
Research Informed Consent Kristin Bialobok, MSN, RN, CCRC, CCRA Director Office of Research...
32
Research Informed Consent Kristin Bialobok, MSN, RN, CCRC, CCRA Director Office of Research Education and Regulatory Management
-
Upload
angeline-leblanc -
Category
Documents
-
view
216 -
download
2
Transcript of Research Informed Consent Kristin Bialobok, MSN, RN, CCRC, CCRA Director Office of Research...

Research Informed Consent
Kristin Bialobok, MSN, RN, CCRC, CCRA
Director
Office of Research Education and Regulatory Management

Purpose of Consent ?Purpose of Consent ?
Prospective Subject Will ..
• Understand nature of research
• Be informed of purpose, risks, and benefits, and alternative therapies
• Make a Voluntary Decision about Participation
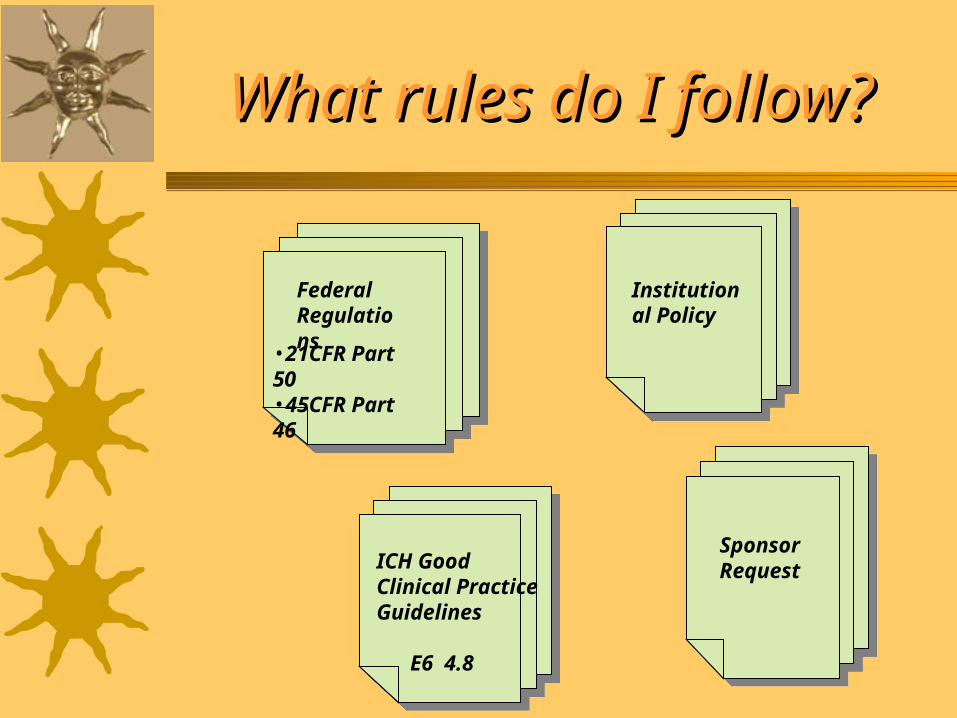
What rules do I follow?What rules do I follow?
Federal Regulations
ICH Good Clinical Practice Guidelines
E6 4.8
Institutional Policy
Sponsor Request
•21CFR Part 50•45CFR Part 46

EXCEPTIONS:• IRB has granted a waiver of consent
• Life-threatening condition and Inability to communicate with the subject and Time is insufficient to obtain consent from subject’s legal
representative and There is no alternative method of approved therapy
Must Be Followed By notification of the IRB within 5 days review and evaluation of test article use by another physician obtain consent ASAP.
Is Informed Consent Is Informed Consent Always Essential?Always Essential?

Research Consent Process Research Consent Process
o Consent DocumentConsent Document
o Consent DiscussionConsent Discussion

THE INFORMED THE INFORMED CONSENT DOCUMENTCONSENT DOCUMENT

Research Consent DocumentResearch Consent Document
21 CFR 50.25 - Basic Elements of IC21 CFR 50.25 - Basic Elements of IC

Research Consent DocumentResearch Consent Document
21 CFR 50.25 - Additional Elements21 CFR 50.25 - Additional Elements

Research Consent Research Consent DocumentDocument
• Language that is easily understood• Language must be appropriate to the
population being studied• Language translators should be qualified and
IRB authorized• Consider comprehension as well as readabilityConsider comprehension as well as readability
• Limit medical terminologyLimit medical terminology
• Avoid informal speechAvoid informal speech
Readability

Research Consent Research Consent DocumentDocument
Informed ConsentInformed Consent
(final version)(final version)
IRBIRB
PIPI SponsorSponsor

INFORMED CONSENT INFORMED CONSENT DISCUSSIONDISCUSSION

Who has the Authority to Obtain Who has the Authority to Obtain Research Consent?Research Consent?
• What do the regulations say?What do the regulations say?• SilentSilent
• What does ICH GCPs say?What does ICH GCPs say?• 4.1.5 – “the investigator should maintain a 4.1.5 – “the investigator should maintain a
list of appropriately qualified persons to list of appropriately qualified persons to whom the investigator has delegated whom the investigator has delegated significant trial-related duties.”significant trial-related duties.”
• What does the institution say?What does the institution say?
Consideration: Nurse Practice ActConsideration: Nurse Practice Act

o Discussion between the physician and the participant.Discussion between the physician and the participant.
o Review of information by research nurse/coordinator.Review of information by research nurse/coordinator.
o All questions and concerns addressed.All questions and concerns addressed.
o No exculpatory languageo Minimal chance of coercion/undue influence
o Allow adequate timeAllow adequate time
o Signatures obtained.Signatures obtained.
Informed Consent ProcessInformed Consent Process

o Informed consent must be obtained prior to Informed consent must be obtained prior to any protocol specific testing being any protocol specific testing being conducted.conducted.
o If protocol specific testing done the same If protocol specific testing done the same day as informed consent document signed, day as informed consent document signed, must be clear documentation of the must be clear documentation of the chronological order in the medical record.chronological order in the medical record.
Informed Consent and Informed Consent and Screening ProceduresScreening Procedures

COMPLETING THE COMPLETING THE INFORMED CONSENT INFORMED CONSENT
DOCUMENTDOCUMENT

Completing the DocumentCompleting the Documento All blank spaces completedAll blank spaces completed
o No additions or deletionsNo additions or deletions
o Signatures obtainedSignatures obtained
FDA 21CFR 50- The patient or authorized representative shall sign and
date the informed consent.
ICH GCP E6 4.8 – …the informed consent should be signed and personally dated by the subject or legally authorized representative.
Institutional PoliciesInstitutional Policies
OHRP 45 CFR 46- ….signed by the patient or legally authorized representative.
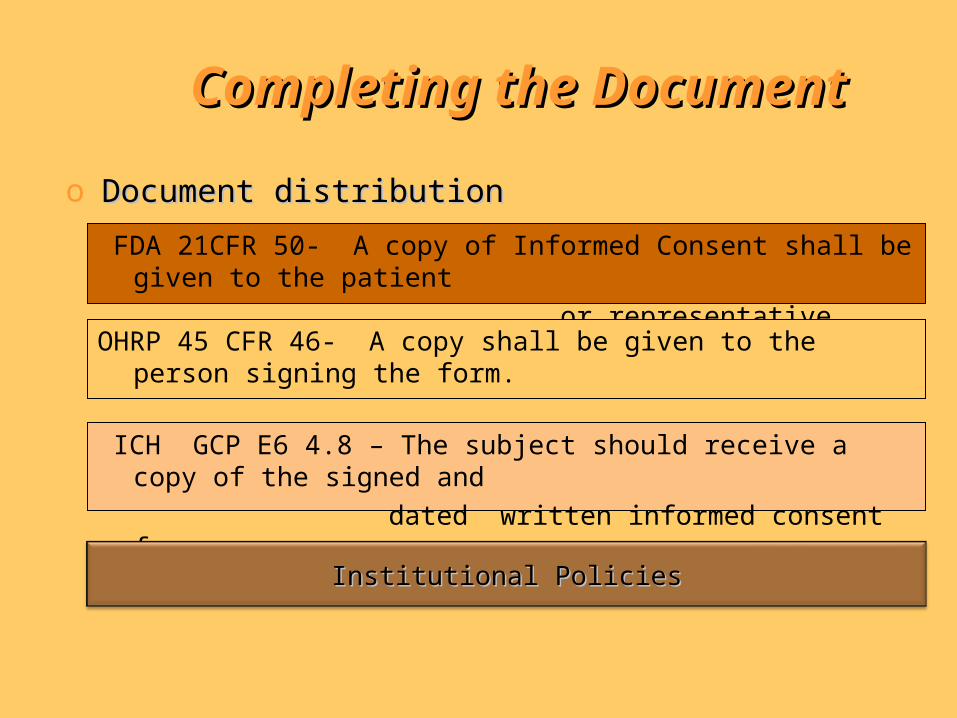
Completing the DocumentCompleting the Document
o Document distributionDocument distribution
FDA 21CFR 50- A copy of Informed Consent shall be given to the patient
or representative.
ICH GCP E6 4.8 – The subject should receive a copy of the signed and
dated written informed consent form…
Institutional PoliciesInstitutional Policies
OHRP 45 CFR 46- A copy shall be given to the person signing the form.

Completing the DocumentCompleting the Document
• When do you need a witness?• When presenting the informed consent document orally• Use of a short form• If required by the IRB
• Who can be the witness?
ICH GCP – Impartial witness – a person who is independent of the trial, who cannot be unfairly influenced by people involved in the trial, who attends the informed consent process…

DOCUMENTING THE DOCUMENTING THE INFORMED CONSENT INFORMED CONSENT
PROCESSPROCESS

DOCUMENTING DOCUMENTING THE PROCESSTHE PROCESS
o Protocol number or name of studyProtocol number or name of study
o Alternative treatment options Alternative treatment options discussed.discussed.
o A statement that the A statement that the protocolprotocol, and the , and the informed consentinformed consent were reviewed with were reviewed with the participant, including the the participant, including the risksrisks and and benefits benefits of the study.of the study.
o Time for questions to be asked and Time for questions to be asked and answered.answered.
o Description of the participant’s Description of the participant’s decisiondecision
o Copy of consent was given to the Copy of consent was given to the participantparticipant

RE-CONSENTINGRE-CONSENTING
Participants must be informed of new Participants must be informed of new information that may affect their information that may affect their willingness to take part in the research.willingness to take part in the research.

CONSENTING MINORSCONSENTING MINORS

CONSENTING MINORSCONSENTING MINORS
MinorMinor Per state law – Texas=Under 18 years oldPer state law – Texas=Under 18 years old
Participant Participant under 18 yunder 18 y
the parent/s or the parent/s or legal guardianlegal guardian
informed consent informed consent processprocess (on the child’s behalf)(on the child’s behalf)

CONSENTING CONSENTING MINORSMINORS
Depending on the type of research, the IRB Depending on the type of research, the IRB
may make provisions for “assent” of childrenmay make provisions for “assent” of children..
Assent Assent
““A child’s affirmative agreement to be a participant in A child’s affirmative agreement to be a participant in research. Mere failure to object should not, absent research. Mere failure to object should not, absent affirmative agreement, be construed as assent.”affirmative agreement, be construed as assent.”
Need to specifically ask for the institution’s policy before auditing.

Yes No
Age >/=18
Subject and Person obtaining consent sign the ICD
Parent/Guardian, witness, and Person obtaining consent sign the ICD
MDACC Flow Chart – Consenting MinorsMDACC Flow Chart – Consenting Minors
Child Age 7-12 – Verbal Assent Only
Child Age 13-17 – Written Assent Required

Non-English Speaking Non-English Speaking ParticipantsParticipants

Non-English Speaking Non-English Speaking ParticipantsParticipants
• Ideal is to use consent translated to participant’s native language
• 45 CFR 46.117(b) - “Short form written consent document…”

What do we look for as auditors?What do we look for as auditors?
• Does ICD contain all required elements?
• Was ICD used for participant the most current IRB approved document at time of enrollment?
• Is a the ICD present in the participant’s medical record?
• Is a copy of the ICD present in the investigator’s file?
• Are all blank spaces completed?

• Does writing appear that participant signed and dated ICD?
• Is witness’s signature and date present, if needed?
• Is person’s who obtained consent signature and date present?
• Was delegation of authority to obtain consent appropriate?
• Do all date on the consent match?
• If dates do not match is explanation present in the on-study progress note?
What do we look for as auditors?What do we look for as auditors?

• Is full informed consent process documented in progress note?
• Is participant a minor?• If minor, is assent portion of the ICD completed?
• Is participant Non-English speaking?
• If Non-English speaking was institutional policy followed?
What do we look for as auditors?What do we look for as auditors?

FDA Inspections and Informed Consent Statistics
• 2007- 11% of the clinical investigators/sites inspected were cited for informed consent related issues.
• 2007- 32% of the investigators/sites who received OAI (Official Action Indicated) letters included informed consent citations.
Good Clinical Practice A Question & Answer Reference Guide Good Clinical Practice A Question & Answer Reference Guide May 2008May 2008

Practice TimePractice Time


















