.12. These peaks were assigned
as tantalum atoms with an initial occupancy set at 0.30 and an initial temperature factor
of 50.00. A further 10 cycles of restrained refinement with isotropic refinement of
temperature factors were performed.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2862 0.3124 0.2250 0.2697
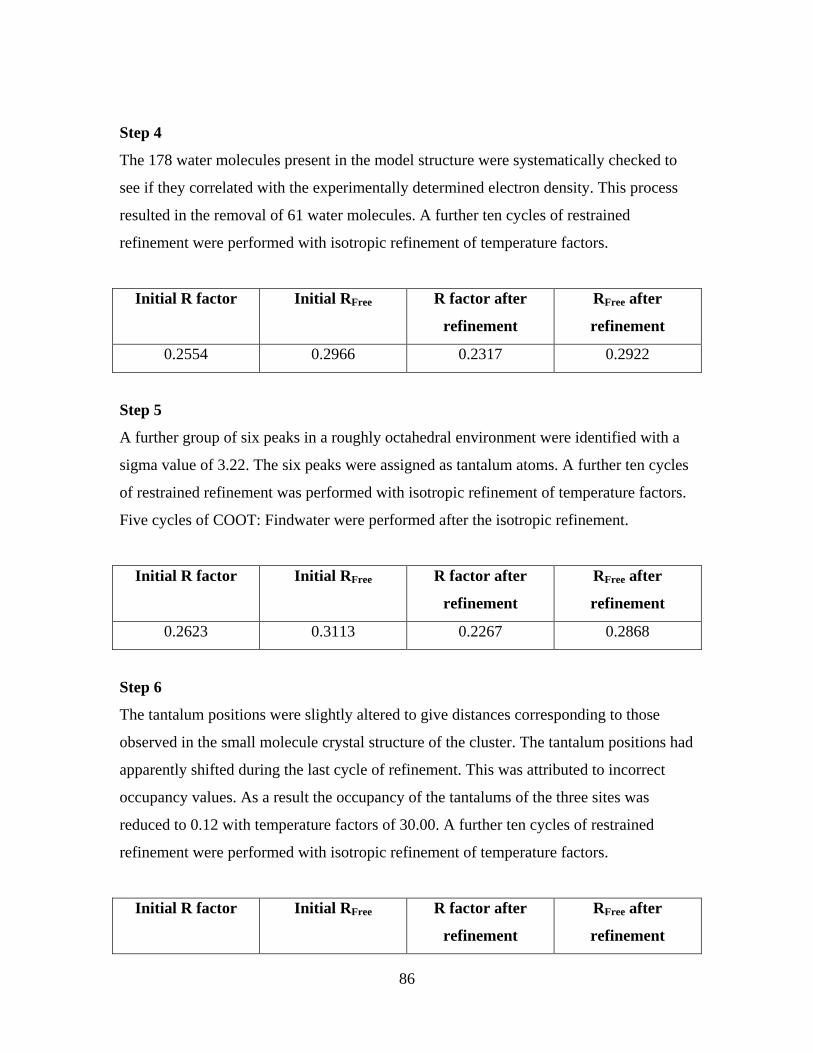
86
Step 4
The 178 water molecules present in the model structure were systematically checked to
see if they correlated with the experimentally determined electron density. This process
resulted in the removal of 61 water molecules. A further ten cycles of restrained
refinement were performed with isotropic refinement of temperature factors.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2554 0.2966 0.2317 0.2922
Step 5
A further group of six peaks in a roughly octahedral environment were identified with a
sigma value of 3.22. The six peaks were assigned as tantalum atoms. A further ten cycles
of restrained refinement was performed with isotropic refinement of temperature factors.
Five cycles of COOT: Findwater were performed after the isotropic refinement.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2623 0.3113 0.2267 0.2868
Step 6
The tantalum positions were slightly altered to give distances corresponding to those
observed in the small molecule crystal structure of the cluster. The tantalum positions had
apparently shifted during the last cycle of refinement. This was attributed to incorrect
occupancy values. As a result the occupancy of the tantalums of the three sites was
reduced to 0.12 with temperature factors of 30.00. A further ten cycles of restrained
refinement were performed with isotropic refinement of temperature factors.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
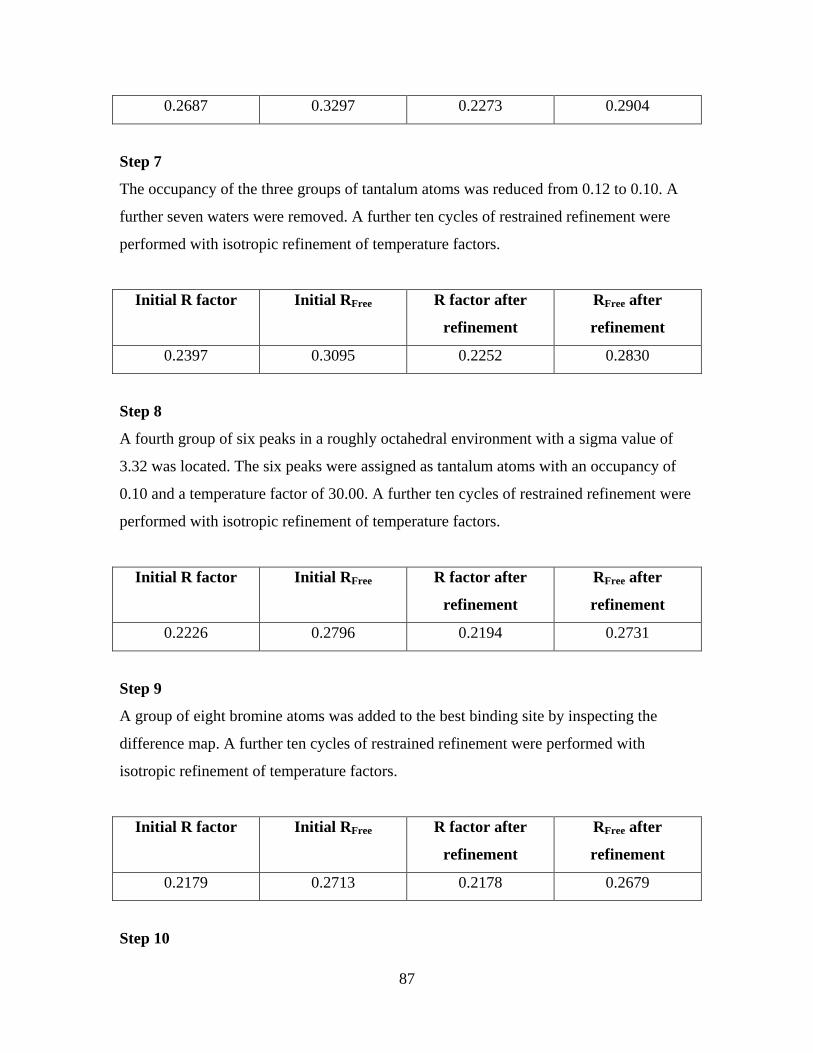
87
0.2687 0.3297 0.2273 0.2904
Step 7
The occupancy of the three groups of tantalum atoms was reduced from 0.12 to 0.10. A
further seven waters were removed. A further ten cycles of restrained refinement were
performed with isotropic refinement of temperature factors.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2397 0.3095 0.2252 0.2830
Step 8
A fourth group of six peaks in a roughly octahedral environment with a sigma value of
3.32 was located. The six peaks were assigned as tantalum atoms with an occupancy of
0.10 and a temperature factor of 30.00. A further ten cycles of restrained refinement were
performed with isotropic refinement of temperature factors.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2226 0.2796 0.2194 0.2731
Step 9
A group of eight bromine atoms was added to the best binding site by inspecting the
difference map. A further ten cycles of restrained refinement were performed with
isotropic refinement of temperature factors.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2179 0.2713 0.2178 0.2679
Step 10
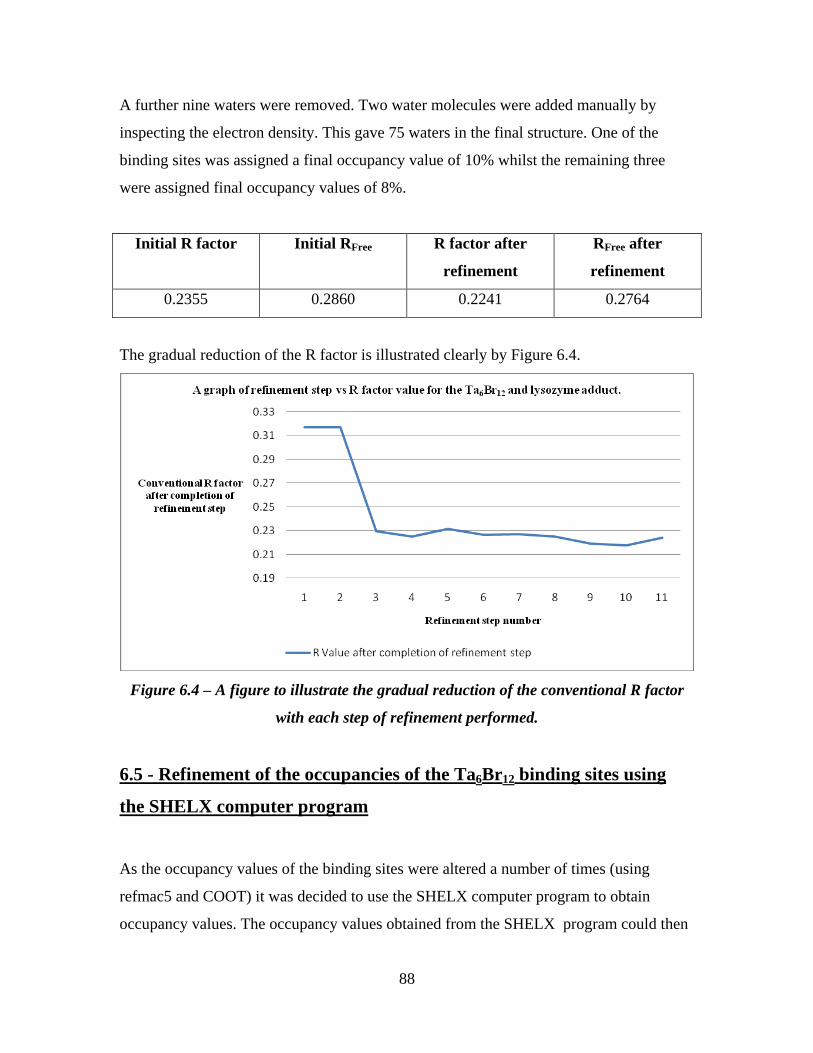
88
A further nine waters were removed. Two water molecules were added manually by
inspecting the electron density. This gave 75 waters in the final structure. One of the
binding sites was assigned a final occupancy value of 10% whilst the remaining three
were assigned final occupancy values of 8%.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2355 0.2860 0.2241 0.2764
The gradual reduction of the R factor is illustrated clearly by Figure 6.4.
Figure 6.4 – A figure to illustrate the gradual reduction of the conventional R factor
with each step of refinement performed.
6.5 - Refinement of the occupancies of the Ta6Br12 binding sites using
the SHELX computer program
As the occupancy values of the binding sites were altered a number of times (using
refmac5 and COOT) it was decided to use the SHELX computer program to obtain
occupancy values. The occupancy values obtained from the SHELX program could then

89
be compared to the occupancy values already being used. It was hoped that this may
provide an indication of the accuracy of the values being used.
The SHELX program required a model structure (known as a fragment). This was
provided in the form of a crystallographically determined structure40 of Ta6Br12.6H2O
from the ICSD database. This structure contained the ideal bond lengths present within
the molecule as well as accurate unit cell dimensions. The coordinated waters molecules
were removed from this cluster as they were not present in the form of the cluster which
is bound to lysozyme. The idea cluster was then fixed one at a time into the four binding
sites within lysozyme. These locations had been determined by inspecting the electron
density map using the COOT computer program. Although all six of the tantalum atoms
in the four binding sites had been located using COOT the bromine atoms had not.
Therefore the bromine atom positions were approximated using the position of a tantalum
atom to which a particular bromine was attached to.
After one Ta6Br12 cluster had been fixed into place, ten cycles of refinement were
performed in the SHELX computer program. After the refinement another Ta6Br12 cluster
was fixed followed by 10 cycles of refinement and so on. After each cycle of refinement
the resulting R factor and occupancy values were recorded.
The SHELX computer program calculates theoretical electron density from the ideal
cluster and attempts to match it and the unit cell dimensions to the experimentally
determined electron density. It refines the position of the cluster and its occupancy value
based on the minimisation of the difference between the observed and calculated electron
density.
After one Ta6Br12 fragment has been placed into a binding site.
R factor Occupancy of binding site
24.61 9.9%
After two Ta6Br12 fragments have been placed into binding sites.
R factor Occupancy of binding site 1 Occupancy of binding site 2
0.2446 8.7% 8.2%

90
After three Ta6Br12 fragments have been placed into binding sites
R factor Occupancy of binding site 1 Occupancy of binding
site 2
Occupancy of binding
site 3
0.2439 9.4% 10.7% 10.2%
After four Ta6Br12 fragments have been placed into binding sites
R factor Occupancy of
binding site 1
Occupancy of
binding site 2
Occupancy of
binding site 3
Occupancy of
binding site 4
0.2431 9.2% 10.0% 10.0% 10.0%
After all four Ta6Br12 clusters had been placed, ten cycles of refinement were carried out
which yielded the results above. However the refinement process did not converge
satisfactorily. As a result the number of refinement cycles was increased from ten to
thirty in the hope of obtaining more accurate results. Convergence was achieved once the
number of cycles was increased, the results of which should be more accurate than
previous results.
After four Ta6Br12 fragments have been placed into binding sites. (30 cycle
refinement)
R factor Occupancy of
binding site 1
Occupancy of
binding site 2
Occupancy of
binding site 3
Occupancy of
binding site 4
0.2318 10.1% 12.4% 9.6% 16.4%
The values of 10.1% and 9.6% are relatively similar to those obtained using the refmac5
and COOT computer programs. However the values of 12.4% and 16.4% are
significantly larger. Inspection of the .res file obtained from SHELX revealed that the
clusters in these positions had been placed too close to the protein. It is possible that this
may account for the high occupancy values observed.
The evolution of the occupancies of each of the four binding sites with each step of
refinement is clearly illustrated in a graphically format by Figure 6.5.
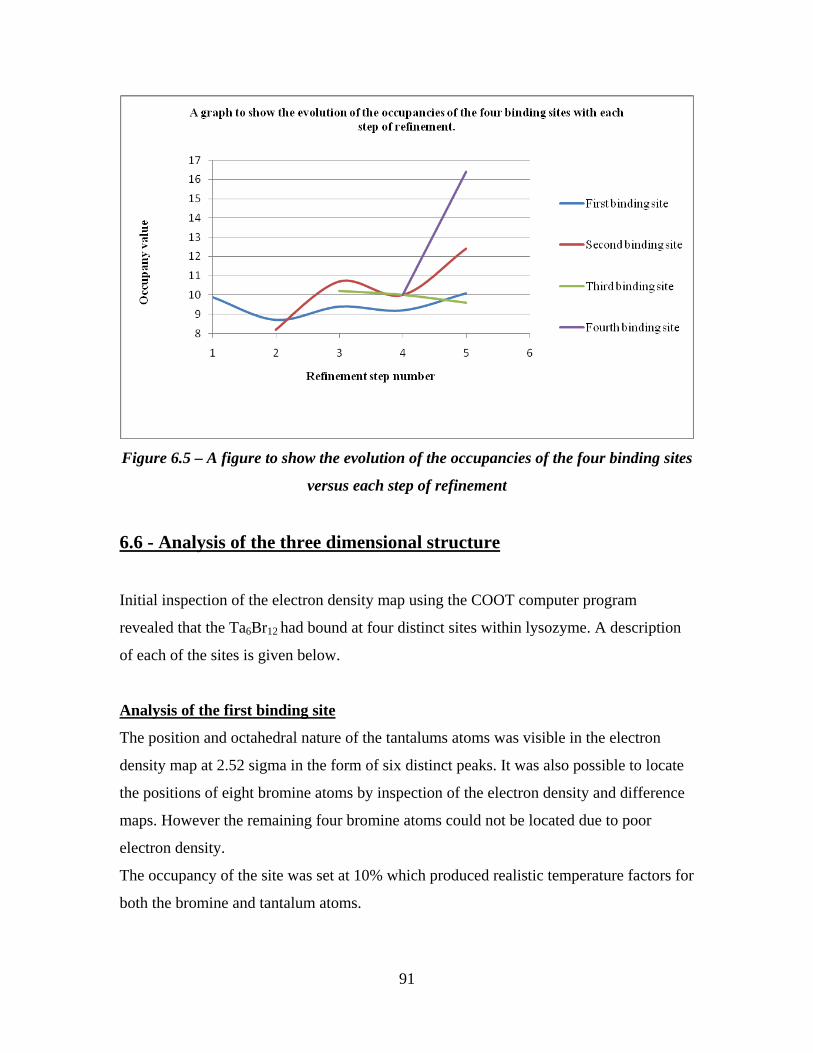
91
Figure 6.5 – A figure to show the evolution of the occupancies of the four binding sites
versus each step of refinement
6.6 - Analysis of the three dimensional structure
Initial inspection of the electron density map using the COOT computer program
revealed that the Ta6Br12 had bound at four distinct sites within lysozyme. A description
of each of the sites is given below.
Analysis of the first binding site
The position and octahedral nature of the tantalums atoms was visible in the electron
density map at 2.52 sigma in the form of six distinct peaks. It was also possible to locate
the positions of eight bromine atoms by inspection of the electron density and difference
maps. However the remaining four bromine atoms could not be located due to poor
electron density.
The occupancy of the site was set at 10% which produced realistic temperature factors for
both the bromine and tantalum atoms.

92
Figure 6.6 – The electron density surrounding the first Ta6Br12 to lysozyme binding
site.
The tantalum atoms are arranged in a fairly regular octahedral environment with the
equatorial tantalums joined by bonds with distances of 2.49 Ǻ , 2.30 Ǻ, 3.11 Ǻ and 3.36
Ǻ. The two axial tantalums are separated by 4.47 Ǻ. These distances compare favourably
with the actual values40 of .2.901(7) Ǻ and 4.096(10) Ǻ respectively.
Figure 6.7 – A figure to show the distances between the tantalum atoms in the first
binding site. Also shown are the distances to the nearest residues.

93
There are two possible residues with which the cluster may interact (as shown in Figure
6.7). The closest residue is arginine 125, the amine groups of which are 1.95 Ǻ and 3.24
Ǻ away from the closest tantalum atoms. It is likely that at the crystallisation pH of 4.7,
arginine would be protonated which would generate a positive charge. As the cluster is
also positively charged it is surprising to find the two so close together.
The next nearest residue is aspartic acid 119 which is 2.67 Ǻ away. It is likely that at the
crystallisation pH of 4.7 aspartic acid would possess a negative charge. As a result it is a
possible that there is a columbic interaction between the cluster and this residue.
The third nearest amino acid is glutamine 121 which is 4.12 Ǻ away which appears to be
too great a distance for an interaction to occur.
Analysis of the second binding site
The position and octahedral environment of the six tantalum atoms was clearly visible at
3.12 sigma. However the position of the bromine atoms could not be determined due to
poor electron density .The tantalum atoms are arranged in a poor octahedral environment
with the equatorial tantalum atoms joined by bonds with distances of 2.70 Ǻ, 3.80 Ǻ, 3.52
Ǻ and 2.09 Ǻ. The two axial tantalum atoms are separated by a distance of 5.54 Ǻ. These
distances display substantial deviation from the ideal values. In addition it was found that
the tantalum positions appeared to shift significantly with each cycle of refinement. This
is probably due to the low occupancy of the binding site, which was set at 8%. The
temperature factors of the bromine atoms were consistent with those of the previous site.

94
Figure 6.8 – The electron density surrounding the second Ta6Br12 to lysozyme binding
site.
The site is most closely located to a carbon atom of a proline residue which is 2.92 Ǻ
away. As the carbon will possess no charge at the crystallisation pH (pH = 4.7) it is
unclear how the cluster interacts at this particular location.
Figure 6.9 – A figure to show the distances between the tantalum atoms in the second
binding site. Also shown is the distance to the nearest residue.

95
Analysis of the third binding site
The position and octahedral environment of the six tantalum atoms was clearly visible at
3.22 sigma. However the position of any of the bromine atoms could not be determined
due to poor electron density .The tantalum atoms are arranged in a poor octahedral
environment with the equatorial tantalums joined by bonds with distances of 1.96 Ǻ, 4.34
Ǻ, 3.05 Ǻ and 3.39 Ǻ. The two axial tantalum atoms are separated by a distance of 5.38
Ǻ. These distances display substantial deviation from the ideal values. In addition it was
found that the tantalum positions appeared to shift significantly with each cycle of
refinement. This is probably due to the low occupancy of the binding site which was set
at 8%. The temperature factors of the tantalum atoms appeared consistent with the
previous binding sites.
It is likely that the cluster interacts with a symmetry related lysozyme molecule that is not
displayed. This is because the closest residues, glycine 71 and arginine 61 are 5.21 and
4.45 Ǻ away respectively. These distances appear to be too great for any significant
interaction to occur.
Figure 6.10 – The electron density surrounding the third Ta6Br12 to lysozyme binding
site.

96
Figure 6.11 – A figure to show the distances between the tantalum atoms in the third
binding site. Also shown are the distances to the nearest residues.
Analysis of the fourth binding site
The position and octahedral environment of the six tantalum atoms was clearly visible at
3.32 sigma. However the position of any of the bromine atoms could not be determined
due to poor electron density. The tantalum atoms are arranged in a poor octahedral
environment with the equatorial tantalum atoms joined by bonds of distances 4.60 Ǻ,
3.11 Ǻ, 3.38 Ǻ and 3.88 Ǻ. The two axial tantalum atoms are separated by a distance of
5.07 Ǻ. The tantalum atoms appeared to shift during the refinement process probably due
to the low occupancy of the site which was set at 8%. The temperature factors of the
tantalum atoms appeared consistent with the previous binding sites.

97
Figure 6.12 – The electron density surrounding the fourth Ta6Br12 to lysozyme binding
site.
There are three possible residues which are close enough for the cluster to interact with
(as shown in Figure 6.13). The closest residue is aspartic acid 18 which is 1.43 Ǻ away.
This would possess a negative charge at the crystallisation ph of 4.7 which would
complement the positive charge of the cluster.
The second closet residue is lysine 13 which is 2.34 Ǻ away. This would possess a
positive charge at ph 4.7 which would be expected to repel the positive charge of the
cluster.
The third nearby residue is leucine 129 which is 3.18 Ǻ away. This side chain will not be
charged at ph 4.7. However this particular residue is relatively far from the cluster which
may prevent the formation of a significant interaction.

98
Figure 6.13 – A figure to show the distances between the tantalum atoms in the fourth
binding site. Also shown are the distances to the nearest residues.
6.7 - Implications of the three dimensional structure
Four Ta6Br12 binding sites to lysozyme were clearly identified at 1.95 Ȧ. The tantalum
atoms present in all four binding sites were located with bond angles and positions
roughly corresponding to those of an octahedral environment. It was possible to locate
eight bromine atoms in only one of the binding sites. The occupancy of one of the
binding sites was around 10% with the remaining three binding sites around 8%.
The high amount of electron density present within the Ta6Br12 cluster allows for it to be
easily located using the electron density map, even at the low occupancies reported here.
In contrast to this the binding of another bromine containing, transition metal cluster,
K2PtBr6 to HEWL has been previously reported41. The cluster was introduced using a
soaking method which resulted in largest occupancies of around 50% for the longest soak
time used of 170 minutes. For the shorter time used of ten minutes this occupancy was
33%. Future work on this cluster may include focusing on its potential susceptibility to
radiation damage with possible loss of the bromine atoms.
The three dimensional structure obtained should provide a model to allow for comparison
with models obtained using new methodologies such a s protein powder X-ray diffraction

99
Chapter 7
Crystal structure determination and model refinement of a co-crystallisation of HEWL and carboplatin
7.1.1 – Introduction to carboplatin
Carboplatin (cis diammine-1,1-cyclobutanedicarboxylate platinum (ΙΙ)) is a second
generation, platinum containing anti cancer medication. A second generation of platinum
anti cancer medications was deemed necessary, owing to the severe side effects attributed
to the administration of the parent compound, cisplatin (cis – diamminedichloro platinum
(ΙΙ)) the most notable of which was nephrotoxicity. Both are commonly used to treat a
variety of cancers including ovarian, testicular and cancers of the head and neck.
The original interest in platinum drugs for anti cancer applications originally arose from a
1965 observation by Rosenberg et al42 who reported that certain transition metal
compounds inhibited bacterial division. The most effective compound was cisplatin and
after successful results in animal models it entered clinical trials in the early 1970`s.
Pt
NH3
NH3
Cl
Cl
Figure 7.1 – The chemical structure of the anti cancer drug cisplatin
Pt
NH3
NH3
O
O
O
O Figure 7.2 – The chemical structure of the anti cancer drug carboplatin

100
The cis isomers of both compounds are used for therapeutic applications as the trans
isomers display no biological activity. Cisplatin contains two labile cis chloride ions
which act as leaving groups. This is in addition to two relatively stable ammonia groups
which in conjugation with the chloride ions are arranged in a square planar geometry
(Figure 7.1).
In contrast carboplatin contains a more stable bidentate dicarboxylate ligand in place of
the chloride ions in addition to the two ammonia groups (Figure 7.2).
It is widely accepted that the labile chlorine ions of cisplatin are exchanged for
nucleophilic groups which result in the formation of chemically stable links.
The administered form of carboplatin reacts with water to form an active hydrated
intermediate. This intermediate is only formed inside cells as the chloride ion
concentration outside cells is sufficiently high enough to prevent hydrolysis. However,
inside cells the chloride ion concentration is low enough for hydrolysis to take place. This
results in the displacement of one chloride ion by a water molecule. The water molecule
is subsequently displaced which allows the platinum atom to coordinate with
nucleophiles present within the DNA helix. The displacement of the second chloride ion
allows the formation of interstrand cross links43.
Cisplatin displays little affinity for the sugars and phosphates but instead reacts with the
purine and pyridimine base pairs of DNA. The primary target of cisplatin at physiological
pH is the N7 atoms of guanine and adenine. The reaction most commonly results in
intrastrand cross linking of two neighbouring guanines which accounts for the majority of
the cross linking seen (around 60%). The cross linking caused by cisplatin causes a major
bending of the DNA helix towards the major groove of DNA44. It is possible that this
structural change is sufficient to inhibit further DNA synthesis resulting in cell death. The
crystal structure of cisplatin and duplex DNA has been determined at 2.6 Ǻ resolution by
Takahara et al45. The structure was solved using the multiple isomorphous derivative
method. This crystal structure support the formation of guanine-guanine cross links
within the DNA.
Carboplatin appears to act via a similar mechanism owing to the more stable nature of the
bidentate carboxylate ligand it acts at a slower rate. In addition for carboplatin the drug

101
passing through the kidneys is not aquated. Therefore it does not react with the kidney
tissue which is the cause of the nephrotoxicity displayed by cisplatin.
7.1.2 – Introduction to work by Casini et al (2006)
The interest in carboplatin originally arose from a paper by Casini et al46 which was
published in 2006. In this paper the authors studied the adducts of anticancer platinum
drugs with hen egg-white lysozyme (HEWL) using both electron spray ionisation mass
spectrometry (ESI-MS) and single crystal X-ray crystallography. The results obtained
using ESI-MS indicated that the protein platination had only partially taken place. This
result was confirmed using ICP-OES (inductively coupled optical emission spectroscopy)
which indicated that platination levels were around 50% for cisplatin and less than 15%
for carboplatin. This proved that the cisplatin and carboplatin had bound to HEWL albeit
at seemingly low binding occupancies.
It was surprising that the platination was of such low levels as high excesses of the
anticancer drugs were used (three fold excess of anticancer drug with respect to
lysozyme) in addition to long soaking times.
In order to gain a more precise idea of the adduct structures and the location of the metal
binding sites single crystal X-ray crystallography was employed. Two sets of lysozyme
crystals were grown using the hanging drop method and separately soaked in solutions
containing excesses of carboplatin and cisplatin. It was found that diffraction quality
crystals were only obtained in the case of cisplatin. The adduct was subsequently solved
at a resolution of 1.9Ǻ.
7.1.3 – Previous work by the Helliwell group
As the soaking method had previously proved to be unsuccessful it was decided by us to
pursue a co-crystallisation of HEWL and carboplatin. The initial attempt was performed
by Joanne Meredith for a MChem dissertation47. For the crystallisation hen egg white
lysozyme (49mg) was transferred to a glass vial to which a 0.04m acetate buffer solution
(1ml) was also added (preparation as described in Chapter. The mixture was gently
stirred for five minutes to ensure the lysozyme powder had fully dissolved. Once
completed carboplatin (3.713mg, 5mM) was added followed by a further five minutes of

102
gentle stirring. Finally 10% salt solution (1ml) was added gradually over a period of five
minutes after which the solution was stirred for a further five minutes. At this point the
glass vial was sealed and left undisturbed at room temperature. The amounts of
carboplatin and lysozyme were chosen to give a three fold excess of carboplatin with
respect to lysozyme (in terms of moles).
After inspection after 72 hours a larger number of colourless block like crystals with a
typical size of 0.3-0.35mm were present. Unfortunately after data collection and
processing it was discovered that the occupancy of the carboplatin was low. There was
insufficient electron density to provide a satisfactory structure so it was decided to find a
method to improve the occupancy. However from this initial wok it appeared that
carboplatin bound to the sugar binding, active site of lysozyme. This was a promising
result as it may indicate the possibility of using carboplatin as an inhibitor of sugar
binding enzymes.
7.2 – Co-crystallisation method and optimisation of the conditions
Following on from the work carried out by Joanne Meredith it was decided to attempt use
a chemical additive to attempt to improve the occupancy of the carboplatin binding. A
search of literature sources indicated that both lysozyme and carboplatin are soluble in
dimethyl sulfoxide (DMSO)48,49. It was envisaged that if the solubility of both
components is increased then the occupancy of the carboplatin binding will also increase.
In addition it was decided to increase the carboplatin concentration to 10mM in the hope
this would promote increased binding.
A paper by Lu et al50 published in 2002 describes the growth of lysozyme crystals from a
binary mixture consisting of 12.5% DMSO and water. It was therefore decided to attempt
to recreate these conditions with the addition of carboplatin.
Therefore, a batch co-crystallisation was attempted using the same method and the
following amounts – lysozyme (24.5mg), 0.04M acetate buffer (0.438ml), 10% salt
solution (0.438ml), DMSO (0.125ml) and carboplatin (3.713mg, 10mM). The amounts of
carboplatin and lysozyme were chosen to give a six fold excess of carboplatin with
respect to lysozyme (in terms of moles). In all the crystallisation attempts carboplatin was

103
obtained from Calbiochem in the form of a white powder. Hen egg white lysozyme was
obtained from Sigma Aldrich in the form of a white powder
The vial was then left undisturbed at room temperature for a one week period.
Unfortunately after this time it was discovered that no crystals had grown. A further
15.5mg of lysozyme was added which again resulted in no crystals. As a final step the
lysozyme concentration was increased to 60 mg/ml. After five days this resulted in
precipitation of the lysozyme suggesting the concentration was too high for crystallisation
to take place.
Initially the lack of crystals was attributed to the presence of carboplatin. In order to test
this theory the crystallisation conditions listed in the Lu et al50 paper were recreated.
These conditions were lysozyme (20mg), 0.04M acetate buffer (0.438ml), 10% salt
solution (0.438ml), DMSO (0.125ml). After two days it was found that no crystals were
present and that the published conditions could not be recreated. As a result it was
decided to systematically vary the lysozyme and DMSO concentrations in order to find
the optimum conditions for crystal growth. This process required multiple attempts as
many cases resulted in a complete absence of crystals. The conditions attempted are listed
in Table 10.
Attempt Crystallisation conditions Result
1 • Lysozyme – 40mg
• DMSO – 0.060ml
• 10% salt solution – 0.470 ml
• Buffer – 0.470 ml
After four days a few extremely
few small crystals were present.
Full magnification of the
microscope was required.
2 • Lysozyme – 45mg
• DMSO – 0.060ml
• 10% salt solution – 0.470 ml
• Buffer – 0.470 ml
After four days only a handful of
crystals had formed. Crystals too
small to be used at around
0.003mm in length.
3 • Lysozyme – 50mg
• DMSO – 0.060ml
• 10% salt solution – 0.470 ml
A large number of square plate
crystals were present that
extinguished well under crossed

104
• Buffer – 0.470 ml polarisers. No precipitated
lysozyme was present. Crystal
dimensions around 0.1mm x
0.1mm.
4 • Lysozyme – 55mg
• DMSO – 0.060ml
• 10% salt solution – 0.470 ml
• Buffer – 0.470 ml
After four days a small number of
crystals were present. Lysozyme
concentrations appears to be too
high as significant amounts have
precipitated.
5 • Lysozyme – 60mg
• DMSO – 0.060ml
• 10% salt solution – 0.470 ml
• Buffer – 0.470 ml
After four days a small number of
crystals were present. Lysozyme
concentrations appears to be too
high as large amounts have
precipitated.
Table 10 – The condition attempted in the crystallisation of HEWL in the presence of
DMSO.
As a result of the findings from the condition optimisation a final crystallisation was set
up. The conditions best suited to crystal growth from Table 10 were used in the presence
of carboplatin (3.713mg, 10mM), lysozyme (50mg), DMSO (0.060ml), 10% salt (0.0470
ml) and 0.04M acetate buffer (0.0470 ml).
After a period of one week it was found that two different crystal morphologies had
formed. A square plate form with dimensions of around 0.1mm in length in addition to
elongated plates of around 0.15mm in length were present (Figure 7.3). Crystals were
present on the liquid surface as well as on the bottom of the vial. An interesting
observation was that after a period of 7 days, it was found that extremely thin needle
shaped crystals were present (Figure 7.3). These crystals were much too thin to be used
and were not observed in the carboplatin free, HEWL and DMSO crystallisations.

105
Figure 7.3 – A picture of the carboplatin & HEWL crystals as viewed under a
microscope after 4 days. Crystals were approximately 0.1mm to 0.15mm in length at
this point in time. The presence of extremely thin needle shaped crystals is also shown.
In addition a number of crystals were found to be grouped together to form an aggregate
(Figure 7.4). These were not found in the carboplatin free, HEWL and DMSO
crystallisations. This suggests that carboplatin has some chemical effect upon the
crystallisation process.

106
Figure 7.4 – An aggregate of lysozyme crystals observed in the carboplatin and HEWL
co-crystallisation in the presence of DMSO.
7.3 – X-ray diffraction data collection procedure
Glycerol (10µl) was used as a cryoprotectant and was added to mother liquor (40µl)
containing the crystals. The mother liquor and glycerol were allowed to mix for a two
minute period before a suitable crystal was selected. A single colourless crystal 0.10mm
in length was selected and fitted into a 50-100 µM loop (Figure 7.5)
Figure 7.5 – A crystal of carboplatin and HEWL mounted onto a loop. Pictured using
high magnification video camera present on diffractometer.

107
and mounted onto an R-Axis imaging plate diffractometer with a rotating copper anode.
The crystal to detector distance was set at 120mm and the data collection temperature at
100K. A full 360º of data were collected with an exposure time of six and a half minutes
per degree. Figure 7.6 is one of the X-ray diffraction images obtained. A summary of the
data collection statics is listed in Table 11.
The resulting data was processed, merged and scaled using the d*trek program36 (part of
the Rigaku suite of programs). An initial model structure was obtained using the model
replacement method. This was done using the PHASER computer program which is part
of the CCP4i suite37. The resolution of the model was solved to 2 Ǻ.
Crystal system Tetragonal
Space group P 43 21 2
Unit cell dimensions a = 77.0869 Ǻ α = 90.00°
b = 77.0869 Ǻ β = 90.00°
c = 36.4220 Ǻ γ = 90.00°
Unit cell volume 216433 Ǻ3
Data collection temperature 100 K
Radiation CuKα rotating anode
Diffractometer R-Axis
Detector Image plate
Crystal size 0.10mm x 0.10mm
Crystal mosaicity 1.737°
Total reflections measured 265965
Independent reflections 21439
Data completeness 82.7% (18.6%)
<I σI> 12.7 (2.4)
Average redundancy 12.41 (4.17)
Rmerge 0.084 (0.512)
Resolution range 54.51 – 1.65 (1.71 – 1.65)
Table 11 – The summary of the X-ray diffraction data collection of HEWL and
carboplatin crystal. Values in parentheses indicate the last resolution shell

108
Figure 7.6 – An X-ray diffraction pattern image from the carboplatin & HEWL data
collection.
7.4 – Model refinement procedure
The following steps were performed to move from an initial model to a final structure.
All the refinement steps were performed in the refmac 5 program which is part of the
CCP4i suite. Map inspection and model building was performed in the COOT program38.
Step 1
A previously reported lysozyme structure was used as an initial model (PDB file
1BWJ51).
To begin with a twenty cycle rigid body refinement was performed on the protein
coordinates with overall refinement of the temperature factor.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.3335 0.3341 0.3335 0.3359

109
Step 2
The COOT program was used to inspect the electron density map. This revealed a good
correlation between the model and the experimentally obtained electron density. Two
peaks of 10.49 sigma and 8.29 sigma were present in the electron density map and were
assigned as platinum atoms. The occupancy of the atoms was set at 30% with a
temperature factor of 50.00. The model was then subjected to 20 cycles of restrained
refinement with isotropic refinement of temperature factors.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.3189 0.3161 0.2030 0.2624
Step 3
Two ammonia groups were added to one of the platinum sites in place of water
molecules. This was because the bond lengths are similar to those reported for the length
of the Pt-NH3. The occupancy of the nitrogen atoms was set at 30% and the temperature
factor at 50.00. A further ten cycles of restrained refinement was performed with
isotropic refinement of temperature factors.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2036 0.2662 0.2016 0.2678
Step 4
One ammonia group was added to the other platinum site in place of a water molecule.
Again the bond length appeared consistent with the reported value of the Pt-NH3 bond.
The occupancy of the nitrogen atom was set at 30% and the temperature factor at 50.00.
A further ten cycles of restrained refinement was performed with isotropic refinement of
temperature factors.

110
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2014 0.2676 0.2012 0.2697
Step 5
The remaining ammonia group was added to the other platinum site in place of a water
molecule. The occupancy of the nitrogen atom was set at 30% and the temperature factor
at 50.00. A further ten cycles of restrained refinement was performed with isotropic
refinement of temperature factors.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2033 0.2728 0.2020 0.2695
Step 6
The electron density around each water molecule was inspected at one sigma to make
sure the waters contained within the starting model correlated with the experimentally
determined electron density. This process resulted in the removal of 52 water molecules.
In addition 5 cycles of COOT : Find water were performed.
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2104 0.2720 0.2069 0.2677
Step 7
A further sixteen water molecules were deleted as well as a sodium atom. This was
because these species displayed no electron density. The final occupancy value of both
sites was set at 30% which is consistent with the cisplatin binding to HEWL as reported
by Casini et al.

111
Initial R factor Initial RFree R factor after
refinement
RFree after
refinement
0.2149 0.2778 0.2133 0.2809
Further optimisation of the structure was not possible and it is noted that the RFree has
increased by around 1% from the last refinement step.
The evolution of the R factor with each step of refinement is illustrated clearly by Figure
7.7.
Figure 7.7 – A figure to illustrate the gradual reduction of the conventional R factor
with each step of refinement performed.
7.5.1 - Analysis of the three dimensional structure
From initial inspection of the electron density map it was observed that carboplatin had
bound at two distinct sites within lysozyme. The two sites were on either side of the

112
histidine 15 residue. Electron density peaks of 12.19 sigma and 9.39 sigma corresponding
to the platinum atom positions were observed on either side of histidine 15.
For the site located on the left hand side of histidine 15 it was possible to locate the
position of the platinum atom and the two amine groups of carboplatin. The ammonia
groups were joined to the platinum atom by bonds of distance 1.89 Ǻ and 1.98 Ǻ. The
angle between the three was 77.30°. The bond length values compare favourably with
those measured in the crystal structure of carboplatin at 100K47 displays (Pt-N bond
lengths of 2.024(3) Ǻ and an N-Pt-N angle of 95.6°(2)).
Similarly, for the site located on the right hand side of histidine 15 it was possible to
locate the position of the platinum atom and the two amine groups of carboplatin. The
ammonia groups were joined to the platinum atom by bonds of distance 1.86 Ǻ and 2.01
Ǻ. The angle between the three was 95.28° (Figure 7.8). This bond angle is almost
identical to that observed in the crystal structure of carboplatin at 100K. The discrepancy
between the values of the two sites may be down to the platinum atom dominating the X-
ray scattering with respect to the relatively light carbon, nitrogen and oxygen atoms.
Figure 7.8 – A figure to show the distances from the platinum atom to the two
ammonia groups in both binding sites.
It is possible that the platinum atom of the left hand site coordinates to the NE atom of
histidine 15 (Figure 7.9). This interaction involves a distance of 3.40 Ǻ.
It is possible that the platinum atom of the right hand side site coordinates to the ND atom
of histidine 15 (Figure 7.9). This interaction involves a similar distance of 3.35 Ǻ.

113
Figure 7.9 - A figure to show the distance from the platinum atom to the nearest
nitrogen atom on the histidine 15 residue for both binding sites.
Both sites displayed significant amounts of electron density (Figure 7.10 & Figure 7.11)
but with shapes that made the location of additional atoms difficult. The binding site on
the left hand side of histidine 15 appeared to more strongly defined suggesting a higher
occupancy than the site on the right hand side of histidine 15.
Figure 7.10- A figure to show the electron density around the binding site on the left
hand side of histidine 15.

114
Figure 7.11 - A figure to show the electron density around the binding site on the right
hand side of histidine 15.
7.5.2 - Comparison with HEWL and cisplatin crystal structure
The structure of HEWL and cisplatin as reported by Casini et al46 was deposited as a
PDB file under the deposition code 216Z. This PDB file was superimposed with respect
to the final version of the HEWL and carboplatin PDB. The superimposition was done
with respect to the alpha carbon atoms of lysozyme and was performed using the Lsqkab
program (which is part of the CCP4i suite). This ensured that both the structures were
arranged in the same orientation within the unit cell. The PDB was then subjected to
twenty cycles of restrained refinement with overall refinement of the temperature factor
using refmac 5. Finally, a FO-FC map was generated with peaks greater than 3sigma listed.
This was done using the FFT program (part of the CCP4i suite).
The superimposition (Figure 7.12) revealed that the positions of the majority of the amino
acid sequences, of the two PDB files closely agreed. However it appears that the histidine
15 residue is noticeably displaced (0.75 Ǻ ) in the HEWL and carboplatin model (with
respect to the HEWL and cisplatin model). It is possible that this displacement allows the
binding of the two carboplatin molecules on either side of histidine 15 as opposed to a
single cisplatin molecule.

115
Figure 7.12 – A figure showing the superimposition of the histidine 15 residue in a
crystal of cisplatin and HEWL (shown in yellow & blue) and a crystal of carboplatin
and HEWL (shown entirely in blue). A noticeable displacement (0.75 Ȧ) in the case
of carboplatin and HEWL is displayed.
The PDB file deposited by Casini et al contains a single DMSO molecule (that is not
mentioned in the paper) in the active site of lysozyme. This molecule is in an almost
identical location to the one observed in the HEWL and carboplatin study described here
(Figure 7.13).

116
Figure 7.13 – A figure displaying the location of the DMSO molecule present within
the lysozyme active site for both the cisplatin and carboplatin models.
7.5.3 - Comparison with previous HEWL and carboplatin crystal structure
This work was conducted by Joanne Meredith for award of an MChem degree47. The
crystals of HEWL and carboplatin were grown using a batch method co-crystallisation in
the absence of DMSO.
In this case it was found that the carboplatin had bound at four distinct sites within
lysozyme with four peaks visible at 3.10 sigma. Interestingly, it appeared one of the
binding sites was in the sugar binding, active site of lysozyme. This is an intriguing result
as it suggests the possibility of using carboplatin to inhibit enzymes where sugar binding
is involved in the catalytic process.
Catalytic residues of lysozyme Top arrow indicates glutamic acid 135. Bottom arrow indicates aspartic acid 52.
Left hand arrow indicates the location of the sulphur atom of DMSO present in the carboplatin and HEWL model. The right hand arrow indicates the location of the DMSO molecule present in the cisplatin and HEWL model.

117
However this result was not observed in the presence of DMSO. This may be because
DMSO has had some kind of chemical effect that has altered the binding behaviour of
carboplatin.
7.5.4 - Comparison with HEWL and NAG (N-acetyl-D-glucosamine) crystal
structure
The crystal structure of HEWL and NAG (N-acetyl-D-glucosamine) was deposited in the
PDB under the deposition code 3A3Q52. This study showed that the NAG trisaccharide
bound to the active site of lysozyme. It was therefore decided to compare how the
location of the trisaccharide compared to the location of the DMSO molecules observed
in the HEWL cisplatin and HEWL carboplatin studies. The 3A3Q PDB file was
superimposed with respect to the final version of the HEWL and carboplatin PDB. The
superimposition was done with respect to the alpha carbon atoms of lysozyme and was
performed using the Lsqkab program (which is part of the CCP4i suite). This ensured that
both the structures were arranged in the same orientation within the unit cell
Figure 7.14 – A figure showing the location of a NAG trisaccharide and the DMSO
molecules present in the cisplatin and carboplatin models. The location of all three
species is almost identical with the DMSO molecules indicated by arrows.

118
The superimposition revealed that one end of the NAG trisaccharide is in the same
position as the DMSO molecule in both the HEWL and cisplatin and HEWL and
carboplatin studies. This may mean that the DMSO prevents the carboplatin from binding
at the active site in the manner observed in the DMSO free work conducted by Joanne
Meredith.
7.6 – Implications of the three dimensional structure
X-ray diffraction analysis of a DMSO free co-crystallisation of carboplatin and HEWL
(conducted by Joanne Meredith) revealed that the carboplatin had bound to the sugar
binding, active site of lysozyme. Binding to this site was thought to be feasible as the
bidentate dicarboxylate ligand possess a reasonable structural resemblance to that of a
sugar ring. This raised the possibility that carboplatin might act as an inhibitor for sugar
binding enzymes such as heparanase.
In an effort to increase the occupancy of the carboplatin, DMSO was introduced into a
co-crystallisation of carboplatin and HEWL. X-ray diffraction analysis revealed that the
presence of DMSO had apparently altered the binding behaviour of carboplatin. It was
found that although DMSO had improved the occupancy of the carboplatin it had also
bound to the active site. This raises the possibility that the presence of DMSO prevented
the carboplatin binding to the sugar binding, active site of lysozyme. Indeed it was found
that the DMSO bound to the same site in lysozyme as a NAG trisaccharide. From this
work it is impossible to verify if carboplatin displays binding behaviour similar to that of
sugars, possibly due to the interference of DMSO.
Instead it was found that the carboplatin bound in a similar manner to that of cisplatin (as
reported by Casini et al). However, where cisplatin had only bound to a single side of
histidine 15, it was found that carboplatin had bound to both sides. A superimposition of
the protein coordinates from the co-crystallisation of carboplatin and HEWL and those of
cisplatin and HEWL reported by Casini et al was performed. This revealed that in the co-
crystallisation of carboplatin and HEWL the histidine 15 residue was noticeably
displaced (displacement of 0.75 Ǻ ) with respect to the histidine 15 observed in the Casini
et al coordinates. This displacement may be essential in allowing the carboplatin to bind

119
at both sides of the residue as opposed to one side. However, Casini et al used a soaking
method as opposed to a co-crystallisation which may have had some bearing upon the
cisplatin and HEWL results. To confirm these apparent differences in carboplatin and
cisplatin binding to HEWL, X-ray diffraction analysis of a co-crystallisation of cisplatin
and HEWL would need to be performed.
In both of the carboplatin binding sites only the platinum atom and the two ammonia
groups could be located (achieving the same level of detail as reported by Casini et al).

120
Chapter 8
Future work
In the HEWL and Ta6Br12 co-crystallisation the location of the tantalum atoms was
relatively easy by inspection of the electron density map. This is owing to the high
amount of electron density possessed by the tantalum atoms. In contrast it was difficult to
locate the bromine positions which have a much lower electron density and scatter X-rays
more weakly. The difficulty in determining the location of the bromine atoms was
probably at least partly due to the low occupancy of the binding sites. A possible solution
would be to perform the co-crystallisation of the Ta6Br12 cluster and HEWL in the
presence of a solvent such as DMSO. This could possibly increase the solubility of the
Ta6Br12 cluster and promote increased binding.
An additional idea would be to perform the co-crystallisation at a different pH. As the
Ta6Br12 cluster is positively charged it may bind to different locations in HEWL. These
derivatives could possibly be used in the multiple isomorphous replacement method in
order to obtain a more detailed three dimensional structure.
In the carboplatin and HEWL co-crystallisation the presence of DMSO apparently
chemically altered the binding behaviour of carboplatin. The obvious suggestion would
be to repeat the co-crystallisation of carboplatin and HEWL in the presence of a different
additive. However as carboplatin is insoluble in solvents such as ethanol and acetone this
may prove difficult. If crystals in the presence of a different additive could be obtained
the possible sugar binding behaviour of carboplatin could be properly assessed.

121
References
1. Bravais lattice types image (2010), [Online] Available at:
www.schielo.cl/fbpe/img/bscq/v46n3/fig04.gif
2. Molloy, C. K., (2004), Group theory for chemists: fundamental theory and applications, Horwood
3. Hahn, T. (Ed)., (1983), International tables for crystallography (volume A), Published for the International Union of Crystallography by Reidel
4. 21 screw axis image (2010), [Online] Available at: www.axxel.iqfr.csic.es/Cristalographica/archives-07/helicodial.jpg
5. Bragg, L. W., (1983), The diffraction of short electromagnetic waves by a crystal, Proc. Camb. Phil. Soc. 17 part 1, 43-57
6. Woolfson, M. M., (1997), An introduction to X-ray crystallography (2nd edition), Cambridge University Press
7. Bragg pictorial representation (2010), [Online] Available at: http://serc.carleton.edu/images/research_education/geochemsheets/braggslaw.jpg
8. Suryanarayna, C., Norton, M. G., (1998), X-ray diffraction: A practical approach, Plenium Press
9. X-ray tube image (2010), [Online] Available at: http://pubs.usgs.gov/of/2001/of01-041/htmldocs/images/xrdtube.jpg
10. Elder, R. F., Gurewitsch, M. A., Langmui,r V. R., Pollock, H. C., (1947),
Radiation from electrons in a synchrotron, Physical review, 71, 829-830
11. Ramakanth, Hebbar, K., (2007), Basics of X-ray diffraction and its applications, I.K. International
12. Clegg, W., Blake, J. A., (2009), Crystal structure analysis: Principles and practice
(2nd edition), Oxford University Press

122
13. Helliwell, R. J., (1992), Macromolecular crystallography with synchrotron
radiation, Cambridge University Press
14. Patterson, L. A., (1935), A direct method for the determination of the component
of interatomic distances in crystals, Z. Krist, 90, 517-542
15. Bruker, (2002), Saint version 6.36a, Bruker AXS Inc.
16. Sheldrick, H. G., Schneider, R. T., (1997), SHELXL: High resolution refinement, Methods in enzymology, 277, 319-343
17. Burla, C. M., Caliandro, R., Camalli, M., Carrozzini, B., Cascarano, L. G., De Caro, L., Giacovazzo, C., Polidori, G., Spagna, R., (2005), SIR2004: An improved tool for crystal structure determination and refinement, J. Appl. Cryst, 38, 381-388
18. Das, A., Dey, B., Jana Dipankar, A., Hemming, J., Helliwell, M., Lee Man, H., Hsiao, T., Suresh, E., Colacio, E., Choudhury, R. S., Mukhopadhyay, S., (2010), Effect of protonated aminopyridines on the structural divergences of M(ΙΙ)-malonate complexes, Polyhedron, 29, 1317-1325
19. Allen H. F, (2002), The Cambridge structural database: a quarter of a million structures and rising, Acta. Cryst. B., 58 ,380-388
20. Bruno, J. I., Cole, C. J., Edgington, R. P., Kessler, M., Macrae, F. C., McCabe, P., Pearson, J., Taylor, R., (2002), New software for searching the Cambridge Structural Database and visualizing crystal structures, Acta. Cryst. B., 58, 389-397.
21. Spek, L. A., (2003), Single crystal structure validation with the program PLATON, J. Appl. Cryst., 36, 7-13.
22. Chieh, C. P., Trotter, J., (1970), Crystal structure of tetraphenyltin, J. Chem. Soc. 6, 911-914.
23. (2010), [Online] Available at: http://nobelprize.org/nobel_prizes/chemistry/laureates/2009/
24. (2010), [Online] Available at www.pdb.org

123
25. Blundell, L. T., Johnson, N. L., (1976), Protein crystallography, Academic Press, 69-77
26. Chayen, N., Comparative studies of protein crystallization by vapour diffusion and micro batch techniques, Acta Cryst D, 54(1), 8-15
27. King, V. M., (1965), A low resolution structural model for cubic glucagon based on the packing of cylinders, J. Mol. Biol, 11, 549-561
28. Evans, P., McCoy, A., (2008), An introduction to molecular replacement, Acta Cryst D, 64, 1-10
29. Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., Walter, P., (2002), Molecular biology of the cell (4th edition), Garland Science
30. Stryer, L., Berg, M. J., Tymoczko, L. J., (2006), Biochemistry (6th edition), W. H. Freeman
31. Yonath, A et al., (2005), Crystallographic studies on the ribosome, a large
macromolecular assembly exhibiting severe nonisomorphoism, extreme beam sensitivity and no internal symmetry, Acta. Cryst A. 54, 945-955
32. Knablein, J et al., (1997), Ta6Br122+, a tool for phase determination of large
biological assemblies by X-ray crystallography, J. Mol. Biol. 270, 1-7.
33. Szcezepanowski, H. R., Filipek, R., Bochtler, M., (2005), Crystal structure of mouse ubiquitin activating enzyme, J. Biol. Chem. 280, 22006-22011
34. (2010) Jena Bioscience tantalum cluster derivatization user guide, [Online] Available at: www.jenabioscience.com
35. Corey, B. R., Stanford, H. R., Marsh, E. R., Leung, C. Y., Kay, M. L., (1962), An
X-ay investigation of wet lysozyme chloride crystals, Acta Cryst, 15, 1157-1163
36. Pflugrath, W. J., (1999), The finer things in X-ray diffraction data collection, Acta Cryst D, 55, 1718-1725
37. Collaborative, (1994), The collaborative computational project number 4:
programs for protein crystallography, Acta. Cryst. D., 50(5), 760-763

124
38. Emsley, P., Lohkamp, P., Scott, W., Cowtan, K., (2010), Features of Coot, Acta.
Cryst. D., In press.
39. PDB ID = 2W1Y Cianci, M., Helliwell R. J., Suzuki, A., (2008), The interdependence of wavelength. Redundancy and dose is sulphur SAD experiments, Acta Cryst D, 64, 1196-1209
40. Vojnovic, M., Brnicevic, N., Basic, I., Planinic, P., Giester, G., (2002), The Co crystallization of the Cubic [Ta6Br12(H2O)6][CuBr2X2]·10H2O and Triclinic [Ta6Br12(H2O)6]X2·trans-[Ta6Br12(OH)4(H2O)2]·18H2O (X = Cl, Br, NO3) Phases with the Coexistence of [Ta6Br12]2+ and [Ta6Br12]4+ in the Latter, ZAAC, 628, 401-408
41. Kumar, Y. S., (2009), X-ray crystal structure analyses of small and large molecules (Msc thesis), The University of Manchester
42. Rosenberg, B., Van Camp, L., Krigas, T., (1965), Inhibition of cell division in
Escherichia Coli by electrolysis products from a platinum electrode, Nature, 205, 698-699
43. Skeel, T. R. (Ed), Handbook of cancer chemotherapy (6th edition), Lippincott Williams and Wilkins.
44. Pratt, B. W., Ruddon, W. R., (1979), The anticancer drugs, Oxford University press.
45. Takahara, T., Rosenweig, C. A., Frederick, A. C., Lippard, J. S., (1995), Crystal
structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin, Nature, 377, 649-652
46. Casini, A., Mastrobuoni, G., Temperini, C., Gabbiani, C., Francese, S., Moneti, G., Supuran, T. C., Scozzfava, A., Messori, L., (2006), ESI mass spectrometry and X-ray diffraction studies of adducts between anticancer platinum drugs and hen egg white lysozyme, Chem. Commun, 2,156-158,
47. Meredith, J., (2010), The University of Manchester (MChem dissertation),
48. Carboplatin (2010), [Online] Available at:

125
www.tocris.com/dispprod.php?ItemId=5205
49. Lehmann, S. M,. Stansfield, D. F. R., (1989), Binding of dimethyl sulfoxide to lysozyme crystals, studied with neutron diffraction, Biochemistry, 28, 7028-7033
50. Lu, J., Wang, J. X,. Ching, B. C., (2002), Effect of additives on the crystallization of lysozyme and chymotrypsinogen A, Crystal growth & design, 3(1), 83-87
51. PDB ID = 1BWJ Dong, J., Boggon, J. T., Chayen, E. N., Raftery, J., Bi, C. R., Helliwell, R. J., (199), Bound solvent structures for microgravity, ground control, gel and microbatch grown hen egg white lysozyme crystals at 1.8Ȧ resolution, Acta Cryst D. 55 745-752
52. PDB ID = 3A3Q
Ose, T., Kuroki, K., Matsushima, M., Maenaka, K., Kumagai, I., (2009), Importance of the hydrogen bonding network including Asp 52 for catalysis, as revealed by Asn 59 mutant hen egg white lysozyme, J Biochem, 146, 651-657

126
Bibliography
• Blow, D., (2002), Outline of crystallography for biologists, Oxford University
Press • Clegg, W., (1998), Crystal structure determination, Oxford University Press • Glusker Pickworth, J., Trueblood, N. K., (1985), Crystal structure analysis,
Oxford University Press • Arndt, W. U., Willis, M. T. B., (1996), Single crystal diffractometery,
Cambridge University Press • Woolfson, W. M., (1997), An introduction to X-ray crystallography,
Cambridge University Press • Blundell, T. L., Johnson, N. L., (1976), Protein crystallography, Academic
Press Inc • McPherson, A., (1982), Preparation and analysis of protein crystals, John
Wiley & Sons • Ramakanth, Hebbar, K., (2007), Basics of X-ray diffraction and its
applications, I.K. International
• Helliwell, R. J., (1992), Macromolecular crystallography with synchrotron
radiation, Cambridge University Press


















