
Seuqence Analysis '18 --lecture 10 · Phyogenetic trees Why build phylogenetic trees? To trace the...
27
Seuqence Analysis '18 --lecture 10 Trees types of trees Newick notation UPGMA Fitch Margoliash Distance vs Parsimony
Transcript of Seuqence Analysis '18 --lecture 10 · Phyogenetic trees Why build phylogenetic trees? To trace the...
Seuqence Analysis '18 --lecture 10
Trees
types of trees
Newick notation
UPGMA
Fitch Margoliash
Distance vs Parsimony

Phyogenetic trees
Why build phylogenetic trees?
To trace the branch order of "taxa" (taxon = a gene, a species, a population, etc.)
To understand the evolution of traits
As part of a multiple sequence alignment algorithm
What is a phylogenetic tree?
A model of evolutionary relationships -- common ancestors and speciation events.
Trees can be "rooted" or "unrooted"
root
no root

Common ancestors (hypothetical)
Lineages
Taxa are observed species or genes.
root
Tree Terminology
A B C D
time(rooted trees only)
taxa
outgroup

Inferring evolutionary relationships between the taxa requires rooting the tree:
To root a tree mentally, imagine that the tree is made of string. Grab the string at the root and tug on it until the ends of the string (the taxa) fall opposite the root:
A
BC
Root D
A B C D
Root
Rooted tree
Unrooted tree

Where the tree is rooted changes its meaning.
C
D
A
B
A
B
C
D
D
C
A
B
A
B
C
D
B
A
C
D
Each of these trees is possible by choosing a different root.
This one says C and D branched
late.
This one says C and D branched
early.

Taxon order doesn't matter.
B
A
C
D
A
B
D
C
B
C
AD
B
D
AC
B
ACD
B
A
C
D
A
B
C
D
Rotating an ancestor node does not change the relationships.
UGENE: “swap siblings”

1. Choose the midpoint between the two most distant branches.
2. Choose one taxon as the "out group." (it branches first.)
Two strategies for rooting a tree: "outgroup" and "midpoint"
A
B C
D
A B D C
A BD
C
cladogram
phylogram A good outgroup is not too distant from the rest of the tree.

(,(,(,)))Trees can be represented in plain text Newick notation.
Each set of parentheses represents a branch-point (split), the comma separates left and right lineages.
Implies a rooted tree.A
B
C
D
(D,(C,(A,B))) =
Newick notation can contain sequence labels or not.
Newick notation

�9
Here is a Newick Tree for 50 taxa(((((((((((((('Phoca caspica':0.07021230607681982,'Halichoerus grypus':0.07021230607681982):0.005901881457347935,'Phoca sibirica':0.07611418753416775):0.0066686738512333615,('Phoca largha':0.04443967284675786,'Phoca vitulina':0.04443967284675786):0.038343188538643255):0.013787136869767208,'Phoca hispida':0.09656999825516832):0.203034327163951,('Phoca fasciata':0.20337512105161756,'Phoca groenlandica':0.20337512105161756):0.09622920436750176):0.10586253190986505,'Cystophora cristata':0.4054668573289844):0.277597277733635,'Erignathus barbatus':0.6830641350626194):0.14657442478466165,((((('Hydrurga leptonyx':0.17446758873283646,'Leptonychotes weddellii':0.17446758873283646):0.11244111751153194,'Lobodon carcinophaga':0.2869087062443684):0.035445887072970195,'Ommatophoca rossii':0.3223545933173386):0.15128664129694624,('Mirounga angustirostris':0.09811944526177002,'Mirounga leonina':0.09811944526177002):0.3755217893525148):0.08981113189163292,('Monachus schauinslandi':0.4601516513954143,'Monachus monachus':0.4601516513954143):0.10330071511050348):0.2661861933413633):0.5780758878993605,((((((('Arctocephalus australis':0.053799989415323844,'Arctocephalus forsteri':0.053799989415323844):0.10499286763782767,'Arctocephalus townsendi':0.15879285705315152):0.050831179967801315,('Neophoca cinerea':0.18323401004037046,'Phocarctos hookeri':0.18323401004037046):0.026390026980582376):0.023746771742663958,('Otaria byronia':0.2137942101190925,'Arctocephalus pusillus':0.2137942101190925):0.019576598644524296):0.06629271789640678,('Eumetopias jubatus':0.21816518989795122,'Zalophus californianus':0.21816518989795122):0.08149833676207235):0.22503546176257072,'Callorhinus ursinus':0.5246989884225943):0.5000522375281334,'Odobenus rosmarus':1.0247512259507277):0.3829632217959138):0.7381691639899293,((((((('Enhydra lutris':0.5669410807408264,'Lontra canadensis':0.5669410807408264):0.18377573103801603,'Mustela vison':0.7507168117788424):0.14938941488894708,(('Martes americana':0.11049911428194073,'Martes melampus':0.11049911428194073):0.48119766656825963,'Gulo gulo':0.5916967808502004):0.3084094458175891):0.12834863391441786,'Meles meles':1.0284548605822073):0.1281351620935769,'Taxidea taxus':1.1565900226757841):0.4597494862810001,'Procyon lotor':1.6163395089567842):0.2702352263259904,(('Mephitis mephitis':0.6741102608383382,'Spilogale putorius':0.6741102608383382):0.9584540267788312,'Ailurus fulgens':1.6325642876171693):0.2540104476656053):0.2593088764537961):0.12001769423538722,(((((('Ursus americanus':0.2041501739476191,'Ursus thibetanus':0.2041501739476191):0.050098425820357534,'Helarctos malayanus':0.25424859976797665):0.05191223248524496,('Ursus arctos':0.05235633974404936,'Ursus maritimus':0.05235633974404936):0.25380449250917225):0.07051918322218598,'Melursus ursinus':0.3766800154754076):0.48977263674241156,'Tremarctos ornatus':0.8664526522178192):0.25036843580810175,'Ailuropoda melanoleuca':1.1168210880259208):1.149080217946037):0.33217551543194546,(('Alopex lagopus':0.34911467288029396,'Vulpes vulpes':0.34911467288029396):0.6087120531261618,('Canis latrans':0.13299259555966594,'Canis lupus':0.13299259555966594):0.8248341304467899):1.6402500953974477):0.3800760543442525,((((('Acinonyx jubatus':0.3268665386781608,'Puma concolor':0.3268665386781608):0.10200923996228234,'Lynx canadensis':0.42887577864044313):0.11666599433183367,'Felis silvestris':0.5455417729722768):0.17421812181062735,((('Panthera pardus':0.2563323902942867,'Uncia uncia':0.2563323902942867):0.10867247005991426,'Panthera tigris':0.365004860354201):0.13697718945179482,'Neofelis nebulosa':0.5019820498059958):0.21777784497690833):1.162879140292626,'Herpestes auropunctatus':1.88263903507553):1.095513840672626):1.4072977330687713,'Manis tetradactyla':4.385450608816927);

Did the Florida Dentist infect his patients with HIV?DENTIST
DENTIST
Patient D
Patient F
Patient CPatient APatient GPatient BPatient EPatient A
Local control 2Local control 3
Local control 9
Local control 35
Local control 3
Yes: The HIV sequences from these patients fall within the clade of HIV sequences found in the dentist.
No
No
From Ou et al. (1992) and Page & Holmes (1998)
Phylogenetic tree of HIV sequences from the DENTIST, his Patients, & Local HIV-infected People:
What good are accurate phylogenetic trees?

Evolutionary time
A
B
C
D
11
1
6
3
5
genetic change
A
B
C
D
time
A
B
C
D
no meaning
Cladogram Phylogram Ultrametric tree
(D:5,(A:1,(C:1,B:6):1):3)Newick format with distances

Neighbor joining: cladogram
Choose the closest neighbors. Add a node between them. Choose the next closest, ad so on.
A B C D E Species A ---- 0.20 0.50 0.45 0.40 Species B 0.23 ---- 0.40 0.55 0.50 Species C 0.87 0.59 ---- 0.15 0.40 Species D 0.73 1.12 0.17 ---- 0.25 Species E 0.59 0.89 0.61 0.31 ----
A B C D E

�13
UPGMA
A B C D E Species A ---- 0.20 0.50 0.45 0.40 Species B 0.23 ---- 0.40 0.55 0.50 Species C 0.87 0.59 ---- 0.15 0.40 Species D 0.73 1.12 0.17 ---- 0.25 Species E 0.59 0.89 0.61 0.31 ----
A B C D E
0.115
0.085
Distance to common ancestor = Average distance between taxa in two clades, divided by two.
2= ____
0.23
0.115
0.0850.145
1) Generate neighbor-joining tree. (NJ)2) For first neighbors, distance to ancestor is dij/23) For next neighbors, distance to ancestor is average pairwise distance between taxa in two clades, divided by two.4) Subtract to get lineage distances.
J-C corrected distances
raw p-distances
fill in the blanks
Unweighted pair group method using averagesAssumes constant evolutionary clock. All sequences have the same distance to the common ancestor. Ultrametric tree.

cladogram to phylogram: Fitch-Margoliash algorithm
A
B
D
E
C0.10
0.05
0.15
0.10
0.10
0.10A B C D E
A B C D E
Species A ---- 0.20 0.50 0.45 0.40 Species B 0.23 ---- 0.40 0.55 0.50 Species C 0.87 0.59 ---- 0.15 0.40 Species D 0.73 1.12 0.17 ---- 0.25 Species E 0.59 0.89 0.61 0.31 ----
0.59
0.14
0.20
Problem statement: Given a tree and a set of sequence distances, derive lineages such that the tree distances maximally match the sequence distances.
sequence distances
lineages
Split the differences to match the differences of the distances

B
Fitch-Margoliash algorithm
1. Find the most closely-related pair of sequences, A and B
2. Calculate the average distance from A to all other sequences, then from B to all other sequences. Average D in and
3. Adjust the position of the common ancestor node for A and B so that the difference between the averaged distances is equal to the difference between the A B branch lengths. bA+bB = DAB bA-bB = - = (DAC+DAE)/2 - (DBC+DBE)/2
Or, in general: bA-bB = Σ DAx/N - Σ DBx/N
x≠A,B x≠A,B
NOTE: If Σ DAx/N - Σ DBx/N > DAB, there is no exact solution.
A B C E
C E
A B C EABCE
xx
x
bA
bB
DAB DAC DAD
DBC DBD DCD
xA

Exercise 8: create a rooted phylogram with 4 taxa
TTGACCAGACCTGTGGTCCG TTGAACAGACCTGCGGTCGG TAGAAAAGACCTGTCGTAGG GTGCAAAGTCCTGTGTATCG
ABCD
Directions:
1.Make a distance matrix. (p-distance, then convert to J-C distance)
2.Use Neighbor-joining to make a tree.
3.Adjust branch lengths using Fitch-Margoliash.
4.Choose the root using the Midpoint method.
5.Write tree in Newick format.
ABCD
A B C D
.15 .3 .3.25 .45
.45
K(A,B) = -3/4 ln [1 - 4/3 pdist(A,B)]
pdist=1-identity
.17
.38
.38.30.69 .69

Orthologs/paralogsOrthologs: homologs originating from a speciation event
Paralogs: homologs originating from a gene duplication event.
clam
duck
crab
fish
clam
A
duck
A
crab
Bfis
h A
duck
B
fish
B
4 species 6 sequences
clam
A
crab
B
duck
Adu
ck B
fish
Afis
h B
duplication
speciation
speciationgene loss
clam A and fish A are orthologs. clam A and crab B are paralogs.
Sequence treeSpecies tree reconciled

Are they paralogs, or are they orthologs?
• If they are paralogs, then at some point in evolutionary history, an ancestor species existed with two identical genes in it.
• One paralog may have been lost since the duplication event, in some lineages. Orthologs are generally conserved.
• Paralogs are likely to have evolved one different function, which is conserved in a lineage.
• Paralogs can have more than the expected sequence divergence.
• Paralogs diverged before speciation. Othologs diverged at speciation.
• Without species information and/or functional information, it’s impossible to tell orthologs from paralogs.

Exercise 8
• Download Para_or_ortho.fa
• Make a tree in UGENE.
• Look at annotations and the tree.
• Which sequences are paralogs? (Send list. Say why.)
• Which sequences are possibly mis-annotated? (Send list, say why.)
due Oct 9
Which sequence is likely to be non-orthologous? (i.e. paralogous)

Maximum parsimony -- it's “character-building”
Optimality criterion: The ‘most-parsimonious’ tree is the one that requires the fewest number of evolutionary events (e.g., nucleotide substitutions, amino acid replacements) to explain the sequences.
A ATGGCTATTCTTATAGTACG B ATCGCTAGTCTTATATTACA C TTCACTAGACCTGTGGTCCA D TTGACCAGACCTGTGGTCCG E TTGACCAGTTCTCTAGTTCG
T
T
T
A
AFor this column, and this tree, one mutation event is required.

character-based tree-building
A ATGGCTATTCTTATAGTACG B ATCGCTAGTCTTATATTACA C TTCACTAGACCTGTGGTCCA D TTGACCAGACCTGTGGTCCG E TTGACCAGTTCTCTAGTTCG
T
T
T
C
For this other column, the same tree requires two mutation events. A different tree would require only one.
C

Finding the minimum number of mutations
Given a tree and a set of taxa, one-letter each (1) choose optional characters for each ancestor. (2) Select the root character that minimizes the number of mutations by selecting each and propagating it through the tree.
T
C
T
C
C
T/C
T/C
T/C
T/C/C C
C
C
C
T
C
T
C
C
minimum 2 mutations minimum 1 mutation

�23
Ignore non-informative sites
• No mismatchs ---> noninformative! Adds 0 mutations in all trees
• One mismatch --> noninformative! Adds 1 mutation in all trees.
• All different --> noninformative! Adds same number of mutation in all trees. Only possible if number of sequences ≤ alphabet
Parsimony tips:

�24
Find noninformative columns
A ATGGCGATTCTTATAGTACG B ATGGCTAGTGTTATATTACA C ATCACAAGATCTGTGGTCCA D ATGACCAGAACTGTGGTCCG
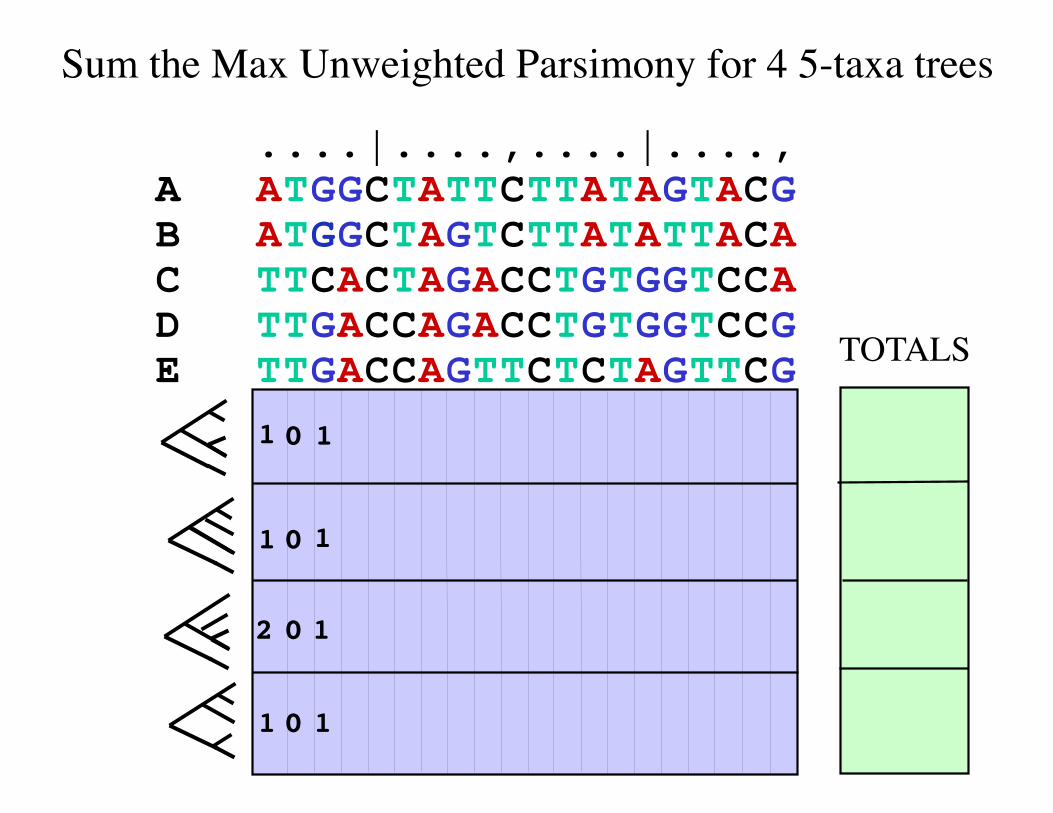
Sum the Max Unweighted Parsimony for 4 5-taxa trees
A ATGGCTATTCTTATAGTACG B ATGGCTAGTCTTATATTACA C TTCACTAGACCTGTGGTCCA D TTGACCAGACCTGTGGTCCG E TTGACCAGTTCTCTAGTTCG TOTALS
1 0
0
0
0
2
1
1
....|....,....|....,
1
1
1
1

Which method do I use?
Sequence similarity Method to use
strong distance
weak parsimony
very weak maximum likelihood

!27
Review
• What do nodes and lineages in a tree represent? • What are the two strategies for rooting a tree? Why root a
tree? • What is maximum parsimony? How do I find the most
parsimonious tree? • What is maximum likelihood? How does it relate to maximum
parsimony? • What kind of MSA positions are not informative of the tree? • What problem does Fitch-Margoliash solve? How? • What is an ortholog? Paralog? How can I tell them apart? • Unroot and re-root this tree: ((A,B),((C,D),E))

















![Phylogeny [phylo = tribe, family; gen = creation, birth] taxonomy: classifying and naming organisms taxon, taxa (pl.): classification unit (e.g. genus,](https://static.fdocuments.in/doc/165x107/56649d0b5503460f949defc7/phylogeny-phylo-tribe-family-gen-creation-birth-taxonomy-classifying.jpg)
