
Rhodium(I) carbonyl complexes of quinoline carboxylic acid: Synthesis, reactivity and catalytic...
5
Journal of Molecular Catalysis A: Chemical 372 (2013) 1–5 Contents lists available at SciVerse ScienceDirect Journal of Molecular Catalysis A: Chemical j our na l ho me p age: www.elsevier.com/locate/molcata Rhodium(I) carbonyl complexes of quinoline carboxylic acid: Synthesis, reactivity and catalytic carbonylation reaction Podma Pollov Sarmah, Dipak Kumar Dutta ∗ Materials Science Division, CSIR-North East Institute of Science and Technology, Jorhat 785006, Assam, India a r t i c l e i n f o Article history: Received 6 October 2012 Received in revised form 22 January 2013 Accepted 5 February 2013 Available online xxx Keywords: Rhodium Carbonyl ligand N-donor ligand Oxidative addition Carbonylation a b s t r a c t The dimeric rhodium precursor [Rh(CO) 2 Cl] 2 reacts with the ligands (L) quinoline-2-carboxylic acid (a) or quinoline-8-carboxylic acid (b) in 1:2 mole ratio to afford complexes of the type cis-[Rh(CO) 2 ClL] (1a and 1b). The complexes have been characterized by elemental analysis, Mass spectrometry, FT-IR and NMR ( 1 H, 13 C) spectroscopy. 1a and 1b undergo oxidative addition (OA) with different electrophiles such as CH 3 I, C 2 H 5 I and I 2 to give Rh(III) complexes of the type [Rh(CO)(COR)ClIL] {R = CH 3 (2a and 2b), R = C 2 H 5 (3a and 3b)} and [Rh(CO)ClI 2 L] (4a and 4b) respectively. OA of the CH 3 I with 1a forms relatively stable acyl intermediate which is evident from IR spectroscopy. The complexes 1a and 1b show higher catalytic activity for carbonylation of methanol to acetic acid and methyl acetate [Turn over number (TON) upto 1775 in 1 h] compared to that of the well known Monsanto’s species [Rh(CO) 2 I 2 ] − (TON = 1000 in 1 h) under the reaction conditions: temperature 130 ± 2 ◦ C, pressure 30 ± 2 bar, 450 rpm and 1 h reaction time. © 2013 Elsevier B.V. All rights reserved. 1. Introduction Acetic acid is one of the vital chemical required for preparation of lots of specialty and important chemicals [1–3]. Methanol is one of the cheap and readily available chemicals, which can be con- verted to acetic acid by carbonylation reaction. About 80% of the total acetic acid is produced annually by methanol carbonylation [4]. The well known Monsanto’s process is the most widely adopted industrial carbonylation technology used for acetic acid production. The Monsanto’s process is based on Rh promoted catalytic car- bonylation where [Rh(CO) 2 I 2 ] − is the active catalytic species and CH 3 I used as co-catalyst [5–7]. But this process requires drastic reaction conditions like high pressure and temperature. There- fore, considerable efforts have been made to improve the catalysts by incorporating different ligands into its coordination sphere for better activity compared to Monsanto’s species [8–14]. So far, phos- phorus containing ligands are most extensively studied, since they can stabilize low valent metal centre by both -bonding and -back bonding. Recently ligands containing N/N∼O donor atoms have also aroused considerable interest because of their structural novelty and catalytic activity [15–24]. N-atom is strong -donor, thereby it imparts more ionic character to metal-ligand bond and makes the metal centre more electron rich. Therefore, the metal centre ∗ Corresponding author. Tel.: +91 376 2370081; fax: +91 376 2370011. E-mail addresses: [email protected], dutta [email protected] (D.K. Dutta). becomes more susceptible to oxidative addition, which is the key step in carbonylation reaction [1,2,14]. Moreover, N-atom is not susceptible to aerial oxidation unlike P-atom and thus N-donor lig- ands may be advantageous over P-donor ligands. In case of N∼O ligands, the oxygen atom, being hard donor, confers stability to metal at high oxidation state in the oxidative addition reaction [15]. Thus, the different hardness and donor properties of N∼O ligands may offer advantages in catalysis. As a part of our continuing research activity [8,9,14–16,23–26], we have chosen two N∼O donor ligand, quinoline-2-carboxylic acid and quinoline-8-carboxylic acid for synthesis of rhodium carbonyl complexes. The reactivities of small molecules like CH 3 I, C 2 H 5 I, and I 2 towards the complexes were evaluated. The kinetics of OA of CH 3 I with the complexes was also carried out. The catalytic activity of the complexes has also been demonstrated in carbonylation of methanol for the production of acetic acid and methyl acetate. 2. Experimental 2.1. General information All operations were carried out under N 2 environment. All solvents were distilled under N 2 prior to use. RhCl 3 ·xH 2 O (Rh con- tent 40%) was purchased from M/S Arrora Matthey Ltd., Kolkata, India. Quinoline carboxylic acid ligands (98%) were purchased from M/S Aldrich, USA and used without further purification. Dichloromethane (99%), diethyl ether (99%), hexane (99%) and methanol (99.5%) were purchased from Ranbaxy fine chemicals 1381-1169/$ – see front matter © 2013 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.molcata.2013.02.005
-
Upload
dipak-kumar -
Category
Documents
-
view
216 -
download
2
Transcript of Rhodium(I) carbonyl complexes of quinoline carboxylic acid: Synthesis, reactivity and catalytic...

Rr
PM
a
ARRAA
KRCNOC
1
oovt[iTbCrfbbpcbaait
d
1h
Journal of Molecular Catalysis A: Chemical 372 (2013) 1– 5
Contents lists available at SciVerse ScienceDirect
Journal of Molecular Catalysis A: Chemical
j our na l ho me p age: www.elsev ier .com/ locate /molcata
hodium(I) carbonyl complexes of quinoline carboxylic acid: Synthesis,eactivity and catalytic carbonylation reaction
odma Pollov Sarmah, Dipak Kumar Dutta ∗
aterials Science Division, CSIR-North East Institute of Science and Technology, Jorhat 785006, Assam, India
r t i c l e i n f o
rticle history:eceived 6 October 2012eceived in revised form 22 January 2013ccepted 5 February 2013vailable online xxx
a b s t r a c t
The dimeric rhodium precursor [Rh(CO)2Cl]2 reacts with the ligands (L) quinoline-2-carboxylic acid (a)or quinoline-8-carboxylic acid (b) in 1:2 mole ratio to afford complexes of the type cis-[Rh(CO)2ClL](1a and 1b). The complexes have been characterized by elemental analysis, Mass spectrometry, FT-IRand NMR (1H, 13C) spectroscopy. 1a and 1b undergo oxidative addition (OA) with different electrophilessuch as CH3I, C2H5I and I2 to give Rh(III) complexes of the type [Rh(CO)(COR)ClIL] {R = CH3 (2a and 2b),
eywords:hodiumarbonyl ligand-donor ligandxidative additionarbonylation
R = C2H5 (3a and 3b)} and [Rh(CO)ClI2L] (4a and 4b) respectively. OA of the CH3I with 1a forms relativelystable acyl intermediate which is evident from IR spectroscopy. The complexes 1a and 1b show highercatalytic activity for carbonylation of methanol to acetic acid and methyl acetate [Turn over number (TON)upto 1775 in 1 h] compared to that of the well known Monsanto’s species [Rh(CO)2I2]− (TON = 1000 in1 h) under the reaction conditions: temperature 130 ± 2 ◦C, pressure 30 ± 2 bar, 450 rpm and 1 h reactiontime.
. Introduction
Acetic acid is one of the vital chemical required for preparationf lots of specialty and important chemicals [1–3]. Methanol is onef the cheap and readily available chemicals, which can be con-erted to acetic acid by carbonylation reaction. About 80% of theotal acetic acid is produced annually by methanol carbonylation4]. The well known Monsanto’s process is the most widely adoptedndustrial carbonylation technology used for acetic acid production.he Monsanto’s process is based on Rh promoted catalytic car-onylation where [Rh(CO)2I2]− is the active catalytic species andH3I used as co-catalyst [5–7]. But this process requires drasticeaction conditions like high pressure and temperature. There-ore, considerable efforts have been made to improve the catalystsy incorporating different ligands into its coordination sphere foretter activity compared to Monsanto’s species [8–14]. So far, phos-horus containing ligands are most extensively studied, since theyan stabilize low valent metal centre by both �-bonding and �-backonding. Recently ligands containing N/N∼O donor atoms have alsoroused considerable interest because of their structural novelty
nd catalytic activity [15–24]. N-atom is strong �-donor, therebyt imparts more ionic character to metal-ligand bond and makeshe metal centre more electron rich. Therefore, the metal centre∗ Corresponding author. Tel.: +91 376 2370081; fax: +91 376 2370011.E-mail addresses: [email protected],
utta [email protected] (D.K. Dutta).
381-1169/$ – see front matter © 2013 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.molcata.2013.02.005
© 2013 Elsevier B.V. All rights reserved.
becomes more susceptible to oxidative addition, which is the keystep in carbonylation reaction [1,2,14]. Moreover, N-atom is notsusceptible to aerial oxidation unlike P-atom and thus N-donor lig-ands may be advantageous over P-donor ligands. In case of N∼Oligands, the oxygen atom, being hard donor, confers stability tometal at high oxidation state in the oxidative addition reaction [15].Thus, the different hardness and donor properties of N∼O ligandsmay offer advantages in catalysis.
As a part of our continuing research activity [8,9,14–16,23–26],we have chosen two N∼O donor ligand, quinoline-2-carboxylic acidand quinoline-8-carboxylic acid for synthesis of rhodium carbonylcomplexes. The reactivities of small molecules like CH3I, C2H5I, andI2 towards the complexes were evaluated. The kinetics of OA ofCH3I with the complexes was also carried out. The catalytic activityof the complexes has also been demonstrated in carbonylation ofmethanol for the production of acetic acid and methyl acetate.
2. Experimental
2.1. General information
All operations were carried out under N2 environment. Allsolvents were distilled under N2 prior to use. RhCl3·xH2O (Rh con-tent 40%) was purchased from M/S Arrora Matthey Ltd., Kolkata,
India. Quinoline carboxylic acid ligands (98%) were purchasedfrom M/S Aldrich, USA and used without further purification.Dichloromethane (99%), diethyl ether (99%), hexane (99%) andmethanol (99.5%) were purchased from Ranbaxy fine chemicals
2 olecul
lMppA
md1
ispcsr
2
a
2L
dosudi
[ı9(((3
[7NP1
3
2R
dCrstoi
[(P(ı
P.P. Sarmah, D.K. Dutta / Journal of M
imited (India) and used after distilled using standard technique.ethyl iodide (99.5%), KBr (IR grade) and CHCl3 (IR grade) were
urchased from M/S Merck, Germany and used without furtherurification. Carbon monoxide gas (99.9%) was purchased fromlchemie gases and chemicals Pvt. Ltd. (India).
Elemental analyses were performed on a Perkin-Elmer 2400 ele-ental analyzer. IR spectra (4000–400 cm−1) were recorded in KBr
iscs and CHCl3 on a Shimadzu IRAffiniry-1 spectrophotometer. TheH and 13C NMR spectra were recorded at room temperature (r.t.)n CDCl3 solution on a Bruker DPX-300 Spectrometer and chemicalhifts were reported relative to SiMe4. Mass spectra of the com-lexes were recorded on ESQUIRE 3000 Mass Spectrometer. Thearbonylation reactions of methanol were carried out in a high pres-ure reactor (Parr-4592, USA) fitted with a pressure gauge and theeaction products were analyzed by GC (Chemito 8510, FID).
.2. Starting materials
[Rh(CO)2Cl]2 was prepared by passing CO gas over RhCl3·3H2Ot 100 ◦C in the presence of moisture [27].
.3. Synthesis of the complexes [Rh(CO)2ClL] (1a and 1b), = quinoline-2-carboxylic acid (a), quinoline-8-carboxylic acid (b)
About 0.257 mmol (100 mg) [Rh(CO)2Cl]2 was dissolved inichloromethane (10 cm3) and to this solution, 0.514 mmol (89 mg)f the appropriate ligand was added. The reaction mixture wastirred at r.t. for 30 min and the solvent was evaporated under vac-um. The dark purple compounds so obtained were washed withiethyl ether, recrystallised and stored over silica gel under vacuum
n a desiccator.Analytical data for 1a and 1b are as follows:1a: yield: 0.156 g, 83%. IR (KBr): 2085, 1999 [�(CO)], 1660
�( COOH)] cm−1. 1H NMR (300 MHz, CDCl3) data (ı in ppm): 7.91–8.28 (4H, m, Ph), ı 8.39 (H-3, d, JH–H = 8.4 Hz, Py), ı.32 (H-4, d, JH–H = 8.4 Hz, Py). 13C NMR (75 MHz, CDCl3) dataı in ppm): ı 125.7–149.3 (m, Ph, Py), ı 169.5 (COOH), ı 186.1CO, 1JRh–C = 67.7 Hz), ı 189 (CO, 1JRh–C = 64.3 Hz). C12H7NClO4Rh367.57): cald. C 39.18, H 1.90, N 3.81; found C 39.01, H 1.81, N.75. MS: m/z = 367.5 [M+].
1b: yield: 0.164 g, 87%. IR (KBr): 2073, 1985 [�(CO)], 1666�( COOH)] cm−1. 1H NMR (300 MHz, CDCl3) data (ı in ppm): ı.81–8.91 (5H, m, Py, Ph), ı 9.23 (H-2, d, JH–H = 7.6 Hz, Py). 13CMR (75 MHz, CDCl3) data (ı in ppm): ı 123.7–151.0 (m, Ph,y), ı 167.2 (COOH), ı 186.4 (CO, 1JRh–C = 64.7 Hz), ı 188.2 (CO,JRh–C = 62.3 Hz). C12H7NClO4Rh (367.57): cald. C 39.18, H 1.90, N.81; found C 38.96, H 1.82, N 3.71. MS: m/z = 367.7 [M+].
.4. Synthesis of [Rh(CO)(COR)ClIL] {R = CH3, (2a and 2b), = C2H5 (3a and 3b)}
[Rh(CO)2ClL] (50 mg) (1a and 1b) was dissolved inichloromethane (5 cm3) and each of RX (3 cm3) (RX = CH3I,2H5I) was added to it. The reaction mixture was then stirred at.t. for about 6–12 h to yield 2a, 2b and 3a, 3b. The color of theolution changed from yellowish red to dark reddish brown andhe solvent was evaporated under vacuum. The compounds sobtained were washed with diethyl ether and stored over silica geln a desiccator.
Analytical data for 2a, 2b, 3a and 3b are as follows:2a: yield: 0.046 g, 67%. IR (KBr): 2073 [�(CO)], 1726
�(acyl)] cm−1. 1H NMR (300 MHz, CDCl3) data (ı in ppm): ı 2.51
3H, s, CH3), ı 7.93–8.39 (4H, m, Ph), ı 8.69 (H-3, d, JH–H = 7.7 Hz,y), ı 9.64 (H-4, d, JH–H = 7.7 Hz, Py). 13C NMR (75 MHz, CDCl3) dataı in ppm): ı 45.0 (CH3), ı 123.9–151.3 (m, Ph, Py), ı 169.9 (COOH),189 (br, CO), ı 204.0 (br, COacyl).
ar Catalysis A: Chemical 372 (2013) 1– 5
2b: yield: 0.053 g, 76%. IR (KBr): 2065 [�(CO)], 1734[�(acyl)] cm−1. 1H NMR (300 MHz, CDCl3) data (ı in ppm): ı 2.43(3H, s, CH3), ı 7.85–8.93 (5H, m, Py, Ph), ı 9.36 (H-2, d, JH–H = 7.6 Hz,Py). 13C NMR (75 MHz, CDCl3) data (ı in ppm): ı 46.0 (CH3), ı129.8–155.0 (m, Ph, Py), ı 170.0 (COOH), ı 188 (br, CO), ı 207.6(br, COacyl).
3a: yield: 0.051 g, 72%. IR (KBr): 2076 [�(CO)], 1731[�(acyl)] cm−1. 1H NMR (300 MHz, CDCl3) data (ı in ppm): ı1.19 (3H, t, JH–H = 6.1 Hz, CH3), ı 2.87 (2H, q, JH–H = 6.1 Hz, CH2), ı7.89–8.37 (4H, m, Ph), ı 8.71 (H-3, d, JH–H = 7.9 Hz, Py), ı 9.59 (H-4,d, JH–H = 7.9 Hz, Py). 13C (75 MHz, CDCl3) data (ı in ppm): ı 22.1(CH3), ı 56.3 (CH2), ı 122.7–159.1 (m, Ph, Py), ı 167.5 (COOH), ı189 (br, CO), 205 (br, COacyl).
3b: yield: 0.049 g, 69%. IR (KBr): 2067 [�(CO)], 1739[�(acyl)] cm−1. 1H NMR (300 MHz, CDCl3) data (ı in ppm): ı 1.27(3H, t, JH–H = 6.3 Hz, CH3), ı 2.98 (2H, q, JH–H = 6.3 Hz, CH2), ı7.89–8.91 (5H, m, Ph), ı 9.28 (H-2, d, JH–H = 7.6 Hz, Py). (75 MHz,CDCl3) data (ı in ppm): ı 25.3 (CH3), ı 56.6 (CH2), ı 125.9–153.9(m, Ph, Py), ı 169.3(COOH), ı 191 (br, CO), ı 208 (br, COacyl).
2.5. Synthesis of [Rh(CO)ClI2L] (4a and 4b)
0.018 mmol (0.05 g) of [Rh(CO)2ClL] (1a and 1b) was dissolvedin dichloromethane (5 cm3). To that solution iodine was added(0.02 mmol, 0.025 g). The reaction mixture was stirred for 6 h togenerate 4a and 4b. The color of the solution changed from yel-lowish red to dark reddish brown and the solvent was evaporatedunder vacuum. Excess iodine was removed by washing several timewith hexane and stored over silica gel in a desiccator.
Analytical data for 4a and 4b are as follows:4a: yield: 0.048 g, 57%. IR (KBr): 2065 [�(CO)] cm−1. 1H NMR
(300 MHz, d6-DMSO) data (ı in ppm): ı 7.81–8.43 (4H, m, Ph), ı8.77(H-3, d, JH–H = 7.2 Hz, Py), ı 9.76 (H-4, d, JH–H = 7.2 Hz, Py). 13CNMR (75 MHz, d6-DMSO) data (ı in ppm): ı 123.7–155.3 (m, Ph,Py), ı 171.2 (COOH), ı 187 (br, CO).
4b: yield: 0.066 g, 81%. IR (KBr): 2068[�(CO)] cm−1. 1H NMR(300 MHz, d6-DMSO) data (ı in ppm): ı 7.81–8.96 (5H, m, Py, Ph),ı 9.29 (H-2, d, JH–H = 7.1 Hz, Py). 13C (75 MHz, d6-DMSO) data (ı inppm): ı 129.1–154.6 (m, Ph, Py), ı 168.7 (COOH), ı 189.2 (br, CO).
2.6. Kinetic experiments
The kinetic experiments of OA reaction of complexes 1a and1b with neat CH3I were monitored using FT-IR spectroscopy in asolution cell (NaCl windows, 1 mm path length). In order to obtainpseudo-first-order condition, excess of CH3I relative to metal com-plex was used. FT-IR spectra (4.0 cm−1 resolution) were scannedin the �(CO) region (2200–1650 cm−1) and saved at regular timeinterval using spectrum software. After completion of experiment,absorbance versus time data for the appropriate �(CO) frequencieswere extracted by subtracting the solvent spectrum and analyzedoff line using OriginPro 8 software. Kinetic measurements weremade by following the decay of lower frequency �(CO) band of thecomplexes 1a and 1b in the region 1980–2000 cm−1. The pseudofirst order rate constants were found from the gradient of the plotof ln(A0/At) versus time, where A0 is the initial absorbance and At
is the absorbance at time, t.
2.7. Carbonylation of methanol using complexes 1a and 1b ascatalyst precursors
CH3OH (0.099 mol, 4 cm3), CH3I (0.016 mol, 1 cm3), H2O
(0.055 mol, 1 cm3) and catalyst (0.0514 mmol) were taken into thereactor. The reactor was then purged with CO for about 5 min andthen pressurized with CO gas (5, 10 and 20 bar) at 25 ◦C. The car-bonylation reactions were carried out at 130 ± 2 ◦C for 1 h with CO
P.P. Sarmah, D.K. Dutta / Journal of Molecular Catalysis A: Chemical 372 (2013) 1– 5 3
RhCl3.xH2O
100oC,moisture
CO
L
2a, 2b 3a, 3b 4a, 4b
N COOH
a
L =
Rh
OC
OC L
Cl
Rh
OC
Cl L
I
COCH3
RhOC
Cl L
I
COC2H5
Rh
OC
I L
I
Cl
1a,1b
stir., 30 min,r.t
[Rh(CO)2Cl]2
N
COOH
guorc
3
3
db1sm1rrctt(1ncnciN
3
gm
2100 200 0 170 0
0.0
0.1
0.2
0.3
0.4
0.5
Ab
sorb
an
ce
Wavenu mber (cm-1)
2085
1999
2073
1726
2026
b
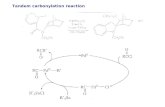
Scheme 1. Synthesis and reactivity of 1a and 1b.
as pressure 14 ± 2, 20 ± 2 and 33 ± 2 bar respectively. The prod-cts were collected and analyzed by GC. The quantitative amountsf the individual components are calculated from the peak area ofespective component and comparing with total peak area of all theomponents.
. Results and discussion
.1. Synthesis and characterization of 1a and 1b
The reaction of the chloro-bridged dimer [Rh(CO)2Cl]2 inichloromethane with two mol equivalents of the ligands, a and
affords the complexes of the type cis-[Rh(CO)2Cl(L)] (la andb) [where L = a and b] (Scheme 1). Elemental analysis and masspectrometric results of the complexes support the observedolecular composition of 1a and 1b. The IR spectra of 1a and
b exhibit two equally intense �(CO) vibrations band in theange 1985–2085 cm−1 indicating the formation of cis-dicarbonylhodium (I) complexes. The 1H NMR spectra of 1a and 1b showharacteristic multiple resonances for the phenylic and pyridyl pro-on in the range ı 7.81–9.32 ppm. In the 13C NMR spectra showwo characteristics doublets at ı 186.1 (1JRh–C = 67.7 Hz) and ı 189.01JRh–C = 64.3 Hz) ppm of 1a and, ı 186.4 (1JRh–C = 64.7 Hz) and ı88.2 (1JRh–C = 62.3 Hz) ppm of 1b for the carbonyl groups coordi-ated to Rh metal. The signals for phenylic, pyridyl and carboxylicarbons are found in the respective ranges. Moreover, no promi-ent shifting of the 13C NMR value of carboxylic group in 1a and 1bompared to the corresponding free ligand were observed, whichndicates that the ligand are coordinated to metal centre through-atom of the ligand only.
.2. Reactivity of 1a and 1b towards different electrophiles.
Activation of small molecules is one of the key steps in homo-eneous catalysis. In this respect, OA of alkyl halides with rhodiumetal complex is very important reaction step in the carbonylation
Fig. 1. Series of IR spectra illustrating the reaction of 1a with CH3I at 25 ◦C andgrowth of 2a.
reaction. Therefore, OA of various electrophiles like CH3I, C2H5I andI2 with 1a and 1b was evaluated.
The complexes 1a and 1b undergo OA with CH3I and C2H5Ito form Rh(III) complexes of the type [RhCl(CO)(COR)IL], whereR = CH3 (2a, 2b) and C2H5 (3a and 3b) (Scheme 1). On theother hand 1a and 1b reacts with I2 in CHCl3 to produce penta-coordinated Rh(III) complexes of the type [RhCl(CO)I2L] (Scheme 1).FT-IR spectra of the oxidative adducts of 1a and 1b show a sin-gle band in the range 2065–2076 cm−1, characteristic of terminalmono-carbonyl group. Moreover, a broad IR band in the range1726–1739 cm−1 for 2a, 2b and 3a, 3b indicates the formation ofacyl group. The single high value of the terminal �(CO) band indi-cates the formation of the oxidized products.
Apart from the characteristics resonances for 2a, 2b and 3a, 3bthe 1H NMR, resonance in the range ı 2.51–2.98 ppm indicates theformation of acyl group. 13C NMR for 2a, 2b and 3a, 3b show sin-glet peak in the range ı 189–191 and a broad peak in the range ı204–208 ppm for terminal carbonyl group and acyl group respec-tively apart from other characteristic resonances. The 1H and 13CNMR of 4a and 4b show characteristic resonance of the phenylicand pyridyl group at slightly downfield compared to the parentcompounds 1a and 1b. In addition, 13C NMR spectra also show char-acteristic resonances in the range ı 187–189 ppm attributable tothe presence of terminal CO group.
Attempts to substantiate the structure of different rhodium(I)and rhodium(III) complexes by single crystal X-ray crystallographywas not possible because, no suitable crystal could be obtained evenafter several attempts.
3.3. The kinetic study of OA reaction of CH3I with 1a and 1b
The kinetic experiments of OA of neat CH3I with complexes 1aand 1b were monitored using FT-IR spectroscopy by monitoringthe decay of the lower �(CO) band of the complexes. Fig. 1 shows atypical series of spectra of 1a during the reaction with CH3I at 25 ◦C,in which the bands at around 1999 and 2085 cm−1 decay and newbands grow at 2073 and a broad band 1720–1745 cm−1 until theequilibrium is attained. Finally, the two terminal �(CO) bands arereplaced by the terminal �(CO) band at 2072 cm−1 and acyl �(CO)band at 1737 cm−1. Absorbance versus time plots for the decay oflower intensity �(CO) bands at 1999 and 1985 cm−1 of 1a and 1b
respectively are shown in Fig. 2. A linear fit of pseudo-first orderwas observed for the entire course of the reaction of CH3I withthe complexes 1a and 1b as is evidenced from the plot of ln(A0/At)versus time, where A0 and At are the absorbance at time t = 0 and t,
4 P.P. Sarmah, D.K. Dutta / Journal of Molecular Catalysis A: Chemical 372 (2013) 1– 5
0 50 0 100 0 150 0 200 0
0.04
0.08
0.12
0.16
0.20
0.24
Ab
sorb
an
ce
Time (sec)
1a
0 50 0 100 0 150 0
0.00
0.01
0.02
0.03
0.04
0.05
0.06
Ab
sorb
an
ce
Tim e (sec )
1b
Fig. 2. Kinetic plot showing the decay of �(CO) bands with time of 1a and 1b during the reaction with neat CH3I at r.t. (∼25 ◦C).
0 50 0 100 0 150 0 200 0
0.0
0.5
1.0
1.5
2.0
ln (
Ao
/At)
Time (sec)
1a
0 50 0 100 0 150 0 200 0
0.0
0.5
1.0
1.5
2.0
2.5
ln (
Ao
/At)
1b
Fig. 3. Plot of ln(A0/At) versus time for the OA reaction of t
Se
rwtc2orto[dd
3
lTa
cheme 2. Carbonylation of methanol to acetic acid and methyl acetate in the pres-nce of catalyst.
espectively (Fig. 3). From the slopes of the plots, the rate constantsere calculated and found as 1.052 × 10−5 and 2.567 × 10−5 s−1 for
he complexes 1a and 1b respectively. The observed values of rateonstants indicate that the rate of OA is higher in case of 1b (about.5 times) than 1a. This may be due to the some short of interactionf carboxylic acid group with the Rh centre which may stericallyestrict the OA of CH3I group with Rh centre [26]. It is interestingo observe additional �(CO) band around 2026 cm−1 after additionf CH3I to 1a, which is assigned to intermediate dicarbonyl speciesRh(CO)2ClIL(CH3)] formed during first step of OA [9,28]. The bandepletes rapidly with the progress of the reaction time and finallyisappears.
.4. Catalytic carbonylation of methanol by 1a and 1b
The catalytic activity of 1a and 1b were evaluated in carbony-ation of methanol to acetic acid and methyl acetate (Scheme 2).he results are shown in Table 1. The precursor complexes show
total conversion of CH3OH in the range 45.1–92.5% at 130 ± 2 ◦C
Tim e (sec )
he complex 1a and 1b with neat CH3I at r.t (∼25 ◦C).
under CO pressure of 14 ± 2 to 33 ± 2 bar for 1 h reaction time withcorresponding TON in the range 866–1768. It has been observedfor 1a, that increase in initial CO pressure from 5 bar to 20 barenhance the conversion from 45.1% to 92.1% with correspondingTON from 866 to 1768. Similar trend has also been observed for 1b.Under the similar reaction conditions, the well known Monsanto’sspecies [Rh(CO)2I2]− shows a total conversion in the range 20.1%to 52.1% (14 ± 2 to 33 ± 2 bar CO pressure at 130 ± 2 ◦C) with corre-sponding TON of 463 to 1000 in 1 h. This indicates that the catalyticefficiency of the complexes is greatly enhanced by the incorpora-tion of the ligands (L) into the coordination sphere of the rhodiumcentre. Again, in our earlier work [15], the rhodium carbonyl com-plex with unsubstituted quinoline showed a maximum TON of 1711under the similar experimental condition. Thus, substitution of car-boxylic acid group in the quinoline ring has increases the catalyticefficiency of the complexes. It is interesting to observe here thatthe both complexes show comparable catalytic efficacy though lessreactivity of complex 1a was observed than 1b during OA of CH3I.This is probably due the drastic reaction condition leading to min-imize the steric restriction imparted by ligand during OA of CH3Igroup with Rh centre of the complex 1a. The formation of methyl
acetate as one of the products (Table 1) is due to the condensa-tion reaction between methanol and acetic acid which is formedduring reaction, and also the acetic acid catalyze simultaneouslythe condensation reaction [29]. The significant higher conversion
P.P. Sarmah, D.K. Dutta / Journal of Molecular Catalysis A: Chemical 372 (2013) 1– 5 5
Table 1Results of carbonylation reaction of methanol to acetic acid and methyl acetate.
Catalyst precursor CO pressure at 25 ◦C (bar) CO pressure at 130 ± 2 ◦C (bar) Total conversion (%) Acetic acid (%)a Methyl acetate (%)a TONb
[Rh(CO)2I2]c 5 14 ± 2 24.1 4 20.1 46310 20 ± 2 41.4 12.3 29.1 79620 33 ± 2 52.1 10.3 41.8 1000
1a 5 14 ± 2 45.1 10.9 34.2 86610 20 ± 2 58.2 20.8 37.4 111720 33 ± 2 92.1 9.8 82.3 1768
1b 5 14 ± 2 47.6 12.2 35.4 91410 20 ± 2 59.4 24.1 35.3 116020 33 ± 2 92.5 18.9 73.6 1775
tpglalw
4
al[[sat1(
A
IfShS3fif
R
[
[
[[
[[
[
[
[[[
[
[[
[
[
[
a Yield of methyl acetate and acetic acid were obtained from GC analyses.b TON = [amount of product (mol)]/[amount of catalyst (Rh mol)].c Formed from added [Rh(CO)2Cl]2 under catalytic condition.
o methyl acetate at 20 bar CO pressure in case of catalyst 1a com-ared to 1b may be due to the chelation tendency of the COOHroup of the ligand a in the catalytic reaction process, which isikely to increase the acidity of carboxylic acid group than in 1b andccordingly enhance the rate of ester formation. The organometal-ic residue of 1a and 1b were recovered after the first catalytic run,
hich were identified as rhodium(III) acyl complexes.
. Conclusion
Two new complexes 1a and 1b have been synthesized and char-cterized. The complexes undergo OA with different electrophilesike CH3I, C2H5I and I2 to afford Rh(III) complexes of the typeRh(CO)(COR)ClIL] [R = CH3 (2a and 2b), R = C2H5 (3a and 3b)] andRh(CO)ClI2L] (4a and 4b). OA of CH3I with 1a formed relativelytable acyl intermediate substantiated by IR spectroscopy. The cat-lytic activities of 1a and 1b for the carbonylation of methanolo acetic acid and its ester exhibit a higher TON (upto 1775) in
h compared to the well known Monsanto’s species [Rh(CO)2I2]−
TON = 1000) under the similar experimental condition.
cknowledgments
The authors are grateful to Dr. P. G. Rao, Director, CSIR-North Eastnstitute of Science and Technology, Jorhat-785006, Assam, Indiaor his kind permission to publish the work. The authors thank Dr P.engupta, Head, Materials Science Division, CSIR-NEIST, Jorhat, foris constant encouragement. Thanks are also due to Department ofcience and Technology (DST), New Delhi (Project File No. SR/S1/IC-4/2011) and CSIR, New Delhi [Project No. MLP-6000/1] for thenancial support. The author P.P. Sarmah thanks to CSIR, New Delhi
or providing the Senior Research Fellowship.
eferences
[1] V.H. Agreda, J.R. Zoellar (Eds.), Acetic Acid and its Derivatives, CRC Press, NewYork, 1992.
[[
[
[2] P.M. Maitlis, A. Haynes, G.J. Sunley, M.J. Howard, J. Chem. Soc. Dalton Trans.(1996) 2187–2196.
[3] C.M. Thomas, G. Süss-Fink, Coord. Chem. Rev. 243 (2003) 125–142.[4] A. Haynes, P.M. Maitlis, G.E. Morris, G.J. Sunley, H. Adams, P.W. Badger, C.M.
Bowers, D.B. Cook, P.I.P. Elliott, T. Ghaffar, H. Green, T.R. Griffin, M. Payne,J.M. Pearson, M.J. Taylor, P.W. Vickers, R.J. Watt, J. Am. Chem. Soc. 126 (2004)2847–2861.
[5] D. Foster, T.C. Singleton, J. Mol. Catal. 17 (1982) 299–314.[6] F.E. Paulik, J.F. Roth, Chem. Commun. (1968) 1578.[7] D. Foster, J. Am. Chem. Soc. 98 (1976) 846–848.[8] D.K. Dutta, J.D. Woollins, A.M.Z. Slawin, D. Konwar, P. Das, M. Sharma, P. Bhat-
tacharyya, S.M. Aucott, Dalton Trans. (2003) 2674–2679.[9] D.K. Dutta, B. Deb, G. Hua, J.D. Woolins, J. Mol. Cat. A: Chem. 353/354 (2012)
7–12.10] G. Lamb, M. Clarke, A.M.Z. Slawin, B. Williams, L. Key, Dalton Trans. (2007)
5582–5589.11] C.M. Thomas, R. Mafua, B. Therrien, E. Rusanov, H.S. Evans, G. Süss-Fink, Chem.
Eur. J. 8 (2002) 3343–3352.12] K.K. Robinson, A. Hershman, J.H. Craddock, J.F. Roth, J. Catal. 27 (1972) 369–389.13] F.E. Paulik, A. Hershman, W.R. Knox, J.F. Roth, Monsanto Company, US Patent
3769329, (1973).14] D.K. Dutta, B. Deb, Coord. Chem. Rev. 255 (2011) 1686–1712.15] P.P. Sarmah, B. Deb, B.J. Borah, A.L. Fullar, A.M.Z. Slawin, J.D. Woollins, D.K.
Dutta, J. Organomet. Chem. 695 (2010) 2603–2608.16] D.K. Dutta, J.D. Woollins, A.M.Z. Slawin, A.L. Fuller, B. Deb, P.P. Sarmah, M.G.
Pathak, D. Konwar, J. Mol. Catal. A: Chem. 313 (2009) 100–106.17] E. Eduardo, M. Angles, A. Huet, A.C. Francisco, J.L. Farnando, L.A. Oro, Inorg.
Chem. 39 (2000) 4868–4878.18] J.G. Haasnoot, Coord. Chem. Rev. 200–202 (2000) 131–185.19] M.H. Klingele, S. Brooker, Coord. Chem. Rev. 241 (2003) 119–132.20] P.R. Ellis, J.M. Pearson, A. Haynes, H. Adams, N.A. Bailey, P.M. Maitlis,
Organometallics 13 (1994) 3215–3226.21] T.R. Griffin, D.B. Cook, A. Haynes, J.M. Pearson, D. Monti, G.E. Morris, J. Am.
Chem. Soc. 118 (1996) 3029–3030.22] A.J. Canty, Acc. Chem. Res. 25 (1992) 83–90.23] B.J. Sarmah, B.J. Borah, B. Deb, D.K. Dutta, J. Mol. Catal. A: Chem. 289 (2008)
95–98.24] B.J. Borah, B. Deb, P.P. Sarmah, D.K. Dutta, J. Mol. Catal. A: Chem. 319 (2010)
66–70.25] B.J. Borah, B. Deb, P.P. Sarmah, K. Saikia, P.P. Khound, D.K. Dutta, Inorg. Chim.
Acta 370 (2011) 117–121.26] M. Sharma, N. Kumari, P. Das, P. Chutia, D.K. Dutta, J. Mol. Catal. A: Chem. 188
(2002) 25–35.27] J.A. McCleverty, G. Wilkinson, Inorg. Synth. 8 (1966) 211–214.28] H.C. Martin, N.H. James, J. Aitken, J.A. Gaunt, H. Adams, A. Haynes,
Organometallics 22 (2003) 4451–4458.29] T. Pöpken, L. Götze, J. Gmehling, Ind. Eng. Chem. Res. 39 (2000) 2601–2611.










![Synthesis of highly functionalized benzo[h]quinoline and ...shodhganga.inflibnet.ac.in/bitstream/10603/39020/17/17...quinoline ) (16 ) and tetracyclic quinoline (3-(epimin omethano)](https://static.fdocuments.in/doc/165x107/606a70077d4f6141007ad728/synthesis-of-highly-functionalized-benzohquinoline-and-quinoline-16.jpg)






