
Regulation of tumor cell migration and invasion by the H19 ... · SHORT COMMUNICATION Regulation of...
9
SHORT COMMUNICATION Regulation of tumor cell migration and invasion by the H19/let-7 axis is antagonized by metformin-induced DNA methylation L Yan 1,2 , J Zhou 2,3 , Y Gao 2,4 , S Ghazal 2 , L Lu 5 , S Bellone 2 , Y Yang 2 , N Liu 2,6 , X Zhao 1 , AD Santin 2 , H Taylor 2 and Y Huang 2 The imprinted, developmentally regulated H19 long noncoding RNA has been implicated in the pathogenesis of diverse human cancers, but the underlying mechanisms have remained poorly understood. Here, we report that H19 promotes tumor cell migration and invasion by inhibiting let-7, a potent tumor suppressor microRNA that functions to posttranscriptionally suppress the expression of oncogenes that regulate cell growth and motility. We show that H19 depletion impairs, whereas its overexpression enhances the motility and invasiveness of tumor cells. These phenomena occur, at least in part through affecting let-7-mediated regulation of metastasis-promoting genes, including Hmga2, c-Myc and Igf2bp3. This H19/let-7-dependent regulation is recapitulated in vivo where co-expressions of oncogenes and H19 exist in both primary human ovarian and endometrial cancers. Furthermore, we provide evidence that the anti-diabetic drug metformin inhibits tumor cell migration and invasion, partly by downregulating H19 via DNA methylation. Our results reveal a novel mechanism underpinning H19-mediated regulation in metastasis and may explain why in some cases increased let-7 expression unexpectedly correlates with poor prognosis, given the widely accepted role for let-7 as a tumor suppressor. Targeting this newly identified pathway might offer therapeutic opportunities. Oncogene (2015) 34, 3076–3084; doi:10.1038/onc.2014.236; published online 4 August 2014 INTRODUCTION The imprinted H19 is among the highest expressed genes during embryogenesis and placental development, but its expression is strongly downregulated in most adult tissues except for skeletal muscle and heart. 1 Although the physiological significance of persistent H19 expression in skeletal muscle and heart has remained unclear, the conserved H19-Igf2 locus, which also contains the reciprocally imprinted Igf2, is known to play an important role in embryo development and growth control. 1 Two human genetic disorders, Beckwith–Wiedemann Syndrome and Silver–Russell Syndrome, have been linked to this locus, with Beckwith–Wiedemann Syndrome also being associated with a predisposition to childhood tumors. 1 H19 encodes a 2.3 kb capped, spliced and polyadenylated noncoding RNA that is predominantly cytoplasmic, with a minor fraction also found in the nucleus. 1 Although H19 is not expressed in most normal adult tissues, its re-expression has been detected in a broad spectrum of malignancies, 2 including ovarian and endometrial cancers. 3,4 Emerging evidence from studies using both cell culture systems and xenogenic mouse models supports H19 being an oncogene, 2,5–8 although a role as a tumor suppressor has also been suggested. 9–11 Further, genome-wide association studies have identified a number of single nucleotide polymorphisms associated with breast cancer susceptibility in H19 and a specific single nucleotide polymorphism has been linked to shorter metastasis-free survival. 12,13 However, how H19 functions as an oncogene is not well understood. Two recent reports have suggested that the oncogenic activity of H19 might be attributed to miR-675, which is processed from full-length H19 long noncoding RNA and which targets the tumor suppressor retinoblastoma Rb. 5,8 On the other hand, Luo et al. 7 have shown that the full-length H19 long noncoding RNA (herein called H19) binds to EZH2 (a key component of Polycomb repressive complex 2) and inhibits transcription of a selective group of genes, thereby promoting bladder cancer metastasis. We have recently discovered that H19 acts as a molecular ‘sponge’ for the microRNA (miRNA) let-7 and that this interaction plays an important role in regulating muscle differentiation. 14 H19 harbors multiple let-7-binding sites. Further, it binds and sequesters let-7, thereby preventing let-7 from inhibiting target gene expression. 14 Let-7 suppresses target gene expression by binding to imperfectly complementary sequences in the mRNA, resulting in translational repression and mRNA degradation. 15,16 As impaired let-7 function (because of its decreased expression and/or bioavailability) has been associated with increased tumor metastasis and poor prognosis, 17–21 we hypothesized that H19 may promote metastasis, in part through reducing the bioavail- ability of let-7. In this report, we demonstrate a positive relationship between H19 expression and tumor cell migration and invasion. We 1 Department of Obstetrics and Gynecology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong, P. R. China; 2 Department of Obstetrics, Gynecology and Reproductive Sciences, Yale Stem Cell Center, Yale University School of Medicine, New Haven, CT, USA; 3 Department of Surgical Oncology, Affiliated Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, P. R. China; 4 Department of Gynecology and Obstetrics, Chinese PLA General Hospital, Beijing, P. R. China; 5 Department of Chronic Diseases Epidemiology, Yale School of Public Health, Yale University School of Medicine, New Haven, CT, USA and 6 Department of Obstetrics and Gynecology, Xiangya Hospital of Central South University, Changsha, Hunan, P. R. China. Correspondence: Professor Y Huang, Department of Obstetrics, Gynecology & Reproductive Sciences, Yale University School of Medicine, Yale Stem Cell Center, 310 Cedar Street LSOG 205C, New Haven, CT 06510, USA. E-mail:[email protected] Received 17 December 2013; revised 16 June 2014; accepted 20 June 2014; published online 4 August 2014 Oncogene (2015) 34, 3076 – 3084 © 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15 www.nature.com/onc
Transcript of Regulation of tumor cell migration and invasion by the H19 ... · SHORT COMMUNICATION Regulation of...

SHORT COMMUNICATION
Regulation of tumor cell migration and invasion bythe H19/let-7 axis is antagonized by metformin-inducedDNA methylationL Yan1,2, J Zhou2,3, Y Gao2,4, S Ghazal2, L Lu5, S Bellone2, Y Yang2, N Liu2,6, X Zhao1, AD Santin2, H Taylor2 and Y Huang2
The imprinted, developmentally regulated H19 long noncoding RNA has been implicated in the pathogenesis of diverse humancancers, but the underlying mechanisms have remained poorly understood. Here, we report that H19 promotes tumor cellmigration and invasion by inhibiting let-7, a potent tumor suppressor microRNA that functions to posttranscriptionally suppress theexpression of oncogenes that regulate cell growth and motility. We show that H19 depletion impairs, whereas its overexpressionenhances the motility and invasiveness of tumor cells. These phenomena occur, at least in part through affecting let-7-mediatedregulation of metastasis-promoting genes, including Hmga2, c-Myc and Igf2bp3. This H19/let-7-dependent regulation isrecapitulated in vivo where co-expressions of oncogenes and H19 exist in both primary human ovarian and endometrial cancers.Furthermore, we provide evidence that the anti-diabetic drug metformin inhibits tumor cell migration and invasion, partly bydownregulating H19 via DNA methylation. Our results reveal a novel mechanism underpinning H19-mediated regulation inmetastasis and may explain why in some cases increased let-7 expression unexpectedly correlates with poor prognosis, given thewidely accepted role for let-7 as a tumor suppressor. Targeting this newly identified pathway might offer therapeutic opportunities.
Oncogene (2015) 34, 3076–3084; doi:10.1038/onc.2014.236; published online 4 August 2014
INTRODUCTIONThe imprinted H19 is among the highest expressed genes duringembryogenesis and placental development, but its expression isstrongly downregulated in most adult tissues except for skeletalmuscle and heart.1 Although the physiological significance ofpersistent H19 expression in skeletal muscle and heart hasremained unclear, the conserved H19-Igf2 locus, which alsocontains the reciprocally imprinted Igf2, is known to play animportant role in embryo development and growth control.1 Twohuman genetic disorders, Beckwith–Wiedemann Syndrome andSilver–Russell Syndrome, have been linked to this locus, withBeckwith–Wiedemann Syndrome also being associated with apredisposition to childhood tumors.1
H19 encodes a 2.3 kb capped, spliced and polyadenylatednoncoding RNA that is predominantly cytoplasmic, with a minorfraction also found in the nucleus.1 Although H19 is not expressedin most normal adult tissues, its re-expression has been detectedin a broad spectrum of malignancies,2 including ovarian andendometrial cancers.3,4 Emerging evidence from studies usingboth cell culture systems and xenogenic mouse models supportsH19 being an oncogene,2,5–8 although a role as a tumorsuppressor has also been suggested.9–11 Further, genome-wideassociation studies have identified a number of single nucleotidepolymorphisms associated with breast cancer susceptibility in H19and a specific single nucleotide polymorphism has been linked to
shorter metastasis-free survival.12,13 However, how H19 functionsas an oncogene is not well understood. Two recent reports havesuggested that the oncogenic activity of H19 might be attributedto miR-675, which is processed from full-length H19 longnoncoding RNA and which targets the tumor suppressorretinoblastoma Rb.5,8 On the other hand, Luo et al.7 have shownthat the full-length H19 long noncoding RNA (herein called H19)binds to EZH2 (a key component of Polycomb repressivecomplex 2) and inhibits transcription of a selective group ofgenes, thereby promoting bladder cancer metastasis.We have recently discovered that H19 acts as a molecular
‘sponge’ for the microRNA (miRNA) let-7 and that this interactionplays an important role in regulating muscle differentiation.14 H19harbors multiple let-7-binding sites. Further, it binds andsequesters let-7, thereby preventing let-7 from inhibiting targetgene expression.14 Let-7 suppresses target gene expression bybinding to imperfectly complementary sequences in the mRNA,resulting in translational repression and mRNA degradation.15,16
As impaired let-7 function (because of its decreased expressionand/or bioavailability) has been associated with increased tumormetastasis and poor prognosis,17–21 we hypothesized that H19may promote metastasis, in part through reducing the bioavail-ability of let-7.In this report, we demonstrate a positive relationship between
H19 expression and tumor cell migration and invasion. We
1Department of Obstetrics and Gynecology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong, P. R. China; 2Department of Obstetrics, Gynecologyand Reproductive Sciences, Yale Stem Cell Center, Yale University School of Medicine, New Haven, CT, USA; 3Department of Surgical Oncology, Affiliated Sir Run Run ShawHospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, P. R. China; 4Department of Gynecology and Obstetrics, Chinese PLA General Hospital, Beijing, P. R. China;5Department of Chronic Diseases Epidemiology, Yale School of Public Health, Yale University School of Medicine, New Haven, CT, USA and 6Department of Obstetrics andGynecology, Xiangya Hospital of Central South University, Changsha, Hunan, P. R. China. Correspondence: Professor Y Huang, Department of Obstetrics, Gynecology &Reproductive Sciences, Yale University School of Medicine, Yale Stem Cell Center, 310 Cedar Street LSOG 205C, New Haven, CT 06510, USA.E-mail:[email protected] 17 December 2013; revised 16 June 2014; accepted 20 June 2014; published online 4 August 2014
Oncogene (2015) 34, 3076–3084© 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15
www.nature.com/onc

determine that the effects are mediated by altered H19/let-7interaction which leads to changes in expression of metastasis-promoting genes targeted by let-7. Finally, we provide evidencethat metformin, an anti-diabetic drug which is increasinglyrecognized as being a potential anti-cancer drug, downregulatesH19, in part through DNA methylation.
RESULTSH19 promotes tumor cell migration and invasionPrevious studies, both in vivo and in vitro, have demonstrated thatH19 overexpression promotes tumor cell growth and metastasiswhereas its downregulation abrogates tumorigenicity of tumorcells.6,7,22 On the basis of our recent finding that H19 reduces thebioavailability of let-7 by acting as a molecular sponge,14 wewished to test the hypothesis that inhibition of let-7 by H19 maycontribute to H19-mediated metastasis. Thus, we first down-regulated H19 using a siRNA specific for H19 (siH1914) in a widelyused human ovarian cancer cell line A2780 and examined effectson cell migration and invasion. Originally established from tumortissue from a patient prior to treatment, this cell line endogen-ously expresses H19, whereas the non-cancerous HEK293 cells donot (Supplementary Figure S1a, compare left column to the right).H19 depletion led to a significant decrease in both migration(Figure 1a) and invasion (Figure 1b) as determined by in vitrotranswell migration and invasion assays. Equal numbers of cellswere loaded into migration chambers and the migration andinvasion assays were carried out for 24 and 36 h, respectively.Thus, it is highly unlikely that the reduced cell migration andinvasion observed were results of decreased cell proliferation.
H19 enhances expression of metastasis-promoting genes targetedby let-7Next, we asked whether the observed effects were results of reliefof H19-dependent inhibition of let-7, given that H19 acts as asponge to sequester let-7.14 In H19-expressing cells, it is theavailability of let-7 rather than its absolute expression level thatdetermines target gene expression.14 We decided to test let-7target gene expression as a read-out for let-7 availability. As aproof of principle, we analyzed the expression of three genes,Hmga2, c-Myc and Igf2bp3 (also called Imp3). We chose these fortwo reasons. First, all three genes contain functional let-7-bindingsites in their 3′-untranslated regions and are validated targets oflet-7.23–27 Second, all three genes have been shown to promotetumor cell migration and invasion using both in vivo and in vitroapproaches. For example, Hmga2 encodes a non-histone chroma-tin-binding protein that promotes tumor cell metastasis throughregulating pro-metastatic genes,19 epithelial–mesenchymal transi-tion (EMT)28–30 and epigenetic modification.31 In the case of c-Myc,it drives tumorigenesis and metastasis by directly stimulating thetranscription of H1922 and Lin2828 (Lin28 blocks processing of let-7precursors to mature forms32,33). The gene product of Imp3belongs to a conserved family of cytoplasmic RNA-bindingproteins that also include Imp1 and Imp2.34 As an oncofetalgene, Imp3 is often re-expressed in many aggressive cancer typesincluding those of female reproductive tissues, and has beenfrequently associated with tumor metastasis34 and poorprognosis.35–37 Mechanistically, Imp3 enhances the motility andinvasiveness of tumor cells by posttranscriptionally regulating theexpression of pro-migration/invasion genes including Hmga2,CD164, MMP9 and CD44.35,37–39 In summary, all three genes,Hmga2, c-Myc and Imp3, have been demonstrated to promotemetastasis via regulating various downstream effector genesdirectly involved in cell migration and invasion.When H19 was downregulated (Figure 1c, first column on the
left, compare red bar to blue bar), the expression of Hmga2, c-Mycand Imp3 were also significantly reduced at both RNA (Figure 1c,
second through fourth columns, compare red bars to blue bars)and protein (Figure 1d, top three blots, compare lanes 2 to lanes 1)levels, consistent with increased availability of let-7, despite thedecreased levels of several let-7 family members tested(Supplementary Figure S1b). The let-7 level decrease was at leastin part a result of decreased expression of Dicer (SupplementaryFigure S1c, middle column, compare red bar with blue bar), aknown let-7 target and which is the key enzyme responsible forprocessing pre-let-7 to mature let-7.40,41 Indeed, a negativefeedback loop between let-7 and Dicer has previously beendocumented.40 Collectively, these results further underscore thenotion that it is the bioavailability of let-7 but not its absolute levelthat controls target gene expression.Next, we performed reciprocal experiments by overexpressing
H19. Thus, an H19-expression plasmid pH1914 or a control emptyvector (Vec) were transfected into A2780. A significant increase inthe expression of Hmga2, c-Myc and Imp3 at both RNA (Figure 1e,second through fourth columns, compare red bars to blue bars)and protein (Figure 1f, top three blots, compare lanes 2 to lanes 1)levels was observed following H19 overexpression (Figure 1e, firstcolumn on the left). Concomitantly, cell migration (Figure 1g) andinvasion (Figure 1h) were also enhanced. These results suggestedthat when H19 levels increased, more let-7 was sequestered andthus unavailable to inhibit target gene expression. The fact thatexogenous H19 overexpression could further increase let-7 targetgene expression suggested that the endogenous let-7 level was inexcess relative to endogenous H19 despite that A2780 alreadyexpressed a high level of endogenous H19 (SupplementaryFigure S1a). Taken together, our results from both H19 loss- andgain-of-function studies support the notion that H19 affects tumorcell migration and invasion through modulating expression ofmetastasis-promoting genes targeted by let-7.EMT has been implicated in the acquisition of metastatic
characters of tumor cells.42 Hmga2 has been shown to play acritical role in EMT.28–30 Thus, we tested the expression of severalEMT markers. Results showed that when H19 was downregulated,there was a concomitant decrease in the expression of Twist, Snail(transcription factors) and Vimentin (a cytoskeletal marker), and anincrease in the expression of E-cadherin (an epithelial cell surfacemarker) (Supplementary Figure S2a), consistent with inhibition ofEMT by H19 depletion.
The H19 effects are mediated by let-7To provide further evidence supporting the H19/let-7 axis inregulating tumor cell migration and invasion, we performed H19knockdown experiments in combination with the let-7-specificinhibitor iLet-7.14 The rationale for this approach was that in thepresence of iLet-7, the effects of H19 downregulation would beabolished. Thus, A2780 cells were transfected with a controlmixture or mixture containing siH19 with or without iLet-7,followed by analysis of gene expression and cell function. H19knockdown (Figure 1i, left panel, first column from left, comparered bar to blue bar) inhibited the expression of Hmga2, c-Myc andImp3 (second through fourth columns, compare red bars to bluebars), whereas co-transfection of iLet-7 relieved this inhibition(second through fourth columns, compare purple bars to redbars). The incomplete rescue of Hmga2 by iLet-7 suggests thatother mechanisms might also be involved in its regulation.Intriguingly, the H19 level slightly increased in the siH19+iLet-7group compared with the siH19+iCon group (Figure 1i, left panel,first column from left, compare purple bar to red bar), suggestinga possible negative feedback regulation between H19 and let-7,which warrants future investigation. Protein analysis showedconsistent results (Figure 1i, right panel, top three blots, comparelanes 3 to lanes 2 and 1). Functional rescue by iLet-7 was furthersupported by the cell migration (Figure 1j) and invasion(Figure 1k) analyses. Taken together, we conclude that the
H19/let-7 axis and cancer prevention and therapyL Yan et al
3077
© 2015 Macmillan Publishers Limited Oncogene (2015) 3076 – 3084
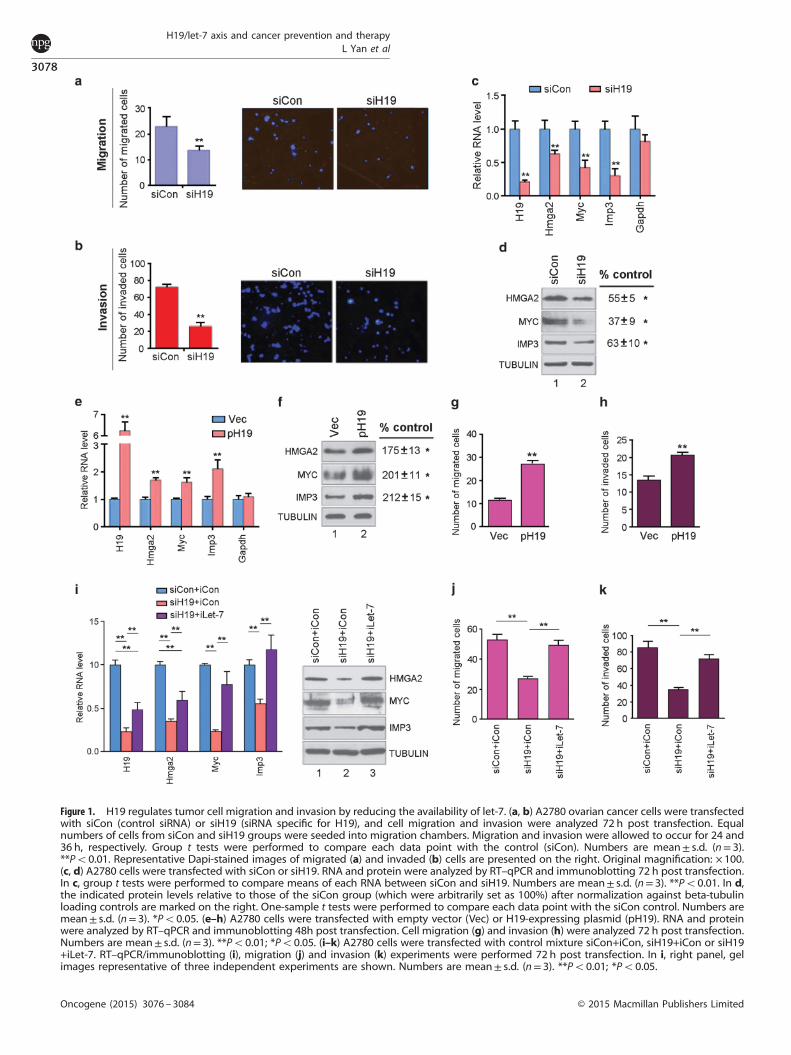
Figure 1. H19 regulates tumor cell migration and invasion by reducing the availability of let-7. (a, b) A2780 ovarian cancer cells were transfectedwith siCon (control siRNA) or siH19 (siRNA specific for H19), and cell migration and invasion were analyzed 72 h post transfection. Equalnumbers of cells from siCon and siH19 groups were seeded into migration chambers. Migration and invasion were allowed to occur for 24 and36 h, respectively. Group t tests were performed to compare each data point with the control (siCon). Numbers are mean± s.d. (n= 3).**Po0.01. Representative Dapi-stained images of migrated (a) and invaded (b) cells are presented on the right. Original magnification: × 100.(c, d) A2780 cells were transfected with siCon or siH19. RNA and protein were analyzed by RT–qPCR and immunoblotting 72 h post transfection.In c, group t tests were performed to compare means of each RNA between siCon and siH19. Numbers are mean± s.d. (n= 3). **Po0.01. In d,the indicated protein levels relative to those of the siCon group (which were arbitrarily set as 100%) after normalization against beta-tubulinloading controls are marked on the right. One-sample t tests were performed to compare each data point with the siCon control. Numbers aremean± s.d. (n= 3). *Po0.05. (e–h) A2780 cells were transfected with empty vector (Vec) or H19-expressing plasmid (pH19). RNA and proteinwere analyzed by RT–qPCR and immunoblotting 48h post transfection. Cell migration (g) and invasion (h) were analyzed 72 h post transfection.Numbers are mean± s.d. (n= 3). **Po0.01; *Po0.05. (i–k) A2780 cells were transfected with control mixture siCon+iCon, siH19+iCon or siH19+iLet-7. RT–qPCR/immunoblotting (i), migration (j) and invasion (k) experiments were performed 72 h post transfection. In i, right panel, gelimages representative of three independent experiments are shown. Numbers are mean± s.d. (n= 3). **Po0.01; *Po0.05.
H19/let-7 axis and cancer prevention and therapyL Yan et al
3078
Oncogene (2015) 3076 – 3084 © 2015 Macmillan Publishers Limited
H19/let-7 axis contributes to regulating tumor cell migration andinvasion.
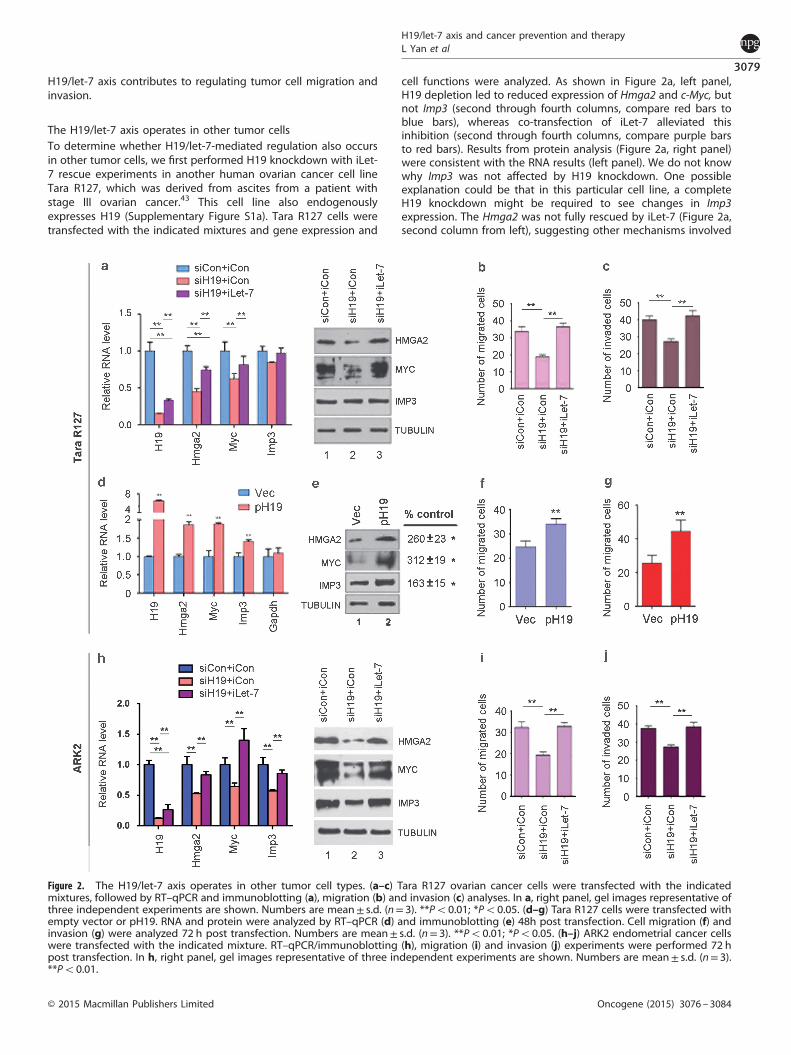
The H19/let-7 axis operates in other tumor cellsTo determine whether H19/let-7-mediated regulation also occursin other tumor cells, we first performed H19 knockdown with iLet-7 rescue experiments in another human ovarian cancer cell lineTara R127, which was derived from ascites from a patient withstage III ovarian cancer.43 This cell line also endogenouslyexpresses H19 (Supplementary Figure S1a). Tara R127 cells weretransfected with the indicated mixtures and gene expression and
cell functions were analyzed. As shown in Figure 2a, left panel,H19 depletion led to reduced expression of Hmga2 and c-Myc, butnot Imp3 (second through fourth columns, compare red bars toblue bars), whereas co-transfection of iLet-7 alleviated thisinhibition (second through fourth columns, compare purple barsto red bars). Results from protein analysis (Figure 2a, right panel)were consistent with the RNA results (left panel). We do not knowwhy Imp3 was not affected by H19 knockdown. One possibleexplanation could be that in this particular cell line, a completeH19 knockdown might be required to see changes in Imp3expression. The Hmga2 was not fully rescued by iLet-7 (Figure 2a,second column from left), suggesting other mechanisms involved
Figure 2. The H19/let-7 axis operates in other tumor cell types. (a–c) Tara R127 ovarian cancer cells were transfected with the indicatedmixtures, followed by RT–qPCR and immunoblotting (a), migration (b) and invasion (c) analyses. In a, right panel, gel images representative ofthree independent experiments are shown. Numbers are mean± s.d. (n= 3). **Po0.01; *Po0.05. (d–g) Tara R127 cells were transfected withempty vector or pH19. RNA and protein were analyzed by RT–qPCR (d) and immunoblotting (e) 48h post transfection. Cell migration (f) andinvasion (g) were analyzed 72 h post transfection. Numbers are mean± s.d. (n= 3). **Po0.01; *Po0.05. (h–j) ARK2 endometrial cancer cellswere transfected with the indicated mixture. RT–qPCR/immunoblotting (h), migration (i) and invasion (j) experiments were performed 72 hpost transfection. In h, right panel, gel images representative of three independent experiments are shown. Numbers are mean± s.d. (n= 3).**Po0.01.
H19/let-7 axis and cancer prevention and therapyL Yan et al
3079
© 2015 Macmillan Publishers Limited Oncogene (2015) 3076 – 3084
in its regulation. Finally, cell migration (Figure 2b) and invasion(Figure 2c) analyses further supported H19/let-7-dependentregulation in this cell line. Next, we performed H19 overexpressionexperiments. A significant increase in the expression of Hmga2,c-Myc and Imp3 at both RNA (Figure 2d) and protein (Figure 2e)levels was evident after H19 overexpression. As expected, cellmigration (Figure 2f) and invasion (Figure 2g) were alsoaugmented. Finally, we provided evidence that the H19/let-7regulatory pathway was active in other cancer types (Figures 2hand j), using the ARK2 cell line derived from human uterine serouscarcinoma (a highly aggressive variant of endometrial cancer)44
and which showed an endogenous expression of H19(Supplementary Figure S1a). Further, the decreased capacity ofARK2 to migrate (Figure 2i) and invade (Figure 2j) could beattributed, at least in part, to inhibition of EMT in response to H19depletion (Supplementary Figure S2b). Collectively, our resultssuggested that the H19/let-7 axis regulates migration and invasionof other tumor cell types with an elevated H19 expression.
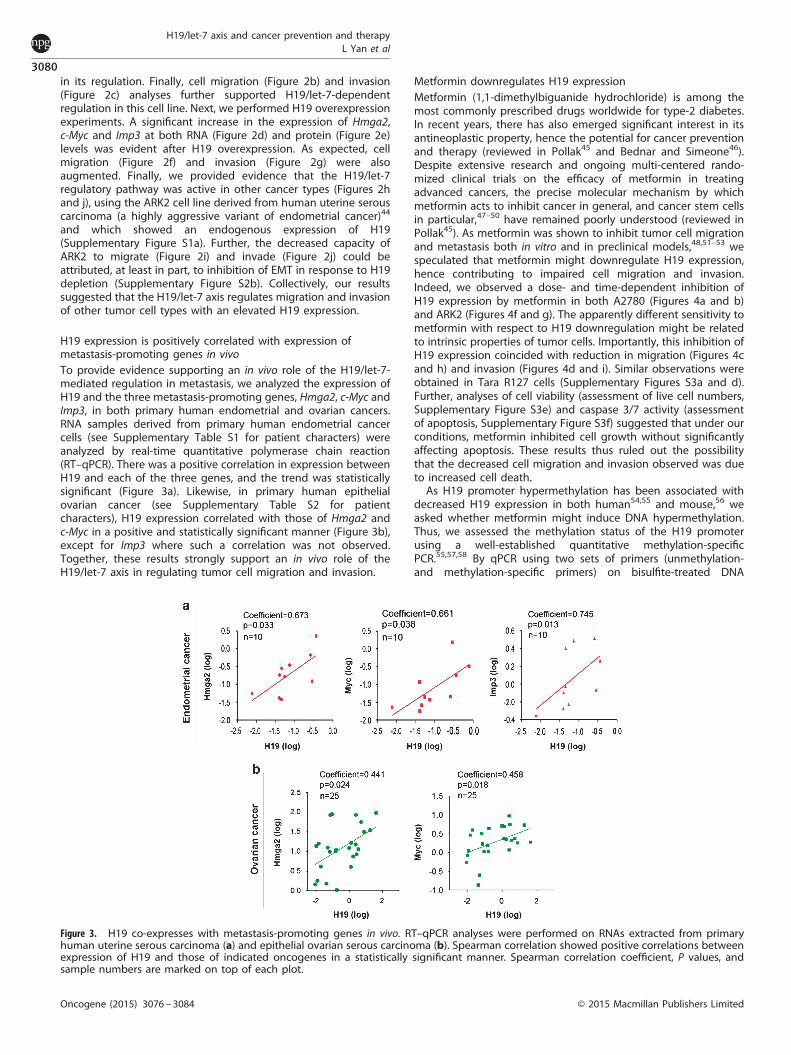
H19 expression is positively correlated with expression ofmetastasis-promoting genes in vivoTo provide evidence supporting an in vivo role of the H19/let-7-mediated regulation in metastasis, we analyzed the expression ofH19 and the three metastasis-promoting genes, Hmga2, c-Myc andImp3, in both primary human endometrial and ovarian cancers.RNA samples derived from primary human endometrial cancercells (see Supplementary Table S1 for patient characters) wereanalyzed by real-time quantitative polymerase chain reaction(RT–qPCR). There was a positive correlation in expression betweenH19 and each of the three genes, and the trend was statisticallysignificant (Figure 3a). Likewise, in primary human epithelialovarian cancer (see Supplementary Table S2 for patientcharacters), H19 expression correlated with those of Hmga2 andc-Myc in a positive and statistically significant manner (Figure 3b),except for Imp3 where such a correlation was not observed.Together, these results strongly support an in vivo role of theH19/let-7 axis in regulating tumor cell migration and invasion.
Metformin downregulates H19 expressionMetformin (1,1-dimethylbiguanide hydrochloride) is among themost commonly prescribed drugs worldwide for type-2 diabetes.In recent years, there has also emerged significant interest in itsantineoplastic property, hence the potential for cancer preventionand therapy (reviewed in Pollak45 and Bednar and Simeone46).Despite extensive research and ongoing multi-centered rando-mized clinical trials on the efficacy of metformin in treatingadvanced cancers, the precise molecular mechanism by whichmetformin acts to inhibit cancer in general, and cancer stem cellsin particular,47–50 have remained poorly understood (reviewed inPollak45). As metformin was shown to inhibit tumor cell migrationand metastasis both in vitro and in preclinical models,48,51–53 wespeculated that metformin might downregulate H19 expression,hence contributing to impaired cell migration and invasion.Indeed, we observed a dose- and time-dependent inhibition ofH19 expression by metformin in both A2780 (Figures 4a and b)and ARK2 (Figures 4f and g). The apparently different sensitivity tometformin with respect to H19 downregulation might be relatedto intrinsic properties of tumor cells. Importantly, this inhibition ofH19 expression coincided with reduction in migration (Figures 4cand h) and invasion (Figures 4d and i). Similar observations wereobtained in Tara R127 cells (Supplementary Figures S3a and d).Further, analyses of cell viability (assessment of live cell numbers,Supplementary Figure S3e) and caspase 3/7 activity (assessmentof apoptosis, Supplementary Figure S3f) suggested that under ourconditions, metformin inhibited cell growth without significantlyaffecting apoptosis. These results thus ruled out the possibilitythat the decreased cell migration and invasion observed was dueto increased cell death.As H19 promoter hypermethylation has been associated with
decreased H19 expression in both human54,55 and mouse,56 weasked whether metformin might induce DNA hypermethylation.Thus, we assessed the methylation status of the H19 promoterusing a well-established quantitative methylation-specificPCR.55,57,58 By qPCR using two sets of primers (unmethylation-and methylation-specific primers) on bisulfite-treated DNA
Figure 3. H19 co-expresses with metastasis-promoting genes in vivo. RT–qPCR analyses were performed on RNAs extracted from primaryhuman uterine serous carcinoma (a) and epithelial ovarian serous carcinoma (b). Spearman correlation showed positive correlations betweenexpression of H19 and those of indicated oncogenes in a statistically significant manner. Spearman correlation coefficient, P values, andsample numbers are marked on top of each plot.
H19/let-7 axis and cancer prevention and therapyL Yan et al
3080
Oncogene (2015) 3076 – 3084 © 2015 Macmillan Publishers Limited
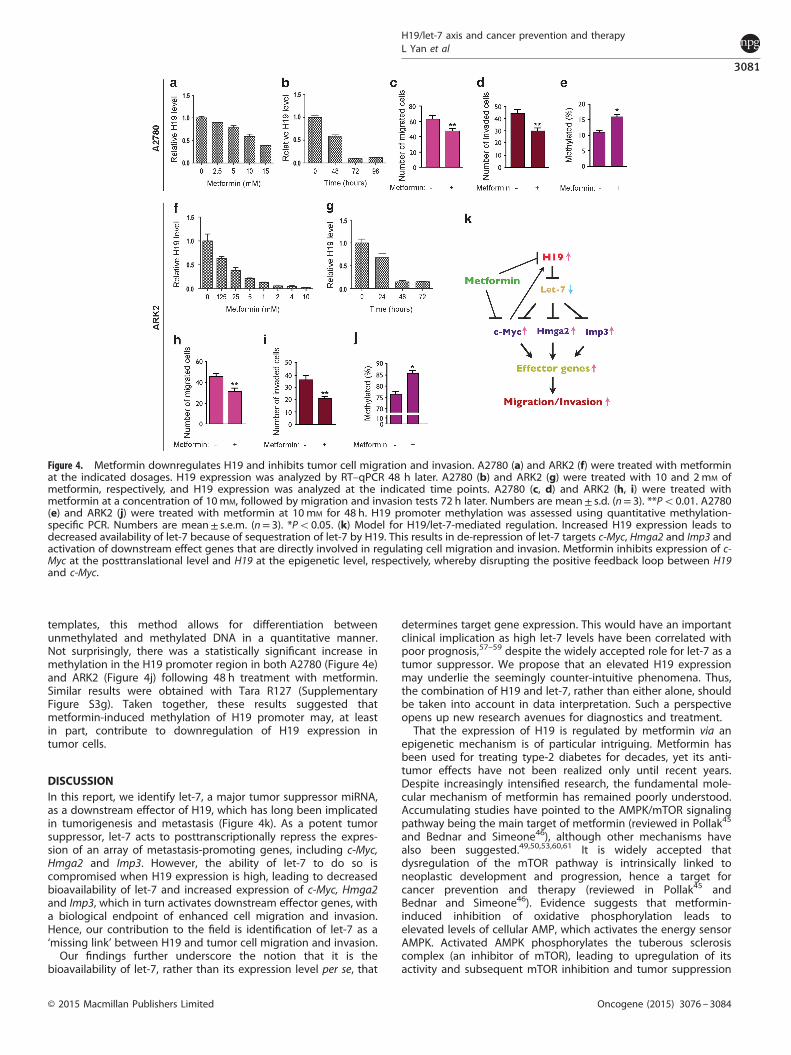
templates, this method allows for differentiation betweenunmethylated and methylated DNA in a quantitative manner.Not surprisingly, there was a statistically significant increase inmethylation in the H19 promoter region in both A2780 (Figure 4e)and ARK2 (Figure 4j) following 48 h treatment with metformin.Similar results were obtained with Tara R127 (SupplementaryFigure S3g). Taken together, these results suggested thatmetformin-induced methylation of H19 promoter may, at leastin part, contribute to downregulation of H19 expression intumor cells.
DISCUSSIONIn this report, we identify let-7, a major tumor suppressor miRNA,as a downstream effector of H19, which has long been implicatedin tumorigenesis and metastasis (Figure 4k). As a potent tumorsuppressor, let-7 acts to posttranscriptionally repress the expres-sion of an array of metastasis-promoting genes, including c-Myc,Hmga2 and Imp3. However, the ability of let-7 to do so iscompromised when H19 expression is high, leading to decreasedbioavailability of let-7 and increased expression of c-Myc, Hmga2and Imp3, which in turn activates downstream effector genes, witha biological endpoint of enhanced cell migration and invasion.Hence, our contribution to the field is identification of let-7 as a‘missing link’ between H19 and tumor cell migration and invasion.Our findings further underscore the notion that it is the
bioavailability of let-7, rather than its expression level per se, that
determines target gene expression. This would have an importantclinical implication as high let-7 levels have been correlated withpoor prognosis,57–59 despite the widely accepted role for let-7 as atumor suppressor. We propose that an elevated H19 expressionmay underlie the seemingly counter-intuitive phenomena. Thus,the combination of H19 and let-7, rather than either alone, shouldbe taken into account in data interpretation. Such a perspectiveopens up new research avenues for diagnostics and treatment.That the expression of H19 is regulated by metformin via an
epigenetic mechanism is of particular intriguing. Metformin hasbeen used for treating type-2 diabetes for decades, yet its anti-tumor effects have not been realized only until recent years.Despite increasingly intensified research, the fundamental mole-cular mechanism of metformin has remained poorly understood.Accumulating studies have pointed to the AMPK/mTOR signalingpathway being the main target of metformin (reviewed in Pollak45
and Bednar and Simeone46), although other mechanisms havealso been suggested.49,50,53,60,61 It is widely accepted thatdysregulation of the mTOR pathway is intrinsically linked toneoplastic development and progression, hence a target forcancer prevention and therapy (reviewed in Pollak45 andBednar and Simeone46). Evidence suggests that metformin-induced inhibition of oxidative phosphorylation leads toelevated levels of cellular AMP, which activates the energy sensorAMPK. Activated AMPK phosphorylates the tuberous sclerosiscomplex (an inhibitor of mTOR), leading to upregulation of itsactivity and subsequent mTOR inhibition and tumor suppression
Figure 4. Metformin downregulates H19 and inhibits tumor cell migration and invasion. A2780 (a) and ARK2 (f) were treated with metforminat the indicated dosages. H19 expression was analyzed by RT–qPCR 48 h later. A2780 (b) and ARK2 (g) were treated with 10 and 2mM ofmetformin, respectively, and H19 expression was analyzed at the indicated time points. A2780 (c, d) and ARK2 (h, i) were treated withmetformin at a concentration of 10mM, followed by migration and invasion tests 72 h later. Numbers are mean± s.d. (n= 3). **Po0.01. A2780(e) and ARK2 (j) were treated with metformin at 10mM for 48 h. H19 promoter methylation was assessed using quantitative methylation-specific PCR. Numbers are mean± s.e.m. (n= 3). *Po0.05. (k) Model for H19/let-7-mediated regulation. Increased H19 expression leads todecreased availability of let-7 because of sequestration of let-7 by H19. This results in de-repression of let-7 targets c-Myc, Hmga2 and Imp3 andactivation of downstream effect genes that are directly involved in regulating cell migration and invasion. Metformin inhibits expression of c-Myc at the posttranslational level and H19 at the epigenetic level, respectively, whereby disrupting the positive feedback loop between H19and c-Myc.
H19/let-7 axis and cancer prevention and therapyL Yan et al
3081
© 2015 Macmillan Publishers Limited Oncogene (2015) 3076 – 3084

(reviewed in Pollak45 and Bednar and Simeone46). Our findingthat metformin downregulates H19 at least in part by alteringmethylation of the H19 promoter represents a new mechanism ofmetformin.CpG methylation of promoters or within gene bodies,
previously thought to be a highly stable epigenetic modification,is now believed to be a rather dynamic process. Rapid cycling ofmethylation and demethylation of CpG dinucleotides in the TFF1/pS2 gene promoter upon estrogen activation has been detectedin human cells.62 The methylation status of a given gene ismaintained not only by the counteracting action of DNAmethyltransferases (which include DNMT1, DNMT3A andDNMT3B) and an active DNA demethylation pathway, but alsoby other protein complexes recruited to the DNA (reviewed inGrayson and Guidotti63). For example, the epilepsy drug valproicacid (2-propylpentanoic acid) silences transcription of a subset ofgenes by inducing promoter CpG methylation and by recruitmentof methyl-CpG binding protein 2 (MeCP2) and histone deacety-lases (HDACs) to the promoters.64 MeCP2 binding and histonedeacetylation generally result in transcription repression.64 How-ever, it is not yet clear whether CpG methylation is a cause orconsequence of MeCP2/HDACs binding which leads to chromo-some structure changes and gene silencing. Metformin may affectH19 expression in tumor cells in a way analogous to that ofvalproic acid, although the exact protein factors or complexesinvolved may differ. Future studies aiming at identifying thefactors and signal transduction pathways involved will benecessary to elucidate the underlying mechanism of metformin-mediated H19 repression.The apparently non-linear correlation between the extent of
H19 downregulation by metformin and the changes in cellmigration/invasion/methylation most likely arose from the differ-ent methods used that have different sensitivities to detectchanges. Our results further highlight the complexity of H19-mediated regulatory network in controlling tumor cell behavior.Disruption of epigenetic modifications, in addition to gene
mutations, is hall marker of cancer (reviewed in Timp andFeinberg65). Indeed, widespread hypomethylation involving nearlyone-third of single-copy genes was first uncovered in humancolorectal cancer, compared with matched normal tissues fromthe same patients.66 Later, similar observations were made withdiverse cancers, including lung, gastric, colon, pancreatic, liver andcervical cancers, with overall global hypomethylation frequentlybeing associated with activation of oncogenes (reviewed in Timpand Feinberg65). As metformin increases methylation of H19, itremains to be determined whether it does so to other oncogenes.If so, what could be the underlying mechanisms of gene selection?Finally, metformin has been found to inhibit c-Myc by acceleratingc-Myc protein degradation.67 Together with the notion that c-Mycdirectly enhances transcription of H19,22 we propose that thereexists a positive feedback loop between H19 and c-Myc, which isdisrupted by metformin (Figure 4k). Future studies aiming atdissecting mechanisms of metformin-mediated epigenetic mod-ification and strategies for targeting the H19/let-7 axis will proveuseful for cancer prevention and therapy.
MATERIALS AND METHODSMaterials and cell culturePlasmids expressing human H19 (pH19) and empty vector werepreviously described.14 Antibodies for HMGA2 (Cell Signaling,5269s, Boston, MA, USA), IMP3 (DAKO, M3626, Carpinteria, CA,USA), c-MYC (Santa Cruz, N-262, Santa Cruz, CA, USA) and β-TUBULIN (Abcam, ab6046, Cambridge, MA, USA) were purchased.Human H19-specific siRNA (siH19), non-targeting siRNA (siCon),let-7 inhibitor (iLet-7) and anti-miR control (iCon) were previouslydescribed.14 Metformin (ALX-270-432-G005) was purchased from
ENZO Life Sciences International Inc. (Uniondale, NY, USA). Quick-gDNA MicroPrep (D3021), EZ DNA Methylation-Gold Kit (D5006)were purchased from ZYMO Research Corporation (Irvine, CA,USA). The A2780,68 Tara R127,43 ARK2,44 and HEK29314 cells werecultured as previously described.
H19 siRNA knockdown and overexpression experimentsCells were transfected in a 48-well plate scale. To prepare siRNAtransfection solution for each well, 16 pmol of siCon or siH19 wasmixed with 50 μl OPTI-MEM by gentle pipetting. In parallel, 0.5 μlLipofectamine 2000 was mixed with 50 μl OPTI-MEM. Following5 min of incubation at room temperature, the two were mixed bygentle pipetting and incubated for 20–30 min at room tempera-ture to allow siRNA/lipid complexes to form. At the end ofincubation, the 100 μl transfection solution was used to re-suspend cell pellet (8 × 104 cells for ARK2; 1.2 × 105 cells for A2780and Tara R127). After incubation at room temperature for 10 min,regular growth medium was added at a ratio of 1:5 (1 volume oftransfection solution/5 volumes of growth medium) and the cellsuspension was transferred to the culture plate. After 24 hincubation at 37 °C in 5% CO2, the medium was replaced withfresh growth medium. RNAs and proteins were extracted andanalyzed at the indicated time points following transfection.Plasmid DNA transfections were carried out as described forsiRNA, except that 0.4 μg DNA and 1 μl Lipofectamine 2000 wereused for each well of cells. For let-7 inhibitor rescue experiments,16 pmol of siCon/siH19 and 8 pmol of iCon/iLet-7 were used foreach well of cells.
Quantitative cell migration and invasion assaysThese were carried out as previously described69 with minormodifications. Briefly, transwell chambers (8-μm pores) placed intoa 24-well plate (Catalog number 07200150, Fisher Scientific,Suwance, GA, USA) were used in the assays. The lower chamberwas filled with 600 μl RPMI 1640 containing 20% FBS. Cells weretrypsinized, counted, and re-suspended in serum-free RPMI. Formigration assays, 3 × 104 cells in 200 μl serum-free RPMI wereadded to the upper chamber. Equal numbers of cells fromexperimental and control groups were loaded into the upperchamber. The cells were allowed to migrate for 24h at 37 °C beforefixing. For invasion assays, 100 μl Matrigel (BD Biosciences,Bedford, MA, USA), 1:3 diluted in serum-free RPMI, was coatedonto the upper chamber and incubated at 37 °C for 60 min. Equalnumbers of cells from experimental and control groups wereseeded into the upper chamber at a concentration of 5 × 104/200 μl and incubated for 36 h at 37 °C before fixing. The non-migrated cells were removed from the upper surface of themembrane by scraping with a cotton swab. Cells on the bottomsurface of the membrane were fixed with 95% ethanol at roomtemperature for 30 min, gently rinsed with phosphate-bufferedsaline, stained with Dapi for 5 min, and photographed using aZeiss (Melville, NY, USA) microscope system. Migration/invasionwas assessed by counting the number of stained cell nuclei fromten random fields per filter in each group at × 100 magnification.The experiments were conducted in triplicate. Cell counts wereexpressed as the mean number of cells per field of view.
RT–qPCR and immunoblot analysesThese were carried out essentially as previously described.14 ThePCR primers for the indicated human genes are listed inSupplementary Table S3.
Quantitative miRNA analysesFor miRNA quantification, total RNAs were extracted from cellsusing PureLink RNA Mini Kit (Ambion, Grand Island, NY, USA,catalog number 12183018A). Levels of mature miRNAs were
H19/let-7 axis and cancer prevention and therapyL Yan et al
3082
Oncogene (2015) 3076 – 3084 © 2015 Macmillan Publishers Limited

determined by RT–qPCR using miScript reverse transcription kit(Qiagen, Valencia, CA, USA, catalog number 218161) and miScriptSYBR Green PCR kit (Qiagen, catalog number 218073) according tothe manufacturers’ instructions. PCR primer sets (miScript primer)specific for let-7a (MS00006482), let-7b (MS00003122), let-7c(MS00003129), let-7e (MS00031227), let-7f (MS00006489 ) andsnRNA U6 (MS00033740) were purchased from Qiagen. Theindicated miRNA levels were normalized against U6.
Cell viability and apoptosisThese were performed as previously described.70 Briefly, cells wereseeded in 96-well plates at a density of 3 × 103/well the nightbefore metformin addition. Cell viability and caspase 3/7 activitywere measured 48 h post metformin treatment using theCellTiterBlue Cell Viability kit (Promega, Madison, WI, USA) andthe Apo-ONE Homogeneous Caspase-3/7 Assay kit (Promega),respectively, according to the manufacturer’s protocols.
Human tumor tissue samplesTen primary type II endometrial cancer cells were obtained asdescribed previously.44 Study approval was obtained from theYale University Human Investigation Committee, and all patientssigned an informed consent form according to the institutionalguidelines. cDNAs were synthesized from RNAs extracted from thetumor cells and used for RT–qPCR analysis. Human ovarian cancertissue cDNA arrays (OriGene, Rockville, MD, USA, Ovarian CancercDNA Array II, HORT302) were purchased. Each array platecontained cDNAs synthesized from total RNAs of pathologist-verified tissues, normalized and validated with beta-actin, andprovided with clinical information.
Methylation analysisThese were performed as described57,58 with minor modifications.Briefly, cells were seeded in 6-well plates at a density of 2.5 × 105
for ARK2 (1 × 106 for A2780 and Tara R127). The next day,metformin (stock concentration at 500mM in water) was diluted ingrowth medium and added to the cells at 10 mM and incubatedfor the indicated time duration. Medium that contained freshmetformin was changed daily. Genomic DNAs were extractedfrom cells using Quick-gDNA MicroPrep according to themanufacturer’s instructions. For bisulfite treatment, 2–3 μg ofDNA was used for each column using EZ DNA Methylation-GoldKit. Fifteen microliter of water was used to elute DNA from eachcolumn. Real-time quantitative PCR was performed in a 15 μlreaction containing 1 μl of the eluant using iQSYBRGreen (Bio-Rad,Hercules, CA, USA) in a Bio-Rad iCycler. PCR was performed byinitial denaturation at 95 °C for 5 min, followed by 40 cycles of 30 sat 95 °C, 30 s at 60 °C, and 30 s at 72 °C. Specificity was verified bymelting curve analysis and agarose gel electrophoresis. Thethreshold cycle (Ct) values of each sample were used in thepost-PCR data analysis. The PCR primers (listed below) forunmethylated and methylated H19 DNA were used at a finalconcentration of 0.3 μM in each PCR reaction.Unmethylated forward: 5′-GTTTATGGGAGTTATATTATGTTTTTGTATTG-3′Unmethylated reverse: 5′-CAAAATTTACTATACTCATCACACAAATAAACA-3′Methylated forward: 5′-GGGAGTTATATTACGTTTTCGTATCG-3′Methylated reverse: 5′-TACTATACTCATCACGCGAATAAACG-3′.
Statistical analysisAll data are presented as mean± s.d. or otherwise indicated. Datawere analyzed using two-tailed Student’s t-test for the averagedifferences. Spearman correlations were performed for gene co-expression analyses. P values of 0.05 or less were consideredsignificant.
CONFLICT OF INTERESTThe authors declare no conflict of interest.
ACKNOWLEDGEMENTSWe thank Gil Mor for A2780 and Tara R127 cells. This work was supported by thefollowing grants: the State of Connecticut Stem Cell grant 09SCAYALE14, AlbertMcKern Scholar Award 1063338, and funds from National Natural Science Foundationof China 81202057 and 81272858.
REFERENCES1 Gabory A, Jammes H, Dandolo L. The H19 locus: role of an imprinted non-coding
RNA in growth and development. Bioessays 2010; 32: 473–480.2 Matouk I, Raveh E, Ohana P, Lail RA, Gershtain E, Gilon M et al. The increasing
complexity of the oncofetal h19 gene locus: functional dissection and therapeuticintervention. Intl J Mol Sci 2013; 14: 4298–4316.
3 Tanos V, Prus D, Ayesh S, Weinstein D, Tykocinski ML, De-Groot N et al. Expressionof the imprinted H19 oncofetal RNA in epithelial ovarian cancer. Eur J ObstetGynecol Reprod Biol 1999; 85: 7–11.
4 Tanos V, Ariel I, Prus D, De-Groot N, Hochberg A. H19 and IGF2 gene expression inhuman normal, hyperplastic, and malignant endometrium. Intl J Gynecol Cancer2004; 14: 521–525.
5 Tsang WP, Ng EK, Ng SS, Jin H, Yu J, Sung JJ et al. Oncofetal H19-derived miR-675regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis 2010;31: 350–358.
6 Matouk IJ, DeGroot N, Mezan S, Ayesh S, Abu-lail R, Hochberg A et al. The H19non-coding RNA is essential for human tumor growth. PLoS ONE 2007; 2: e845.
7 Luo M, Li Z, Wang W, Zeng Y, Liu Z, Qiu J. Long non-coding RNA H19 increasesbladder cancer metastasis by associating with EZH2 and inhibiting E-cadherinexpression. Cancer Lett 2013; 333: 213–221.
8 Wang G, Lunardi A, Zhang J, Chen Z, Ala U, Webster KA et al. Zbtb7a suppressesprostate cancer through repression of a Sox9-dependent pathway for cellularsenescence bypass and tumor invasion. Nat Genet 2013; 45: 739–746.
9 Hao Y, Crenshaw T, Moulton T, Newcomb E, Tycko B. Tumour-suppressor activityof H19 RNA. Nature 1993; 365: 764–767.
10 Yoshimizu T, Miroglio A, Ripoche MA, Gabory A, Vernucci M, Riccio A et al. TheH19 locus acts in vivo as a tumor suppressor. Proc Natl Acad Sci USA 2008; 105:12417–12422.
11 Zhang L, Yang F, Yuan J-h, Yuan S-x, Zhou W-p, Huo X-s et al. Epigenetic activationof the MiR-200 family contributes to H19-mediated metastasis suppression inhepatocellular carcinoma. Carcinogenesis 2013; 34: 577–586.
12 Fanale D, Amodeo V, Corsini LR, Rizzo S, Bazan V, Russo A. Breast cancer genome-wide association studies: there is strength in numbers. Oncogene 2011; 31:2121–2128.
13 Riaz M, Berns EM, Sieuwerts AM, Ruigrok-Ritstier K, de Weerd V, Groenewoud Aet al. Correlation of breast cancer susceptibility loci with patient characteristics,metastasis-free survival, and mRNA expression of the nearest genes. Breast CancerRes Treat 2011; 133: 12.
14 Kallen AN, Zhou XB, Xu J, Qiao C, Ma J, Yan L et al. The imprinted H19 lncRNAantagonizes let-7 microRNAs. Mol Cell 2013; 52: 101–112.
15 Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol 2008; 18:505–516.
16 Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: alook under the hood of miRISC. Nat Struct Mol Biol 2012; 19: 586–593.
17 Liu Q, Lv G-D, Qin X, Gen Y-H, Zheng S-T, Liu T et al. Role of microRNA let-7 andeffect to HMGA2 in esophageal squamous cell carcinoma. Mol Biol Rep 2012; 39:1239–1246.
18 Qian P, Zuo Z, Wu Z, Meng X, Li G, Wu Z et al. Pivotal role of reduced let-7gexpression in breast cancer invasion and metastasis. Cancer Res 2011; 71:6463–6474.
19 Yun J, Frankenberger CA, Kuo W-L, Boelens MC, Eves EM, Cheng N et al. Signallingpathway for RKIP and Let-7 regulates and predicts metastatic breast cancer. EMBOJ 2011; 30: 4500–4514.
20 Fu X, Meng Z, Linag W, Tian Y, Wang X, Han W et al. miR-26a enhances miRNAbiogenesis by targeting Lin28B and Zcchc11 to suppress tumor growth andmetastasis. Oncogene 2014; 33: 4296–4306.
21 Zhao B, Han H, Chen J, Zhang Z, Li S, Fang F et al. MicroRNA let-7c inhibitsmigration and invasion of human non-small cell lung cancer by targeting ITGB3and MAP4K3. Cancer Lett 2014; 342: 43–51.
22 Barsyte-Lovejoy D, Lau SK, Boutros PC, Khosravi F, Jurisica I, Andrulis IL et al. Thec-Myc oncogene directly induces the H19 noncoding RNA by allele-specificbinding to potentiate tumorigenesis. Cancer Res 2006; 66: 5330–5337.
H19/let-7 axis and cancer prevention and therapyL Yan et al
3083
© 2015 Macmillan Publishers Limited Oncogene (2015) 3076 – 3084

23 Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2oncogene. Genes Dev 2007; 21: 1025–1030.
24 Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2enhances oncogenic transformation. Science 2007; 315: 1576–1579.
25 Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P et al. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkittlymphoma cells. Cancer Res 2007; 67: 9762–9770.
26 Zhu H, Shyh-Chang N, Segre AV, Shinoda G, Shah SP, Einhorn WS et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011; 147: 81–94.
27 Toledano H, D'Alterio C, Czech B, Levine E, Jones DL. The let-7-Imp axis regulatesageing of the Drosophila testis stem-cell niche. Nature 2012; 485: 605–610.
28 Dangi-Garimella S, Yun J, Eves EM, Newman M, Erkeland SJ, Hammond SM et al.Raf kinase inhibitory protein suppresses a metastasis signalling cascade involvingLIN28 and let-7. EMBO J 2009; 28: 347–358.
29 Morishita A, Zaidi MR, Mitoro A, Sankarasharma D, Szabolcs M, Okada Y et al.HMGA2 is a driver of tumor metastasis. Cancer Res 2013; 73: 4289–4299.
30 Guo L, Chen C, Shi M, Wang F, Chen X, Diao Det al. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-drivenepithelial-mesenchymal transition. Oncogene 2013; 32: 5272–5282.
31 Sun M, Song C-X, Huang H, Frankenberger CA, Sankarasharma D, Gomes S et al.HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth andmetastasis. Proc Natl Acad Sci USA 2013; 110: 9920–9925.
32 Thornton JE, Gregory RI. How does Lin28 let-7 control development and disease?Trends Cell Biol 2012; 22: 474–482.
33 Huang Y. A mirror of two faces: Lin28 as a master regulator of both miRNAand mRNA. Wiley Interdiscip Rev RNA 2012; 3: 483–494 29.
34 Bell JL, Wachter K, Muhleck B, Pazaitis N, Kohn M, Lederer M et al. Insulin-likegrowth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers ofcancer progression? Cell Mol Life Sci 2013; 70: 2657–2675.
35 Jeng Y-M, Chang C-C, Hu F-C, Chou H-YE, Kao H-L, Wang T-H et al. RNA-bindingprotein insulin-like growth factor II mRNA-binding protein 3 expression promotestumor invasion and predicts early recurrence and poor prognosis in hepatocel-lular carcinoma. Hepatology 2008; 48: 1118–1127.
36 Walter O, Prasad M, Lu S, Quinlan RM, Edmiston KL, Khan A. IMP3 is a novelbiomarker for triple negative invasive mammary carcinoma associated with amore aggressive phenotype. Hum Pathol 2009; 40: 1528–1533.
37 Samanta S, Sharma VM, Khan A, Mercurio AM. Regulation of IMP3 by EGFR sig-naling and repression by ERbeta: implications for triple-negative breast cancer.Oncogene 2012; 31: 4689–4697.
38 Vikesaa J, Hansen TVO, Jonson L, Borup R, Wewer UM, Christiansen J et al. RNA-binding IMPs promote cell adhesion and invadopodia formation. EMBO J 2006;25: 1456–1468.
39 Suvasini R, Shruti B, Thota B, Shinde SV, Friedmann-Morvinski D, Nawaz Z et al.Insulin growth factor-2 binding protein 3 (IGF2BP3) is a glioblastoma-specificmarker that activates phosphatidylinositol 3-kinase/mitogen-activated proteinkinase (PI3K/MAPK) pathways by modulating IGF-2. J Biol Chem 2011; 286:25882–25890.
40 Tokumaru S, Suzuki M, Yamada H, Nagino M, Takahashi T. let-7 regulates Dicerexpression and constitutes a negative feedback loop. Carcinogenesis 2008; 29:2073–2077.
41 Forman JJ, Legesse-Miller A, Coller HA. A search for conserved sequences incoding regions reveals that the let-7 microRNA targets Dicer within its codingsequence. Proc Natl Acad Sci USA 2008; 105: 14879–14884.
42 Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J ClinInvest 2009; 119: 1429–1437.
43 Green JM, Alvero AB, Kohen F, Mor G. Targeting the mitochondria activates twoindependent cell death pathways in ovarian cancer stem cells. Mol Cancer Ther2009; 8: 1385–1393.
44 Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E et al. Landscape ofsomatic single-nucleotide and copy-number mutations in uterine serous carci-noma. Proc Natl Acad Sci USA 2013; 110: 2916–2921.
45 Pollak M. Potential applications for biguanides in oncology. J Clin Invest 2013; 123:3693–3700.
46 Bednar F, Simeone DM. Metformin and cancer stem cells: old drug, new targets.Cancer Prev Res (Phila) 2012; 5: 351–354.
47 Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancerstem cells, and acts together with chemotherapy to block tumor growth andprolong remission. Cancer Res 2009; 69: 7507–7511.
48 Bao B, Wang Z, Ali S, Ahmad A, Azmi AS, Sarkar SH et al. Metformin inhibits cellproliferation, migration and invasion by attenuating CSC function mediated by
deregulating miRNAs in pancreatic cancer cells. Cancer Prev Res (Phila) 2012; 5:355–364.
49 Gou S, Cui P, Li X, Shi P, Liu T, Wang C. Low concentrations of metformin selec-tively inhibit CD133+ cell proliferation in pancreatic cancer and haveanticancer action. PloS One 2013; 8: e63969.
50 Wurth R, Pattarozzi A, Gatti M, Bajetto A, Corsaro A, Parodi A et al.Metformin selectively affects human glioblastoma tumor-initiating cellviability: A role for metformin-induced inhibition of Akt. Cell Cycle 2013; 12:145–156.
51 Wu B, Li S, Sheng L, Zhu J, Gu L, Shen H et al. Metformin inhibits the developmentand metastasis of ovarian cancer. Oncol Rep 2012; 28: 903–908.
52 Cerezo M, Tichet M, Abbe P, Ohanna M, Lehraiki A, Rouaud F et al. Metforminblocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol Cancer Ther 2013; 12: 1605–1615.
53 Sarfstein R, Friedman Y, Attias-Geva Z, Fishman A, Bruchim I, Werner H. Metformindownregulates the insulin/IGF-I signaling pathway and inhibits different uterineserous carcinoma (USC) cells proliferation and migration in p53-dependent or-independent manners. PLoS ONE 2013; 8: e61537.
54 Steenman MJ, Rainier S, Dobry CJ, Grundy P, Horon IL, Feinberg AP. Loss ofimprinting of IGF2 is linked to reduced expression and abnormal methylation ofH19 in Wilms' tumour. Nat Genet 1994; 7: 433–439.
55 Gao Z-H, Suppola S, Liu J, Heikkila P, Janne J, Voutilainen R. Association of H19promoter methylation with the expression of H19 and IGF-II genes inadrenocortical tumors. J Clin Endocrinol Metabol 2002; 87: 1170–1176.
56 Srivastava M, Hsieh S, Grinberg A, Williams-Simons L, Huang SP, Pfeifer K. H19 andIgf2 monoallelic expression is regulated in two distinct ways by a shared cisacting regulatory region upstream of H19. Genes Dev 2000; 14: 1186–1195.
57 Lu L, Katsaros D, de la Longrais IA, Sochirca O, Yu H. Hypermethylation of let-7a-3in epithelial ovarian cancer is associated with low insulin-like growth factor-IIexpression and favorable prognosis. Cancer Res 2007; 67: 10117–10122.
58 Lu L, Katsaros D, Zhu Y, Hoffman A, Luca S, Marion CE et al. Let-7a regulation ofinsulin-like growth factors in breast cancer. Breast Cancer Res Treat 2011; 126:687–694.
59 Tang Z, Ow GS, Thiery JP, Ivshina AV, Kuznetsov VA. Meta-analysis of tran-scriptome reveals let-7b as an unfavorable prognostic biomarker and predictsmolecular and clinical subclasses in high-grade serous ovarian carcinoma. Int JCancer 2013; 134: 306–318.
60 Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B et al. Metformin, inde-pendent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. CellMetabol 2010; 11: 390–401.
61 Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger Pet al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cyclearrest through REDD1. Cancer Res 2011; 71: 4366–4372.
62 Kangaspeska S, Stride B, Metivier R, Polycarpou-Schwarz M, Ibberson D,Carmouche RP et al. Transient cyclical methylation of promoter DNA. Nature 2008;452: 112–115.
63 Grayson DR, Guidotti A. The dynamics of DNA methylation in schizophrenia andrelated psychiatric disorders. Neuropsychopharmacology 2013; 38: 138–166.
64 Reid G, Metivier R, Lin C-Y, Denger S, Ibberson D, Ivacevic T et al. Multiplemechanisms induce transcriptional silencing of a subset of genes, includingoestrogen receptor alpha, in response to deacetylase inhibition by valproic acidand trichostatin A. Oncogene 2005; 24: 4894–4907.
65 Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellulargrowth advantage at the expense of the host. Nature Rev Cancer 2013; 13:497–510.
66 Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some humancancers from their normal counterparts. Nature 1983; 301: 89–92.
67 Akinyeke T, Matsumura S, Wang X, Wu Y, Schalfer ED, Saxena A et al. Metformintargets c-MYC oncogene to prevent prostate cancer. Carcinogenesis 2013; 34:2823–2832.
68 Behrens BC, Hamilton TC, Masuda H, Grotzinger KR, Whang-Peng J, Louie KG et al.Characterization of a cis-diamminedichloroplatinum(II)-resistant human ovariancancer cell line and its use in evaluation of platinum analogues. Cancer Res 1987;47: 414–418.
69 Yin G, Alvero AB, Craveiro V, Holmberg JC, Fu HH, Montagna MK et al. Constitutiveproteasomal degradation of TWIST-1 in epithelial-ovarian cancer stem cellsimpacts differentiation and metastatic potential. Oncogene 2013; 32: 39–49.
70 Feng C, Neumeister V, Ma W, Xu J, Lu L, Bordeaux J et al. Lin28 regulates HER2 andpromotes malignancy through multiple mechanisms. Cell Cycle 2012; 11: 13.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
H19/let-7 axis and cancer prevention and therapyL Yan et al
3084
Oncogene (2015) 3076 – 3084 © 2015 Macmillan Publishers Limited


















