
Sequence information - lecturesbio.lundberg.gu.se/courses/bio2/lecture1.pdf · Sequence information...
28
Sequence information - lectures Pairwise alignment Alignments in database searches Multiple alignments Profiles Patterns RNA secondary structure / Transformational grammars Genome organisation / Gene prediction / HMMs Phylogeny
Transcript of Sequence information - lecturesbio.lundberg.gu.se/courses/bio2/lecture1.pdf · Sequence information...

Sequence information - lectures
Pairwise alignmentAlignments in database searchesMultiple alignmentsProfiles
Patterns
RNA secondary structure / Transformational grammars
Genome organisation / Gene prediction / HMMs
Phylogeny

Dates in the history of sequence information
Experimental Sequence analysis1953 Amino acid sequence of insulin.1965 Base sequence of a tRNA molecule,75 bases
1970 Global alignment Needleman-Wunsch
1976 Sequence of MS2 RNA, 3569 bases
1970s Birth of cloning1975 PAM matrix (Dayhoff)
1977 DNA sequencing (Sanger,F)1978 Bacteriophage genome fd
(6408 bases)1981 Local alignment
Smith-Waterman
1982 EMBL database1985 FASTA1985 Multiple sequence alignment
1st attempts1988 Neural networks(protein secondary structure)1989 Clustal1989 Profilesearch1991 BLAST1994 Profile HMMs1994 Context-free grammars
(RNA secondary structure)1995 Genomes of H influenzae & M genitalium
(1.8 / 0.6 million bases)
1997 PSI-BLAST2000 >95% of human genome
(3000 million bases)

Sequences - The analysis of raw data fromthe laboratory
Base-calling (sequence from rawdata) :
PhredSequence assembly:
PhrapCAP3 / CAP4
Cleaning up sequences:
Quality filteringVector filtering

Sequence formats / ASCII-Binary formats:
In ASCII text format (= human readable) each character is stored as a byte, for instance the ASCII code of 'A' = 65 as a decimal number = 01000001 as a binary number
However, sequence data is often stored in a binary format:
For instance in a binary system the three bases may be stored as:
A = 00 T = 01 C = 10 G = 11
In this way there is room in one byte (= 8 bits) for 4 bases.
Typically databases are downloaded by the bioinformatician in ASCII format but then reformatted in a binary format for use with different sequence analysis tools. One example is databases for blast searches that may be formatted by the NCBI utility 'formatdb’
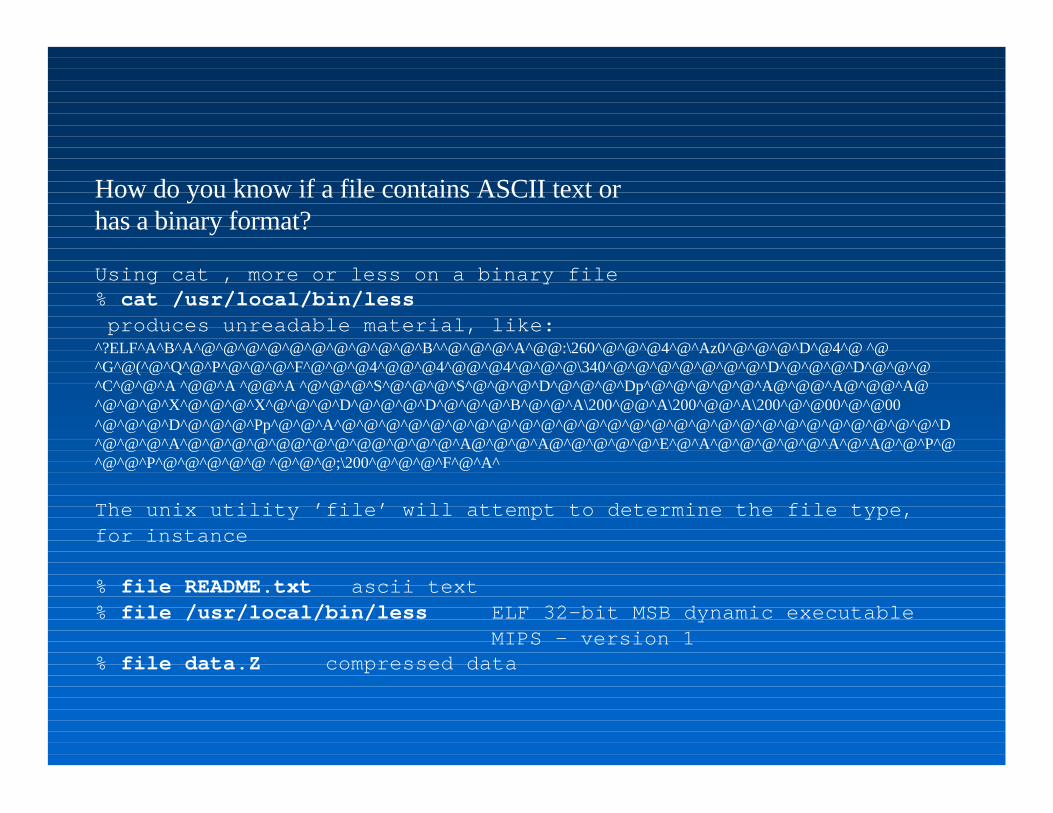
How do you know if a file contains ASCII text or has a binary format?
Using cat , more or less on a binary file% cat /usr/local/bin/less produces unreadable material, like:^?ELF^A^B^A^@^@^@^@^@^@^@^@^@^@^B^^@^@^@^A^@@:\260^@^@^@4^@^Az0^@^@^@^D^@4^@ ^@^G^@(^@^Q^@^P^@^@^@^F^@^@^@4^@@^@4^@@^@4^@^@^@\340^@^@^@^@^@^@^@^D^@^@^@^D^@^@^@^C^@^@^A ^@@^A ^@@^A ^@^@^@^S^@^@^@^S^@^@^@^D^@^@^@^Dp^@^@^@^@^@^A@^@@^A@^@@^A@^@^@^@^X^@^@^@^X^@^@^@^D^@^@^@^D^@^@^@^B^@^@^A\200^@@^A\200^@@^A\200^@^@00^@^@00^@^@^@^D^@^@^@^Pp^@^@^A^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^@^D^@^@^@^A^@^@^@^@^@@^@^@^@@^@^@^@^A@^@^@^A@^@^@^@^@^E^@^A^@^@^@^@^@^A^@^A@^@^P^@^@^@^P^@^@^@^@^@ ^@^@^@;\200^@^@^@^F^@^A^
The unix utility ’file’ will attempt to determine the file type, for instance
% file README.txt ascii text% file /usr/local/bin/less ELF 32-bit MSB dynamic executable MIPS - version 1% file data.Z compressed data

Sequence formats
Examples
1. Embl
ID LISOD standard; DNA; PRO; 756 BP.XXAC X64011; S78972;XXSV X64011.1XXDT 28-APR-1992 (Rel. 31, Created)DT 30-JUN-1993 (Rel. 36, Last updated, Version 6)XXDE L.ivanovii sod gene for superoxide dismutase........SQ Sequence 756 BP; 247 A; 136 C; 151 G; 222 T; 0 other; cgttatttaa ggtgttacat agttctatgg aaatagggtc tatacctttc gccttacaat 60 gtaatttctt ttcacataaa taataaacaa tccgaggagg aatttttaat gacttacgaa 120 ttaccaaaat taccttatac ttatgatgct ttggagccga attttgataa agaaacaatg 180........

2. Fasta
>LISOD L.ivanovii sod gene for superoxide dismutasecgttatttaa ggtgttacat agttctatgg aaatagggtc tatacctttc gccttacaatgtaatttctt ttcacataaa taataaacaa tccgaggagg aatttttaat gacttacgaattaccaaaat taccttatac ttatgatgct ttggagccga attttgataa agaaacaatg........
3. GCG
lisod.seq Length: 756 October 27, 2000 13:17 Type: N Check: 5188 ..
1 cgttatttaa ggtgttacat agttctatgg aaatagggtc tatacctttc
51 gccttacaat gtaatttctt ttcacataaa taataaacaa tccgaggagg
101 aatttttaat gacttacgaa ttaccaaaat taccttatac ttatgatgct
....
....

Multiple sequence format of GCG
1.msf MSF: 44 Type: P October 24, 2002 12:41 Check: 9117 ..
Name: ftsy_bucai Len: 44 Check: 4221 Weight: 1.00 Name: ftsy_ecoli Len: 44 Check: 2326 Weight: 1.00 Name: ftsy_aquae Len: 44 Check: 2339 Weight: 1.00 Name: ftsy_bacsu Len: 44 Check: 6177 Weight: 1.00 Name: sr54_aciam Len: 44 Check: 6296 Weight: 1.00 Name: sr54_aerpe Len: 44 Check: 7291 Weight: 1.00 Name: sr54_arcfu Len: 44 Check: 122 Weight: 1.00 Name: sr54_aquae Len: 44 Check: 345 Weight: 1.00
//
1 44ftsy_bucai KNS.EKLYFL LKRKMFNILK KVEIP...LE ISSHSPFVIL VVGVftsy_ecoli RDA.EALYGL LKEEMGEILA KVDEP...LN VEGKAPFVIL MVGVftsy_aquae KEG.EKIKEL LKKELKELLK NCQ...GELK IPEKVGAVLL FVGVftsy_bacsu QDP.KEVQSV ISEKLVEIYN SGDEQISELN IQDGRLNVIL LVGVsr54_aciam PTYIERREWF IKIVYDELSN LFGGDKEPKV IPDKIPYVIM LVGVsr54_aerpe PPGVTRRDWM IKIVYEELVK LFGGDQEPQV DPPKTPWIVL LVGVsr54_arcfu LPALNAKEQI LKIVYEELLR GVGEGLEIPL KKAK....IM LVGLsr54_aquae PKNLSPAEFV IKTVYEELVD ILGGEK.... .ADLKKGTVL FVGL

FASTA multiple sequence format
>ftsy_bucai, 44 bases, 52FF1180 checksum.KNS-EKLYFLLKRKMFNILKKVEIP---LEISSHSPFVILVVGV>ftsy_ecoli, 44 bases, B14506B6 checksum.RDA-EALYGLLKEEMGEILAKVDEP---LNVEGKAPFVILMVGV>ftsy_aquae, 44 bases, 27571BEE checksum.KEG-EKIKELLKKELKELLKNCQ---GELKIPEKVGAVLLFVGV>ftsy_bacsu, 44 bases, FA023A4F checksum.QDP-KEVQSVISEKLVEIYNSGDEQISELNIQDGRLNVILLVGV>sr54_aciam, 44 bases, 2FC13632 checksum.PTYIERREWFIKIVYDELSNLFGGDKEPKVIPDKIPYVIMLVGV>sr54_aerpe, 44 bases, 37AFB895 checksum.PPGVTRRDWMIKIVYEELVKLFGGDQEPQVDPPKTPWIVLLVGV>sr54_arcfu, 44 bases, 8294461 checksum.LPALNAKEQILKIVYEELLRGVGEGLEIPLKKAK----IMLVGL>sr54_aquae, 44 bases, 794768A2 checksum.PKNLSPAEFVIKTVYEELVDILGGEK-----ADLKKGTVLFVGL

CLUSTAL multiple sequence format
CLUSTAL W (1.81) multiple sequence alignment
ftsy_bucai KNS-EKLYFLLKRKMFNILKKVEIP---LEISSHSPFVILVVGVftsy_ecoli RDA-EALYGLLKEEMGEILAKVDEP---LNVEGKAPFVILMVGVftsy_aquae KEG-EKIKELLKKELKELLKNCQ---GELKIPEKVGAVLLFVGVftsy_bacsu QDP-KEVQSVISEKLVEIYNSGDEQISELNIQDGRLNVILLVGVsr54_aciam PTYIERREWFIKIVYDELSNLFGGDKEPKVIPDKIPYVIMLVGVsr54_aerpe PPGVTRRDWMIKIVYEELVKLFGGDQEPQVDPPKTPWIVLLVGVsr54_arcfu LPALNAKEQILKIVYEELLRGVGEGLEIPLKKAK----IMLVGLsr54_aquae PKNLSPAEFVIKTVYEELVDILGGEK-----ADLKKGTVLFVGL .:. :: ::.**:
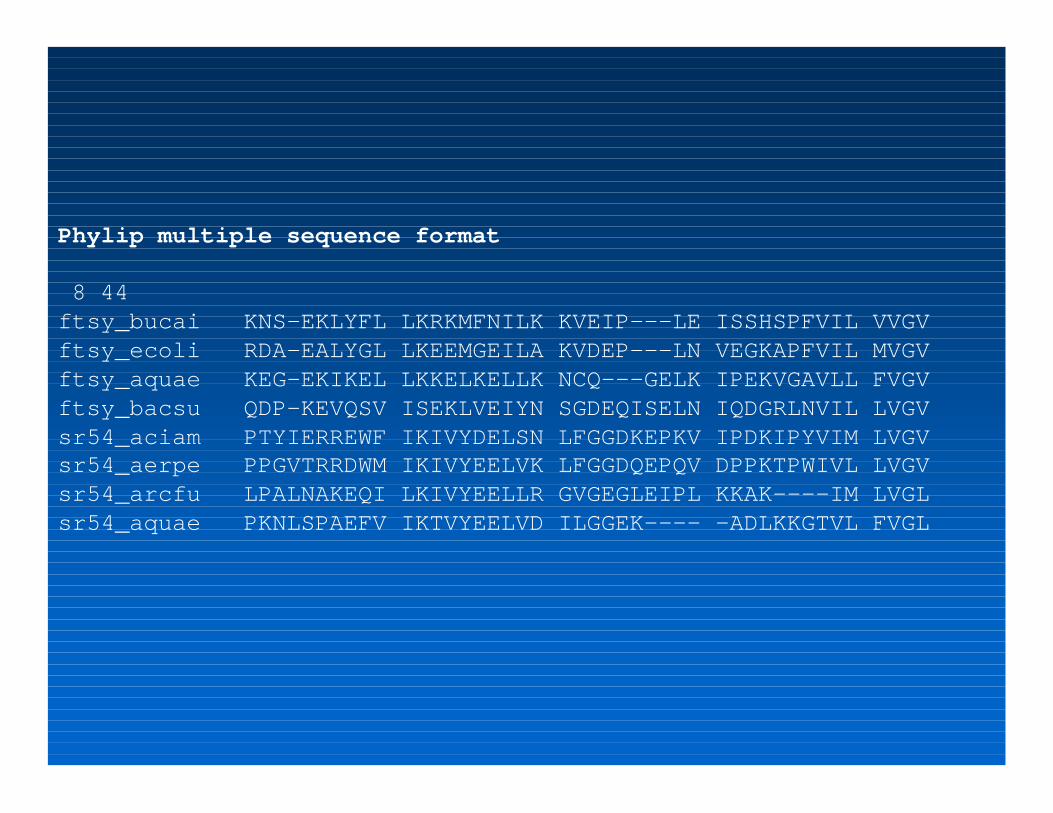
Phylip multiple sequence format
8 44ftsy_bucai KNS-EKLYFL LKRKMFNILK KVEIP---LE ISSHSPFVIL VVGVftsy_ecoli RDA-EALYGL LKEEMGEILA KVDEP---LN VEGKAPFVIL MVGVftsy_aquae KEG-EKIKEL LKKELKELLK NCQ---GELK IPEKVGAVLL FVGVftsy_bacsu QDP-KEVQSV ISEKLVEIYN SGDEQISELN IQDGRLNVIL LVGVsr54_aciam PTYIERREWF IKIVYDELSN LFGGDKEPKV IPDKIPYVIM LVGVsr54_aerpe PPGVTRRDWM IKIVYEELVK LFGGDQEPQV DPPKTPWIVL LVGVsr54_arcfu LPALNAKEQI LKIVYEELLR GVGEGLEIPL KKAK----IM LVGLsr54_aquae PKNLSPAEFV IKTVYEELVD ILGGEK---- -ADLKKGTVL FVGL

Sequence editors
SeqLab / GCG

Genedoc

Conversion of sequence formats - Readseq
(one useful version of readseq is part of the SAM package http://www.cse.ucsc.edu/research/compbio/sam2src/)
Readseq can convert between the following formats:
1. IG/Stanford 10. Olsen (in-only) 2. GenBank/GB 11. Phylip3.2 3. NBRF 12. Phylip 4. EMBL 13. Plain/Raw 5. GCG 14. PIR/CODATA 6. DNAStrider 15. MSF 7. Fitch 16. ASN.1 8. Pearson/Fasta 17. PAUP/NEXUS 9. Zuker (in-only) 18. Pretty (out-only)

GCG package utilities:Convert from to
* Reformat text GCG * FromEMBL EMBL GCG * FromGenBank Genbank GCG * FromFasta Fasta GCG * ToFastA GCG Fasta
Clustal:
clustalw my_alignment -convert -output=gcgclustalw my_alignment -convert -output=phylip

Downloading bioinformatics data and software for local use


Downloading bioinformatics data andsoftware for local useData and software downloaded from the net are typically compressed in oneway or the other. The most common formats are *.Z and *.gz
These are binary files that cannot be read with normal editorsTo uncompress a *.Z file :
% uncompress [file]To uncompress a *.gz file :
% gunzip [file]
A set of files is often downloaded as a set of compressed tar archive. Forinstance if you download a file called dna.tar.gz you can do
% gunzip dna.tar.gz
This will yield a file called dna.tar. Then do
% tar -xvf dna.tar
This will unpack all files contained in dna.tar

Alignments and database searches
Common biological problem: We have a novel protein sequence. What can we inferfrom this sequence about the biological function of theprotein ??
* Pattern search - PROSITE* Profile search - Pfam* Prediction of transmembrane domains ( ~ 25 % of all proteins are membrane bound!) * Sequence homology - BLAST, FASTA, SSEARCH
Simple example: unknown human protein is highly homologous to aprotein with known function from another organism => The human protein has the same function (it’s anortholog or a paralog)

BLAST
1. Heuristic step where short word matches are identified
2. Extending matches using dynamic programming methodas for local pairwise alignments. (Substitution matrix, gap penalties)
-The nature of protein sequence and structure evolution -what is the sensitivity of BLAST searches?
-General principles of database searches

ATGGCAAAACTTGAAAAACTGAATCAAGCAGGCCTGATGGTCGCTGGT M A K L E K L N Q A G L M V A G
60%ATGGCTAGGTTGGAGAAGAUAAACCAAGCTGGGATAATAGTTGCAGGA M V R L E K I N Q A G L L V A G69%
M V R I Q K I N E K G A L L A G38%
Q V R I Q K I Y E K G A L L A A19% (‘twilight zone’)
Q V R I Q K I Y E K T A L L F A6% (‘midnight zone’)
Evolution of protein genes

Blast report
Sequences producing significant alignments: (bits) Value
pir||F69494 (R)-hydroxyglutaryl-CoA dehydratase activator (hgdC)... 462 e-129gb|AAD31675.1| (AF123384) (R)-2-hydroxyglutaryl-CoA dehydratase ... 233 1e-060sp|P39383|YJIL_ECOLI HYPOTHETICAL 27.4 KD PROTEIN IN IADA-MCRD I... 184 9e-046emb|CAA67409.1| (X98916) orf6 [Methanopyrus kandleri] 170 1e-041gb|AAF13150.1|AF156260_1 (AF156260) unknown [Methanosarcina bark... 143 2e-033pir||A69117 activator of (R)-2-hydroxyglutaryl-CoA - Methanobact... 132 4e-030pir||A72369 (R)-2-hydroxyglutaryl-CoA dehydratase activator-rela... 129 4e-029gb|AAC23928.1| (U75363) benzoyl-CoA reductase subunit [Rhodopseu... 117 1e-025pir||S04476 hypothetical protein (hdgA 5' region) - Acidaminococ... 104 1e-021sp|P27542|DNAK_CHLPN DNAK PROTEIN (HEAT SHOCK PROTEIN 70) (HSP70... 42 0.005gb|AAC15473.1| (AF016711) heat shock protein 70 [Burkholderia ps... 39 0.036pir||F75029 o-sialoglycoprotein endopeptidase (gcp) PAB1159 - Py... 38 0.082pir||F72514 probable glucokinase APE2091 - Aeropyrum pernix (str... 37 0.18sp|P42373|DNAK_BURCE DNAK PROTEIN (HEAT SHOCK PROTEIN 70) (HSP70... 37 0.18emb|CAA10035.1| (AJ012470) mitochondrial-type hsp70 [Encephalito... 36 0.31sp|P56836|DNAK_CHLMU DNAK PROTEIN (HEAT SHOCK PROTEIN 70) (HSP70... 36 0.41gb|AAF39496.1| (AE002336) dnaK protein [Chlamydia muridarum] 36 0.41pir||B70189 rod shape-determining protein (mreB-1) homolog - Lym... 36 0.41sp|O57716|GCP_PYRHO PUTATIVE O-SIALOGLYCOPROTEIN ENDOPEPTIDASE (... 36 0.54sp|O33522|DNAK_ALCEU DNAK PROTEIN (HEAT SHOCK PROTEIN 70) (HSP70... 36 0.54ref|NP_012874.1| Ykl050cp >gi|549677|sp|P35736|YKF0_YEAST HYPOTH... 36 0.54emb|CAA53420.1| (X75781) D513 [Saccharomyces cerevisiae] >gi|158... 36 0.54sp|P30722|DNAK_PAVLU DNAK PROTEIN (HEAT SHOCK PROTEIN 70) >gi|99... 36 0.54pir||A40158 dnaK-type molecular chaperone - Chlamydia trachomati... 34 1.2gb|AAF07742.1|AE001584_39 (AE001584) hypothetical protein [Borre... 34 1.6gb|AAF07521.1|AE001577_35 (AE001577) hypothetical protein [Borre... 34 1.6gb|AAF38963.1| (AE002276) cell shape-determining protein MreB [C... 34 2.1gb|AAG08147.1|AE004889_10 (AE004889) DnaK protein [Pseudomonas a... 33 2.7dbj|BAB03215.1| (AB017035) dnaK [Bacillus thermoglucosidasius] 33 2.7sp|P43736|DNAK_HAEIN DNAK PROTEIN (HEAT SHOCK PROTEIN 70) (HSP70... 33 2.7sp|P45554|DNAK_STAAU DNAK PROTEIN (HEAT SHOCK PROTEIN 70) (HSP70... 33 2.7sp|Q58303|FLA3_METJA FLAGELLIN B3 PRECURSOR 32 4.7gb|AAG08239.1|AE004898_10 (AE004898) phosphoribosylaminoimidazol... 32 6.1

Conclusions / comments
If two proteins have significant sequencehomology it is highly likely that the two proteinshave the same 3D structure (and same function)
If two proteins have the same 3D structure isdoes not necessarily mean that the sequences arerelated
Very remote evolutionary relationships aredifficult or impossible to detect with normalBLAST / pairwise alignment
Database sequence searches involving proteinsshould be carried out at the protein level and notat the DNA level

More rules of database searches
?Compare sequences as proteins and not as DNA*?Use of smallest possible database (not too small though)?Sequence statistics should be used rather than percent identity/similarity as criterion for homology?Consider different scoring matrices and gap penalties
* 1) DNA sequences encoding the same protein sequence can be verydifferent, due to the degeneracy of the genetic code.
TTTCGATTCTCAACAAGAAGC** * ** ** * *TTCAGGTTTAGCACGCGGTCC F R F S T R S
2) Amino acid substitution matrices may be taken into account.

PAM matrices
Starting point of PAM matrices are closely related orthologs, full sequences are taken into account.In the calculation of the PAM matrix extrapolation is done to more distant relationships
Neurospora_crassa GSVDGYAYTD ANKQKGITWD ENTLFEYLEN PKKYIPGTKM AFGGLKKDKD Stellaria_longipes GSVEGFSYTD ANKAKGIEWN KDTLFEYLEN PKKYIPGTKM AFGGLKKDKD Thermomyces_lanuginosus GSVEGYSYTD ANKQAGITWN EDTLFEYLEN PKKFIPGTKM AFGGLKKNKD Arabidopsis_thaliana GSVAGYSYTD ANKQKGIEWK DDTLFEYLEN PKKYIPGTKM AFGGLKKPKD Aspergillus_niger GQSEGYAYTD ANKQAGVTWD ENTLFSYLEN PKKFIPGTKM AFGGLKKGKE Debaryomyces_occidentalis GQAAGYSYTD ANKKKGVEWT EQTMSDYLEN PKKYIPGTKM AFGGLKKPKD Schizosaccharomyces_pombe GQAEGFSYTE ANRDKGITWD EETLFAYLEN PKKYIPGTKM AFAGFKKPAD Fagopyrum_esculentum GTTAGYSYSA ANKNKAVTWG EDTLYEYLLN PKKYIPGTKM VFPGLKKPQE Sesamum_indicum GTTPGYSYSA ANKNMAVIWG ENTLYDYLLN PKKYIPGTKM VFPGLKKPQE Haematobia_irritans GQAAGFAYTN ANKAKGITWQ DDTLFEYLEN PKKYIPGTKM IFAGLKKPNE Lucilia_cuprina GQAPGFAYTN ANKAKGITWQ DDTLFEYLEN PKKYIPGTKM IFAGLKKPNE Ceratitis_capitata GQAAGFAYTD ANKAKGITWN EDTLFEYLEN PKKYIPGTKM IFAGLKKPNE Sarcophaga_peregrina GQAPGFAYTD ANKAKGITWN EDTLFEYLEN PKKYIPGTKM IFAGLKKPNE Manduca_sexta GQAPGFSYSD ANKAKGITWN EDTLFEYLEN PKKYIPGTKM VFAGLKKANE Samia_cynthia GQAPGFSYSN ANKAKGITWG DDTLFEYLEN PKKYIPGTKM VFAGLKKANE Schistocerca_gregaria GQAPGFSYTD ANKSKGITWD ENTLFIYLEN PKKYIPGTKM VFAGLKKPEE Apis_mellifera GQAPGYSYTD ANKGKGITWN KETLFEYLEN PKKYIPGTKM VFAGLKKPQE Macaca_mulatta GQAPGYSYTA ANKNKGITWG EDTLMEYLEN PKKYIPGTKM IFVGIKKKEE Pan_troglodytes GQAPGYSYTA ANKNKGIIWG EDTLMEYLEN PKKYIPGTKM IFVGIKKKEE Anas_platyrhynchos GQAEGFSYTD ANKNKGITWG EDTLMEYLEN PKKYIPGTKM IFAGIKKKSE Aptenodytes_patagonicus GQAEGFSYTD ANKNKGITWG EDTLMEYLEN PKKYIPGTKM IFAGIKKKSE
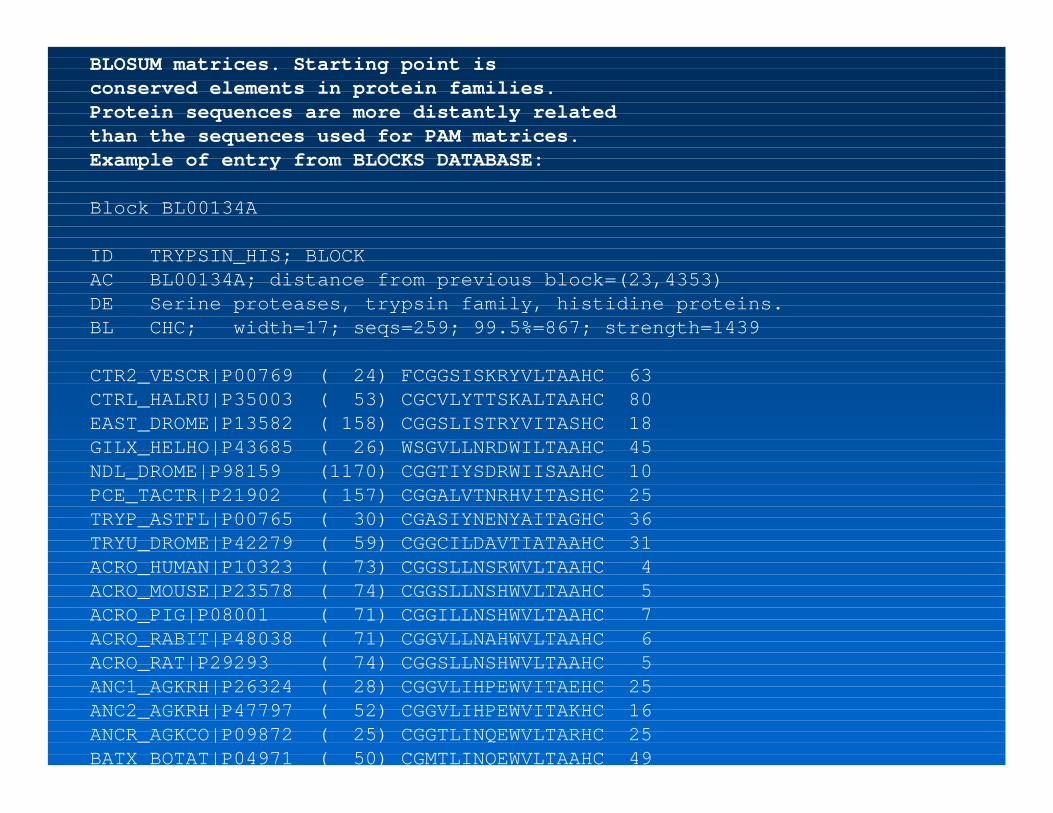
BLOSUM matrices. Starting point is conserved elements in protein families.Protein sequences are more distantly related than the sequences used for PAM matrices.Example of entry from BLOCKS DATABASE:
Block BL00134A
ID TRYPSIN_HIS; BLOCKAC BL00134A; distance from previous block=(23,4353)DE Serine proteases, trypsin family, histidine proteins.BL CHC; width=17; seqs=259; 99.5%=867; strength=1439
CTR2_VESCR|P00769 ( 24) FCGGSISKRYVLTAAHC 63CTRL_HALRU|P35003 ( 53) CGCVLYTTSKALTAAHC 80EAST_DROME|P13582 ( 158) CGGSLISTRYVITASHC 18GILX_HELHO|P43685 ( 26) WSGVLLNRDWILTAAHC 45NDL_DROME|P98159 (1170) CGGTIYSDRWIISAAHC 10PCE_TACTR|P21902 ( 157) CGGALVTNRHVITASHC 25TRYP_ASTFL|P00765 ( 30) CGASIYNENYAITAGHC 36TRYU_DROME|P42279 ( 59) CGGCILDAVTIATAAHC 31ACRO_HUMAN|P10323 ( 73) CGGSLLNSRWVLTAAHC 4ACRO_MOUSE|P23578 ( 74) CGGSLLNSHWVLTAAHC 5ACRO_PIG|P08001 ( 71) CGGILLNSHWVLTAAHC 7ACRO_RABIT|P48038 ( 71) CGGVLLNAHWVLTAAHC 6ACRO_RAT|P29293 ( 74) CGGSLLNSHWVLTAAHC 5ANC1_AGKRH|P26324 ( 28) CGGVLIHPEWVITAEHC 25ANC2_AGKRH|P47797 ( 52) CGGVLIHPEWVITAKHC 16ANCR_AGKCO|P09872 ( 25) CGGTLINQEWVLTARHC 25BATX_BOTAT|P04971 ( 50) CGMTLINQEWVLTAAHC 49


Variants of BLAST / FASTA
DNA/DNA P/P DNA/P P/DNA DNA/DNA
blastall -p blastn -p blastp -p blastx -p tblastn -p tblastxfasta fasta fasta fastx,fasty* tfasta tfastx, tfasty
*Compare a DNA sequence to a protein sequence database, by comparing the translated DNA sequence in three frames and allowing gaps and frameshifts. fastx3 uses a simpler, faster algorithm for alignments that allows frameshifts only between codons; fasty3 is slower but produces better alignments with poor quality sequences because frameshifts are allowed within codons.


















