
Stochastic modeling of molecular reaction networks Daniel Forger University of Michigan.
description

Stochastic modeling of molecular reaction networks
Daniel Forger
University of Michigan
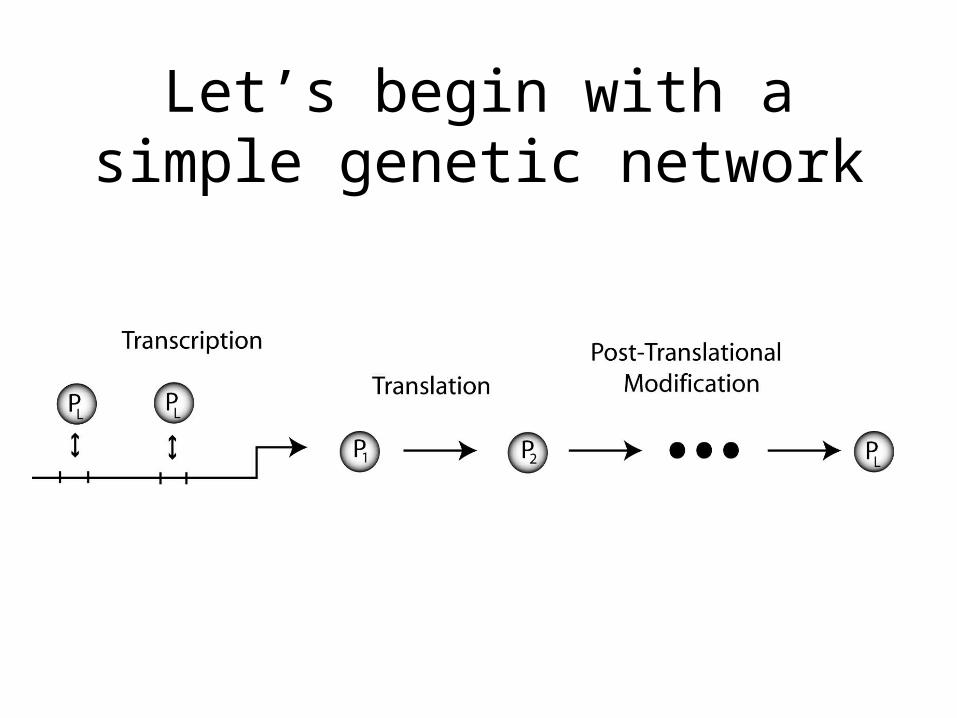
Let’s begin with a simple genetic network

We can list the basic reaction rates and stochiometry
numsites = total # of sites on a gene, G = # sites bound M = mRNA, Po = unmodified protein, Pt = modified protein
Transcription trans or 0 +MTranslation tl*M +PoProtein Modification conv*Po -Po, +PtM degradation degM*M -MPo degradation degPo*Po -PoPt degradation degPt*Pt -PtBinding to DNA bin(numsites - G)*Pt -Pt, +GUnbinding to DNA unbin*G -G

We normally track concentrationLet’s track # molecules instead
• Let M, Po, Pt be # molecules• First order rate constants (tl, unbin, conv,
degM, degPo and degPt) have units 1/time and stay constant
• Zero order rate constant (trans) has units conc/time, so multiply it by volume
• 2nd order rate constant (bin) has units 1/(conc*time), so divide it by volume

numsites = total # of sites on a gene, G = # sites bound M = mRNA, Po = unmodified protein, Pt = modified proteinV = Volume
Transcription trans*V or 0 +MTranslation tl*M +PoProtein Modification conv*Po -Po, +PtM degradation degM*M -MPo degradation degPo*Po -PoPt degradation degPt*Pt -PtBinding to DNA bin/V(numsites - G)*Pt -Pt, +GUnbinding to DNA unbin*G -G

How would you simulate this?
• Choose which reaction happens next– Find next reaction– Update species by stochiometry of next
reaction– Find time to this next reaction

How to find the next reaction
• Choose randomly based on their reaction rates
trans*V tl*M conv*Po degM*M degPo*Po degPt*Pt bin/V(numsites - G)*Ptunbin*G
Random #

Now that we know the next reaction modifies the protein
• Po = Po - 1
• Pt = Pt + 1
• How much time has elapsed– a0 = sum of reaction rates
– r0 = random # between 0 and 1
⎟⎟⎠
⎞⎜⎜⎝
⎛=
00
1ln
1
raτ

This method goes by many names
• Computational Biologists typically call this the Gillespie Method– Gillespie also has another method
• Material Scientists typically call this Kinetic Monte Carlo

Myth 1:“Mass Action Formulations do not account for Stochasticity”

Consider a simple model inspired by the circadian clock
in Cyanobacteria
A B
C
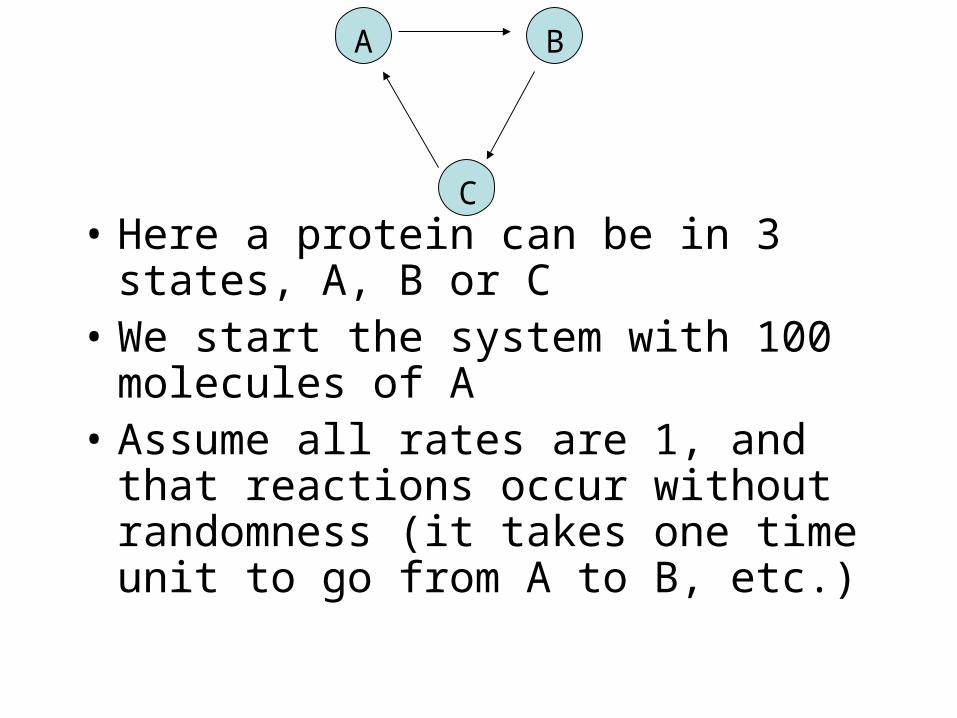
• Here a protein can be in 3 states, A, B or C
• We start the system with 100 molecules of A
• Assume all rates are 1, and that reactions occur without randomness (it takes one time unit to go from A to B, etc.)
A B
C


Mass Action Representation
€
dA
dt= C − A,
dB
dt= A − B,
dC
dt= B − C
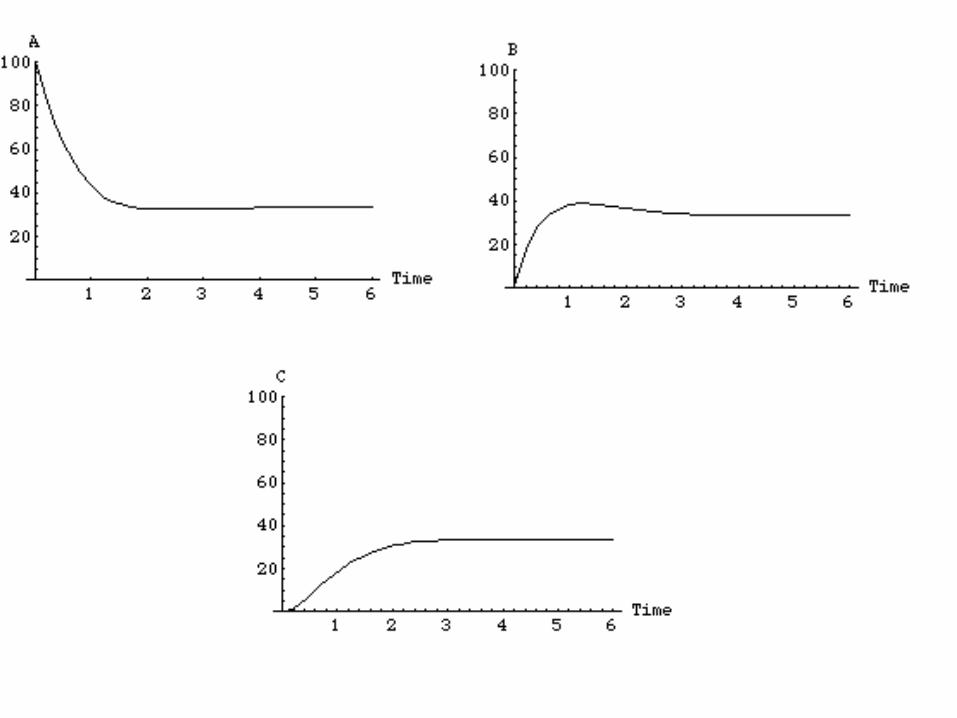

Matlab simulation

Mass Action represents a limiting case of Stochastics
• Mass action and stochastic simulations should agree when certain “limits” are obtained
• Mass action typically represents the expected concentrations of chemical species (more later)

Myth 2:Stochastic and Mass Action
Approaches agree only if there are enough molecules


What matters is the number of reactions
• This is particularly important for reversible reactions
• By the central limit theorem, fluctuations dissapear like n-1/2
• There are almost always a very limited number of genes, – Ok if fast binding and unbinding

There are several representations in between Mass Action and Gillespie
• Chemical Langevin Equations
• Master Equations
• Fokker-Planck
• Moment descriptions

We will illustrate this with an exampleKepler and Elston Biophysical Journal 81:3116
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

Master Equations describe how the probability of being in
each state
€
dpm0
dt= −Kk0 + δm + α 0( ) pm
0 + Kk1pm1 + δ(m +1)pm +1
0 + α 0 pm−10
dpm1
dt= −Kk1 + δm + α 1( ) pm
1 + Kk0 pm0 + δ(m +1)pm +1
1 + α 1pm−11
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

Sometimes we can solve for the mean and variance
€
moments = m j
s≡ m j pm
s
m
∑
at steady state
mean =α 0k1 + α 1k0
δ
var iance = mean + k0k1
α 0 −α 1
δ
⎛
⎝ ⎜
⎞
⎠ ⎟2
δ
δ + K
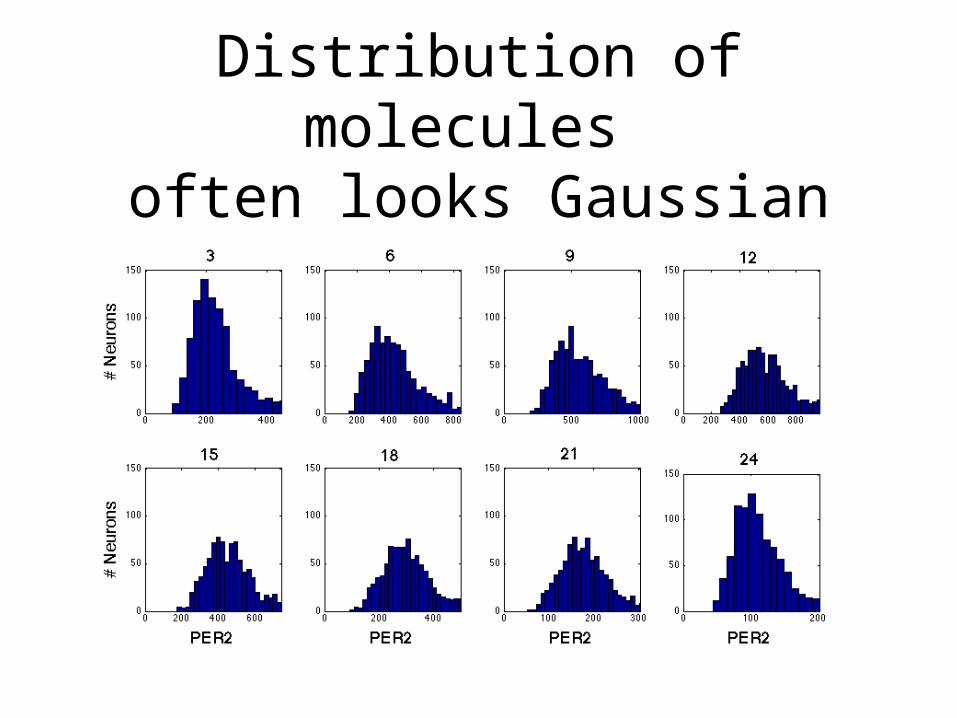
Distribution of molecules often looks Gaussian

Moment Descriptions
• Gaussian Random Variables are fully characterized by their mean and standard deviation
• We can write down odes for the mean and standard deviation of each variable
• However, for bimolecular reactions, we need to know the correlations between variables (potentially N2)

Towards Fokker Planck
• Let’s divide the master equation by the mean m*.
• Although this equation described many states, we can smooth the states to make a probability distribution function
€
pms (t) ≡ dxps(x, t)
(m−1/ 2)/ m*
(m +1/ 2)/ m*
∫

Note
€
ps x +1
m*
⎛
⎝ ⎜
⎞
⎠ ⎟=
1
j!∂x( )
jps(x)
1
m*
⎛
⎝ ⎜
⎞
⎠ ⎟j
= e1
m*∂ x
ps(x)j
∑
If 1/m* is small, we can then derive a simplifedVersion of the Master equations
€
∂t ps(x) = −∂x
α s
m*−δx
⎛
⎝ ⎜
⎞
⎠ ⎟ps(x)
⎡
⎣ ⎢
⎤
⎦ ⎥+
1
2m*∂x
2 α s
m*+ δx
⎛
⎝ ⎜
⎞
⎠ ⎟ps(x)
⎡
⎣ ⎢
⎤
⎦ ⎥+ K[k ˆ s pˆ s (x) − ks ps(x)]

QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

Chemical Langevin Equations
• If we don’t want the whole probability distribution, we can sometimes derive a stochastic differential equation to generate a sample
€
dX
dt= A(X) + B(X)ξ (t)

Adalsteinsson et al. BMC Bioinformatics 5:24
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

Examples
• Transcription Control
• Lac Operon
• Oscillations
• Accounting for diffusion

Rossi et al. Molecular Cell
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
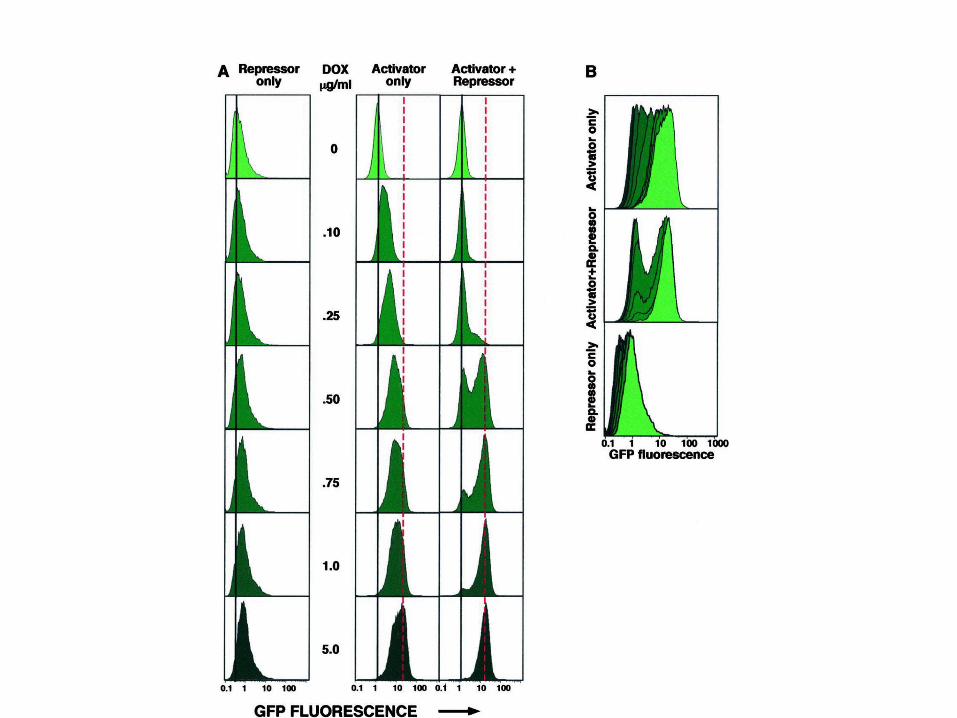

Ozbudak et al. Nature 427:737
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

Guantes and Poyatos PLoS Computational Biology 2:e30
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

SNIC BifurcationSNIC Bifurcation
Invariant Circle
Limit Cycle
x2
p1
node
saddle
Saddle-Node on anInvariant Circle
max
min
max
SNIC

Hopf BifurcationHopf Bifurcation
x2
p1
stable limit cycle
sss
uss
slc max
min

Noise Induced oscillations

Liu et al. Cell 129:605
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

3-D Gillespie
http://www.math.utah.edu/~isaacson/3dmodel.html


















