
Vendor Update: Penguin Computing Arend Dittmer, Penguin Computing.
Upload
norma-moodyCategory
view
214download
1
SHRiMP: Accurate Mapping of Short Reads in Letter- and Colour-spaces
Stephen Rumble, Phil Lacroute, …, Arend Sidow, Michael Brudno

How SHRiMP works:
Stage 1: Map reads to target genome
Stage 2: Compute statistics

Read Mapping
Three phases
Very fast k-mer scan (index reads, scan genome)
Fast, vectorized Smith-Waterman to confirm
Slow, complete backtracking S-W for top ‘n’ hits

Read Mapping: Phase 1
Create a hash table of size 4^(k-mer length) 4 bases – ignore all else (‘N’, ‘X’, wobble
codes…)
This becomes our kmer to read index
…
AACTGTACCAGTGAG

Read Mapping: Phase 1
Create a hash table of size 4^(k-mer length) 4 bases – ignore all else (‘N’, ‘X’, wobble
codes…)
This becomes our kmer to read index
…
AACTGTaccagtgag
AACTGT

Read Mapping: Phase 1
Create a hash table of size 4^(k-mer length) 4 bases – ignore all else (‘N’, ‘X’, wobble
codes…)
This becomes our kmer to read index
…
aACTGTAccagtgag
AACTGTACTGTA

Read Mapping: Phase 1
Create an index of size 4(k-mer length) 4 bases – ignore all else (‘N’, ‘X’, wobble
codes…)
This is our k-mer to read index
…
aaCTGTACcagtgag
AACTGTACTGTACTGTAC
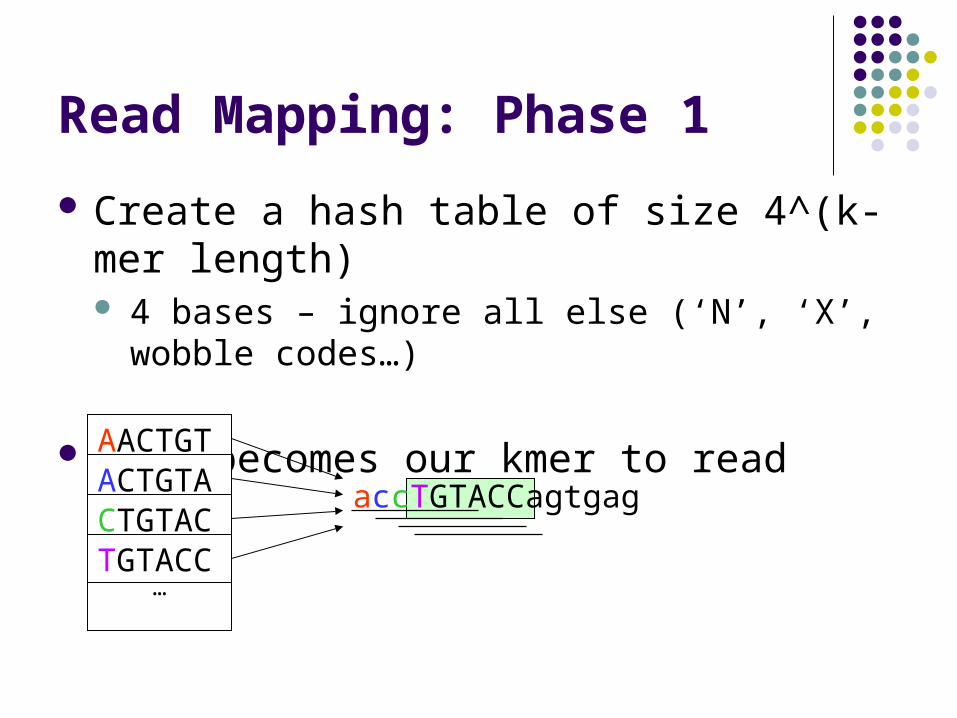
Read Mapping: Phase 1
Create a hash table of size 4^(k-mer length) 4 bases – ignore all else (‘N’, ‘X’, wobble
codes…)
This becomes our kmer to read index
…
accTGTACCagtgag
AACTGTACTGTACTGTACTGTACC

Read Mapping: Phase 1
Create a hash table of size 4^(k-mer length) 4 bases – ignore all else (‘N’, ‘X’, wobble
codes…)
This becomes our kmer to read index
…
AACTGTACTGTACTGTACTGTACC
Read 7
Read 32
Read 18
Read 12
Read 13
Read 12
Read 7
Read 15
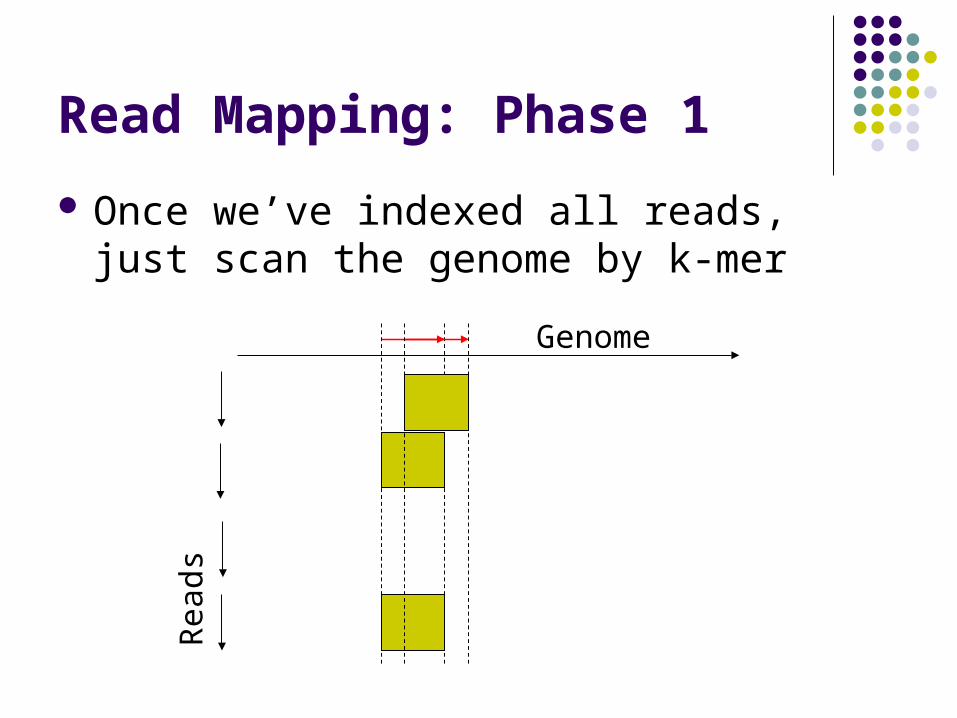
Read Mapping: Phase 1
Once we’ve indexed all reads, just scan the genome by k-mer
Genome
Rea
ds

Read Mapping: Phase 1
Remember the k-mer hits within a given interval (window)
When sufficient hits, look more closely
“Look more closely” means calculate a fast Smith-Waterman score

Technicalities
We don’t always use full k-mers (q-grams).
We actually support ‘spaced seeds’, but the algorithm doesn’t change much. For each spaced seed, ‘compress out’ the k-mer
and use it as the hash index

Read Mapping: Phase 2
Smith-Waterman is very expensive
NxM matrix isn’t too big for short reads and windows, but…
We call the vectorized code millions of times
We don’t want a bottleneck – aim for no more than 50% of the total runtime
We only want one score as quickly as possible
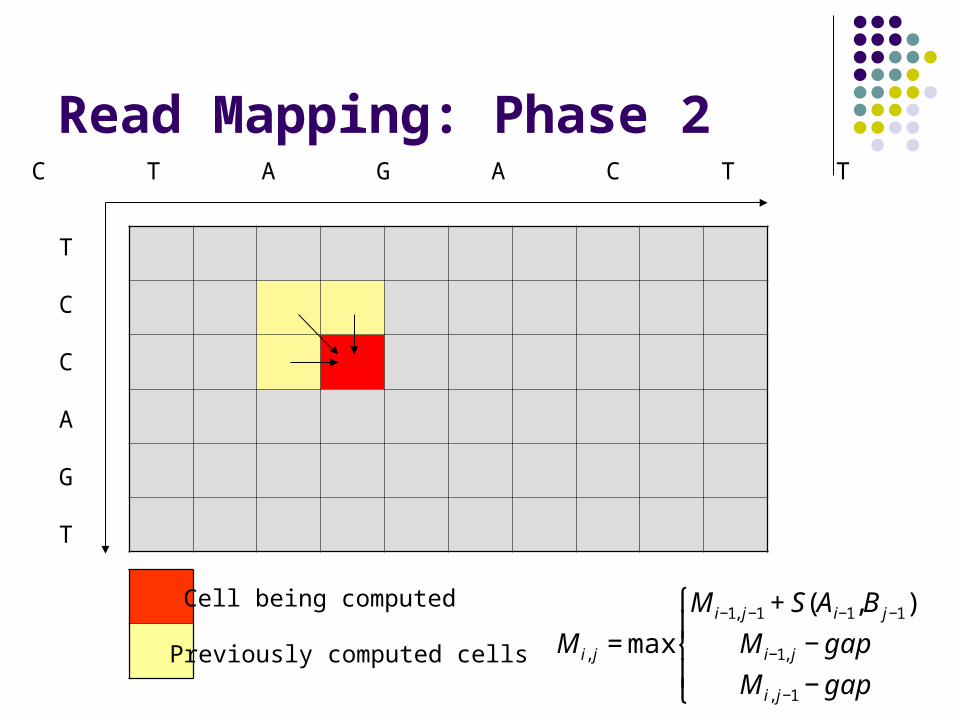
Read Mapping: Phase 2
Cell being computed
Previously computed cells
A C T A G A C T T G
T
C
C
A
G
T
€
M i, j = max
M i−1, j−1 + S(Ai−1,B j−1)
M i−1, j − gap
M i, j−1 − gap
⎧
⎨ ⎪
⎩ ⎪

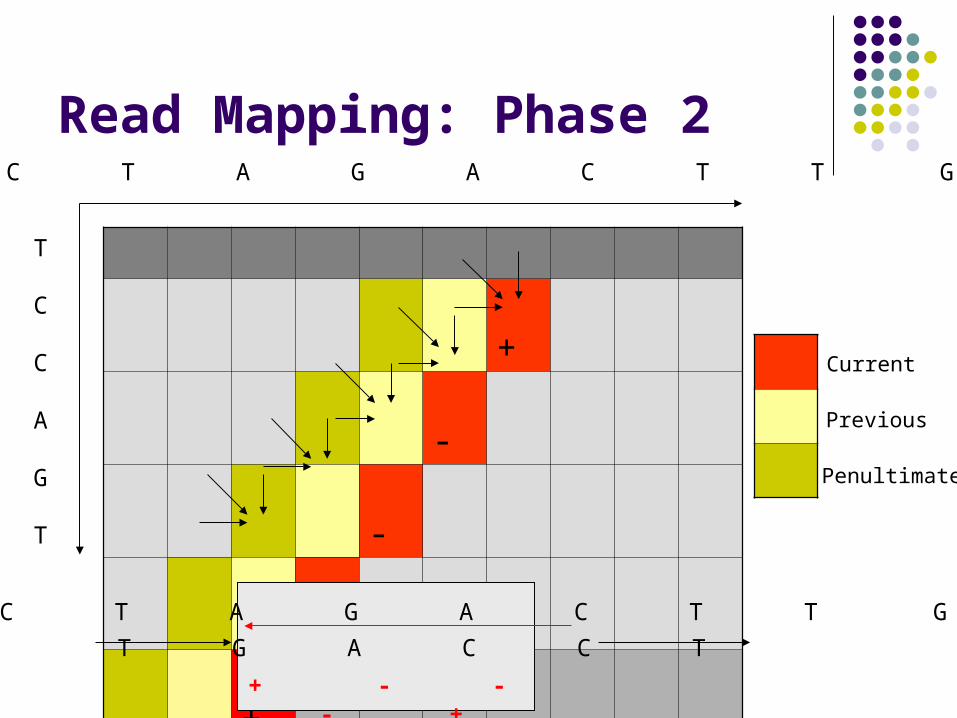
Read Mapping: Phase 2
Each forward-facing diagonal in S-W matrix depends on: Small constant # of previous diagonals Small constant # of scalars
We can compute entire diagonals in parallel Our speed-up is proportional to the diagonal size
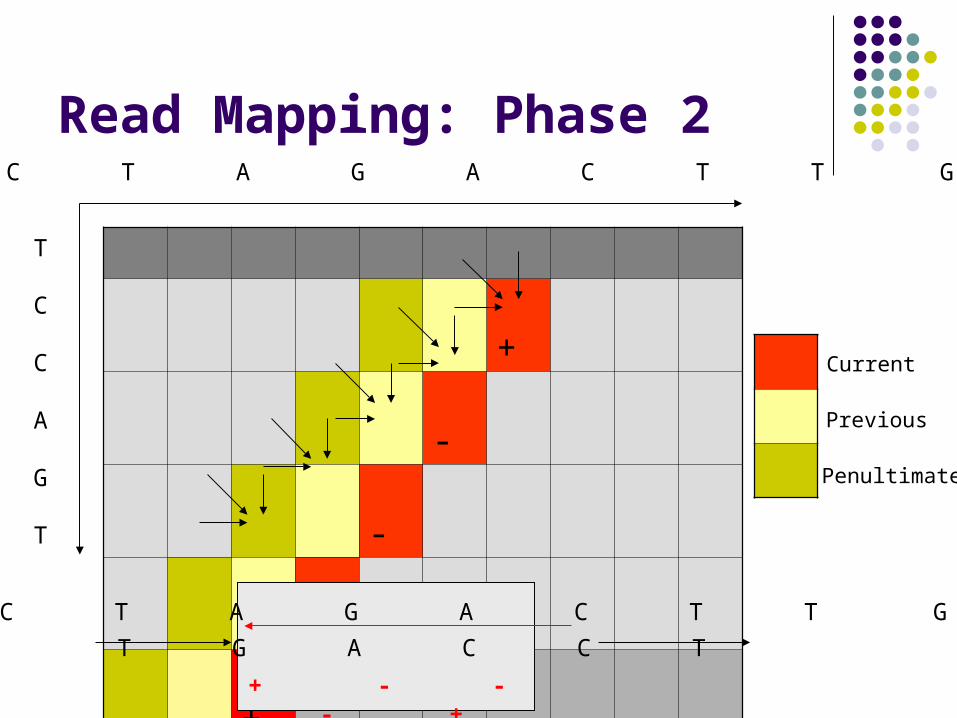
Read Mapping: Phase 2
+
-
-
-
+
Current
Previous
Penultimate
A C T A G A C T T G
T
C
C
A
G
T
A C T A G A C T T G
T G A C C T
+ - - - +
Read Mapping: Phase 2
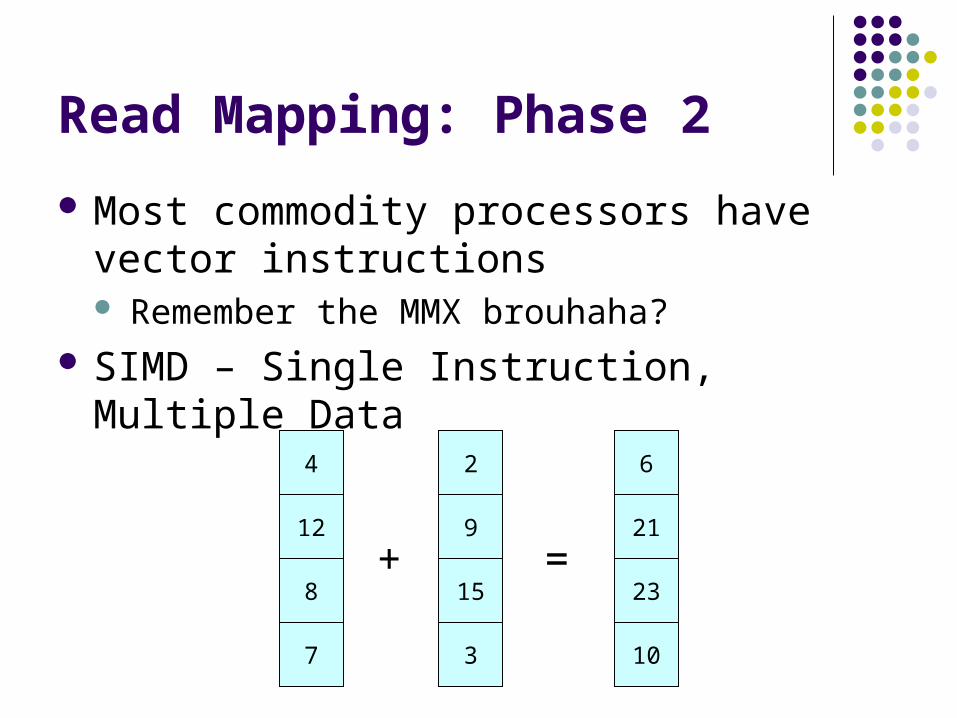
Most commodity processors have vector instructions Remember the MMX brouhaha?
SIMD – Single Instruction, Multiple Data
4
12
8
7
2
9
15
3
6
21
23
10
+ =
Read Mapping: Phase 2
+
-
-
-
+
Current
Previous
Penultimate
A C T A G A C T T G
T
C
C
A
G
T
A C T A G A C T T G
T G A C C T
+ - - - +

Read Mapping: Phase 2
Match scores typically use a scoring matrix ScoringMatrix[SeqA[i]][SeqB[j]] But this doesn’t scale: Individual cell scores become a
bottleneck
Can precompute a ‘query profile’ (expensive), or…
If we only care about strict match/mismatch we can use logical bit-wise operations SIMD instructions work here (fully parallel)

Read Mapping: Phase 2
Results: Our vectorized S-W is as fast, or faster than other
very complicated SIMD implementations
500 million+ matrix cells/second on Core 2 machines
Even with small seeds, S-W accounts for at most half of the total run time

Read Mapping: Phase 3
Recap: K-mer scan selects areas of reasonable similarity
Vectorized S-W (dis)confirms similarity
Best ‘n’ hits per read are given a full alignment with backtrace

Read Mapping: Phase 3
Letter-space alignments are simple: K-mer scan, Vectorized S-W, Full S-W in letters,
give user pretty output
What about AB SOLiD colour-space? Biologists want to see A,C,G,T, not 0,1,2,3… Dealing with strange SOLiD properties… Our solution:
K-mer scan, Vectorized S-W in colour-space Full S-W in letter-space, but we can’t just convert
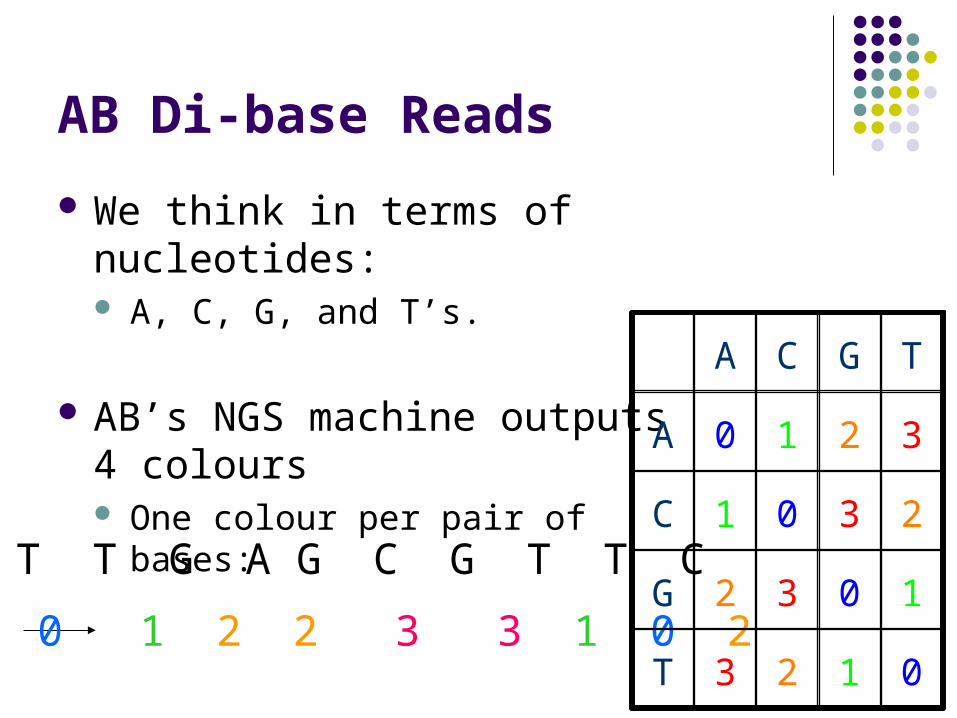
AB Di-base Reads
We think in terms of nucleotides: A, C, G, and T’s.
AB’s NGS machine outputs 4 colours One colour per pair of bases:
T T G A G C G T T C
T 0 1 2 2 3 3 1 0 20123T
1032G
2301C
3210A
TGCA
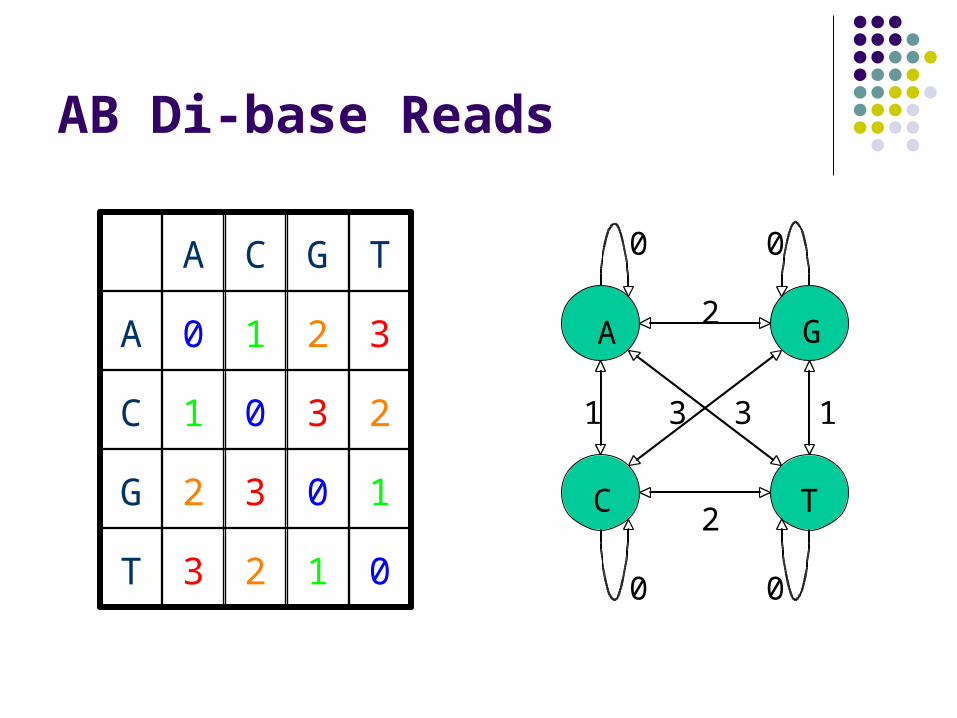
AB Di-base Reads
A G
C T
0 0
0 0
1 1
2
2
3 3
0123T
1032G
2301C
3210A
TGCA
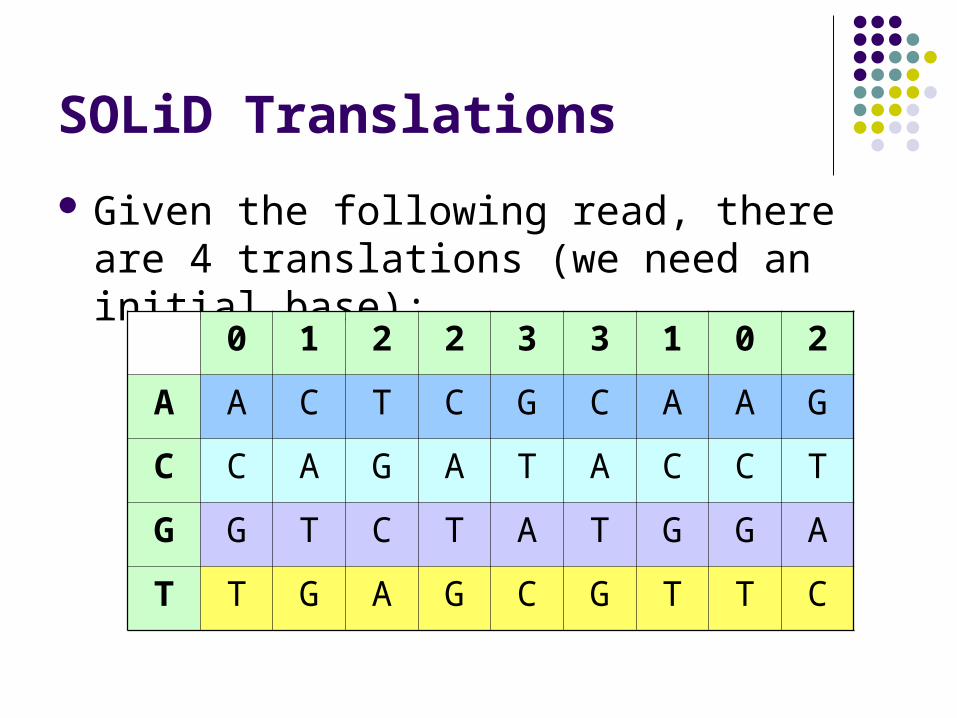
SOLiD Translations
Given the following read, there are 4 translations (we need an initial base):
0 1 2 2 3 3 1 0 2
A A C T C G C A A G
C C A G A T A C C T
G G T C T A T G G A
T T G A G C G T T C
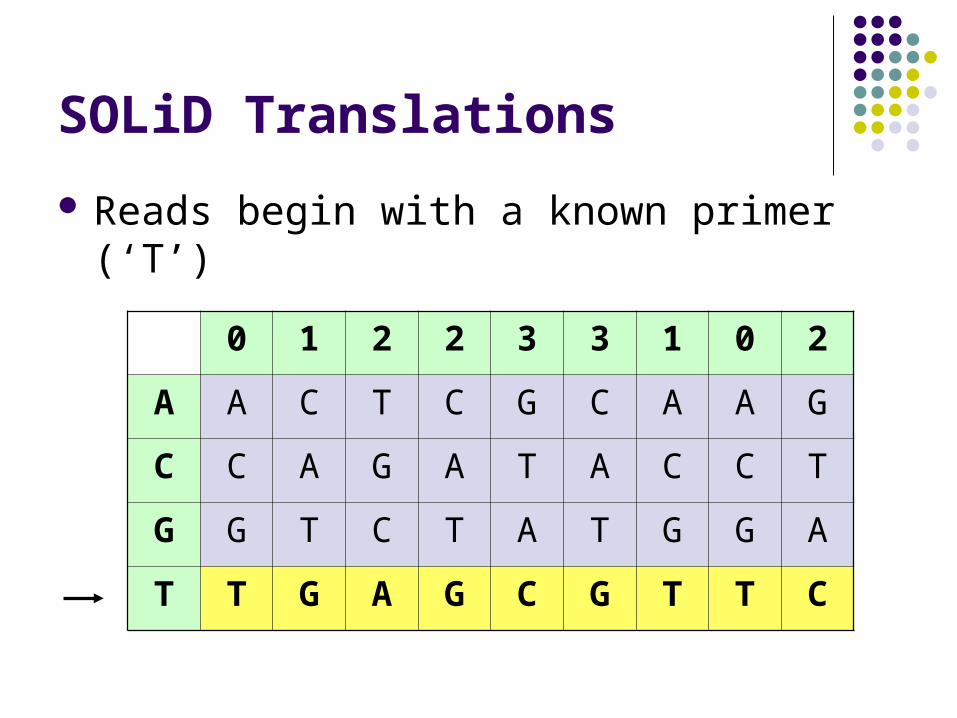
SOLiD Translations
Reads begin with a known primer (‘T’)
0 1 2 2 3 3 1 0 2
A A C T C G C A A G
C C A G A T A C C T
G G T C T A T G G A
T T G A G C G T T C

SOLiD Translations
What happens if a read error occurs? The right translation was: T T G A G C G T T C
0 1 0 2 3 3 1 0 2
A A C C T A T G G A
C C A A G C G T T C
G G T T C G C A A G
T T G G A T A C C T

Colour-space Smith-Waterman
There are four unique translations for every read
An error will cause us to change frames (different translation)
Why not do a S-W across all four letter-space translations with some error penalty?
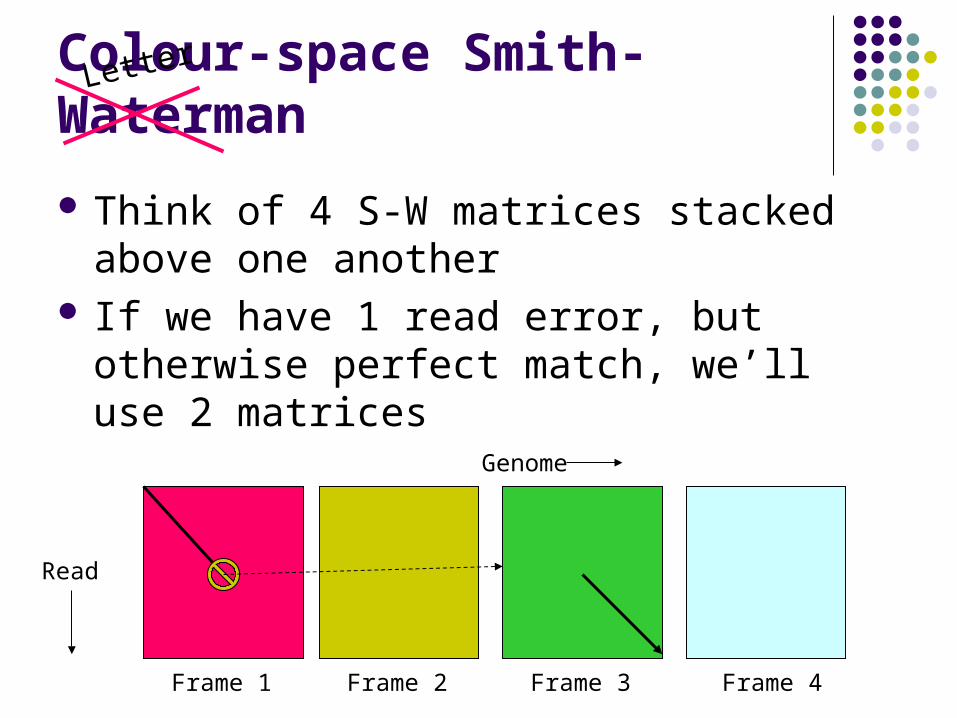
Colour-space Smith-Waterman
Think of 4 S-W matrices stacked above one another
If we have 1 read error, but otherwise perfect match, we’ll use 2 matrices
Genome
Read
Frame 1 Frame 2 Frame 3 Frame 4
Letter

Colour-space Smith-Waterman
End result:
G: 1123724 TA-ACCACGGTCACACTTGCATCAC 1123701 || |||||||||| |||X|||||||T: TACACCACGGTCAGACTtGCATCACR: 0 T0311101130121221211313211 24
Should be ‘0’

Statistics
After reads are mapped, mull over the results For each read:
P(hit by pure chance – not a valid hit)
P(hit generated by genome – valid hit)
P(hit is best of all for particular read)

Results
Speed Simple k-mer scan is very fast
Important when seeds are bigger (less S-W)
Vectorized S-W is fast Important when seeds are smaller (more S-W)
Generally well-balanced run time Big seeds make k-mer scan the bottleneck (this is good -
it’s really fast)
Easily parallelised – just divide the reads over CPUs

Results
C. Savingyi 22M 25bp reads 173Mb genome
S-W would take at least a few thousand CPU days SHRiMP runs in about 50 CPU days with fairly small
seeds (length 8, weight 7)
SNP, indel, error rates correspond well to known averages for this organism


















