
ROADMAP TO MARKET FOR CELL AND GENE THERAPIES
24
1 ROADMAP TO MARKET FOR CELL AND GENE THERAPIES 20 NOVEMBER 2020 PRESENTED BY EWAN CAMPBELL, KEVIN HENNEGAN, & RACHEL SMITH
Transcript of ROADMAP TO MARKET FOR CELL AND GENE THERAPIES

1
ROADMAP TO MARKET FOR CELL AND GENE THERAPIES
20 NOVEMBER 2020
PRESENTED BY EWAN CAMPBELL,KEVIN HENNEGAN, & RACHEL SMITH

2 VERISTAT.COMVERISTAT.COM
AGENDA
CELL & GENE THERAPIES LANDSCAPE
CONSIDERATIONS & CASE STUDIES FOR CELL & GENE TRIALS
• PLANNING
• EXECUTION
• SUBMISSION
• POST-MARKETING
OUR ROADMAP TO MARKET
QUESTIONS

3
CELL AND GENE THERAPY PRODUCTS
Tissue engineeringCells or tissues that are modified for repair, regeneration or replacement
Cell therapyThe injection or infusion of whole
cells into the patient
Gene therapyGene addition or gene editing in vivo, ex vivo or viral vector
CELL AND GENE THERAPIES REGENERATIVE MEDICINE
ADVANCED THERAPIES MEDICINAL PRODUCT (ATMP EU)REGENERATIVE MEDICINE ADVANCED THERAPY (RMAT US)

4 VERISTAT.COM
POLL QUESTION

5
Only a Handful of Cell & Gene Approvals in the Past Five Years
SMALL MOLECULE VS CELL & GENE THERAPY
1. https://www.ema.europa.eu/en/about-us/what-we-do/authorisation-medicines/medicine-evaluation-figures#section12. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products3. https://www.fda.gov/media/134493/download
65
81
90
79
93
1
3
2
2
2
0 20 40 60 80 100
2019
2018
2017
2016
2015
EMA Approvals1
ATIMP Non-ATIMP
• 2015: Imlygic, Holoclar• 2016: Zalmoxis & Strimvelis• 2017: Spherox & Alofisel• 2018: Kymriah, Yescarta & Luxturna• 2019: Zynteglo
• 2015: Imlygic• 2016: Clevecord, HPC Cord Blood (Bloodworks), Maci• 2017: Kymriah, Yescarta & Luxturna• 2018: HPC Cord Blood (MD Anderson)• 2019: Zolgensma
48
59
46
22
45
1
1
3
3
1
0 10 20 30 40 50 60 70
2019
2018
2017
2016
2015
FDA Approvals2,3
Cell/Gene Drug

6
Well Defined Route to Market
TRADITIONAL SMALL MOLECULE ROAD MAP
› Well defined requirements for in vitro / in vivo data
› Healthy Volunteers
› 10’s of subjects
› Patients
› 10’s to 100’s patients
› “Real-world”
› Patients
› 1000’s patients
PRE-CLINICAL
› Defined NDA/MAA pathways
› Accepted data requirements clear
› Real-world studies
› Global post-marketing safety
› Data-focused
PHASE I PHASE II PHASE IIIMARKETING APPROVAL
PHASE IV

7
ROADMAP FOR CELL AND GENE THERAPIES?
There isn’t one!
Every cell or gene therapy has its own unique challenges – welcome to the world of personalized medicine
No clear pre-clinical requirements
Unique indication challenges, especially in those for rare diseases including small patient numbers and definition of suitable endpoints
Poorly defined regulatory pathways and in some cases, no regulatory pathway for cell and gene therapies
Post-marketing commitments are more extensive than traditional therapies

8 VERISTAT.COM
KEY CONSIDERATIONS: PLANNING

9
COLLABORATION WITH GLOBAL REGULATORY AGENCIES
Global regulators are becoming more familiar with advanced therapies e.g. EMA and FDA
BUT many countries still do not have defined ATIMP CTA/IND pathways
First gene therapy to be approved in Europe
▪ Lesson learnt, early engagement and collaboration with regulatory agencies is key!
10
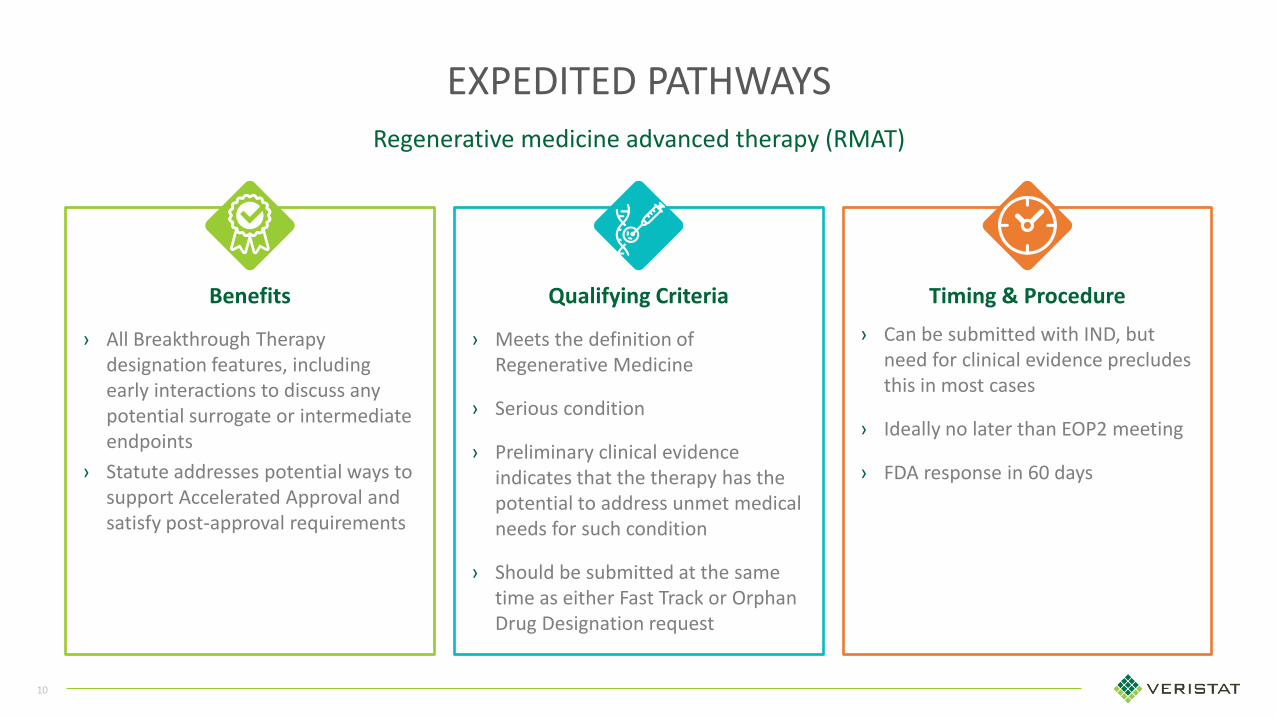
Benefits Qualifying Criteria Timing & Procedure
EXPEDITED PATHWAYS
› All Breakthrough Therapy designation features, including early interactions to discuss any potential surrogate or intermediate endpoints
› Statute addresses potential ways to support Accelerated Approval and satisfy post-approval requirements
› Meets the definition of Regenerative Medicine
› Serious condition
› Preliminary clinical evidence indicates that the therapy has the potential to address unmet medical needs for such condition
› Should be submitted at the same time as either Fast Track or Orphan Drug Designation request
› Can be submitted with IND, but need for clinical evidence precludes this in most cases
› Ideally no later than EOP2 meeting
› FDA response in 60 days
Regenerative medicine advanced therapy (RMAT)

11
REGULATORY CASE STUDIES
Case Study: CNS Disease Novel Gene Therapy Study
Collaborated with US FDA to refine nonclinical and clinical study designs to support initiation of US clinical trials
Case Study: Melanoma Biologic Fast Track Designation
Company received negative feedback from FDA to their preliminary Breakthrough Designation inquiry
Veristat supported a change in pathway, preparing an application for Fast Track, resulting in successfully obtaining designation
Case Study: Graft vs Host Disease Novel Cell Therapy Study
TGA had not approved a CTA for this type of cell therapy previously
Gained TGA buy-in to issue approval based on MHRA approval

12 VERISTAT.COM
KEY CONSIDERATIONS: EXECUTION

13
NOVEL CLINICAL TRIAL DESIGN
Natural History Arms
Natural history data are crucial for drug development, many advanced therapies target rare diseases with limited natural history data available
▪ Used to identify patient population, biomarkers, clinical outcomes/assessments and standard of care processes
Case Study: Orphan Disease Gene Therapy Program
Natural history arm introduced into Phase I/IIa trial, allowed collection of standard of care data in parallel to FIH use of a gene therapy
Separate natural history trial in US collecting historical data
Data used to refine Phase II/III trials and comparator dataset for BLA

14
NOVEL CLINICAL TRIAL DESIGN
Synthetic Control Arms
Using "Real World Data" as a control arm from disease registries, electronic health records, historical clinical trials, etc.
Case Study: Rare Cancer Study
Utilized historical databases from clinical sites as comparator arm
Standard of Care data from Standard of Care patients
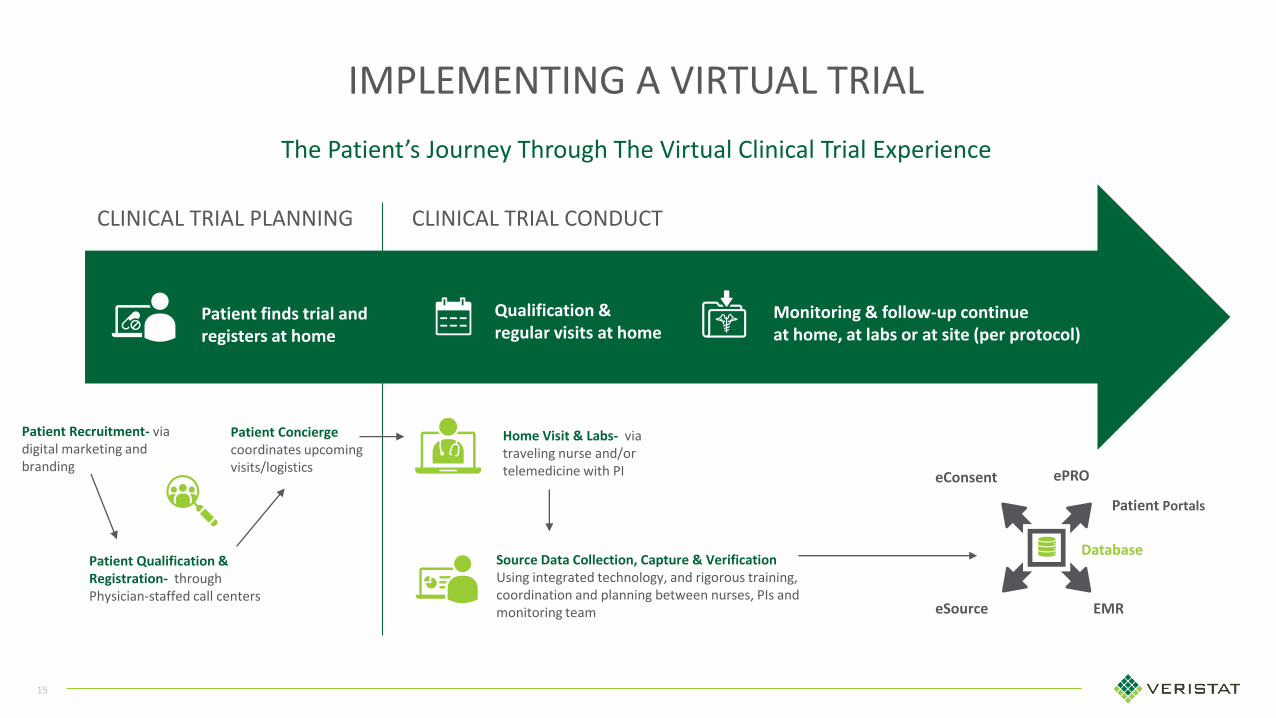
15
The Patient’s Journey Through The Virtual Clinical Trial Experience
IMPLEMENTING A VIRTUAL TRIAL
CLINICAL TRIAL PLANNING CLINICAL TRIAL CONDUCT
Patient Concierge coordinates upcoming visits/logistics
Qualification & regular visits at home
Patient finds trial and registers at home
Monitoring & follow-up continueat home, at labs or at site (per protocol)
Source Data Collection, Capture & VerificationUsing integrated technology, and rigorous training, coordination and planning between nurses, PIs and monitoring team
Patient Recruitment- via digital marketing and branding
Patient Qualification & Registration- through Physician-staffed call centers
Home Visit & Labs- via traveling nurse and/or telemedicine with PI
Database
EMReSource
ePRO
Patient Portals
eConsent

16
Allows patients to be treated at one or two central sites, minimizing the travel burden on patients and caregivers
CENTRAL SITE MODEL
Secure, controlled, and timely
delivery of study therapies
Electronic CRFs, study diaries,
and telemedicine visits
Conducted by mobile
health care providers
Local Healthcare Providers
Engagement with central site to act
as a data source, providing standard
of care as a non-study site
Couriers / Shipping
Secure, controlled, and timely
delivery of critical lab samples
In-Home Follow-up Visits
Conducted by mobile healthcare
providers
Information Technology
Electronic CRFs, study diaries, and
telemedicine visits

17
CASE STUDIES: OVERCOMING COMPLEX CHALLENGES & LOGISTICS
Case Study: Intracranial Injection of a Stem Cell Therapy in Stroke Patients
Patients were hesitant to sign up so soon after stroke. Media advertising and TV documentary exposure helped to raise the profile of the program to meet recruitment goals.
Stem cells arrived frozen with a 60-min expiration time after thaw, patients had to be under anesthetic and the surgeon to be ready. Pharmacy-Surgeon communications were critical. Dummy runs were essential in successful treatment.
Case Study: Commercialization of a Gene Therapy in Pediatric Patients
Stem cells were harvested from patients on the US West Coast, transported to Europe for manufacture within 48 hours & final product was sent back to the US. Training, dummy runs & exceptionally close communication between site, lab processing teams, couriers, GMP facility & Sponsors were crucial.
As manufacturing slots were bi-monthly, an airplane seat assignment strategy was employed to ensure slots were achieved
Chain of custody was unique and required a bespoke tracking & documentation for compliance with GCP for advanced therapies

18
LONG-TERM FOLLOW-UP
Long term effects of cell and gene therapy – we have more data now than ever, but still lacking long-term impact data
Regulatory requirement to follow patient safety long-term, generally up to 15 years
Long term data can be obtained in several ways using national registries, standard of care or formal LTFU studies

19 VERISTAT.COM
KEY CONSIDERATIONS: SUBMISSION & POST-MARKETING

20
KEY CONSIDERATIONS: SUBMISSION
Initial Marketing Applications (authoring, publishing, submission)
Additional Marketing Applications in other regions - e.g. Japan FDA (authoring, publishing, submission)
Support of inspection readiness activities in preparation for MA regulatory inspection

21
KEY CONSIDERATIONS: POST-MARKETING
Realizing a therapy’s full potential
Management of Phase IV studies
Ongoing support and/or management for long-term follow-up studies and post-marketing commitments
Pharmacovigilance and safety monitoring
Payer and reimbursement submissions
Post-marketing study commitments (PASS, REMS)

22
VERISTAT ROADMAP TO MARKET
Expertise in pre-clinical strategy
Pre-IND/Pre-CTA
▪ Advice Meetings with Regulators
o Pre-clinical data package review
o CMC Process
o Phase I Design
▪ Comprehensive Clinical Strategy – Work from Marketing Strategy backwards
Post-IND/CTA
▪ Maintain regulatory engagement
▪ Utilize novel trial designs & central site models
Post Marketing
▪ Regulatory commitments e.g. LTFU, additional analyses

23 VERISTAT.COM
QUESTIONS?

24
SPEAKER CONTACT INFORMATION
Rachel SmithProject Director
Veristat
Kevin HenneganSenior Regulatory Strategist
Veristat
Ewan CampbellAdvanced Therapy &
Biotech Director, Veristat


















