
Spinal Muscular Atrophy Carrier Frequency and Estimated Prevalence of the Disease in Moroccan...
4
Spinal Muscular Atrophy Carrier Frequency and Estimated Prevalence of the Disease in Moroccan Newborns Jaber Lyahyai, 1,2 Aziza Sbiti, 2 Amina Barkat, 3 Ilham Ratbi, 1,2 and Abdelaziz Sefiani 1,2 Spinal muscular atrophy (SMA) is one of the most common autosomal recessive diseases caused by homozygous deletion of exon 7 of the survival motor neuron 1 (SMN1) gene in approximately 95% of SMA patients. Carrier frequency studies of SMA have been reported for various populations. The aim of our study was to estimate the carrier frequency of the common SMN1 exon 7 deletion in the Moroccan population to achieve an insight into the prevalence of SMA in Morocco. In this study, we used a reliable quantitative real-time polymerase chain reaction assay with SYBR Green I dye to determine the copy number of the SMN1 gene. Analysis of 150 Moroccan newborns predicts a carrier frequency of approximately 1:25, which would mean a calculated SMA prevalence of 1:1800 after correction due to consanguinity. These results show as expected that the SMA carrier frequency in Morocco is higher than in the European populations and is close to those of Middle Eastern countries. Genetic carrier testing for genetic counseling should be recommended particularly to families with a clear clinical history of SMA. Introduction S pinal muscular atrophy (SMA) is an autosomal reces- sive severe neuromuscular disease characterized by de- generation of the anterior horn of the spinal cord, which results in progressive muscle weakness and paralysis (Biros and Forrest, 1999; Talbot, 1999). Childhood SMA is sub- divided into three clinical groups on the basis of age of onset and disease progression (Munsat and Davies, 1992; Zerres and Rudnik-Schoneborn, 1995). Type I SMA, also known as Werdnig-Hoffman disease, is the most severe form charac- terized by generalized muscle weakness and hypotonia at birth or within the first 3 months. Death from respiratory failure usually occurs within the first 2 years. In Type II SMA, children are able to sit, although they cannot stand or walk unaided, and often survive beyond 4 years. Type III SMA (Kugelberg-Welander) is a milder form, with onset during infancy or youth: patients learn to walk unaided and have prolonged survivals. Type III SMA is further subdivided into two groups, type IIIa (onset before 3 years) and type IIIb (onset at age 3 years). Adult form of SMA type IV, while less frequent, has also been reported (Prior, 2010a). All types of SMA are associated with homozygous deletion or mutations in the survival motor neuron (SMN) gene. In a majority of normal individuals in the population, SMN genes are present in at least one telomeric (SMN1) and one centromeric (SMN2) copy per chromosome. The two SMN genes are highly homologous, but nucleotide variations in exon 7 and exon 8 of SMN1 and SMN2 genes are responsible for functional differences (Rochette et al., 2001; Yoon et al., 2010). Approximately 95% of SMA patients carry homozygous deletion of the SMN1 gene, and the remaining 5% of the af- fected individuals may be compound heterozygote for a de- leted gene and an intragenic mutation on the other SMN1 gene (Lefebvre et al., 1995; Rodrigues et al., 1995; Kang et al., 2009; Yoon et al., 2010; Morikawa et al., 2011). SMA is reported to have an overall worldwide incidence of 1 in 10,000 live births; however, because of the prevailing custom of consanguineous marriage in Morocco, the inci- dence, prevalence, and carrier frequency of SMA in Moroccan population could be higher than the European. Treatment and prevention of SMA are complementary re- sponses to the challenges presented by SMA. Even though a specific therapy for SMA is not currently available, a newborn screening test may allow the child to be enrolled in a clinical trial before irreversible neuronal loss occurs and enable pa- tients to obtain more proactive treatments. Until an effective treatment is found to cure or arrest the progression of the disease, prevention of new cases through accurate diagnosis and carrier and prenatal diagnosis is of utmost importance and allows at-risk family members to make informed repro- ductive choices (Prior, 2010b). In the present study, to estimate the prevalence of SMA in Moroccan newborns by indirect genetic methods, we used a 1 Centre de Ge ´nomique Humaine, Faculte ´ de Me ´decine et Pharmacie, Universite ´ Mohamed V Souissi, Rabat, Morocco. 2 De ´partement de Ge ´ne ´tique Me ´dicale, Institut National d’Hygie `ne, Rabat, Morocco. 3 Centre National de Re ´fe ´rence en Ne ´onatologie et en Nutrition, Rabat, Morocco. GENETIC TESTING AND MOLECULAR BIOMARKERS Volume 16, Number 3, 2012 ª Mary Ann Liebert, Inc. Pp. 215–218 DOI: 10.1089/gtmb.2011.0149 215
Transcript of Spinal Muscular Atrophy Carrier Frequency and Estimated Prevalence of the Disease in Moroccan...

Spinal Muscular Atrophy Carrier Frequency and EstimatedPrevalence of the Disease in Moroccan Newborns
Jaber Lyahyai,1,2 Aziza Sbiti,2 Amina Barkat,3 Ilham Ratbi,1,2 and Abdelaziz Sefiani1,2
Spinal muscular atrophy (SMA) is one of the most common autosomal recessive diseases caused by homozygousdeletion of exon 7 of the survival motor neuron 1 (SMN1) gene in approximately 95% of SMA patients. Carrierfrequency studies of SMA have been reported for various populations. The aim of our study was to estimate thecarrier frequency of the common SMN1 exon 7 deletion in the Moroccan population to achieve an insight into theprevalence of SMA in Morocco. In this study, we used a reliable quantitative real-time polymerase chain reactionassay with SYBR Green I dye to determine the copy number of the SMN1 gene. Analysis of 150 Moroccannewborns predicts a carrier frequency of approximately 1:25, which would mean a calculated SMA prevalenceof 1:1800 after correction due to consanguinity. These results show as expected that the SMA carrier frequencyin Morocco is higher than in the European populations and is close to those of Middle Eastern countries.Genetic carrier testing for genetic counseling should be recommended particularly to families with a clear clinicalhistory of SMA.
Introduction
Spinal muscular atrophy (SMA) is an autosomal reces-sive severe neuromuscular disease characterized by de-
generation of the anterior horn of the spinal cord, whichresults in progressive muscle weakness and paralysis (Birosand Forrest, 1999; Talbot, 1999). Childhood SMA is sub-divided into three clinical groups on the basis of age of onsetand disease progression (Munsat and Davies, 1992; Zerresand Rudnik-Schoneborn, 1995). Type I SMA, also known asWerdnig-Hoffman disease, is the most severe form charac-terized by generalized muscle weakness and hypotonia atbirth or within the first 3 months. Death from respiratoryfailure usually occurs within the first 2 years. In Type II SMA,children are able to sit, although they cannot stand or walkunaided, and often survive beyond 4 years. Type III SMA(Kugelberg-Welander) is a milder form, with onset duringinfancy or youth: patients learn to walk unaided and haveprolonged survivals. Type III SMA is further subdivided intotwo groups, type IIIa (onset before 3 years) and type IIIb(onset at age 3 years). Adult form of SMA type IV, while lessfrequent, has also been reported (Prior, 2010a). All types ofSMA are associated with homozygous deletion or mutationsin the survival motor neuron (SMN) gene.
In a majority of normal individuals in the population, SMNgenes are present in at least one telomeric (SMN1) and onecentromeric (SMN2) copy per chromosome. The two SMN
genes are highly homologous, but nucleotide variations in exon7 and exon 8 of SMN1 and SMN2 genes are responsible forfunctional differences (Rochette et al., 2001; Yoon et al., 2010).
Approximately 95% of SMA patients carry homozygousdeletion of the SMN1 gene, and the remaining 5% of the af-fected individuals may be compound heterozygote for a de-leted gene and an intragenic mutation on the other SMN1gene (Lefebvre et al., 1995; Rodrigues et al., 1995; Kang et al.,2009; Yoon et al., 2010; Morikawa et al., 2011).
SMA is reported to have an overall worldwide incidence of1 in 10,000 live births; however, because of the prevailingcustom of consanguineous marriage in Morocco, the inci-dence, prevalence, and carrier frequency of SMA in Moroccanpopulation could be higher than the European.
Treatment and prevention of SMA are complementary re-sponses to the challenges presented by SMA. Even though aspecific therapy for SMA is not currently available, a newbornscreening test may allow the child to be enrolled in a clinicaltrial before irreversible neuronal loss occurs and enable pa-tients to obtain more proactive treatments. Until an effectivetreatment is found to cure or arrest the progression of thedisease, prevention of new cases through accurate diagnosisand carrier and prenatal diagnosis is of utmost importanceand allows at-risk family members to make informed repro-ductive choices (Prior, 2010b).
In the present study, to estimate the prevalence of SMA inMoroccan newborns by indirect genetic methods, we used a
1Centre de Genomique Humaine, Faculte de Medecine et Pharmacie, Universite Mohamed V Souissi, Rabat, Morocco.2Departement de Genetique Medicale, Institut National d’Hygiene, Rabat, Morocco.3Centre National de Reference en Neonatologie et en Nutrition, Rabat, Morocco.
GENETIC TESTING AND MOLECULAR BIOMARKERSVolume 16, Number 3, 2012ª Mary Ann Liebert, Inc.Pp. 215–218DOI: 10.1089/gtmb.2011.0149
215
reliable quantitative real-time polymerase chain reaction(PCR) assay with SYBR Green I dye to determine the carrierrate for SMA in the Moroccan population.
Materials and Methods
Samples and DNA extraction
Blood samples were collected from 150 unrelated new-borns’ umbilical cords. They originated from different regionsof Morocco and the Moroccan origin of their parents andgrandparents was confirmed. Thus, informed consent forDNA analysis was obtained from the parents. To validate ourresults, 10 carrier parents with at least a child with the ho-mozygous deletion of exon 7 were included in this study.Genomic DNA was extracted from 3 mL using the salting-outmethod (Sambrook et al., 1989). The quality and quantity ofthe DNA were checked by A260/A280 using a Nanodropspectrophotometer (Nanodrop Technologies).
Real-time PCR
The SMA carrier test determines the SMN1 gene copy numberby comparing the levels of amplified products generated from
exon 7 of SMN1 and a two-copy control gene following PCRamplification. As a reference gene we used the albumin gene.Carrier and unrelated normal samples were analyzed togetherwith the calibrator sample on every assay plate. The quantitativereal-time PCR assay with SYBR green I dye employed previouslypublished primers that specially amplify theSMN1 gene (Lee et al.,2004). Each sample was run in duplicates in separate tubes topermit quantification of the SMN1 gene normalized to albumin.
Data analysis
All data were calculated by the comparative Ct method todetect the relative gene copy (Vandesompele et al., 2002; Leeet al., 2004) using a sample of normal control DNA as a cali-brator. The relative gene copy numbers were determined bythe expression 2 -DDCt, with DDCt calculated by the followingformula: DDCt = [DCt albumin (calibrator sample) -DCtSMN1 (calibrator sample)] - [DCt albumin (unknown sam-ple) -DCt SMN1 (unknown sample)]. The [2 -DDCt] ratio wasexpected to be about 1 in normal controls, about 0.5 in carriers,and 0 in patients with SMA.
The effect of inbreeding on the prevalence of SMA was esti-mated according to the formula of Wright: P(MM) = q2 + Fq(1 - q),
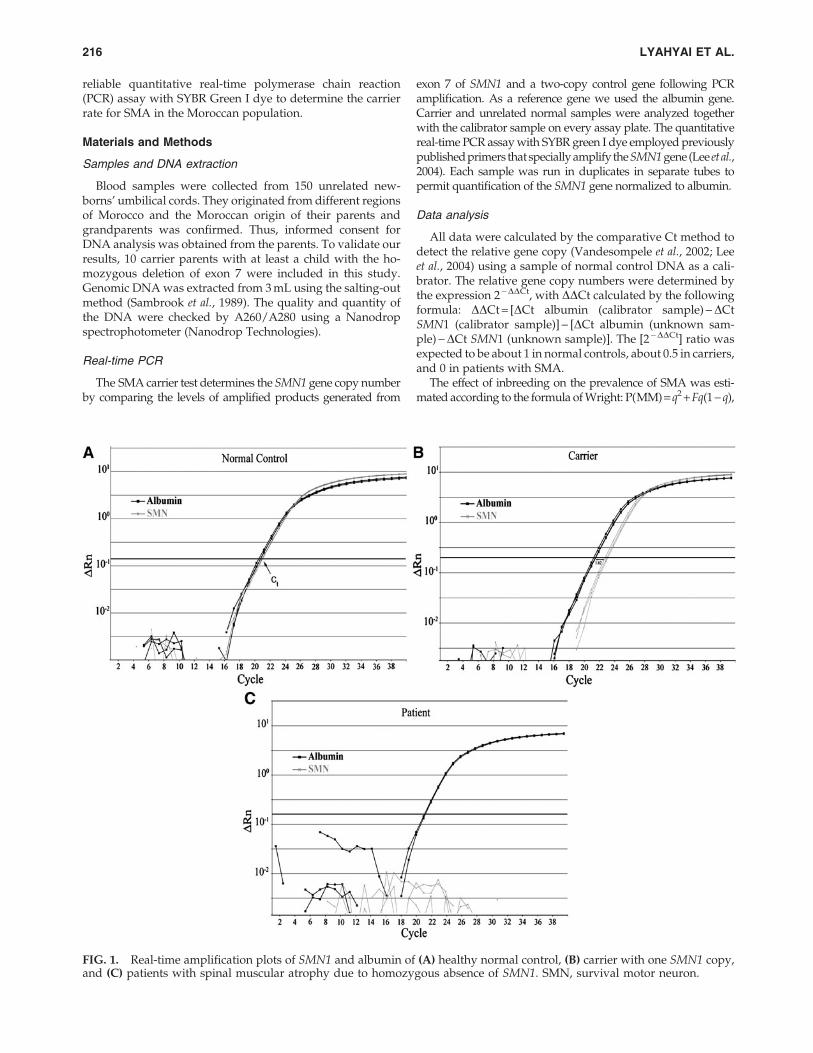
FIG. 1. Real-time amplification plots of SMN1 and albumin of (A) healthy normal control, (B) carrier with one SMN1 copy,and (C) patients with spinal muscular atrophy due to homozygous absence of SMN1. SMN, survival motor neuron.
216 LYAHYAI ET AL.

where q is the frequency of the mutated allele (q = 1:25) and F isthe mean inbreeding coefficient of the Moroccan population(Wright, 1921).
Results
The Ct values of SMN1 and albumin duplicates were al-most identical at the amplification plots of a normal control(Fig. 1A). In the parents of SMA, the Ct values of SMN1 in-creased about 1.62 compared with albumin (Fig. 1B). In theSMA patients, the albumin gene was amplified as in normalcontrols, but the SMN1 gene was not amplified as expected(Fig. 1C). The relative gene copy numbers were detected bythe expression 2 -DDCt.
To validate the analysis, we determined the range of carriergene dosage in 10 parents of SMA patients in which homo-zygous deletion of the SMN1 gene were previously identifiedby PCR–restriction fragment length polymorphism (Table 1).Among the 150 newborns analyzed, 6 were classified as het-erozygous for exon 7 deletion. This carrier frequency of 1:25allowed us to estimate an SMA prevalence of 1:2500 in Mor-occan newborns. If we take into account the mean inbreedingcoefficient of 0.0065 in the Moroccan general population, theprevalence of SMA would be increased to 1:1900 according tothe formula of Wright. With the prevalence of 1:1900 and if wealso consider that 95% of SMA patients carry homozygousdeletion of the SMN1 gene, our results predict a prevalence of1:1800 for all types of SMA.
Discussion
With a worldwide prevalence of 1 in 10,000 live births,SMA represents the second common fatal autosomal re-cessive disorder after cystic fibrosis. SMA is one of the mostfrequent heredity disorders for which patients and familiesare referred to our genetic center, but to date no data areavailable on the prevalence of SMA in Morocco. Therefore,we calculate the SMA carrier frequency in Morocco using areliable quantitative real-time PCR to quantify the SMN1gene, as homozygous deletion of SMN1 exon 7 is a com-mon variant.
Six of 150 newborns tested were classified as heterozygousfor exon 7 deletion, which enabled us to estimate a carrierfrequency for SMN1 homozygous deletion at approximately 1in 25 Moroccan invidious. These results show that the SMAcarrier frequency in Morocco is higher than in the Europeanpopulations (from 1:50 to 1:80). Reports of carrier frequenciesin other countries vary from lower in the United Kingdom(from 1:60 to 1:80), intermediate in North Dakota, UnitedStates (1:41), Korea (1:47), and Australia (1:49), to higher fre-
quencies (1:20) in Iran and Saudi Arabia (Burd et al., 1991;Mostacciuolo et al., 1992; Thieme et al., 1994; Ludvigsson et al.,1999; Lee et al., 2004; Hasanzad et al., 2010; Goncalves-Rochaet al., 2011).
Assuming Hardy–Weinberg balance, this carrier frequency(1:25) would mean a calculated SMA prevalence of 1:2500.The consanguinity rate among married couples is high inMorocco (15.25%–19.90%) and in some regions it exceeds> 28% (Bouazzaoui, 1994; Cherkaoui et al., 2005; Hami et al.,2006; Talbi et al., 2007; Jaouad et al., 2009), resulting in a highincidence and prevalence of autosomal recessive genetic dis-orders. The mean inbreeding coefficient in the Moroccangeneral population is 0.0065; taking into account this factor,the prevalence of SMA is increased to 1:1900 according to theFormula of Wright. Moreover, SMA patients carry homozy-gous deletion of the SMN1 gene, representing 95% of the totalforms of the disease, which means that our results predict aprevalence of 1:1800 for all types of SMA. This figure confirmsthat SMA is one the most prevalent genetic diseases in theMoroccan population.
In conclusion, this is the first report addressing the esti-mation of SMA carrier frequency in Morocco. With this highprevalence of SMA linked to the SMN1 gene deletion, healthmeasures should be implemented. The carrier testing could bea helpful tool for genetic counseling to individuals with apositive family history. This cost-effective genetic test couldbe essentially useful in a developing country with a high rateof consanguineous marriage.
Disclosure Statement
No competing financial interests exist.
References
Biros I, Forrest S (1999) Spinal muscular atrophy: untangling theknot? J Med Genet 36:1–8.
Bouazzaoui N (1994) Consanguinity and public health in Mor-occo. Bull Acad Natl Med 178:1013–1025.
Burd L, Short SK, Martsolf JT, et al. (1991) Prevalence of type Ispinal muscular atrophy in North Dakota. Am J Med Genet41:212–215.
Cherkaoui M, Baali A, Larrouy G, et al. (2005) Consanguinity,fertility of couples and mortality of children in the high Atlaspopulation (commons of Anougal and Azgour, Marrakesh,Morocco). Int J Anthropol 20:199–206.
Goncalves-Rocha M, Oliveira J, Rodrigues L, et al. (2011) Newapproaches in molecular diagnosis and population carrierscreening for spinal muscular atrophy. Genet Test Mol Bio-markers 15:319–326.
Hami H, Soulaymani A, Mokhtari A (2006) Endogamy, isonymyand consanguinity in the region of the Gharb-Chrarda-BeniHssen (Morocco). Antropo Revista de Antropologıa fısica 11:223–233.
Hasanzad M, Azad M, Kahrizi K, et al. (2010) Carrier frequencyof SMA by quantitative analysis of the SMN1 deletion in theIranian population. Euro J Neurol 17:160–162.
Jaouad IC, Elalaoui SC, Sbiti A, et al. (2009) Consanguineousmarriages in Morocco and the consequence for the incidenceof autosomal recessive disorders. J Biosoc Sci 41:575–581.
Kang SH, Cho SI, Chae JH, et al. (2009) False homozygous de-letions of SMN1 exon 7 using Dra I PCR-RFLP caused by anovel mutation in spinal muscular atrophy. Genet Test MolBiomarkers 13:511–513.
Table 1. Validation of SMN1 Gene Dosage for Spinal
Muscular Atrophy Carrier Testing
DDCt ratio of SMN1 gene
Subjects Mean Range Standard deviation
Patient (n = 5) 0.00 — —Parent (n = 10) 0.48 0.33–0.6 0.1Normal (n = 150) 1.23 0.5–2.39 0.42
SMN, survival motor neuron.
MOROCCAN SMA PREVALENCE 217

Lee TM, Kim SW, Lee KS, et al. (2004) Quantitative analysis ofSMN1 gene and estimation of SMN1 deletion carrier fre-quency in Korean population based on real-time PCR. J Ko-rean Med Sci 19:870–873.
Lefebvre S, Burglen L, Reboullet S, et al. (1995) Identification andcharacterization of a spinal muscular atrophy-determininggene. Cell 80:155–165.
Ludvigsson P, Olafsson E, Hauser WA (1999) Spinal muscularatrophy. Incidence in Iceland. Neuroepidemiology 18:265–269.
Morikawa S, Harahap IS, Kaszynski RH, et al. (2011) Diagnosisof spinal muscular atrophy via high-resolution melting anal-ysis symmetric polymerase chain reaction without probe: ascreening evaluation for SMN1 deletions and intragenic mu-tations. Genet Test Mol Biomarkers. [Epub ahead of print];DOI: 10.1089/gtmb.2010.0237.
Mostacciuolo ML, Danieli GA, Trevisan C, et al. (1992) Epide-miology of spinal muscular atrophies in a sample of the Italianpopulation. Neuroepidemiology 11:34–38.
Munsat TL, Davies KE (1992) International SMA Consortiummeeting. (26–28 June 1992, Bonn, Germany). NeuromusculDisord 2:423–428.
Prior TW (2010a) Perspectives and diagnostic considerations inspinal muscular atrophy. Genet Med 12:145–152.
Prior TW (2010b) Spinal muscular atrophy: newborn and carrierscreening. Obstet Gynecol Clin North Am 37:23–36, Table ofContents.
Rochette CF, Gilbert N, Simard LR (2001) SMN gene duplicationand the emergence of the SMN2 gene occurred in distincthominids: SMN2 is unique to Homo sapiens. Hum Genet108:255–266.
Rodrigues NR, Owen N, Talbot K, et al. (1995) Deletions in thesurvival motor neuron gene on 5q13 in autosomal recessivespinal muscular atrophy. Hum Mol Genet 4:631–634.
Sambrook J, Fritsh EF, Maniatis T (1989) Isolation of DNA frommammalian cells. In: Sambrook J, Fritsh EF, Maniatis T (eds)
Molecular Cloning-A Laboratory Manual. Cold Spring HarborLaboratory Press, New York, pp 917–919.
Talbi J, Khadmaoui A, Soulaymani A, et al. (2007) Study ofconsanguinity in Moroccan population. Influence on the pro-file of health. Antropo Revista de Antropologıa fısica 15:1–11.
Talbot K (1999) Spinal muscular atrophy. J Inherit Metab Dis22:545–554.
Thieme A, Mitulla B, Schulze F, et al. (1994) Chronic childhoodspinal muscular atrophy in Germany (West-Thuringen)—anepidemiological study. Hum Genet 93:344–346.
Vandesompele J, De Preter K, Pattyn F, et al. (2002) Accuratenormalization of real-time quantitative RT-PCR data by geo-metric averaging of multiple internal control genes. GenomeBiol 3:RESEARCH0034.
Wright S (1921) Systems of mating. II. the effects of inbreeding onthe genetic composition of a population. Genetics 6:124–143.
Yoon S, Lee CH, Lee KA (2010) Determination of SMN1 andSMN2 copy numbers in a Korean population using multiplexligation-dependent probe amplification. Korean J Lab Med30:93–96.
Zerres K, Rudnik-Schoneborn S (1995) Natural history in prox-imal spinal muscular atrophy. Clinical analysis of 445 patientsand suggestions for a modification of existing classifications.Arch Neurol 52:518–523.
Address correspondence to:Jaber Lyahyai, Ph.D.
Centre de Genomique HumaineFaculte de Medecine et Pharmacie
Universite Mohamed V SouissiRabat 6203
Morocco
E-mail: [email protected]
218 LYAHYAI ET AL.


















