
Review: CMV Escapes! - Annals of Clinical & Laboratory Science
8
123 Address correspondence to M. Kent Froberg, DVM, MD, Department of Pathology and Laboratory Medicine, University of Minnesota–Duluth School of Medicine, 1035 University Drive, Duluth, MN 55812, USA; tel 218 726 7223; fax 218 726 7559; e-mail [email protected]. Review: CMV Escapes! M. Kent Froberg Department of Pathology and Laboratory Medicine, School of Medicine, University of Minnesota–Duluth, Duluth, Minnesota Abstract. Cytomegalovirus (CMV) is an opportunistic pathogen that establishes life-long latent infection without clinical disease in immunocompetent individuals, but can cause severe illness in newborns, transplant recipients, and patients with HIV. CMV has evolved complex molecular mechanisms to avoid host immune detection and destruction. Collectively these mechanisms have been termed “immunoevasion” or “escapology.” Perhaps the most essential mechanism of virus survival within the host is latency, a form of reversible nonproductive infection of host cells by replication-competent virus. During periods of active virus replication, however, there are multiple strategies by which CMV evades host defenses. These include methods referred to as camouflage, which aid the virus in hiding from immune defenses, and those referred to as sabotage, whereby the virus disrupts or manipulates host inflammatory or immune responses. The ultimate pathogen survival strategy, host cell transformation, has been demonstrated in vitro for CMV, but to date has not been demonstrated in vivo. This review surveys the current literature on CMV immunoevasion and suggests a paradigm whereby CMV survives host defenses and contributes to atherogenesis. (received 20 November 2003; accepted 28 November 2003) Keywords: cytomegalovirus, escapology, immunoevasion, cytokines, atherosclerosis Introduction Cytomegalovirus is a ubiquitous beta-herpesvirus which causes most adult persons to become sero- positive by the age of 35 yr [1]. Most CMV infect- ons are mild or asymptomatic. However, like other herpes viruses, CMV establishes life-long latent infection with the potential of causing clinical disease following reactivation. Acute clinical illness is most often seen in neonates, solid organ or bone marrow transplant patients, or patients with AIDS. While the extremes of infection, latent virus without clinical disease vs life threatening disease in immuno- compromised patients, are well recognized, the in vivo battle where neither virus or host appears to win, is less clearly defined. There is substantial evidence that CMV infection plays a role in atherosclerosis, restenosis after angioplasty, and the de novo atherosclerosis that may arise following heart transplantation [2-5]. Since atherosclerosis is a chronic inflammatory disease of blood vessel walls, and latent CMV infection occurs in tissues at these sites, one may reason that reactivation of the virus could contribute to the chronic inflammatory process and exacerbate or accelerate vascular lesions. Understanding how CMV survives in these tissues and the mechanisms of reactivation and immunoevasion could establish the extent to which CMV contributes to atherogenesis and could lead to new methods to suppress the reactivation of CMV and its accom- panying inflammatory cascade. CMV has a large genome compared to many viruses and appears to have acquired several host genes over a long period of co-evolution with its host [6-9]. Many of these genes allow CMV to modulate both the host immune response and virus Annals of Clinical & Laboratory Science, vol. 34, no. 2, 2004 0091-7370/04/0200-0123 $2.00. © 2004 by the Association of Clinical Scientists, Inc.
Transcript of Review: CMV Escapes! - Annals of Clinical & Laboratory Science

123
Address correspondence to M. Kent Froberg, DVM, MD,Department of Pathology and Laboratory Medicine, Universityof Minnesota–Duluth School of Medicine, 1035 UniversityDrive, Duluth, MN 55812, USA; tel 218 726 7223; fax 218726 7559; e-mail [email protected].
Review: CMV Escapes!
M. Kent FrobergDepartment of Pathology and Laboratory Medicine, School of Medicine, University of Minnesota–Duluth,Duluth, Minnesota
Abstract. Cytomegalovirus (CMV) is an opportunistic pathogen that establishes life-long latent infectionwithout clinical disease in immunocompetent individuals, but can cause severe illness in newborns, transplantrecipients, and patients with HIV. CMV has evolved complex molecular mechanisms to avoid host immunedetection and destruction. Collectively these mechanisms have been termed “immunoevasion” or“escapology.” Perhaps the most essential mechanism of virus survival within the host is latency, a form ofreversible nonproductive infection of host cells by replication-competent virus. During periods of activevirus replication, however, there are multiple strategies by which CMV evades host defenses. These includemethods referred to as camouflage, which aid the virus in hiding from immune defenses, and those referredto as sabotage, whereby the virus disrupts or manipulates host inflammatory or immune responses. Theultimate pathogen survival strategy, host cell transformation, has been demonstrated in vitro for CMV, butto date has not been demonstrated in vivo. This review surveys the current literature on CMV immunoevasionand suggests a paradigm whereby CMV survives host defenses and contributes to atherogenesis. (received 20
November 2003; accepted 28 November 2003)
Keywords: cytomegalovirus, escapology, immunoevasion, cytokines, atherosclerosis
Introduction
Cytomegalovirus is a ubiquitous beta-herpesviruswhich causes most adult persons to become sero-positive by the age of 35 yr [1]. Most CMV infect-ons are mild or asymptomatic. However, like otherherpes viruses, CMV establishes life-long latentinfection with the potential of causing clinical diseasefollowing reactivation. Acute clinical illness is mostoften seen in neonates, solid organ or bone marrowtransplant patients, or patients with AIDS. Whilethe extremes of infection, latent virus withoutclinical disease vs life threatening disease in immuno-compromised patients, are well recognized, the invivo battle where neither virus or host appears towin, is less clearly defined.
There is substantial evidence that CMVinfection plays a role in atherosclerosis, restenosisafter angioplasty, and the de novo atherosclerosis thatmay arise following heart transplantation [2-5].Since atherosclerosis is a chronic inflammatorydisease of blood vessel walls, and latent CMVinfection occurs in tissues at these sites, one mayreason that reactivation of the virus could contributeto the chronic inflammatory process and exacerbateor accelerate vascular lesions. Understanding howCMV survives in these tissues and the mechanismsof reactivation and immunoevasion could establishthe extent to which CMV contributes toatherogenesis and could lead to new methods tosuppress the reactivation of CMV and its accom-panying inflammatory cascade.
CMV has a large genome compared to manyviruses and appears to have acquired several hostgenes over a long period of co-evolution with itshost [6-9]. Many of these genes allow CMV tomodulate both the host immune response and virus
Annals of Clinical & Laboratory Science, vol. 34, no. 2, 2004
0091-7370/04/0200-0123 $2.00. © 2004 by the Association of Clinical Scientists, Inc.

124
replication in the face of host cell mitogenic orproinflammatory activation. Collectively these genesand proteins have been termed “immunoevasins”and the overall process termed viral “escapology” [7,8]. CMV avoids immune detection throughcamouflage, the art of hiding from host defenses, aswell as subverting the immune system by sabotage,the synthesis of molecules that disrupt or manipulatehost immune/inflammatory responses. Virallyproduced molecules involved in these processesinclude cytokine homologs (virokines), solublecytokine scavengers that sequester free chemokines,and cell–surface receptor homologs (viroceptors) [7].Most studies of CMV escapology have been cond-ucted on human CMV (hCMV) or murine CMV(mCMV) using in vitro techniques. In combination,these immunoevasive activities may allow CMV tocontribute to atherosclerotic disease by surviving theonslaught of host defenses and contributing tochronic inflammation during periods of reactivation.Herein, we review the numerous and complex waysin which CMV interacts with host defenses and weattempt to formulate a hypothesis to define CMV’srole in atherogenesis.
Primary infection/latency
Like other herpes viruses, CMV establishes life-longlatent infection once the host is exposed [10,11].Replication is controlled by three groups of genestermed immediate early (IE), early (E), and late (L)phase genes. IE genes are under the control of themajor IE promoter (MIEP), which controls theexpression of two genetic elements IE-1 and IE-2.IE genes, in turn, regulate expression of E and Lgenes. MIEP is a potent transactivator of severalviral and host genes and along with IE genes appearsto play an important role in viral pathogenesis [12].IE gene products appear to be particularly importantin the proinflammatory response seen in CMVinfection and may (a) increase levels of adhesionmolecules such as ICAM, (b) increase productionof cytokines such as IL-1 and TNF-α; (c) upregulateseveral chemokines like MCP-1, MIP-1β, RANTES,and IL-8; and (d) stimulate expression of growthfactors and mitogens including IL-6, TGF-β andGM-CSF [13-16].
Control of latency is defined as the reversiblenon-productive infection of cells by replicative-competent virus and probably presents the best wayfor a virus to camouflage itself from immunedetection. It implies that either no or relatively fewviral genes are expressed and that no virions areproduced. CMV may infect and be transferredbetween monocytes, endothelial cells, and smoothmuscle cells, which are all central to atherogenesis.These cells may be activated by CMV infection andproduce proinflammatory cytokines and chemo-kines that attract inflammatory cells to sites ofvascular injury, adhesion molecules that enhancebinding of inflammatory cells to the vessel wall,mitogens that may stimulate replication of smoothmuscle cells or fibroblasts, and agents that stimulatelipid oxidation and free radical accumulation indamaged vascular walls [17-19]. The exact mechan-isms of latency and reactivation of CMV are notwell understood, but recent studies suggestreactivation may be more common than previouslybelieved and may occur with minor stress-relatedevents such as work overload, the occurrence of oralherpes, or alcohol ingestion [20]. These events,which are relatively common in many patients, couldprovide the CMV virus with multiple opportunitiesto contribute to the chronic inflammatory processwe know as atherogenesis.
Apoptosis/mitogenesis
A potential way by which CMV may prevent virionsfrom being exposed to host defenses would be toreduce apoptosis of CMV infected cells, thuslimiting the amount of extra-cellular virus releasedfrom these cells. Virally infected or damaged cellsmay undergo apoptosis as a host defense mechanismto maintain stable cell populations and perhaps limittissue injury or oncogenesis. Apoptotic cells arepresent in atherosclerotic lesions and may play asignificant role in restenosis following coronaryangioplasty [3,4,21]. Angioplasty induces excessivesmooth muscle cell proliferation, with rapidrestenosis occurring in 25–50% of patients under-going coronary angioplasty [4]. CMV appears toblock TNF-α induced apoptosis through the actionof IE1 and IE2 genes [22-25]. p53 is a nucleoprotein
Annals of Clinical & Laboratory Science

125
that directs cells to programmed cell death, and thehCMV protein IE2-84 binds to and inhibits p53transcriptional activity. p53 is sequestered withinthe cytoplasm of CMV-infected human endothelialcells in vitro [26]. By inhibiting apoptosis, CMVmay contribute to the excessive proliferation of SMCseen in restenosis.
CMV genes also induce proliferation of hostcells and cause transformation of several cell lines invitro. The MIEP of CMV transactivates NFκB,which in turn activates several host and viral genesenhancing viral replication and infectivity [27,28].Indeed, inflammation and activation of macrophagescan lead to upregulation of PGE2, TNF-α, and IL-1β, which can alone or synergistically upregulateMIEP in vitro [29,30]. Thus, it is possible thatactivation of macrophages can lead to reactivationof CMV from the latent state facilitating viralpathogenesis within atherosclerotic lesions. Otherfactors that appear to activate CMV include reactiveoxygen species (ROS) and nitric oxide (NO) [18,19,31]. ROS are generated during atherogenesis, andNO is produced by activated macrophages andendothelium, suggesting that renewal of inflam-mation within an atheromatous plaque couldreactivate latent virus within the lesion and escalatethe degree of tissue injury. These in vitro obser-vations are supported by data indicating cyclo-oxygenase inhibitors may reduce the thrombogenictendency of atheromatous lesions and may reduceCMV-induced production of ROS as well as CMVreplication in smooth muscle cells [32].
Blocking antigen presentation
While latency may be the ultimate method by whicha virus may survive within the host without exposureof viral antigens to host defenses, it has the distinctdisadvantage of preventing spread of the virus toother tissues or transmission to new hosts. CMV,in part, overcomes this by establishing latentinfection in most host tissues, but maintainingchronic persistent infection of salivary glands tofacilitate transmission to susceptible hosts. If hCMVis reactivated readily from relatively commonstresses, there may be frequent opportunities for thehost immune system to recognize and destroy the
virus. The CMV genome contains several openingreading frames (ORFs) that produce proteins thatblock or subvert viral antigen expression.
hCMV uses several strategies to inhibitpresentation of viral antigens in conjunction withMHC class I and MHC class II complexes. MHCclass I proteins are ubiquitously expressed andpresent antigen to CD8+ T lymphocytes, whileMHC II expression is limited to antigen presentingcells, B cells, and monocytes/macrophages, whichpresent antigen to CD4+ T lymphocytes. hCMVencodes several unique short (US) segments thatinhibit multiple steps of the antigen presentationpathway [33,34] (Fig. 1). US2 causes degradationof the MHC class I complex [34]. US6 inhibitspeptide translocation to the endoplasmic reticulumby the MHC-encoded TAP peptide transporter [35].US3 binds to MHC class I complexes and leads toretention of peptide-loaded complexes within theendoplasmic reticulum (ER) [36]. US11 dislocatesclass I complexes to the cytosol, where they arerapidly degraded [37]. Taken together, theseglycoproteins produce significant reduction of viralantigen presentation at the cell surface with MHCclass I complexes. This could potentially lead to anincrease in cell killing of CMV-infected cells bynatural killer (NK) cells, since these cells areinhibited from activation by the expression of MHCclass I antigens on the surface of host cells. hCMVmay overcome this problem by production of anMHC class I homolog, a product of the UL18 geneof hCMV, which can bind endogenous peptide, beexpressed on cell surfaces, and may protect virus-infected cells against NK cell cytotoxicity byengaging inhibitory receptors on the surface of NKcells [38-40]. Similar sequential disruption of MHCclass I processing has been demonstrated for mCMV[33,41,42].
CMV also interferes with antigen presentationby MHC class II complexes to CD 4+ lymphocytes.In vitro studies indicate that CMV inhibits MHCclass II expression through the Jak/Stat pathway bydecreasing levels of Janus kinase 1 [43]. The US2 ofhCMV destroys two essential proteins in the MHCclass II antigen presentation pathway [44]. MCMVinduces early host IL-10 production, which down-regulates MHC class II expression on macrophages
Cytomegalovirus escapology and immunoevasion
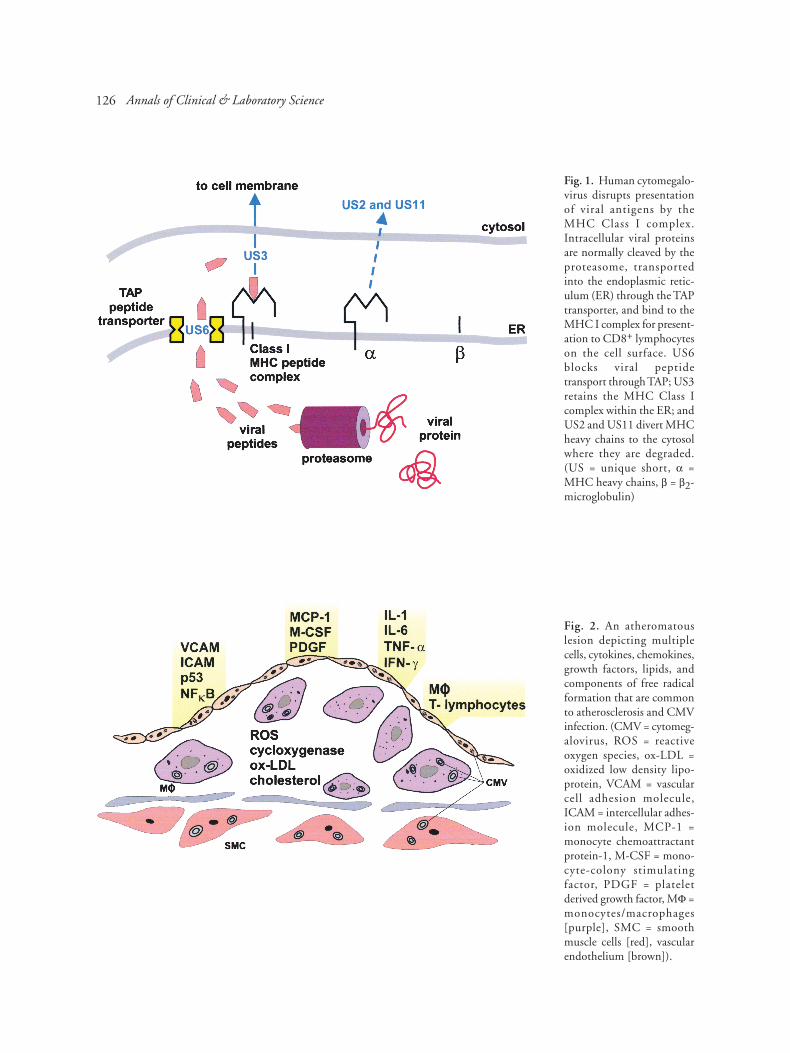
126
Fig. 1. Human cytomegalo-virus disrupts presentationof viral antigens by theMHC Class I complex.Intracellular viral proteinsare normally cleaved by theproteasome, transportedinto the endoplasmic retic-ulum (ER) through the TAPtransporter, and bind to theMHC I complex for present-ation to CD8+ lymphocyteson the cell surface. US6blocks viral peptidetransport through TAP; US3retains the MHC Class Icomplex within the ER; andUS2 and US11 divert MHCheavy chains to the cytosolwhere they are degraded.(US = unique short, α =MHC heavy chains, β = β2-microglobulin)
Fig. 2. An atheromatouslesion depicting multiplecells, cytokines, chemokines,growth factors, lipids, andcomponents of free radicalformation that are commonto atherosclerosis and CMVinfection. (CMV = cytomeg-alovirus, ROS = reactiveoxygen species, ox-LDL =oxidized low density lipo-protein, VCAM = vascularcell adhesion molecule,ICAM = intercellular adhes-ion molecule, MCP-1 =monocyte chemoattractantprotein-1, M-CSF = mono-cyte-colony stimulatingfactor, PDGF = plateletderived growth factor, MΦ =monocytes/macrophages[purple], SMC = smoothmuscle cells [red], vascularendothelium [brown]).
Annals of Clinical & Laboratory Science

127
[45,46]. In addition, CMV produces its own IL-10homolog, which may inhibit host production ofcytokines including TNF-α, IFN-γ, IL-2, andgranulocyte-monocyte colony stimulating factor[47]. This could lead to a TH1/TH2 switch, sinceIL-10 is known to be inhibitory to the TH1 pathway.This may be important for virus survival, becauseprotection against CMV, which, like many intra-cellular pathogens, depends more on cell-mediatedimmune mechanisms. Extracellular viral antigensmay be neutralized by specific antibody recognition;however, both hCMV and mCMV produce an Fcreceptor that binds IgG [48,49]. Other viruses andsome bacteria also produce Fc receptors that mayreduce the efficacy of neutralizing antibodies,complement fixation, or interfere with antibody-mediated immune cytotoxicity.
G Protein-coupled receptor homologs
Both murine and human CMV contain ORFs thatencode G protein-coupled receptor homologs,which bind host chemokines and may sequesterchemokines from the extracellular environment [50].HCMV contains UL33, US27, and US28, whichare all transcribed during CMV infection [51].Unlike wild type virus, US28-deleted virus failed todownregulate either of the chemokines RANTESor MCP-1 in the medium of cultured fibroblasts[50]. Moreover, US28 mediates vascular smoothmuscle cell migration in vitro [52], which is animportant facet of atherogenesis. An mCMV ORFtermed M33 encodes a G protein-coupled receptorwith homology to UL33 [53]. An M33-deletedMCMV grows normally in vitro, but shows atten-uated growth in vivo. MCP-1 is a C-C chemokineproduced in vascular lesions; it causes chemotaxisof monocytes and T lymphocytes, which are majorcells found in atheromatous lesions. mCMVinfection accelerates inflammation in vascular tissuethat overexpress MCP-1 [54]. The present authoris investigating whether wild type mCMV is moreeffective than an M33-depleted strain of mCMV inreducing serum chemokine levels in MCP-1transgenic mice. If so, it would indicate that M33may sequester chemokines in vivo and modulate thehost inflammatory response.
Lipid metabolism and CMV
Hyperlipidemia, especially of low density lipoprotein(LDL), is a risk factor for atherosclerosis. The earliesthistologically recognizable lesion of atherogenesisconsists of a few lipid laden macrophages (foam cells)beneath the vascular endothelium. Oxidative modif-ication of LDL activates endothelial cells, macro-phages, and smooth muscle cells, and hasproinflammatory, proatherogenic and prothrombo-genic effects. A possible connection between CMVand lipid metabolism was first recognized whenMarek’s Disease Virus (MDV), a herpesvirus, wasshown to induce atherosclerotic lesions in the aortasof chickens as well as to alter lipid metabolism andlead to accumulation of cholesterol and cholesterolester within these lesions [55]. hCMV also increaseslevels of neutral lipids in cultured human saphenousvein smooth muscle cells [56]. In CMV-infectedmonocytes, the presence of oxidized LDL andendothelial cells within the culture mediumincreased MIEP activity over seven-fold [57].hCMV infection of human aortic smooth musclecells in vitro stimulated the expression of scavengerreceptor mRNA and the uptake of oxidized andacetylated LDL [18]. We reported that CMV-seropositvity is associated with higher serumcholesterol levels in young females, although theclinical significance of this finding is unclear [58].
Conclusions
Cytomegalovirus infection has been linked toatherosclerosis through epidemiologic studies, thefinding of viral DNA within atheromatous lesions,and studies demonstrating increased developmentof atherosclerosis in animal models [13,59].Together, these studies provide strong circumstantialevidence that CMV may be a risk factor foratherosclerosis. However, a causal relationship hasnot been proven and it is possible that themicroenvironment of atheromatous lesions is simplymore suitable to CMV survival. Even if CMV doesnot initiate vascular lesions, it may be an importantcontributing factor to atherogenesis by acceleratingor exacerbating existing lesions. hCMV infectionhas been shown to produce in vitro many of the
Cytomegalovirus escapology and immunoevasion

128
inflammatory responses that are hallmarks ofatherosclerosis (Fig. 2). These responses are commonto many chronic inflammatory conditions, and arenot unique to either CMV infection or athero-sclerosis. Although CMV antigens and DNA canbe found in atheromatous plaques, other organisms,such as Chlamydia pneumoniae, may also be present[60]. However, these infections may be largely latentwith little or no inflammatory response attributableto the presence of the microorganism.
There is broad agreement that atherosclerosis isa chronic inflammatory response of the vascular wallto injury and may progress through multiple periodsof acute inflammation followed by relativequiescence [2]. Several important factors may allowCMV infection to contribute to formation of theatherogenic lesion: (a) reactivation from latencyappears to be a common event giving the virus ampleopportunity to contribute to atherogenesis; (b)CMV is often present in atheromatous plaques andvirus replication is induced by several factorsactivated during inflammatory episodes known tooccur during atherogenesis, again amplifying theopportunity for CMV to contribute to theinflammatory damage; (c) CMV may be capable ofcontributing to the lipid perturbations seen withinaffected vascular walls, and in particular may increasethe oxidation of LDL and affect cholesterolaccumulation (or conversely, CMV may simplythrive in the particular lipid microenvironmentfound in atheromatous lesions); (d) once activated,the IE genes of CMV in turn activate many pro-inflammatory host genes creating the probability ofa cyclic succession of inflammatory damage tovascular walls, no matter what the initiating factormay be; and (e) CMV dampens the inflammatoryassault by immunoevasive techniques that allowthe virus to survive in inflamed tissues untilreactivated by factors directly involving the arterialwall, or more distant factors that may elicit virusreactivation.
CMV infection or reactivation elicits and thendampens the inflammatory response. For example,IFN-γ and TNF-α are both upregulated inmonocytes following infection. However, viralinfection also induces differentiation of monocytesinto hCMV-permissive macrophages, which are
resistant to the antiviral effects of these cytokines[61], thus protecting viruses from the detrimentalaction of these cytokines. Despite recurrent orpersistent inflammation of the artery wall, CMVmay be able to survive within these vascular lesionsand persist through multiple episodes of reactivationand viral replication. In this manner CMV maycontribute to the chronic fibro-inflammatoryprogression of atheroma formation and vesselstenosis. Finding reactivated hCMV within earlyactive atheromatous lesions in young patients wouldsupport the concept that CMV could contribute toatherogenesis during lesion development. It wouldalso be useful to demonstrate virus reactivation inhumans or animal models in concert withexacerbations by other known risk factors, such asperiods of hypertension or hyperlipidemia.
It is likely that CMV infection or reactivationis simply one of many environmental factors thatmay contribute to atherogenesis. However, it maybe possible to control or prevent that contributionby controlling or eliminating the infection. If thatis the case, clinical improvement and prevention ofthe sequelae of atherosclerosis should be possiblewith adequate antiviral therapy.
References
1. Bruggeman CA. Cytomegalovirus and latency: anoverview. Virchows Archiv B Cell Pathol 1993;64:325-333.
2. Ross R. Atherosclerosis—an inflammatory disease. NEngl J Med 1999;340:115-126.
3. Hendrix MGR, Dorman PHJ, Kitslaar P, Bosman F,Bruggeman CA. The presence of cytomegalovirus nucleicacids in arterial walls of atherosclerotic and nonathero-sclerotic patients. Am J Pathol 1989;134:1151-1157.
4. Zhou YF, Leon MB, Waclawiw MA, Popma JJ, Yu ZX,Finkel T, Epstein SE. Association between prior cyto-megalovirus infection and the risk of restenosis aftercoronary atherectomy. N Engl J Med 1996;335:624-630.
5. Gratten MT, Moreno-Cabral CE, Starnes VA, Oyer PE,Stinson EB, Shumway NE. Cytomegalovirus infection isassociated with cardiac allograft rejection and athero-sclerosis. JAMA 1989;261:3561-3566.
6. Hengel H, Brune W, Koszinowski UH. Immune-evasionby cytomegalovirus—survival strategies of a highlyadapted opportunist. Trends Microbiol 1998;6;190-197.
7. Lucas M, Karrer U, Lucas A, Klenerman P. Viral escapemechanisms—escapology taught by viruses. Int J Exp Path2001;82:269-286.
Annals of Clinical & Laboratory Science

129
8. Reddehase MJ. Antigens and immunoevasins: opponentsin cytomegalovirus immune surveillance. Nature ReviewsImmunology 2002;2:831-844.
9. Michelson S. Human cytomegalovirus escape fromimmune detection. Intervirology 1999;42:301-307.
10. Jarvis MA, Nelson JA. Mechanisms of human cytomeg-alovirus persistence and latency. Frontiers Bioscience2002;7:1575-1582.
11. Sissons JGP, Bain M, Wills MR, Sinclair JH. Latencyand reactivation of human cytomegalovirus. J Infection2002;44:73-77.
12. Sweet C. The pathogenicity of cytomegalovirus. FEMSMicrobiol Rev 1999;23:457-482.
13. Iwamotom GK, Monick MM, Clark BD, Auron PE,Stinski MR, Hunninghake GW. Modulation of inter-leukin 1 beta gene expression by the immediate early genesof human cytomegalovirus. J Clin Invest 1990;85:1853-1857.
14. Yurochko AD, Huang E-S. Human cytomegalovirusbinding to human monocytes induces immunoregulatorygene expression. J Immunol 1999;162:4806-4816.
15. Cinatl J, Voegl J-U, Kotchetkov R, Scholz M, Doerr HW.Proinflammatory potential of cytomegalovirus infection.Intervirology 1999;42:419-424.
16. Geist LJ, Monick MM, Stinski MF, Hunninghake GW.The immediate early genes of human cytomegalovirusupregulate tumor necrosis factor-α gene expression. J ClinInvest 1994;93:474-478.
17. Speir E, Shibutani T, Yu Z-X, Ferrans V, Epstein SE. Roleof reactive oxygen intermediates in cytomegalovirus geneexpression and in the response of human smooth musclecells to viral infection. Circ Res 1996;79:1143-1152.
18. Zhou YF, Guetta E, Yu Z-X, Finkel T, Epstein SE. Humancytomegalovirus increases modified low density lipo-protein uptake and scavenger receptor mRNA expressionin vascular smooth muscle cells. J Clin Invest 1996;98:2129-2138.
19. Shibutani T, Johnson TM, Yu, Z-X, Ferrans VJ, Moss J,Epstein SE. Pertussis toxin-sensitive G proteins asmediators of the signal transduction pathways activatedby cytomegalovirus infection of smooth muscle cells. JClin Invest 1997;100:2054-2061.
20. Toro AI, Ossa J. PCR activity of CMV in healthy CMV-positive individuals: does latency need redefinition? ResVirol 1996;147:233-238.
21. Epstein SE, Speir E, Zhou YF, Guetta E, Leon M, FinkelT. The role of infection in restenosis and atherosclerosis:focus on cytomegalovirus. Lancet 1996;348:s13-s17.
22. Zhu H, Shen Y, Shenk T. Human cytomegalovirus IE1and IE2 proteins block apoptosis. J Virol 1995;69:7960-7970.
23. Speir E, Modali R, Huang E-S, Leon MB, Shawl F, FinkelT, Epstein SE. Potential role of human cytomegalovirusand p53 interaction in coronary restenosis. Science1994;265:391-394.
24. Tsai H-L, Kou G-H, Chen S-C, Wu C-W, Lin Y-S.Human cytomegalovirus immediate-early protein IE2
tethers a transcriptional repression domain to p53. J BiolChem 1996;271:3534-3540.
25. Muganda P, Mendoza O, Hernandez J, Qian Q. Humancytomegalovirus elevates levels of the cellular protein p53in infected fibroblasts. J Virol 1994;68:8028-8034.
26. Kovacs A, Weber ML, Burns LJ, Jacob HS, VercellottiGM. Cytoplasmic sequestration of p53 in cytomegalo-virus-infected human endothelial cells. Am J Pathol 1996;149:1531-1539.
27. Yurochko AD, Kowalik TF, Huong S-M, Huang E-S.Human cytomegalovirus upregulated NF-kB activity bytransactivating the NF-kB p105/p50 and p65 promoters.J Virol 1995;69:5391-5400.
28. Sambucetti LC, Cherrington JM, Wilkinson GWG,Mocarski ES. NF-κB activation of the cytomegalovirusenhancer is mediated by a viral transactivator and by Tcell stimulation. EMBO J 1989;9:4251-4258.
29. Kline JN, Hunninghake GM, He B, Monick MM,Hunninghake GW. Synergistic activation of the humancytomegalovirus major immediate early promoter byprostaglandin E2 and cytokines. Exp Lung Res 1998;24:3-14.
30. Mocarski ES. Virus self-improvement through inflam-mation: no pain, no gain. PNAS 2002;99:3362-3364.
31. Okada K, Tanaka K, Noda S, Okazaki M, Koga Y. Nitricoxide increases the amount of murine cytomegalovirus-DNA in mice latently infected with the virus. Arch Virol1999;144:2273-2290.
32. Speir E, Yu Z-X, Ferrans VJ, Huang E-S, Epstein SE.Aspirin attenuates cytomegalovirus infectivity and geneexpression mediated by cyclooxygenase-2 in coronaryartery smooth muscle cells. Circ Res 1998;83:210-216.
33. Koszinowski UH, Hengel H. (Eds) Viral ProteinsCounteracting Host Defenses. Springer-Verlag, Berlin2002.
34. Loenen WAM, Bruggeman CA, Wiertz EJHJ. Immuneevasion by human cytomegalovirus: lessons in immun-ology and cell biology. Immunology 2001;13:41-49.
35. Ahn K, Gruhler A, Galocha B, Jones TR, Wiertz EJHJ,Ploegh HL, Peterson PA, Yang Y, Früh K. The ER-luminaldomain of the HCMV glycoprotein US6 inhibits peptidetranslocation by TAP. Immunity 1997;6:613-621.
36. Ahn K, Angulo A, Ghazal P, Peterson PA, Yang Y, FrühK. Human cytomegalovirus inhibits antigen presentationby a sequential multistep process. PNAS 1996;93:10990-10995.
37. Wiertz EJHJ, Jones TR, Sun L, Bogyo M, Geuze HJ,Ploegh HL. The human cytomegalovirus US11 geneproduct dislocates MHC class I heavy chains from theendoplasmic reticulum to the cytosol. Cell 1996;84:769-770.
38. Reyburn HT, Mandelboim O, Valés-Gomez M, DavisDM, Pazmany L, Strominger JL. The class I MHC homo-logue of human cytomegalovirus inhibits attack by naturalkiller cells. Nature 1997;386:514-517.
39. Lee SH, Zafer A, de Repentigny Y, Kothary R, TremblayML, Gros P, Duplay P, Webb JR, Vidal SM. Transgenic
Cytomegalovirus escapology and immunoevasion

130
expression of the activating natural killer receptor Ly49Hconfers resistance to cytomegalovirus in geneticallysusceptible mice. J Exp Med 2003;197:515-526.
40. Lee S-H, Girard S, Macina D, Busa M, Zafer A, BelouchiA, Gros P, Vidal SM. Susceptibility to mouse cytomegalo-virus is associated with deletion of an activating naturalkiller cell receptor of the C-type lectin superfamily. NatureGenetics 2001;28:42-45.
41. Ziegler H, Thäle R, Lucin P, Muranyi W, Flohr T, HengelH, Farrell H, Rawlinson W, Koszinowski UH. A mousecytomegalovirus glycoprotein retains MHC class I comp-lexes in the ERGIC/cis-golgi compartments. Immunity1997;6:57-66.
42. Reusch U, Muranyi W, Lucin P, Burgert H-G, HengelH, Koszinowski UH. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes fordegradation. EMBO J 1999;18:1081-1091.
43. Miller DM, Rahill BM, Boss JM, Lairmore MD, DurbinJE, Waldman WJ, Sedmak DD. Human cytomegalovirusinhibits major histocompatibility complex class IIexpression by disruption of the jak/stat pathway. J ExpMed 1998;187:675-683.
44. Tomazin R, Boname J, Hegde NR, Lewinsohn DM,Altschuler Y, Jones TR, Cresswell P, Nelson JA, RiddellSR, Johnson DC. Cytomegalovirus US2 destroys twocomponents of the MHC class II pathway, preventingrecognition by CD4+ T cells. Nature Medicine 1999;5:1039-1043.
45. Redpath S, Angulo A, Gascoigne NRJ, Ghazal P. Murinecytomegalovirus infection down-regulates MHC class IIexpression on macrophages by induction of IL-10. JImmunol 1999;162:6701-6707.
46. Redpath S, Ghazal P, Gascoigne NRJ. Hijacking andexploitation of IL-10 by intracellular pathogens. TrendsMicrobiol 2001;9:86-92.
47. Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV,Pestka S. Human cytomegalovirus harbors its own uniqueIL-10 homolog (cmvIL-10). PNSA 2000;97:1695-1700.
48. Furukawa T, Hornberger E, Sakuma S, Plotkin SA.Demonstration of immunoglobulin G receptors inducedby human cytomegalovirus. J Clin Microbiol 1975;2:332-336.
49. Thale R, Lucin P, Schneider K, Eggers M, KoszinowskiUH. Identification and expression of a murine cytomeg-alovirus early gene coding for an Fc receptor. J Virol 1994;68:7757-7765.
50. Bodaghi B, Jones TR, Zipeto D, Vita C, Sun L, LaurentL, Arenzana-Seisdedos F, Virelizier J-L, Michelson S.Chemokine sequestration by viral chemoreceptors as a
novel viral escape strategy: withdrawal of chemokinesfrom the environment of cytomegalovirus-infected cells.J Exp Med 1998;188:855-866.
51. Welch AR, McGregor LM, Gibson W. Cytomegalovirushomologs of cellular G protein-coupled receptor genesare transcribed. J Virol 1991;65:3915-3918.
52. Streblow DN, Soderberg-Naucler C, Vieira J, Smith P,Wakabayashi E, Ruchti F, Mattison K, Altschuler Y,Nelson JA. The human cytomegalovirus chemokinereceptor US28 mediates vascular smooth muscle cellmigration. Cell 1999;99:511-520.
53. Davis-Poynter NJ, Lynch DM, Vally H, Shellam GR,Rawlinson WD, Barrell BG, Farrell HE. Identificationand characterization of a G protein-coupled receptorhomolog encoded by murine cytomegalovirus. J Virol1997;71:1521-1529.
54. Froberg MK, Adams A, Seacotte N, Parker-Thornberg J,Kolattukudy P. Cytomegalovirus infection acceleratesinflammation in vascular tissue overexpressing monocytechemoattractant protein-1. Circ Res 2001;89:1224-1230.
55. Hajjar DP, Fabricant CG, Minick CR, Fabricant J. Virus-induced atherosclerosis: herpesvirus infection alters aorticcholesterol metabolism and accumulation. Am J Pathol1986;122:62-70.
56. Abrahamsen LH, Clay MJ, Lyle JM, Zink JM, FredricksonLJ, DeSierevo AJ, Jerkofsky M. The effects of cyto-megalovirus infection on polar lipids and neutral lipidsin cultured human cells. Intervirology 1996;39:223-229.
57. Guetta E, Guetta V, Shibutani T, Epstein SE. Monocytesharboring cytomegalovirus: interactions with endothelialcells, smooth muscle cells, and oxidized low-densitylipoprotein. Circ Res 1997;81:8-16.
58. Froberg MK, Seacotte N, Dahlberg E. Cytomegalovirusseropositivity and serum total cholesterol levels in youngpatients. Ann Clin Lab Sci 2001;31:157-161.
59. Hsich E, Zhou YF, Paigen B, Johnson TM, Burnett MS,Epstein SE. Cytomegalovirus infection increasesdevelopment of atherosclerosis in apolipoprotein-Eknockout mice. Atherosclerosis 2001;156:23-28.
60. Chiu B, Viira E, Tucker W, Fong IW. Chlamydiapneumoniae, cytomegalovirus, and herpes simplex virusin atherosclerosis of the carotid artery. Circulation 1997;96:2144-2148.
61. Soderberg-Naucler C, Fish KN, Nelson JA. Interferon-γand tumor necrosis factor-α specifically induce formationof cytomegalovirus-permissive monocyte-derived macro-phages that are refractory to the antiviral activity of thesecytokines. J Clin Invest 1997;100:3154-3163.
Annals of Clinical & Laboratory Science


















