
Stimuli‐Responsive Metal‐Organic Frameworks with ...
15
REVIEW 1700239 (1 of 14) © 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.mrc-journal.de Stimuli-Responsive Metal-Organic Frameworks with Photoswitchable Azobenzene Side Groups Anemar Bruno Kanj, Kai Müller, and Lars Heinke* DOI: 10.1002/marc.201700239 1. Introduction Smart, stimuli-responsive molecules change their proper- ties as a response to external signals such as light, heat, and pH. [1] Since light is a handy and usually non-invasive signal, photochromic molecules like azobenzene attract particular attention. Upon UV irradiation, azobenzene can isomerize from the nonpolar, planar trans form to the nonplanar cis form with a dipole moment of 3 D. [2] By irradiation with visible light or thermal relaxation, the cis azobenzene goes back to its ther- modynamically stable trans form; see Figure 1a. Various func- tionalizations enable the azobenzene switching with light of different wavelengths. [2a] Even azobenzene that can be switched with visible light only [3] and bridged azobenzene having cis as thermodynamically stable form [4] have been synthesized. Such photoswitchable azobenzene molecules have been used as smart moieties for various single molecular machines, like molecular rotors [5] or single-molecule “nanocars” [6] on a solid substrate. The preparation of macroscopic smart materials that are based on the light-induced changes of the molecular com- ponents is a current major aim. Polymers and liquid crystals which incorporate azobenzene were used as actuators [7] and Metal-Organic Frameworks Metal-organic frameworks (MOFs) are nanoporous, crystalline hybrid mate- rials, which enable various functionalities by incorporating functional organic molecules. By using organic linker molecules that possess photoswitchable azobenzene side groups, the remote control over certain properties was introduced to MOFs. Different MOF materials in the form of powders and thin films have been used to demonstrate the photoswitching. The applica- tions of these stimuli-responsive nanoporous solids range from switching the adsorption capacity of various gases over remote-controlled release of guest molecules to continuously tunable membrane separation of molecular mixtures. A particular focus of this review is the effect of the azobenzene photoswitching on the host-guest interaction, enabling smart applications of the material. Steric hindrance, which may suppress the photoswitching in some MOF structures, is also discussed. A. B. Kanj, K. Müller, Dr. L. Heinke Karlsruhe Institute of Technology (KIT) Institute of Functional Interfaces (IFG) Hermann-von-Helmholtz-Platz 1 76344 Eggenstein-Leopoldshafen, Germany E-mail: [email protected] light-driven motors. [8] Another class of materials that enable the incorporation of photochromic molecules are metal- organic frameworks (MOFs). [9] MOFs are nanoporous, crystalline solids, com- posed of metal nodes and organic linker molecules. These materials possess many unique properties like well-defined, peri- odic nanoporous structures with very large specific areas. Large structural and chemical variety is enabled by altering the metal nodes and/or the linker molecules. These unique properties make MOFs very attractive to various applications, such as for storage of light gases like H 2 , CH 4 or CO 2 , for separation of gases, as well as for catalysis. [9] The option of incorporating various functional organic molecules in the MOF structure may enable the prepa- ration of functional, nanoporous materials. MOF materials in the form of nanoporous crystalline powders usually are pre- pared by solvothermal synthesis. For many applications, MOFs in the form of thin films are desirable, which can be fabricated by various techniques, [10] for instance by controlled layer-by- layer techniques. [11] Here, we review and critically discuss MOFs with photos- witchable azobenzene side groups. The potential of switching the adsorption capacity and the diffusion properties in MOF materials, in form of powders or thin films, is a focus, in addition to being an update to previous reviews on the photoswitchable MOFs. [12] Since the switching of the host-guest interaction as a result of the trans–cis photoisomerization of the azobenzene side groups is crucial for the smart applications, we will pay particular attention to this. Due to many structural similarities, covalent-organic frameworks (COFs) [13] with photo- switchable azobenzene side groups will also be discussed. It should be noted that, in addition to photoswitchable MOFs with azobenzene side groups, further smart functionalizations of MOFs also exist. For instance, embedding photoswitchable azobenzene molecules in the MOF pores results in a straight- forward functionalization. [14] The azobenzene moiety can also be implemented in the backbone of the MOF scaffold. [15] Unlike the azobenzene side groups in MOFs, which (ideally) isomerizes similarly to azobenzene molecules in solution, the trans–cis isomerization of the azobenzene moiety in the back- bone significantly deviates from the isomerization of isolated azobenzene molecules. Further research based on precise structural and spectroscopic investigations are desirable to illu- minate the isomerization behavior of the azobenzene moiety in the MOF backbone. It may be assumed that the trans–cis Macromol. Rapid Commun. 2017, 1700239
Transcript of Stimuli‐Responsive Metal‐Organic Frameworks with ...

REVIEW
1700239 (1 of 14) © 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
www.mrc-journal.de
Stimuli-Responsive Metal-Organic Frameworks with Photoswitchable Azobenzene Side Groups
Anemar Bruno Kanj, Kai Müller, and Lars Heinke*
DOI: 10.1002/marc.201700239
1. Introduction
Smart, stimuli-responsive molecules change their proper-ties as a response to external signals such as light, heat, and pH.[1] Since light is a handy and usually non-invasive signal, photochromic molecules like azobenzene attract particular attention. Upon UV irradiation, azobenzene can isomerize from the nonpolar, planar trans form to the nonplanar cis form with a dipole moment of 3 D.[2] By irradiation with visible light or thermal relaxation, the cis azobenzene goes back to its ther-modynamically stable trans form; see Figure 1a. Various func-tionalizations enable the azobenzene switching with light of different wavelengths.[2a] Even azobenzene that can be switched with visible light only[3] and bridged azobenzene having cis as thermodynamically stable form[4] have been synthesized. Such photoswitchable azobenzene molecules have been used as smart moieties for various single molecular machines, like molecular rotors[5] or single-molecule “nanocars”[6] on a solid substrate.
The preparation of macroscopic smart materials that are based on the light-induced changes of the molecular com-ponents is a current major aim. Polymers and liquid crystals which incorporate azobenzene were used as actuators[7] and
Metal-Organic Frameworks
Metal-organic frameworks (MOFs) are nanoporous, crystalline hybrid mate-rials, which enable various functionalities by incorporating functional organic molecules. By using organic linker molecules that possess photoswitchable azobenzene side groups, the remote control over certain properties was introduced to MOFs. Different MOF materials in the form of powders and thin films have been used to demonstrate the photoswitching. The applica-tions of these stimuli-responsive nanoporous solids range from switching the adsorption capacity of various gases over remote-controlled release of guest molecules to continuously tunable membrane separation of molecular mixtures. A particular focus of this review is the effect of the azobenzene photoswitching on the host-guest interaction, enabling smart applications of the material. Steric hindrance, which may suppress the photoswitching in some MOF structures, is also discussed.
A. B. Kanj, K. Müller, Dr. L. HeinkeKarlsruhe Institute of Technology (KIT)Institute of Functional Interfaces (IFG)Hermann-von-Helmholtz-Platz 176344 Eggenstein-Leopoldshafen, GermanyE-mail: [email protected]
light-driven motors.[8] Another class of materials that enable the incorporation of photochromic molecules are metal-organic frameworks (MOFs).[9] MOFs are nanoporous, crystalline solids, com-posed of metal nodes and organic linker molecules. These materials possess many unique properties like well-defined, peri-odic nanoporous structures with very large specific areas. Large structural and chemical variety is enabled by altering the metal nodes and/or the linker molecules. These unique properties make MOFs very attractive to various applications, such as for storage of light gases like H2, CH4 or CO2, for separation of gases, as well as for catalysis.[9] The option of incorporating various functional organic molecules in the MOF structure may enable the prepa-
ration of functional, nanoporous materials. MOF materials in the form of nanoporous crystalline powders usually are pre-pared by solvothermal synthesis. For many applications, MOFs in the form of thin films are desirable, which can be fabricated by various techniques,[10] for instance by controlled layer-by-layer techniques.[11]
Here, we review and critically discuss MOFs with photos-witchable azobenzene side groups. The potential of switching the adsorption capacity and the diffusion properties in MOF materials, in form of powders or thin films, is a focus, in addition to being an update to previous reviews on the photoswitchable MOFs.[12] Since the switching of the host-guest interaction as a result of the trans–cis photoisomerization of the azobenzene side groups is crucial for the smart applications, we will pay particular attention to this. Due to many structural similarities, covalent-organic frameworks (COFs)[13] with photo-switchable azobenzene side groups will also be discussed.
It should be noted that, in addition to photoswitchable MOFs with azobenzene side groups, further smart functionalizations of MOFs also exist. For instance, embedding photoswitchable azobenzene molecules in the MOF pores results in a straight-forward functionalization.[14] The azobenzene moiety can also be implemented in the backbone of the MOF scaffold.[15] Unlike the azobenzene side groups in MOFs, which (ideally) isomerizes similarly to azobenzene molecules in solution, the trans–cis isomerization of the azobenzene moiety in the back-bone significantly deviates from the isomerization of isolated azobenzene molecules. Further research based on precise structural and spectroscopic investigations are desirable to illu-minate the isomerization behavior of the azobenzene moiety in the MOF backbone. It may be assumed that the trans–cis
Macromol. Rapid Commun. 2017, 1700239

© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (2 of 14)
www.advancedsciencenews.com www.mrc-journal.de
isomerization decreases the crystallinity and destroys the mate-rial, as observed for COFs with azobenzene in the backbone[16] or for molecular frameworks on the surface.[17] Although addi-tional interesting options to prepare stimuli-responsive MOFs exist (e.g., other photochromic molecules like spiropyran or diarylethene can be incorporated in the MOF structure),[18] this review focuses solely on the structure, functionality, and poten-tial applications of MOFs with azobenzene side groups.
2. Metal-Organic Frameworks with Azobenzene Side Groups in Form of Powders
The idea of photoswitchable MOFs where photoisomeriza-tion of azobenzene is enabled by a dangling side group is sketched in Figure 1b. The first MOF made from photo-switchable linker molecules was published by N. Stock et al. in 2011.[19] 3-(phenyldiazenyl)-4,4′-bipyridine, that is, 4,4′-bipyri-dine with an azobenzene side group (AzoBiPy), was used to prepare a pillared-layer MOF of type Zn2(NDC)2(AzoBiPy), also referred to as CAU-5 (NDC: 2,6-naphthalenedicarboxylic acid). The proposed MOF structure was verified by X-ray dif-fraction (XRD) and Raman spectroscopy. It was found that the scaffold is two-fold interpenetrated. The adsorption iso-therms of nitrogen, argon, and carbon dioxide (without UV or visible light irradiation) show the permanent porosity. UV-vis absorption spectroscopy of the linker molecule in ethanolic solution clearly shows reversible intensity shifts of the π–π* (at 320 nm) and n–π* (at approximately 450 nm) bands upon UV (365 nm) or visible light (440 nm) irradiation. This is a clear indication that the azobenzene side groups of the linker
Anemar Bruno Kanj received his B.Sc. (2011) and M.Sc. (2015) in chemistry from Al-Baath University, Syria. He joined the group led by Lars Heinke at the Karlsruhe Institute of Technology (KIT) and started his PhD in January 2017. His research focuses on stimuli-responsive films of metal-organic frameworks (MOF).
Kai Müller received his B.Sc. (2014) and M.Sc. (2015) in chemistry from KIT, where he began his PhD studies in January 2016. His research concentrates on defect struc-tures in MOFs and smart MOF materials.
Lars Heinke studied physics in Greifswald and Leipzig and obtained his Diploma in 2006. In his Ph.D. thesis (2009) under the supervision of Jörg Kärger, he investigated mass transfer in nanoporous mate-rials. After his postdoctoral positions at Hans-Joachim Freund at the Fritz-Haber Institute, Berlin (2009–2010), and at Gabor A. Somorjai
at the Lawrence Berkeley National Laboratory, California (2011), he joined the Institute of Functional Interfaces (IFG) at the KIT as group leader. In 2015, he finished his habilitation mentored by Christof Wöll. His research focuses on functional nanoporous films and their physical properties.
molecules in solution can be reversibly switched by light. UV-vis spectroscopy of the Zn2(NDC)2(AzoBiPy) MOF crystals in a BaSO4 matrix shows small, reversible intensity changes of the π–π* band and of the n–π* band upon UV and visible light irradiation. The authors stated that the switching effi-ciency of the azobenzene moiety in the MOF is much lower than of the molecules in solution and attribute this to struc-tural aspects, such as the two-fold interpenetration which hinders the isomerization.[20] The result of the light irradia-tion on the adsorption isotherm (or the switching of another
Macromol. Rapid Commun. 2017, 1700239
Figure 1. Photoswitchable azobenzene side groups. a) Azobenzene in the planar trans form undergoes isomerization to the nonplanar cis form by irradiation with UV light. Irradiation with visible light or thermal relaxation of the cis azobenzene results in isomerization to the thermodynami-cally stable trans form. b) Introducing the photoswitchable azobenzene moiety into the MOF: A) as part of the backbone, and B) as azobenzene side group covalently attached to the inner pore wall. Reproduced with permission.[19] Copyright 2011, Royal Society of Chemistry.

© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (3 of 14)
www.advancedsciencenews.com www.mrc-journal.de
functionality) of the MOF material is not demonstrated in the first paper with MOFs possessing photoswitchable linker molecules.
In a further publication, N. Stock et al. synthesized four varia-tions of Zn2(NDC)2(AzoBiPy) MOF using the same azobenzene linker molecule: Co2(NDC)2(AzoBiPy), Cu2(NDC)2(AzoBiPy), Zn2(BPDC)2(AzoBiPy) and Zn2(CCA)2(AzoBiPy) (BPDC: 4,4′-biphenyl-dicarboxylic acid; CCA: 4-carboxy-cinnamic acid).[20] An XRD investigation of the structures shows a two-fold interpenetration of all four structures. Since Zn2(BPDC)2(AzoBiPy) and Zn2(CCA)2(AzoBiPy) show limited stability, the investigations focused on Co2(NDC)2(AzoBiPy) and Cu2(NDC)2(AzoBiPy), also referred to as Co-CAU-5 and Cu-CAU-5. Both MOFs are stable and show permanent porosity. UV-vis spectroscopy of the photoswitching with UV and blue (455 nm) light was carried out and demonstrated a partially reversible trans–cis isomerization. Only small changes in the intensity of the π–π* and n–π* band were found, which was attributed to the limited pores space, i.e., the isomerization is sterically hindered. The switching properties of all M-CAU-5 with MZn, Co and Cu are very similar and the authors con-clude that the metal nodes have no significant influence on the photoisomerization.
In ref. [21], a ZIF-8 derivative with azobenzene side groups (azo-ZIF-8) was prepared and carefully investigated via XRD, nuclear magnetic resonance spectroscopy (NMR), and Raman spectroscopy. The permanent porosity was demonstrated by measuring the N2 absorption isotherm, where the surface area of azo-ZIF-8 was found to be significantly lower than that of ZIF-8. The switching properties were investigated using UV-vis spectroscopy, where partially reversible intensity changes of the π–π* band (here at 390 nm) and n–π* band were found. The fact that the initial UV-vis spectra cannot be retained after only three cis–trans-switching cycles might be attributed to photobleaching or steric hindrance. Stock et al. also showed that the thermal cis-to-trans isomerization of the azobenzene side groups in these MOF structures is very slow and the amount of cis azobenzene only slightly decreases overnight.[20,21]
By means of post-synthetic modification (PSM), N. Stock et al. functionalized Cr-MIL-101 MOF with azobenzene side groups.[22] Cr-MIL-101-NH2 was chosen as the primary MOF due to its high surface area, its large accessible pores, as well as its chemical and thermal stability. The Cr-MIL-101-NH2 MOF mate-rial was modified by p-phenylazobenzoylchloride, resulting in Cr-MIL-101-amide-azobenzene (see Figure 2a) and by 4-(phenyl-azo) phenylisocyanate, resulting in Cr-MIL-101-urea-azoben-zene. The MOF structures and the PSM were examined using XRD, infrared spectroscopy (IR), and NMR spectroscopy. UV-vis absorption spectroscopy of the azobenzene-modified MOF in BaSO4 showed reversible intensity shifts of the π–π* and n–π* band upon UV (365 nm) and visible (455 nm) light irradiation. The indication that the azobenzene side groups in the MOF can be photoswitched was verified by measuring the methane adsorption isotherms of the photoswitchable MOF, Figure 2b. UV irradiation, resulting in a trans-to-cis isomerization of the azobenzene side groups, causes an increase of the methane adsorption capacity. Visible light, resulting in the isomerization back to the trans state, decreased the adsorption capacity again. Although the initial adsorption amount was not exactly obtained,
two complete trans–cis–trans cycles of the adsorption isotherm demonstrate the reversible photoswitching behavior.
H. C. Zhou et al. used 2-(phenyldiazenyl)terephthalic acid (AzoBDC) to prepare a MOF-5 derivative with azobenzene side groups, referred to as PCN-123 (also referred to as Azo-MOF-5).[23] The functionality of the photoswitching was demon-strated by switching the adsorption capacity. Due to its relatively high adsorption capacity at room temperature, CO2 was chosen as the probe molecule. The CO2 uptake by the pristine PCN-123 sample at room temperature after activation was 22.9 cm3 g−1 at 1 bar, while it decreased by approximately 27% after UV irradiation for 1 h. After storing the sample for another 5 h in the dark, the CO2 adsorption amount decreased to 46% of the initial value; see Figure 3. The authors suggest that the effect of the continuing decrease of the adsorption amount in the dark was caused by a very slow azobenzene isomerization process in the MOF. (This azobenzene switching mechanism would significantly deviate from the switching of azobenzene molecules in solutions or in polymer matrixes, which occur in a few nanoseconds.)[2a] Instead of light irradiation, the cis-to-trans isomerization was performed by thermal relaxation at 60 °C for 20 h. The thermal relaxation of the azobenzene groups increased the CO2 adsorption amount again. Two switching cycles show reversible behavior. Based on the detailed analysis of the XRD data of the pristine sample (trans) and of the sample after UV irradiation (cis), it is shown that the electron density
Macromol. Rapid Commun. 2017, 1700239
Figure 2. Photoswitching in Cr-MIL-101-amide-azobenzene MOFs. a) Sketch of light-induced switching of the azobenzene side group in Cr-MIL-101-amide-azobenzene. b) Methane adsorption isotherms after irradiating with UV and visible light for two cycles. The adsorp-tion amount increases upon trans-to-cis and decrease upon cis-to-trans photoisomerization. Reproduced with permission.[22] Copyright 2012, Royal Society of Chemistry.

© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (4 of 14)
www.advancedsciencenews.com www.mrc-journal.de
close to the metal centers of the MOF is higher in the cis than in the trans state. The authors explain that this electron density stems from the phenyl rings of the cis azobenzene, which shield the metal centers. It is explained that the shielding of the attrac-tive CO2 adsorption sites at the metal centers by the phenyl ring in the cis conformation results in the observed decrease of the CO2 uptake amount. This is the only crystallographic data of the azobenzene switching in the MOF pores published so far. The significance of these data would have been even higher if the switching yield, i.e., the amount of cis azobenzene, had been determined (e.g., by UV-vis or IR spectroscopy).
Using density functional theory (DFT) and grand canonical Monte Carlo (GCMC), N. V. Medhekar et al. simulated the CO2 adsorption in PCN-123 (Azo-MOF-5) and in Azo-IRMOF-10, which is topologically similar to PCN-123 but has a longer 2-azobenzene-4,4′-biphenyldicarboxylic acid linker.[24] In the simulations, the CO2 uptake isotherms of the MOFs with 100 % trans azobenzene is compared with the isotherms of the MOFs with 100% of the azobenzene groups in cis state. (Due to the photoexcitation and switching properties of azobenzene,[2a] MOFs with 100% cis azobenzene have not yet been experimen-tally realized. The switching yield, i.e., the amount of cis azoben-zene obtained in the experiments, is not stated in ref. [23]). The simulation results show a reduction of the CO2 uptake amount upon trans-to-cis isomerization for both MOF structures. Moreover, the simulations could reproduce the
experimentally observed uptake isotherms for the PCN-123 in the pristine state (trans) and after UV irradiation for 1 h (cis state) on a quantitative level. The large CO2 uptake decrease observed for the sample that was stored in the dark for 5 h after UV irradiation was not quantitatively reproduced by the simu-lations. Careful analyses of the simulation data by the authors show small changes of the accessible pore volume and surface area and changes of the CO2 density in the pores. The simula-tion results were interpreted that the change of the interaction (long-range dispersion and electrostatic interactions), rather than shielding effects, are responsible for changing the CO2 uptake amount in PCN-123 upon trans-cis isomerization.
Macromol. Rapid Commun. 2017, 1700239
Figure 3. Photoswitching in PCN-123. a) UV-induced trans-to-cis and heat-induced cis-to-trans isomerization in the PCN-123 and an illustration that shows the CO2 uptake. b) CO2 adsorption isotherms of PCN-123 at 295 K. The trans-to-cis photoisomerization decreased the uptake amount (half-filled blue), repeating the measurement after 5 h while the sample was stored in the dark resulted in a further decrease of the uptake mount (fully filled blue). Thermal relaxation increased the uptake amount again (orange spheres). Reproduced with permission.[23] Copyright 2012, Royal Society of Chemistry.
Figure 4. Dye-molecule release from Azo-IRMOF-74-III. a) Azo-IRMOF-74-III structure has one-dimensional pores with azobenzene side groups projecting into the pores. The pore aperture changes when all azobenzene groups are in trans configuration (left) or all azobenzene side groups in the cis configuration (right). b) Propidium iodide released from azo-IRMOF-74-III under sequential exposure to 676 nm and 403 nm lasers. The power of the 403 nm laser is 50 mW; the power of the 676 nm laser is not stated in ref. [25]. The irradiation with light close to the isosbestic wavelength causes simultaneous trans-to-cis and cis-to-trans isomerization. The authors explain that this wagging motion of the azobenzene side groups results in the release of the guest molecules. Reproduced with permission.[25] Copyright 2013, Royal Society of Chemistry.

© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (5 of 14)
www.advancedsciencenews.com www.mrc-journal.de
Azo-IRMOF-74-III, an isoreticular expansion of MOF-74, was prepared by O. Yaghi et al.[25] The cell parameters were obtained from XRD and further characterization of the struc-ture was done by cross-polarization magic-angle spinning (CP/MAS) and 13C NMR. While the pore diameter is 0.83 nm for the Azo-IRMOF-74-III with the azobenzene side groups in the trans state, the pore diameter would increase up to 1.03 nm when all azobenzene moieties could be switched to cis. This means that the pore aperture size could be increased by up to 20% by trans-to-cis photoswitching; see Figure 4a. N2 adsorption isotherms were measured to confirm its porosity and a spe-cific BET surface of 2410 m2 g−1 was determined. The revers-ible photoswitching of the azobenzene linker in solution was shown by UV-vis spectroscopy. While no spectroscopy data of the photoswitching of the Azo-IRMOF-74-III was shown, the functionality of the MOF upon light irradiation was demon-strated by controlling the release of the guest molecules. The one-dimensional pores were loaded with propidium iodide, a luminescent dye with a size of 0.8 nm × 1.1 nm × 1.6 nm. The sample acted as a container storing the dye molecules, which were not (or only very slowly) released from the channels until the sample was irradiated by light. In Figure 4b, the release of the dye molecule from the MOF, first under irradiation with 676 nm and then under irradiation with 403 nm, is shown. The authors discussed that the light irradiation with the 403 nm laser (50 mW or 408 nm; see ref. [25]), which has a wavelength close to the isosbestic point, stimulated cis–trans wagging motions of the azobenzene side groups and the dye molecules were released from the MOF channels. The slow release, which took significantly longer than 1 day (see Figure 4b), was attri buted to the long and narrow one-dimensional pores
and to the interactions between the guest molecules and the host MOF.
The optically triggered release of guest molecules from powder MOFs was also demonstrated by C. Wang et al.[26] A water-stable UiO-68 MOF with azobenzene side groups was used to store rhodamine B. In the pristine MOF, cyclodextrin capping hindered the release of the guest molecules. UV irra-diation resulted in the release of the guest molecules.
D. Bléger, S. Hecht, and co-workers reported the synthesis of o-fluoroazobenzene that can be reversibly switched with visible light only.[3] The ortho-fluoro substituents on the phenyl ring result in a separation of the n–π* bands due to the electron-withdrawing effect on the central NN bond by the introduction of fluorine atoms. Thus, the band of the trans isomer of the fluorinated azobenzene is redshifted in comparison to the band of the cis isomer, unlike to plain azobenzene (C12H10N2) where the n–π* bands of both isomers are at an identical wavelength. Based on this work, MOF linker molecules with fluorinated azobenzene side groups were prepared, namely 2-[(2,6-difluo-rophenyl)diazenyl]terephthalic acid (F2AzoBDC). UV-vis spec-troscopy of the linker molecules in DMSO solution show that irradiation with green light (>500 nm) results in trans-to-cis isomerization, while violet light (405 nm) results in cis-to-trans isomerization (Figure 5a,b). The photostationary states (PSS) of the linker molecules in solution were determined to 88% trans (12% cis) upon violet light irradiation and 86% cis upon green light irradiation.
The F2AzoBDC molecules were used to prepare two terephthalic-acid-based MOFs, F-azo-MIL-53(Al) and F-azo-UiO-66(Zr). The crystalline structures of both prepared MOFs, which are identical to MIL-53(Al) and UiO-66(Zr),
Macromol. Rapid Commun. 2017, 1700239
Figure 5. Photoisomerization of ortho-fluoroazobenzene MOFs. a) Sketch of the F2AzoBDC molecule in the trans and cis configuration. b) UV-vis absorption spectra of F2AzoBDC in its thermodynamically stable trans (E) form (black line) and the photostationary state (PSS) mixtures of different wavelengths in DMSO. Representation of the proposed structures after cell-parameter refinement of F-azo-UiO-66(Zr) (c,d) and F-azo-MIL-53(Al) (e,f). c) Octahedral supercages and d) tetrahedral smaller cages of F-azo-UiO-66(Zr). e) Disordered F-azo-groups located in the pores of F-azo-MIL-53(Al). f) Perspective view of the spatial arrangement in F-azo-MIL-53(Al) and conformation of F-azo-groups with potentially stabilizing edge-to-face aromatic interactions (red dotted line). Reproduced with permission.[27] Copyright 2015, Wiley-VCH.
© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (6 of 14)
www.advancedsciencenews.com www.mrc-journal.de
respectively, were verified by XRD. The structural differences are the fluorinated azobenzene side groups which point into the pores (Figure 5c–f). While UV-vis and infrared spectroscopy of the F-azo-UiO-66(Zr) show trans-cis isomerization upon green and violet light irradiation; only a drastically reduced efficiency of the photoswitching was found for F-azo-MIL-53(Al). This is explained by the steric hindrance due to the small pore size and the close packing of the azobenzene side groups in F-Azo-MIL-53(Al). UV-vis spectroscopy of F-azo-UiO-66(Zr) show that the position of the n–π* band was blue-shifted by 22 nm upon green light irradiation and its original position could be almost recovered by irradiation with violet light. The initial UV-vis spectra, i.e., the pristine trans state, could be obtained by thermal relaxation. When irradiating with UV light with a wavelength of 310 nm or 365 nm, the n–π* bands experienced blue-shifts by only 10 nm and 13 nm, respectively, indicating the higher trans-to-cis switching efficiency by green light. The green-violet-photoswitching cycle was repeated several times, showing the reversible switching behavior in F-azo-UiO-66(Zr). The light-induced alteration of CO2 uptake was investigated at 273 K. Both azobenzene-functionalized MOF structures have a significantly reduced CO2 loading capacity in comparison to the corresponding frameworks without azobenzene moieties. This is a result of the partial occupation of the pores by the pendant azobenzene moieties. It was found that the CO2 uptake by the F-azo-UiO-66(Zr) MOF was not affected by green light irra-diation, although the trans-to-cis isomerization was shown by UV-vis spectroscopy. On the other hand, green light irradiation results in a decrease of the uptake by the F-azo-MIL-53(Al) by approximately 10%. When the samples were measured again in the dark, the initial CO2 adsorption isotherms were observed. Based on these data, the reduction of the CO2 uptake amount by F-azo-MIL-53(Al), which has a drastically reduced photo-switching efficiency, is interpreted as thermal effects due to the light irradiation. The F2AzoBDC linker was also used for photo-switchable MOF films and membranes (see Figure 9 later).
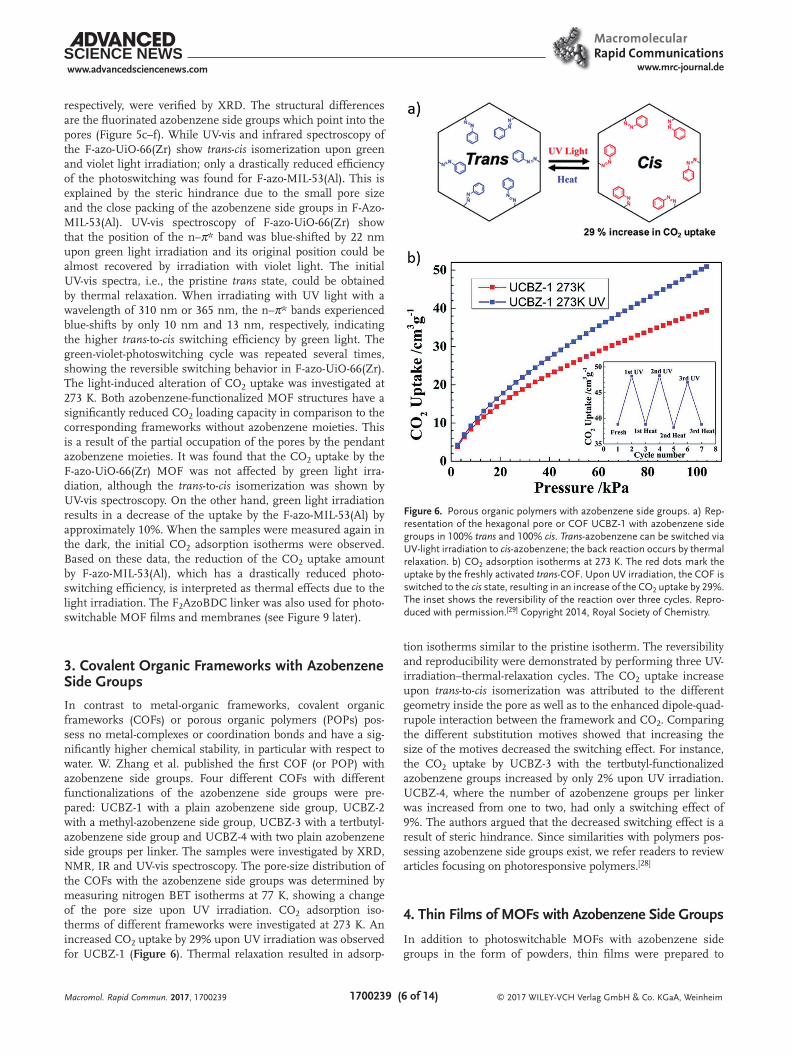
3. Covalent Organic Frameworks with Azobenzene Side Groups
In contrast to metal-organic frameworks, covalent organic frameworks (COFs) or porous organic polymers (POPs) pos-sess no metal-complexes or coordination bonds and have a sig-nificantly higher chemical stability, in particular with respect to water. W. Zhang et al. published the first COF (or POP) with azobenzene side groups. Four different COFs with different functionalizations of the azobenzene side groups were pre-pared: UCBZ-1 with a plain azobenzene side group, UCBZ-2 with a methyl-azobenzene side group, UCBZ-3 with a tertbutyl-azobenzene side group and UCBZ-4 with two plain azobenzene side groups per linker. The samples were investigated by XRD, NMR, IR and UV-vis spectroscopy. The pore-size distribution of the COFs with the azobenzene side groups was determined by measuring nitrogen BET isotherms at 77 K, showing a change of the pore size upon UV irradiation. CO2 adsorption iso-therms of different frameworks were investigated at 273 K. An increased CO2 uptake by 29% upon UV irradiation was observed for UCBZ-1 (Figure 6). Thermal relaxation resulted in adsorp-
tion isotherms similar to the pristine isotherm. The reversibility and reproducibility were demonstrated by performing three UV-irradiation–thermal-relaxation cycles. The CO2 uptake increase upon trans-to-cis isomerization was attributed to the different geometry inside the pore as well as to the enhanced dipole-quad-rupole interaction between the framework and CO2. Comparing the different substitution motives showed that increasing the size of the motives decreased the switching effect. For instance, the CO2 uptake by UCBZ-3 with the tertbutyl-functionalized azobenzene groups increased by only 2% upon UV irradiation. UCBZ-4, where the number of azobenzene groups per linker was increased from one to two, had only a switching effect of 9%. The authors argued that the decreased switching effect is a result of steric hindrance. Since similarities with polymers pos-sessing azobenzene side groups exist, we refer readers to review articles focusing on photoresponsive polymers.[28]
4. Thin Films of MOFs with Azobenzene Side Groups
In addition to photoswitchable MOFs with azobenzene side groups in the form of powders, thin films were prepared to
Macromol. Rapid Commun. 2017, 1700239
Figure 6. Porous organic polymers with azobenzene side groups. a) Rep-resentation of the hexagonal pore or COF UCBZ-1 with azobenzene side groups in 100% trans and 100% cis. Trans-azobenzene can be switched via UV-light irradiation to cis-azobenzene; the back reaction occurs by thermal relaxation. b) CO2 adsorption isotherms at 273 K. The red dots mark the uptake by the freshly activated trans-COF. Upon UV irradiation, the COF is switched to the cis state, resulting in an increase of the CO2 uptake by 29%. The inset shows the reversibility of the reaction over three cycles. Repro-duced with permission.[29] Copyright 2014, Royal Society of Chemistry.

© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (7 of 14)
www.advancedsciencenews.com www.mrc-journal.de
take advantage of switching the adsorption and diffusion properties of the guest molecules. Although various options to prepare MOF films on solid substrates exist,[10] all films of MOFs with azobenzene side groups were prepared in a layer-by-layer fashion using liquid phase epitaxy so far (Figure 7a). These MOF films are referred to as surface-mounted MOF (SURMOFs).[11,30] The thickness of SURMOFs is controlled by the number of synthesis cycles and the crystalline orientation can be controlled by substrate functionalization.[31] SURMOFs are usually strongly bonded to the substrate[32] and heteroepi-taxy allows the preparation of different SURMOF films on top of each other.[33] SURMOF films can be grown to be very smooth[34] and pin-hole free.[35] Thus, SURMOFs in combina-tion with a quartz crystal microbalance (QCM) are well-defined model systems for adsorption[36] and diffusion[37] studies in MOFs. As a result of their small thickness and the fact that SURMOFs can be grown without diffusion-hindering surface barriers,[37b,38] the uptake and release of guest molecules is typi-cally very fast. Further details of SURMOFs and their synthesis can be found in ref. [11,30,39].
The remote control over the SURMOF properties was intro-duced by using linkers with azobenzene side groups. To take advantage of switching the diffusion properties, a two-layer MOF film was prepared: a bottom layer with a pillared-layer Cu2(BPDC)2(BiPy) MOF structure was prepared on a MUD-functionalized gold substrate (BPDC: biphenyl-4,4′-dicarboxylic acid, BiPy: 4,4′-bipyridine, MUD: 11-mercapto-1-undecanol). This layer has no photoswitchable groups and acts as a passive container, which can store molecules. On top of this bottom layer, a pillared-layer MOF of type Cu2(AzoBPDC)2(BiPy) with the same lattice parameters was prepared (AzoBPDC: 2-azobenzene-4,4′-biphenyldicarboxylic acid). This top layer has photoswitchable azobenzene side groups (Figure 7a). UV-vis spectroscopy showed small reversible intensity changes of the π–π* and n–π* bands upon UV and visible light irradiation. However, the switching yield was not determined. By using a QCM, the uptake of 1,4-butanediol by the two-layered SURMOF was investigated. It was found that the uptake rate and the dif-fusion coefficient decreased by more than one order of magni-tude when switching the top-layer from trans to cis (Figure 7b).
Macromol. Rapid Commun. 2017, 1700239
Figure 7. Optically triggered release from a two-layered MOF film. a) Sketch of the synthesis of two-layered SURMOF. The MOF film is prepared in a layer-by-layer fashion by successively exposing the functionalized substrate surfaces to solutions of the metal complexes and of the organic linker molecules. By exchanging the organic linker molecule during the synthesis, a different SURMOF is grown on top, resulting in a two-layered structure. The pore windows of the Cu2(BPDC)2(BiPy) bottom layer and the Cu2(AzoBPDC)2(BiPy) top layer in the [001] direction are shown. The azobenzene side groups in the top layer can be reversibly switched by light. b) Butanediol uptake by the two-layer SURMOF in trans and in cis state measured by QCM. By switching the top layer from trans to cis, the uptake rate was reduced by a factor of 15. c) The optically triggered release from a molecular container was demonstrated for butanediol. The two-layered SURMOF was loaded with guest molecules and the top layer was closed by irradiation with UV light. Although the release was started, only a small leakage rate was observed by QCM until the top layer was opened by irradiation with visible light. Reproduced with permission.[40] Copyright 2012, American Chemical Society.

© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (8 of 14)
www.advancedsciencenews.com www.mrc-journal.de
Switching the mass transfer properties was employed to dem-onstrate the remote-controlled release of guest molecules from a nanoporous container (Figure 7c). Before the experiment, the two-layered SURMOF was loaded with butanediol and the top layer was switched by UV light to its cis state. Since the mass transfer in the cis state is very slow, this state is referred to as “closed”. Then, the gas atmosphere above the SURMOF was switched to pure argon and the release of guest molecules was actually started. A small release rate of the butanediol (of less than 0.5 µg cm−2 h−1) was measured by QCM. After approxi-mately 2 h, the sample was irradiated with visible light and the top layer was switched to its trans form resulting in an increase of the mass transfer (i.e., release) rate, that is, the top layer was opened. The increase of the release rate was measured by QCM and the remote-controlled release of guest molecules from a nanoporous container was experimentally demonstrated.
MOF membranes possess enormous potential for an effi-cient, energy-saving separation of molecular mixtures.[41] Using membranes made of MOFs with azobenzene side groups could enable the dynamic control of permeation and separation of the molecular mixture. For this purpose, MOF thin films with a pillared-layer structure of type Cu2(AzoBPDC)2(AzoBiPyB) were prepared on mesoporous aluminum oxide substrates (Figure 8a) (AzoBiPyB: 4,4′-(2-(phenyldiazenyl)-1,4-phenylene)dipyridine (or dipyridylazobenzene)[42]). Photoswitching of the azobenzene side groups was investigated by UV-vis and IR spec-troscopy. UV-vis spectroscopy shows small reversible intensity changes of the π–π* and n–π* bands upon UV- and blue-light irradiation. IR spectroscopy enabled the determination of the photoisomerization yield. A minimum cis ratio of approxi-mately 0% was determined after irradiation with blue light (trans state) and a maximum cis ratio of approximately 63% was
Macromol. Rapid Commun. 2017, 1700239
Figure 8. Tunable molecular separation by photoswitchable MOF membranes. a) Schematic illustration of continuously tunable, remote-controllable molecular selectivity by a photoswitchable MOF membrane. The nanoporous membrane separates the molecular feed mixture (red and blue molecules; left-hand side). The molecular separation factor, giving the composition of the permeation flux (right-hand side), can be continuously tuned by light irradiation. Structures of Cu2(AzoBPDC)2(AzoBiPyB) MOF with all azobenzene groups in trans state and in cis state are shown. The photoisomeriza-tion from trans to cis is performed by irradiating with UV light of 365 nm, while cis-to-trans isomerization is performed by irradiating with blue light of 455 nm. b) The separation of H2:CO2 mixture is shown where the membrane was irradiated with 365 nm and 455 nm for 5 min each. Permeances of H2 and CO2 are shown as black solid squares and black open squares, respectively, with logarithmic scale on the right-hand side. The molecular selectivities (or separation factors) are shown as red spheres with the scale on the left-hand side. c) Correlation of the H2:CO2 separation factor with the ratio of cis azobenzene. The adjustment of the cis-azobenzene ratio, which is realized by short illumination times or by simultaneous irradiation with UV and blue light, results in a continuous tuning of the separation factor. Reproduced with permission.[43] Copyright 2016, Nature Publishing Group.

© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (9 of 14)
www.advancedsciencenews.com www.mrc-journal.de
determined after UV irradiation (cis state). The separation of a molecular hydrogen–carbon dioxide mixture was investigated using a Wicke-Kallenbach setup where the membrane can be illuminated in situ.[43] This allows the trans-cis photoswitching of the MOF membrane during the separation experiment. While the separation factor of hydrogen versus carbon dioxide was approximately 3 for the trans membrane, it increased to 8 when the azobenzene moieties were switched to cis. By irra-diating with 365 nm and 455 nm, i.e., by switching the azoben-zene side groups between trans and cis, the separation factor can be reversibly switched (Figure 8b). This shows that the molecular membrane separation factor was controlled by photo switching the azobenzene side groups.
The membrane separation by photoswitchable MOF films offers a further interesting feature. The irradiation of the azobenzene-containing MOF film for short durations or the simultaneous irradiation with light of different wavelengths enables the adjustment of the cis azobenzene ratio between the minimum (≈0%) and maximum (≈63%) value (Figure 8c). It is shown that adjusting the cis:trans azobenzene ratio enables the adjustment of the separation factor. Thus, the molecular composition of the permeate can be continuously tuned in a dynamic, remote-controlled manner.
By using F2AzoBDC linker molecules, Cu2(F2AzoBDC)2(dabco) SURMOFs could be prepared.[44] The reversible trans-to-cis photo isomerization upon irradiation with green (530 nm) light and cis-to-trans photoisomerization upon irradiation with violet (400 nm) light were shown by UV-vis (Figure 9b) and IR spec-troscopy. The switching yields were determined by IR spectro-scopy to 13% cis azobenzene after violet light irradiation (trans state) and 86% cis azobenzene after green light irradiation (cis state). The switching yield is very similar to the switching yield obtained in DMSO solution; thus, the authors concluded that there is no hindrance to the isomerization. QCM uptake experiments using butanediol as a probe molecule showed that the uptake amount increases by 47 ± 20% upon trans-to-cis isomerization. The Cu2(F2AzoBDC)2(dabco) SURMOFs were prepared on mesoporous substrates to investigate the membrane separation properties. While no switching effect was found for the H2:CO2 mixture, the separation of H2:C2H4 and H2:C3H6 (Figure 9c) mixtures could be photoswitched. For both mixtures, the separation factor reversibly decreased/increased upon trans-to-cis and cis-to-trans isomerization. The authors explained that, unlike switching in the large pore Cu2(AzoBPDC)2(AzoBiPyB) membrane, the switching effect is not dominated by switching the azobenzene dipole moment. It is based on the changing pore structure. In the trans MOF, the azobenzene side groups point into the pore and hinder the diffusion of the guest molecules (ethene or propene). In the cis state, the azobenzene side groups are bent, do not point into the pore, and leave larger pore diam-eters, as sketched in Figure 9a. This enables a faster diffusion of
Macromol. Rapid Commun. 2017, 1700239
Figure 9. Switching thin films of azobenzene-containing MOFs with vis-ible light. a) Structure of Cu2(F2AzoBDC)2(dabco), in which the fluorinated azobenzene side groups can be reversibly switched from trans to cis by irra-diation with green light (530 nm) and from cis to trans by irradiation with violet light (400 nm). A view of the pore window along the [001] direction is shown. b) UV-vis absorbance spectra of the Cu2(F2AzoBDC)2(dabco) SURMOF on quartz. The black line shows the pristine SURMOF after thermal relaxation to its trans state, the green line shows the spectrum after 15 min green light irradiation, and the violet line after irradiation with violet light. The black and violet spectra are virtually identical. The inset shows the absorbance at 320 nm measured during five switching cycles. The large SURMOF band at approximately 250 nm can be assigned
to the Cu node in the framework.[45] c) H2:C3H6 membrane separation. The membrane was illuminated in situ by 400 nm and 530 nm light for 20 min each. Permeances of H2 (solid square) and C3H6 (open squares) are shown on the left-hand side of the graphs, whereas the resulting sepa-ration factors (red spheres) are shown on the right-hand side. Reproduced with permission.[44] Copyright 2017, Wiley-VCH.

© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (10 of 14)
www.advancedsciencenews.com www.mrc-journal.de
the guest molecules. Diffusion of H2 is not significantly affected by the changes of the pore opening, since it is much smaller. Theoretical simulations and calculations proving or disproving this hypothesis would be desirable.
5. MOFs as Model-System for Azobenzene Isomerization
The well-defined, crystalline structure of MOFs can be used as an advantage in comparison to other materials incorporating photoswitchable molecules. For instance, it enables meaningful calculations by advanced theoretical tools like density func-tional theory (DFT) calculations.[45,46]
The experimental data based on UV-vis spectroscopy and on QCM uptake experiments indicate that azobenzene moieties in Cu2(AzoBPDC)2(BiPy) SURMOFs undergo trans-cis isomeri-zation upon UV or visible light irradiation; however, there is no photoswitching in Cu2(NDC)2(AzoBiPy) SURMOFs. For a detailed understanding, time-dependent DFT calculations were performed to investigate the isomerization of the azobenzene moieties in the pores of these two MOFs (Figure 10). The cal-culated potential energy landscape shows that the azoben-zene isomerization in Cu2(AzoBPDC)2(BiPy) is similar to the isomerization of free azobenzene. However, the azobenzene isomerization in Cu2(NDC)2(AzoBiPy) is sterically hindered.[42] The calculations show that a relaxation of the framework during the isomerization process would be required to enable trans-cis switching. This explains the experimental findings.
Cu2(NDC)2(AzoBiPy), also referred to as Cu-CAU-5, has the same structure as Zn-CAU-5.[19,20] In ref. [19], the trans-cis photoswitching of the linker in solution is clearly visible from the change of the π–π* and n–π* azobenzene bands.
When the powder MOF was prepared with this linker, only small changes of the n–π* and even smaller changes of the π–π* were observed. The authors explain this by the steric hindrance, potentially caused by the two-fold interpenetra-tion in these powder MOFs.[19] Please note that the interpen-etration in SURMOFs is usually suppressed by the synthesis process.[47] It might be speculated that only a small amount of the azobenzene moieties can be switched in the Cu-CAU-5 powder MOFs, possibly only at defect sites. This results in the small observed changes in the UV-vis spectrum. On the other hand, it is also acknowledged that in other MOF or SURMOF structures the photoswitchable functionality is demonstrated by uptake or membrane experiments and IR spectroscopy clearly shows the photoisomerization, although the observed intensities of the π–π* and n–π* azobenzene bands as well as their light-induced changes are very small. One example is SURMOF Cu2(AzoBPDC)2(AzoBiPyB), in which the rela-tively small azobenzene bands are a result of the large absorp-tion bands of the copper metal nodes in the MOF.[43] Thus, it requires further investigation whether the minor changes of the UV-vis spectra indicate that powder Cu2(NDC)2(AzoBiPy) MOF is photoswitchable or if the photoswitching is (fully) sterically hindered as in Cu2(NDC)2(AzoBiPy) SURMOFs. For instance, different experimental conditions, such as the different synthesis solvents (ethanol vs DMF and methanol), the different synthesis temperatures (60 °C vs 120 °C), the dif-ferent setups, and the different light sources, might potentially result in different photoswitching behavior. It could be argued that a higher light intensity stimulating the trans-to-cis isomer-ization might result in successful photoswitching in the MOF.
Taking advantage of the nanoporous character of MOF, thermal cis-to-trans relaxation of the azobenzene moieties can be investigated. SURMOFs of type Cu2(DMTPDC)2(AzoBiPyB)
Macromol. Rapid Commun. 2017, 1700239
Figure 10. Photoswitching of two similar azobenzene-containing linkers and MOFs. a) AzoBPDC and b) AzoBiPy linkers in the trans state can be switched to the cis state by UV light and vice versa by visible light. Side views of the structures of c) Cu2(AzoBPDC)2(BiPy) MOF and d) Cu2(NDC)2(AzoBiPy) MOF are shown with the azobenzene side groups in trans state. Cu atoms are plotted in orange, O red, C grey, N blue, and H white. While the photoi-somerization in MOF Cu2(AzoBPDC)2(BiPy) is enabled (e), the photoisomerization in MOF Cu2(NDC)2(AzoBiPy) is sterically hindered (f). Reproduced with permission.[42] Copyright 2015, The PCCP Owners’ Society.

© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (11 of 14)
www.advancedsciencenews.com www.mrc-journal.de
(DMTPDC: 2,2″-dimethyl-[1,1′:4′,1″-terphenyl]-4,4″-dicarboxylic acid) and Cu2(BDC)2(AzoBiPyB) were prepared and studied in ultrahigh vacuum (UHV) to ensure that no guest molecules are inside the pores.[48]
In Cu2(DMTPDC)2(AzoBiPyB), the azobenzene moiety points into the pore and has no interaction with other parts of the MOF framework; thus, it may serve as a model system for an isolated azobenzene moiety. It should be stressed that pure plain azobenzene is a crystalline solid at room temperature, which cannot undergo trans-to-cis isomerization. The photo-switching of azobenzene in solution or embedded in polymer matrices is usually enabled; however, the switching parameters are significantly influenced by the molecular environment.
The trans-cis isomerization of the isolated azobenzene moi-eties in the Cu2(DMTPDC)2(AzoBiPyB) SURMOF was inves-tigated by using IR spectroscopy. After irradiating the sample with UV light, i.e., after partial trans-to-cis isomerization, the azobenzene moieties thermally relaxed to the trans state in the dark. The time evolution of the intensities of the IR vibration bands of the trans and cis azobenzene moieties enabled the determination of the rate constants. By repeating the experi-ments at different temperatures and using an Arrhenius-plot, the activation energy for the thermal cis-to-trans isomerization was determined (Figure 11). The determined activation energy of 1.09 eV is in very good agreement with the activation energy that was calculated for the isolated azobenzene.[49] Moreover, a thermal cis-to-trans relaxation time constant of about 3 weeks at room temperature can be extrapolated from the Arrhenius plot.
6. Impact of the Photoswitching on the Host-Guest Interaction
For virtually all applications of MOFs, the host-guest interac-tion is crucial. Thus, the alteration of host-guest interaction is
essential for the smart application of photoswitchable MOFs. Although significantly more research is required for gaining a detailed understanding, a few key preliminary conclusions can be drawn.
The switching effect on the guest-host interaction was investigated for Cu2(BDC)2(AzoBiPyB) SURMOFs using dif-ferent alkanes, alcohols and diols as guest molecules.[50] It was found that the adsorption amount of polar molecules increases upon trans-to-cis photoswitching. No significant change of the adsorption capacity was found for nonpolar molecules. For the investigated molecules, the interaction increases with increasing dipole moment of the guests (Figure 12). Based on the uptake data, the interaction between guest molecules and the azobenzene moiety can be described as molecular dipole-dipole interaction, also referred to as the Keesom interaction. This means the change of the azobenzene dipole moment (0 D in trans and 3 D in cis) causes the switching effect. Steric effects seem to have only a minor effect, which is a result of the large pore size and the flexibility of the investigated MOF structure.
Macromol. Rapid Commun. 2017, 1700239
Figure 11. Arrhenius plot of thermal cis-to-trans isomerization of the azobenzene side group. Rate constants of the cis-to-trans isomerization of azobenzene side groups in Cu2(DMTPDC)2(AzoBiPyB) is plotted versus the inverse temperature. The activation energy is determined from the slope of a linear fit to the data points (dotted lines). The AzoBiPyB linker molecule with the azobenzene side groups is sketched in cis and trans state. Repro-duced with permission.[48] Copyright 2015, Royal Society of Chemistry.
Figure 12. Impact of the photoswitching on the host-guest interaction. a) The azobenzene side group of the AzoBiPyB linker undergoes photo-isomerization from trans to cis when exposed to UV light. cis-AzoBiPyB isomerizes to the trans state due to irradiation with visible light or thermal relaxation. b) One unit cell of Cu2(BDC)2(AzoBiPyB) SURMOF with all azobenzene side groups in their trans state. c) The ratio of the uptake amounts of guest molecules by Cu2(BDC)2(AzoBiPyB) in the cis and trans states, mcis:mtrans, versus the dipole moment of the guest molecules. The dotted line shows the result of the molecular dipole–dipole (Keesom) interaction. The amount of cis azobenzene was determined by IR spec-troscopy to approximately 32% in the cis-rich state (referred to as cis) and 0% in the trans-rich state (referred to as trans). Reproduced with permis-sion.[50] Copyright 2015, Wiley-VCH.

© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (12 of 14)
www.advancedsciencenews.com www.mrc-journal.de
Macromol. Rapid Commun. 2017, 1700239
The dipole-dipole interaction also explains the increase of butanediol uptake upon trans-to-cis isomerization as observed in refs. [40,42,43]. The stronger attractive interaction of butan-ediol with cis azobenzene than with trans azobenzene is also the reason for the decrease of the diffusion coefficient upon trans-to-cis isomerization, as found in ref. [40]. The switching of the azobenzene dipole moment also changes the interaction with CO2, which possesses a large quadrupole moment. This means the attractive interaction between CO2 and the azoben-zene moiety significantly increases by trans-to-cis isomerization, reducing the diffusion coefficient. Since nonpolar molecules are not affected by the dipole moment switching, their diffu-sion coefficients are not affected by the azobenzene switching. This results in the switching of the separation factors of the H2:CO2 or N2:CO2 mixtures as observed in ref. [43]. For UCBZ COFs, the CO2 adsorption capacity increases upon trans-to-cis isomerization, presumably as result of the azobenzene dipole moment switching.[29]
On the other hand, an increase of the adsorption capacity upon trans-to-cis isomerization was observed for methane in Cr-MIL-101-urea-azobenzene and Cr-MIL-101-amide-azobenzene.[22] Since methane is nonpolar, this capacity increase cannot be explained by dipole-dipole interaction and further effects must have an influence. In PCN-123 MOFs
(i.e. Azo-MOF-5), the CO2 adsorption capacity decreases by trans-to-cis switching.[23] The authors explain the observed capacity decrease by a blocking of the attractive CO2 adsorption sites close to the metal node by the cis azobenzene. The decrease is also verified by DFT and MC calculations; however, the calcu-lations are interpreted that there is no shielding effect, but a decrease of the attractive interaction upon trans-to-cis isomeri-zation.[24] The switching of the separation factor of H2:CO2, H2:C2H4 and H2:C3H6 mixtures by Cu2(F2AzoBDC)2(dabco) membranes can also not be explained by dipole-dipole inter-action.[44] It seems that steric effects result in the observed switching of the separation factor.
Based on these results, we believe that the effect of trans-cis isomerization in large MOF pores on the guest-host interaction is based on the switching of the azobenzene dipole moment, resulting in switching the dipole-dipole or dipole-quadrupole interaction. However, this effect can be superimposed by steric effects, in particular in MOFs with small pores.
7. Conclusion
Photoswitchable materials offer a huge potential with respect to many smart applications. Here, nanoporous MOFs
Table 1. Overview of MOFs with photoswitchable azobenzene side groups. The frameworks are listed according to their date of publication.
MOF Effect of trans-to-cis Photoisomerization Comment Ref.
Zn2(NDC)2(AzoBiPy) – First MOF made of linkers with photoswitchable azoben-
zene side groups
[19]
Azo-ZIF-8 – – [21]
PCN-123 Decrease of CO2 adsorption capacity cis-to-trans by thermal relaxation [23]
Co2(NDC)2(AzoBiPy), Cu2(NDC)2(AzoBiPy),
Zn2(BPDC)2(AzoBiPy), Zn2(CCA)2(AzoBiPy)
– Comparison of four pillared-layer MOFs with azoben-
zene side groups
[20]
Cr-MIL-101-urea-azobenzene,
Cr-MIL-101-amide-azobenzene
Increase of methane adsorption capacity Azobenzene introduced by PSM; Reversible photo-
switching by UV and blue light
[22]
Azo-IRMOF-74-III – Release of dye upon illumination with isosbestic wave-
length (408 nm, 50 mW)
[25]
SURMOF Cu2(AzoBPDC)2(BiPy) Increase of adsorption capacity and decrease of diffu-
sion coefficient of butanediol
Two-layered MOF thin film [40]
COF UCBZ-1, 2, 3 and 4 Increase of CO2 adsorption capacity cis-to-trans by thermal relaxation;
different functionalizations change switching effect
[29]
SURMOF Cu2(AzoBPDC)2(BiPy),
Cu2(NDC)2(AzoBiPy)
– Investigation of steric hindrance for trans-cis
isomerization
[42]
SURMOF Cu2(BDC)2(AzoBiPyB),
Cu2(DMTPDC)2(AzoBiPyB)
– Investigation of thermal cis-to-trans relaxation [48]
SURMOF Cu2(BDC)2(AzoBiPyB) Increase of adsorption capacity of various alkanes,
alcohols, and glycols
Investigation of switching effect [50]
F-Azo-UiO-66(Zr), F-Azo-MIL-53(Al) No effect on CO2 adsorption Azobenzene switchable with visible light only; steric
hindrance suppresses switching in F-Azo-MIL-53(Al)
[27]
UiO-68-azo Release of rhodamine B by release of β-cyclodextrin-
capping upon UV irradiation
[26]
SURMOF Cu2(AzoBPDC)2(AzoBiPyB) Increase of H2:CO2 and N2:CO2 membrane separa-
tion factor
Separation factor continuously tunable [43]
SURMOF Cu2(F2AzoBDC)2(dabco) Decrease of H2:C3H6 and H2:C2H4 membrane separa-
tion factor; Increase of butanediol adsorption capacity
Azobenzene switchable with visible light only [44]

© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (13 of 14)
www.advancedsciencenews.com www.mrc-journal.de
Macromol. Rapid Commun. 2017, 1700239
with photoswitchable azobenzene side groups that enable the remote control of the key properties of the material are dis-cussed. Pioneered by the work of N. Stock, followed by H. C. Zhou and others, more than 20 different MOF structures have been published, see Table 1.
While UV light is usually required for the photoswitching, functionalization of the azobenzene moiety with fluorine ena-bles the preparation of MOF materials that can be switched with visible light only. The applications of the smart MOF materials, which are prepared in the form of powders or thin films, range from photoswitching the uptake amount of the guest molecules to remote-controlled release of dye and guest molecules. Mem-branes of these photoswitchable MOFs enable the dynamic control of the permeation of gases and of the separation factor of molecular mixtures. In addition, the selection factor can be continuously tuned by adjusting the trans-cis azobenzene ratio.
Although a detailed understanding of the photoswitching in the MOF pores had been a minor priority in some ground-breaking articles, precise spectroscopic data of the photo-switching in the MOF is crucial for understanding and optimizing the switching effect. This is particularly impor-tant because detailed investigations have shown that the isomerization of the azobenzene side groups can be sterically hindered in many MOF structures, although the corresponding azobenzene-containing linker molecules in solution are revers-ibly photoswitchable.
We believe that MOFs possessing azobenzene side groups, but also other photoswitchable MOF structures, will allow fur-ther fascinating experiments and applications. In addition to the switching of the adsorption and diffusion behavior, other properties like the conduction of the guest molecules or the cat-alytic activity of the MOF might be remote-controllable by the azobenzene moiety.
AcknowledgementsThe authors gratefully acknowledge funding by the Volkswagen Foundation (“Smart Materials with Remote-Controllable, Functional Molecules”), the German Research Foundation DFG (SFB1176-C6), the Fonds der Chemischen Industrie (Lecturer-Award) and the Baden-Württemberg-Foundation.
Keywordsazonbenzene, covalent organic frameworks, metal–organic frameworks, photoswitching, stimuli-sensitive polymers
Received: April 15, 2017Revised: May 31, 2017
Published online:
[1] B. L. Feringa, W. R. Browne, Molecular Switches, Wiley, 2011.[2] a) H. M. D. Bandara, S. C. Burdette, Chem. Soc. Rev. 2012, 41, 1809;
b) G. S. Hartley, R. J. W. Le Fevre, J. Chem. Soc. (Resumed) 1939, 0, 531.
[3] D. Bleger, J. Schwarz, A. M. Brouwer, S. Hecht, J. Am. Chem. Soc. 2012, 134, 20597.
[4] R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Nather, F. Renth, F. Temps, J. Am. Chem. Soc. 2009, 131, 15594.
[5] H. W. Kim, M. Han, H. J. Shin, S. Lim, Y. Oh, K. Tamada, M. Hara, Y. Kim, M. Kawai, Y. Kuk, Phys. Rev. Lett. 2011, 106, 146101.
[6] G. Vives, J. M. Tour, Acc. Chem. Res. 2009, 42, 473.[7] S. Iamsaard, S. J. Asshoff, B. Matt, T. Kudernac,
J. J. L. M. Cornelissen, S. P. Fletcher, N. Katsonis, Nature Chemistry 2014, 6, 229.
[8] M. Yamada, M. Kondo, J.-i. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett, T. Ikeda, Angew. Chem. Int. Ed. 2008, 47, 4986.
[9] S. Kaskel, The Chemistry of Metal-Organic Frameworks: Synthesis, Characterization, and Applications, Vol. 2, Wiley, 2016.
[10] P. Falcaro, R. Ricco, C. M. Doherty, K. Liang, A. J. Hill, M. J. Styles, Chem. Soc. Rev. 2014, 43, 5513.
[11] L. Heinke, M. Tu, S. Wannapaiboon, R. A. Fischer, C. Wöll, Micro-porous Mesoporous Mater. 2015, 216, 200.
[12] a) S. Castellanos, F. Kapteijn, J. Gascon, Crystengcomm 2016, 18, 4006; b) R. D. Mukhopadhyay, V. K. Praveen, A. Ajayaghosh, Mater. Horiz. 2014, 1, 572; c) C. Jones, A. J. Tansell, T. L. Easun, J. Mater. Chem., A 2016, 4, 6714.
[13] A. P. Cote, A. I. Benin, N. W. Ockwig, M. O’Keeffe, A. J. Matzger, O. M. Yaghi, Science 2005, 310, 1166.
[14] a) D. Hermann, H. Emerich, R. Lepski, D. Schaniel, U. Ruschewitz, Inorg. Chem. 2013, 52, 2744; b) N. Yanai, T. Uemura, M. Inoue, R. Matsuda, T. Fukushima, M. Tsujimoto, S. Isoda, S. Kitagawa, J. Am. Chem. Soc. 2012, 134, 4501; c) A. Knebel, L. Sundermann, A. Mohmeyer, I. Strauss, S. Friebe, P. Behrens, J. Caro, Chem. Mater. 2017, 29, 3111; d) K. Müller, J. Wadhwa, J. Malhi, L. Schöttner, H. Schwartz, D. Hermann, U. Ruschewitz, L. Heinke, Chem. Commun. 2017, 53, 8070.
[15] R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, C. J. Kepert, M. R. Hill, Angew. Chem. Int. Ed. 2013, 52, 3695.
[16] J. Zhang, L. B. Wang, N. Li, J. F. Liu, W. Zhang, Z. B. Zhang, N. C. Zhou, X. L. Zhu, Crystengcomm 2014, 16, 6547.
[17] C. H. Liu, W. Zhang, Q. D. Zeng, S. B. Lei, Chem. – Eur. J. 2016, 22, 6768.
[18] I. M. Walton, J. M. Cox, J. A. Coppin, C. M. Linderman, D. G. Patel, J. B. Benedict, Chem. Commun. 2013, 49, 8012.
[19] A. Modrow, D. Zargarani, R. Herges, N. Stock, Dalton Trans. 2011, 40, 4217.
[20] A. Modrow, M. Feyand, D. Zargarani, R. Herges, N. Stock, Zeitschrift für anorganische und allgemeine Chemie 2012, 638, 2138.
[21] S. Bernt, M. Feyand, A. Modrow, J. Wack, J. Senker, N. Stock, Eur. J. Inorg. Chem. 2011, 2011, 5378.
[22] A. Modrow, D. Zargarani, R. Herges, N. Stock, Dalton Trans. 2012, 41, 8690.
[23] J. Park, D. Q. Yuan, K. T. Pham, J. R. Li, A. Yakovenko, H. C. Zhou, J. Am. Chem. Soc. 2012, 134, 99.
[24] R. Huang, M. R. Hill, R. Babarao, N. V. Medhekar, J. Phys. Chem. C 2016, 120, 16658.
[25] J. Brown, B. L. Henderson, M. D. Kiesz, A. C. Whalley, W. Morris, S. Grunder, H. Deng, H. Furukawa, J. I. Zink, J. F. Stoddart, O. M. Yaghi, Chemical Science 2013, 4, 2858.
[26] X. Meng, B. Gui, D. Yuan, M. Zeller, C. Wang, Science Advances 2016, 2, e1600480.
[27] S. Castellanos, A. Goulet-Hanssens, F. Zhao, A. Dikhtiarenko, A. Pustovarenko, S. Hecht, J. Gascon, F. Kapteijn, D. Bleger, Chem. – Eur. J. 2016, 22, 746.
[28] a) A. Natansohn, P. Rochon, Chem. Rev. 2002, 102, 4139; b) F. D. Jochum, P. Theato, Chem. Soc. Rev. 2013, 42, 7468.
[29] Y. Zhu, W. Zhang, Chemical Science 2014, 5, 4957.[30] O. Shekhah, H. Wang, S. Kowarik, F. Schreiber, M. Paulus,
M. Tolan, C. Sternemann, F. Evers, D. Zacher, R. A. Fischer, C. Wöll, J. Am. Chem. Soc. 2007, 129, 15118.

© 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1700239 (14 of 14)
www.advancedsciencenews.com www.mrc-journal.de
[31] D. Zacher, K. Yusenko, A. Betard, S. Henke, M. Molon, T. Ladnorg, O. Shekhah, B. Schupbach, T. de los Arcos, M. Krasnopolski, M. Meilikhov, J. Winter, A. Terfort, C. Wöll, R. A. Fischer, Chem. – Eur. J. 2011, 17, 1448.
[32] Z. B. Wang, P. G. Weidler, C. Azucena, L. Heinke, C. Wöll, Micro-porous Mesoporous Mater. 2016, 222, 241.
[33] Z. Wang, J. Liu, S. Grosjean, D. Wagner, W. Guo, Z. Gu, L. Heinke, H. Gliemann, S. Bräse, C. Wöll, ChemNanoMat 2015, 1, 338.
[34] Z.-G. Gu, A. Pfriem, S. Hamsch, H. Breitwieser, J. Wohlgemuth, L. Heinke, H. Gliemann, C. Wöll, Microporous Mesoporous Mater. 2015, 211, 82.
[35] J. Liu, M. Paradinas, L. Heinke, M. Buck, C. Ocal, V. Mugnaini, C. Woell, Chemelectrochem 2016, 3, 713.
[36] a) M. Cakici, Z.-G. Gu, M. Nieger, J. Burck, L. Heinke, S. Bräse, Chem. Commun. 2015, 51, 4796; b) Z. Gu, S. Grosjean, S. Bräse, C. Wöll, L. Heinke, Chem. Commun. 2015, 51, 8998.
[37] a) W. Zhou, C. Wöll, L. Heinke, Materials 2015, 8, 3767; b) L. Heinke, Z. Gu, C. Wöll, Nat Commun 2014, 5, 4562; c) L. Heinke, C. Wöll, Phys. Chem. Chem. Phys. 2013, 15, 9295; d) L. Heinke, J. Phys. D: Appl. Phys. 2017, 50, 193004.
[38] a) L. Heinke, P. Kortunov, D. Tzoulaki, J. Karger, Phys. Rev. Lett. 2007, 99; b) C. Chmelik, F. Hibbe, D. Tzoulaki, L. Heinke, J. Caro, J. Li, J. Karger, Microporous Mesoporous Mater. 2010, 129, 340; c) F. Hibbe, C. Chmelik, L. Heinke, S. Pramanik, J. Li, D. M. Ruthven, D. Tzoulaki, J. Karger, J. Am. Chem. Soc. 2011, 133, 2804.
[39] J. L. Zhuang, A. Terfort, C. Wöll, Coord. Chem. Rev. 2016, 307, 391.[40] L. Heinke, M. Cakici, M. Dommaschk, S. Grosjean, R. Herges,
S. Bräse, C. Wöll, ACS Nano 2014, 8, 1463.[41] a) T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma,
F. Kapteijn, F. X. Llabrés i Xamena, J. Gascon, Nat Mater 2015, 14,
48; b) R. Ameloot, F. Vermoortele, W. Vanhove, M. B. J. Roeffaers, B. F. Sels, D. De Vos, Nature Chemistry 2011, 3, 382; c) Y. S. Li, F. Y. Liang, H. Bux, A. Feldhoff, W. S. Yang, J. Caro, Angew. Chem. Int. Ed. 2010, 49, 548; d) S. Hurrle, S. Friebe, J. Wohlgemuth, C. Wöll, J. Caro, L. Heinke, Chem. – Eur. J. 2017, 23, 2294.
[42] Z. Wang, L. Heinke, J. Jelic, M. Cakici, M. Dommaschk, R. J. Maurer, H. Oberhofer, S. Grosjean, R. Herges, S. Bräse, K. Reuter, C. Wöll, Phys. Chem. Chem. Phys. 2015, 17, 14582.
[43] Z. Wang, A. Knebel, S. Grosjean, D. Wagner, S. Bräse, C. Wöll, J. Caro, L. Heinke, Nature Communications 2016, 7, 13872.
[44] K. Müller, A. Knebel, F. Zhao, D. Bléger, J. Caro, L. Heinke, Chem. – Eur. J. 2017, 23, 5434.
[45] Z. Gu, L. Heinke, C. Wöll, T. Neumann, W. Wenzel, Q. Li, K. Fink, O. D. Gordan, D. R. T. Zahn, Appl. Phys. Lett. 2015, 107, 183301.
[46] a) L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud, C. Lamberti, Chem. Mater. 2011, 23, 1700; b) A. Kuc, T. Heine, G. Seifert, H. A. Duarte, Theor. Chem. Acc. 2008, 120, 543.
[47] O. Shekhah, H. Wang, M. Paradinas, C. Ocal, B. Schupbach, A. Terfort, D. Zacher, R. A. Fischer, C. Wöll, Nat. Mater. 2009, 8, 481.
[48] X. Yu, Z. Wang, M. Buchholz, N. Fullgrabe, S. Grosjean, F. Bebensee, S. Bräse, C. Wöll, L. Heinke, Phys. Chem. Chem. Phys. 2015, 17, 22721.
[49] A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi, G. Orlandi, J. Am. Chem. Soc. 2004, 126, 3234.
[50] Z. Wang, S. Grosjean, S. Braese, L. Heinke, ChemPhysChem 2015, 16, 3779.
Macromol. Rapid Commun. 2017, 1700239

本文献由“学霸图书馆-文献云下载”收集自网络,仅供学习交流使用。
学霸图书馆(www.xuebalib.com)是一个“整合众多图书馆数据库资源,
提供一站式文献检索和下载服务”的24 小时在线不限IP
图书馆。
图书馆致力于便利、促进学习与科研,提供最强文献下载服务。
图书馆导航:
图书馆首页 文献云下载 图书馆入口 外文数据库大全 疑难文献辅助工具


















