
REVERSE SYNTHESIS AND 3’-MODIFICATION OF RNA 2-Medicinal Chemistry of... · REVERSE SYNTHESIS AND...
45
REVERSE SYNTHESIS AND 3’-MODIFICATION OF RNA Lucas Bethge, 1 Stefan Vonhoff 1 and Sven Klußmann 1* 1 NOXXON Pharma AG, Max-Dohrn-Str. 8-10, 10589 Berlin, Germany. * Correspondence to: [email protected] ABSTRACT A D-enantiomer of the Spiegelmer® mNOX-E36 was synthesized both in 3’->5’ direction as well as the 5’->3’ direction using normal and reverse RNA synthesis. Fol- lowing the synthesis the crude product was pegylated at its 3’ end. Reverse RNA synthesis showed superior puri- ty and yield of the final 3’ modified oligonucleotide. INTRODUCTION, RESULTS AND DISCUSSION, CONCLUSION Spiegelmers® are chemical entities based on synthetic mirror-image oligonucleotides that bind and efficiently in- hibit target molecules in a manner conceptually similar to monoclonal antibodies. [1] They are highly selective for their pharmacological target and potent inhibitors of target function with affinities in the low nano- to picomolar range. Spiegelmers® combine the benefits of small molecule drugs and biopharmaceuticals. As a chemical scaffold their prop- erties, like affinity, pharmacokinetic or bio-distribution are tunable through site-specific modifications. One of NOXXON’s lead compounds, NOX-E36 [2] is pegylated at the 5’ end to prolong plasma half-life. [3] However, the murine specific variant mNOX-E36 was found to be most potent if pegylated at the 3’ end. Direct pegylation of crude RNA synthesis products is desirable to reduce overall process steps. If this is done with a modifica- tion at the 5’ end, the changed properties of the resulting conjugate can be easily exploited to purify the conjugated full length oligonucleotide from unconjugated truncated sequences. If the modification is placed at the 3’ end the full length oligonucleotide and its truncated failure sequences will be conjugated, so that this purification approach will be more difficult. Srivastava and co-workers recently showed the use of re- versed RNA phosphoramidites carrying a 3’-DMT and a 5’- cyanoethylphosphoamidite group in RNA synthesis. [4] Such synthetic strategy would place the desired modifica- tion site at the 3’ end to the synthetic terminus and therefore again allow for direct modification of crude RNA synthesis products. Such reversed phosphoamidites are commercially availa- ble only for D-RNA. Therefore, we decided to evaluate the potential of reverse RNA synthesis with the D-enantiomer of mNOX-E36. The synthesis was carried out with conven- tional TBDMS chemistry in 3’->5’ direction as well as in 5’->3’ direction at a ~60 µmol scale. The syntheses were both completed by terminal coupling of a C6-amino-MMT modifier to allow for the conjugation of further modifica- tions. Both strategies yielded a comparable crude product quality after cleavage from the solid support and removal of the 2’-TBDMS groups. Pegylation of the respective crude products was achieved using a 40kDa PEG NHS ester. Reversely synthesized mNOX-E36 showed superior in purity and yield over con- ventionally synthesized mNOX-E36. REFERENCES 1. Klußmann, S., Nolte, A., Bald, R., Nat. Biotechnol. 1996, 14, 1112-1115. 2. Wlotzka, B., Leva, S., Eschgfaller, B., Proc. Natl. Acad. Sci. USA 2002, 99, 8898-8902. 3. Kulkarni, O., Pawar, R. D., Purschke, W., J. Am. Soc. Nephrol. 2007, 18, 2350-2358. 4. Srivastava, S. C., Pandey D., Srivastava N. P., Nucleic Acids Symp. Ser., 2008, 52, 103-104. Figure 1. Conventional (A) and Reverse (B) Chemical RNA Synthesis, Conjugation of 40kDa PEG NHS-Ester (C). 64
-
Upload
phunghuong -
Category
Documents
-
view
218 -
download
0
Transcript of REVERSE SYNTHESIS AND 3’-MODIFICATION OF RNA 2-Medicinal Chemistry of... · REVERSE SYNTHESIS AND...

REVERSE SYNTHESIS AND 3’-MODIFICATION OF RNA
Lucas Bethge,1 Stefan Vonhoff
1 and Sven Klußmann
1*
1NOXXON Pharma AG, Max-Dohrn-Str. 8-10, 10589 Berlin, Germany. * Correspondence to:
ABSTRACT
A D-enantiomer of the Spiegelmer® mNOX-E36 was
synthesized both in 3’->5’ direction as well as the 5’->3’
direction using normal and reverse RNA synthesis. Fol-
lowing the synthesis the crude product was pegylated at
its 3’ end. Reverse RNA synthesis showed superior puri-
ty and yield of the final 3’ modified oligonucleotide.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
Spiegelmers® are chemical entities based on synthetic
mirror-image oligonucleotides that bind and efficiently in-
hibit target molecules in a manner conceptually similar to
monoclonal antibodies. [1] They are highly selective for
their pharmacological target and potent inhibitors of target
function with affinities in the low nano- to picomolar range.
Spiegelmers® combine the benefits of small molecule drugs
and biopharmaceuticals. As a chemical scaffold their prop-
erties, like affinity, pharmacokinetic or bio-distribution are
tunable through site-specific modifications.
One of NOXXON’s lead compounds, NOX-E36 [2] is
pegylated at the 5’ end to prolong plasma half-life. [3]
However, the murine specific variant mNOX-E36 was
found to be most potent if pegylated at the 3’ end. Direct
pegylation of crude RNA synthesis products is desirable to
reduce overall process steps. If this is done with a modifica-
tion at the 5’ end, the changed properties of the resulting
conjugate can be easily exploited to purify the conjugated
full length oligonucleotide from unconjugated truncated
sequences. If the modification is placed at the 3’ end the full
length oligonucleotide and its truncated failure sequences
will be conjugated, so that this purification approach will be
more difficult.
Srivastava and co-workers recently showed the use of re-
versed RNA phosphoramidites carrying a 3’-DMT and a 5’-
cyanoethylphosphoamidite group in RNA synthesis. [4]
Such synthetic strategy would place the desired modifica-
tion site at the 3’ end to the synthetic terminus and therefore
again allow for direct modification of crude RNA synthesis
products.
Such reversed phosphoamidites are commercially availa-
ble only for D-RNA. Therefore, we decided to evaluate the
potential of reverse RNA synthesis with the D-enantiomer of
mNOX-E36. The synthesis was carried out with conven-
tional TBDMS chemistry in 3’->5’ direction as well as in
5’->3’ direction at a ~60 µmol scale. The syntheses were
both completed by terminal coupling of a C6-amino-MMT
modifier to allow for the conjugation of further modifica-
tions. Both strategies yielded a comparable crude product
quality after cleavage from the solid support and removal of
the 2’-TBDMS groups.
Pegylation of the respective crude products was achieved
using a 40kDa PEG NHS ester. Reversely synthesized
mNOX-E36 showed superior in purity and yield over con-
ventionally synthesized mNOX-E36.
REFERENCES
1. Klußmann, S., Nolte, A., Bald, R., Nat. Biotechnol.
1996, 14, 1112-1115.
2. Wlotzka, B., Leva, S., Eschgfaller, B., Proc. Natl. Acad.
Sci. USA 2002, 99, 8898-8902.
3. Kulkarni, O., Pawar, R. D., Purschke, W., J. Am. Soc.
Nephrol. 2007, 18, 2350-2358.
4. Srivastava, S. C., Pandey D., Srivastava N. P., Nucleic
Acids Symp. Ser., 2008, 52, 103-104.
Figure 1. Conventional (A) and Reverse (B) Chemical RNA Synthesis, Conjugation of 40kDa PEG NHS-Ester (C).
64

Figure 3. a) No transfection. b) Trans-
fection with 5:1 lipofectamine to 3-insert
siRNA.
a)
b)
Figure 2. Cell transfected with 2:1
lipofectamine to 3-insert siRNA.
Vinculin Cy-3
NOVEL SIRNA-AMINO ACID CONJUGATES EXHIBIT IMPROVEMENTS IN CELLULAR
UPTAKE AND REDUCE DRR EXPRESSION IN TUMOR CELLS
Jovanka Bogojeski,1 Richard Johnsson,
1 Ken Yamada,
1 Phuong U. Le,2 Kevin Petrecca,2 Masad J Damha*
1McGill Department of Chemistry, 801 Sherbrooke St. West, Montreal, Canada and
2McGill Department of Neurology
and Neurosurgery, 3801 University St., Montreal, Canada. * Correspondence to: [email protected]
ABSTRACT
We have synthesized siRNA conjugates containing 1, 2
and 3 N-L-phenylalanine modifications at C2ꞌ of the sug-
ar ring. These siRNAs were successfully transfected
with as little as 10% of the standard lipofectamine
amount generally used in transfection experiments. Si-
lencing was demonstrated under these conditions with
morphological changes in glial cells (primary brain can-
cer cells) that corresponded to DRR (down regulated in
renal cell carcinoma) protein reduction and thus re-
duced tumor malignancy.
INTRODUCTION
The invasion of glial cells into brain tissue is a multistep
process resulting in the spreading of the malignant cancer.
Cellular attachment to focal adhesions (FAs), one of the first
steps in invasion, is eventually followed by FA disassembly
after the extracellular matrix upstream the cell is degraded
and the cell has translocated forward.1 It is possible to re-
duce FA disassembly via DRR protein reduction in vitro and
in vivo with siRNA encapsulated in a positively charged
liposome (i.e., lipofectamine).1 This standard siRNA deliv-
ery vehicle is generally used in a ratio of 20:1 lipofectamine
to siRNA duplex.
Non-invasive cellular delivery of therapeutic siRNA is a
major hurdle due to its highly hydrophobic and negatively
charged character; however, therapeutic liposomal delivery
has proven disadvantages such as cellular toxicity. We have
synthesized 5ꞌ cyanine-3 labelled siRNA conjugates with 2ꞌ-
N-L-phenylalanine modifications at
different positions, reducing the net
negative charge on the duplexes and
increasing the lipophilic character in
order to enhance cellular uptake.
RESULTS AND DISCUSSION The conjugates were formed by annealing the modified
sense strands with the non-modified antisense strand target-
ing drr mRNA and transfected into glial cells with 25 and
10% standard lipofectamine amount. DRR reduction was
seen with 1 (when the single insert was moved from inside
the duplex to an overhang, 1a1b), 2 and 3 modifications
(Fig. 1). We have visualized uptake of the siRNA as well as
cellular morphology that is consistent with DRR reduction
and cell proliferation; the red cyanine dye is seen within the
cytoplasm of the stained cells, with the siRNA concentrating
around the nuclei. Furthermore, the once spindle-like elon-
ganted cells become rounded and are surrounded by large
FAs (arrow) (Fig. 2). The fluorescence intensity as well as
the effect on cell morphology and FA size is greatest with
the most modified conjugates for a given amount of lipofec-
tamine.
Delicate tumor
spheroids were
doped with the
conjugates into the
center of their
glial cell mass.
The distance that
the cells travelled
from their invasion
front into the sur-
rounding medium
compared to non-
transfected sphe-
roids is signifi-
cantly less (Fig. 3, surrounding invasive DRR+ cells best
seen when greatly enlarged).
CONCLUSION
We have developed siRNA amino acid conjugates that re-
quire less transfection agent relative to native siRNAs. The
ability of the 2ꞌ-amino acid modification to increase cellular
uptake of siRNA with minimal tranfecting agent (lipofec-
tamine) amount gives merit to the biolable 2ꞌ-O-L-
phenylalanine acetal ester strategy recently described by our
group.2
REFERENCES 1. Le, P.U., Angers-Loustau, A., de Oliveira, R.M.W., Ajlan, A.,
Brassard, C.L., Dudley, A., Brent, H., Siu, V., Trinh, G., Möl-
enkamp, G., Wang, J., Seyed Sadr M., Bedell, B., Del Maes-
tro, R.F., Petrecca, K. Oncogene, 2010, 29, 4636-4647.
2. Johnsson, R., Lackey, J.G., Bogojeski, J.J., Damha, M.J.
Bioorg. Med. Chem. Lett., 2011, 21, 3721-3725.
Figure 1. DRR reduction by siRNA with 1 (a & b), 2 and 3 inserts.
65

DEVELOPMENT OF LNA-MODIFIED APTAMERS AGAINST CANCER TARGETS
Ida Coordt Elle*1, Torben Højland*1, Henrik Ditzel2, Jan Mollenhauer2 and Jesper Wengel1
1Nucleic Acid Center, University of Southern Denmark, Campusvej 55, DK-5230 Odense M, Denmark; 2Institute of Molecular Medicine, University of Southern Denmark, JB Winsløwsvej 25, DK-5000 Odense C, Denmark.
* Correspondence to: [email protected] or [email protected]
ABSTRACT
In recent years, aptamers have attracted great scien-tific attention as possible drugs and drug delivery agents. Herein, we present different strategies for the develop-ment of LNA-containing aptamers against cancer-relevant targets.
INTRODUCTION, METHODS AND CONCLUSION
Aptamers are short, single-stranded nucleic acids with defined three-dimensional shapes, which enable them to bind to a wide range of targets, including cell surface proteins, with high specificity and affinity. Aptamers against a molecule of interest can be generated through systematic evolution of ligands by exponential enrichment (SELEX)1,2. Aptamers have been dubbed “chemical antibodies”, but possess a number of advantages over traditional antibodies such as smaller size, low immunogenic potential, higher stability, and ease of synthesis and manipulation. However, chemical modification of aptamers is necessary to ensure bioavailabilty and -stability. LNAs (locked nucleic acids) are RNA analogues containing a 2’-O,4’-C-methylene bridge, which locks the furanose in a C3’-endo conformation. LNA improves thermal stability and confers nuclease-resistance when incorporated into nucleic acids. So far, LNA has only been introduced into aptamers “post-SELEX”3, but we wish to employ LNA-modified sequence libraries to ensure that affinity is retained in the LNA-containing aptamers.
Figure 1: Structural representation of LNA monomers.
Due to their high degree of specificity, aptamers show great promise in targeting sub-populations of cells displaying changes in the expression of certain proteins; e.g. cancer cells. The ecto-5’-nucleotidase CD73 is a cell surface protein that hydrolyzes extracellular AMP to adenosine and phosphate and is over-expressed in many solid tumors. Extracellular adenosine is a potent immunosuppressor, and tumor-derived CD73 has therefore been suggested as a mediator of tumor immune escape.
Studies have shown that anti-CD73 antibody therapy inhibits tumor growth and metastasis, while RNAi-mediated CD73 suppression induces cell-cycle arrest and apoptosis in breast cancer cells4,5. We are employing traditional SELEX to produce an LNA-modified aptamer, which will bind and inhibit the activity of CD73. The aptamer(s) generated will be tested for their ability to inhibit CD73 activity in MDA-MB-231 cancer cells. The aptamers will then be modified for in vivo use and tested for their ability to inhibit metastasis in mice. CD44 is a transmembrane glycoprotein belonging to the family of cell adhesion molecules6. CD44 regulates growth, survival, differentiation and migration, and is involved in the metastatic processes of certain tumors. The extracellular domain is highly variable giving rise to many different CD44 isoforms. Expression of certain isoforms correlates with poor prognosis for several tumors. Targeting aptamers against specific isoforms may provide improved treatment of cancer. We perform cell-SELEX to develop aptamers against CD44v3,8-10 and CD44is4. For this purpose, MCF7 cell lines expressing CD44v3,8-10 and CD44is4, in addition to an MCF7 cell line that does not express CD44, are produced. The developed aptamers will be used as targeting devices against breast cancer stem cells expressing CD44. To combat malignant cell growth and avoid adverse side effects, cancer drugs must be highly specific. Aptamers offer this specificity. We hope to show that different strategies can be employed in developing aptamers for drug delivery or direct inhibition of cancer cell growth and metastasis.
REFERENCES
1. Ellington A. D., Szostak J. W., Nature, 1990, 346, 818-822.
2. Tuerk, C., Gold, L., Science, 1990, 249, 505-510.
3. Veedu, R., Wengel, J., Chemistry and Biodiversity, 2010, 7, 536-542
4. Stagg, J. et al., PNAS, 2010, 107, 1547-1552.
5. Zhi et al., Cancer Sci, 2010, 101, 5261-2569.
6. Orian-Rousseau, V., Eur. J. Cancer, 2010, 46, 1271-1277.
66

SYNTHESIS OF 5’-CAPPED RNA (7M
GPPPNNNN) USING SOLID-PHASE CHEMISTRY
COUPLED WITH ENZYMATIC (GUANINE-N7) METHYLATION
Thillier Y.,1 Decroly E.,
2 Morvan F.,
1 Canard B.,
2 Vasseur, J.-J.,
1 and Debart, F.
1*
1 IBMM, UMR 5247 CNRS-UM1-UM2, Université Montpellier 2, Pl. E. Bataillon, 34095 Montpellier Cedex 05, France.
2AFMB, UMR 6098 CNRS-Universités d’Aix-Marseille I et II, ESIL Case 925, 163 Avenue de Luminy, 13288 Marseille
Cedex 9, France. * Correspondence to: [email protected]
ABSTRACT
Availability of 5’-capped RNA is an important bottle-
neck for many biological studies. In the present work, we
combined a chemical synthesis method on solid support
and an enzymatic methylation assay in order to produce
large amounts of RNA carrying different cap structures.
INTRODUCTION
The RNA processing consists in a cap structure which is
a N7-methylguanosine linked to the 5’-terminal nucleoside
of the pre-mRNA via a 5’-5’ triphosphate linkage. This cap
moiety (7m
Gppp) is an essential RNA structural modification
to allow its efficient translation, limiting its degradation by
cellular 5’-exonucleases and avoiding its recognition as
« nonself » by the innate immunity machinery. The lack of
methods allowing the synthesis of large amounts of 5’-
capped RNAs have hampered biological and structural stud-
ies of proteins recognizing the cap structure or involved in
the capping pathway. In vitro enzymatic synthesis of capped
RNA remains challenging since it is difficult to produce 5’-
capped RNAs of defined sequences in great amounts. In
particular, while the cellular machinery is able to cap any
5’-RNA sequence, it is known that viral enzymes cap de-
fined and specific RNA sequences present at the RNA 5’-
end. To overcome this bottleneck, we have developed a
straightforward strategy for the synthesis of 5’-capped
RNAs with high yields and without any limitation concern-
ing the nucleotides present at 5’ of the substrate RNA.
RESULTS AND DISCUSSION
Most of the chemical methods for synthesis of capped
oligoribonucleotides reported in the literature, produce a
very limited amount of capped RNA [1]. Therefore our aim
was to synthesize RNA carrying various cap structures:
GpppN (cap), 7m
GpppN (cap-0) and 7m
GpppN2’-Om (cap-1) in
great quantities using the combination of chemical synthesis
of Gppp-RNAs followed by enzymatic methylation. The
RNA assembly was performed with the pivaloyloxymethyl
(PivOM) technology recently introduced by our group for
RNA synthesis on solid support [2]. Its major feature is to
use base-labile protecting groups exclusively removed under
basic conditions without RNA damage. The mild ammonia
treatment applied for deprotection prevents degradation of
the triphosphate moiety of the cap structure. Our present
approach involves the triphosphate motif formation by the
reaction of commercially available GDP with an activated
phosphate group at the 5’-terminus of the solid-supported
protected RNA (Figure 1). After RNA assembly, the 5’-OH
of the oligomers was converted with >95% yield to its H-
phosphonate monoester which was simply activated by ami-
dative oxidation to give the solid-supported 5’-
phosphoroimidazolide RNAs with quantitative yield [3].
The conversion of RNAs into the desired capped Gpp-
pRNAs reached a satisfactory average yield between 40%
and 50% [4].
Figure 1. General course of synthesis of 5’-7mGppp-RNAs
Once deprotected and released from support, GpppRNAs
were quantitatively methylated into 7m
Gppp-RNAs using
purified human N7 guanine-methyltransferase.
CONCLUSION
The easier access to capped RNAs and their greater
availability are very attractive and promising for structural
and mechanistic studies of their complexes with RNA cap-
ping enzymes.
REFERENCES
1. Kadokura, M.; Wada, T.; Seio, K.; Moriguchi, T.;
Huber, J.; Luhrmann, R.; Sekine, M. Tetrahedron
Lett. 2001, 42, 8853-8856; Jemielity, J.; Heinonen,
P.; Lonnberg, H.; Darzynkiewicz, E. Nucleosides
Nucleotides Nucleic Acids 2005, 24, 601-605.
2. Lavergne, T.; Bertrand, J. R.; Vasseur, J. J.; Debart,
F. Chem. Eur. J. 2008, 14, 9135-9138.
3. Zlatev, I.; Lavergne, T.; Debart, F.; Vasseur, J.-J.;
Manoharan, M.; Morvan, F. Org. Lett. 2010, 12,
2190-2193.
4. Thillier, Y.; Decroly, E.; Morvan, F.; Canard, B.;
Vasseur, J. J.; Debart, F. RNA 2012, 18, 856-868.
Protected RNA
3'5'
OP
O
O-
N
N
O P O
O
O-
P
O
O-
O-
3'5'
OP
O
O-
O P O
O
O-
P
O
O-
OGuanosine
RNA deprotection and release from solid support
5'-RNA capping
Guanosine
Protected RNA
3'5'
OP
O
O-
O P O
O
O-
P
O
O-
OGuanosine RNA
3'5'
OP
O
O-
O P O
O
O-
P
O
O-
O7mN-Guanosine RNA
Enzymatic N 7-guanine methylation
OH
OH
Solid Support
ZnCl2, DMF
DBU, CH3CN
NH4OH
Solid Support
Figure 1. Caption. (Font: Times or Times New Roman, 9 pt)
67

FLUORINATED OLIGONUCLEOTIDE ANALOGUES (2′F-ANA, 2′F-RNA): COMPATIBILITY
WITH AGO2 AND APPLICATIONS IN GENE SILENCING
Glen F. Deleavey,1 Filipp Frank,
2 Phuong U. Le,
3 Dianna Chan,
4 Naira Souleimanian,
5 Cy Stein,
5
Molly Shoichet,4 Kevin Petrecca,
3 Bhushan Nagar,
2 and Masad J. Damha
1*
1Department of Chemistry, McGill University, 801 Sherbrooke Street West, Montréal, QC, Canada,
2Department of
Biochemistry, McGill University, Montréal, QC, Canada, 3Department of Neurology and Neurosurgery, McGill Universi-
ty, Montréal, QC, Canada, 4Department of Chemistry, University of Toronto, Toronto, ON, Canada, and
5Department of
Medical Oncology, City of Hope Medical Center, Duarte, CA, USA. *Correspondence to: [email protected]
ABSTRACT
The relative binding of native and modified 5′-pUpG
dimers for the MID domain of hAGO2 will be discussed
in relation to the gene silencing applications of fluorinat-
ed nucleic acid analogues. Strategies for siRNA and
AON modification with fluorinated nucleotide analogues
(i.e. 2′-deoxy-2′-fluoroarabinonucleic acid, 2′F-ANA) to
silence therapeutically relevant targets will be presented,
along with recent developments regarding the delivery of
and enzyme interactions with modified oligonucleotides.
INTRODUCTION
Targeted gene silencing directed by small interfering
RNAs (siRNAs) or antisense oligonucleotides (AONs) has
numerous applications in genomics, therapeutic target vali-
dation, and drug discovery. Oligonucleotide-based therapeu-
tics must overcome four main obstacles: (1) Oligonucleo-
tides have poor nuclease stability; (2) Targeted delivery and
cellular uptake of oligonucleotides is difficult to achieve; (3)
Oligonucleotides can cause “off-target” effects, and (4) Oli-
gonucleotides can cause immunostimulation effects.[1]
Chemically modified oligonucleotides are being investigat-
ed as a potential solution to some of these challenges.
2′F-ANA is an example of a modified oligonucleotide
analogue that can offer advantages over native gene silenc-
ing agents. 2′F-ANA can impart nuclease resistance to siR-
NAs and AONs, reduce immunosimulation effects of siR-
NAs, improve the potency of gene silencing in some cases,
and together with RNA-like chemical modifications (i.e.
2′F-RNA), allow for tuning of local siRNA duplex thermo-
dynamics in rational siRNA design.[2] Importantly, when
applied properly, 2′F-ANA oligonucleotides are fully com-
patible with RNase H and RISC-mediated gene silencing.
RESULTS AND DISCUSSION
Chemically altered AONs and siRNAs, composed of
fluorinated nucleotide analogues, are potent gene silencing
agents when directed towards DRR, a regulator of invasion
in malignant gliomas. 2′F-ANA oligonucleotides silence
DRR expression, and cause phenotypic changes reducing
cellular invasion in cell-based assays (Figure 1(a)). In other
anticancer applications, phosphorothioated 2′F-ANA-
modified AONs targeting Bcl-2 or the androgen receptor
(AR)
are able to silence gene expression with high efficiency
when delivered in the absence of transfection reagent using
a gymnotic delivery strategy.
Fluorinated oligonucleotides can clearly offer advantages
in gene silencing applications. To probe the tolerance of
RISC towards these modifications, binding affinities of 5′-
pUpG dimers of RNA, DNA, and fluorinated analogues for
the 5′ binding pocket (in the MID domain) of hAGO2 (the
core endonuclease of RISC) were determined and compared
with corresponding in vitro gene silencing potencies. Data
suggests that 5′ recognition by hAGO2 is not significantly
perturbed by sugar modifications (Figure 1(b)).
CONCLUSION
Oligonucleotide-based therapeutics can benefit from
chemical modifications. Here, we demonstrate the utility of
modified oligonucleotides in the silencing of therapeutically
relevant genes, advances in oligonucleotide delivery in cell-
based assays, and the tolerance of the hAGO2 for 5′ chemi-
cal modifications.
REFERENCES
1. Deleavey, G.F., Watts, J.K., and Damha, M.J. Curr.
Protoc. in Nucl. Acids Chem., 2009, 16.3.1–16.3.22.
2. Deleavey, G.F., Watts, J.K., Alain, T., et al., Nucleic
Acids Res., 2010, 38, 4547–4557.
Figure 1. (a) Untreated (left) and treated (right) malignant glioma cells. (b) Binding affinities of native and sugar-modified 5′-pUpG di-mers for the MID do-main of hAGO2.
68

RESTORATION OF DYSTROPHIN BY TRICYCLO-DNA
Branislav Dugovič,1 Damian Ittig,
1 Aurelie Goyenvalle,
2 Luis Garcia
2 and Christian J. Leumann
1*
1Department of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland and
2Institut
de Myologie, Faculté de Médecine Pierre et Marie Curie, 105 Boulevard de l´Hopital, 75634 Paris, France. *Correspondence to: [email protected]
ABSTRACT
Antisense oligonucleotides designed as splice modula-
tors are promising tools for the treatment of genetic dis-
orders such as Duchenne muscular dystrophy (DMD).
We have evaluated tricyclo-DNA phosphorothioate oli-
gonucleotides as a potential molecular platform for the
treatment of DMD in the dystrophic mdx mouse and the
dystrophin/utrophin deficient dKO mouse.
INTRODUCTION
Duchenne muscular dystrophy is a lethal muscle degen-
erative disease caused by mutations in the dystrophin gene.
In the vast majority of cases these mutations disrupt the
open reading frame leading to abnormal translation and,
therefore, to the absence of dystrophin. Most DMD patients
are wheelchair dependent in their early teens and dying
prematurely before they reach their third decade of age. The
milder allelic form of the disease, Becker muscular dystro-
phy (BMD) is caused by mutations maintaining the open
reading frame and permitting the production of partially
deleted but still functional dystrophin. This offers the possi-
bility of an antisense mediated exon-skipping therapy of
DMD, where the removal of exons leads to the restoration
of the open reading frame and therefore production of short-
ened, yet functional, dystrophin. The principle of the exon-
skipping therapy for DMD has been demonstrated in the late
nineties [1]. Since then, numerous studies utilizing various
modified oligonucleotides such as 2´OMe phosphorothioate
RNA, phosphorodiamidate morpholino oligomers (PMO)
and LNA have provided evidence of therapeutic potential of
this approach in several animal models as well as in humans
[2].
Tricyclo-DNA (tc-DNA, Figure 1) has been developed
earlier in our group [3] and possesses excellent antisense
properties [4]. In this study we aimed at skipping exon 23 in
the mdx mouse as well as in the dystrophin and utrophin
deficient dKO mouse with systemic administration of phos-
phorothioate tricyclo-DNA (PS-tc-DNA).
RESULTS AND DISCUSSION
We selected a 15-mer antisense oligonucleotide (PS
M23D [+2-13]) targeting the exon 23 donor splice site. It is
designed to anneal with the last two bases of the exon 23
and the first thirteen nucleotides of the intron 23. Introduc-
tion of thiophosphate into the sugar backbone of tc-DNA
significantly improved the uptake of oligonucleotides when
administered intravenously or subcutaneously. The analysis
of mRNA encoding the dystrophin by reverse transcription
polymerase chain reaction (RT-PCR) showed the presence
of the transcripts with deleted exon 23 in all skeletal mus-
cles as well as in the heart (Figure 2). Quantification of the
amount of dystrophin by Western blot revealed 25 and 15 %
of normal level in skeletal muscles and 5 % in the heart for
mdx and dKO mice, respectively.
CONCLUSION
The tc-DNA platform shows high potential for splice cor-
rection therapy of DMD.
REFERENCES
1. For review see: Wood, M.A., Gait, M.J., Yin, H. Brain,
2010, 133, 957-972.
2. For review see: Goyenvalle, A., Davies, K.E. Skeletal
Muscle, 2011, 1:8.
3. Rennenberg, D., Leumann, C.J. J. Am. Chem. Soc.,
2002, 124, 5993-6002.
4. Ittig, D., Liu, S., Rennenberg, D., Schümperli, D.,
Leumann, C.J. Nucleic Acids Res., 2004, 32, 346-353.
Figure 2. Restoration of the dystrophin of mdx mouse after 8
weeks of systemic treatment with the dose 100 mg/kg/week of PS
M23D [+2-13]. RT-PCR analysis of tissues, the 903 base-pairs (bp)
product represents the full-length transcript and 688 bp represents
the transcript that excludes the exon 23.
Figure 1. Repetition motif in PS-tc-DNA
69
THE SYNTHESIS AND CELL-BASED ACTIVITY OF TRIAZOLE-MODIFIED SIRNAS
Tim Efthymiou,1* Vanthi Huynh, Jaymie Oentoro, Brandon Peel, Jean-Paul Desaulniers2
1,2University of Ontario Institute of Technology, 2000 Simcoe St. N., Oshawa, Canada. * Correspondence to: [email protected]
ABSTRACT
Short interfering RNAs (siRNAs) were modified with
novel triazole-linked nucleoside dimer anlalogs at vari-
ous positions of the duplex. All triazole-modified siRNAs
were capable of silencing their targets within HeLa cells
in a dose-dependent manner, with a noticeable en-
hancement in potency when modifications are within the
3’ end region of the sense strand. In addition, a decrease
in susceptibility to exonuclease-mediated degradation
was imparted to siRNAs modified at the 3’ overhangs.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
For over a decade, the use of double-stranded short inter-fering RNAs (siRNAs) to silence the expression of genes associated with disease at the translational level has gained much attention. Using a highly conserved endogenous pathway within cells, siRNA technology displays a high degree of target specificity and potency [1]. However, traits required for the successful design of siRNA-based therapeu-tics such as resistance to nuclease-mediated degradation, improved cell membrane permeability and reduced off-target toxicity, are compromised by the native structure of duplex RNA’s charged backbone [2]. We have therefore synthesized novel and neutrally-charged triazole-linked nu-cleoside dimer analogs which were incorporated throughout siRNA duplexes using DMT-phosphoramidite chemistry, in order to attenuate the negative contributions of RNA’s na-tive backbone.
The design was inspired by the popular peptide nucleic acid (PNA) scaffold [3], which has seen great utility in the modification of several nucleic acid-based constructs. Through the robust Copper(I)-catalyzed Huisgen (3+2) cycloaddition [4], azide and alkyne nucleic acid monomers
are joined through the heterocyclic linkage in nearly quanti-tative yields (Figure 1).
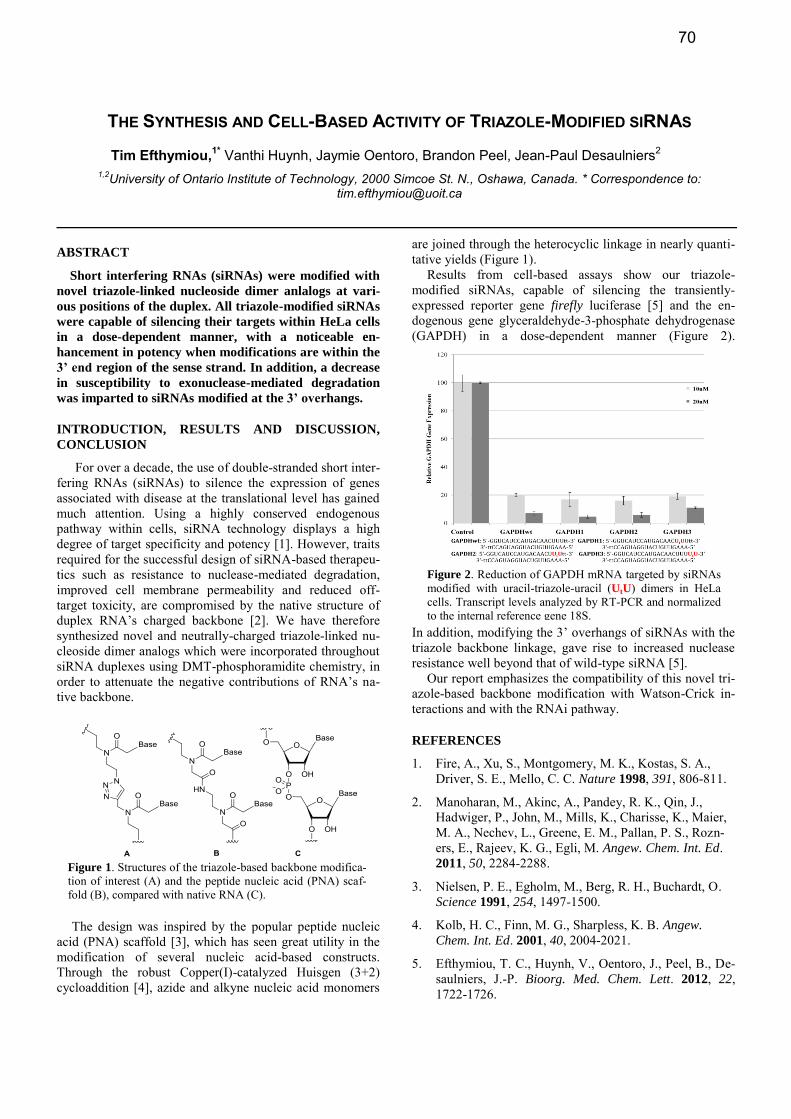
Results from cell-based assays show our triazole-modified siRNAs, capable of silencing the transiently-expressed reporter gene firefly luciferase [5] and the en-dogenous gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in a dose-dependent manner (Figure 2).
In addition, modifying the 3’ overhangs of siRNAs with the triazole backbone linkage, gave rise to increased nuclease resistance well beyond that of wild-type siRNA [5].
Our report emphasizes the compatibility of this novel tri-azole-based backbone modification with Watson-Crick in-teractions and with the RNAi pathway.
REFERENCES
1. Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E., Mello, C. C. Nature 1998, 391, 806-811.
2. Manoharan, M., Akinc, A., Pandey, R. K., Qin, J., Hadwiger, P., John, M., Mills, K., Charisse, K., Maier, M. A., Nechev, L., Greene, E. M., Pallan, P. S., Rozn-ers, E., Rajeev, K. G., Egli, M. Angew. Chem. Int. Ed. 2011, 50, 2284-2288.
3. Nielsen, P. E., Egholm, M., Berg, R. H., Buchardt, O. Science 1991, 254, 1497-1500.
4. Kolb, H. C., Finn, M. G., Sharpless, K. B. Angew.
Chem. Int. Ed. 2001, 40, 2004-2021.
5. Efthymiou, T. C., Huynh, V., Oentoro, J., Peel, B., De-saulniers, J.-P. Bioorg. Med. Chem. Lett. 2012, 22, 1722-1726.
Figure 2. Reduction of GAPDH mRNA targeted by siRNAs modified with uracil-triazole-uracil (UtU) dimers in HeLa cells. Transcript levels analyzed by RT-PCR and normalized to the internal reference gene 18S.
Figure 1. Structures of the triazole-based backbone modifica-tion of interest (A) and the peptide nucleic acid (PNA) scaf-fold (B), compared with native RNA (C).
70
NON-TOXIC INTRACELLULAR DELIVERY AND EFFICIENT GENE SILENCING BY FUNCTIONAL FUSION OF SIRNA AND PEPTIDES
Shutaro Fujiaki and Masayuki Fujii*
Department of Biological & Environmental Chemistry, School of Humanity Oriented Science and Technology, Kinki University, 11-6 Kayanomori, Iizuka, Fukuoka 820-8555, Japan
ABSTRACT
In the present study, we investigated the intracellular delivery of siRNA using some hybrid peptides as trans-fection reagents and the silencing effect of siRNA target-ing hTERT mRNA in 3 human cancer cell lines, Jurkat, HeLa and K562. The complex of siRNA and a specific amphiphilic peptide or its hybrid with an intracellular transport signal peptides could be effectively taken up into cells. The complex also showed a high silencing ef-fect against hTERT mRNA. Moreover, the combination of siRNA-NES conjugates and the amphiphilic peptides improved silencing effects up to 95.2 %.
INTRODUCTION
Recently, small interfering RNA (siRNA), one kind of RNA interference (RNAi) technology represent the most common and, to date, the most effective method to inhibit target gene expression in human cells. It is also a common recognition that non-toxic delivery of siRNA is an urgent problem for the therapeutic application of siRNA. For the efficient gene silencing in vivo, prolonged circulation of siRNA with take efficient and non-toxic cellular uptake and resistance against enzymatic degradation are indispensably required.1)
Telomerase activity has been regarded as a critical step in cellular immortalization and carcinogenesis and because of this, regulation of telomerase represents an attractive target for anti-tumor specific therapeutics.
In this paper, we present the efficient and non-toxic cellu-lar uptake of siRNA using novel amphiphilic peptides and the application to silencing of hTERT in human cancer cell lines.
RESULTS AND DISCUSSION
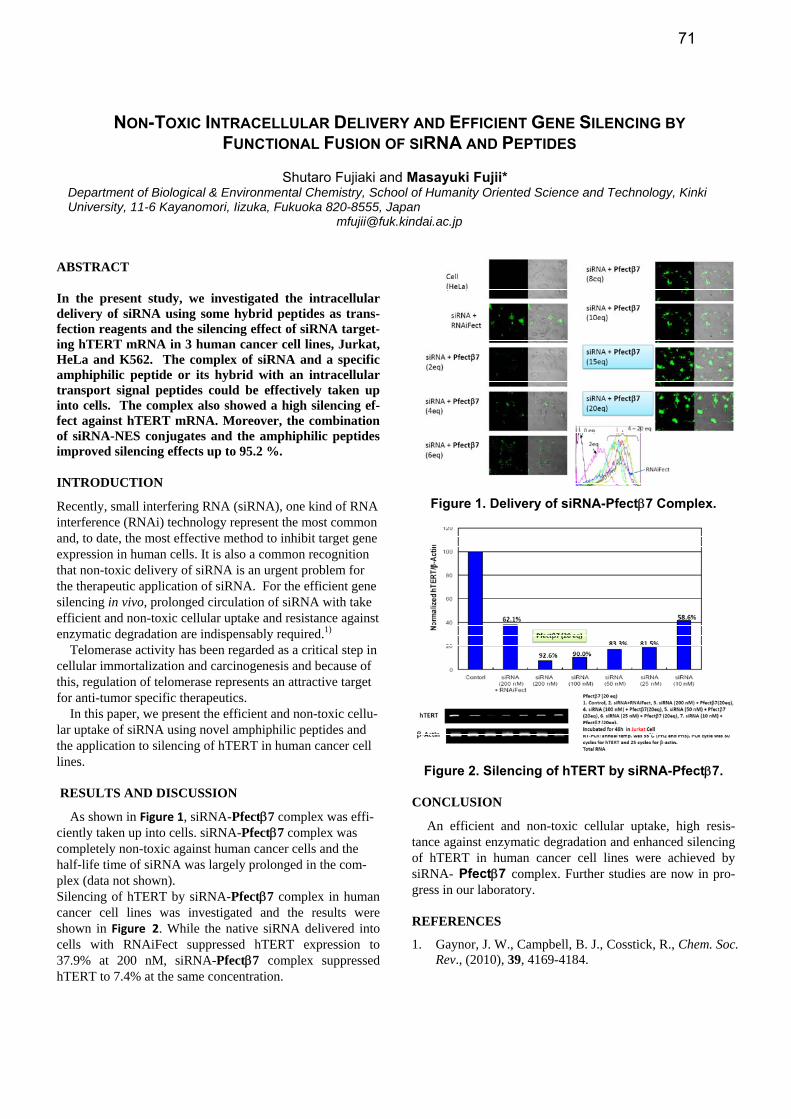
As shown in Figure 1, siRNA-Pfectβ7 complex was effi-ciently taken up into cells. siRNA-Pfectβ7 complex was completely non-toxic against human cancer cells and the half-life time of siRNA was largely prolonged in the com-plex (data not shown). Silencing of hTERT by siRNA-Pfectβ7 complex in human cancer cell lines was investigated and the results were shown in Figure 2. While the native siRNA delivered into cells with RNAiFect suppressed hTERT expression to 37.9% at 200 nM, siRNA-Pfectβ7 complex suppressed hTERT to 7.4% at the same concentration.
Figure 1. Delivery of siRNA-Pfectβ7 Complex.
Figure 2. Silencing of hTERT by siRNA-Pfectβ7.
CONCLUSION
An efficient and non-toxic cellular uptake, high resis-tance against enzymatic degradation and enhanced silencing of hTERT in human cancer cell lines were achieved by siRNA- Pfectβ7 complex. Further studies are now in pro-gress in our laboratory.
REFERENCES
1. Gaynor, J. W., Campbell, B. J., Cosstick, R., Chem. Soc. Rev., (2010), 39, 4169-4184.
71

SYNTHESIS AND PROPERTIES OF ISO-BICYCLO DNA
Anna-Barbara Gerber, Christian Leumann*
Department of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. * Corre-spondence to: [email protected]
ABSTRACT
We have synthesized novel iso-bicyclo nucleosides con-taining the all four bases A, C, G and T and incorpo-rated them into DNA. Tm measurements showed that the modifications can have stabilizing effects on DNA duplexes.
INTRODUCTION
Oligonucleotides for therapeutic purposes gained additional interest in the last years due to the discovery of RNA inter-ference.
1 The best results to cover these features were
shown with conformationally constrained oligonucleotides. A well known example is LNA.
2 In our group we developed
different types of constrained oligonucleotides in recent years, such as bicyclo
3 or tricyclo DNA
4 (Fig. 1). In still
ongoing experiments we are investigating their structure-affinity relationship in complementary binding to DNA and RNA. In this context we currently became interested in the iso-bicyclo-DNA (Fig. 1B).
Figure 1. A bc-DNA, B. iso-bc-DNA, C. tc-DNA
RESULTS AND DISCUSSION
The synthesis starts from the known intermediate 15. The
sugar unit 2 is obtained in 5 steps via hydroboration and
defunctionalization of the 5’ hydroxy group.
Nucleosides were obtained via Vorbrüggen conditions. In
further steps the 6’ hydroxy group was inverted and the nu-
cleosides were DMT protected and the 3’ phosphoramidite
function was introduced.
O
O
OH
TBSO
O
O
AcO
OAc
O
O
AcO
OAc
O
B
AcO
OAc
O
B
DMTO
O P
N
O
CN
1 2
2 B = ABz (3)B = T (4)B = CBz (5)B = 2-aminopurine (6) B = ABz (7)
B = T (8)B = CBz (9)B = GiBu (10)
5 steps
Base, BSA, TMSOTf, MeCN 7 steps
Figure 2. Synthetic route of iso-bicyclo nucleosides A, C, G and T
Oligonucleotide synthesis was performed using standard
phosphoramidite chemistry. The following sequences con-
taining iso-bicyclo T residues were synthesized. Table 1
shows the Tm data of the oligonucleotides with complemen-
tary DNA or RNA.
Sequences Tm vs. DNA [°C] Tm vs. RNA [°C]
S1: 5'-d(GGAtGTTCTCGA)-3' 48 (+1) 49 (0)
S2: 5'-d(GGATGttCTCGA)-3' 49 (+2) 47.5 (-1.5)
S3: 5'-d(GGATGTTCtCGA)-3' 48 (+1) 50 (+1)
S4: 5'-d(GGAtGttCtCGA)-3' 51 (+4) 49.5 (+0.5)
S5: 5'-d(ataatttaataa)-3' 25 (+2) ‹10
S6: 5'-d(ttattaaattat)-3' 26 (+3) ‹10
S7: 6'-d(cctacaagagct)-3' 51 (+4) 30 (-8.5) Table 1. Synthesized DNA sequences with iso-bicyclo modifica-
tions (small letter t) and their corresponding Tm values. In paren-
theses the deviation in Tm of the corresponding duplex to the natu-
ral duplex (DNA/DNA or DNA/RNA resp.) is shown.
CONCLUSION
The iso-bicyclo modification stabilizes duplexes with com-
plementary DNA. This is shown with isolated or consecu-
tive modifications in S1-S4 and with fully modified oligo-
nucleotides S5 and S7.
In the case of RNA, single incorporations seem to be stabi-
lizing whereas consecutive modifications and fully modified
oligos destabilize the duplex. The fully modified strand S7
containing all four bases confirms this findings
Investigations into the biological properties of iso-bicyclo
DNA, such as RNase H activity and serum stabilty, are cur-
rently in progress.
REFERENCES
1. J. Kurreck, Angew. Chem. Int. Ed., 2009, 48, 1378-1398
2. S.K. Singh, P. Nielsen, A.A. Koshkin, J. Wengel, J.
Chem. Soc. Chem. Commun, 1998, 4, 455-456
3. M. Tarköy, M. Bolli, B. Schweizer, C.J. Leumann, Helv.
Chim. Acta, 1993, 76, 481-510.
4. D. Renneberg, C.J. Leumann, J. Am. Chem. Soc., 2002,
124, 5993-6002.
5. R. Steffens, C. Leumann, Helv. Chim. Acta., 1997, 80,
2426-2439
O
O
O
B
H
DNA
DNA
O
O
BO
H
DNA
DNA O
O
O
B
H
DNA
DNA
A B C
72

FURAN-MODIFIED OLIGONUCLEOTIDES FOR TRIPLEX DNA CROSS-LINKING
Ellen Gyssels,1 Emma Vercruysse,1 Marieke Op de Beeck1 and Annemieke Madder1*
1University of Ghent, Laboratorium for Organic and Biomimetic Chemistry, Krijgslaan 281 S4, 9000 Ghent, Belgium * Correspondence to: [email protected]
ABSTRACT
Recently, an efficient and selective nucleic acid duplex cross-link strategy was developed in our laboratory [1, 2]. A furan-modified oligonucleotide is synthesized and af-ter hybridization, the furan moiety is selectively oxidized upon which an interstrand cross-link (ICL) is immedi-ately formed. This strategy has now been further inves-tigated for cross-linking in a triplex DNA context.
INTRODUCTION
Triplex forming oligonucleotides (TFO) recognize a dou-ble helix in a highly specific way. For this reason TFOs of-fer applications in gene therapy and diagnostics [3, 4]. A short polypyrimidine oligonucleotide can bind into the ma-jor groove of a purine rich part of a duplex by Hoogsteen hydrogen bonds, forming a parallel triplex. To enhance the stability of the resulting triplex, an interstrand cross-link can be introduced. A selective and efficient strategy to induce nucleic acidcross-linking is through incorporation of a fu-ran-modified building block in an oligonucleotide. This strategy has proven his efficiency in duplex DNA cross-linking and is now investigated for triplex cross-linking.
RESULTS AND DISCUSSION
A furan-modified oligonucleotide is synthesized and after hybridization with the complementary duplex the furan moiety can be oxidized selectively using N-bromosuccinimide (NBS). The oxidation product is a 4-oxo-enal functionality which is susceptible to nucleophilic attack of one of the opposite nucleobases and in this way a stable covalent bond is formed. In this project we further illustrate the applicability of this cross-link strategy in a triplex DNA context.
First, a furan-modified building block was synthesized and introduced into triplex forming oligonucleotides. It could be observed that across-linked adduct is only formed with a cytosine or adenine near the modification in one of the target duplex strands. Furthermore, cross-linking is only observed if the furan modification is introduced at the 3’-end of the TFO and no cross-linking occurs with internally modified TFOs
As cross-link yields are rather low in comparison with duplex cross-linking, we attempted to optimize the reaction by introducing a mismatch in the duplex at the position of the furan moiety and by working at higher temperatures, in an attempt to destabilize the Watson-Crick base pairing and enhance the nucleophility of the bases.
Therefore, experiments were performed at higher tem-
peratures. Indeed, in this way it was shown that the duplex is disturbed and the nucleophiles of the bases are more available for the cross-link reaction. In a further attempt to improve the yield, a mismatch was introduced in the duplex exactly across the furan-modification. We have been able to clearly demonstrate that in this way, the desired cross-linked adduct is indeed formed in higher yields. Alternative furan containing building blocks have further been designed to improve upon triplex cross-linking.
CONCLUSION
The applicability of an efficient and selective furan-oxidation cross-link strategy has been illustrated in a triplex DNA context. Cross-linked adducts are selectively formed with cytosine or adenine. Yields of the cross-link reaction are improved by increasing the dynamics in the target du-plex.
REFERENCES 1. Op de Beeck, M. and Madder, A., J. Am. Chem. Soc.,
2011, 133(4), 796-807 2. Stevens, K., Claeys, D., Chem. Eur. J., 2011, 17(25),
6940-6953 3. Guntaka R.V., Varma B.R., Int. J. of Biochemistry and
Cell Biology, 2003, 35, 22-31 4. Duca, M., Vekhoff, P., Nucleic Acids Research, 2008,
36(16), 5123-5138
ACKNOWLEDGMENT
E. Gyssels is indebted to the Agency for Innovation by Science and Technology in Flanders (IWT). We further thank Jos Van den Begin and Jan Goeman for technical support and mass analysis.
Figure 1. Interstrand cross-link formation in triplex DNA
73

CROSSLINK FORMING OLIGONUCLEOTIDE AS A STERIC TERMINATOR OF TRANSLATION
Shinya Hagihara,* Shuhei Kusano, Nao Iwamoto and Fumi Nagatsugi
1Institute of Multidisciplinary Research for Advanced Materials, Tohoku University,
2-1-1 Katahira, Aoba-ku, Sendai-shi, Miyagi 980-8577, Japan. * Correspondence to: [email protected]
ABSTRACT
The development of convenient methods for control-
ling the protein expression is an important challenge in
the postgenomic era. In this study, we applied the cross-
link forming oligonucleotide (CFO) as a terminator of
the ribosomal translation. In vitro and in cell translation
experiments revealed that the crosslinked mRNA can
produce the truncated proteins in which the translation
terminates at the desired position.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
Artificial regulation of the protein expression is an essential technology for the effective utilization of genomic information in the biological science and clinical use. For this purpose, oli-gonucleotides (ONs) complementary to target mRNAs have been widely used to disrupt the gene expression via RNase H-dependent or -independent manner. However, the production of a truncated protein using antisense ONs has been difficult due to the instability of the RNase H-digested mRNA and the inef-ficient steric blockage of the antisense ONs. As of now, few studies using poly-pyrimidine PNA, which forms the PNA2:RNA triplex, have shown that ONs can halt elongating ribosomes.
One of the ways to achieve the steric blockage without a se-quence limitation is to create a crosslink; i.e., an irreversible complex between the mRNA and ONs. We have reported the synthesis and evaluation of the 2’-OMe oligonucleotide con-taining 2-amino-6-vinylpurine (AVP, Figure 1a), in which AVP is a crosslink-forming nucleobase possessing a hydrogen-bonding pattern complementary to thymine (T) or uridine (U). The base-pairing of AVP with T or U in a duplex induces the interstrand covalent bond formation. We demonstrated that 2’-OMe ON bearing AVP forms a crosslink with the complemen-tary DNA at the T residue across from AVP under physiologi-cal condition; however, the crosslink formation with the U res-idue in RNA at neutral pH proceeded with a slightly low yield.
In this paper, we designed two AVP-containing 2’-OMe ONs (CFO1 and CFO2), which are complementary to the cod-ing region of firefly luciferase mRNA and consist of two dif-ferent sequence contexts around AVP, 5’-UXU-3’ and 5’-CXG-3’, respectively (Figure 1b). The crosslink reactivity of these CFOs to their complementary oligoribonucleotides was analyzed by denaturing PAGE and plotted versus the incuba-tion time (Figure 1c). It is apparent that the reactivity of CFO1 was quite low at neutral pH. This is consistent with our previ-ous results using CFO composed of the 5’-UXA-3’ context. In contrast, CFO2 showed a much higher reactivity at pH 7 than CFO1 did. This enhancement of the crosslink reactivity sug-
gests that the high-order structure of CFO–RNA duplex strong-ly influences the crosslink reactivity of AVP.
The firefly luciferase mRNA incubated with CFO1 or CFO2 at pH 7.0 was applied to in vitro translation using a rabbit retic-ulocyte extract. The luciferase activity of the translation prod-uct was plotted versus the incubation time for the crosslink formation (Figure 1d). The incubation of mRNA with CFO1 resulted in almost no effect on the translation. In contrast, CFO2 showed the time-dependent inhibition of the ribosomal translation. The time course of the translational inhibition with CFO2 was identical to that of the crosslink formation, indicat-ing that CFO2 could bind to the long mRNA as efficiently as to the short ORN. These data suggest that covalent bond between CFO and mRNA resists the helicase activity of the translating ribosome, and thereby, inhibits the translation.
In order to confirm the expression of the truncated proteins from the crosslinked mRNA, the translation products were ana-lyzed by SDS-PAGE. The fluorescent band at 60 kDa and 30 kDa represents the translation product of the full-length firefly luciferase and the back ground band derived from rabbit reticu-locyte lysate, respectively. Upon formation of the crosslink with CFO2, the full-length luciferase was completely dimin-ished with the concomitant appearance of a new band at 28 kDa. Taking into account the sequence information, this band is con-sidered to be a truncated protein, at which the translation was stalled at the crosslink.
In conclusion, our results clearly show that our CFOs can be considered to be a reliable approach for synthesizing truncated proteins in cells without any genetic engineering. Our current objective is to improve the crosslink reactivity inside the cells.
Figure 1. Production of truncated proteins using CFOs. (a) Schematic illustration of the truncated protein expression with CFO. (b) CFO sequences that targeted the coding region of the firefly luciferase mRNA. (c) The reactivity of the CFO1 (dia-mond) or CFO2 (square) toward fluorescently labeled ORN (d) suppression of luciferase activity by the crosslink formation. (e) The in vitro translation product was analyzed by SDS-PAGE.
74

SYNTHESIS AND HYBRIDIZATION PROPERTY OF OLIGONUCLEOTIDES CONTAINING 2’,4’-BNA-7-DEAZAGUANINE ANALOG
Takashi Hara1, Tetsuya Kodama
1,2*, Yumi Takegaki
1, Kunihiko Morihiro
1, Kosuke Ramon Ito
1, and
Satoshi Obika1*
1Graduate School of Pharmaceutical Sciences, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan.
2Guraduate School of Pharmaceutical Sciences, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8601,
Japan.*Correspondence to: [email protected]; [email protected]
ABSTRACT
2’,4’-BNA-7-deazaguanine (BNA-7c
G) and 2’,4’-BNA-8-
aza-7-deazaguanine (BNA-8n7c
G), which evade extra hy-
drogen bond formation at the 7 position, were synthe-
sized and incorporated in oligonucleotide. BNA-7c
G has
high duplex-forming ability with complementary DNA
and RNA. In contrast, BNA-8n7c
G has low binding affin-
ity compared to natural guanine base.
INTRODUCTION
Guanine-rich segments on nucleic acid are able to create
inter- and intrastrand hydrogen bonding in itself to form
higher-order structure, which are held by Hoogsteen hydro-
gen bonding at the N7 position of guanine. Therefore, the
handling of nucleic acid containing guanine-rich segments
become highly problematical. Base modified guanine nucle-
osides have been used to overcome these problems. 7-
Deazaguanosine analogs1,2
are some of such nucleosides.
These nucleosides do not form Hoogsteen hydrogen bond-
ing because they do not have hydrogen-bond acceptor at the 7 position, but are able to form Watson-Crick hydro-
gen bonding. Meanwhile, we have developed bridged nucle-
ic acid 2’,4’-BNA whose sugar conformation is locked into
a N-type3,4
. Because of this conformation, 2’,4’-BNA exhib-
its high binding affinity for complementary ssRNA. Due to
this fact, we designed BNA-7c
G and 8n7c
G as nucleic acid
units to be used in place of guano-
sine at guanine rich oligonucleo-
tides (Figure 1).
In this study, we synthesized
novel 2’,4’-BNA monomers with
7-deazaguanine and 8-aza-7-
deazaguanine, incorporated them
into oligonucleotide, and
evaluated the duplex-forming
ability with ssDNA and
ssRNA.
RESULTS AND DISCUSSION
The novel BNA with 7-deazaguanine was synthesized as
shown in Scheme 1. Compound 35 was treated with suger 4
6,
BSA and TMSOTf to give 5. Ring-closing reaction, deben-
zylation and deiodination afforded 2’,4’-BNA nucleoside 6.
Tritylation and the following phosphitylation yielded phos-
phoroamidite 7. Compound 7 was successfully introduced
into oligonucleotide ON-1 using an automated DNA synthe-
sizer with a conventional phosphoramidite protocol. oligo-
nucleotide ON-2 containing BNA-8n7c
G was synthesized by
similar way. The duplex-forming ability of the oligonucleo-
tides (ON-1 and 2) with ssDNA and ssRNA was evaluated
by Tm measurement. As a result, BNA-7c
G exhibited a great
increased stability of duplex formed with ssRNA. However,
BNA-7c8n
G significantly decreased in the Tm value with
ssDNA and ssRNA compared with BNA-7c8n
G.
Scheme 1. Reagents: a) BSA, TMSOTf; b) 3-hydroxypropionitrile;
c) BCl3; d) Pd/C, H2; e) DMTrCl; f) i-Pr2NP(Cl)OCH2CH2CN, i-
Pr2NEt
CONCLUSION
We have achieved the synthesis of BNA-7c
G and BNA-8n7c
G phosphoramidites and introduction into oligonucleo-
tides. UV melting experiments demonstrate that BNA-7c
G
show high binding affinity with complementary DNA and
RNA. On the other hand, BNA-7c8n
G decreased in the bind-
ing affinity with ssDNA and ssRNA.
REFERENCES
1. Seela, F., Kaiser, K. Helv. Chim. Acta, 1988, 71, 1813-
1823.
2. Seela, F., Winker, H.-D. J. Org. Chem., 1983, 48, 3119-
3122.
3. Obika, S., Nanbu, D., Hari, Y., Morio, K., In, Y., Ishida,
T., Imanishi, T. Tetrahedron Lett., 1997, 38, 8735-8738.
4. Singh, S. K., Nielsen, P., Koshkin, A. A., Wengel, J.
Chem, Commun., 1998, 455-456.
5. Klepper, F., Jahn, E.-M., Hickmann, V., Carell, T. An-
gew. Chem. Int. Ed., 2007, 46, 2325-2327.
6. Rahman S. M. A., Seki S., Obika S., Yoshikawa H.,
Miyashita K., Imanishi T., J. Am. Chem. Soc., 2008,
130, 4886-4896.
Figure 1. Structure of BNA-7cG (1) and BNA-8n7cG (2)
75

NUCLEASE RESISTANT OLIGONUCLEOTIDES WITH CELL PENETRATING PROPERTIES
Dmytro Honcharenko*, Stefan Milton, Jyotirmoy Maity and Roger Strömberg*
Department of Biosciences and Nutrition, Karolinska Institute, Huddinge, S-14183, Sweden.* Correspondence to: [email protected], [email protected]
ABSTRACT
Cell penetrating oligonucleotides (CPOs) have been
developed. These modified oligonucleotides are also nu-
clease resistant and give stabilised duplexes with target
RNA.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
Oligonucleotide therapy is still strongly limited by ineffi-
cient in vivo delivery and in many cases by the stability of
potential drug in extra- and intracellular fluids. Delivery of
oligonucleotides for the splice-switching or other therapy
where the process takes place in the cell nucleus means to
that the oligonucleotide has to enter not only into the cell
but also to accumulate in the nucleus.
We present recently developed cell penetrating oligonu-
cleotides (CPOs) that have high potential to be used as ther-
apeutic oligonucleotides. Because of their nature these
CPOs are capable of entering cells without any assistance,
such as cationic lipids or cell penetrating peptides that often
come with toxicity issues. Confocal microscopy reveals that
a similarly fluorescein-labelled non-modified oligodeox-
ynucleotide is not taken up at all while the modified CPOs
are spontaneously taken up into the cellular interior. Se-
quences containing modified nucleotide units also demon-
strate high resistance towards degradation by Snake Venom
and Spleen phosphodisterases and high stability in human
serum. Oligonucleotides containing CPO modifications also
displayed enhanced stability of the corresponding duplexes
formed with target RNA.
Initial studies were made on oligomers containing modi-
fied adenosine monomers. For the preparation of fully modi-
fied splice-switching CPOs methods for synthesis of mono-
mers from all natural nucleosides has also been developed.
Synthesis of building blocks with modified pyrimidine and
purine bases carrying the CPO functionality has been per-
formed. Fully modified oligonucleotides (with a sequence
known to cause splice-switching in an in vitro luciferase
reporter cell assay) are in the process of being made and
tested for splice-correction. To increase efficacy the CPOs
are also being equipped with other entities that further pro-
mote uptake or localisation.
76

G
G
G
G
G
G C
C
C
C
C
C
CC
T
T
T
TT
T
T A
A
A
A
A
A
A
A5ʹ 3ʹ
POST-SELEX MODIFICATION OF A STREPTAVIDIN-BINDING APTAMER BY INTRODUCTION OF LNA
Anna S. Jørgensen* and Jesper Wengel
Nucleic Acid Center, Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense M, Denmark. * Correspondence to [email protected]
ABSTRACT
To increase the biostability of aptamers they are
usually subjected to chemical post-SELEX-modifications.
A well-established streptavidin-binding aptamer has
been post-SELEX-modified by introducing single LNA
nucleotides at various positions.
INTRODUCTION
Aptamers are small single-stranded oligonucleotides
composed of either DNA or RNA. They fold into sequence-
dependent tertiary structures which give them high affinity
and specificity for their target. Aptamers are applicable for
several purposes, in particular in the fields of diagnostics
and therapeutics.1 Aptamers are prone to nuclease degrada-
tion and therefore need to be stabilized for in vivo use. This
is usually done by chemical modification. LNA (Locked
Nucleic Acid), a 2ʹ-O,4ʹ-C-methylene linked bicyclic ribo-
nucleoside locked in the 3ʹ-endo conformation, represents
an obvious modification for increasing biostability. LNA-
containing oligonucleotides have high affinity and specifici-
ty towards DNA and RNA, display excellent mismatch dis-
crimination, increase the nuclease resistance, and are non-
toxic. To date LNA has found widespread use in antisense
oligonucleotides, DNAzymes, siRNA, and anti-microRNA.2
LNA stabilizes duplex regions and consequently has
great potential for being incorporated into stem regions of
aptamers, hereby improving the thermo- and biostability of
the aptamers. The LNA-modified ricin RNA aptamer is the
most recent example of an aptamer being post-SELEX mod-
ified by the incorporation of LNA.3
Streptavidin is a tetrameric protein widely used in bio-
chemistry due to its extremely tight binding to biotin. Strep-
tavidin has also been used as a model system for aptamer
evolution. This has resulted in different streptavidin-binding
aptamers (SBAs) all predicted to have the same secondary
motif.4 The predicted secondary motif is a stem-bulge-stem-
loop structure in which the nucleotides in the bulge and the
loop are conserved. The nucleotides in the stem regions can
be varied without affecting the affinity making them ideal
for LNA-substitutions (Figure 1).
The SBA St-2-1 developed by Bing et al.5 was chosen as
a starting point for the LNA-optimization study, since this
sequence already has been studied in detail.4,5
Figure 1. A) Structure of LNA. B) Structure of the streptavidin binding aptamer St-2-1 developed by Bing et al.5
RESULTS AND DISCUSSION
13 LNA modified SBAs have been synthesized each with
a single LNA-substitution, except for a truncated version
having three consecutive LNAs in the terminal stem.
Surface plasmon resonance binding affinity studies are on-
going, and the LNA modified SBAs will be compared with
the parent unmodified St-2-1 DNA aptamer.
The expected outcome of this study is a LNA-containing
streptavidin-binding aptamer with minimum size, high bio-
stability, and high binding affinity.
CONCLUSION
To examine the influence of LNA-substitution in stem re-
gions of aptamers, 13 LNA-modified streptavidin-binding
aptamers have been synthesized. Binding affinity studies are
ongoing and will be presented.
REFERENCES
1. Mayer, G. Angew. Chem. Int. Ed. 2009, 48, 2672 –
2689.
2. Vester, B., Wengel, J. Biochemistry 2004, 43, 13233-
13241
3. Förster, C., Zydek, M., Rothkegel, M., Wu, Z., Gallin,
C., Geßner, R., Lisdat, F., Fürste, J. P., Biochem. Bio-
phys. Res. Commun., 2012, 419, 60-65
4. Ruigrok V.J.B., van Duijn, E., Barendregt, A., Dyer, K.,
Tainer, J.A., Stoltenburg, R., Strehlitz, B., Levisson, M.,
Smidt, H., van der Oost, J., ChemBioChem 2012, 13,
829-836
5. Bing, T., Yang, X., Mei, H., Cao, Z., Shangguan, D.
Bioorg. Med. Chem. 2010, 18, 1798-1805
A) B)
77

SYNTHESIS OF CYCLIC AZOBENZENE ANALOGUES FOR INCORPORATION INTO
OLIGONUCLEOTIDES
Dhruval K. Joshi,a Doug Bruce,
b Hongbin Yan
a*
aDepartment of Chemistry, Brock University,500 Glenridge Ave. St. Catharines, ON, L2R 3A1, Canada
bDepartment of Biological Sciences, Brock University, Glenridge Ave, St. Catharines, ON, L2R 3A1, Canada
* Correspondence to: [email protected]
ABSTRACT
Five analogues (chloro-, bromo-, cyano-, carboxyl-, and
amido) of cyclic azobenzene were synthesized from 2,2’-
dinitro dibenzyl. The amido analogue readily undergoes
reversible photoisomerization.
INTRODUCTION
Photo- and thermoisomerizable aromatic azo
compounds have been extensively explored, and have found
applications as dyes, pigments, radical initiators, and thera-
peutic agents.1 Among the different aromatic azo
compounds, cyclic azobenzene have shown characteristic
photoisomerization property (ZE and EZ) when excited
by light using selected wavelength.2 Due to the ethylene
bridge the (Z)-isomer of cyclic azobenzene is thermodynam-
ically more stable than the (E)-isomer, which is in contrast
to linear azobenzene.2 The (Z)-isomer of the cyclic azoben-
zene can be switched to (E)-isomer with an efficiency of
90% by irradiation with blue light (370-400 nm), whereas
the (E)-isomer can be switched back to (Z)-isomer in ~100%
efficiency by green light (480-550 nm).2 Since the irradia-
tion wavelengths used for photo-swtiching of cyclic azoben-
zene are away from the UV region, this system is more like-
ly to be tolerated in biological systems compared to linear
azobenzene, where irradiation with UV light is required.
RESULTS AND DISCUSSION
A series of cyclic azobenzene analogues 2 (chloro-
bromo- cyano-, and carboxyl-) as well as unsubstituted
cyclic azobenzene were synthesized from 2,2'-
dinitrodibenzyl 1 (Scheme 1).
Scheme 1. Synthesis of cyclic azobenzene analogues from 2,2'-
dinitrodibenzyl.
The carboxylic acid analogue was then treated with D-
threoninol to give the corresponding amide 3.
The amide substituted cyclic azobenzene 3 readily
undergoes photoswitching upon irradiation in a fashion sim-
ilar to unsubstituted cyclic azobenzene2 with light at select-
ed wavelengths. The extent of photoisomerization of amide
substituted cyclic azobenzene as well as unsubstituted cyclic
azobenzene was studied by HPLC. The UV/Vis spectra of
the (Z)- and (E)-isomers of the amide substituted cyclic azo-
benene 3 were documented.
This D-threoninol modified cyclic azobenzene 3 is cur-
rently being incorporated into oligonucleotides (Scheme 2)
for spatiotemporal control of nucleic acid functions.
Scheme 2. Synthesis of cyclic azobenzene phosphoramidite.
CONCLUSION
Five analogues of cyclic azobenzene (unsubtituted, chlo-
ro-, bromo-, cyano-, and carboxyl) were synthesized from
2,2'-dinitrodibenzyl. Reaction of carboxyl-substituted cyclic
azobenzene with D-threoninol gave corresponding amide
substituted cyclic azobenzene 3 which readily undergoes
photoisomerization upon illumination with light in the visi-
ble region. Incorporation of amide linked cyclic azobenzene
3 into oligonucleotides using the phosphoramidite chemistry
is in progress. These oligonucleotides will be evaluated in
spatiotemporal control of nucleic acid functions.
REFERENCES 1. Merino, E. Chem. Soc. Rev. 2011, 40, 3835-3853.
2. Bandarab, H. M. D., Burdette, S. C. Chem. Soc. Rev.
2012, 41, 1809-1825.
3. Siewertsen, R., Neumann, H., Buchheim-Stehn, B.,
Herges, R., Näther, C., Renth, F., Temps, F. J. Am.
Chem. Soc. 2009, 131, 15594-15595.
4. Asanuma, H., Liang, X., Nishioka, H., Matsunaga, D.,
Liu, M., Komiyama, M. Nat. Protocols 2007, 2, 203-
212.
78

CHEMICALLY MODIFIED SIRNAS WITH NOVEL RIBOSUGAR MODIFICATIONS
Gopalan Rajeev Kallanthottathil,1* Sudhakar R. Takkellapati,1 Klaus Charisse,1 Satya Kuchimanchi,1
Martin A. Maier,1 Kondi Santhoshi,2 Subir Sabui,2 Shyamapada Banerjee,2 Yogesh S. Sanghvi3 and Muthiah Manoharan1
1Alnylam Pharmaceuticals, 300 Third Street, Cambridge, MA 02142, USA; 2Sapala Organics Pvt. Ltd., Plot No. 146B & 147, IDA Mallapur, Hyderabad 500 076 (A.P.), India; 3Rasayan Inc., 2802 Crystal Ridge Road, Encinitas, CA 92024-
6615, USA. * Correspondence to: [email protected]
ABSTRACT
Chemical modifications are known to offer “drug-like”
properties and modulate therapeutic characteristics such
as biostability, immune stimulation and pharmacology of
short interfering RNAs (siRNA). The acceptance of ex-
tent of chemical modification on sense (or passenger)
and antisense (or guide) strands are determined by the
nature and placement of the chemical modification in
the oligonucleotide sequence in each strand. In the pre-
sent work, we describe design and synthesis of xylo-
sugar modified nucleoside phosphoramidites with meth-
oxy and fluoro modification at the 3′-postion on the sug-
ar moiety. Incorporation of these novel modifications at
desired positions on sense and antisense strands of siR-
NA, impact of these modifications on nuclease stability
and gene silencing activity will also be presented.
79

SYNTHESIS OF OLIGONUCLEOTIDE GLYCOCONJUGATES USING SEQUENTIAL CLICK AND
OXIMATION LIGATIONS
Marika Karskela,* Mia Helkearo, Pasi Virta and Harri Lönnberg
Department of Chemistry, University of Turku, FIN-20014 Turku, Finland. * Correspondence to: [email protected]
ABSTRACT
Oligodeoxyribonucleotide glycoconjugates bearing two trivalent glycoclusters have been synthesized by two alternative methods based on solid-supported oximation of aminooxy functionalized oligonucleotides with gly-coclusters constructed by click chemistry.
INTRODUCTION
The applicability of oligonucleotide-based drugs is lim-
ited, in particular, by poor cellular uptake. A possible way to
enhance cell penetration and to provide the oligonucleotide
with cell-type or organ selectivity is to conjugate the oligo-
nucleotide with an agent known to be actively transported
into the cell. [1] Multiantennary glycoconjugates show po-
tential as agents with which oligonucleotides, including
siRNA and antisense oligonucleotides, could be enriched on
the surface of desired cell-types. [2] High affinity binding to
membrane-anchored lectins may require simultaneous inter-
action with several appropriately situated sugar ligands with
the protein, a phenomenon known as a glyco-cluster effect.
RESULTS AND DISCUSSION
In the present study [3], click chemistry has been used to
synthesize glycoclusters which have been further ligated to
oligonucleotides using on-support oximation. Two diverse
phosphoramidite reagents (1 and 2, Figure 1) bearing
masked aminooxy functions enable the attachment of alde-
hyde functionalized glycoclusters (3 and 4) either to the 5’-
end or to a desired position in the sequence. In addition, the
latter method enables the combination of diverse glycoclus-
ters within a single oligonucleotide.
Figure 1. Prepared phosphoramidite reagents and glycoclusters.
Tripodal glycolusters 3 and 4 were assembled using cop-
per-catalyzed 1,3-dipolar cycloaddition reaction to attach
azido-functionalized gluco- and mannopyranoside deriva-
tives on an aldehyde functionalized core.
Phosphoramidate 1 with two phthaloyl protected ami-
nooxy functions was used to synthesize an oligodeoxyribo-
nucleotide with two glucose clusters in the 5'-end (5, Figure
2). After chain assembly and oximation step on support, the
conjugate was released and globally deprotected by ammo-
nolysis, followed by RP HPLC purification.
Phosphoramidate 2 was used to synthesize a glycoconju-
gate with two different glycoclusters in the 3'-end (6). After
coupling of the first aminooxy unit, oximation with man-
nose cluster 4 was performed. Thymidine was inserted be-
tween the adjacent aminooxy units and second oximation
step was carried out using glucose cluster 3. The rest of the
oligodeoxyribonucleotide chain was synthesized and the
product was obtained after ammonolysis and RP HPLC pu-
rification.
Figure 2. Synthesized oligonucleotide glycoconjugates.
REFERENCES
1. Manoharan, M. Antisense Nucleic Acids Drug Dev.
2002, 12, 103-108.
2. Yan, H., Tram, K. Glycoconj. J. 2007, 24, 107-123.
3. Karskela, M., Helkearo, M., Virta, P., Lönnberg, H.
Bioconjugate Chem. 2010, 21, 748-755.
80

Synthesis of urea-substituted microRNAs possessing nuclease-resistance
and their anticancer activities
Y. Kitade,1,2
S. Ogawa,2 Y. Masegi,
2 R. Nakashima,
1 M. Kandeel,
1 Y. Kitamura,
2 M. Ikeda,
2 Y. Akao,
1
1United Graduate School of Drug Discovery and Medical Information Sciences, Gifu University,
2Department of Bio-
molecular Science, Faculty of Engineering, Gifu University, Yanagido, Gifu 501-1193, Japan: [email protected]
ABSTRACT
The chemically modified micro(mi)RNA-143s having
urea or thiourea substituents at the 3’-overhang position
of RNA were synthesized. These urea or thiourea substi-
tuted miRNAs showed greater growth inhibitory effect
on human colon cancer than wild-type miRNA-143 and
moderate nuclease resistance.
INTRODUCTION
MicroRNAs (miRNAs), short non-coding RNAs, regulate
gene expression by controlling mRNA translation or degra-
dation. The roles of miRNAs in carcinogenesis have been
documented in many literatures. The 3’-overhang region of
miRNA duplex was recognized by the PAZ domain in the
Ago2, and the 2-nucleotide overhang at 3’-end of RNA se-
quence was accommodated into a binding pocket composed
of hydrophobic amino acids in the PAZ domain.
We have recently found that mature type of miRNA-143s
modified at the 3’-overhang region exhibits greater growth
inhibitory effect on human colon cancer than wild-type
miRNA-143.1-3
In order to enhance anticancer activity, we
designed artificial miRNA-143 possessing urea or thiourea
groups at the 3’-overhang position of RNA molecules and
evaluated their properties.
RESULTS AND DISCUSSION The chemically modified microRNA-143s having urea or
thiourea substituent at the 3’-overhang position of RNA
were synthesized in good yield by using a DNA/RNA syn-
thesizer. These urea or thiourea substituted miRNAs
showed greater growth inhibitory effect on human cancer
cells than wild-type miRNA-143. These modified miRNA-
143s also exhibited moderate nuclease resistance.
CONCLUSION We demonstrated the structure-activity relationship between
chemical modification on miRNAs having urea or thiourea
moieties at the 3’-overhang position of RNA and their bio-
logical properties such as anticancer activity and nuclease-
resistance. Thus, the urea or thiourea modification at the 3’-
overhang position of miRNAs may hold promise as a meth-
od to improve the anticancer activity and nuclease-
resistance of RNA medicines.
REFERENCES
1. Akao, Y., Nakagawa, Y., Kitade, Y., Kinoshita, T.,
and Naoe, T., Down-regulation of microRNAs-143
and -145 in B-cell malignancies, Cancer Science, 98,
1914-1920 (2007).
2. Akao, Y., Nakagawa, Y., Hirata, I., Iio, A., Itoh, T.,
Kojima, K., Nakashima, R., Kitade, Y., and Naoe, T.,
Role of anti-oncomirs miR-143 and -145 in human
colorectal Tumors, Cancer Gene Therapy, 17, 398-
408 (2010).
3. Taniho K, Nakashima R, Kandeel M, Kitamura Y,
Kitade Y, Synthesis and biological properties of
chemically modified siRNAs bearing 1-deoxy-D-
ribofuranose in their 3'-overhang region, Bioorg. Med.
Chem. Lett., 22, 2518-2521 (2012).
X
O HN C
Y
NH
X
OHRNA'5
X = CH or NY = O or S
Figure 1. Structure of urea or thiourea substituted microRNAs.
81

INTRACHAIN CLICK-CONJUGATION OF OLIGONUCLEOTIDES
Anu Kiviniemi,1* Pasi Virta
1, Mikhail S. Drenichev
2, Sergey N. Mikhailov
2 and Harri Lönnberg
1
1Department of Chemistry, University of Turku, FIN-20014 Turku, Finland and
2Engelhardt Institute of Molecular Biolo-
gy, Russian Academy of Sciences, Vavilov Street 32, Moscow 119991, Russia. * [email protected]
ABSTRACT
Altogether five different nucleosidic building blocks
that allow the development of post-synthetic click-
conjugation strategies were synthesized in high yield: 4´-
C-azidomethylthymidine 3´-(H-phosphonate) (1), 4´-C-
[N,N-di(4-pentyn-1-yl)aminomethyl]thymidine 3´-
phosphoramidite (2), 4´-C-[N-methyl-N-(4-pentyn-1-
yl)aminomethyl]thymidine 3´-phosphoramidite (3), 2´-O-
[(2-azidoethoxy)methyl]cytidine 3´-(H-phosphonate) (4)
and 2´-O-[(2-bromoethoxy)methyl]cytidine 3´-
phosphoramidite (5). Their utility particularly in gly-
coconjugation was demonstrated with oligonucleotides
bearing one to four sugar ligands.
INTRODUCTION
Click chemistry, i.e. Cu(I)-catalyzed 1,3-dipolar cy-
cloaddition between alkynes and azides provides an efficient
and orthogonal conjugation method for oligonucleotides. To
apply a post-synthetic click conjugation strategy on oligo-
nucleotides, four different 4´- and 2´-modified building
blocks 1-4 bearing azido or alkynyl moiety was introduced
(Scheme 1).1-3
Nucleoside 2 bearing two 4-pentyn-1-yl
groups is designed for high density functionalization of
modified oligonucleotides whereas nucleosides 1, 3 and 4
allow one-armed conjugations.
Scheme 1.
RESULTS AND DISCUSSION
Because of the intramolecular Staudinger reaction be-
tween azido and phosphoramidite groups H-phosphonate
coupling was used for monomers 1 and 4 and otherwise
phosphoramidite method was applied for oligonucleotide
synthesis.
The parallel use of monomers 1-4 with monomer 5 hav-
ing 2´-O-[(2-bromoethoxy)methyl]-branch enables double
click conjugation strategy. That is, two different ligands can
be attached to the oligonucleotide when after first click reac-
tion of monomers 1-4, the same reaction is utilized to con-
jugate the second ligand after conversion of the bromo sub-
stituent in monomer 5 to azido group on solid support
(Scheme 2).
Scheme 2.
Scheme 3.
Scheme 3 presents some of the prepared 4´-conjugates.
The melting temperature studies revealed that 2´- and 4´-
modified nucleosides were well accommodated to the du-
plexes formed with complementary DNA and 2´-O-Me
RNA targets. The 4´-modified oligonucleotides containing
one to four mannose ligands in central part of the chain
formed equally stable duplexes with compl. 2´-O-Me RNA
as the corresponding unmodified DNA sequence. Especially
the conjugates of 4´-C-alkynyl monomers 2 and 3 showed
encouraging hybridization properties and all amino group
bearing conjugates stabilized the DNA∙DNA duplex signifi-
cantly and the DNA∙2´-O-methyl RNA duplex notably.
REFERENCES
1. Kiviniemi, A., Virta, P., Lönnberg, H. Bioconjugate
Chem. 2008, 19, 1726-1734.
2. Kiviniemi, A., Virta, P., Lönnberg, H. Bioconjugate
Chem. 2010, 21, 1890-1901.
3. Kiviniemi, A., Virta, P., Drenichev, M. S., Mikhailov, S.
N., Lönnberg, H. Bioconjugate Chem. 2011, 22, 1249–
1255.
82

Synthesis and Characterization of Cross-Link Induced by N-
nitrosomorpholine in DNA and Oligonucleotides
Niangoran Koissi*, James C. Fishbein
University of Maryland Baltimore County. Department of Chemistry and Biochemistry. 1000 Hilltop Circle, MD 21250 USA. * Correspondence to: [email protected]
ABSTRACT
Synthesized DNA cross-link Ade-ethoxyethyl-Gua has
been used as standard for the study on site-specific Oligo
deoxynucleotides and Calf thymus DNA.
INTRODUCTION
Nitrosomorpholine (NMOR, 1) is a potent liver and esopha-
geal carcinogen1to which there is human exposure through
foods2, groundwater
3, personal care products
4, as well as in
certain workplace environs such as the rubber industry5,6
.
DNA interstrand and intrastrand crosslinks are highly cyto-
toxic lesions formed by a variety of important anti-tumor
agents. Synthetically derived DNA cross-link Ade-
ethoxyethyl-Gua (A-G) has been used as standard for the
study on the characterization and the kinetics of N-
nitrosomorpholine reaction in DNA and site specific oligo-
nucleotides.
RESULTS AND DISCUSSION
We developed a good mass spectrometric method to moni-
tor and characterized the synthesized cross-linked adducts
(Figure 1) in calf thymus DNA treated with N-nitroso-
morpholine and modified oligodeoxynucleoides. To have a
stable structure, we trapped the adduct with a reducing
agent. The A-G crosslinked adducts were measured using
LC-MS/MS. We studied the effect of various conditions
such as temperature, salt concentration, exposure time, and
reducing agents on the crosslink’s yield of formation. The
results have shown that these factors have an effect on the
observed yields.
Preliminary studies have been undertaken based on se-
quence context on oligonucleotides. It has been possible to
estimate the yield of crosslink formed in 6 different site-
specifically modified with O6-Ethoxyacetaldehyde dGuo
phosphoramidite in oligonucleotides. The sequence has an
impact on the observed yield of crosslink.
These results have shown the first established crosslink
from NMOR in DNA and oligonucleotides.
REFERENCES
1. Lijinsky, W. Chemistry and Biology of N-Nitroso
Compounds, 1992, Cambridge University Press, Cambridge,
UK.
2. Sanches-Filho, P.J., Rios, A., Valcarcel, M., Zanin, K.D.,
Caramao, E.B. J. Chromatogr. 2003, A 985, 503-512.
3. Charrois, J.W., Arend, M.W., Froese, K.L., Hrudey,
S.E. Environ. Sci. Technol. 2004, 38, 4835-4841.
4. Spiegelhalder, B. Preussmann, R. J. Cancer Res. Clin.
Oncol. 1984, 108, 160-163.
5. Fajen, J.M., Carson, G.A., Rounbehler, D.P., Fan, T.Y.,
Vita, R. Science 1979, 205, 1262-1264.
6. Oury, B., Limasset, J.C., Protois, J.C. Int. Arch. Occup.
Environ. Health. 1997, 70, 261-271.
1. Lijinsky, W. Chemistry and Biology of N-Nitroso
Compounds, 1992, Cambridge University Press, Cambridge,
UK.
2. Sanches-Filho, P.J., Rios, A., Valcarcel, M., Zanin, K.D.,
Caramao, E.B. J. Chromatogr. 2003, A 985, 503-512.
3. Charrois, J.W., Arend, M.W., Froese, K.L., Hrudey,
S.E. Environ. Sci. Technol. 2004, 38, 4835-4841.
4. Spiegelhalder, B. Preussmann, R. J. Cancer Res. Clin.
Oncol. 1984, 108, 160-163.
5. Fajen, J.M., Carson, G.A., Rounbehler, D.P., Fan, T.Y.,
Vita, R. Science 1979, 205, 1262-1264.
6. Oury, B., Limasset, J.C., Protois, J.C. Int. Arch. Occup.
Environ. Health. 1997, 70, 261-271.
Figure 1. CrossLink Standard used for LC-MS/MS characterization and quantification
O
N
N N
NH
H2N
ONH
N
NN
NH
83
2’-GUANIDINO-FUNCTIONALIZED OLIGONUCLEOTIDES
Venubabu Kotikam* and Vaijayanti A. Kumar
Division of Organic Chemistry, National Chemical Laboratory, Pune 411008, India
* Correspondence to: [email protected] [email protected]
ABSTRACT
Guanidine, a pervasive chemical functional group in
the biological activity/applications is substituted at the
2’-position of pyrimidine nucleosides. The ring pucker-
ing for this novel nucleoside analogue was found to be N-
type as studied by NMR. Its substitution in duplex- and
triplex-forming oligonucleotides, consequent effects on
stability of the modified complexes formed with DNA
and RNA as studied by UV Tm experiments and their
resistance to nucleases is presented in this poster.
INTRODUCTION
Intensive research in the area of modified oligonu-
cleotides for a better antisense candidate has resulted in
some promising modifications at 2’-position of the sugar
ring. The basic chemical difference between DNA and RNA
is the 2’-substituion of the ribose sugar unit. The electro-
negative substituents such as oxygen or fluorine can shift
the ribose conformational equilibrium towards the C3’-endo
pucker that can form stable duplexes with target RNA. Alt-
hough the 2’-OMe1 and 2’-F
2 modifications formed stable
duplexes with the target RNA, they could not confer ade-
quate metabolic stability to the antisense oligonucleotides.
We present here oligonucleotides tethered with guanidino
(Gn) function at 2’-position (Figure 1). The guanidinium
group has a special feature to remain protonated over a wide
pH range. Like 2’-OMe and 2’-F modifications, in 2’-Gn
substituted nucleosides, protonated guanidine could act as
an electron-withdrawing group and may shift the ribose con-
formation towards C3’-endo pucker. The guanidinium
groups are known to play crucial roles biologically, such as
for cell-penetration.3 The guanidinium group may also con-
fer nuclease stability to the antisense oligonucleotide like
2’-O-GE modifications.4
RESULTS AND DISCUSSION
Towards the synthesis of 2’-Gn nucleosides, we
envisaged guanidinylation of 2’-deoxy-2’-amino-
ribonucleoside using commercially available reagents.
Successful guanidinylation of the 2’-amino group could be
achieved with N,N′-bis(benzyloxycarbonyl)-1H-pyrazole-1-
carboxamidine. After -Cbz deprotection, the 1H NMR of 2’-
Gn-nucleoside revealed a significant conformational change
when 2’-amino (82 % S conformation) group is converted to
the 2’-Gn (18 % S conformation) group (Figure 2, a). We
used -Cbz protecting groups on 2’-Gn to prepare the phos-
phoramidite (Figure 2, b) after confirming that the Cbz
group could be completely deprotected in final oligomer
deprotection conditions. The 2’-GnCbz
-U was incorporated
in DNA oligomers using an automated DNA synthesizer to
get the 18mer sequences containing one or two 2’-Gn-U
units respectively. Subsequent HPLC and MALDI-ToF
mass analysis revealed that Cbz-deprotection was incom-
plete under the conditions used and also gave monoprotect-
ed oligomers along with the required completely deprotect-
ed oligomers. Preliminary biophysical studies of the duplex
and triplex forming oligonucleotides are currently in pro-
gress.
CONCLUSIONS
We have successfully synthesized the 2’-guanidino substi-
tuted nucleoside analogues and incorporated them into oli-
gonucleotides. The UV-Tm studies for duplex and triplex
stability are currently in progress and will be presented in
the poster.
REFERENCES
1. Inoue, H., Hayase, Y., Imura, A., Iwai, S., Miura, K.,
Ohtsuka, E. Nucleic Acids Res. 1987, 15, 6131–6148.
2. Kawasaki, A.M., Casper, M.D., Freier, S.M., Lesnik,
E.A., Zounes, M.C., Cummins, L.L., Gonzalez, C., Cook
P.D. J.Med. Chem. 1993, 36, 831–841.
3a. Deglane, G., Abes, S., Michel, T., Prévot, P., Vives, E.,
Debart, F., Barvik, I., Lebleu, B., Vasseur, J-J. ChemBio-
Chem. 2006, 7, 684-692. b. Patil, K.M., Naik, R.J., Rajpal,
Fernandes, M., Ganguli, M., Kumar, V.A. J. Am. Chem.
Soc. dx.doi.org/10.1021/ja210026m.
4. Prakash, T. P.; Puschl, A., Lesnik, E., Mohan, V., Te-
reshko, V., Egli, M., Manoharan, M. Org. Lett. 2004, 6,
1971-1974.
Figure 1. Rationale behind the design
a b
Figure 2. a) Change of %S conformation based on 1H NMR and
b) Structure of protected phosphoramidite
84
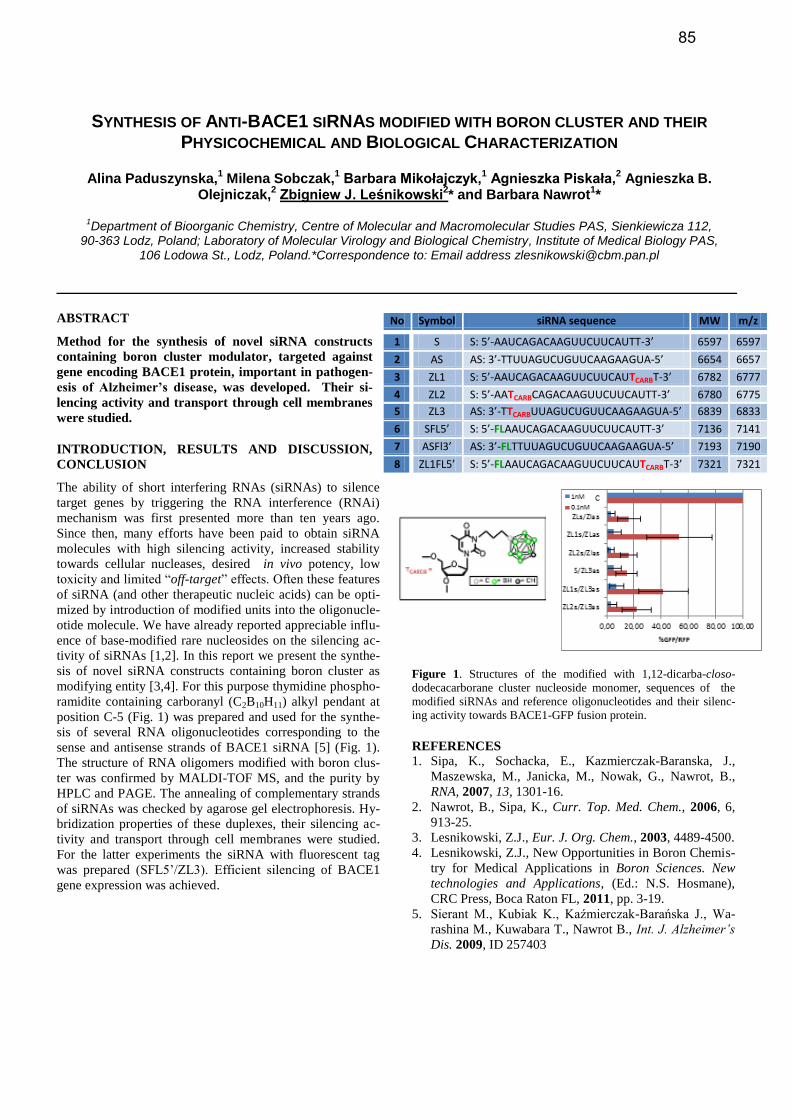
SYNTHESIS OF ANTI-BACE1 SIRNAS MODIFIED WITH BORON CLUSTER AND THEIR
PHYSICOCHEMICAL AND BIOLOGICAL CHARACTERIZATION
Alina Paduszynska,1 Milena Sobczak,
1 Barbara Mikołajczyk,
1 Agnieszka Piskała,
2 Agnieszka B.
Olejniczak,2 Zbigniew J. Leśnikowski
2* and Barbara Nawrot
1*
1Department of Bioorganic Chemistry, Centre of Molecular and Macromolecular Studies PAS, Sienkiewicza 112,
90-363 Lodz, Poland; Laboratory of Molecular Virology and Biological Chemistry, Institute of Medical Biology PAS, 106 Lodowa St., Lodz, Poland.*Correspondence to: Email address [email protected]
ABSTRACT
Method for the synthesis of novel siRNA constructs
containing boron cluster modulator, targeted against
gene encoding BACE1 protein, important in pathogen-
esis of Alzheimer’s disease, was developed. Their si-
lencing activity and transport through cell membranes
were studied.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
The ability of short interfering RNAs (siRNAs) to silence
target genes by triggering the RNA interference (RNAi)
mechanism was first presented more than ten years ago.
Since then, many efforts have been paid to obtain siRNA
molecules with high silencing activity, increased stability
towards cellular nucleases, desired in vivo potency, low
toxicity and limited “off-target” effects. Often these features
of siRNA (and other therapeutic nucleic acids) can be opti-
mized by introduction of modified units into the oligonucle-
otide molecule. We have already reported appreciable influ-
ence of base-modified rare nucleosides on the silencing ac-
tivity of siRNAs [1,2]. In this report we present the synthe-
sis of novel siRNA constructs containing boron cluster as
modifying entity [3,4]. For this purpose thymidine phospho-
ramidite containing carboranyl (C2B10H11) alkyl pendant at
position C-5 (Fig. 1) was prepared and used for the synthe-
sis of several RNA oligonucleotides corresponding to the
sense and antisense strands of BACE1 siRNA [5] (Fig. 1).
The structure of RNA oligomers modified with boron clus-
ter was confirmed by MALDI-TOF MS, and the purity by
HPLC and PAGE. The annealing of complementary strands
of siRNAs was checked by agarose gel electrophoresis. Hy-
bridization properties of these duplexes, their silencing ac-
tivity and transport through cell membranes were studied.
For the latter experiments the siRNA with fluorescent tag
was prepared (SFL5’/ZL3). Efficient silencing of BACE1
gene expression was achieved.
Figure 1. Structures of the modified with 1,12-dicarba-closo-
dodecacarborane cluster nucleoside monomer, sequences of the
modified siRNAs and reference oligonucleotides and their silenc-
ing activity towards BACE1-GFP fusion protein.
REFERENCES
1. Sipa, K., Sochacka, E., Kazmierczak-Baranska, J.,
Maszewska, M., Janicka, M., Nowak, G., Nawrot, B.,
RNA, 2007, 13, 1301-16.
2. Nawrot, B., Sipa, K., Curr. Top. Med. Chem., 2006, 6,
913-25.
3. Lesnikowski, Z.J., Eur. J. Org. Chem., 2003, 4489-4500.
4. Lesnikowski, Z.J., New Opportunities in Boron Chemis-
try for Medical Applications in Boron Sciences. New
technologies and Applications, (Ed.: N.S. Hosmane),
CRC Press, Boca Raton FL, 2011, pp. 3-19.
5. Sierant M., Kubiak K., Kaźmierczak-Barańska J., Wa-
rashina M., Kuwabara T., Nawrot B., Int. J. Alzheimer’s
Dis. 2009, ID 257403
No Symbol siRNA sequence MW m/z
1 S S: 5’-AAUCAGACAAGUUCUUCAUTT-3’ 6597 6597
2 AS AS: 3’-TTUUAGUCUGUUCAAGAAGUA-5’ 6654 6657
3 ZL1 S: 5’-AAUCAGACAAGUUCUUCAUTCARBT-3’ 6782 6777
4 ZL2 S: 5’-AATCARBCAGACAAGUUCUUCAUTT-3’ 6780 6775
5 ZL3 AS: 3’-TTCARBUUAGUCUGUUCAAGAAGUA-5’ 6839 6833
6 SFL5’ S: 5’-FLAAUCAGACAAGUUCUUCAUTT-3’ 7136 7141
7 ASFl3’ AS: 3’-FLTTUUAGUCUGUUCAAGAAGUA-5’ 7193 7190
8 ZL1FL5’ S: 5’-FLAAUCAGACAAGUUCUUCAUTCARBT-3’ 7321 7321
85

TOWARDS A PRODRUG OF TRICYCLO-DNA: SYNTHESIS AND CELLULAR UPTAKE
PROPERTIES OF C(6’)-FUNCTIONALIZED TC-DNA
Jory Lietard and Christian J. Leumann
Department of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern. Correspondence to : [email protected]
ABSTRACT
Tricyclo-DNA (tc-DNA) analogs were functionalized
at position C(6’) with various groups in an attempt to
improve the cellular uptake properties of tc-DNA.
INTRODUCTION
The development of antisense and siRNA strategies has
been slowed down by the inability of oligonucleotides
(ODN) to cross the cellular membrane1. Chemical modifica-
tions to the structure of oligonucleotides offer an attractive
opportunity to improve their cell penetration efficiencies as
well as to address the issues of RNA affinity and resistance
to degradation. In this context, we have developed an oligo-
nucleotide analog, tc-DNA (Figure 1), which binds to RNA
with high affinity and is stable in serum2. tc-DNA has been
recently evaluated as a therapeutic agent in antisense and
siRNA approaches3 but, like most oligonucleotides, does not
readily enter cells.
We wished to bring modifications to the tricyclic core
structure so as to ameliorate the cellular uptake of tc-DNA.
A promising strategy resides in temporarily masking func-
tional groups with biolabile moieties which are expected to
be cleaved once the oligonucleotide is internalized4. There-
fore, we set out to prepare tc-DNA analogs bearing a biola-
bile ester function at position 6’.
RESULTS AND DISCUSSION
Two new building blocks were obtained, tcee
-T and tchd
-T
(Figure 1), carrying an ethyl and a hexadecyl ester, respec-
tively. Their synthesis was performed in 8 and 10 steps
starting from an already known ketone5 by taking advantage
of a -selective NIS-mediated nucleosidation and a late-
stage hydrolysis which granted access to ester derivatives
without altering a major part of the synthetic route. The
phosphoramidites of tcee
-T and tchd
-T were then incorpo-
rated into oligonucleotides and the reactivity of the ester
group was explored by applying various ODN deprotection
conditions. Hydrolysis and aminolysis afforded the corre-
sponding carboxylic acid and amide of tcee
-T. Furthermore,
conditions that maintain the integrity of the ester groups
were developed.
ODNs containing tcee
-T units in their amide, ester or car-
boxylic acid forms were increasing the thermal stabilities of
the corresponding duplexes. However, tchd
-T residues were
found to be destabilizing, most likely due to aggregation of
the hydrophobic C16 chains as evidence by fluorescence
emission properties.
Decathymidylates substituted with five tc-T, tcee
-T or tc-hd
-T units and labelled with fluorescein at their 3’-end were
evaluated for their cellular uptake properties in two human
cell lines without the use of any transfection agent. While
tc-T, tcee
-T and the control dT10 were not internalized, tchd
-T
containing ODN was efficiently taken up.
CONCLUSION
In summary, these results show that 6’-modified tc-DNA
with lipophilic esters are promising analogs for the unassist-
ed delivery of oligonucleotides and represent a first step
towards the preparation of a tc-DNA prodrug.
REFERENCES
1. R. Juliano, J. Bauman, H. Kang, X. Ming, Mol.
Pharmaceutics 2009, 6, 686-695.
2. D. Renneberg, E. Bouliong, U. Reber, D.
Schumperli, C. J. Leumann, Nucleic Acids Res.
2002, 30, 2751-2757.
3. D. Ittig, S. Liu, D. Renneberg, D. Schümperli, C. J.
Leumann, Nucleic Acids Res. 2004, 32, 346-353; D.
Ittig, S. Luisier, J. Weiler, D. Schumperli, C.
Leumann, Artificial DNA: PNA & XNA 2010, 1, 9-
16.
4. G. Tosquellas, K. Alvarez, C. Dell'Aquila, F.
Morvan, J.-J. Vasseur, J.-L. Imbach, B. Rayner,
Nucleic Acids Res. 1998, 26, 2069-2074.
5. S. Luisier, C. J. Leumann, Heterocycles 2010, 82,
775-790.
Figure 1. Chemical structure of tc-DNA and 6’-modified tc- DNA analogs.
86

OPTIMIZING DNA DETECTION USING AMPLIFICATION BY DESTABILIZATION
Katie Mitran*, Abu Kausar, Yimeng Li, Julianne Gibbs-Davis
University of Alberta, Department of Chemistry, Edmonton, Canada. * Correspondence to: [email protected]
ABSTRACT
We have developed a method for isothermal DNA self-
replication that can be applied to the development of
point-of-care diagnostic systems. Herein, we report a
strategy that enables sensitive detection, temperature
tunability and simplified result read-out methods.
INTRODUCTION, RESULTS AND
DISCUSSION, CONCLUSION
Rapid and accurate, yet cost-effective diagnostic
methods are urgently needed to curb the spread of infectious
diseases in developing countries. The challenge is that often,
the most reliable way to diagnose many of these infectious
diseases is to detect the presence of a DNA sequence unique
to a pathogen.1 However, the target sequence must first be
amplified, in order to create a large enough quantity of DNA
for most instruments to detect. There are a number of tech-
niques, such as the Polymerase Chain Reaction (PCR), that
can replicate the target sequence using temperature cycling.
However, these techniques require expensive instrumenta-
tion and temperature-stable enzymes, which make it an im-
probable technique for point-of-care diagnostics.
We have developed a system of amplification that
allows for turnover numbers of greater than 1 million to be
obtained at a single temperature, often close to room tem-
perature.2 This system incorporates a chemical modification
into the DNA, which encourages the product to dissociate
from the target, without an increase in temperature. For this
system to be applied to point-of-care diagnostics, three re-
quirements must be met: first, it has to be very sensitive;
second, the optimal temperature for the reaction needs to be
tunable and third, the read-out methods need to be simple.
The first requirement was met by introducing destabilization
into the system and the second was met by altering the na-
ture of this destabilization. Finally, the read-out method was
simplified from a two-step process consisting of target sepa-
ration with polyarcrylamide gel electrophoresis and detec-
tion with a computer-controlled imager, to a one-step meth-
od using a molecular beacon3, an LED flashlight, and a sim-
ple excitation filter.
REFERENCES
1. Boehme, C. C.; Nabeta, P.; Hillemann, D. et al. New
Engl J Med. 2010, 363, 1005–1015.
2. Kausar, A.; McKay, R. D.; Lam, J.; Bhogal, R. S.; Tang,
A. Y.; Gibbs-Davis, J. M. "Tuning DNA Stability to
Achieve Turnover in Template for an Enzymatic Liga-
tion Reaction" Angew. Chem. 2011, 50, 8922-8926.
3. Tyagi, S.; Kramer, F. R. Nat. Biotechnol. 1996, 14,
303-308.
Figure 1. iPhone image of samples containing molecular beacon (1.25 µM) and differing amounts of target sequence, from left to right; 1.25 µM, 0.63 µM and no target, illuminat-ed with a keychain LED flashlight.
87

Fluorescence enhancement in G-quadruplex folding
Synthesis, structure and stability of a novel dansyl-tagged, conjugated TBA
F. Morvan1, A. Meyer1, J. J. Vasseur1, A. Cummaro2, L. Petraccone2, B. Pagano3, S. De Tito3, E. Novellino3, A. Randazzo3, C. Giancola2 and D. Montesarchio2
1Institut des Biomolécules Max Mousseron, Université de Montpellier 2, Place E. Bataillon, 34095 Montpellier Cedex, France, 2Dipartimento di Scienze Chimiche, via Cintia 4, and 3Dipartimento di Chimica Farmaceutica e Tossicologica, via D. Montesano 49, Università di Napoli Federico II, Napoli, Italy. *Correspondence to: [email protected]
ABSTRACT
A novel fluorescent TBA15 aptamer, conjugated with the environment-sensitive dansyl probe at the 3’-end and a cyclodextrin residue at the 5’-end, has been realized. NMR, CD, DSC and fluorescence studies have been car-ried out to determine its conformation and stability. Remarkably, relevant fluorescence enhancement is ob-served when the 15-mer is folded in the G-quadruplex structure.
INTRODUCTION
Thrombin is a well known target for anticoagulants and antithrombotics, playing vital roles in the coagulation cas-cade. [1] The 15-mer Thrombin Binding Aptamer (TBA15) is able to inhibit thrombin-mediated coagulation. Due to its intrinsic instability in physiological media, large amounts of it are needed to achieve effective anticoagulant responses. To overcome these drawbacks, several modified TBA15 have been proposed. [2] Here we describe a novel bis-conjugated TBA15, exhibiting relevant fluorescence en-hancement in the conversion single strand → G-quadruplex. RESULTS AND DISCUSSION
A variety of fluorescent TBAs have been prepared, typi-cally showing dramatic fluorescence decrease in response to the G-quadruplex folding process. [2] In these systems, TBAs are conjugated with a fluorescent probe and a quencher at the termini, which produce strong fluorescence quenching if the system is folded in a G-quadruplex struc-ture. In alternative, in a pyrene-bis-conjugated TBA broad excimer emission is observed when the terminal pyrene moieties interact as a result of G-quadruplex formation. [3]
Our approach is based on the well known inclusion prop-erties of cyclodextrins, able to capture the environment-sensitive dansyl probe, which in an apolar milieu exhibits a large fluorescence enhancement. In this synthetic scheme, a suitable N -derivative of dansyl was immobilized by a first 3
CuAAC on a CPG support on which the TBA sequence 15
was assembled and 5’-functionalized with a linker carrying a terminal alkyne, allowing the coupling with N -3
derivatized -cyclodextrin using a second CuAAC deriva
Detailed NMR studies have proved that the dansyl tag can be efficiently lodged in the -cyclodextrin cavity, with the chair-type G-quadruplex of TBA not distorted by the pres-ence of the conjugating agents. Calorimetric and spectro-scopic analyses of modified vs. natural TBA confirmed that the conjugating agents affect only to a minor extent G-quadruplex stability. Fluorescence-monitored melting curves showed a relevant fluorescence enhancement upon single strand to G-quadruplex conversion.
OHOGGT TGG TGT GGT TGG
O
CH3
N
SNH
O
O
H3C
CH3
N N
N
O
P
OO
O
NN
N
3'5'
O
OHHOO
O
OH
HO
OHO
OOH
HOOH
O
OOH
OH
OH
O
O
OHOH
HO
O
O
OH
OHHO
O
O OH
HO
HO
O
PO
O
O
Figure 1. Bis-conjugated TBA15 described in this study. CONCLUSION
A modified TBA15, carrying a dansyl group and a -cyclodextrin residue at its extremities, has been efficiently synthesized on a mg scale. An in-depth NMR analysis, in association with CD and DSC studies, showed that the con-jugating agents do not hamper G-quadruplex formation, essential for thrombin recognition. The transition: single strand-→G-quadruplex cause relevant fluorescence en-hancement, as a result of inclusion of dansyl into cyclodex-trin. Future efforts will be directed to exploiting this system for early thrombin detection in biological fluids.
REFERENCES [1] a) Di Cera, E. Mol. Aspects Med. 2008, 29, 203 – 254; b) Wolberg, A. S. Blood Rev. 2007, 21, 131 –142. [2] For a recent review: Aviñó, A., Fàbrega, C., Tintoré, M., Eritja, R. Curr. Pharm. Des., 2012, 18, 2036-2047. [3] Nagatoishi, S., Nojima, T., Juskowiak, B., Takenaka, S. Angew. Chem. Int. Ed., 2005, 117, 5195 –5198. [4] Pourceau, G., Meyer, A., Vasseur, J. J., Morvan, F. J. Org. Chem., 2009, 74, 1218–1222.
ti-zation. [4]
88

INVESTIGATION OF THE FACTOR AFFECTING THE DUPLEX-FORMING ABILITY OF BOAT-SHAPED GLUCOPYRANOSYL NUCLEIC ACID
Kazuto Mori1, Tetsuya Kodama
1,2* and Satoshi Obika
1*
1Graduate School of Pharmaceutical Sciences, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan and 2Graduate School of Pharmaceutical Sciences, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8601,
Japan. * Correspondence to: [email protected]; [email protected]
ABSTRACT
A boat-shaped glucopyranosyl nucleic acid (BsNA)
was synthesized and introduced into oligonucleotides.
The oligonucleotides containing BsNA formed unstable
duplexes with complementary DNA and RNA. From the
crystal structure of BsNA nucleoside, it was found that
the lean of the nucleobase and the rotation angle of the
glycosidic bond axis could affect the duplex-forming
ability.
INTRODUCTION
Several clinical studies using antisense oligonucleotides
(ONs) containing 2’,4’-bridged nucleic acid (BNA)/locked
nucleic acid (LNA) are currently under way.1 We have de-
veloped numerous BNA analogues bearing various bridged
structures, but most of them exhibits lower affinities for
RNA than the original BNA/LNA. Thus, there is a necessity
to design a novel type of artificial nucleic acid based on a
new concept.
When internucleotidic phosphodiester groups are kept
immobile, the nucleobases of BNA analogues lean in the
direction of Figure 1 with decreasing the size of the bridged
moiety. In BNA analogues, the duplex-forming ability for
complementary RNA tends to be more enhanced with de-
creasing size of the bridge. We assumed the lean of the nu-
cleobase could affect the binding affinity and newly de-
signed a boat-shaped glucopyranosyl nucleic acid (BsNA)
bearing a pyranose ring as the basic skeleton.2 The nucleo-
base will lean more than that of the original BNA/LNA due
to this skeleton. In this study, we synthesized BsNA and
evaluated the duplex-forming ability for complementary
DNA and RNA.
Figure 1. Design of BsNA.
RESULTS AND DISCUSSION
BsNA phosphoramidite building block was synthesized
in 12 steps from known glucopyranoside3 and successfully
introduced into ONs using an automated DNA synthesizer
(Scheme 1).
Scheme 1. Synthesis of BsNA
We evaluated the binding affinity of the ONs with com-
plementary DNA and RNA using UV melting experiments.
As a result, introduction of BsNA destabilized the duplex
with complementary DNA and RNA.
We also performed X-ray
crystallographic analysis of
BsNA nucleoside and com-
pared the structure to BNA
analogues. The thymine
base of BsNA leans at larger
angle than that of BNA ana-
logues (Figure 2, left). The
lean of BsNA nucleobase
may be too large to form the
stable duplexes. However, other factors can be the cause of
the destabilization. The axial H3’ can inhibit - interaction
between neighboring nucleobases. In addition, nucleobase
orientation of BsNA nucleoside is different from those of
BNA analogues (Figure 2, right).
The details regarding the investigation of these possibili-
ties will be also presented.
CONCLUSION
We successfully synthesized BsNA and incorporated it
into oligonucleotides. From the thermal stability of duplex
oligonucleotides and the X-ray structure of BsNA nucleo-
side, it was found that the lean of the nucleobase and the
rotation angle of the glycosidic bond axis could impact on
the duplex-forming ability.
REFERENCES
1. Yamamoto, T., Nakatani, M., Narukawa, K., Obika, S.
Future Med. Chem., 2011, 3, 339-365.
2. Mori, K., Kodama, T., Obika, S. Org. Lett., 2011, 13,
6050-6053.
3. Blériot, Y., Vadivel, S. K., Herrera, A. J., Greig, I. R.,
Kirby A. J., Sinaÿ, P. Tetrahedron, 2004, 60, 6813-6828.
Figure 2. Comparison of X-ray structures of BsNA thy-midine (black) with that of BNA analogues (gray)
89

ZINC COMPLEXES OF BIS(AZACROWN)CONJUGATED OLIGONUCLEOTIDES AS ARTIFICIAL RNASES: THE EFFECT OF URIDINE SELECTIVITY ON NUCLEASE ACTIVITY
Teija Niittymäki,* Ekaterina Burakova, Anna Leisvuori, Pasi Virta and Harri Lönnberg
Department of Chemistry, University of Turku, FIN-20014 Turku, Finland. * Correspondence to: [email protected]
ABSTRACT
Zn2+
complexes of 3,5-bis[(1,5,9-triazacyclododecane-
3-yl)oxymethyl]phenyl conjugates of 2´-O-methyl oli-
goribonucleotides were synthesized and their nuclease
activity were tested. The goal is to elucidate if two aza-
crowns attached in the vicinity act co-operatively, the
one recognizing the uracil base of the target and the oth-
er one cleaving the phosphodiester bond of the target.
INTRODUCTION
Artificial ribonucleases, i.e., oligonucleotide conjugates that sequence-selectively cleave complementary RNA tar-gets have attracted considerable attention. Such man-made catalysts are believed to find applications as artificial re-striction enzymes with which large RNA molecules could be tailored in a pre-design manner in vitro and hopefully also as chemotherapeutic agents capable of silencing an over-expressed gene by recognizing and destroying the re-spective mRNA. We have previously shown that 2´-O-methyl oligoribonucleotides bearing two 3-(3-hydroxypropyl)-1,5,9-triazacyclododecane ligands cleave oligoribonucleotide targets at a single phosphodiester bond and show turnover [1]. Our group has also previously stud-ied base moiety selectivity in cleavage of short oligoribonu-cleotides by the dinuclear Zn2+ complex of 1,3-bis[(1,5,9-triazacyclododecane-3-yl)oxymethyl]benzene and the trinu-clear Zn2+ complex of 1,3,5-tris[(1,5,9-triazacyclododecane-3-yl)oxymethyl]benzene [2]. These ligands are interesting because they, besides their marked cleaving activity, recog-nize uracil and thymine bases. The results show that anchor-ing of one of the Zn2+ azacrown moieties to the uracil base keeps the other moiety in the vicinity of the neighboring phosphodiester bonds, resulting in site-selective cleavage.
The present study is aimed at exploiting this anchoring
ability in synergy with the sequence recognition by a 2´-O-methyl oligoribonucleotide probe. To achieve this goal, two azacrowns were attached in the vicinity in oligonucleotide, and it was studied whether these conjugates really enhances the catalytic efficiency and, hence, ensures cleavage precise-ly at a single phosphodiester bond.
RESULTS AND DISCUSSION
The two cleaving agents synthesized are illustrated in Figure 1. They both have dinuclear Zn2+ complex of 3,5-bis[(1,5,9-triazacyclododecane-3-yl)oxymethyl]phenyl at-tached at the 3´-end of the 2´-O-methyl oligoribonucleotide. Table 1 shows rate constants for the cleavage of the chimer-ic 2´-O-methylribo/ribooligonucleotide targets by these con-jugates. Targets contain either uridine or some other nucleo-side at the potential cleaving site.
The results show that the potential anchoring of the aza-crown moiety to the uridine near the cleaving site did not enhance the cleavage. On the contrary, target containing no uridine was cleaved even more efficiently than the uridine containing targets. It may be that the tethering of bis(azacrown) moiety to the oligonucleotide results in con-strain that prevents it to interact simultaneously with the uracil base and the adjacent phosphodiester linkage.
Table 1. Rate constants for the cleavage of targets by zinc ion chelates of conjugates 1 and 2 in 0.10 mol L-1 HEPES buffer at pH 7.3 and 35 ˚C (I=0.1 mol L-1 with NaNO3). The concentrations of cleaving agent and target were 18 μmol L-1 and Zn2+ was 90 μmol L-1. Bold letters in the target sequences refer to ribonucleotides, the rest to 2´-O-methylribonucleotides. Target k/10-6 s-1
Cleaving agent 1 2
3´-UGU GUC UGU GCG GAC AAC AA-5´ 2.44 ± 0.07 1.66 ± 0.08 3´-UGU GUC UGU GCG GUA AAC AA-5´ 1.09 ± 0.04 0.19 ± 0.03 3´-UGU GUC UGU GCG GAU AAC AA-5´ 2.7 ± 0.1 0.44 ± 0.03 3´-UGU GUC UGU GCG GAA UAC AA-5´ 0.39 ± 0.04 0.25 ± 0.02 3´-UGU GUC UGU GCG GAA AUC AA-5´ 0.82 ± 0.05 3´-UGU GUC UGU GCG GAA AAU AA-5´ 0.79 ± 0.08
REFERENCES
1. Niittymäki, T., Virta, P., Ketomäki, K., Lönnberg, H. Bioconjugate Chem. 2007, 18, 1583-1592.
2. Laine, M., Ketomäki, K., Poijärvi-Virta, P., Lönnberg, H. Org. Biomol. Chem. 2009, 7, 2780-2787.
Figure 1.
90

EFFICIENT SYNTHESIS OF OLIGONUCLEOTIDES CONSISTING OF PHOSPHODIESTER AND PHOSPHOTRIESTER LINKAGES
Naoki Sagawa, Takahito Tomori, Itaru Okamoto and Akira Ono*
Kanagawa University, 3-27-1 Rokkakubashi, Kanagawa-ku, Yokohama, Kanagawa, 221-8686 Japan.
* Correspondence to: [email protected]
ABSTRACT
A new and efficient method for preparing oligonucleo-tides consisting of phosphodiester and phosphotriester linkages has been developed. A new nucleoside 3’-O phosphoramidite unit having a dioxolenone structure was synthesized and used for oligonucleotide syntheses on a DNA synthesizer with a solid support having a light sensitive linker. Oligonucleotide synthesized using a gen-eral protocol was cleaved from the solid support by photo-irradiation, without NH4OH treatment, oligonu-cleotide consisting phosphodiester linkages were ob-tained. The dioxolenone groups were expected to be cleaved in the oxidation step with an I2 solution.
INTRODUCTION, RESULTS AND DISCUSSION, CONCLUSION
Chimeric oligonucleotides consisting of both phosphodi-ester and phosphotriester linkages have been expected to be efficiently applied for antisense strategy, since such chi-meric oligonucleotides are sufficiently nuclease resistant and soluble in aqueous solutions. In one of the strategies for synthesizing chimeric oligonucleotides, a fully protected oligonucleotide having two kinds of protecting groups on phosphotriester linkages is synthesized, then one kind of the protecting groups is selectively deprotected to generate phosphodiester linkages but the other protecting groups are remained, thus a chimaric oligonucleotide is generated .
In this report, we describe a new strategy for preparing the chimeric oligonucleotides. We synthesized a new syn-thetic unit, thymidine 3’-O-phosphoramidite 1 which has a
dioxolenone structure in a protecting group for the phos-phoramidite moiety (Figure 1) [1]. Using 1, a commercially available methyl-phosphoramidite unit 2, and a solid sup-port having a light sensitive linker 3 [2], an oligodeoxyribo-nucleotide was synthesized on DNA synthesizer using a general protocol. Without NH4OH treatment, the synthe-sized oligonucleotide was released from the solid support by photo-irradiation and the structure of the oligonucleotide was analyzed by HPLC and MALDI-TOF MS. In the syn-thetic cycle, the dioxolenone protecting groups were depro-tected, but the methyl groups were maintained through the synthesis and deprotection procedures, thus a chimeric oli-gonucleotide consisting of phosphodiester linkages and methyl-phosphotriester linkages was obtained.
Dioxolenone group have been used for protection of car-boxyl group and phosphodiester groups, and the di-oxolenone groups are deprotected by alkaline solutions and enzymatic activities such as esterases [3]. In above synthetic procedure, no alkaline condition and no enzymatic treatment were used. Namely, the dioxolenone groups are able to be deprotected in condition(s) in the synthetic protocol. At pre-sent, we expected that the dioxolenone groups are depro-tected in an I2 solution used for oxidization step.
Correspondingly, the new and efficient strategy for pre-paring chimeric oligonucleotides consisting of phosphodi-ester and phosphotriester linkages is described. Since no alkaline condition is used, the strategy could be used for preparing oligonucleotide having alkaline sensitive func-tional groups.
REFERENCES
1. S. Ito, et al., PACIFICHEM 2010, Honolulu.
2. D. L. McMinn, M. M. Greenberg Tetrahedron Lett., 1998, 39, 4155-4158
3. J. Alexander et al., J. Med. Chem., 1996, 39, 480-486.
Figure 1.
P
T
HO
O
OHP
T
OO
OMeP
T
OO
OHOH
4
TODMTrO
N PO
O
OO
O
1
TODMTrO
N PO
OMe
2
O O O
2
DMTrO
O NH
NO2
MeO O
3
1) DNA synthesis2) hv
91

ROS-ACTIVATED DNA CROSS-LINKING AGENTS AS TAGETED ANTICANCER DRUGS
Xiaohua Peng,1* Yunyan Kuang1, and Sheng Cao1
1 Department of Chemistry and Biochemistry, University of Wisconsin-Milwaukee, 3210 N. Cramer St., Milwaukee, 53211, U.S.A. * Correspondence to: [email protected]
ABSTRACT
We have developed two types of ROS-inducible DNA cross-linking agents that act as targeted anticancer prodrugs. These agents can be activated by the high level of H2O2 found in tumor cells to release active drugs, such as nitrogen mustard or biquinone methide that cross-link or alkylate DNAs leading to cancer cell death. How-ever, they are non-toxic or less toxic to normal cells as the toxicity of the effectors is masked in the prodrugs.
INTRODUCTION. Many anticancer drugs kill cancer cells by inducing DNA interstrand crosslink (ICL) formation. However, most of them are also toxic to normal cells as they lack of good selectivity. One approach to reduce the toxicity of the cross-linking agents for normal cells is to trigger the prodrugs in tumor cells. Cancer cells are known to exhibit increased intrinsic oxidative stress, such as high level of hydrogen peroxide (H2O2), superoxide anions (O2
·-), and hydroxyl radical.1,2 We have exploited the differences in redox state between cancer and normal cells for creating prodrugs that can undergo tumor-specific activation.3,4 Two types of novel ROS-activated anticancer prodrugs 1a,b and 2 have been designed and synthesized. Their biological activites have been studied.
BOO
1a
B
NCl
Cl
BrMe
HO OH
1b
NCl
Cl
BrMe
2
B
B
OO
O O
NN
Br
Br
RESULTS AND DISCUSSION. NMR studies showed that compounds 1a,b reacted with H2O2 to form boronate inter-mediates 3a,b. Spontaneously, deboronation occured to re-lease nitrogen mustard HN2 that produced DNA ICLs and/or alkylations (Scheme 1). In the absence of H2O2, no ICLs were observed. The toxicity of nitrogen mustard was masked in the prodrugs 1a,b because the positive charge developed on the nitrogen strongly decreased its electron density required for alkylation. The positive charge devel-oped on the nitrogen also made the amino group a better leaving group. Compounds 1a,b showed about 90% inhibi-tion toward SR cells (Leukemia cell), 85% inhibition toward NCI-H460 (Non-small Cell Lung Cancer cells), and 66% inhibition toward CAKI-1, and 57% toward SN12C (Renal Cancer cells). However, normal lymphocytes were not af-fected.
BORRO
N
ClCl
O
Br
N
Cl
OHH
3a,b a: R = H or b: 2R = MeMeMe
Me
aziridinium ion
BRO OR
N
ClCl
Br
1a, 1b
Tumorcell
H2O2
NMe
Cl Cl
Active drugHN2
Activation Releasing effectors DNA damage
Normal DNA
No reaction
Scheme 1. The activation of 1a,b by H2O2 found in cancer cells and induced DNA damage.
In a similar way, compound 2 can be selectively activated by H2O2 to generate a powerful and reversible DNA alkylat-ing agent 4 (Scheme 2). The presence of two electron-donating OH groups in 4 greatly facilitate the generation of biquinone methide under physiological conditions.4, 5 The ICL formation was determined by gel electrophoresis. The mechanism of H2O2-activation and the QM formation was determined by NMR analysis and a QM trapping experi-ment, respectively.
OH
DNA
OH
DNA
DNA
O
OH
Me3N
OH
DNA
O
OH
OH
Me3NDNA
DNA
o-quinone methide
o-quinone methide 65
2
1
2
2'
34
55'
6
B
B
OO
O O
NMe3Me3N
4
OH
OH
NMe3Me3N
Br
BrTumor
cell
H2O2
BrBr
Br
Br
Scheme 2. Activation of quinone methide prodrugs.
CONCLUSION. We are, for the first time, to develop ROS-activated antitumor agents that contain efficient ROS-responsive triggers and multiple potent effectors, e.g. DNA cross-linking and alkylating agents. These compounds showed selective toxicity toward cancer cells, while are less toxic to normal cells.
References 1. Szatrowski, T. P.; Nathan, C. F. Cancer Res. 1991, 51, 794-
798. 2. Trachootham, D.; Alexandre, J.; Huang, P. Nature Rev. 2009,
8, 579-591. 3. Kuang, Y.; Balakrishnan, K.; Gandhi, V.; Peng, X. J.
Am. Chem. Soc. 2011, 133, 19278-19231 4. Cao, S.; Wang, Y.; Peng, X. Chem. Eur. J. 2012, 18,
3850-3854. 5. Weinert, E. E.; Dondi, R.; Colloredo-Melz, S.; Frankenfield,
K. N.; Mitchell, C. H.; Freccero, M.; Rokita, S. E. J. Am. Chem. Soc. 2006, 128, 11940-11947.
92

SYNTHESES OF NOVEL 2-5A TRIMERS WITH METHYLPHOSPHONATE INTERNUCLEOTIDE LINKAGE AS POTENTIAL RNASE L ACTIVATORS
Vivek K. Rajwanshi*, Guangyi Wang, Antitsa D. Stoycheva, Roopa Rai, Jerome Deval, Julian Symons, Lawrence Blatt and Leonid Beigelman*
Alios BioPharma Inc., 260 E. Grand Ave., South San Francisco, CA-94080, USA
* Correspondence to: [email protected] and/or [email protected]
ABSTRACT
A series of novel 2-5A trimers with methylphosphonate backbone were synthesized, and evaluated for activation of RNase L
INTRODUCTION
The 2-5A system is one of the major pathways induced by the interferon, and has been implicated in some of its antivi-ral activities. This system has been described as comprising three enzymatic activities, including 2-5A-synthetases, 2-5A-phosphodiesterase, and RNaseL. In the presence of dou-ble-stranded viral RNA, IFN induces expression of the 2-5A-oligoadenylate synthetase that utilizes ATP as a sub-strate for the synthesis of 5’-O-phosphorylated 2-5A oli-gomers. RNase L is normally inactive within the cell. 5’-O-Phosphorylated 2-5As bind to inactive monomer of RNase L and subsequently activate to its dimer form. Activation by 2-5A leads to the degradation of viral RNA within the cell. The effects of 2-5A in cells are transient since natural 2-5A is unstable in cells due to the activities of phosphodiesterase and phosphatase [1]. These findings offer an excellent op-portunity to design and synthesize non charged and stable 2-5A analogues for the exogenous activation of RNase L as a strategy for broad spectrum antiviral drugs. RESULTS Syntheses of 2-5A trimers with methylphosphonate back-bone 4-7 are described in this report. For this purpose, 3’-deoxyadenosine-methylphosphonate monomer 3 was syn-thesized from 3’-deoxy-A [2] (1) through a sequence of re-actions (Scheme-1) consisting of (a) 5’-O-dimethoxytritylation, (b) 3’-O-silylation with TBDMSCl, (c) protection of exocyclic NH2 functionality at N6 position by phenoxyacetyl (Pac) group using PacCl/ 1-HOBT/ CH3CN: Py, (d) removal of 3’-O-TBDMS protecting group with TBAF/THF, and finally methylphosphonamidite 3 was synthesized by reacting intermediate 2 with chloro-N,N-diisopropylamino methylphosphine/ DIPEA in dry THF.
Scheme-1
2-5A trimers 4-7 (Figure 1) were synthesized through the stepwise coupling of monomer unit 3 to a solid support loaded with 5’-O-DMT-A(NH-Bz)-2’-O-acetyl-3’-succinyl-CPG (1.0 mol scale), and deprotection was done at room temperature with ethylenediamine: C2H5OH (1:1) for 5h, followed by treatment with aq. ammonium hydroxide at room temperature for 1-3 h.
(Figure 1)
The synthesized trimer analogs were evaluated for activa-tion of RNase L using a FRET-based enzymatic assay [3]. The EC50 values of the trimers 4-7 were 200, >1000, >1000, and 20 nM respectively. CONCLUSION We have demonstrated the synthesis of the novel 2–5A ana-logs containing methylphosphonate backbone starting from 3’-deoxy-A.Trimers 4-6 containing one or two methylphos-phonate internucleotide linker resulted in significant loss in RNase L activation ability. Interestingly, trimer 7 containing ribose sugar instead of 3’-deoxy sugar in the middle position showed good activation, which suggests importance of ri-bose sugar in middle position of trimers for better activation ability. Trimers design, synthesis and RNase L activation results will be presented. REFERENCES
1. Silverman, R.H., J. Virology, 1997, 81, 12720-12729.
2. Robins, M.J., Wilson, J.S., Madej, D., Low, N.H., Hansske, F., Wnuk, S.F., J.O.C, 1995, 60, 7902-7908.
3. Thakur, C., Xu, Z., Wang, Z., Novince, Z., Silverman, R. Methods for Mol. Med., 2005, 116, 103-113.
93

ENZYMATIC SYNTHESIS OF PHOSPHONOMETHYL OLIGONUCLEOTIDES
Marleen Renders1*, Roel Lievrouw, Gert Emmerechts, Jef Rozenski
2, Marcela Krecmerová
3,
Antonin Holý3 and Piet Herdewijn
2
1Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver V6T 1Z1, Canada
2Laboratory for Medicinal Chemistry, Rega Institute for Medical Research, Minderbroedersstraat 10, 3000 Leuven,
Belgium
3Institute of Organic Chemistry & Biochemistry, Academy of Sciences of the Czech Republic, Prague, Czech Republic
*Correspondence to: Email address [email protected]
ABSTRACT
Therminator is the hero when it comes to the
enzymatic production of phosphonomethyl–
threosyl oligonucleotides with a non-natural
3′–2′ linkage. The ability of Therminator pol-
ymerase to catalyze the condensation of di-
phosphate derivatives of nucleosides to give
modified oligomers promises to be quite gen-
eral. Such oligonucleotides are useful tools in
synthetic biology because of the innate stability
of the phosphonate linkage.
INTRODUCTION, RESULTS AND
DISCUSSION, CONCLUSION
We have studied the enzymatically catalyzed
polymerization of the diphosphate of the phos-
phonomethyl derivative of L-threose nucleosides
because we are interested in the use of these en-
zymatically stable phosphonate oligonucleotides
in synthetic biology. We have selected a wide
spectrum of polymerases for the synthesis of such
‘exotic’ oligonucleotides. Of the enzymes tested
only the members of the B-family polymerases,
i.e. Vent exo- DNA polymerase and Therminator
polymerase, could extend the primer with more
than two phosphonate nucleosides in a row (+6
and +10, respectively) with formation of the unu-
sual (3′-2′) connected oligonucleotides. Regarding
the chemical synthesis, this is an alternative but
straightforward way to obtain small amounts of
these hypermodified oligonucleotides. Degrada-
tion of the primer with snake venom phos-
phodiesterase yields an end-phosphorylated oli-
gomer. Phosphonate oligonucleotides with three
to five phosphonate adenine nucleoside analogs
and with a deoxyguanosine monophosphate at the
5′ end were detected with LC-ESI/MS.
An enzymatic
route for the synthesis
and isolation of 3′-2′
phoshonate oligomers
can be regarded as an
important step to
make this unnatural
informative polymer
available for further
studies. The fact that
after digestion with snake venom phosphodiester-
ase a phosphorylated guanosine was left at the 5′
end of the phosphonate oligomer could provide a
solution for the ligation problems often encoun-
tered with chimer construction of mixed DNA-
modified nucleotides sequences (i.e. due to the
presence of modified nucleosides at the 5′ end,
synthetic oligonucleotides are mostly bad sub-
strates for kinases) Enzymatic approaches to ob-
tain oligonucleotides with an enhanced stability to
nucleolytic degradation implies that the synthetic
nucleotide monomers are substrates for polymer-
ases and that the obtained oligomers are not sub-
strates for nucleases. This methodology may be
useful to obtain modified oligonucleotides as po-
tential therapeutic agents for the antisense and the
antigene approach. In the field of synthetic biolo-
gy, the obtained 5′ phosphorylated synthetic oli-
gonucleotides may be used directly for incorpora-
tion in plasmids and investigate the translation of
this enzymatic stable information system.
Figure 1 The structure of
a phosphonomethyl-
threosyl-adenin oligomer
94

LONG LIVED LUMINOGENIC DNA PROBE FOR DNA/RNA DETECTION
Hisao Saneyoshi,1 Hiroshi Abe
1* and Yoshihiro Ito
1*
1RIKEN, Advanced Science Institute, Nano Medical Engineering Laboratory, 2-1, Hirosawa, Wako-shi, Saitama,
JAPAN, * Correspondence to: Email address for [email protected] or [email protected]
ABSTRACT
Long lived luminogenic probe for DNA/RNA detection
was developed. Luminogenic system is based on the con-
struction of light harvesting molecule on the oligonucleo-
tide strand with target nucleic acid sequences. Properties
of long live luminogenic DNA probe were described
herein.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
Fluorescence technique for detection and imaging of
DNA/RNA is very important tool in the field of life science
and recent therapy-directed diagnostics.1 However there is
critical problem of auto fluorescence from crude sample
such as cell, blood, lysate etc. to disturb sensitive detection
of DNA/RNA sequences.2 The main problem is overlap of
photophysical property between autofluorescence and con-
ventional organic fluorophorce in the probe. If the fluores-
cence life time of oligonucleotide probe extends to millisec-
ond, the issue raised here should be avoided by using time
gated spectroscopy technique. One promising candidate is
the use of lanthanide complex that has long lived lumines-
cence property and application of time gated luminescence
spectroscopy to eliminate short-lived fluorescence.5 Howev-
er there is few design methodology of OFF-ON light har-
vesting molecule (antenna) for lanthanide complex in the
oligonucleotide probe.
In this study, we report the design, synthesis and
DNA/RNA detection of long lived luminogenic DNA probe
having new antenna molecule with OFF-ON switch.
Our probe is built with three components (OFF-antenna,
lanthanide chelate, and oligonucleotide). Off-antenna is in-
corporated into oligonucleotide for switching and light har-
vesting device. Chemical reaction catalyzed by target
DNA/RNA converts On-antenna structure to emit lantha-
nide ion.
At first, we designed and synthesized a new antenna mol-
ecule built on a bi phenyl ring skeleton carrying two reac-
tive functional group. The synthesized OFF-antenna mole-
cule was converted into ON-antenna structure under reduc-
tive condition to give a large red shift of UV absorption.
On-antenna molecule can emit lanthanide ion to generate
long lived luminescence. Conjugation with oligonucleotide
to lanthanide complex with OFF-antenna molecule was per-
formed by conventional mean. According to the photophys-
ical analysis, luminogenic DNA probes have long lived and
two colored luminescence property depending on the anten-
na structure and lanthanide ion. The detection ability was
assessed to detect Ras gene and 23S rRNA in E.coli cell by
time gated luminescence analysis.
In conclusion, we synthesized long lived luminogenic
DNA probe based on the construction of light harvesting
molecule promoted by target nucleic acid sequences. The
combination of new probe and time gated luminescence
technique was effective for elimination of short lived auto-
fluorescence species.
REFERENCES
1. Stender, H. Expert Rev. Mol. Diagn, 2003, 3, 649-655.
2. Billinton, N., Knight, A. W. Analytical Biochemistry,
2001, 291, 175–197.
3. Yang, L, Zhou, Y, Zhu, S, Huang, T, Wu, L, Yan, X.
Anal chem., 2012, 84, 1526-1532.
4. Berezin, M. Y.; Achilefu, S. Chem.Rev. 2010, 110,
2641–2684.
5. Bu¨nzli, J.-C. G. Chem. Rev. 2010, 110, 2729–2755.
95

SULFONAMIDE BEARING NUCLEOSIDES: SIMPLE SYNTHESIS, EFFICIENT RNA RECOGNITION
Pawan K. Sharma,*1 Pawan Kumar,1,2 Navneet Chandak,1 and Poul Nielsen2
1Department of Chemistry, Kurukshetra University, Kurukshetra-136 119, India and 2Nucleic Acid Center, Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, 5230 Odense M, Denmark. * Correspondence
to: Email: [email protected]
ABSTRACT
Four pyrimidine nucleoside monomers W, X, Y and Z wherein a benzensulfonamide group is linked to C5 posi-tion of the uracil nucleobase through a triazolyl or an alkynyl linker were prepared and successfully incorpo-rated into oligonucleotides. C5-triazole nucleosides in the major groove were found to increase the thermal stabil-ity of a DNA:RNA duplex significantly due to more effi-cient π-π-stacking, whereas C5-alkynyl nucleosides were less potent in terms of duplex stability.
INTRODUCTION
The idea of synthetically increasing the stability of a DNA:RNA duplex, and hence target mRNA in the so called antisense approach, by increasing the aromatic stacking in-teractions has been exploited by us1 and others2. We ob-served that a triazole group attached to the C5 position of dU projecting in the major groove of DNA:DNA or DNA:RNA duplex lead to increased thermal stabilities through efficient π-π-stacking.1 Furthermore, only two con-secutive incorporations were found to be sufficient to give significant DNA:RNA duplex stabilization, as compared to a DNA:DNA duplex, due to stacking of triazoles and the aromatic substituents attached. The most pronounced effect was given by the monomer containing a phenyltriazole with a distal sulfonamide moiety. In the present study, we have explored the scope of this stacking effect concerning the positional orientation of the sulfonamide group as well as comparing the stacking potential of the triazole vs the al-kynyl linker.
RESULTS AND DISCUSSION
Nucleoside monomers W,1a X, Y and Z were synthesized using either CuAAC or Sonogashira reactions and were suc-cessfully incorporated into 9-mer ologonucleotides which were studied in DNA:DNA and DNA:RNA duplexes with one to four consecutive incorporations (ON1-ON4). Incor-poration of either of the modified monomers W, X, Y or Z lead to significant decrease in DNA:DNA duplex stability by one incorporation, levelling off with four incorporations in case of W and X while still destabilizing in case of Y and Z. In case of DNA:RNA duplex, stabilizing effect is more pronounced as compared to DNA:DNA with decrease in duplex stability by one incorporation to increase in stability up to ∼+6.1 °C per modification for four consecutive incor-
porations of W and X, while up to ∼+3.0 °C in Y and Z. A direct comparison of triazolyl and alkynyl nucleosides clear-ly demonstrates that stacking efficiency of triazole is better than that of alkynyl groups. For mismatch studies, ON4 with either W, X, Y or Z was mixed with RNA-sequences containing a single central mismatch. Fine mismatch dis-crimination from A to C, U and G, respectively, was ob-served by large decreases in Tm.
NH
O
ON
O
O
O
NN
N
P OO
R'R
W R = H, R' = SO2NH2X R = SO2NH2, R' = H
NH
O
ON
O
O
O
P OO
Y R = H, R' = SO2NH2Z R = SO2NH2, R' = H
R'
R
CONCLUSION
The stacking of sulfonamide substituted phenyltriazoles in the major groove leads to very stable DNA:RNA du-plexes. C5-alkynylnucleosides are less potent than the tria-zolenucleosides in terms of duplex stability, but still show-ing discrimination for recognizing RNA in preference to DNA. The study indicates that triazole nucleus has a better stacking potential as compared to the alkynyl moiety.
REFERENCES
1. (a) Andersen, N.K, Chandak, N., Brulikova, L., Kumar, P., Jensen, M.D., Jensen, F., Sharma, P.K., Nielson, P. Bioorg. Med. Chem. 2010, 18, 4702-4710. (b) Andersen, N. K., Døssing, H., Jensen, F., Vester, B., Nielsen, P. J. Org. Chem. 2011, 76, 6177-6187.
2. (a) Gutierrez, A. J., Terhorst, T. J., Matteucci, M. D., Froehler, B. C. J. Am. Chem. Soc. 1994, 116, 5540-5544; (b) Sinkeldam, R. W., Greco, N. J., Tor, Y. ChemBioChem 2008, 9, 706-709.
96

DEVELOPMENT OF A NOVEL PROTECTING GROUP FOR OLIGONUCLEOTIDE SYNTHESIS
Adriaan W. Tuin,1* Galyna V. Peterburgska,
1 Rebecca Krijt,
1, Jarno G. ter Beek,
1 and
Peter C. de Visser1
1Prosensa Therapeutics B.V.,J.H. Oortweg 21, 2333 CH Leiden, the Netherlands
* Correspondence to: [email protected]
ABSTRACT
A novel protecting group for the exocyclic amines on
nucleobases is being developed.
INTRODUCTION
Synthetic antisense oligonucleotides (AONs) are rapidly
finding their way into the pharmaceutical industry [1]. The-
se developments have led to continuous efforts to improve
the synthesis of AONs on multigram scale. Protecting group
properties, such as ease of removal, prevention of the for-
mation of sideproducts during synthesis and upon cleavage
still remain optimizable parameters in ON synthesis.
In an ongoing effort to optimize the large scale synthesis
of therapeutic AONs, a program is dedicated to developing
a novel base-sensitive nucleobase protecting group that is
compatible with current phosphoramidite based synthesis
procedures and possesses favorable deprotection character-
istics compared to the currently employed protecting groups.
The newly designed protection group has been installed
using only a limited number steps onto the guanosine phos-
phoramidite and showed excellent cleavage characteristics
during a mild and short deprotection protocol with a t1/2 of
less than 2 minutes in concentrated ammonia at 55°C.
This novel proprietary protecting group is currently being
evaluated in AON synthesis to assess its stability during
automated synthesis. In addition, this protecting group will
also be evaluated as universal protecting group on other
(modified) nucleoside monomers.
REFERENCES
1. Goemans N.M., Tulinius M., van den Akker J.T., Burm
B.E., Ekhart P.F., Heuvelmans N., Holling T., Janson
A.A., Platenburg G.J., Sipkens J.A., Sitsen J.M.,
Aartsma-Rus A., van Ommen G.J., Buyse G., Darin N.,
Verschuuren J.J., Campion G.V., de Kimpe S.J., van
Deutekom J.C., N. Engl. J. Med., 2011, 364, 16, 13 – 22.
97

STEREOCONTROLLED SYNTHESIS OF BORANOPHOSPHATE DNA BY THE BORANOPHOSPHOTRIESTER APPROACH
Naoki Uchiyama1 and Takeshi Wada
1*
1Department of Medical Genome Sciences, Graduate School of Frontier Sciences, The University of Tokyo,
Bioscience Building 702, 5-1-5 Kashiwanoha, Kashiwa, Chiba 277-8562, Japan.
* Correspondence to: [email protected]
ABSTRACT
Diastereoselective synthesis of dithymidineborano-
phosphate was achieved by the boranophosphotriester
method using a diasteromixture of thymidine 3’-
boranophosphate derivatives. Chiral auxilialies intro-
duced at the phosphate moiety induced the high stere-
oselectivity for the condensation reaction.
INTRODUCTION
Boranophosphate DNA is regarded as a potentially useful
antisense molecule because of its high resistance to nucleas-
es [1]. Another remarkable feature of this DNA analogue is
its ability to induce RNase H-mediated hydrolysis of the
complementary mRNA strand [1]. Boranophosphate DNA
has chiral internucleotidic linkages, and it has been reported
that the chirality of phosphorus atom affects the biological
and physicochemical properties of boranophophate DNA [2].
However, there has been no efficient method to synthesize
the P-stereoregulated boranophosphate DNA with all possi-
ble nucleobases, A, G, C and T.
To overcome this problem, we tried to synthesize P-
stereoregulated boranophosphate DNA by the boranophos-
photriester approach which is an efficient method for the
synthesis of boranophosphate DNA including all kinds of
nucleobases [3].
RESULTS AND DISCUSSION
We hypothesized that the stereoselectivity of condensation
reaction would be induced by a chiral auxiliary attached to
the phosphorous moiety in the nucleoside 3’-
boranophosphate monomer unit, which would act as an in-
tramolecular chiral nucleophilic catalyst for condensation.
First, a diasteromixture of the monomer unit 1 was synthe-
sized from a thymidine 3’-H-phosphonate derivative and
amino alcohol (Scheme 1). The amino alcohols were syn-
thesized by the 7-step raction from L-proline. Then the
boronation was performed with TMSCl and the thymidine
3’-boranophosphate monomer unit was obtained.
Next, the monomer unit 1 was condensed with a nucleo-
side 2 in the presence of condensing reagent and a nucleo-
philic catalyst (Scheme 2). Introduction of bulky alkyl
groups such as i-Pr or t-Bu in the chiral auxiliary (R = i-Pr
or t-Bu) significantly affected on the stereoselectivity.
Moreover, the use of 4-cyanotriazole (CT) and 4-
nitroimidazole (NI), which have relatively high nucleo-
philicity and low acidity, improved the stereoselectivity.
CONCLUSION
We achieved the diastereoselective synthesis of dithymi-
dine boranophasphate by a boranophosphotriester approach
using chiral auxiliaries. We will try to apply this method for
the synthesis of more complicated nucleotide derivatives
bearing other nucleobases, A, G and C.
REFERENCES
1. Zinaida, P. L.; Dobrikov, S. M.; Shaw, B. R. Chem. Rev.
2007, 107, 4746-4796.
2. Sergueeva, Z, A.; Sergueev, D. S.; Ribeiro, A. A.;
Summers, J. S.; Shaw, B. R. Helv. Chim. Act. 2000, 83,
1377-1391.
3. Shimizu, M.; Saigo, K.; Wada, T. J. Org. Chem. 2006,
71, 4262-4269.
O
O
TBDPSO Tbz
PO-HN+Et3
H
O
N OH
R
O
O
TBDPSO Tbz
PO
N
R OHNEt3
H3B
O
O
TBDPSO Tbz
PO
H
O
N
Rcondensation
boronation
1 Scheme 1. Synthesis of monomer unit.
O
O
TBDPSO Tbz
PO
N
R OHNEt3
H3B
1
condensing reagent
nucleophilic catalyst
O
O
TBDPSO Tbz
PO N
H3B
R
O
OTBDMS
HO Tbz
stereoselective
condensation
O
O
TBDPSO Tbz
O
OTBDMS
TbzPOO
H3B
R
N
2
Scheme 2. Diastereoselective condensation using mono-
mer unit 1
98

OLIGONUCLEOTIDE CHELATE COMPLEXES AND THEIR CLINICAL APPLICATION IN
REDUCING ADMINISTRATION-RELATED SIDE EFFECTS
Andrew Vaillant* and Michel Bazinet
REPLICor Inc., 6100 Royalmount, Suite D-101, Montreal, Quebec, Canada H8Y 3E6. * Correspondence to: [email protected]
ABSTRACT
Oligonucleotide chelate complexes are novel solution
structures occurring in the presence of divalent metal
cations. Their characterization and application in a clin-
ical setting is presented.
INTRODUCTION
The parenteral administration of phosphorothioate oligo-
nucleotides (PS-ONs) is typically associated with side-
effects consisting of fever, chills, shivering and weakness
with intravenous infusion and inflammation, induration and
pain with subcutaneous injection. Many of these symptoms
are consistent with a transient reduction of calcium levels in
the serum or in the subcutaneous space during PS-ON ad-
ministration. In solution, oligonucleotides in the presence
of divalent metal cations form multimeric complexes which
effectively sequester calcium from solution interactions.
The characterization of these complexes and their applica-
tion to mitigate administration-related side effects of oligo-
nucleotides was examined.
RESULTS
Monitoring of the solution structure of oligonucleotides
by fluorescence polarization revealed that in the presence of
any divalent metal cation with a 2+ charge, a large increase
in solution mass occurred with oligonucleotides which were
not capable of intra- or intermolecular hybridization. This
change in solution mass occurred with a wide range of 2+
metal salts including alkali earths and transition metals and
was not observed in the presence of monovalent cations
such as sodium, potassium and NH4+. This increase in so-
lution mass also occurred with double stranded oligonucleo-
tides and with single stranded oligonucleotides of diverse
sequences and lengths except for poly A and poly G. Solu-
tion mass increases were observed for oligonucleotides with
phosphodiester or phosphorothioate linkages and also in the
presence of a 2’ O methyl ribose modification. These in-
creased solution mass “complexes” were temporally stable
and remained soluble in aqueous solutions.
These results suggested divalent metals could be seques-
tered in solutions containing oligonucleotides and also sug-
gested that the administration side effects of oligonucleo-
tides were consistent with calcium chelation. The blood
anticoagulation induced by a 40mer PS-ON could be com-
pletely neutralized in vitro by the addition of supplemental
calcium. Additionally, the tolerability of two different RNA
and DNA PS-ONs when administered by SC injection was
significantly improved in human patients when these PS-
ONs were “pre-complexed” with calcium prior to injection.
Finally, in human proof of concept studies with the antiviral
PS-ON REP 9AC’, its administration after being pre-
complexed with calcium did not compromise its activity in
vivo but resulted in a dramatic improvement in IV infusion
tolerability compared to an unchelated predecessor.
DISCUSSION
The solution mass changes observed in oligonucleotide
solutions containing divalent metal cations are likely due to
the formation of an “oligonucleotide chelate complex” con-
sisting of two or more oligonucleotides linked by divalent
metal cations. The ability of double stranded PS-ONs to
form chelate complexes also suggests that this linkage likely
consists of a metal bridge formed between the non-bridging
oxygen atoms from the phosphodiester linkages of two ad-
jacent ON strands. This metal bridge may also form with
the non-bridging sulfur atoms present in phosphorothioate
linkages. It appears that any divalent metal will suffice to
form chelate complexes and it may also be possible that
trivalent metals such as aluminium could also form chelate-
complexes. These chelate complexes also appear to require
multiple lateral interactions as poly G and poly A, which are
known to form thermodynamically stable intra-
molecularinteractions, did not form chelate complexes.
This novel chelation property of ONs in general may under-
lie many of the administration-related side effects observed
with PS-ON administration. These studies suggest that oli-
gonucleotides should be viewed as divalent metal chelators
and this property should be taken into consideration when
oligonucleotides are formulated for parenteral administra-
tion. Finally, the administration of any PS-ON as a calcium
chelate complex may be a novel formulation which can
greatly improve its tolerability.
99
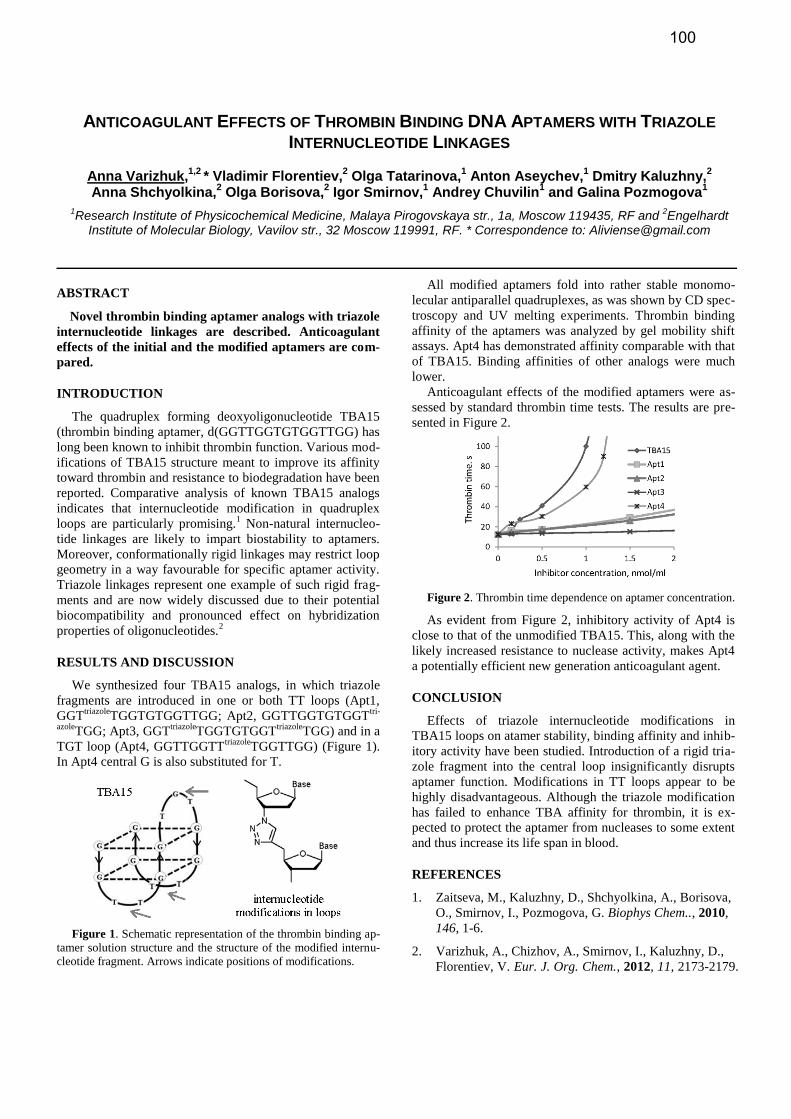
ANTICOAGULANT EFFECTS OF THROMBIN BINDING DNA APTAMERS WITH TRIAZOLE
INTERNUCLEOTIDE LINKAGES
Anna Varizhuk,1,2
* Vladimir Florentiev,2 Olga Tatarinova,
1 Anton Aseychev,
1 Dmitry Kaluzhny,
2
Anna Shchyolkina,2 Olga Borisova,
2 Igor Smirnov,
1 Andrey Chuvilin
1 and Galina Pozmogova
1
1Research Institute of Physicochemical Medicine, Malaya Pirogovskaya str., 1a, Moscow 119435, RF and
2Engelhardt
Institute of Molecular Biology, Vavilov str., 32 Moscow 119991, RF. * Correspondence to: [email protected]
ABSTRACT
Novel thrombin binding aptamer analogs with triazole
internucleotide linkages are described. Anticoagulant
effects of the initial and the modified aptamers are com-
pared.
INTRODUCTION
The quadruplex forming deoxyoligonucleotide TBA15
(thrombin binding aptamer, d(GGTTGGTGTGGTTGG) has
long been known to inhibit thrombin function. Various mod-
ifications of TBA15 structure meant to improve its affinity
toward thrombin and resistance to biodegradation have been
reported. Comparative analysis of known TBA15 analogs
indicates that internucleotide modification in quadruplex
loops are particularly promising.1 Non-natural internucleo-
tide linkages are likely to impart biostability to aptamers.
Moreover, conformationally rigid linkages may restrict loop
geometry in a way favourable for specific aptamer activity.
Triazole linkages represent one example of such rigid frag-
ments and are now widely discussed due to their potential
biocompatibility and pronounced effect on hybridization
properties of oligonucleotides.2
RESULTS AND DISCUSSION
We synthesized four TBA15 analogs, in which triazole
fragments are introduced in one or both TT loops (Apt1,
GGTtriazole
TGGTGTGGTTGG; Apt2, GGTTGGTGTGGTtri-
azoleTGG; Apt3, GGT
triazoleTGGTGTGGT
triazoleTGG) and in a
TGT loop (Apt4, GGTTGGTTtriazole
TGGTTGG) (Figure 1).
In Apt4 central G is also substituted for T.
Figure 1. Schematic representation of the thrombin binding ap-
tamer solution structure and the structure of the modified internu-
cleotide fragment. Arrows indicate positions of modifications.
All modified aptamers fold into rather stable monomo-
lecular antiparallel quadruplexes, as was shown by CD spec-
troscopy and UV melting experiments. Thrombin binding
affinity of the aptamers was analyzed by gel mobility shift
assays. Apt4 has demonstrated affinity comparable with that
of TBA15. Binding affinities of other analogs were much
lower.
Anticoagulant effects of the modified aptamers were as-
sessed by standard thrombin time tests. The results are pre-
sented in Figure 2.
Figure 2. Thrombin time dependence on aptamer concentration.
As evident from Figure 2, inhibitory activity of Apt4 is
close to that of the unmodified TBA15. This, along with the
likely increased resistance to nuclease activity, makes Apt4
a potentially efficient new generation anticoagulant agent.
CONCLUSION
Effects of triazole internucleotide modifications in
TBA15 loops on atamer stability, binding affinity and inhib-
itory activity have been studied. Introduction of a rigid tria-
zole fragment into the central loop insignificantly disrupts
aptamer function. Modifications in TT loops appear to be
highly disadvantageous. Although the triazole modification
has failed to enhance TBA affinity for thrombin, it is ex-
pected to protect the aptamer from nucleases to some extent
and thus increase its life span in blood.
REFERENCES
1. Zaitseva, M., Kaluzhny, D., Shchyolkina, A., Borisova,
O., Smirnov, I., Pozmogova, G. Biophys Chem.., 2010,
146, 1-6.
2. Varizhuk, A., Chizhov, A., Smirnov, I., Kaluzhny, D.,
Florentiev, V. Eur. J. Org. Chem., 2012, 11, 2173-2179.
100

APPLICATION OF "CLICK CHEMISTRY" FOR DESIGN AND SYNTHESIS OF SEQUENCE-SPECIFIC LIGANDS AND PROBES TARGETING DOUBLE-STRANDED DNA
Svetlana Vasilyeva1,2*
, Viacheslav Filichev3, Vladimir Silnikov
1,2, Alexandre Boutorine
4
1 Institute of Chemical Biology & Fundamental Medicine, 2“NanoTech-S” LLC, Lavrent’ev Ave 8, Novosibirsk,
630090, Russia; 3 Institute of Fundamental Sciences, Massey University, Private Bag 11-222, 4442 Palmerston North,
New Zealand 4 Muséum National d'Histoire Naturelle, CNRS UMR 7196, INSERM U565, 57 rue Cuvier, C.P. 26 75231
Paris cedex 05 France
* Correspondence to: Email address for [email protected]
ABSTRACT
A simple method of conjugate synthesis using Huisgen
1,3-cycloaddition ("click chemistry") was elaborated
and applied for conjugation of polyamide minor groove
binders to triplex-forming oligonucleotides of different
nature and structure. A series of diverse bifunctional
linker was synthesized and applied for insertion of azide
or alkyne groups into oligonucleotides and minor groove
binders. All the conjugates formed a triple helix with the
target DNA. The presence of MGB in conjugate did not
greatly affect the binding parameters such as affinity
and triplex stability.
INTRODUCTION
Recognition and detection of specific regions in genomic
DNA is an important problem of gene therapy and imaging.
Two classes of synthetic molecules are able to recognize
specifically native double-stranded DNA sequences: triplex-
forming oligonucleotides (TFO) [1] that bind to the major
groove of DNA polypurine tracts, and minor groove binders
(MGB) [2], polyamides composed of the N-methylpyrrole
and N-methylimidazole carboxamides. In order to increase
the recognition sequence and profit from binding properties
of both components, the conjugates of TFO and MGB were
synthesized. Application of Huisgen 1,3-cycloaddition
("click chemistry") could simplify conjugation in physiolog-
ical conditions. Fluorescently labeled conjugates could be
used as fluorescent probes for native double-stranded DNA
detection in vitro, in chromatin and finally in living cells.
RESULTS AND DISCUSSION
We have developed a new method of conjugation of se-
quence–specific polyamide minor groove binders and tri-
plex-forming oligonucleotides (including fluorescent twist-
ed intercalating nucleic acids – TINA [3]) using the Huisgen
1,3-dipolar cycloaddition of azides and alkynes. Nine new
bifunctional chemical linkers containing azide or alkyne
groups were synthesized and attached either to a terminal
phosphate of oligonucleotide or to N-terminal amine of
MGB by classical chemistry methods or directly upon solid-
phase synthesis of the components. Their structures were
proved by mass spectrometry. The pairs "alkyne-TFO" and
"azide-MGB" (or "azide-TFO" and "alkyne-MGB") react
easily in aqueous conditions in the presence of copper (I)
ions and form a triazole cycle that links two molecules in
one conjugate. 7 conjugates were obtained using different
linkers for their coupling. The electrophoresis analysis
demonstrated quantitative yield and high purity of the con-
jugates. However, rapid and strong oligonucleotide degrada-
tion was observed due to presence of copper ions. A special
procedure of copper ions sorption on a chelating resin
Chelex-100 was introduced. HPLC purification of TINA
derivatives was difficult due to high hydrophobicity of py-
rene moiety. Preparative electrophoresis was applied. Inter-
action of conjugates with target DNA was studied by gel
retardation and thermal denaturation methods. We demon-
strated that all the conjugates form a triple helix with target
DNA, but the presence of MGB in conjugate did not affect
such binding parameters as affinity and triplex stability.
CONCLUSION
A series of diverse bifunctional linkers bearing azide or
alkyne groups was synthesized and used for oligonucleo-
tide-MGB conjugation using Huisgen 1,3-cycloaddition
("click chemistry"). Triplex-forming oligonucleotides of
different nature and structure were coupled to polyamide
minor groove binders, purified and their interaction with
dsDNA was characterized by physico-chemical methods.
The method is more simple and effective compared to "clas-
sic" chemical synthesis. However, additional optimization is
needed to improve DNA-binding affinity of conjugates.
REFERENCES
1. Praseuth, D., Guieysse, A.L. and Hélène, C. Biochim
Biophys Acta - Gene Struct Express, 1999, 1489, 181.
2. Dervan, P.B., Edelson, B.S. Curr. Opin. Struct. Biol.,
2003, 13, 284.
3. Doluca, O., Boutorine, A.S. and Filichev, V.V. Chem-
BioChem, 2011, 12, 2365-2374
L1 p-Oligonucleotide
N
O
N
HN
N
O
H NN
O
H
HN
O
NN
O
HNN
O
H
HN
NN
H
O
NH
OC
O
NHNH
N3 L2
OH2O/DMSO
CuSO4, THPTA
NaAscorbate
TEAAc
L1p-Oligonucleotide
N
O
N
HN
N
O
H NN
O
H
HN
O
NN
O
HNN
O
H
HN
NN
H
O
NH
OC
O
NHNH
N L2
ONN
Figure 1. General scheme of synthesis of the MGB-Oligonucleotide conjugates by Huisgen 1,3-dipolar cycloaddition of azides and alkynes.
101

NUCLEIC ACID-TRIGGERED AMINOACYL AND PEPTIDYL TRANSFER FOR THE SYNTHESIS
OF BIOACTIVE PEPTIDES FROM INACTIVE FRAGMENTS
Olalla Vázquez,1
Anne Adams1 and Oliver Seitz
1*
1Department of Organic and Bioorganic Chemistry of Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, D-12489,
Berlin, Germany. *Correspondence to: [email protected]
ABSTRACT
Our group has successfully introduced a DNA-
triggered reaction, which involves the transfer of only
one amino acid residue to form peptide-PNA-conjugates
with biological activity. Based on this discovery, our cur-
rent work is directed to an extension of this concept to
other bioactive peptides.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
The proper functioning of cells involves accurate control
of DNA and the expressed RNA molecules. Diseases are
frequently caused by changes in the genetic infrastructure.
Therefore, the information obtained from nucleic acids
might be used to direct molecular therapies only to diseased
cells and tissues. In this context, the in situ generation of
drugs on the basis of nucleic acid-templated reactions
should provide a unique strategy to induce cell-specific re-
sponses.
Recently, it was shown that nucleic acid hybridization
processes can control the release of model drugs such as
pnitrophenol, [1] coumarin, [2] biotin, or benzoic acid, [3],
or singlet oxygen [4]. Unlike these reported release systems,
which relied upon ester cleavage or DNA-strand exchange,
we assumed that the synthesis of bioactive agents by tem-
plate-directed bond-forming reactions should provide im-
proved specificity.
Our strategy involves the hybridization of two short
PNA-oligomers equipped with fragments of the peptide to a
complementary nucleic acid-template (Figure 1). This prox-
imity of both probes allows the aminoacyl group transfer
from the donor conjugate to the acceptor through a templat-
ed native chemical ligation [5]. Remarkably, this reaction
occurred even in presence of substoichiometric amounts of
complementary nucleic acid template and this method is
highly sensitive and selective. This allows us the possibility
of discriminating single base mismach DNA [6].
Herein we introduce the design of a template-controlled
aminoacyl-coupling reaction that transduces the nucleic acid
information into the generation of a peptide conjugate that
acts as antagonist to the X-linked inhibitor of apoptosis pro-
tein (XIAP) [7]. In the previous work we have studied the
DNA-triggered transfer of an aminoacyl group, now we
present our most recent step forward in this field in order to
achieve transfer of longer peptide fragments for the genera-
tion of other bioactive peptides. Particularly, the cytotoxic
KLAK peptide (KLAKLAKKLAKLAK) has been success-
fully synthesized by a nucleic acid triggered reaction. Our
initial studies of the transfer reaction show the first example
of such a fast and effective transfer reaction of long peptide
fragments. Importantly, this was just observed in presence
of the nucleic acid-template.
Figure 1. General scheme of the nucleic acid triggered reaction.
REFERENCES
1. Jacobsen, M., F., Cló, E., Mokhir, A., Gothelf, K., V.,
ChemMedChem 2007, 2, 793 – 799.
2. a) Ma, Z., Taylor, J., S., Proc. Natl. Acad. Sci. USA
2000, 97, 11159 – 11163; b) Ma, Z., Taylor, J., S.,
Bioconjugate Chem. 2003, 14, 679 – 683.
3. Okamoto, A., Tanabe, K., Inasaki, T., Saito, I., Angew.
Chem. Int. Ed. 2003, 42, 2502 – 2504.
4. Arian, D., Cló, E., Gothelf, K., V., Mokhir, A., Chem.
Eur. J. 2010, 16, 288 – 295.
5. Dawson, P., E., Muir, T., W., Clark-Lewis, I., Kent, S.
B. Science 1994, 266, 776 – 779.
6. Grossmann, T., N., Röglin, L., Seitz, O.
Angew. Chem. Int. Ed 2008, 47, 7119 – 7122.
7. Erben. A., Grossmann, T., N., Seitz, O. Angew.
Chem. Int. Ed. 2011, 50, 2828-2832.
102

NOTA- AND DOTA-DERIVED CPG-SUPPORTS FOR THE SYNTHESIS OF 3’-RADIOMETALLATED OLIGONUCLEOTIDES
Pasi Virta,1* Anu Kiviniemi,
1 Joonas Mäkelä,
1 Tiina Saanijoki,
2 Harri Lönnberg,
1 Tiina Laitala-
Leinonen2 and Anne Roivainen
2
1Department of Chemistry, University of Turku, FIN-20014 Turku and
2Turku PET Centre, Turku University Hospital,
FIN-20521 turku, Finland
ABSTRACT
Esterified NOTA- and DOTA-ligands were immobi-
lized to LCAA-CPG and oligonucleotide chains on these
supports were automatically assembled. A two step
cleavage with aqueous alkali and ammonia released the
desired 3’-conjugated oligonucleotides. After conversion
to their 68
Ga chelates, applicability of the conjugates for
in vivo imaging with PET was verified.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
Imaging by positron emission tomography (PET) is an
emerging technique for quantitative visualization of molecu-
lar binding in vivo.1 The positron emitter may be introduced
into the biomolecule of interest either covalently or by bind-
ing an appropriate radiometal (e.g. 89
Zr, 68
Ga, 64
Cu, 86
Y) to a
chelating structure. The latter approach usually involves two
post-synthetic steps. An appropriate ligand precursor is first
attached to the biomolecule and then the resulting conjugate
is complexed with the radiometal. Although the conjugation
itself by this bifunctional chelator approach (BFC) may be
carried out in a high yield, chromatographic behaviour of
the desired product may be difficult, owing to insufficient
difference in the chromatographic behaviour between the
conjugate and the parent biomolecule.
Our interest has been to conjugate other biomolecules to
oligonucleotides2 and to study their biodistribution in vivo
by PET imaging. In many cases, the 5´-terminus of the oli-
gonucleotide chain bears the desired biomolecule and, hence,
the chelator should be introduced in the 3´-terminus. Con-
struction of such biomolecular conjugates is not trivial and
preliminary attempts to radiolabel them by post synthetic
BFC approach has proven inefficient.
An efficient solid-supported technique for the introduc-
tion of 3´-terminal NOTA/DOTA-chelators, leaving the 5´-
OH for further conjugation, is described in the present study.
Appropriate NOTA and DOTA precursors were immobi-
lized to long chain alkyl amine derivatized controlled pore
glass (LCAA-CPG) and automatic oligonucleotide chain
elongation on these supports was carried out (Figure 1). A
two-step cleavage3
(first 0.1 mol L-1
aq NaOH, then ammo-
nolysis) released virtually pure 3’-NOTA and 3´-DOTA
conjugated oligonucleotides (according to HPLC and ESI-
MS). Apart the synthesis of these universal supports, no
profound synthetic skill is required. 3’-NOTA/DOTA-
conjugated dT6 and 2’-O-methyl oligoribonucleotide 5’-
UGU AAA CCA UGA UGU GCU GCU A-3’(anti-miR15b)
were synthesized. These conjugates were radiolabelled with 68
Ga and their applicability to in vivo imaging by PET was
verified. It was clearly seen that differences in biodistribu-
tion were mainly caused by oligonucleotide sequence and
marginally by the chelators. Each ligand resulted well-
behaving and stable complex with 68
Ga.
The financial support from the Academy of Finland is
gratefully acknowledged
REFERENCES
1. a) Tanaka, K. and Fukase, K. (2008) Org. Biomolec.
Chem., 2008, 6, 815-828. b) Wadas, T. J. et al. Chem.
Rev. 2010, 110, 2858-2902. c) Zeglis, B. M. and Lewis,
J. S. Dalton Trans. 2011, 40, 6168-6195.
2. a) Katajisto, J. et al. Bioconjugate Chem. 2004, 15, 890-
896. b) Karskela, M. et al. Bioconjugate Chem. 2008,
19, 2549-2558. c) Karskela, M. et al. Bioconjugate
Chem. 2010, 21, 748-755. d) Kiviniemi, A. et al. Bio-
conjugate Chem. 2008, 19, 1726-1734. e) Kiviniemi, A.
et al. Bioconjugate Chem. 2010. 21, 1890-1901. f) Ki-
viniemi, A. et al. Bioconjugate Chem. 2011, 22, 1249-
1255.
3. Jaakkola L. et al. Bioconjugate Chem. 2006, 17, 1105-
1107.
Figure 1. i) 1: Automated DNA/RNA-synthesis, 2: Two step cleavage: first aq NaOH, then ammonia
103

STUDIES ON THE EFFECTS OF ARGININE RESIDUES INTRODUCTION TO PEPTIDE RIBONUCLEIC ACIDS (PRNA) ON
THE COMPLEX STABILITIES WITH DNA/RNA AND CELL MEMBRANE PERMEABILITY
Takehiko Wada,1* Ryohei Uematsu,
1 Tatsuya Mizutani,
1 Akira Nagami,
2 Seiji, Sakamoto,
1
Yasuyuki Araki,1Ikuhiko Nakase,3 Shiroh Futaki,3 and Yoshihisa Inoue
2
1Institute of Multidisciplinary Research for Advanced Materials(IMRAM), Tohoku University, 2-1-1, Katahira, Aoba-ku,
Sendai, 980-8577, Miyagi, Japan, 2Graduate School of Engineering, Osaka University, 2-1, Yamadaoka,Suita, Osaka
565-0871, Japan and 3 Institute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan.
*Correspondence to: [email protected]
ABSTRACT
In this study, a new series of PRNAs with alternating
PRNA-arginine and serine sequences were prepared in
order to stabilize the PRNA-RNA complex through
electrostatic interactions and also to enhance the cell
membrane permeability. It was revealed that these
PRNAs form stable complexes with the complementary
RNAs or DNAs in a high sequence-specific manner.
Moreover, these PRNAs show high cell membrane
permeability without showing any toxicity.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
We have recently proposed a novel category of nucleic acid
model, i.e., peptide ribonucleic acid (PRNA), which
possesses an isopoly-L-glutamine or poly-L-glutamine
backbone, instead of the phosphate sugar backbone.1
Possessing improved solubility in water, longer ribosetether,
and matched helical pitch, PRNA with an isopoly
(L-glutamic acid) backbone form a stable complex with
complementary DNAs/RNA’s. Furthermore, the recognition
and complexation behavior of PRNA with target RNAs can
be controlled by borax added as an external factor through
the borate formation. Such an active control of DNA
recognition by external factor has not been achieved and
hence the PRNA approach is certainly useful as a unique
method to realize the ‘on-demand,’ such as cancer specific
gene therapeutics in the siRNA/antisense strategy and may
find various biochemical and pharmaceutical applications as
powerful and versatile tools.
In order to extend this methodology, we have designed
and synthesized a series of PRNAs, in which the
5’-amino-5’-deoxypyrimidine ribonucleosides moiety was
appended to a -(L-glutamic acid) backbone through the
5'-amino group at carbonyl group of L-glutamic acid, and
PRNA derivatives. In this study, a series of novel
-peptide ribonucleic acid (PRNA) oligomers, possessing
alternative -PRNA - arginine sequences were newly
designed, synthesized, and evaluated, as the third-generation
PRNA.
As expected, these PRNAs formed highly stable
sequence-specific complexes with the complementary
RNAs, for which both the conventional hydrogen-bonding
interactions between the complementary nucleobase pairs
and the electrostatic interactions between the arginine’s
guanidinium cation and the RNA’s phosphate anion on the
backbone are jointly responsible. Moreover, in the cases of
mismatched RNA’s mixing systems, appreciable Tm could
not be observed, thus PRNAs containing Arg were
expected to have high nucleobase sequence discrimination
abilities. Additionally, It was demonstrated that the
recognition behaviour of Arg introduced PRNA with
complementary RNA can be controlled externally through
the orientation change of pyrimidine nucleobase induced by
borate ester formation of the ribose's 2',3'-cis-diol.
Furthermore, these PRNAs show high cell membrane
permeability without showing any toxicity.
FERENCES
1. Wada, T.; Minamimoto, N.; Inaki, Y.; Inoue, Y. Chem.
Lett., 1998, 27, 1025-26; J. Am. Chem. Soc., 2000, 122,
6900-10; Sato, H.; Wada, T.; Inoue, Y. Tetrahedron,
2003, 59, 7871-08.
2. Futaki, S. Bioplym., 2006, 84, 241-08.
Chart 1. Structure of RNA, - PRNA, and - PRNA.
Figure 1. Fluorescence images of
HeLa cell followed incubation in
fluorophore labelled PRNAs in
MEM (+) buffer at 37˚C for 1h.
104

Clicking the m3G-CAP
Malgorzata Wenska-Honcharenko1*, Joanna Romanowska2, Martina Jezowska1,
Margarita Alvira1, Pedro Moreno3, C. I. Edvard Smith3, Roger Strömberg1*
1 Department of Biosciences and Nutrition, Karolinska Institutet, Novum, SE-141 57 Huddinge, Sweden;
2 Institute of
Bioorganic Chemistry, Polish Academy of Sciences, Noskowskiego 12/14, 61-704 Poznań, Poland; 3Department of
Laboratory Medicine, Karolinska Institutet, Novum, SE-141 57 Huddinge, Sweden; * Correspondence to: [email protected]; [email protected]
ABSTRACT
Accessing the nucleus through the surrounding mem-
brane poses one of the major obstacles for therapeutic
molecules large enough to be excluded due to nuclear
pore size limits. In some therapeutic applications the
large size of some nucleic acids, like plasmid DNA,
hampers their access to the nuclear compartment. We
report on the synthesis and possible applications of a
natural RNA 5’-end nuclear localization signal com-
posed of a 2,2,7-trimethylguanosine cap (m3G-CAP)1.
We demonstrate the use of the m3G signal as an adaptor
that can be attached to different oligonucleotides as well
as proteins, thereby conferring nuclear targeting capa-
bilities with capacity to transport large-size cargo mole-
cules.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
Nucleic acid technologies, including siRNA (short
interfering RNA)2 and antisense technologies
3, have in
common a number of limitations for use in vivo for therapy.
Limiting factors are stability of oligonucleotides in biologi-
cal fluids and delivery to the site of action.
Over the last decade several groups have contribut-
ed to the development of methods for splice-switching, with
the aim of converting DMD into the milder Becker form by
removing exons with mutations causing frame-shifts. 4
The route of delivery is a limitation for this kind of
treatment, the oligonucleotides have to enter cells and ac-
cumulate in cell nucleus where the splice switching takes
place. We have reported on the synthesis and possible appli-
cations of a natural RNA 5’-end nuclear localization signal
composed of a 2,2,7-trimethylguanosine cap (m3G-CAP, Fig
1)1. Here we demonstrate the use of the m3G signal ana-
logues as adaptors that can be attached to different oligonu-
cleotides as well as proteins.
For visualization of transport to the nucleus a pro-
tocol using fluorescent STV-Alexa is used. Streptavidin
binds biotin residue tightly and this complex is readily de-
tected by confocal microscopy. We are developing a general
method for convenient and efficient attachment of the m3G-
Cap to different biological cargos for which modified m3G-
Caps were synthesized. With these efficient building blocks
it is possible to synthe-
size the conjugates of
m3G-CAP, by Huisgen
[3+2] dipolar cycloaddi-
tion, at 0.5 mM concen-
tration. These are m3G-
CAP structures
equipped with azido-
groups that then can be
readily attached to oli-
gonucleotides or other
cargos that carry triple
bond functionalities.
Biological evaluations
of these conjugates are
ongoing to see which linker length and position will be ac-
cepted by the transport proteins.
For the purpose of studying transport of large car-
gos (i.e., Steptavidin) we decided to synthesize two biotin
linkers, with different lengths, equipped with a reactive
group that can be reacted with the m3G-CAPs above. This
group is an activated triple bond donor p-(N-
propynoylamino)toluic acid (PATA)6 that we have recently
developed and it allows the click reaction to be efficient also
at low concentration, where standard triple bonds give virtu-
ally no reaction.
REFERENCES 1. Moreno P.#, Wenska M.#, Lundin K.E., Wrange Ö.,
Strömberg R., and Smith C. I. E., Nuc. Acids Res., 2009; 37:
2867 - 2881.
2. Dorsett, Y.; Tuschl, T. Nature Rev. Drug Disc. 2004, 3, 318-
329.; Manoharan, M. Curr. Opin. Chem. Biol. 2004, 8, 570-
579
3. Crooke, S. T. Annu. Rev. Med. 2004, 55, 61-95.; Crooke, S. T.
Curr. Molec. Med. 2004, 4, 465-487
4. Van Deutekom J., Janson A., et al. N Engl. J. Med., 2007,
357: 2677-86,
5. Arechavala-Gomeza, K.M. at all; Lancet Neur. 2009, 8(10),
918-928.
6. Wenska, M., Alvira, M., Steunberg, P., Stenberg, A., Murtola,
M., Strömberg, R., Nuc. Acids Res., 2011; 39: 9047 - 9060.
Figure 1. Schematic representation of
the 2,2,7-trimethyl-guanosine-cap (m3G-
cap)
105

EVIDENCE FOR C-H...F-C PSEUDOHYDROGEN BONDING IN SHORT 2'F MODIFIED NUCLEIC ACID DUPLEXES
M. Yahyaee Anzahaee 1,N. Martin-Pintado 2 ,C. Gonzalez 2 and
M.J. Damha 1*. 1 Department of Chemistry, McGill University, Montreal, QC, Canada;
2Instituto de Química Física Roca solano,
CSIC, C/. Madrid, Spain. * Correspondence to:[email protected]; [email protected]
ABSTRACT
We describe the solution (NMR) structure of
several single stranded tetra-nucleotides as well as
their hybrid duplexes with complementary
RNA.We also establish the role of C-2'F...H8-C
non-covalent interactions on modulating π-π
stacking and pre-organization of these short 2'F-
modified hybrid duplexes.
We previously showed that short H...F distances exist
in duplexes comprising oligo-2'F-arabinucleotides
(2F-ANA) bound to a target RNA strand [1].
Comparison between the solution structure of 2'F-
ANA:RNAandANA:RNA hybrids revealed that sugar
and glycosidic angle conformations provoke a
systematic steric clash between the 2'-OH and their
own H6/H8 base protons in ANA nucleotides.
However, when 2'-OH is replaced with 2'-F,
favorable electrostatic interactions are provoked, due
to the smaller radii of the fluorine and its higher
electronegativity, compared to the 2'-OH group. This
noncovalent interaction appears to be more favorable
in pyrimidine-purine (Py-Pu) steps.
In a more recent study, comparisons of the melting of
duplexes with identical sequence composition but a
rearranged sequence confirmed that energetically
important fluorine-mediated pseudohydrogen
bonding is indeed in operation [2]. It was observed
that depending on the geometry of the C2'-F/H8C
network, the C2'F...H8C interaction occurs either in a
sequential pattern (Py-Pu steps), or in an intra-
residual pattern i.e. within the same 2'F-ANA
modified purine (Fig. 1). While in most cases, an
intra-residual pseudo-hydrogen bonding is expected
as a result of the close proximity of the hydrogen
bond acceptor and donor, C2'F and H8C respectively,
in some other cases, where the base stacking
geometry of duplex can adjust without incurring a
steric penalty,a sequential interaction may occur; and
this inter-residual interaction is of particular
importance when the H-bond donor (purine H8) is
activated by the presence of fluorine at its own 2'-
position.
.
Fig. 1 Inter-residual Hydrogen bond Intra-residual Hydrogen bond
Independently, Egli and co-workers have obtained
crystal structures of fluorine-modified RNA (2F-
RNA) that are also suggestive of an attractive H...F-C
interaction [3].
To further characterize fluorine-modified nucleic
acids and Energetically important C-2'F...H8-C
pseudohydrogen bonds, in which organic fluorine
acts as hydrogen bond acceptor, we conducted
detailed NMR, UV melting and CD experiments on
short duplexes designed to better characterize the
putative C-H...F-C bonding. Specifically, we have
combined 1H and
19F NMR spectroscopy to study the
solution structure of several single stranded
oligonucleotides (tetramers) as well as their hybrid
duplexes with complementary RNA. The sequences
include different patterns of DNA, RNA, 2'F-ANA
and 2'F-RNA nucleotides. We further establish the
role of C-2'F...H8-C non-covalent interactions on
modulating π-π stacking and pre-organization of
single strands and compare them with their RNA
hybrid duplexes.
This work was funded in part by CIHR (MJD& MY).
REFERENCES
[1] Watts et al., Nucleic Acids Res.; 2010, 38, 2498-
2511.
[2] Yahyaee et al., J. Am. Chem. Soc.; 2011, 133,
728-731.
[3]Manoharan et al. Angew. Chem. Int. Ed.; 2010, 50,
2284-2288.
106

Enhanced Gene Silencing Activity Using Phosphorodithioated siRNAs Xianbin Yang1*, Barbara Nawrot2, Malgorzata Sierant2, Magdalena Janicka2, and Na Li1
1AM Biotechnologies LLC, 12521 Gulf Freeway, Houston, Texas 77034, USA;
2Department of Bioorganic Chemistry,
Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Lodz, Poland. * Correspondence to: [email protected]
ABSTRACT
Appropriately protected ribonucleoside thiophosphorami-dite monomers were prepared for the synthesis of siRNA containing phosphorodithioate (PS2) substitutions in which the two non-bridging oxygen atoms are replaced by sulfur atoms. Incorporating the PS2 modification at certain posi-tions significantly enhanced gene silencing activity when compared with unmodified siRNA.
INTRODUCTION
RNA interference (RNAi) has emerged as a novel me-chanism that is activated in mammalian cells by small inter-fering RNAs (siRNAs), which hold great potential in pro-viding a new class of therapeutics. Although unmodified siRNAs have been used with success for gene silencing, chemical modifications of one or both strands are desired for research and pharmaceutical applications in order to enhance potency and to improve pharmacokinetic properties. Like natural phosphorodiester linkages of RNA, PS2-RNA is achiral at phosphorus. The PS2 substitution is thus a very attractive RNA analog because it closely mimics the struc-ture of natural RNA and should have other biochemical and biophysical properties similar to phosphorodiester linkages of RNA. In this presentation, we report the synthesis of pro-tected ribonucleoside thiophosphoramidite monomers and
their use for the solid-phase preparation of RNA molecules containing PS2 substitutions. We anticipated that the addi-tion of PS2 modifications to certain positions of the siRNA duplex would increase nuclease resistance and enhance RNAi activity. To the best of our knowledge these are the first studies evaluating biological properties of siRNA dup-lexes containing PS2 substitutions.
RESULTS AND DISCUSSION
Protected ribonucleoside thiophosphoramidite monomers were devised based on a sulfur-linked, base-labile β-(benzoylmercapto)ethyl protecting group used in the Ca-ruthers laboratory for the solid-phase synthesis of PS2-DNA (1). The PS2-RNAs were synthesized on an Expedite 8909 DNA/RNA Synthesizer using commercial 5′-DMT-2′-O-TBDMS nucleoside (ABz, CAc, GAc, and U) phosphoramidite monomers as well as thiophosphoramidites. The average stepwise coupling efficiency of all phosphoramidites includ-ing thiophosphoramidites was about 97% as estimated by the DMT-cation assay. After deprotection, all of the mod-ified RNAs were isolated by FPLC according to a previous-ly described protocol for purifying PS2-DNAs (2). The
structures of the PS2-RNAs were confirmed by ESI-MS. The purity of all RNAs and correctness of double strand structure of PS2-siRNAs were assessed by denaturing 20% polyacrylamide (PAGE) and agarose gel electrophoresis analyses, respectively.
Gene silencing activity of the PS2-siRNAs was tested us-ing a previously developed dual fluorescence reporter sys-tem based on measurement of the relative fluorescence in-tensity of the enhanced green fluorescent protein (EGFP) expressed from a BACE1-EGFP fusion or EGFP plasmid, versus red fluorescent protein (RFP) expressed from a non-target plasmid serving as a transfection control (3). The en-hanced gene silencing experiments performed on several PS2-siRNA duplexes containing the PS2 linkages either in the sense or antisense strand were demonstrated. In addition, PS2-siRNAs have also showed statistically significant in-creased in gene silencing activity (p<0.001) when compared with their counter-parts containing phosphoromonothioate linkages, which may have variable biological effects. More-over, 5′-phosphorylation had little or no effect on the activi-ty of any of these PS2-siRNAs. These results suggest that the free 5′-hydroxyl of the wild-type siRNA or PS2-siRNA duplex is first phosphorylated by endogenous kinases, and then enters the RNAi pathway. Finally, PS2 modifications added to siRNA increase serum stability (4).
CONCLUSION
The enhanced gene silencing activity observed in this work indicates that PS2-siRNAs can improve gene silencing in vivo. Given the straightforward synthesis of PS2 linkages as well as their known nuclease resistance and lack of chi-rality, PS2 modifications appear promising for use in siRNA research and therapeutic applications.
REFERENCES
1. Wiesler, W.T. and Caruthers, M.H. (1996) J. Org.
Chem., 61, 4272-4281. 2. Yang, X., Hodge, R.P., Luxon, B.A., Shope, R. and
Gorenstein, D.G. (2002) Anal. Biochem., 306, 92-99.
3. Sipa, K., Sochacka, E., Kazmierczak-Baranska, J., Maszewska, M., Janicka, M., Nowak, G. and Nawrot, B. (2007) RNA, 13, 1301-1316.
4. Yang X., Sierant M., Janicka M., Peczek L., Marti-nez C., Hassell T., Li N., Li X., Wang T. and Naw-rot B. ACS Chem. Biol. DOI: 10.1021/cb300078e.
107

PEPTIDE NUCLEIC ACIDS (PNAS) IN CANCER DIAGNOSTICS
Eylon Yavin1*, Abraham Rubinstein1, Aviram Nissan2 and Yossi Kam1
1Institute for Drug Research, School of Pharmacy, Faculty of Medicine, The Hebrew University of Jerusalem,
Ein Kerem 12065, Jerusalem 91120, Israel. * Correspondence to: [email protected] 2Department of Surgery, Rabin Medical Center, Beilinson Campus, 39 Jabotinski Street, Petah Tikvah. 49100, Israel.
ABSTRACT
Two case studies are shown where Peptide Nucleic Ac-ids (PNAs) are used for the detection of cancer. The former is based on the detection of endogenous mRNA (kRAS) as a biomarker for cancer. The latter demon-strates the detection of a non-coding RNA as a novel biomarker in a variety of cancer cell lines.
INTRODUCTION
Peptide nucleic acids (PNA) are DNA analogs that avidly bind to DNA and RNA and are highly resistant to degrada-tion in a biological medium. (1) In addition, such molecules have been shown to readily penetrate cells by the covalent attachment of cell penetrating peptides. (2)
RESULTS AND DISCUSSION
Detection of mRNA alterations is a promising approach for identifying biomarkers as means of differentiating be-nign from malignant lesions. By choosing the K-ras onco-gene as a target gene, two types of molecular beacons (MBs) based on either phosphothioated DNA (PS-DNA-MB) or peptide nucleic acid (TO-PNA-MB, where TO = thiazole orange (3)) were synthesized and compared in vitro and in-vivo. (4) Their specificity was examined in wild-type K-Ras (HT29) or codon 12 point mutations (Panc-1, SW480) cell lines. Incubation of both beacons in-vitro with total RNA, extracted from the three cell lines, showed high-er fluorescent signal detected for target (fully complement, Panc-1) than in single mismatch mRNA transcripts. Trans-fection of TO-PNA-MB with PEI resulted in fluorescence in cells expressing the fully complementary RNA transcript (Panc-1) but no detectable fluorescence in cells expressing the K-RAS mRNA that has a single mismatch to the de-signed TO-PNA-MB. In contrast, PS-DNA-MB showed no fluorescence in all cell lines tested post PEI transfection.
CCAT-1 (colon cancer associated transcript 1) is a non-coding RNA abundantly expressed in many cancers (5) but not in somatic cells. We have recently synthesized TO-PNA-MB with a cell penetrating peptide that targets this endogenous RNA bio-marker. Preliminary data shows that TO-PNA-MB detects this non-coding RNA in a variety of cancer cells after adding the PNA’s in medium without the need of a transfection agent. Interestingly, we find that a cancer cell line (Melanoma, SK-Mel) that does not express
CCAT-1 shows negligible fluorescence after adding the PNA probe.
Finally, fluorescence in-situ hybridization (FISH) of the TO-PNA-MB (targeting CCAT-1) with a biopsy (tissue) from a colon cancer patient shows high fluorescence in comparison to a slice taken from the same patient from a non-malignant region (Figure 1).
Figure 1. Detection of CCAT-1 by TO-PNA-MB in malignant
and normal tissue.
CONCLUSION
These studies highlight the medical potential of using PNAs as diagnostic molecules for the detection of cancer with high sensitivity and excellent specificity.
REFERENCES
1. Egholm, M., Buchardt, O., Christensen, L., Behrens, C., Freier, S.M., Driver, D.A., Berg, R.H., Kim, S.K., Nor-den, B., Nielsen, P. E. Nature 1993, 365, 566−568.
2. Turner, J.J., Ivanova, G.D., Verbeure, B., Williams, D., Arzumanov, A.A., Abes, S., Lebleu, B. Gait, M.J. Nucl. Acids Res. 2005, 33, 6837-6849.
3. Kohler, O., Jarikote, D.V., Seitz, O. ChemBioChem 2005, 6, 69−77.
4. Kam, Y., Rubinstein, A., Nissan, A., Halle, D., Yavin, E. Mol. Pharm. 2012, 9, 685-693
5. Nissan, A., Stojadinovic, A., Mitrani-Rosenbaum, S., Halle, D., Grinbaum, R., Roistacher, M., Bochem, A., Dayan, B.E., Ritter, G., Gomceli, I., Bostanci, E.B., Akoglu, M., Chen, Y-T., Old, L.J., Gure, A.O. Int. J. Cancer 2011, 130, 1598-1606.
108


















![RNA N6-methyladenosine reader IGF2BP3 regulates cell ......RNA methylation modification information and partici-pate in the translation and degradation of downstream RNA [4]. Colon](https://static.fdocuments.in/doc/165x107/60e95ed35f0c0378245d0e57/rna-n6-methyladenosine-reader-igf2bp3-regulates-cell-rna-methylation-modification.jpg)