
Scaling relationships for adsorption energies of C2 hydrocarbons on transition metal surfaces
6
Scaling relationships for adsorption energies of C 2 hydrocarbons on transition metal surfaces Glenn Jones a , Felix Studt a,b , Frank Abild-Pedersen a,b , Jens K. Nørskov a,b,c , Thomas Bligaard a,n a Center for Atomic-scale Materials Design (CAMD), Department of Physics, Building 307, Technical University of Denmark, DK-2800 Kgs. Lyngby, Denmark b Center for Interface Science and Catalysis, SLAC National Accelerator Laboratory, 2575 Sand Hill Road, Menlo Park, CA 94025, USA c Department of Chemical Engineering, Stanford University, Stanford, CA 94305, USA article info Article history: Received 3 November 2010 Received in revised form 9 February 2011 Accepted 23 February 2011 Available online 11 March 2011 Keywords: Adsorption Catalysis Computational chemistry Reaction engineering Selectivity abstract Using density functional theory calculations we show that the adsorption energies for C 2 H x -type adsorbates on transition metal surfaces scale with each other according to a simple bond order conservation model. This observation generalizes some recently recognized adsorption energy scaling laws for AH x -type adsorbates to unsaturated hydrocarbons and establishes a coherent simplified description of saturated as well as unsaturated hydrocarbons adsorbed on transition metal surfaces. A number of potential applications are discussed. We apply the model to the dehydrogenation of ethane over pure transition metal catalysts. Comparison with the corresponding full density functional theory calculations shows excellent agreement. & 2011 Elsevier Ltd. All rights reserved. 1. Introduction Modern society is heavily dependent on fossil hydrocarbons, both as an energy resource and perhaps almost equally impor- tantly as a provider of the basic building blocks for a multitude of derived chemicals and materials. Most of the important industrial processes for the selective transformations of hydrocarbons are dependent on catalysts to facilitate the reactions. Significant examples of such processes include the catalytic steam cracking of hydrocarbons (Bodke et al., 1999), alkene hydrogenation (Somorjai, 1994; Horiuti and Polanyi, 1934), steam and CO 2 reforming (Rostrup-Nielsen et al., 2002), and Fischer–Tropsch synthesis (Dry, 2002). To describe such reactions in detail, knowl- edge is required of the involved hydrocarbon species, both in the reaction mixture and adsorbed on the catalytic surface. To acquire this knowledge of the adsorbed species over a single catalyst surface represents a significant challenge, and to achieve this over a larger range of interesting materials requires an even greater effort and is in many cases simply intractable. In order to build a molecular level understanding of surface catalyzed processes, it is essential to have the surface reaction energetics from which thermodynamic and kinetic studies can progress. To this end, the adsorption energy is a key quantity, describing the strength of the interaction between a molecule and a surface. Density functional theory (DFT) offers the possibility of calculating adsorption energies with reasonable accuracy at an acceptable computational cost, and over the past two decades this approach has enjoyed considerable success (van Santen and Neurock, 2006; Hammer and Nørskov, 2000; Liu and Hu, 2003; Greeley and Mavrikakis, 2004; Nørskov et al., 2006). DFT thus provides a method by which analysis of reaction pathways concerning a specific reaction over a single surface can be obtained within reasonable time. In a few cases this has led to the full understanding of all relevant reaction steps for a given catalytic reaction over a given catalytic surface, with the Haber– Bosch ammonia synthesis over ruthenium surfaces as the perhaps most elaborate example (Hellman et al., 2006). Such detailed calculations at present cannot however, be performed for a large number of potentially interesting catalytic surfaces for a particu- lar reaction. Whereas a limited number of pure transition metal surfaces are now accessible, the computational screening of for example alloy catalysts exhibits a complexity requiring orders of magnitude more computational time than what is currently accessible practically. Simple models to estimate adsorption and reaction energies with good accuracy and with the ability to reproduce the chemical trends observed with higher level com- putational methods (or careful experiments) are therefore highly desirable (Shustorovich and Baetzold, 1985; Shustorovich, 1986, 1988; Shustorovich and Bell, 1988). We here present a step in this direction for the case of hydrocarbon species. Contents lists available at ScienceDirect journal homepage: www.elsevier.com/locate/ces Chemical Engineering Science 0009-2509/$ - see front matter & 2011 Elsevier Ltd. All rights reserved. doi:10.1016/j.ces.2011.02.050 n Corresponding author. Fax: þ45 4593 2399. E-mail address: [email protected] (T. Bligaard). Chemical Engineering Science 66 (2011) 6318–6323
-
Upload
glenn-jones -
Category
Documents
-
view
212 -
download
0
Transcript of Scaling relationships for adsorption energies of C2 hydrocarbons on transition metal surfaces

Chemical Engineering Science 66 (2011) 6318–6323
Contents lists available at ScienceDirect
Chemical Engineering Science
0009-25
doi:10.1
n Corr
E-m
journal homepage: www.elsevier.com/locate/ces
Scaling relationships for adsorption energies of C2 hydrocarbonson transition metal surfaces
Glenn Jones a, Felix Studt a,b, Frank Abild-Pedersen a,b, Jens K. Nørskov a,b,c, Thomas Bligaard a,n
a Center for Atomic-scale Materials Design (CAMD), Department of Physics, Building 307, Technical University of Denmark, DK-2800 Kgs. Lyngby, Denmarkb Center for Interface Science and Catalysis, SLAC National Accelerator Laboratory, 2575 Sand Hill Road, Menlo Park, CA 94025, USAc Department of Chemical Engineering, Stanford University, Stanford, CA 94305, USA
a r t i c l e i n f o
Article history:
Received 3 November 2010
Received in revised form
9 February 2011
Accepted 23 February 2011Available online 11 March 2011
Keywords:
Adsorption
Catalysis
Computational chemistry
Reaction engineering
Selectivity
09/$ - see front matter & 2011 Elsevier Ltd. A
016/j.ces.2011.02.050
esponding author. Fax: þ45 4593 2399.
ail address: [email protected] (T. Bligaard
a b s t r a c t
Using density functional theory calculations we show that the adsorption energies for C2Hx-type
adsorbates on transition metal surfaces scale with each other according to a simple bond order
conservation model. This observation generalizes some recently recognized adsorption energy scaling
laws for AHx-type adsorbates to unsaturated hydrocarbons and establishes a coherent simplified
description of saturated as well as unsaturated hydrocarbons adsorbed on transition metal surfaces.
A number of potential applications are discussed. We apply the model to the dehydrogenation of
ethane over pure transition metal catalysts. Comparison with the corresponding full density functional
theory calculations shows excellent agreement.
& 2011 Elsevier Ltd. All rights reserved.
1. Introduction
Modern society is heavily dependent on fossil hydrocarbons,both as an energy resource and perhaps almost equally impor-tantly as a provider of the basic building blocks for a multitude ofderived chemicals and materials. Most of the important industrialprocesses for the selective transformations of hydrocarbons aredependent on catalysts to facilitate the reactions. Significantexamples of such processes include the catalytic steam crackingof hydrocarbons (Bodke et al., 1999), alkene hydrogenation(Somorjai, 1994; Horiuti and Polanyi, 1934), steam and CO2
reforming (Rostrup-Nielsen et al., 2002), and Fischer–Tropschsynthesis (Dry, 2002). To describe such reactions in detail, knowl-edge is required of the involved hydrocarbon species, both in thereaction mixture and adsorbed on the catalytic surface. To acquirethis knowledge of the adsorbed species over a single catalystsurface represents a significant challenge, and to achieve this overa larger range of interesting materials requires an even greatereffort and is in many cases simply intractable.
In order to build a molecular level understanding of surfacecatalyzed processes, it is essential to have the surface reactionenergetics from which thermodynamic and kinetic studies canprogress. To this end, the adsorption energy is a key quantity,
ll rights reserved.
).
describing the strength of the interaction between a molecule anda surface. Density functional theory (DFT) offers the possibility ofcalculating adsorption energies with reasonable accuracy at anacceptable computational cost, and over the past two decades thisapproach has enjoyed considerable success (van Santen andNeurock, 2006; Hammer and Nørskov, 2000; Liu and Hu, 2003;Greeley and Mavrikakis, 2004; Nørskov et al., 2006). DFT thusprovides a method by which analysis of reaction pathwaysconcerning a specific reaction over a single surface can beobtained within reasonable time. In a few cases this has led tothe full understanding of all relevant reaction steps for a givencatalytic reaction over a given catalytic surface, with the Haber–Bosch ammonia synthesis over ruthenium surfaces as the perhapsmost elaborate example (Hellman et al., 2006). Such detailedcalculations at present cannot however, be performed for a largenumber of potentially interesting catalytic surfaces for a particu-lar reaction. Whereas a limited number of pure transition metalsurfaces are now accessible, the computational screening of forexample alloy catalysts exhibits a complexity requiring orders ofmagnitude more computational time than what is currentlyaccessible practically. Simple models to estimate adsorption andreaction energies with good accuracy and with the ability toreproduce the chemical trends observed with higher level com-putational methods (or careful experiments) are therefore highlydesirable (Shustorovich and Baetzold, 1985; Shustorovich, 1986,1988; Shustorovich and Bell, 1988). We here present a step in thisdirection for the case of hydrocarbon species.

G. Jones et al. / Chemical Engineering Science 66 (2011) 6318–6323 6319
Recently, it was shown that the adsorption energies of hydro-genated sulfur, oxygen, nitrogen, and carbon atoms on a number oftransition metal surfaces scale approximately with the adsorptionenergy of the surface-bonded atom alone (Abild-Pedersen et al.,2007). It has also been shown that the scaling relations can beextended from transition metal surfaces to the surfaces ofcompounds such as nitrides, oxides, and sulfides (Fernandez et al.,2008). Furthermore, it has also been demonstrated that the use ofthese scaling relationships in combination with a thermodynamicanalysis can lead to considerable insight into a number ofindustrially important catalytic processes (Jones et al., 2008). Suchuse of the scaling relations relies on the fact that if we havecalculated the energy, DEAHx
M1 , of the reaction intermediate AHx onone metal, M1, we can estimate the energy, DEAHx
M2 , of the sameintermediate on another metal, M2, from the adsorption energies ofatom A on the two metals as
DEAHx
M2 ¼DEAHx
M1 þgðxÞðDEAM2�DEA
M1Þ, ð1Þ
where
gðxÞ ¼ ðxmax�xÞ=xmax, ð2Þ
is the valence parameter describing the slope of the scaling relationbetween AHx and A, and xmax is the maximum number of hydrogenatoms required to provide sufficient electrons to fulfill the octet rulefor A. The simple expression for the valence parameter can beviewed as a result of AHx obtaining full bond order when interactingwith a metal surface. Even though the full adsorption energy of AHx
on a given transition metal is not proportional to the surface–adsorbate bond order, the variations between adsorption on differ-ent transition metals are (Abild-Pedersen et al., 2007). The modelcircumvents the need to carry out a full calculation for everyreaction intermediate on every metal. Originally, it was suggestedthat scaling relations are not limited to only the class of hydro-genated atom adsorbates, but that they are also valid for othersaturated compounds. For example hydrogenation or dehydrogena-tion reaction energies of hydrocarbons, alcohols, amino acids, andthiols with one adsorbate–surface bond were shown to be estimatedwell based on the simple hydrogenated atom model (Abild-Pedersenet al., 2007). In the present paper we extend the model fromhydrogenated atoms to adsorbates with one or more internal doubleand triple bonds and explicitly show that the scaling relations holdfor a broad class of C2 intermediates, namely C2Hx (x¼0, 1, 2, 3, 4, 5,and 6) species on different transition metal surfaces. This opens upthe possibility for using scaling relations for the study of trendsunderlying a number of important industrial hydrogenation anddehydrogenation reactions involving unsaturated hydrocarbonspecies.
The study has been carried out on close-packed fcc(1 1 1),hcp(0 0 0 1), and bcc(1 1 0) surfaces and the stepped fcc{2 1 1}and hcp{101 5} surfaces. After presentation of the model we willdemonstrate an application to the dehydrogenation of ethane,where the only input is the carbon binding energy on each metalin question. In order to verify the obtained results, the modelpredictions are tested against full DFT calculations.
2. Computational details
The surface DFT calculations were carried out with the Dacapocode (The Dacapo plane-wave/pseudopotential code) which usesa plane wave basis to describe the valence electrons and Vander-bilt ultrasoft pseudo-potentials (Vanderbilt, 1990) to representthe ionic cores. The plane wave expansion of the valence stateswas cut-off at an energy of 340 eV. All calculations were per-formed with the Revised Perdew–Burke–Ernzerhof (RPBE) gen-eralized gradient approximation functional (Hammer et al., 1999).
The self-consistent electron density was determined by iterativediagonalization of the Kohn–Sham Hamiltonian, with the occupa-tion of the Kohn–Sham states being smeared according to aFermi–Dirac distribution with a smearing factor of kBT¼0.1 eV,and Pulay mixing of the resulting electron density (Kresse andFurthmuller, 1996). All energies have been extrapolated to anelectronic temperature of kBT¼0 eV. Slabs consisting of threeclose-packed layers and separated by 11 A of vacuum wereperiodically repeated with a 2�2 surface unit cell for the closedpacked and a 1�2 unit cell for the stepped surfaces, respectively.A Monkhorst–Pack k-point sampling of 4�4�1 was applied(Monkhorst and Pack, 1976). In the case of Ni, Co, and Fe thecalculations were spin polarized. The reported adsorption ener-gies (E�x) are defined with reference to a neutral molecule orfragment of species x in isolation (Eisolated
x ) and the clean metalsurface (Emetal) in question
E�x ¼ Ex=metal�Eisolatedx �Emetal: ð3Þ
3. The model
In the following we describe the bond order conservation modelfor hydrocarbon species on transition metals and how the model isparameterized. Subsequently, results obtained with the model arecompared to full density functional theory calculations.
The bond order parameters, li, of the individual bonds that theatom A participates in can be normalized, gi ¼ li=xmax, such that thesum is unity when A has reached its maximal bond order (satisfiesthe octet rule):
XK
i
gi ¼ 1, ð4Þ
where K is the number of covalent bonds that atom A forms. Thenormalized bond orders, gi, we also refer to as the individual bondvalence parameters, since they resemble the valence parameter ofthe adsorbate–surface bond defined in Eq. (2). For an atom A withbonds saturated by hydrogen AHxmax Eq. (4) implies that
Xxmax
i
gi ¼ xmax gH ¼ 13gH ¼ 1=xmax: ð5Þ
When the molecule AHxmax approaches a metal surface then Eq. (4)implies that no covalent bonds between the surface and themolecule will form. In the following we will work with thehypothesis that any under-coordinated atom or molecular fragmentbound to a surface, will form a bond with the surface such thatEq. (4) will be satisfied, corresponding to the surface bond leading tofull saturation of A. Hence, for the adsorbed species AHx, wherexrxmax we have for the surface–adsorbate valence parameter, gS
gsþxgH ¼ 13gs ¼ 1�xgH ¼xmax�x
xmax, ð6Þ
which is the bond order conservation derivation of Eq. (2).In general for each atom A, which is bound to K other atoms or
fragments in addition to the surface, we find
gS ¼ 1�XK
i ¼ 1
gi: ð7Þ
Now for any molecule with L surface-bonded atoms, we wouldexpect that
gS ¼XL
j ¼ 1
1�XKj
i ¼ 1
gi
!: ð8Þ
Hence for hydrocarbon species, CnHm, mA ½0;2nþ2� with onlys intra-molecular bonds, and where all the carbon atomsattain their full bond order by interacting with the surface the
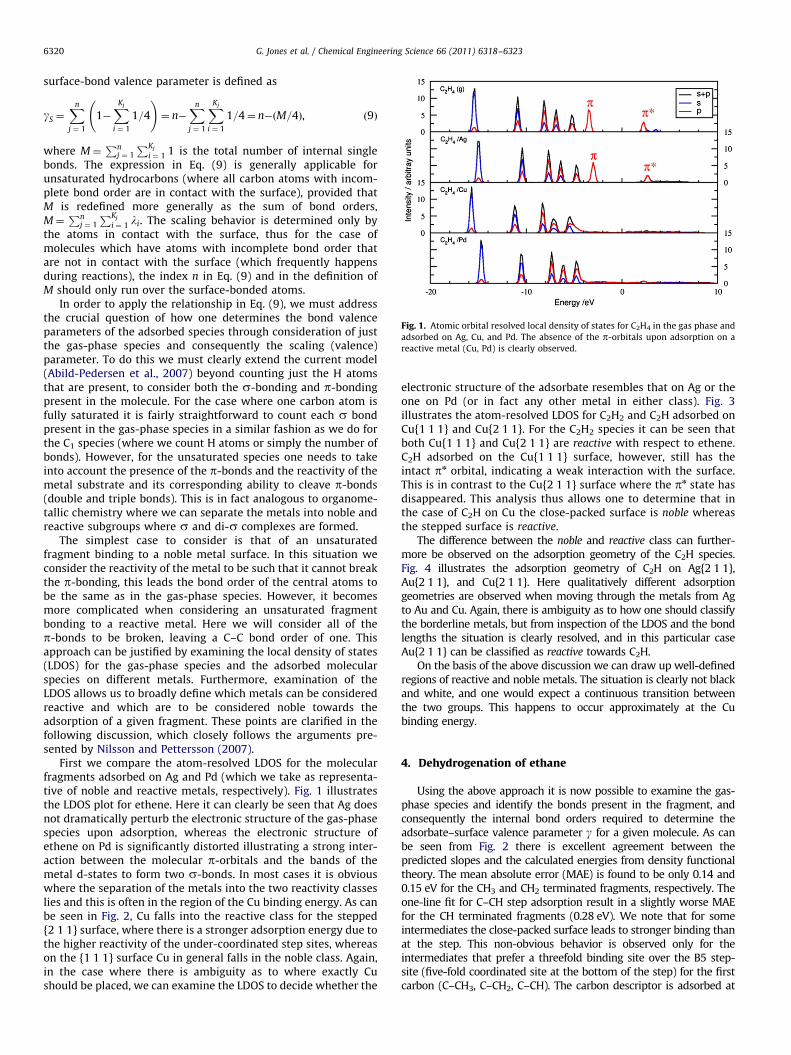
Fig. 1. Atomic orbital resolved local density of states for C2H4 in the gas phase and
adsorbed on Ag, Cu, and Pd. The absence of the p-orbitals upon adsorption on a
reactive metal (Cu, Pd) is clearly observed.
G. Jones et al. / Chemical Engineering Science 66 (2011) 6318–63236320
surface-bond valence parameter is defined as
gS ¼Xn
j ¼ 1
1�XKj
i ¼ 1
1=4
!¼ n�
Xn
j ¼ 1
XKj
i ¼ 1
1=4¼ n�ðM=4Þ, ð9Þ
where M¼Pn
j ¼ 1
PKj
i ¼ 1 1 is the total number of internal singlebonds. The expression in Eq. (9) is generally applicable forunsaturated hydrocarbons (where all carbon atoms with incom-plete bond order are in contact with the surface), provided thatM is redefined more generally as the sum of bond orders,M¼
Pnj ¼ 1
PKj
i ¼ 1 li. The scaling behavior is determined only bythe atoms in contact with the surface, thus for the case ofmolecules which have atoms with incomplete bond order thatare not in contact with the surface (which frequently happensduring reactions), the index n in Eq. (9) and in the definition ofM should only run over the surface-bonded atoms.
In order to apply the relationship in Eq. (9), we must addressthe crucial question of how one determines the bond valenceparameters of the adsorbed species through consideration of justthe gas-phase species and consequently the scaling (valence)parameter. To do this we must clearly extend the current model(Abild-Pedersen et al., 2007) beyond counting just the H atomsthat are present, to consider both the s-bonding and p-bondingpresent in the molecule. For the case where one carbon atom isfully saturated it is fairly straightforward to count each s bondpresent in the gas-phase species in a similar fashion as we do forthe C1 species (where we count H atoms or simply the number ofbonds). However, for the unsaturated species one needs to takeinto account the presence of the p-bonds and the reactivity of themetal substrate and its corresponding ability to cleave p-bonds(double and triple bonds). This is in fact analogous to organome-tallic chemistry where we can separate the metals into noble andreactive subgroups where s and di-s complexes are formed.
The simplest case to consider is that of an unsaturatedfragment binding to a noble metal surface. In this situation weconsider the reactivity of the metal to be such that it cannot breakthe p-bonding, this leads the bond order of the central atoms tobe the same as in the gas-phase species. However, it becomesmore complicated when considering an unsaturated fragmentbonding to a reactive metal. Here we will consider all of thep-bonds to be broken, leaving a C–C bond order of one. Thisapproach can be justified by examining the local density of states(LDOS) for the gas-phase species and the adsorbed molecularspecies on different metals. Furthermore, examination of theLDOS allows us to broadly define which metals can be consideredreactive and which are to be considered noble towards theadsorption of a given fragment. These points are clarified in thefollowing discussion, which closely follows the arguments pre-sented by Nilsson and Pettersson (2007).
First we compare the atom-resolved LDOS for the molecularfragments adsorbed on Ag and Pd (which we take as representa-tive of noble and reactive metals, respectively). Fig. 1 illustratesthe LDOS plot for ethene. Here it can clearly be seen that Ag doesnot dramatically perturb the electronic structure of the gas-phasespecies upon adsorption, whereas the electronic structure ofethene on Pd is significantly distorted illustrating a strong inter-action between the molecular p-orbitals and the bands of themetal d-states to form two s-bonds. In most cases it is obviouswhere the separation of the metals into the two reactivity classeslies and this is often in the region of the Cu binding energy. As canbe seen in Fig. 2, Cu falls into the reactive class for the stepped{2 1 1} surface, where there is a stronger adsorption energy due tothe higher reactivity of the under-coordinated step sites, whereason the {1 1 1} surface Cu in general falls in the noble class. Again,in the case where there is ambiguity as to where exactly Cushould be placed, we can examine the LDOS to decide whether the
electronic structure of the adsorbate resembles that on Ag or theone on Pd (or in fact any other metal in either class). Fig. 3illustrates the atom-resolved LDOS for C2H2 and C2H adsorbed onCu{1 1 1} and Cu{2 1 1}. For the C2H2 species it can be seen thatboth Cu{1 1 1} and Cu{2 1 1} are reactive with respect to ethene.C2H adsorbed on the Cu{1 1 1} surface, however, still has theintact pn orbital, indicating a weak interaction with the surface.This is in contrast to the Cu{2 1 1} surface where the pn state hasdisappeared. This analysis thus allows one to determine that inthe case of C2H on Cu the close-packed surface is noble whereasthe stepped surface is reactive.
The difference between the noble and reactive class can further-more be observed on the adsorption geometry of the C2H species.Fig. 4 illustrates the adsorption geometry of C2H on Ag{2 1 1},Au{2 1 1}, and Cu{2 1 1}. Here qualitatively different adsorptiongeometries are observed when moving through the metals from Agto Au and Cu. Again, there is ambiguity as to how one should classifythe borderline metals, but from inspection of the LDOS and the bondlengths the situation is clearly resolved, and in this particular caseAu{2 1 1} can be classified as reactive towards C2H.
On the basis of the above discussion we can draw up well-definedregions of reactive and noble metals. The situation is clearly not blackand white, and one would expect a continuous transition betweenthe two groups. This happens to occur approximately at the Cubinding energy.
4. Dehydrogenation of ethane
Using the above approach it is now possible to examine the gas-phase species and identify the bonds present in the fragment, andconsequently the internal bond orders required to determine theadsorbate–surface valence parameter g for a given molecule. As canbe seen from Fig. 2 there is excellent agreement between thepredicted slopes and the calculated energies from density functionaltheory. The mean absolute error (MAE) is found to be only 0.14 and0.15 eV for the CH3 and CH2 terminated fragments, respectively. Theone-line fit for C–CH step adsorption result in a slightly worse MAEfor the CH terminated fragments (0.28 eV). We note that for someintermediates the close-packed surface leads to stronger binding thanat the step. This non-obvious behavior is observed only for theintermediates that prefer a threefold binding site over the B5 step-site (five-fold coordinated site at the bottom of the step) for the firstcarbon (C–CH3, C–CH2, C–CH). The carbon descriptor is adsorbed at
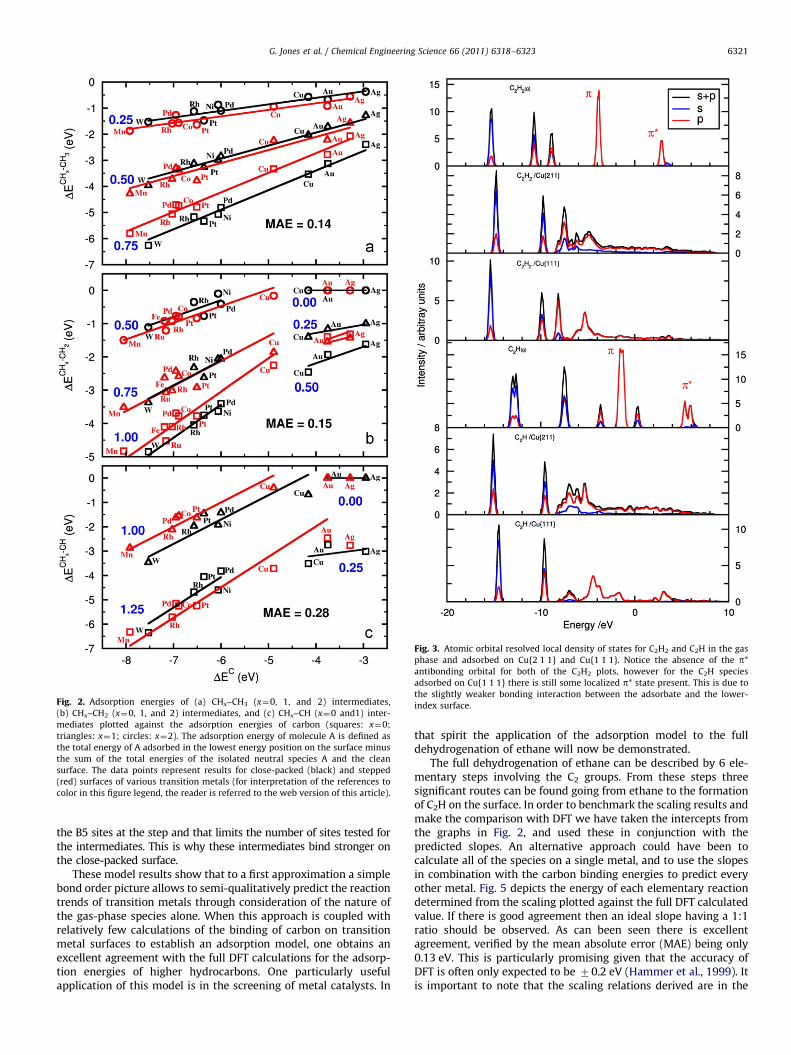
Fig. 3. Atomic orbital resolved local density of states for C2H2 and C2H in the gas
phase and adsorbed on Cu{2 1 1} and Cu{1 1 1}. Notice the absence of the p*
antibonding orbital for both of the C2H2 plots, however for the C2H species
adsorbed on Cu{1 1 1} there is still some localized p* state present. This is due to
the slightly weaker bonding interaction between the adsorbate and the lower-
index surface.Fig. 2. Adsorption energies of (a) CHx–CH3 (x¼0, 1, and 2) intermediates,
(b) CHx–CH2 (x¼0, 1, and 2) intermediates, and (c) CHx–CH (x¼0 and1) inter-
mediates plotted against the adsorption energies of carbon (squares: x¼0;
triangles: x¼1; circles: x¼2). The adsorption energy of molecule A is defined as
the total energy of A adsorbed in the lowest energy position on the surface minus
the sum of the total energies of the isolated neutral species A and the clean
surface. The data points represent results for close-packed (black) and stepped
(red) surfaces of various transition metals (for interpretation of the references to
color in this figure legend, the reader is referred to the web version of this article).
G. Jones et al. / Chemical Engineering Science 66 (2011) 6318–6323 6321
the B5 sites at the step and that limits the number of sites tested forthe intermediates. This is why these intermediates bind stronger onthe close-packed surface.
These model results show that to a first approximation a simplebond order picture allows to semi-qualitatively predict the reactiontrends of transition metals through consideration of the nature ofthe gas-phase species alone. When this approach is coupled withrelatively few calculations of the binding of carbon on transitionmetal surfaces to establish an adsorption model, one obtains anexcellent agreement with the full DFT calculations for the adsorp-tion energies of higher hydrocarbons. One particularly usefulapplication of this model is in the screening of metal catalysts. In
that spirit the application of the adsorption model to the fulldehydrogenation of ethane will now be demonstrated.
The full dehydrogenation of ethane can be described by 6 ele-mentary steps involving the C2 groups. From these steps threesignificant routes can be found going from ethane to the formationof C2H on the surface. In order to benchmark the scaling results andmake the comparison with DFT we have taken the intercepts fromthe graphs in Fig. 2, and used these in conjunction with thepredicted slopes. An alternative approach could have been tocalculate all of the species on a single metal, and to use the slopesin combination with the carbon binding energies to predict everyother metal. Fig. 5 depicts the energy of each elementary reactiondetermined from the scaling plotted against the full DFT calculatedvalue. If there is good agreement then an ideal slope having a 1:1ratio should be observed. As can been seen there is excellentagreement, verified by the mean absolute error (MAE) being only0.13 eV. This is particularly promising given that the accuracy ofDFT is often only expected to be 70.2 eV (Hammer et al., 1999). Itis important to note that the scaling relations derived are in the

Fig. 4. Atomic orbital resolved local density of states for C2H in the gas phase and adsorbed on the (2 1 1) surfaces of Ag, Au, and Cu. To the right the relaxed geometries for
this species are shown with the C–C bond length indicated in Angstrom. It is observed that C2H adsorbed on Cu and Au have the same binding motif and qualitatively the
same electronic structure, whereas on Ag it has an internal bond length closer to that of the gas-phase and a less delocalized electronic structure.
Fig. 5. Calculated reaction energies for a number of dehydrogenation reactions
(relevant for the full dehydrogenation of ethane) plotted against the model
predictions. The data points represent results for the close-packed (circles) and
stepped (triangles) surfaces on a number of transition metal surfaces. The model
data has been generated using the model gradients and calculated intercepts
from Fig. 2.
G. Jones et al. / Chemical Engineering Science 66 (2011) 6318–63236322
low coverage regime and that any adsorbate–adsorbate interac-tions will lead to shifts in the binding energies and hence thescaling relations. However, ethane dehydrogenation is run at fairlyhigh temperatures and a mean-field approach should therefore beadequate to describe the process. In this limit, the accuracy of theadsorption scaling model is within a useful tolerance-level, andsuggests that the model may be accurate enough to be used in thesearch for new catalysts (Studt et al., 2008).
5. Conclusions
We have shown in this work how the scaling relationshipspreviously identified for AHx species can be extended to the C2Hx
species. Whereas the adsorption of these species is clearly compli-cated by the presence of unsaturated bonds it is shown that the levelof internal saturation can be determined directly from an electronicstructure analysis which classifies surfaces into a reactive (p-bonddestroying) group and a noble group (on which the internal p-bondsremain). This enables the extension of the adsorption scaling modelfrom exclusively s-bonded adsorbates to general saturated as well asunsaturated hydrocarbons. The model has been verified to be inexcellent agreement with the energetics obtained from full DFTcalculations. Furthermore, the example of the dehydrogenation ofethane on both the {1 1 1} and {2 1 1} surfaces shows that theapproach can be generally applied to a full reaction scheme compris-ing a number of elementary steps.
The general applicability of this model is not just limited to the C2
species, it applies equally well to larger Cn species, provided thatsome knowledge of the adsorption geometries is available. Forexample, if one extends the saturated chain then the scaling willcontinue to follow the trends determined for the CHx species.Furthermore, the presented analysis of the adsorption of unsaturatedfragments defines the general scaling for transition metal bonding ofunsaturated species, and again if we substitute a hydrogen atom onthese species with a saturated chain the scaling is expected to persistwith unchanged slope. The intercept, however, might be different andwill need to be determined in each specific case. Some complicationsdo arise when multiple unsaturated bonds are present in the samemolecule and multiple bonding points to the surface are present. Theessential element, however, is still to determine the bond orderparameter of each surface-bonded atom from consideration of thegas-phase species alone; once this parameter is determined thecorresponding scaling relation can be predicted.
Acknowledgments
The authors wish to acknowledge support from the Centerfor Interface Science and Catalysis, funded by DOE, the Center for

G. Jones et al. / Chemical Engineering Science 66 (2011) 6318–6323 6323
Atomic-scale Materials Design, funded by the Lundbeck Founda-tion, the Catalysis for Sustainable Energy (CASE) initiative, fundedby the Danish Ministry of Science, Technology, and Innovation,the Danish Research Agency through Grant 26-04-0047, and theDanish Center for Scientific Computing.
References
Abild-Pedersen, F., Greeley, J., Studt, F., Rossmeisl, J., Munter, T.R., Moses, P.G.,Skulason, E., Bligaard, T., Nørskov, J.K., 2007. Phys. Rev. Lett. 99, 016105.
Bodke, A.S., Olschki, D.A., Schmidt, L.D., Ranzie, E., 1999. Science 285, 712.Dry, M.E., 2002. Catal. Today 71, 227.Fernandez, E.M., et al., 2008. Angew. Chem. Int. Ed. 47, 4683.Greeley, J., Mavrikakis, M., 2004. Nat. Mater. 3, 810.Horiuti, I., Polanyi, M., 1934. Trans. Faraday Soc. 30, 1164.Hammer, B., Nørskov, J.K., 2000. Adv. Catal. 45, 71.Hellman, A., et al., 2006. J. Phys. Chem. B 110, 17719.Hammer, B., Hansen, L.B., Nørskov, J.K., 1999. Phys. Rev. B 59, 7413.Jones, G., Bligaard, T., Abild-Pedersen, F., Nørskov, J.K., 2008. J. Phys.: Condens
Matter 20, 064239.
Kresse, G., Furthmuller, J., 1996. Comput. Mater. Sci. 6, 15.
Liu, Z.P., Hu, P., 2003. J. Am. Chem. Soc. 125, 1958.
Monkhorst, H.J., Pack, J.D., 1976. Phys. Rev. B 13, 5188.
Nørskov, J.K., Scheffler, M., Toulhoat, H., 2006. MRS Bull. 31, 669.
Nilsson, A., Pettersson, L.G.M., 2007. Adsorbate electronic structure and bondingon metal surfaces. In: Nilsson, A., Pettersson, L.G.M., Nørskov, J.K. (Eds.),Chemical Bonding at Surfaces and Interfaces. Elsevier (Chapter 2).
Rostrup-Nielsen, J.R., Sehested, J., Nørskov, J.K., 2002. Adv. Catal. 47, 65.
Somorjai, G.A., 1994. Introduction to Surface Chemistry and Catalysis. WileyNew York.
Shustorovich, E., Baetzold, R.C., 1985. Science 227, 876.
Shustorovich, E., 1986. Surf. Sci. 176, L863.
Shustorovich, E., 1988. Acc. Chem. Res. 21, 183.
Shustorovich, E., Bell, A.T., 1988. Surf. Sci. 205, 492.
Studt, F., Abild-Pedersen, F., Bligaard, T., Sørensen, R.Z., Christensen, C.H., Nørskov,J.K., 2008. Science 320, 1320.
The Dacapo plane-wave/pseudopotential code is available as open-sourcesoftware from the ‘‘CAMPOS’’ web site: /https://wiki.fysik.dtu.dk/dacapoS.
Vanderbilt, D., 1990. Phys. Rev. B 41, 7892.
van Santen, R.A., Neurock, M., 2006. Molecular Heterogeneous Catalysis—A Concep-tual and Computational Approach. Wiley-VCH, Weinheim.


















