
Screening a human population sample for DNA repair gene deficiencies utilizing the protein...
7
Screening a Human Population Sample for DNA Repair Gene Deficiencies Utilizing the Protein Truncation Test Jian Chen, Zhe Yu, Barry N. Ford, Moyra E. Brackley, Roderick J.C. Haesevoets, Magomed Khaidakov, and Barry W. Glickman* Centre for Environmental Health and the Department of Biology, University of Victoria, Victoria, British Columbia, Canada A significant fraction of human cancers are thought to have a genetic component and several lines of evidence suggest that deficiencies in DNA repair may be a contributing factor. Little is known, however, about the frequency and distri- bution of variants of DNA repair genes in the general human population. The protein trunca- tion test (PTT) was used to screen 136 healthy volunteers for protein-truncating variants of 10 DNA repair genes: APE, CDK7, ERCC1, WAF1, HOGG1, MGMT, POLB, UNG, HAAG, and CCNH. This sample consisted of 41males (30%) and 95 females (70%) with an average age of 25.3 years, ranging from 17 to 60 years of age. No truncating mutations were found in the 10 genes examined in any of the subjects. The 95% confidence interval for a proportion of 0 over the 272 alleles examined per locus is 0 – 0.01. The calculated frequency of truncating mutations in each of these genes, among the general popula- tion, is thus less than 1%. Among the 10 genes tested in 136 people, a single sample had no PCR product for HAAG, even though PCR prod- ucts were obtained on all other genes. Total RNA dot hybridization confirmed the presence of HAAG mRNA transcripts in this sample. De- spite identification of this single DNA repair vari- ant, these results indicate a low frequency of truncating mutations in DNA repair genes in the general population. Environ. Mol. Mutagen. 36: 228 –234, 2000. © 2000 Wiley-Liss, Inc. Key words: human DNA repair genes; protein truncation test; population study INTRODUCTION DNA repair is fundamental in protecting genetic material against accumulation of mutations. It is thus no surprise that an increasing number of human hereditary cancers and cancer-prone syndromes are related to deficiencies in DNA repair [Takebe, 1984, 1995; Wei et al., 1995; Abrahams et al., 1998]. Increasing evidence suggests that even the ap- parently sporadic cancers may involve DNA repair gene deficiencies [Lynch et al., 1997; Cvitkovic, 1998; Krokan and Slupphaug, 1998]. The potential involvement of DNA repair gene deficien- cies in human cancer [Mohrenweiser and Jones, 1998] makes the identification of cancer-predisposing DNA repair genes extremely relevant. Identification of candidate repair genes depends on obtaining preliminary knowledge of fre- quencies of DNA repair gene variants in the general popu- lation. Considering that more than 70 human DNA repair genes are now known, along with their homologs in bacte- ria, yeast, and rodents, we still know little about variation in humans. The functions and involvement of some of these genes in human disease have been reported [Yu et al., 1999]. Knowledge of the cDNA sequences of many of these genes makes it possible to screen for human repair gene deficiencies in the general population. One approach to investigating the prevalence of DNA repair deficiency is the protein truncation test (PTT). PTT is an assay that detects truncating mutations based on in vitro transcription and translation of genes of interest. PTT pro- vides a powerful approach, as a large fraction of mutations associated with human disorders would be detected using this assay [Roest et al., 1993; Heim et al., 1994; Liu et al., 1994; Papadopoulos et al., 1994; van der Luijt et al., 1994; Gardner et al., 1995; Hogervorst et al., 1995; Eccles et al., 1996; Lancaster et al., 1996; FitzGerald et al., 1997]. This suggests that PTT would be a very useful technique for screening mutation. Additionally, as the effects of truncat- ing mutations are generally quite significant [den Dunnen et al., 1996], PTT provides a good practical option for the detection of functionally relevant mutations. PTT also of- fers the additional advantage of detecting heterozygotes [den Dunnen et al., 1996]. PTT is based on the detection of altered proteins pro- duced by the in vitro translation of PCR products amplified from mRNA transcripts of the target genes by RT-PCR. Grant sponsor: Medical Research Council of Canada; Grant sponsor: Canadian Space Agency. *Correspondence to: Barry W. Glickman, University of Victoria, Centre for Environmental Health and the Department of Biology, P.O. Box 3020 STN CSC, Victoria, BC V8W 3N5, Canada. E-mail: [email protected] Received 1 December 1999; provisionally accepted 8 March 2000; and in final form 25 June 2000 Environmental and Molecular Mutagenesis 36:228 –234 (2000) © 2000 Wiley-Liss, Inc.
Transcript of Screening a human population sample for DNA repair gene deficiencies utilizing the protein...

Screening a Human Population Sample for DNA RepairGene Deficiencies Utilizing the Protein Truncation Test
Jian Chen, Zhe Yu, Barry N. Ford, Moyra E. Brackley,Roderick J.C. Haesevoets, Magomed Khaidakov, and Barry W. Glickman*
Centre for Environmental Health and the Department of Biology, University ofVictoria, Victoria, British Columbia, Canada
A significant fraction of human cancers arethought to have a genetic component and severallines of evidence suggest that deficiencies inDNA repair may be a contributing factor. Little isknown, however, about the frequency and distri-bution of variants of DNA repair genes in thegeneral human population. The protein trunca-tion test (PTT) was used to screen 136 healthyvolunteers for protein-truncating variants of 10DNA repair genes: APE, CDK7, ERCC1, WAF1,HOGG1, MGMT, POLB, UNG, HAAG, andCCNH. This sample consisted of 41males (30%)and 95 females (70%) with an average age of25.3 years, ranging from 17 to 60 years of age.No truncating mutations were found in the 10genes examined in any of the subjects. The 95%
confidence interval for a proportion of 0 over the272 alleles examined per locus is 0–0.01. Thecalculated frequency of truncating mutations ineach of these genes, among the general popula-tion, is thus less than 1%. Among the 10 genestested in 136 people, a single sample had noPCR product for HAAG, even though PCR prod-ucts were obtained on all other genes. TotalRNA dot hybridization confirmed the presenceof HAAG mRNA transcripts in this sample. De-spite identification of this single DNA repair vari-ant, these results indicate a low frequency oftruncating mutations in DNA repair genes in thegeneral population. Environ. Mol. Mutagen. 36:228–234, 2000. © 2000 Wiley-Liss, Inc.
Key words: human DNA repair genes; protein truncation test; population study
INTRODUCTION
DNA repair is fundamental in protecting genetic materialagainst accumulation of mutations. It is thus no surprise thatan increasing number of human hereditary cancers andcancer-prone syndromes are related to deficiencies in DNArepair [Takebe, 1984, 1995; Wei et al., 1995; Abrahams etal., 1998]. Increasing evidence suggests that even the ap-parently sporadic cancers may involve DNA repair genedeficiencies [Lynch et al., 1997; Cvitkovic, 1998; Krokanand Slupphaug, 1998].
The potential involvement of DNA repair gene deficien-cies in human cancer [Mohrenweiser and Jones, 1998]makes the identification of cancer-predisposing DNA repairgenes extremely relevant. Identification of candidate repairgenes depends on obtaining preliminary knowledge of fre-quencies of DNA repair gene variants in the general popu-lation. Considering that more than 70 human DNA repairgenes are now known, along with their homologs in bacte-ria, yeast, and rodents, we still know little about variation inhumans. The functions and involvement of some of thesegenes in human disease have been reported [Yu et al.,1999]. Knowledge of the cDNA sequences of many of thesegenes makes it possible to screen for human repair genedeficiencies in the general population.
One approach to investigating the prevalence of DNArepair deficiency is the protein truncation test (PTT). PTT is
an assay that detects truncating mutations based on in vitrotranscription and translation of genes of interest. PTT pro-vides a powerful approach, as a large fraction of mutationsassociated with human disorders would be detected usingthis assay [Roest et al., 1993; Heim et al., 1994; Liu et al.,1994; Papadopoulos et al., 1994; van der Luijt et al., 1994;Gardner et al., 1995; Hogervorst et al., 1995; Eccles et al.,1996; Lancaster et al., 1996; FitzGerald et al., 1997]. Thissuggests that PTT would be a very useful technique forscreening mutation. Additionally, as the effects of truncat-ing mutations are generally quite significant [den Dunnen etal., 1996], PTT provides a good practical option for thedetection of functionally relevant mutations. PTT also of-fers the additional advantage of detecting heterozygotes[den Dunnen et al., 1996].
PTT is based on the detection of altered proteins pro-duced by the in vitro translation of PCR products amplifiedfrom mRNA transcripts of the target genes by RT-PCR.
Grant sponsor: Medical Research Council of Canada; Grant sponsor:Canadian Space Agency.
*Correspondence to: Barry W. Glickman, University of Victoria, Centrefor Environmental Health and the Department of Biology, P.O. Box 3020STN CSC, Victoria, BC V8W 3N5, Canada. E-mail: [email protected]
Received 1 December 1999; provisionally accepted 8 March 2000; and infinal form 25 June 2000
Environmental and Molecular Mutagenesis 36:228 –234 (2000)
© 2000 Wiley-Liss, Inc.

This is a sensitive technique since RT-PCR permits thedetection of even small amounts of mRNA [Telatar et al.,1996]. Although mutations that cause premature truncationmay produce unstable RNA molecules [Baserga and Benz,1988; Jones et al., 1992; Dietz et al., 1993], RT-PCR–basedmutation screening methods, such as restriction endonucle-ase fingerprinting (REF), PTT, conformation-sensitive gelelectrophoresis (CSGE), and single-strand conformationalpolymorphism (SSCP), have all been generally effective inidentifying mutated genes. PTT has been efficiently appliedto detect mutations involved in many diseases, includingadenomatous polyposis coli (APC) [van der Luijt et al.,1994], neurofibrosis [Heim et al., 1995], and breast cancer[Hogervorst et al., 1995]. It has been previously used in ourlab to screen forATM mutations in breast cancer patients[Bebb et al., 1999].
Here, we report PTT examination of 10 DNA repairgenes in 136 healthy individuals. These genes encodeO6-methylguanine DNA methyl-transferase (MGMT), 3-alky-
ladenine DNA glycosylase (HAAG), 8-oxoguanine DNAglycosylase (HOGG1), uracil DNA glycosylase (UNG),apurinic endonuclease (APE), DNA polymeraseb (POLB),cyclin-dependent kinase 7 (CDK7), cyclin H (CCNH), ex-cision repair cross-complementing rodent repair deficiency,complementation group 1 (ERCC1), and wild-type P53activated fragment-1 (WAF1) (Table I).
MATERIALS AND METHODS
Sample Collection and Cell Culturing
Blood samples were collected under a protocol approved by the HumanSubjects Committee at the University of Victoria. Participants completed aquestionnaire requesting information concerning their age, sex, ancestry,and family history of cancer. Samples were anonymized, that is, theycannot be traced back to individual participants. Peripheral blood (10 ml)was taken from each of the volunteers using heparinized Vacutainer tubes(Becton Dickinson, Franklin Lakes, NJ). Lymphocytes were isolated fromthe blood using Ficoll-Paque (Pharmacia Biotech, Uppsala, Sweden), ac-
TABLE I. Human DNA Repair Genes Examined by PTTa
Human enzyme and geneSize and gene
locationAssociated cancer or potential
involvement in cancerRepairsystem References
O6-methylguaine DNA methyltransferase (MGMT)
22 kDa10q24.33-qter
Brain and esophageal cancers Direct repair Tano et al., 1990; Silberet al., 1996; Wang etal., 1997
Human 3-alkyladenine DNAglycosylase (HAAG)
32.9 kDachromosome 16
Inducibility by carcinogen Baseexcisionrepair
Samson et al., 1991;Lefebvre et al., 1993
8-Oxoguanine DNA glycosylase(HOGG1)
39 kDa3p25/26
Lung and kidney cancers Baseexcisionrepair
Rosenquist et al., 1997;Chevillard et al., 1998;Kohno et al., 1998;Ishida et al., 1999
Uracil DNA glycosylase (UNG) 33.8–25.8 kDa12q23-q24.1
Glioblastoma Baseexcisionrepair
Olsen et al., 1989; Moonet al., 1998
Apurinic endonuclease (APE) 36 kDa14q11.2–14q12
Inducibility by carcinogen Baseexcisionrepair
Demple et al., 1991;Fung et al., 1998
DNA polymeraseb (POLB) 39 kDa8p11-12
Breast cancer Baseexcisionrepair
SenGupta et al., 1986;Canitrot et al., 1998;Bhattacharyya et al.,1999
Cyclin-dependent kinase 7(CDK7)
41 kDa2q15-2cen
Cell-cycle control; transcriptionregulation; component ofexcinuclease; elevation in tumor cells
Nucleotideexcisionrepair
Wu et al., 1994; Bartkovaet al., 1996; Yankulovand Bentley, 1997
Cyclin H (CCNH) 38 kDa5q13.3-14
Cell-cycle control; transcriptionregulation; component ofexcinucleose
Nucleotideexcisionrepair
Fisher and Morgan, 1994;Park et al., 1995;Yankulov and Bentley,1997
Excision repair cross-complementing rodent repairdeficiency, complementationgroup 1 (ERCC1)
33 kDa19q13.3
Leukemia Nucleotideexcisionrepair
van Duin et al., 1986; Linet al., 1998
Wild-type P53 activatedfragment-1 (WAF1)
21 kDa6p21.2
CDK7 inhibitor; control cellproliferation
Cell-cycleregulation
el-Deiry et al., 1993;McDonald et al., 1996;Cayrol andDucommun, 1998
aAdapted from Yu et al., 1999.
Protein Truncation Test Screen for DNA Repair 229

cording to the instructions provided. The recovered lymphocytes wereresuspended and cultured in lymphocyte growth medium (LGM). LGMconsisted of 40% HL-1 (Biowhittaker, Walkersville, MD), 38% RPMI(Gibco-BRL, Grand Island, NY), 10% CBS (Hyclone, Logan, UT), 8% 41[0.0015 g/ml penicillin G (Sigma, St. Louis, MO), 0.0025 g/ml streptomy-cin sulfate (Sigma), 0.0073 g/ml GlutaMax (GibcoBRL), and 0.00545 g/mlpyruvic acid (Sigma)], 1% Fungizone (Gibco-BRL), 2% human serum(Sigma), 10mg/ml PHA (Sigma), and 5mg/ml of IL-2 (Gibco-BRL). After3–7 days culturing, cells were ready for total RNA extraction.
Total RNA Extraction
Total RNA was extracted from up to 106 cultured lymphocytes, accord-ing to the instructions of Trizol (Gibco-BRL). Culture medium was re-moved by centrifugation and the cell pellets were immediately washed in1 ml Trizol. Phase separation, conducted in a 1.5-ml microtube, wasaccomplished by adding 200ml chloroform and centrifuging at 12,000g for15 min. Total RNA was precipitated from the aqueous phase by adding 4/5to 1 volume of isopropyl alcohol. After being washed with room-temper-ature 70% ethanol, the RNA pellet was dissolved in 25ml RNase-freewater and digested at 37°C for 20 min with 2 U DNase I (BoehringerMannheim, Mannheim, Germany) in a 50-ml reaction containing 50 mMTris–HCl (pH 7.5), 10 mM MgCl2, and 4 U/ml RNasin (Promega, Madison,WI). After phenol:chloroform (1:1) extraction, RNA was precipitated againby adding 15ml 3 M Na-acetate (pH 4.8–5.0) and 1 ml 96% ethanol at270°C. RNA was recovered by centrifugation at 12,000g for 10 min at4°C. The RNA pellet was washed with 200ml cold 80% ethanol anddissolved in RNase-free water to get a final concentration of 0.5mg/ml.RNA quality was checked by running 1mg of the sample on 1% agarosegel.
RT-PCR
First-strand cDNA was generated by reverse transcription of 3mg totalRNA in reactions containing 20 ng poly(T) primers, 4 mM dNTP, 4 nMDTT, 0.8 U/ml RNasin, and 8 U/ml M-MuLV reverse transcriptase at 37°Cfor 1 hr. A 0.6-ml sample of the reaction was amplified by 30 cycles of PCR(94°C for 30 sec, 60–65°C for 1 min, and 72°C for 1.5 min) in 15-mlreactions containing 10 mM Tris–HCl (pH 8.8), 1.5 mM MgCl, 25 mMKCl, 0.17 U/ml Taqpolymerase, and 0.8mM each of forward and reverseprimers. Specific primers were designed to target each of the 10 cDNAsequences (Table II). The sense primer (forward) for each gene contains a59 extension, specifically designed for PTT. It contains, from 59, a T7promoter sequence, a spacer sequence, and a eukaryotic translation initia-tion sequence (Kozak sequence). The 39 end of the elongation is connectedwith the target gene sequence in-frame with the ATG codon from theKozak sequence [den Dunnen et al., 1996; Telatar et al., 1996; Hogervorst,1997] (Fig. 1).
Transcription and Translation
The total amount of PCR product used for the TnT T7 Quick CoupledTranscription/Translation System (Promega) was 4.4ml. The in vitrotranscription and translation reactions were carried in 12.5ml with theconcentration of 0.52mCi/ml for 35S-methionine (Amersham, Bucking-hamshire, U.K.). Proteins were resolved by 14% SDS–PAGE and visual-ized by autoradiography [Telatar et al., 1996]. PTT was repeated whentruncations were suspected.
Sequencing
FreshCCNH RT-PCR products of a suspected truncation sample werepurified using a mini-PCR preparation kit (Promega), following Promega
protocol. After cloning the purified PCR product using the pBAD TOPOTA Cloning kit (Invitrogen, Carlsbad, CA), according to the manufactur-er’s protocol, 14 colonies were sequenced. Sequencing analysis was per-formed by simultaneous bidirectional cycle sequencing on an LI-COR4200 DNA Sequencer (LI-COR Inc., Lincoln, NE) using a SequiThermEXEL II Sequencing kit (Epicentre Technologies, Madison, WI).
Total RNA Dot Hybridization
Total RNA dot hybridization was done according to the protocol onmolecular cloning [Sambrook et al., 1989]. Total RNA, with serial dilu-tions of 5, 0.5, 0.05, and 0.005mg of the sample withoutHAAG PCRproduct, and of two normal positive control samples, was used for totalRNA dot hybridization. Probe-correspondent PCR product, with serialdilutions of 50, 5, 0.5, and 0.05 ng of theHAAG,was set up for positivecontrol as well. Total RNA and DNA samples were immobilized on0.45-mm HyBond N1 nylon membrane (Amersham Pharmacia Biotech,Buckinghamshire, U.K.). The probe was prepared using purified freshHAAG PCR product, CTP-32P, and a Random Primed DNA Labeling kit(Boehringer Mannheim). After hybridization, the membrane was storedovernight in an exposure cassette and analyzed on a PhosphorImager 860(Molecular Dynamics, Sunnyvale, CA).
RESULTS
A total of 164 healthy volunteers without a personalhistory of cancer agreed to anonymous genetic analysis. Ofthese, 136 were successfully cultured and screened. Thetested samples consisted of 41 males (30%) and 95 females(70%). Average age was 25.3 years, with a range of 17 to 60years. Ninety-five volunteers had a relative who had hadcancer, whereas thirty-nine did not. Family cancer historydata were missing for two volunteers. Ancestry for thisgroup of people was predominantly European (including theBritish Isles), but included some individuals of Asian de-scent.
This study screened in vitro transcribed-translated pro-teins for truncated segments to investigate the truncating
TABLE II. Gene-Specific Primer Sequences
APE forward: GTGGCGGAAGACGGGGATGAGreverse: ATGGTAGTTGAGGGGGCTTATTTC
ERCC1 forward: GGGCCGCCAGCAAGGAAGAreverse: CTGGGAGGACGATTTATTATTACA
CCNH forward: GCAAGACTGCGGGCTGACGreverse: AATGACATCGCTCCAACTTCTG
CDK7 forward: GGCTGGAGTCGGGCTTTACGreverse: CCTTTTTGGCTATTTCCCTCAG
POLB forward: GCCATGAGCAAACGGAAGGreverse: TGCGCCAGGGAGGATACAGG
HOGG1 forward: GCGGTGCCTGCTGTGGAreverse: GAAGTGGGGAATGGAGGGGAAGGT
WAF1 forward: GAACCGGCTGGGGATGTCreverse: AGGCGGGGTGGTCTGCTC
UNG forward: GCCCTGGGCTCTTACTGTCCreverse: CATCTCCCCTGTTTCACCATA
MGMT forward: GAAATGAAACGCACCACACTGreverse: GCCTCCACGCCCCGCATCC
HAAG forward: GGGTGTTTGTGCCTCATAAreverse: CTGCCCCGGACATAGAAGC
230 Chen et al.
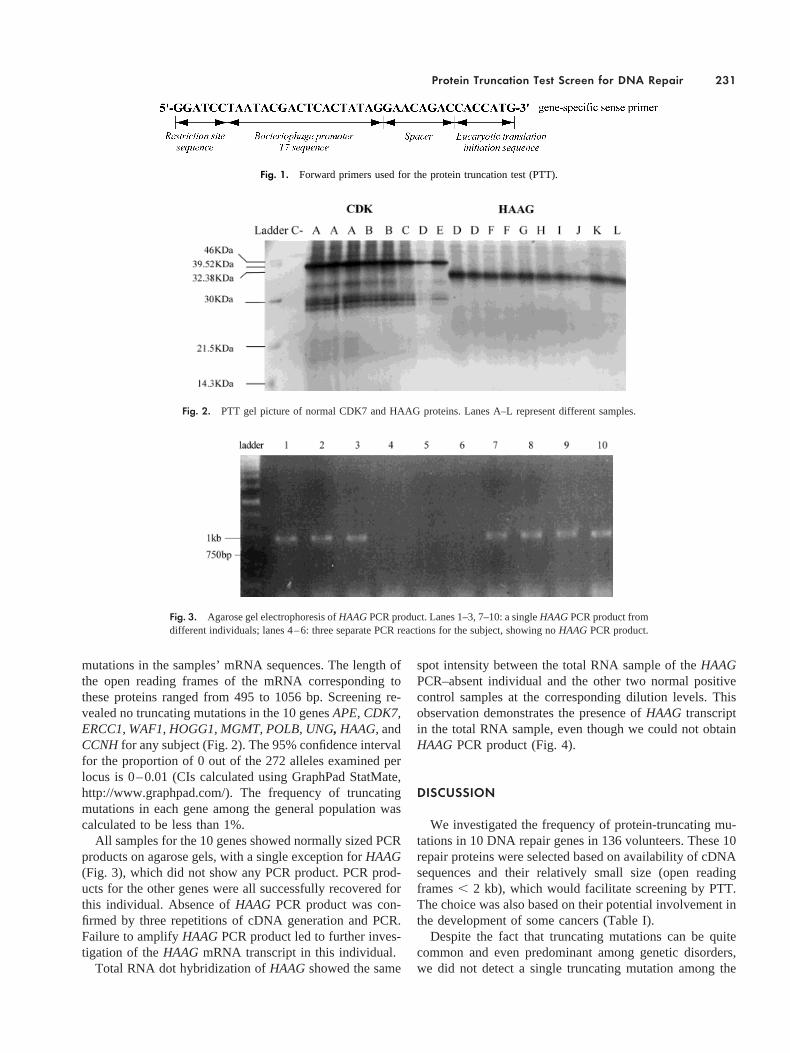
mutations in the samples’ mRNA sequences. The length ofthe open reading frames of the mRNA corresponding tothese proteins ranged from 495 to 1056 bp. Screening re-vealed no truncating mutations in the 10 genesAPE, CDK7,ERCC1, WAF1, HOGG1, MGMT, POLB, UNG, HAAG,andCCNHfor any subject (Fig. 2). The 95% confidence intervalfor the proportion of 0 out of the 272 alleles examined perlocus is 0–0.01 (CIs calculated using GraphPad StatMate,http://www.graphpad.com/). The frequency of truncatingmutations in each gene among the general population wascalculated to be less than 1%.
All samples for the 10 genes showed normally sized PCRproducts on agarose gels, with a single exception forHAAG(Fig. 3), which did not show any PCR product. PCR prod-ucts for the other genes were all successfully recovered forthis individual. Absence ofHAAG PCR product was con-firmed by three repetitions of cDNA generation and PCR.Failure to amplifyHAAGPCR product led to further inves-tigation of theHAAG mRNA transcript in this individual.
Total RNA dot hybridization ofHAAGshowed the same
spot intensity between the total RNA sample of theHAAGPCR–absent individual and the other two normal positivecontrol samples at the corresponding dilution levels. Thisobservation demonstrates the presence ofHAAG transcriptin the total RNA sample, even though we could not obtainHAAG PCR product (Fig. 4).
DISCUSSION
We investigated the frequency of protein-truncating mu-tations in 10 DNA repair genes in 136 volunteers. These 10repair proteins were selected based on availability of cDNAsequences and their relatively small size (open readingframes, 2 kb), which would facilitate screening by PTT.The choice was also based on their potential involvement inthe development of some cancers (Table I).
Despite the fact that truncating mutations can be quitecommon and even predominant among genetic disorders,we did not detect a single truncating mutation among the
Fig. 1. Forward primers used for the protein truncation test (PTT).
Fig. 2. PTT gel picture of normal CDK7 and HAAG proteins. Lanes A–L represent different samples.
Fig. 3. Agarose gel electrophoresis ofHAAGPCR product. Lanes 1–3, 7–10: a singleHAAGPCR product fromdifferent individuals; lanes 4–6: three separate PCR reactions for the subject, showing noHAAGPCR product.
Protein Truncation Test Screen for DNA Repair 231

approximately 2700 alleles examined. Indeed, the only ob-servation of an aberrant situation was the case of one subjectfrom whom PCR product could not be generated. We con-clude that the frequency of truncating mutations in DNArepair genes is quite low.
The low carrier frequency of truncating DNA repairgenes in the general population is consistent with the ideathat these genes play critical roles in survival. However, theexperimental system might also have contributed some fac-tors to this result. PTT is based on RT-PCR, and thustheoretically might not detect unstable RNA moleculescaused by truncating mutations.
The absence ofHAAG RT-PCR product for one subject,despite total RNA dot hybridization evidence for the exis-tence of its mRNA transcript, indicates that theHAAGmRNA sequence for this subject is somehow aberrant. Al-though the observation is intriguing, lack of a knowngenomic DNA sequence forHAAG, the limited material oftotal RNA extracted, and the inability to return to the subjectmake it difficult to further investigate the mechanism re-sponsible. HAAG, human 3-alkyadenine DNA glycosylase,is responsible for the removal of 3-alkyadenine by the baseexcision repair pathway. In higher organisms, the existenceof multiple repair pathways leads to functional redundancyso that failure of one enzyme of a system does not totallyabolish the repair of a lesion [Yu et al., 1999].
An important issue of using the PTT is to distinguishgenuine truncating mutations from artifacts. For example,one sample was initially thought to be a carrier of a trun-cating mutation inCCNH.However, this result could not beconfirmed by PTT, and the absence of any mutation wasconfirmed by the sequencing of the cloned PCR products.The source of this false positive result could not be ascer-
tained. The RT-PCR process can be error-prone as a resultof the low accuracy ofTaqDNA polymerase [Cariello et al.,1991], which is 6–10 times higher than that ofPfu usingcDNAs as a template [Cline et al., 1996; Bracho et al.,1998]. In addition, the high pH in our experiment, pH 8.8,may be another contributing factor to the false positive PTTresult, since an increase in error rate with pH has also beennoted for the exonuclease-deficientTaq DNA polymerase[Cline et al., 1996]. It is likely that by usingPfu andoptimizing PCR reaction conditions, the occurrence of falsepositive PTT results could be minimized.
Mutations other than truncating mutations can also obvi-ously affect function. The disruption of gene function bymissense and nonsense mutations is well documented, buteven apparently silent mutations can influence function [Yuet al., 2000]. Indeed, whereas truncating mutations were notobserved in this study, considerable variation on humanDNA repair genes has been reported [Mohrenweiser andJones, 1998; Shen et al., 1998; Lunn et al., 1999]. Thus,other approaches capable of revealing a broader range ofmutational classes will be required to better investigate therelationship between variation in DNA repair genes andcancer predisposition.
ACKNOWLEDGMENTS
We thank the volunteer donors for their generous partic-ipation, without which this study could not be done. Thanksgo to Sherry Swanson, RLT, and her colleagues, for volun-teering to draw blood for this study. We also thank Ms.Pauline Tymchuk for her help in editing the manuscript.
REFERENCES
Abrahams PJ, Houweling A, Cornelissen-Steijger PD, Jaspers NG, Dar-roudi F, Meijers CM, Mullenders LH, Filon R, Arwert F, PinedoHM, Natarajan AP, Terleth C, Van Zeeland AA, van der Eb AJ.1998. Impaired DNA repair capacity in skin fibroblasts from vari-ous hereditary cancer-prone syndromes. Mutat Res 407:189–201.
Bartkova J, Zemanova M, Bartek J. 1996. Expression of CDK7/CAK innormal and tumor cells of diverse histogenesis, cell-cycle positionand differentiation. Int J Cancer 66:732–737.
Baserga SJ, Benz EJJ. 1988. Nonsense mutations in the human beta-globingene affect mRNA metabolism. Proc Natl Acad Sci USA 85:2056–2060.
Bebb DG, Yu Z, Chen J, Telatar M, Gelmon K, Phillips N, Gatti RA,Glickman BW. 1999. Absence of mutations in the ATM gene inforty-seven cases of sporadic breast cancer. Br J Cancer 80:1979–1981.
Bhattacharyya N, Chen HC, Grundfest-Broniatowski S, Banerjee S. 1999.Alteration of hMSH2 and DNA polymerase beta genes in breastcarcinomas and fibroadenomas. Biochem Biophys Res Commun259:429–435.
Bracho MA, Moya A, Barrio E. 1998. Contribution ofTaq polymerase-induced errors to the estimation of RNA virus diversity. J Gen Virol79:2921–2928.
Canitrot Y, Cazaux C, Frechet M, Bouayadi K, Lesca C, Salles B, Hoff-mann JS. 1998. Overexpression of DNA polymerase beta in cell
Fig. 4. Total RNA dot hybridization. C1: PCR product ofHAAG; C2:negative control; a, c: total RNA of two samples withHAAGPCR product;b: total RNA of the sample withoutHAAGPCR product; lanes 1–4: serialdilution of samples. Lane 1: 5mg for total RNA, 50 ng for DNA; lane 2:0.5 mg for total RNA, 5 ng for DNA; lane 3: 0.05mg for total RNA, 0.5ng for DNA; lane 4: 0.005mg for total RNA, 0.05 ng for DNA.
232 Chen et al.

results in a mutator phenotype and a decreased sensitivity to anti-cancer drugs. Proc Natl Acad Sci USA 95:12586–12590.
Cariello NF, Thilly WG, Swenberg JA, Skopek TR. 1991. Deletion mu-tagenesis during polymerase chain reaction: dependence on DNApolymerase. Gene 99:105–108.
Cayrol C, Ducommun B. 1998. Interaction with cyclin-dependent kinasesand PCNA modulates proteasome-dependent degradation of p21.Oncogene 17:2437–2444.
Chevillard S, Radicella JP, Levalois C, Lebeau J, Poupon MF, Oudard S,Dutrillaux B, Boiteux S. 1998. Mutations in OGG1, a gene in-volved in the repair of oxidative DNA damage, are found in humanlung and kidney tumours. Oncogene 16:3083–3086.
Cline J, Braman JC, Hogrefe HH. 1996. PCR fidelity ofPfu DNA poly-merase and other thermostable DNA polymerases. Nucleic AcidsRes 24:3546–3551.
Cvitkovic E. 1998. Ongoing and unsaid on oxaliplatin: the hope. Br JCancer 77(Suppl 4):8–11.
Demple B, Herman T, Chen DS. 1991. Cloning and expression of APE, thecDNA encoding the major human apurinic endonuclease: definitionof a family of DNA repair enzymes. Proc Natl Acad Sci USA88:11450–11454.
den Dunnen JT, Roest PA, van der Tuijn AC, Hogervorst FB. 1996. Theprotein truncation test (PTT) for rapid detection of translation-terminating mutations. p 323–431.
Dietz HC, Valle D, Francomano CA, Kendzior RJJ, Pyeritz RE, CuttingGR. 1993. The skipping of constitutive exons in vivo induced bynonsense mutations. Science 259:680–683.
Eccles DM, van der Luijt R, Breukel C, Bullman H, Bunyan D, Fisher A,Barber J, du Boulay C, Primrose J, Burn J, Fodde R. 1996. Hered-itary desmoid disease due to a frameshift mutation at codon 1924 ofthe APC gene. Am J Hum Genet 59:1193–1201.
el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM,Lin D, Mercer WE, Kinzler KW, Vogelstein B. 1993. WAF1, apotential mediator of p53 tumor suppression. Cell 75:817–825.
Fisher RP, Morgan DO. 1994. A novel cyclin associates with MO15/CDK7to form the CDK-activating kinase. Cell 78:713–724.
FitzGerald MG, Bean JM, Hegde SR, Unsal H, MacDonald DJ, Harkin DP,Finkelstein DM, Isselbacher KJ, Haber DA. 1997. HeterozygousATM mutations do not contribute to early onset of breast cancer.Nat Genet 15:307–310.
Fung H, Kow YW, Van Houten B, Taatjes DJ, Hatahet Z, Janssen YM,Vacek P, Faux SP, Mossman BT. 1998. Asbestos increases mam-malian AP-endonuclease gene expression, protein levels, and en-zyme activity in mesothelial cells. Cancer Res 58:189–194.
Gardner RJ, Bobrow M, Roberts RG. 1995. The identification of pointmutations in Duchenne muscular dystrophy patients by using re-verse-transcription PCR and the protein truncation test. Am J HumGenet 57:311–320.
Heim RA, Silverman LM, Farber RA, Kam-Morgan LN, Luce MC. 1994.Screening for truncated NF1 proteins. Nat Genet 8:218–219.
Heim RA, Kam-Morgan LN, Binnie CG, Corns DD, Cayouette MC, FarberRA, Aylsworth AS, Silverman LM, Luce MC. 1995. Distribution of13 truncating mutations in the neurofibromatosis 1 gene. Hum MolGenet 4:975–981.
Hogervorst FB. 1997. The protein truncation test (PTT). Promega NotesMagazine (Madison, WI: Promega). p 7–10.
Hogervorst FB, Cornelis RS, Bout M, van Vliet M, Oosterwijk JC, OlmerR, Bakker B, Klijn JG, Vasen HF, Meijers-Heijboer H. 1995. Rapiddetection of BRCA1 mutations by the protein truncation test. NatGenet 10:208–212.
Ishida T, Takashima R, Fukayama M, Hamada C, Hippo Y, Fujii T,Moriyama S, Matsuba C, Nakahori Y, Morita H, Yazaki Y, Ko-dama T, Nishimura S, Aburatani H. 1999. New DNA polymor-phisms of human MMH/OGG1 gene: prevalence of one polymor-
phism among Japanese lung-adenocarcinoma patients.. Int J Cancer80:18–21.
Jones CT, McIntosh I, Keston M, Ferguson A, Brock DJ. 1992. Threenovel mutations in the cystic fibrosis gene detected by chemicalcleavage: analysis of variant splicing and a nonsense mutation.Hum Mol Genet 1:11–17.
Kohno T, Shinmura K, Tosaka M, Tani M, Kim SR, Sugimura H, NohmiT, Kasai H, Yokota J. 1998. Genetic polymorphisms and alternativesplicing of the hOGG1 gene, that is involved in the repair of8-hydroxyguanine in damaged DNA. Oncogene 16:3219–3225.
Krokan HE, Slupphaug G. 1998. DNA repair enzymes and their genes.Tidsskr Nor Laegeforen 118:2037–2042.
Lancaster JM, Wooster R, Mangion J, Phelan CM, Cochran C, Gumbs C,Seal S, Barfoot R, Collins N, Bignell G, Patel S, Hamoudi R,Larsson C, Wiseman RW, Berchuck A, Iglehart JD, Marks JR,Ashworth A, Stratton MR, Futreal PA. 1996. BRCA2 mutations inprimary breast and ovarian cancers. Nat Genet 13:238–240.
Lefebvre P, Zak P, Laval F. 1993. Induction of O6-methylguanine-DNA-methyltransferase and N3-methyladenine-DNA-glycosylase in hu-man cells exposed to DNA-damaging agents. DNA Cell Biol 12:233v241.
Lin YW, Kubota M, Koishi S, Sawada M, Usami I, Watanabe K, AkiyamaY. 1998. Analysis of mutations at the DNA repair genes in acutechildhood leukaemia. Br J Haematol 103:462–466.
Liu B, Parsons RE, Hamilton SR, Petersen GM, Lynch HT, Watson P,Markowitz S, Willson JK, Green J, de la Chapelle A. 1994. hMSH2mutations in hereditary nonpolyposis colorectal cancer kindreds.Cancer Res 54:4590–4594.
Lunn RM, Langlois RG, Hsieh LL, Thompson CL, Bell DA. 1999. XRCC1polymorphisms: effects on aflatoxin B1-DNA adducts and glyco-phorin A variant frequency. Cancer Res 59:2557–2561.
Lynch HT, Fusaro RM, Lynch JF. 1997. Cancer genetics in the new era ofmolecular biology. Ann N Y Acad Sci 833:1–28.
McDonald ER, Wu GS, Waldman T, el-Deiry WS. 1996. Repair defect inp21 WAF1/CIP12/2 human cancer cells. Cancer Res 56:2250–2255.
Mohrenweiser HW, Jones IM. 1998. Variation in DNA repair is a factor incancer susceptibility: a paradigm for the promises and perils ofindividual and population risk estimation? Mutat Res 400:15–24.
Moon YW, Park WS, Vortmeyer AO, Weil RJ, Lee YS, Winters TA,Zhuang Z, Fuller BG. 1998. Mutation of the uracil DNA glycosy-lase gene detected in glioblastoma. Mutat Res 421:191–196.
Olsen LC, Aasland R, Wittwer CU, Krokan HE, Helland DE. 1989.Molecular cloning of human uracil-DNA glycosylase, a highlyconserved DNA repair enzyme. EMBO J 8:3121–3125.
Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, RosenCA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD.1994. Mutation of a mutL homolog in hereditary colon cancer.Science 263:1625–1629.
Park CH, Mu D, Reardon JT, Sancar A. 1995. The general transcription-repair factor TFIIH is recruited to the excision repair complex bythe XPA protein independent of the TFIIE transcription factor.J Biol Chem 270:4896–4902.
Roest PA, Roberts RG, van der Tuijn AC, Heikoop JC, van Ommen GJ,den Dunnen JT. 1993. Protein truncation test (PTT) to rapidlyscreen the DMD gene for translation terminating mutations. Neu-romuscul Disord 3:391–394.
Rosenquist TA, Zharkov DO, Grollman AP. 1997. Cloning and character-ization of a mammalian 8-oxoguanine DNA glycosylase. Proc NatlAcad Sci USA 94:7429–7434.
Sambrook J, Fritsch EF, Maniatis T. 1989. Synthetic oligonucleotideprobes. In: Molecular cloning: a laboratory manual (2nd ed.). ColdSpring Harbor, NY: Cold Spring Harbor Laboratory Press. p 11.55–11.57.
Samson L, Derfler B, Boosalis M, Call K. 1991. Cloning and character-
Protein Truncation Test Screen for DNA Repair 233

ization of a 3-methyladenine DNA glycosylase cDNA from humancells whose gene maps to chromosome 16. Proc Natl Acad Sci USA88:9127–9131.
SenGupta DN, Zmudzka BZ, Kumar P, Cobianchi F, Skowronski J, WilsonSH. 1986. Sequence of human DNA polymerase beta mRNAobtained through cDNA cloning. Biochem Biophys Res Commun136:341–347.
Shen MR, Jones IM, Mohrenweiser H. 1998. Nonconservative amino acidsubstitution variants exist at polymorphic frequency in DNA repairgenes in healthy humans. Cancer Res 58:604–608.
Silber JR, Blank A, Bobola MS, Mueller BA, Kolstoe DD, Ojemann GA,Berger MS. 1996. Lack of the DNA repair protein O6-methylgua-nine-DNA methyltransferase in histologically normal brain adja-cent to primary human brain tumors. Proc Natl Acad Sci USA93:6941–6946.
Takebe H. 1984. DNA repair and carcinogenesis. Gan No Rinsho 30:1433–1438.
Takebe H. 1995. Mechanisms of hereditary tumorigenesis. Nippon Rinsho53:2634–2639.
Tano K, Shiota S, Collier J, Foote RS, Mitra S. 1990. Isolation andstructural characterization of a cDNA clone encoding the humanDNA repair protein for O6-alkylguanine. Proc Natl Acad Sci USA87:686–690.
Telatar M, Wang Z, Udar N, Liang T, Bernatowska-Matuszkiewicz E,Lavin M, Shiloh Y, Concannon P, Good RA, Gatti RA. 1996.Ataxia-telangiectasia: mutations in ATM cDNA detected by pro-tein-truncation screening. Am J Hum Genet 59:40–44.
van der Luijt R, Khan PM, Vasen H, van Leeuwen C, Tops C, Roest P, denDunnen J, Fodde R. 1994. Rapid detection of translation-terminat-ing mutations at the adenomatous polyposis coli (APC) gene bydirect protein truncation test. Genomics 20:1–4.
van Duin M, de Wit J, Odijk H, Westerveld A, Yasui A, Koken HM,Hoeijmakers JH, Bootsma D. 1986. Molecular characterization ofthe human excision repair gene ERCC-1: cDNA cloning and aminoacid homology with the yeast DNA repair gene RAD10. Cell44:913–923.
Wang L, Zhu D, Zhang C, Mao X, Wang G, Mitra S, Li BF, Wang X, WuM. 1997. Mutations of O6-methylguanine-DNA methyltransferasegene in esophageal cancer tissues from Northern China. Int JCancer 71:719–723.
Wei Q, Matanoski GM, Farmer ER, Hedayati MA, Grossman L. 1995.DNA repair capacity for ultraviolet light-induced damage is re-duced in peripheral lymphocytes from patients with basal cellcarcinoma. J Invest Dermatol 104:933–936.
Wu L, Yee A, Liu L, Carbonaro-Hall D, Venkatesan N, Tolo VT, Hall FL.1994. Molecular cloning of the human CAK1 gene encoding acyclin-dependent kinase-activating kinase. Oncogene 9:2089–2096.
Yankulov KY, Bentley DL. 1997. Regulation of CDK7 substrate specific-ity by MAT1 and TFIIH. EMBO J 16:1638–1646.
Yu JJ, Lee KB, Mu C, Li Q, Abernathy TV, Bostick-Bruton F, Reed E.2000. Comparison of two human ovarian carcinoma cell lines(A2780/CP70 and MCAS) that are equally resistant to platinum,but differ at codon 118 of the ERCC1 gene. Int J Oncol 16:555–560.
Yu Z, Chen J, Ford BN, Brackley ME, Glickman BW. 1999. Human DNArepair systems: an overview. Environ Mol Mutagen 33:3–20.
Accepted by—J. Felton
234 Chen et al.


















