Team 6 Milestone 3 ● Matt Bouchard, Paras Metha, Andrew Butcher.

Simuulation
De
R
of a lab
egree Proje
Seyedeh
Prof.
PhD stu
Energy pr
Royal Insti
1
b‐scale
ect in Chem
Guilnaz M
Supervis
. Mats Wes
udent Farza
rocesses dep
tute of Tech
June 201
e metha
mical Engine
Mirmoshtag
sor
stermark
ad Mohsen
partment (E
hnology (K
11
anation
eering
ghi
ni
EP)
KTH)
n reactoor

2
Preface
This is the degree project in chemical engineering which is held in Energy processing section since June 2010. It has started as a project like a complimentary part to a PhD work and then continued as master thesis.

3
Abstract
By the everyday increasing enthusiasm for using renewable‐sustainable sources in energy production area, focusing on one and optimizing it in the best possible way should be of much interest.
Biogas production from anaerobic digestion of wastes is a well known energy source which could be applied more efficiently if the CO2 portion of it would be upgraded to CH4 as well. There is a methanation reaction which could convert carbon dioxide to methane with the use of hydrogenation.
In this report, the effort is to simulate this methanation reactor which is a catalytic bed of ruthenium on alumina base. The temperature change and its’ effect on reaction kinetics and equilibrium, also deriving designing parameters for the catalyst bed are different tasks which was tried to be covered in this thesis work.
Based on calculations, the reactor can operate isothermally or adiabatically. The point is that each method has its own cons and pros. For the isothermal case finally the optimum temperature to run the reaction is decided to be 600 K in 10 bar total pressure. In adiabatic case then it is understood to work on interstage cooling strategy which in given conditions came to the number of 6 for reactors and 5 for interstage cooling devices.
Afterwards it is thought to apply some technical changes to conventional adiabatic method and recycle some part of the product to the entrance of the reactor and assist the conversion. In this method number of reactors would be reduced to 2 and one heat exchanger in the middle.
Selecting the best process in large scale treatment, needs lots of economical analysis and detail design while in small scale condition the most preferred method to run the reaction is isothermal.

4
Notation
P Partial pressur of component i, atm
T Temperature, °R
T : Temperature, °K
t time, hr
k reaction rate constant, hr atm
E activation energy,Btu
lbmolof reactant
n: catalyst coeffiecient, dimensionless
K Equilibrium constant, atm
H enthalpy, J/ mol. K
R gas constant,J
mol. K
θ characteristic vibration temperatures of molecule i, K
Q Heat generated or released , kW
H / Enthalpy of input and output stream, kJ/mol
F / Molar flowrate of reactants and products, mol/sec
h Heat transfer coefficient from gas to wall,W
m K
A Surface area of element exposed to coolant, m
D Diameter of the reactor, m
length of element which is exposed to coolant

5
Table of contents
Preface ........................................................................................................................................................... 1
Abstract ......................................................................................................................................................... 3
Notation ........................................................................................................................................................ 4
Table of contents ........................................................................................................................................... 5
Introduction .................................................................................................................................................. 6
The reaction ................................................................................................................................................... 6
Aim of the project .......................................................................................................................................... 7
Methodology ................................................................................................................................................. 7
1 Discussion and results ............................................................................................................................... 9
1.1 Simulation and calculation data ..................................................................................................... 9
1.1.1 Input data ................................................................................................................................... 9
1.1.2 Division of bed into 20 elements ........................................................................................... 10
1.1.3 Cases for simulation ............................................................................................................... 11
1.1.4 Kinetic equation ...................................................................................................................... 11
1.1.5 Equilibrium considerations ................................................................................................... 12
1.2 Results of adiabatic case ................................................................................................................ 13
1.3 Results of Real case ........................................................................................................................ 20
1.3.1 Theoretical formulation .......................................................................................................... 20
1.3.2 Analytical calculations and results ....................................................................................... 22
1.3.2.1 Trial at 550 Kelvin as initial temperature ...................................................................... 25
1.3.2.2 Trial at 600 Kelvin as initial temperature ...................................................................... 26
1.3.2.2 Trial at 650 Kelvin as initial temperature ...................................................................... 26
1.4Comparison of adiabatic case with isothermal case ................................................................... 28
2 Conclusion ............................................................................................................................................... 31
Bibliography ................................................................................................................................................ 33

6
Introduction
Analysis shows that fossil fuel sources are under the threat of fast decrease. Although these sources are highly efficient and economically beneficial, they have high level of environmental risks and hazards as well. Considering these important facts, encourages different countries to change their energy policy and to focus more on non‐fossil fuels. To have more energy security and less environmental problems, lead to finding as much renewable sources as possible.
One of the sustainable sources in the renewable category is biogas. Some authors define biogas as those gases which are produced through anaerobic digestion. But there is a wider definition that considers biogas as all combustible gases from biological sources (Taglia P.G 2010) and also includes gases from pyrolysis or thermal gasification. Biogas is mainly methane and carbon dioxide together with some other inert gases. (Violeta Bescós 2008) Methane is the main part of biogas which could be used as fuel and energy source, so companies are more interested in optimizing the biogas production process in the way to have as much methane as possible.
The reaction
In order to increase the biogas supply, different methods can be used. One of them is to use Sabatier reaction. It is called “Sabatier” reaction after its investigator who was a Belgian chemist. He discovered this reaction to hydrogenise carbon compounds in a catalytic bed at ~480‐650 K which is called methanation. In this reaction the CO2 which exists in biogas stream reacts with H2 and produces CH4 again. Therefore the product would have more CH4 than the biogas process itself (Lunde 1974)
Hydrogen in this reaction could be prepared through different methods such as steam reforming and water electrolysis. In order to have a complete renewable pathway for methane production, water electrolysis using electricity provided by wind farms, would be a better choice.
Methanation is a highly exothermal reaction which produces ‐165 kJ/mol produced CH4 .This parameter should be considered in design of the reactor, so that the accumulation of the heat does not destroy the catalysts. The equilibrium is more favorable at low temperature, but the reaction rate is higher at high temperature so controlling the temperature in an applicable

7
range is important. In industry, using heat exchanger around the reactor wall is a method to control temperature fluctuation in bed.
Aim of the project
In this thesis, a lab‐scale Sabatier reactor is simulated. Studying different parameters such as reactant temperature, coolant temperature and pressure in this project gives the optimum point to run the reaction. Kinetics of reaction through the bed and temperature profile are final results.
Methodology
The studied input gas contains 60 vol% CH4 and 40 vol% CO2 which is typical for the gas leaves an anaerobic digester. Hydrogen is added then at stoichiometric ratio (4 H2 per CO2)
In order to simulate the reaction, the methodology is to divide the bed in 20 hypothetical elements in which a conversion of 4.5%/element is assumed. The final yield is thus assumed to be 90% (expressed as reacted hydrogen/inlet hydrogen). The temperature in each cross section is considered to be the same and so there is just temperature profile along the length of the bed. This simulation has been done all in excel, since the method was numerical based.
Fig 1. Schematic figure of one element in bed
H2 CO2 CH4 H2O
T1
H2 CO2 CH4 H2O T2
Q First Element

8
In fig 1, there is a schematic view of one element of the 20 ones in the reactor tube. As it is clear, the streams with temperature T1 and with different volumetric flows enter the element. The heat which is produced in the reaction can either increase the temperature of the effluent stream to T2 or be released from the wall to the cooling media. In reality both of these mechanisms happen in parallel, but in our calculations we simulate them individually. The results of the isothermal and the adiabatic simulation then can be compared with one another.
As reactor tubes, the inner diameter of 35 mm was chosen since this is a standard diameter that is easily available and is used in laboratory reactor. Tube length up to 6 m is available and it seems logical to design the reactor for single pass. A load of 1.94 mmol/sec CO2 and 90% conversion was assumed for tube and 10 bar pressure was chosen.

9
1 Discussion and results
1.1 Simulation and calculation data
1.1.1 Input data
There are some initial values used as the basis of calculations. These data were taken mainly from Lunde work as the major source of information in this task. Based on Lunde paper on modeling and simulation of Sabatier reactor, the estimated inlet flow in atmospheric pressure is 0.638 ft3/min (18.1 lit/min) (Lunde 1974). This value could be changed in 10 atm case easily, so the values for volumetric flow rate of each component in influent become as in table 1.
Input parameter Unit value Reaction pressure atm 10 Inlet temp K 550 inlet flow of CO2 (20 ◦C, 10 atm) lit/ min 0.28 inlet flow of CH4(20 ◦C, 10 atm) lit/ min 0.42 inlet flow of H2(20 ◦C, 10 atm) lit/ min 1.12 inlet flow of vapor mol/ min 0 Reacted CO2 per stage % 4.5 Inner diameter of reactor tube mm 35
Table 1. Input data
The strategy is that hydrogen and carbon dioxide enter the reactor in stoichiometric ratio. In each hypothetical element 4.5% of the initial CO2 should be converted. For instance, in the first element 4.5% of 0.28 lit/min converts and the rest flows to the second element. In the second element another 4.5% of 0.28 lit/min would convert and the rest gets to the third element and this process continues till the 20th element. This assumption then gives us a fixed conversion of 90% in all different cases under consideration
The described mechanism is the same for all other compounds in influent. The only thing which is necessary to mention is considering the existence of CH4 from the beginning in the reactor. This stream is injected to the reactor separately just to simulate the real biogas stream which consists of both CH4 and CO2 together. The inlet concentration of methane does not change the equilibrium constant, but it is increasing the flow and the heat capacity.
We can think of the conversion as the fixed parameter and size of the bed reactor as the unknown parameter which should be calculated for different cases. Temperature profile along the reactor could be the other unknown parameter which depends on changes in size of the bed and other initial parameters.

10
In order to use the correlations in different articles for further calculation it is needed to change the flow rate unit from lit/min to mol/min. This is easily done by using the ideal gas equation. Pressure term in the correlation is equal to 10 atm which is the total pressure of the system and temperature is equal to 293 K.
1.1.2 Division of bed into 20 elements
In table 2, there are molar changes along the bed from top to bottom. The first row in this table is the input flow of the whole reactor and first element as well, but the other rows could be interpreted as the outflow of the element which is mentioned in the same row of the table. As it is shown, the total number of moles is reducing through the reaction. This is because of the non‐equimolar characteristics of the reaction. Also the relation between molar flows of different components is based on stoichiometric ratio.
No. of elements
n CO2 (mmol/min)
n H2 (mmol/min)
n CH4 (mmol/min)
delta CH4 (mmol/min)
nH2O (mmol/min)
ntot (mmol/min)
0 116.45 465.8 174.7 0 0 756.95
1 111.21 444.84 179.92 5.25 10.5 746.50
2 106.00 424.00 185.16 10.50 21.00 736.16
3 100.73 403.00 190.40 15.72 31.44 725.60
4 95.50 382.00 195.64 21.00 41.92 715.06
5 90.25 361.00 201.00 26.20 52.40 704.65
6 85.00 340.00 206.12 31.44 63.00 694.12
7 80.00 319.10 211.36 36.7 73.40 683.90
8 74.53 298.11 216.60 41.92 83.85 673.10
9 69.30 277.15 221.84 47.16 94.32 662.61
10 64.05 256.20 227.10 52.40 104.80 652.15
11 58.81 235.23 232.32 57.64 115.30 641.70
12 53.56 214.30 237.56 62.88 125.80 631.122
13 48.32 193.30 242.80 68.12 136.25 620.70
14 43.08 172.35 248.04 73.36 146.73 610.20
15 37.85 151.40 253.30 78.60 157.21 599.76
16 32.60 130.42 258.50 84.00 167.70 589.22
17 27.36 109.50 263.8 89.08 178.17 578.83
18 22.12 88.50 269.00 94.32 188.67 568.30
19 17.00 67.54 274.25 99.56 199.13 557.92
20 11.65 46.60 279.50 104.80 209.61 547.40
Table 2. Mole fractions in each element (4.5% conversion in each)

11
1.1.3 Cases for simulation
In this report there are two main cases considered. The first case is the adiabatic one in which all the generated heat is used to increase the temperature of the flows downward and nothing can be released to the surrounding. The second case then is known as the real case which happens in reality. The attempt in this case is to control temperature in the bed on the constant amount by cooling the bed in a heat exchanging mechanism with a coolant flows around the reactor.
1.1.4 Kinetic equation
Emmett did a complete research on carbon dioxide methanation which was published as a book named “catalysis”. He believed that the methanation process can be described as two consecutive reactions (Emmett 1951):
(a)
3 (b)
Since the first reaction is slow at the used reaction temperature (207‐371◦C) and the second one is more rapid than the (a), more in equilibrium form, then there is no carbon monoxide in the output stream. (Kester 1974)
Knowing the real reactions which are happening helps finding the catalytic mechanism and then formulating a model for the reaction. Since the reaction is in gas phase and reversible, reaction rate is dependent both on partial pressure of reactants and products. Lunde has found a correlation for calculating this parameter (Lunde 1974).
(1)
Lunde and Kester in the process of finding a well‐suited correlation for rate of reaction did some experiments in laboratory reactors. Through these tests, they could estimate values for k, Ea and n with a rather good liability. In their investigation the following values were found:
k= 0.1769× 1010 Ea= 16.84 kcal/gmol (30.320 btu/lbmol) n= 0.225
They derived these values from curves and lines of experimental results in different H2/CO2 ratios and also in different temperatures. (Lunde, Peter J.,Kester, Frank L 1973)

12
1.1.5 Equilibrium considerations
The rate of reaction is also depending on the equilibrium constant (Ke ) and Ke itself is temperature dependent. The extended equation for equilibrium constant calculation is as following (Lunde, 1974):
.
. . . (2)
In this equation, Tk stands for Temperature in Kelvin unit. This equation is derived based on thermodynamic data by Wagman and et al in 1945 according to methods described by Brewer and Pitzer in 1961. The heat capacities that were used in this equation derived from Bureau of Mines Bulletins in 1949, 1950 and 1960 (Lunde, Peter J., Kester, Frank L. 1974)

13
1.2 Simulation result for the adiabatic case with interstage cooling
In this case, it is assumed that the heat which is generated through the reaction in each element is not released from the reactor, but increases the temperature of the outflow instead.
First of all it is needed to have some strategy for bed sizing, so then it could be modified or completed with temperature and enthalpy considerations. The following steps are used to calculate the length of bed:
I. Calculating the rate of reaction by equation (1) II. Changing the unit atm./hr to mol/m3.min by simple using of ideal gas correlation III. Calculating volume of bed by dividing produced CH4 molar flow rate in each
element with rate of reaction in that specific element. IV. Calculating length of each element by considering the fixed cross sectional area V. The strategy in simulating this case is based on enthalpy changes along the bed.
The enthalpy transformations occur because of temperature increase due to the exothermic nature of reaction. The total enthalpy is constant, but chemical enthalpy is converted to thermal enthalpy. In order to relate the enthalpy of components to temperature changes, there are some polynomial correlations in different articles. Constants in these correlations are experimentally determined for different gases. Frolov and et.al (2009) in an article derived different experimental correlations for enthalpy calculations of real gases.
The molar enthalpy for CO2 with reference temperature of 0 K in ideal‐gas case is given as follows in Frolov article:
. . ∑ / (3)
In equation (3), θi for CO2 has different values. θ1 = 3380 K, θ2 = 1995 K and θ3 = θ4=960 K.
In the same article, there is another correlation which would be applied for the thermal enthalpy of H2 as an ideal gas. In this one the assumptions are the same as before and θ = 6323.26 K.
. . / (4)
The given correlations are both for reactants, but in order to do the calculations for the adiabatic case it is needed to have functions for product enthalpies as well. For H2O, there is a correlation in the same article as well; therefore the assumptions are nearly similar with two previous components.

14
. . . ∑ / (5)
In equation 5, the new parameter xi is just a simple θi to T ratio : ( xi = θi/T). For H2O the values for θi are as follows: θ1 = 5260.73 K, θ2 = 2294.37 K, θ3 = 5403.36 K.
To calculate the thermal enthalpy for CH4 there is another type of polynomial which is given by Passut and Danner (1972).
(6)
A,B,C,D,E and F are coefficients regarding to enthalpy in Btu/lb and temperature in ◦R . For CH4 the values for A to F are as following:
A = ‐5.58114, B = 0.564834, C = ‐0.282973 × 10‐3, D = 0.417399 × 10‐6 E = ‐1.525576 × 10‐10, F = 1.958857 × 10‐14
It is necessary to mention that since all the values in this report is in SI system, the enthalpy unit from equation (5) should be changed into SI
Now having the enthalpy function of all components in the reaction, the calculations could be done properly and knowing the fact that in Sabatier reaction there is ‐165 kJ/mol produced CH4 heat production, the exit temperature of each element can be calculated by the following equations.
(7)
. % (8)
Based on equation (8), the value for Qgenerated is a fixed amount for all elements which in this case is equal to 14.4 W per element. In equation (7) then, the left side is the same as equation (8), the F values are changing through the bed from top to bottom and enthalpies are changing due to the temperature changes, as well. Hin is a function of Tin and Hout is Tout dependent. The output temperature of each element is the input one for the next element. The other point which is necessary to mention is to assume the element temperature equal to input flow temperature. For example, in element number 1, the input flow temperature is 550K and it is assumed that the reaction in this element happens in 550 K as well.
In calculating the output temperature of each element, the temperature level increases to the point that the equilibrium conversion is reached in the middle of reactor and so the reaction could not go further and get to the assumed conversion of 90%. In table 3 there are results of adiabatic simulation on temperature and enthalpy aspects. As it is clearly shown the critical temperature happens somewhere around the 11thelement. The equilibrium conversion (Xe) in this

15
case is 47% which is happening somewhere around the 11th element. The highest possible temperature which could be reached in adiabatic trial is 907.4469 K and in temperatures higher than that, equation (1) gets to zero or negative values and so it means that there could not be any reaction in temperatures higher than this. Therefore the 12th element and so forth ones do not play any role in this reaction!
Next step is to size each element and the whole bed respectively. The strategy has been already discussed, but there is some more to consider and it is about the steep increase of reaction rate by temperature rise. This fact then results in a very small volume for each element and so almost 50% conversion happens just in the first 1cm of the bed!
In table 3, there are calculation results of the 11 elements which reaction happens in them. It is clear enough that the volume of each element is decreasing sharply and also the rate of reaction is increasing in big steps even though the conversion is the fixed value of 4.5% in each element.
The graphical view of results in table 4 and 5 are given in following graphs. In figure 2, there are changes of yield and equilibrium conversion changes through the bed. It is clearly shown that in conversion around 50% the equilibrium and yield curve intercept one another. This means that the reaction gets to equilibrium at this point and so it cannot go further and increase the conversion of the reactants.
No. of elements
Tgas K Yield H(kW) r CO2
(mol/lit.hr) L (mm)
1 550 0.045 0.222 43.09 7.30 2 586.4 0.09 0.237 100.72 3.12 3 621.6 0.135 0.251 204.86 1.53 4 658.3 0.18 0.265 389.40 0.81 5 696 0.225 0.281 687.52 0.46 6 731.5 0.27 0.295 1077.38 0.29 7 767.1 0.315 0.310 1561.98 0.20 8 803.6 0.36 0.325 2095.01 0.15 9 838.6 0.405 0.340 2480.90 0.13 10 873.2 0.45 0.353 2497.60 0.126 11 907.4 0.495 0.368 1764.25 0.18 12 941.3 0.54 0.382 ‐266.58 ‐1.18
Table 3. Temperature changes along the bed and reactor sizing in the adiabatic case

16
14.30, 0.47
0
0.2
0.4
0.6
0.8
1
1.2
0 5 10 15 20
Conversion
Length of the bed(mm)
yield equilibriumconversion
In figure 3 then, there are the changes in temperature through bed. The sudden increase in temperature in the very beginning of the reactor is something which is obvious in this figure.
Fig 2. Conversion versus length of the bed in the adiabatic case
There is another point in figure 3 which shows the point which the curve is somehow turning back to the left. This is just to show that the reaction gets to equilibrium and stops progressing further more.
In adiabatic case the temperature increase occurs which has negative impact on the efficiency and catalyst activity, therefore engineers thought of some solutions; an Inter stage cooling system which is processing after each two or three elements.
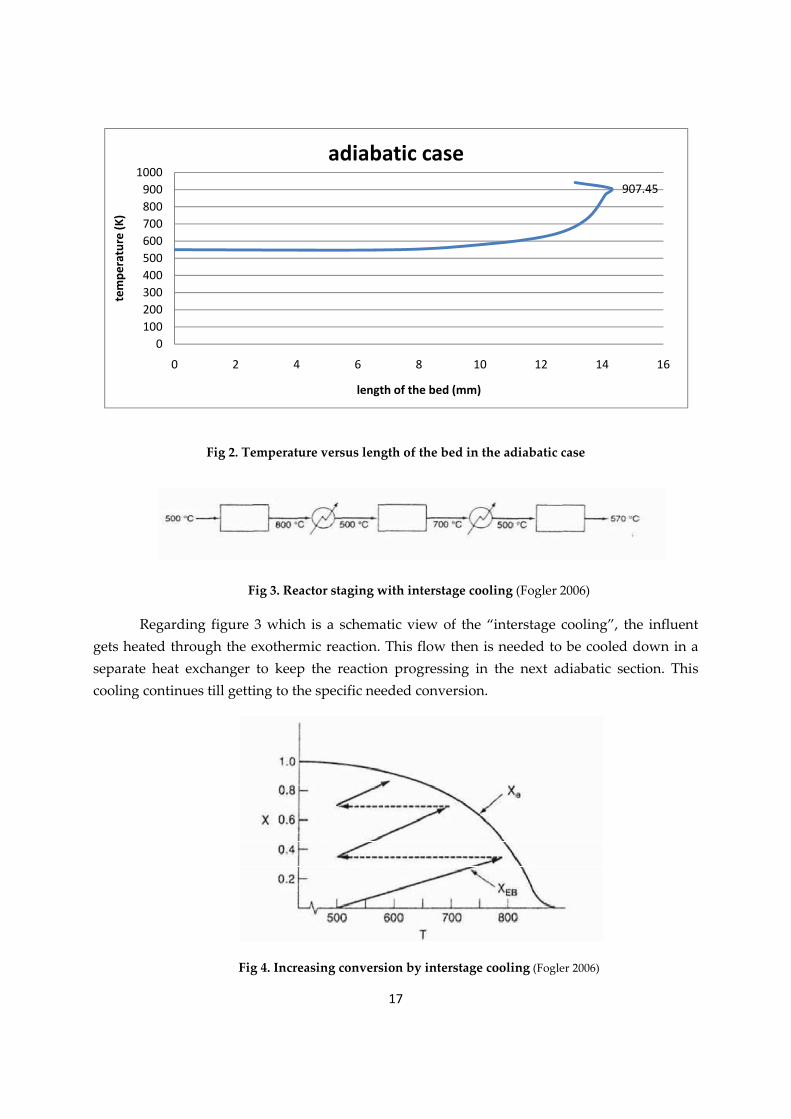
17
907.45
0100200300400500600700800900
1000
0 2 4 6 8 10 12 14 16
tempe
rature (K
)
length of the bed (mm)
adiabatic case
Fig 2. Temperature versus length of the bed in the adiabatic case
Fig 3. Reactor staging with interstage cooling (Fogler 2006)
Regarding figure 3 which is a schematic view of the “interstage cooling”, the influent gets heated through the exothermic reaction. This flow then is needed to be cooled down in a separate heat exchanger to keep the reaction progressing in the next adiabatic section. This cooling continues till getting to the specific needed conversion.
Fig 4. Increasing conversion by interstage cooling (Fogler 2006)

18
As it is shown in figure 4, the interstage cooling increases the conversion in a certain temperature range by preventing the reaction to get to equilibrium conversion.
In the current study the reaction is highly exothermic, so in the adiabatic case without any interstage cooling the reaction gets to equilibrium conversion somewhere in the middle of the reactor which stops reaction to move forward more. This phenomenon could be controlled then by applying a strategy of intercooling.
Fig 5. Increasing conversion by interstage cooling
Referred to Lunde and Kester work which was published on 1974 and also their separate efforts on this, there is a suitable temperature range for carbon dioxide methanation from 400‐700°F (200‐371 °C or 473‐644 K). Sticking to this temperature range should be always considered for temperature controlling. (Lunde, Peter J.,Kester, Frank L 1973) (Kester 1974)
In figure 5 the interstage cooling mechanism is shown by black arrows and dashes which could be interpreted as using 5 heat exchangers in the reactor. In other words it is needed to divide the reactor in 6 reactions which are working in series and the reactant flow passes through the heat exchangers during the reaction period.
Based on figure 5, 6 reactors are needed for this type of operating the reaction. The sizing parameters of these reactors are the same as before. The inner diameter of all of them is 35 mm, but their lengths are varying which is given in table 4.
0
0.2
0.4
0.6
0.8
1
1.2
550 650 750 850 950 1050
Conversion
Temperature, K
equilibrium conversionyield
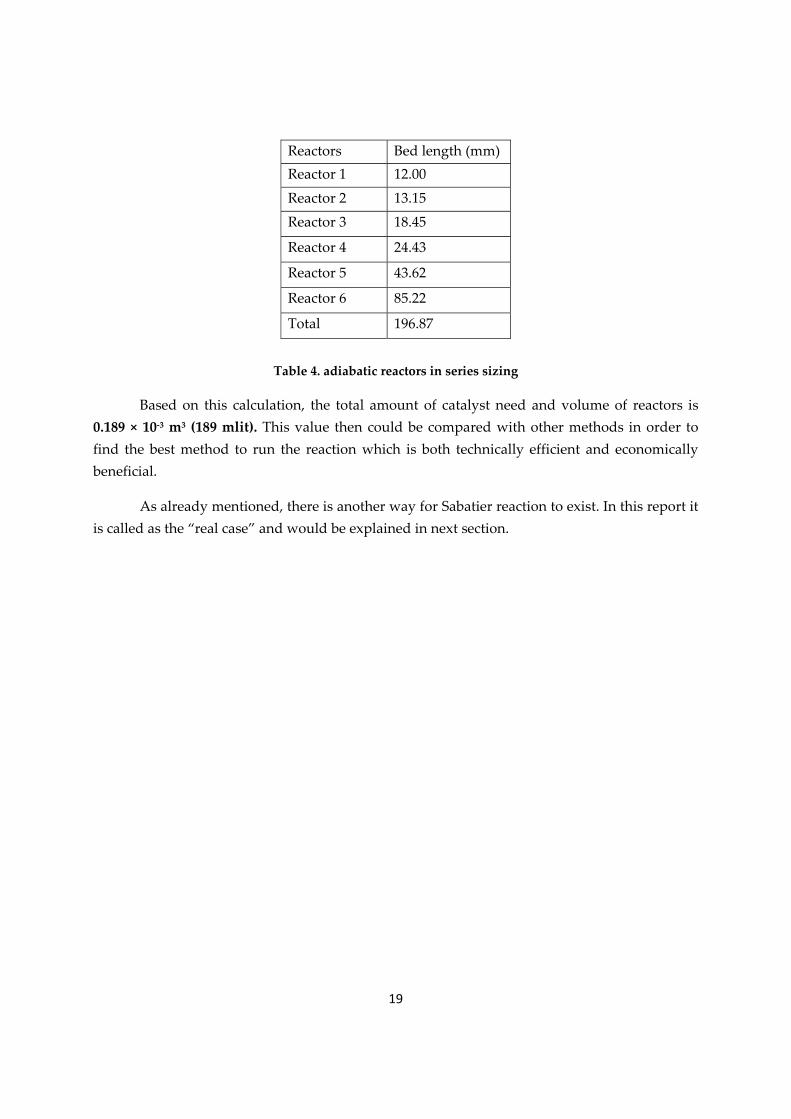
19
Reactors Bed length (mm) Reactor 1 12.00
Reactor 2 13.15
Reactor 3 18.45
Reactor 4 24.43
Reactor 5 43.62
Reactor 6 85.22
Total 196.87
Table 4. adiabatic reactors in series sizing
Based on this calculation, the total amount of catalyst need and volume of reactors is 0.189 × 10‐3 m3 (189 mlit). This value then could be compared with other methods in order to find the best method to run the reaction which is both technically efficient and economically beneficial.
As already mentioned, there is another way for Sabatier reaction to exist. In this report it is called as the “real case” and would be explained in next section.

20
1.3 Results of Real case
1.3.1 Theoretical formulation
Next strategy for simulating the reactor is to consider a cooling system around it. In this way of thinking, the heat which is produced through reaction could be released and so keep the temperature on a suitable level.
The energy balance for one element considering both exothermic reaction and a coolant flow around the reactor wall is as follows:
(9)
This is almost the same as equation (8), but with an additional parameter which is a symbol for the amount of heat that is released to coolant. Qreleased should be calculated based on heat transfer through reactor wall and coolant media.
Fig 6. Schematic heat transfer from catalyst surface to coolant media
Generally talking, there are three main heat transfer mechanism in this reactor: convection, radiation and conduction. Based on fig 6 the produced heat in the reaction is transferred to the coolant media in different steps. First, the reaction heat from catalyst surface is transferred to the reactor wall by conduction, convection and radiation. The next step is convection from the wall outer surface to the coolant media (Lunde 1974). The heat transfer to ambient for the coolant media is not regarded, in this analysis.
The heat transfer coefficient between wall and external coolant is chosen as 164.5 W/m2.K according to the article by Lunde (1974), so the heat transfer resistance within the catalyst side is dominating. External cooling temperature in all trials in this case is 500 K.
Reactant
Gas bulk
Catalyst surface
Reactor w
all
Coolant media
Convection, conduction, radiation
Conduction Convection

21
Regarding the existence of a cooling system, the released heat should be calculated through some correlations directly. It could be assumed that heat transfer mechanism which is more dominant in this case is the convection from the gas to the wall because it could be assumed that there is a same temperature in all parts of the wall and also the fixed temperature in all cooling media. These assumptions then can result in following equation for Qreleased .
, , (10)
In this equation, the heat transfer coefficient hwall is considered as a known value and the reference for it is the article about Sabatie reactor modeling and simulation by Lunde (1974). In this article the value for hw is 14 Btu/hr.ft2.◦F which is equivalent to 79.5 W/m2.K. The parameter A then is heat transfer area which is the area of the element wall exposed to the coolant and could be calculated as follows:
(11)
On the other hand it is important to have some idea about the cooling mechanism as well. The heat exchanging process around the reactor wall could be either co‐current or counter‐current. The difference between these two categories could be seen qualitatively in different literatures and references. For example there are some typical graphs in “Elements of chemical reaction engineering” by Fogler which is about heat effects in PFR/PBR s.
(a) (b)
Fig 7. Schematic graphs for exothermic reactions with cooling system (a).The exothermic reaction with counter‐current cooling,(b) The exothermic co‐current exchanger (Fogler 2006)

22
1.3.2 Analytical calculations and results
Considering all formulations in section 1.3.1, there should be some analysis prior to simulation. Actually size of each element is one of the important parameters since it directly affects the amount of heat release through reactor wall. Therefore, element length has key influence on the gases temperature flowing through the bed.
If the reactor should not be overheated, it is needed to have enough heat transfer area to let the heat be released. To provide cooling surface, using some inert packings such as catalyst support material are definitely needed. Therefore in each element the inert packings can be added to catalyst packings and to increase the length of element for achieving sufficient cooling.
In this analysis, there are two different lengths, active length and physical length. The active length is just referred to the catalyst amount, but physical length is the total size of element which is the combination of both active catalysts and inert packings. In figure 8, there is the schematic picture of what is thought to be done.
To explain the strategy it could be started with describing figure 8 in detail. The process starts at the top of reactor with the first element. The active length of this element has the smallest one among the other elements if the temperature needed to be controlled in a fixed value. In the top elements which the conversion is around 50%, the cooling process is rate determining and the more cooling happens the better rate controlling could be done. Therefore in order to eliminate temperature increase it is needed to increase heat transfer area. This is the reason for two different sizing (Active and physical lengths) for top elements of the reactor
On the other hand, the more reactants go further down in reactor the less would be the impact of cooling for rate determining and instead the reaction itself would be the rate determining parameter. In this strategy for the last two elements in which the reactants have the least concentration, full active size is considered. In other words, these final elements do not have any inert packings and are filled only by active catalysts.
According to what has been mentioned above, the most important part of the reactor design is the last two elements. They have the major load of catalyst in them and so in order to optimize technically and economically it is needed to optimize the amount of catalysts needed for the whole process.
If the reactor operates in high temperature, then the rate of the reaction increases and the active length would be reduced so the higher temperature gets the less amount of catalyst would be needed.

23
Fig 8. Schematic picture of the strategy in simulating real case
On the other hand, it is important to consider the negative impacts of very high temperature. First of all the reaction cannot go forward in high temperatures and the equilibrium occurs earlier than the expected conversion. In equilibrium conditions, as already discussed, there are reactions at both directions and it could be interpreted as “reaction stops”. The other side effect is sintering and deactivation of catalysts. Sintering happens in the case that catalyst has too long exposure to high temperature and the support gets soft and flows, so the active pores of catalyst get narrow and close inside catalyst pellet (Fogler 2006). At high temperatures the catalysts are needed to be regenerated or renewed earlier and this is a fact which should be thought of in designing the reaction conditions.
Regarding stated parameters, there should be an operating range for the reaction temperature in which there is definitely one point which could cover all aspects of concern. In order to determine this specific point, calculations should be done for the whole bed, but before that the operating range should be derived by focusing on last two elements. The procedure is based on trial and error method. Sizing of the last element would be done in different temperatures and X‐Y coordinance of temperature‐size curve would be saved then. The initial temperature in this trial is 550 K in the case that coolant has the fixed temperature of 500 K in all the tests. The results are shown in table 6 and figure 8 respectively.
1st element physical length 1st element active size
T °K
2nd element physical length 2nd element active size
T °K
3rd element physical length 3rd element active size
T °K
20th element physical length 20th element active size
T °K
T °K

24
Initial temperature
(K)
Last element temperature (K)
Last element size (mm)
550 537 104.57 560 550.85 76.61 600 606 27.72 650 664.5 23.74 656 670.86 27.97 662 677.11 37.32 670 685.15 95.94 675 690.06 ‐354.88 700 709 ‐11.2
Table 5. Different trials to find operating temperature range for the last element
Fig 9. Changes on last element length by operating temperature
According to table 5 and figure 9, the temperature increase would result in length decrease. This temperature rise change its’ effect more than an amount and also from a level on there is no more reaction happening. This fact could be viewed in the negative values appeared in “last element size” column in table 6 and points on the right side of the red line in figure 9.
Regarding these results, the smallest size for the last element would be resulted in 650K as the initial temperature which has a bit of increase before the last element and gets to 664.5 K. The question is then if 650K is the optimum temperature for running the reactor in? In order to find a proper answer for this one, it is needed to consider some other facts as well. In following
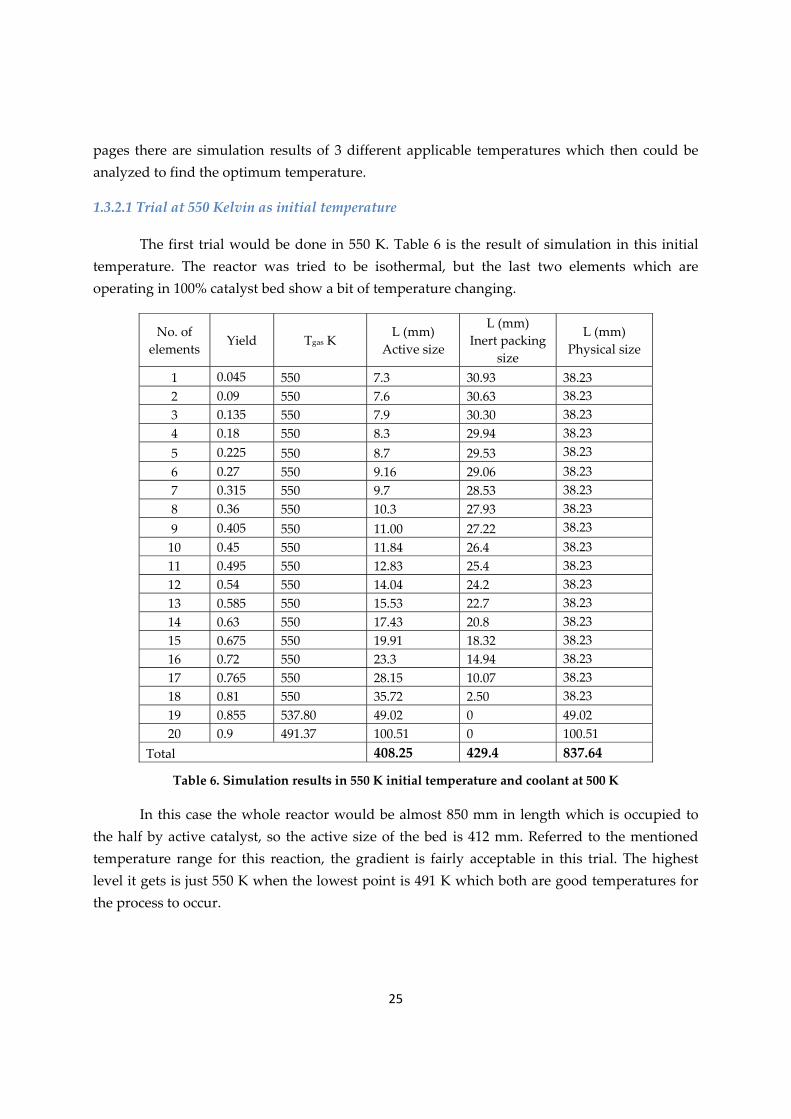
25
pages there are simulation results of 3 different applicable temperatures which then could be analyzed to find the optimum temperature.
1.3.2.1 Trial at 550 Kelvin as initial temperature
The first trial would be done in 550 K. Table 6 is the result of simulation in this initial temperature. The reactor was tried to be isothermal, but the last two elements which are operating in 100% catalyst bed show a bit of temperature changing.
No. of elements
Yield Tgas K L (mm)
Active size
L (mm) Inert packing
size
L (mm) Physical size
1 0.045 550 7.3 30.93 38.23 2 0.09 550 7.6 30.63 38.23 3 0.135 550 7.9 30.30 38.23 4 0.18 550 8.3 29.94 38.23 5 0.225 550 8.7 29.53 38.23 6 0.27 550 9.16 29.06 38.23 7 0.315 550 9.7 28.53 38.23 8 0.36 550 10.3 27.93 38.23 9 0.405 550 11.00 27.22 38.23 10 0.45 550 11.84 26.4 38.23 11 0.495 550 12.83 25.4 38.23 12 0.54 550 14.04 24.2 38.23 13 0.585 550 15.53 22.7 38.23 14 0.63 550 17.43 20.8 38.23 15 0.675 550 19.91 18.32 38.23 16 0.72 550 23.3 14.94 38.23 17 0.765 550 28.15 10.07 38.23 18 0.81 550 35.72 2.50 38.23 19 0.855 537.80 49.02 0 49.02 20 0.9 491.37 100.51 0 100.51
Total 408.25 429.4 837.64
Table 6. Simulation results in 550 K initial temperature and coolant at 500 K
In this case the whole reactor would be almost 850 mm in length which is occupied to the half by active catalyst, so the active size of the bed is 412 mm. Referred to the mentioned temperature range for this reaction, the gradient is fairly acceptable in this trial. The highest level it gets is just 550 K when the lowest point is 491 K which both are good temperatures for the process to occur.
26
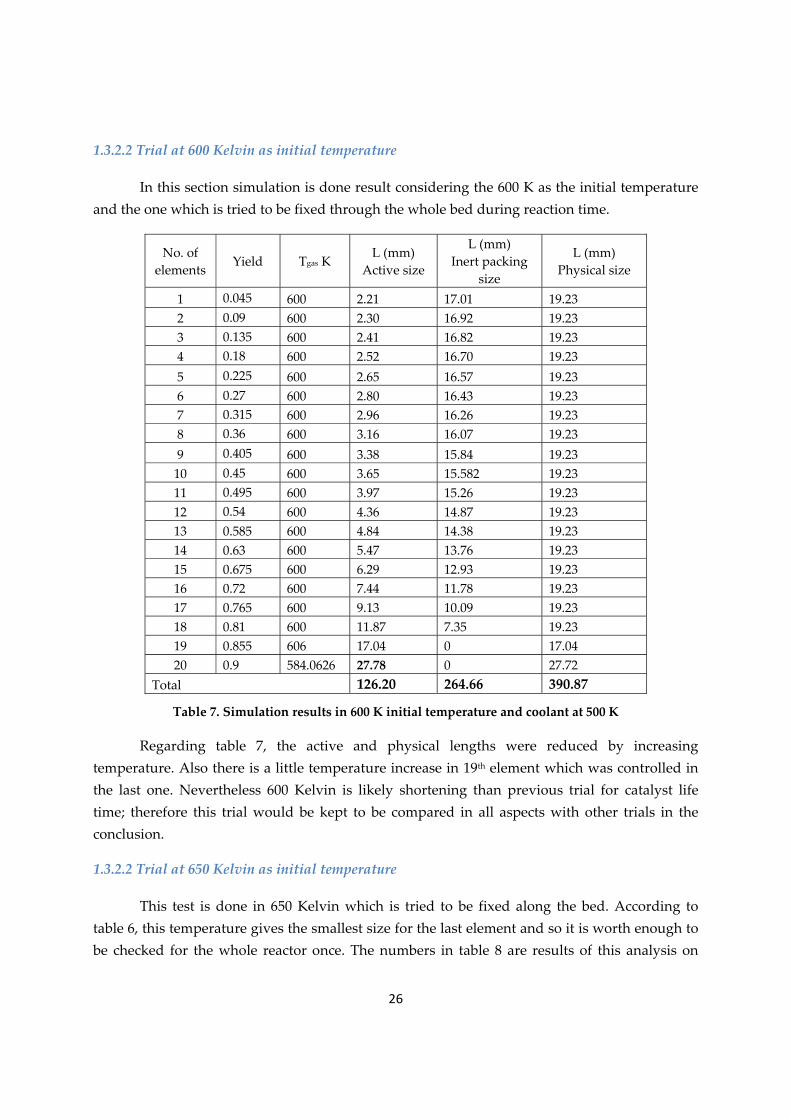
1.3.2.2 Trial at 600 Kelvin as initial temperature
In this section simulation is done result considering the 600 K as the initial temperature and the one which is tried to be fixed through the whole bed during reaction time.
No. of elements
Yield Tgas K L (mm)
Active size
L (mm) Inert packing
size
L (mm) Physical size
1 0.045 600 2.21 17.01 19.23 2 0.09 600 2.30 16.92 19.23 3 0.135 600 2.41 16.82 19.23 4 0.18 600 2.52 16.70 19.23 5 0.225 600 2.65 16.57 19.23 6 0.27 600 2.80 16.43 19.23 7 0.315 600 2.96 16.26 19.23 8 0.36 600 3.16 16.07 19.23 9 0.405 600 3.38 15.84 19.23 10 0.45 600 3.65 15.582 19.23 11 0.495 600 3.97 15.26 19.23 12 0.54 600 4.36 14.87 19.23 13 0.585 600 4.84 14.38 19.23 14 0.63 600 5.47 13.76 19.23 15 0.675 600 6.29 12.93 19.23 16 0.72 600 7.44 11.78 19.23 17 0.765 600 9.13 10.09 19.23 18 0.81 600 11.87 7.35 19.23 19 0.855 606 17.04 0 17.04 20 0.9 584.0626 27.78 0 27.72
Total 126.20 264.66 390.87
Table 7. Simulation results in 600 K initial temperature and coolant at 500 K
Regarding table 7, the active and physical lengths were reduced by increasing temperature. Also there is a little temperature increase in 19th element which was controlled in the last one. Nevertheless 600 Kelvin is likely shortening than previous trial for catalyst life time; therefore this trial would be kept to be compared in all aspects with other trials in the conclusion.
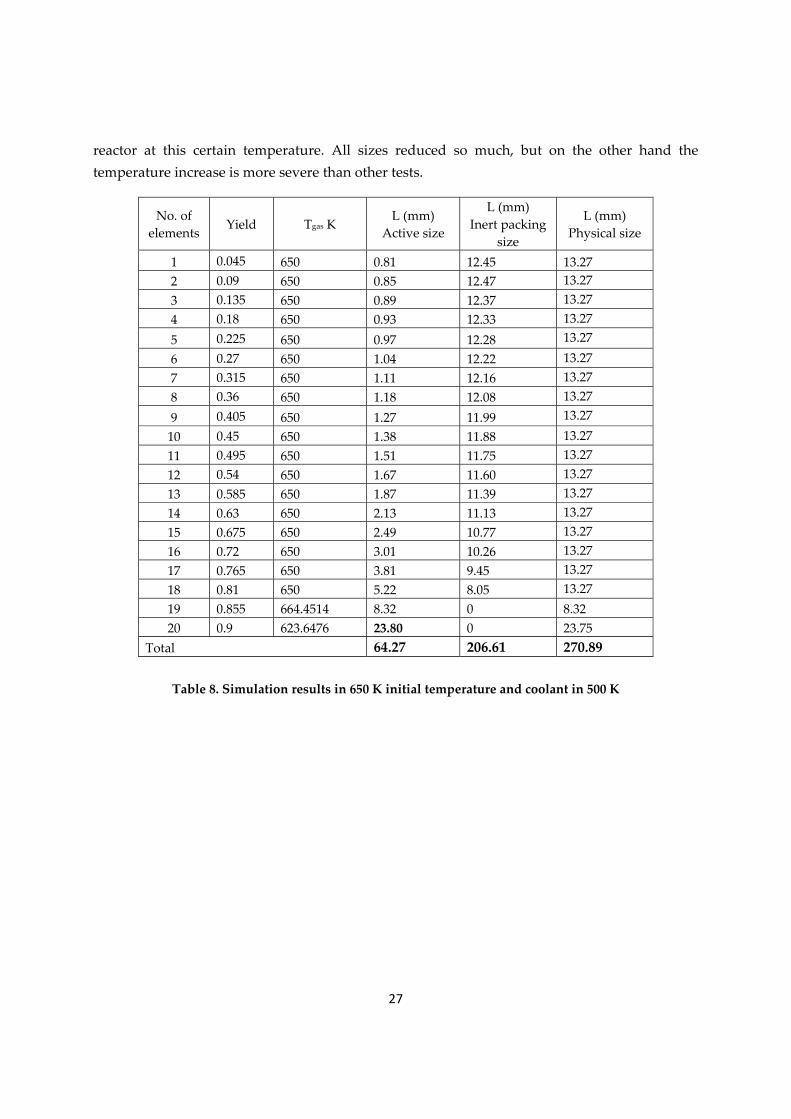
1.3.2.2 Trial at 650 Kelvin as initial temperature
This test is done in 650 Kelvin which is tried to be fixed along the bed. According to table 6, this temperature gives the smallest size for the last element and so it is worth enough to be checked for the whole reactor once. The numbers in table 8 are results of this analysis on
27
reactor at this certain temperature. All sizes reduced so much, but on the other hand the temperature increase is more severe than other tests.
No. of elements Yield Tgas K
L (mm) Active size
L (mm) Inert packing
size
L (mm) Physical size
1 0.045 650 0.81 12.45 13.27 2 0.09 650 0.85 12.47 13.27 3 0.135 650 0.89 12.37 13.27 4 0.18 650 0.93 12.33 13.27 5 0.225 650 0.97 12.28 13.27 6 0.27 650 1.04 12.22 13.27 7 0.315 650 1.11 12.16 13.27 8 0.36 650 1.18 12.08 13.27 9 0.405 650 1.27 11.99 13.27 10 0.45 650 1.38 11.88 13.27 11 0.495 650 1.51 11.75 13.27 12 0.54 650 1.67 11.60 13.27 13 0.585 650 1.87 11.39 13.27 14 0.63 650 2.13 11.13 13.27 15 0.675 650 2.49 10.77 13.27 16 0.72 650 3.01 10.26 13.27 17 0.765 650 3.81 9.45 13.27 18 0.81 650 5.22 8.05 13.27 19 0.855 664.4514 8.32 0 8.32 20 0.9 623.6476 23.80 0 23.75
Total 64.27 206.61 270.89
Table 8. Simulation results in 650 K initial temperature and coolant in 500 K

28
1.4Comparison of adiabatic case with isothermal case
Regarding all the challenges about the adiabatic and isothermal cases, each of them has some cons and pros. Adiabatic method is technically possible, but there are some economical and operational concerns which make it a bit difficult.
i. Maintenance cost: Using small heat exchangers instead of one big cooling system around the whole reactor increases the risk of operation and maintenance. They have their piping systems each which add up to controlling systems of the process on the plant and make the whole package more complicated than isothermal case.
ii. Occupied space: The other point is about size of the whole “reaction package”. The adiabatic case consists of five heat exchangers and six small reactors which surely occupy more space than one big reactor with one heat exchanger. This makes the method critical in the situation with “limited space” issue.
iii. Energy cost: Running five small heat exchangers needs more energy than a big
one. This is categorized in economical concerns about adiabatic method. iv. Operation cost: The adiabatic case consumes more catalyst in comparison with
almost all of the runs in isothermal case. This is considered surely in economical concerns. Also in the adiabatic method each reactor works in high temperature which can reduce the lifetime of catalyst after some hours of operating, when in isothermal case the temperature control in a fairly low temperature keeps the life time span of catalyst in a better condition. This parameter leads to more catalyst regenerating or changing during the life time of the reactor which has its own cost effects as well.
On the other hand adiabatic case has some advantages compared to the isothermal case. For example, the reaction could go on even if one or more reactors do not work. Of course in this situation the final conversion is not satisfying, but anyhow the reaction does not shutdown completely. This is the same with heat exchangers. They could cover one another role in the system if there would be a fault in any of them, since in isothermal case there is just one heat exchanger.
In order to increase the possibility of using adiabatic method, there is a modification which could be done. This system is designed more to reduce the number of reactors and heat exchangers in series, therefore it consists of a big reactor instead of first 5 ones, and one small

29
reactor as the last one. The amount of conversion in this system is the same as conventional adiabatic and also the temperature limit for catalyst activity is considered similar to the adiabatic method.
The mechanism in modified adiabatic system, as in figure 9, is that some part of the cooled product stream would be recycled to the entrance of the reactor. This would help converting of influent before entering to the reactor to some extent.
Fig 10. Modified adiabatic method
The flows in fig. 10 are marked with letters. The amount and concentration of each line is shown in table 9.
Total flowrate (mmol/min)
CO2 (mmol/min)
H2 (mmol/min)
CH4 (mmol/min)
H2O (mmol/min)
Tgas (K)
min = 757 116.45 465.8 174.7 0 550 mʺ= 4167 252.85 1011.40 1777.40 1125.30 550 mʹ= 3410 136.40 545.6 1602.7 1125.30 550 m= 3978 159.13 636.53 1869.81 1312.84 653.23 mout = 568 22.73 90.93 267.11 187.54 550
Table 9. Modified adiabatic reaction design parameters
The stream mout, then would be processed in the second reactor to get the conversion of 90%. The first reactor, which is a substitute for 5 first reactors, has the bigger size while the second one is exactly similar to reactor 6 in conventional adiabatic method. The sizing result is shown in table 10 as follows.
Recycled stream
Cooled product stream Influent
Reactor
m
m'
m"
min
mout

30
Table 10. Modified adiabatic reactor sizing
Although reactor size through this method shows an increase in the amount of catalyst use about 3.5 times more than conventional adiabatic, reducing the risk of operation and maintenance and number of heat exchangers needed for reaction would balance the advantages and disadvantages. Also the life time of catalysts in this method could be longer than adiabatic method, because in that case the temperature for all reactors is ranging from 550‐650 K and this is repeated 6 times while in current method temperature increase happens 2 times and so the catalysts are less under the risk of sintering and deactivation. Also the system simplicity of modified method in comparison with conventional adiabatic is the other important parameter. This would help in constructing and running the unit easier.
Lbed
(mm)
First reactor 609.46 Second reactor 85.22 total 694.70

31
2 Conclusion
The present use of Sabatier reaction is CO2 elimination in syngas for ammonia plants. This reaction can increase the efficiency of biogas industry on methane production. In this report the focus is more on temperature controlling of this system and optimizing different possible methods for running the reactor.
Considering all results, discussions and analyses in this report and according to the comparison of adiabatic reactor with the isothermal one, it is clear now that running the reaction in isothermal condition is more beneficial and optimized than conventional adiabatic. It is also considerable that for a small laboratory reactor a single tube that combines reaction and cooling surfaces is much cheaper and easier to run than six adiabatic beds and five separate coolers while for a large reactor with a large cooling demand it may be more economic to choose interstage coolers and adiabatic reactor sections.
Even though applying some changes in adiabatic run, would lead us to a commercially feasible method which could be competitive with isothermal method to some extent. In modified adiabatic method, construction and maintenance is less complicated and costly while the amount of active catalyst is a bit more than isothermal case. On the other hand, up scaling these cases 10,000 times than the lab scale adds some extra considerations and points to analyze which would be more important than the amount of active catalyst use. The possibilities of system upgrading, temperature controlling, substituting deactivated catalysts with active ones, constructing easily the plant from scratch, are some parameters which make the choice a bit difficult.
Since all these parameters have their own effect and cost to the total observation of the system, selecting the best one is so dependent and needs different detail designs and economical calculations.
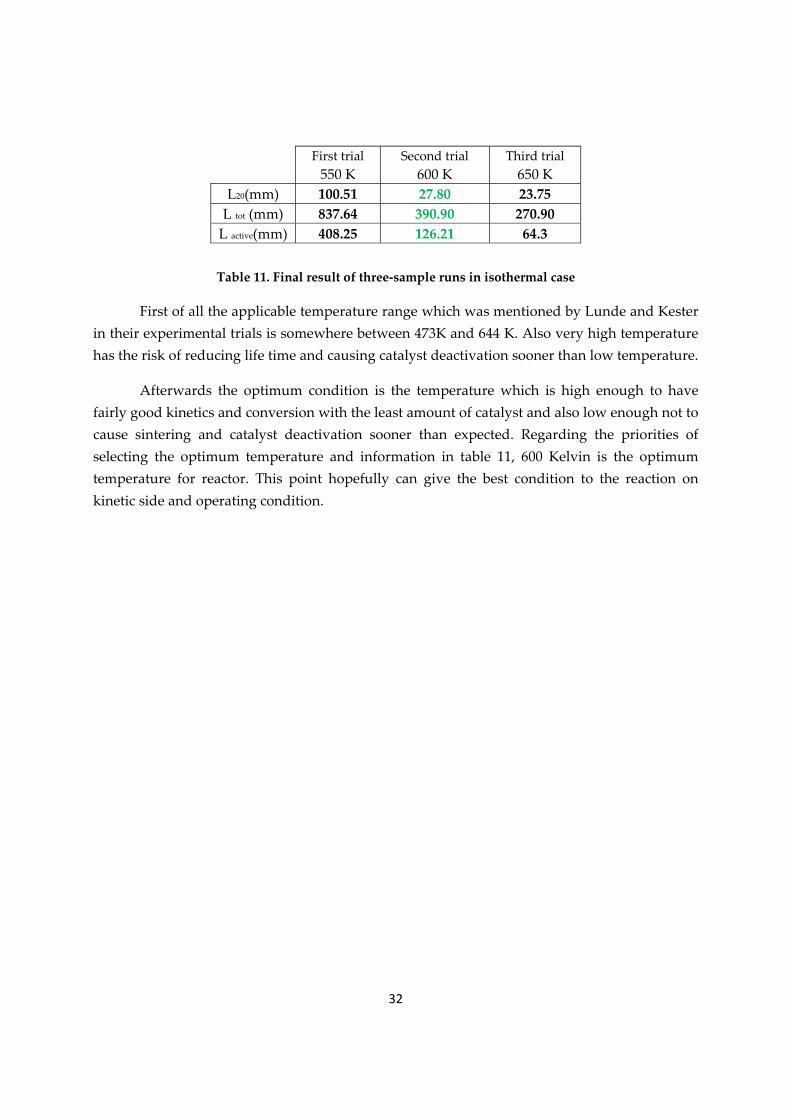
Regarding all above, for the lab scale design there is less doubt of selecting the isothermal run. This method should be optimized and the best condition would be selected to operate the reactor on that. In table 12 there are results of isothermal runs in three different temperatures. The sample runs are in 550 K, 600 K and 650 K respectively. There are also the length of final element, total physical length and total active length in table 11. The more temperature increases the shorter would be these lengths for reactor, but the optimum temperature is not the highest possible one.
32
Table 11. Final result of three‐sample runs in isothermal case
First of all the applicable temperature range which was mentioned by Lunde and Kester in their experimental trials is somewhere between 473K and 644 K. Also very high temperature has the risk of reducing life time and causing catalyst deactivation sooner than low temperature.
Afterwards the optimum condition is the temperature which is high enough to have fairly good kinetics and conversion with the least amount of catalyst and also low enough not to cause sintering and catalyst deactivation sooner than expected. Regarding the priorities of selecting the optimum temperature and information in table 11, 600 Kelvin is the optimum temperature for reactor. This point hopefully can give the best condition to the reaction on kinetic side and operating condition.
First trial 550 K
Second trial 600 K
Third trial 650 K
L20(mm) 100.51 27.80 23.75 L tot (mm) 837.64 390.90 270.90 L active(mm) 408.25 126.21 64.3

33
Bibliography Emmett, PH 1951, Catalysis, Reinhold, New York.
Fogler, HS 2006, Elements of chemical reaction engineering, 4th edn, Pearson education, United state.
Frolov, S.M., Kuznetsov, N.M., Krueger, C 2009, ʹ Real‐gas properties of n‐alkanes O2,N2,H2O,CO,CO2 and H2 for diesel engine operation conditionsʹ, Russian journal of physical chemistry B, vol 3, no. 8, pp. 1191‐1252.
Kester, FL 1974, ʹHydrogenation of carbon dioxide over a supported Ruthenium catalystʹ, Am Chem Soc, Div Fuel Chem, vol 19, no. 1, pp. 146 ‐156.
Lunde, PJ 1974, ʹModeling, simulation. and operation of a Sabatier Reactorʹ, Industrial Engineering Chemical Process Design Develop., vol 13, no. 3, pp. 226‐233.
Lunde, Peter J., Kester, Frank L. 1974, ʹ Carbon dioxide methanation on a Ruthenium catalystʹ, Industrial Engineering Chemical Process Design Develop., vol 13, no. 1, pp. 27‐33.
Lunde, Peter J.,Kester, Frank L 1973, ʹRates of Methane formation from carbon dioxide and hydrogen over a ruthenium catalystʹ, Journal of catalysis, no. 30, pp. 423‐429.
Passut, Charles A., Danner, Ronaldo P. 1972, ʹCorrelation of ideal gas enthalpy, heat capacity and entropyʹ, Industrial Engineering Chemical Process Design Develop., vol 11, no. 4, pp. 543‐544.
Taglia P.G, P 2010, ʹBiogas: rethinking the mid west’s potentialʹ, clean Wisconsin.
Violeta Bescós, LCSC 2008, ʹIntegration of synthetic methane into the existing biogas production of Henriksdalʹ, Report for the design course, p. 3.


















