
Redox regulation of angiotensin II preconditioning of the myocardium requires MAP kinase signaling
8
Original article Redox regulation of angiotensin II preconditioning of the myocardium requires MAP kinase signaling Samarjit Das a , Hajime Otani b , Nilanjana Maulik a , Dipak K. Das a, ⁎ a Cardiovascular Research Center, University of Connecticut School of Medicine, Farmington, CT 6030-1110, USA b Department of Cardiothoracic Surgery, Kansai Medical University, Moriguchi, Osaka, Japan Received 4 February 2006; received in revised form 7 March 2006; accepted 14 March 2006 Available online 11 May 2006 Abstract A recent study documented reactive oxygen species (ROS), generated through NADPH oxidase by angiotensin II (Ang II) with the activation of NADPH oxidase subunits, p22phox and gp91phox, to be responsible for the preconditioning effect of Ang II. The present study was designed to determine if similar to ischemic preconditioning (PC), mitogen-activated protein (MAP) kinases are also involved in Ang II PC of the heart. Isolated working rat hearts were perfused for 15 min with KHB (Krebs–Henseleit bicarbonate) buffer containing Ang II in the absence or presence of an Erk (1/2) inhibitor, PD 098059, a p38MAPK inhibitor, SB 202190, a JNK inhibitor, SP 600125 or a ROS scavenger, N-acetyl cysteine (NAC). All hearts were subsequently subjected to 30 min global ischemia followed by 2 h reperfusion with KHB buffer only. Cardioprotection was examined by determining infarct size, cardiomyocyte apoptosis and ventricular recovery. Redox and MAP kinase regulation were studied by determining the survival signaling mediated by Akt and Bcl-2. In consistent with previous results, Ang II preconditioned the heart as evidenced by improved postischemic ventricular recovery and reduced infarct size and decreases cardiomyocyte apoptosis. Ang II phosphorylated both Akt, Bcl-2 and Bad, which was blocked by NAC, PD 098059 or SP 600125, but not by SB 202190. NAC, PD 098059 and SP600125, but not SB202190, also abolished the cardioprotective effect of Ang II preconditioning. The results indicate that Ang II preconditioning is potentiated through MAP kinases that are regulated by redox signaling. © 2006 Elsevier Inc. All rights reserved. Keywords: Heart; ROS; MAPK; Erk (1/2); JNK; p38MAPK; Akt; Bcl-2; Preconditioning 1. Introduction Although angiotensin II (Ang II) has been shown to precondition mammalian heart against ischemia–reperfusion injury [1–8], the precise mechanism of preconditioning remains unknown. In one of the earlier studies, protein kinase C was implicated in the cardioprotective effects of angiotensin II preconditioning [9]. In another study, activation of Na + /H + exchanger was found to be involved in such preconditioning [6]. Several studies demonstrated a role of angiotensin II type I (AT1) receptor in preconditioning against infarction [3,5], suggesting that activation of AT1 receptors by angiotensin II produced locally in the heart contributes to the limitation of infarct size by preconditioning. Consistent with these results, selective block- ade of AT1 angiotensin II receptors abolished preconditioning effect in isolated rabbit hearts [9]. Another group of studies provided evidence for the role of bradykinin B2 receptor in angiotensin II preconditioning [10]. In a related study, captopril potentiated the myocardial infarct size-limiting effect of preconditioning through bradykinin B2 receptor activation [11]. A recent study showed that Ang II preconditioning is triggered by redox cycling of reactive oxygen species (ROS), which are generated by both NADPH oxidase-dependent, by the increase activity of p22phox and gp91phox, and NADPH oxidase-independent pathways [12]. A previous study showed the phosphorylation of p47phox by Ang II with concomitant induction of ROS formation via NADPH oxidase [13]. NADH/ NADPH oxidases are considered to be the major sources of ROS in VSMCs responsible for redox signaling [14]. The mitogen-activated protein (MAP) kinases have been shown to play a crucial role in ischemic preconditioning Journal of Molecular and Cellular Cardiology 41 (2006) 248 – 255 www.elsevier.com/locate/yjmcc ⁎ Corresponding author. Tel.: +1 860 679 3687; fax: +1 860 679 4606. E-mail address: [email protected] (D.K. Das). 0022-2828/$ - see front matter © 2006 Elsevier Inc. All rights reserved. doi:10.1016/j.yjmcc.2006.03.009 RETRACTED
-
Upload
samarjit-das -
Category
Documents
-
view
215 -
download
1
Transcript of Redox regulation of angiotensin II preconditioning of the myocardium requires MAP kinase signaling

Journal of Molecular and Cellular Cardiology 41 (2006) 248–255www.elsevier.com/locate/yjmcc
Original article
Redox regulation of angiotensin II preconditioning of the myocardiumrequires MAP kinase signaling
Samarjit Das a, Hajime Otani b, Nilanjana Maulik a, Dipak K. Das a,⁎
a Cardiovascular Research Center, University of Connecticut School of Medicine, Farmington, CT 6030-1110, USAb Department of Cardiothoracic Surgery, Kansai Medical University, Moriguchi, Osaka, Japan
Received 4 February 2006; received in revised form 7 March 2006; accepted 14 March 2006Available online 11 May 2006
D
AbstractA recent study documented reactive oxygen species (ROS), generated through NADPH oxidase by angiotensin II (Ang II) with the activationof NADPH oxidase subunits, p22phox and gp91phox, to be responsible for the preconditioning effect of Ang II. The present study was designedto determine if similar to ischemic preconditioning (PC), mitogen-activated protein (MAP) kinases are also involved in Ang II PC of the heart.Isolated working rat hearts were perfused for 15 min with KHB (Krebs–Henseleit bicarbonate) buffer containing Ang II in the absence or presenceof an Erk (1/2) inhibitor, PD 098059, a p38MAPK inhibitor, SB 202190, a JNK inhibitor, SP 600125 or a ROS scavenger, N-acetyl cysteine(NAC). All hearts were subsequently subjected to 30 min global ischemia followed by 2 h reperfusion with KHB buffer only. Cardioprotectionwas examined by determining infarct size, cardiomyocyte apoptosis and ventricular recovery. Redox and MAP kinase regulation were studied bydetermining the survival signaling mediated by Akt and Bcl-2. In consistent with previous results, Ang II preconditioned the heart as evidenced byimproved postischemic ventricular recovery and reduced infarct size and decreases cardiomyocyte apoptosis. Ang II phosphorylated both Akt,Bcl-2 and Bad, which was blocked by NAC, PD 098059 or SP 600125, but not by SB 202190. NAC, PD 098059 and SP600125, but notSB202190, also abolished the cardioprotective effect of Ang II preconditioning. The results indicate that Ang II preconditioning is potentiatedthrough MAP kinases that are regulated by redox signaling.© 2006 Elsevier Inc. All rights reserved.
ACTE
Keywords: Heart; ROS; MAPK; Erk (1/2); JNK; p38MAPK; Akt; Bcl-2; PreconditioningR
1. IntroductionAlthough angiotensin II (Ang II) has been shown toprecondition mammalian heart against ischemia–reperfusioninjury [1–8], the precise mechanism of preconditioning remainsunknown. In one of the earlier studies, protein kinase C wasimplicated in the cardioprotective effects of angiotensin IIpreconditioning [9]. In another study, activation of Na+/H+
exchanger was found to be involved in such preconditioning [6].Several studies demonstrated a role of angiotensin II type I (AT1)receptor in preconditioning against infarction [3,5], suggestingthat activation of AT1 receptors by angiotensin II producedlocally in the heart contributes to the limitation of infarct size bypreconditioning. Consistent with these results, selective block-
RET
⁎ Corresponding author. Tel.: +1 860 679 3687; fax: +1 860 679 4606.E-mail address: [email protected] (D.K. Das).
0022-2828/$ - see front matter © 2006 Elsevier Inc. All rights reserved.doi:10.1016/j.yjmcc.2006.03.009
ade of AT1 angiotensin II receptors abolished preconditioningeffect in isolated rabbit hearts [9]. Another group of studiesprovided evidence for the role of bradykinin B2 receptor inangiotensin II preconditioning [10]. In a related study, captoprilpotentiated the myocardial infarct size-limiting effect ofpreconditioning through bradykinin B2 receptor activation [11].
A recent study showed that Ang II preconditioning istriggered by redox cycling of reactive oxygen species (ROS),which are generated by both NADPH oxidase-dependent, bythe increase activity of p22phox and gp91phox, and NADPHoxidase-independent pathways [12]. A previous study showedthe phosphorylation of p47phox by Ang II with concomitantinduction of ROS formation via NADPH oxidase [13]. NADH/NADPH oxidases are considered to be the major sources ofROS in VSMCs responsible for redox signaling [14].
The mitogen-activated protein (MAP) kinases have beenshown to play a crucial role in ischemic preconditioning

249S. Das et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 248–255
A
[15,16]. In addition, several MAP kinases including Erk (1/2),JNK and p38MAPK are main targets for ROS signaling. UnlikeErk (1/2), which is activated by growth signal via Ras-dependent signal transduction pathway [17], the activation ofJNKs and p38MAPK is potentiated by diverse stresses andproinflammatory cytokines [12].
Among the known cell survival pathways, the MEK(mitogen-activated protein kinase [MAPK] kinase)/Erk (1/2)/PI-3-kinase (phosphatidylinositol 3-kinase)/Akt (PKB; p57Akt)/Bcl-2 pathway has received a considerable amount ofimportance [18–20]. In many survival signaling pathways, theBcl-2 family of proteins are considered as the key determinantof apoptotic cell death [21]. There are at least 15 family memberof this group been identified so far, either functioning asantiapoptotic proteins (e.g., Bcl-2, Bcl-XL) or proapoptotic(e.g., Bax, Bad, Bak) regulators. Interestingly, ischemicpreconditioning upregulates Bcl-2 and downregulates Bax[22], and Bcl-2 plays an important role in preconditioning-mediated cell survival [23–25].
The present study was undertaken to determine if similar toischemic preconditioning, Ang II preconditioning also involvesMAP kinase signaling. The results of our study demonstrate thatredox regulation of Ang II preconditioning occurs by activatingErk (1/2) and JNK, but not p38MAPK.
2. Materials and methods
2.1. Angiotensin II
Angiotensin II, the Erk (1/2) inhibitor, PD 098,059 (2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one) and
Fig. 1. Experimental protocol. KHB, Krebs–Henseleit bicarbonate buffer; PD 098,Fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)-1H-imidazole; SP600125, 1,9-Pykinase B.
RETR
p38MAPK inhibitor, SB 202190 (4-(4-Fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)-1H-imidazole) and free radicalscavenger, N-acetyl cysteine (NAC) were obtained fromSigma Chemical Co. (St. Louis, MO, USA). A highly specificblocker of JNK, SP 600125 (1,9-Pyrazoloanthrone), waspurchased from Calbiochem, La Jolla, CA. The drugs weredissolved in DMSO, and the aliquots were frozen at 4 °C.Control experiments contained the vehicle, dimethyl sulfoxide(DMSO) only.
2.2. Animals
All animals used in this study received humane care incompliance with the principles of the laboratory animal careformulated by the National Society for Medical Researchand Guide for the Care and Use of Laboratory Animalsprepared by the National Academy of Sciences andpublished by the National Institutes of Health (PublicationNumber NIH 85-23, revised 1985). Sprague–Dawley malerats weighing between 225 and 250 g were fed ad libitumregular rat chow with free access to water until the start ofthe experimental procedure. The rats were randomly assignedto one of the following groups (Fig. 1): perfused for 15 minwith KHB with (i) vehicle (DMSO) only; (ii) KHBcontaining 100 nM Ang II; (iii) 20 μM PD 098,059; (iv)10 μM SB 202190; (v) 10 μM SP 600125; (vi) 1 μM NAC;(vii) 100 nM Ang II + 20 μM PD 098,059; (viii) 100 nMAng II + 10 μM SB 202190; (ix) 100 nM Ang II + 10 μMSP 600125 and (x) 100 nM Ang II + 1 μM NAC. All heartswere then subjected to 30 min ischemia followed by 2 hreperfusion.
CTED
059, 2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one; SB202190, 4-(4-razoloanthrone; Ang II, Angiotensin II; NAC, N-acetyl cysteine; Akt, Protein

250 S. Das et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 248–255
A
2.3. Isolated working heart preparation
Rats were anesthetized with sodium pentobarbital (80 mg/kg, i.p.) (Abbott Laboratories, North Chicago, IL, USA) andanticoagulant with heparin sodium (500 IU/kg., i.v.) (Elkins-Sinn Inc., Cherry Hill, NJ, USA). After ensuring sufficientdepth of anesthesia thoracotomy was performed, hearts wereperfused in the retrograde Langendorff mode at 37 °C at aconstant perfusion pressure of 100 cm of water (10 kPa) for a5 min washout period. The perfusion buffer used in this studyconsisted of a modified Krebs–Henseleit bicarbonate buffer(KHB) (in mM: sodium chloride 118, potassium chloride 4.7,calcium chloride 1.7, sodium bicarbonate 25, potassiumbiphosphate 0.36, magnesium sulfate 1.2 and glucose 10).The Langendorff preparation was switched to the working modefollowing the washout period as previously described [26].
At the end of 10 min, after the attainment of steady statecardiac function, baseline functional parameters were recorded.The circuit was then switched back to the retrograde mode andhearts were perfused either KHB with vehicle or any of theMAPK's inhibitor or NAC (Control), Ang II at a concentrationof 100 nM or a combination of Ang II and any of the MAPK'sinhibitor or NAC for a duration of 15 min. This was followed bya 5-min washout with KHB buffer, and then the hearts weresubjected to global ischemia for 30 min and then 2 h ofreperfusion. The first 10 min of reperfusion was in theretrograde mode to allow for postischemic stabilization andthere after, in the antigrade working mode to allow forassessment of functional parameters, which were recorded at10-, 30-, 60- and 120-min reperfusion.
2.4. Cardiac function assessment
Aortic pressure was measured using a Gould P23XLpressure transducer (Gould Instrument Systems Inc., ValleyView, OH, USA) connected to a side arm of the aortic cannula,the signal was amplified using a Gould 6600 series signalconditioner and monitored on a CORDAT II real-time dataacquisition and analysis system (Triton Technologies, SanDiego, CA, USA) [26]. Heart rate (HR), left ventriculardeveloped pressure (LVDP) (defined as the difference of themaximum systolic and diastolic aortic pressures) and the firstderivative of developed pressure (dp/dt) were all derived orcalculated from the continuously obtained pressure signal.Aortic flow (AF) was measured using a calibrated flow-meter(Gilmont Instrument Inc., Barrington, IL, USA) and coronaryflow (CF) was measured by timed collection of the coronaryeffluent dripping from the heart.
2.5. Infarct size estimation
At the end of 2 h reperfusion, a 10% (w/v) solution oftriphenyl tetrazolium in phosphate buffer was infused intoaortic cannula for 20 min at 37 °C [27]. The hearts wereexcised and stored at −70 °C. Sections (0.8 mm) of frozenheart were fixed in 2% paraformaldehyde, placed betweentwo cover slips and digitally imaged using a Microtek
RETR
ScanMaker 600 z. To quantitate the areas of interest in pixels,an NIH image 5.1 (a public-domain software package) wasused. The infarct size was quantified and expressed in pixels.
2.6. TUNEL assay for assessment of apoptotic cell death
Immunohistochemical detection of apoptotic cells wascarried out using TUNEL [18]. The sections were incubatedagain with mouse monoclonal antibody recognizing cardiacmyosin heavy chain to specifically recognize apoptoticcardiomyocytes. The fluorescence staining was viewed with aconfocal laser microscope. The number of apoptotic cells wascounted randomly at 100× magnification and at least four fieldsper sample and expressed as a percent of total myocytepopulation.
2.7. Western blot analysis
Left ventricles from the hearts were homogenized in abuffer containing 25 mM Tris–HCl, 25 mM NaCl, 1 mMorthovanadate, 10 mM NaF, 10 mM pyrophosphate, 10 mMokadaic acid, 0.5 mM EDTA and 1 mM PMSF [23]. 100 μgof protein of each heart homogenate was incubated with 1 μgof antibody against the phospho-Akt, Bad or Bcl-2 (SantaCruz Biotechnology, Inc., Santa Cruz, CA) for 1 h at 4 °C.The immune complexes were precipitated with protein ASepharose, immunoprecipitates separated by SDS-PAGE andimmobilized on polyvinylidene difluoride membrane. Themembrane was immunoblotted with PY20 to evaluate thephosphorylation of the compounds. The membrane was strippedand reblotted with specific antibodies against Akt, Bad and Bcl-2. The resulting blots were digitized and subjected todensitometric scanning using a standard NIH image program.
2.8. Statistical analysis
The values for myocardial functional parameters, total andinfarct volumes and infarct sizes, and cardiomyocyte apoptosisare all expressed as the mean ± SEM. Analysis of variance testfollowed by Bonferroni's correction was first carried out to testfor differences between the mean values of all groups. Ifdifferences were established, the values of the treated groupswere compared with those of the control group by a modifiedt test. The results were considered significant if P < 0.05.
3. Results
3.1. Effects of PD 098,059, SB 202190, SP 600125 and NACon Ang II preconditioning
There were no differences in baseline functions among the10 groups. In general, there were no significant differencesbetween Ang II vs. control and also Ang II + PD, Ang II + SB orAng II + SP or Ang II + NAC vs. Ang II on heart rate andcoronary flow (Table 1). In accordance to the previous studies,on reperfusion, the absolute values of all functional parameterswere decreased in all the groups as compared with the
CTED

Table 1Effects of angiotensin II, NAC and the inhibitors of Erk (1/2), p38 MAPPK and JNK on ventricular functions
Group Baseline 10 min R 30 min R 60 min R 120 min R
Heart rate (beats/min) Control 345 ± 31 316 ± 26 365 ± 17 344 ± 35 414 ± 24Angiotensin II 369 ± 17.4 372 ± 14 389 ± 10.4 411 ± 10.8 444 ± 12.6PD 098,059 383 ± 17 371 ± 17 392 ± 6.5 405 ± 5.7 439 ± 5.9SB 202190 390 ± 20 372 ± 20 388 ± 18 408 ± 8.7 422 ± 8.5SP 600125 384 ± 24 366 ± 16 402 ± 22 422 ± 24 453 ± 11NAC 327 ± 15 330 ± 12 356 ± 14 395 ± 12 421 ± 20Ang II + PD 398 ± 23 372 ± 18 386 ± 14.3 406 ± 9.8 426 ± 10Ang II + SB 392 ± 15.5 404 ± 14 395 ± 7.8 401 ± 5.8 424 ± 7.2Ang II + SP 413 ± 12.1 369 ± 13.6 380 ± 7.3 408 ± 3.7 435 ± 8.4Ang II + NAC 299 ± 13 318 ± 15 320 ± 12 366 ± 17 423 ± 7
LVDP (mm Hg) Control 127 ± 3.2 107 ± 5.4 104 ± 7.1 88 ± 9.6 53 ± 4.6Angiotensin II 128 ± 1.6 108 ± 3.5 116 ± 4 101 ± 3.5 81 ± 3.4 a
PD 098,059 127 ± 3.2 109 ± 4 111 ± 4.4 92 ± 3 57 ± 5.1SB 202190 126 ± 3.4 104 ± 3.4 102 ± 3.5 84.3 ± 5.6 56.6 ± 5.8SP 600125 127 ± 4.4 99 ± 6.8 104 ± 4.4 82 ± 6.1 49 ± 4.2NAC 129 ± 7 110 ± 8 115 ± 7 95 ± 6 68 ± 7Ang II + PD 129 ± 2.5 115 ± 4.4 111 ± 4 98 ± 3.7 65 ± 5 b
Ang II + SB 131 ± 2.1 111 ± 4.1 113 ± 3.8 102 ± 5.7 72 ± 6.3Ang II + SP 130 ± 3.6 118 ± 3.7 117 ± 3.8 96 ± 3.2 63 ± 4.4 b
Ang II + NAC 133 ± 3 119 ± 3 120 ± 2 101 ± 2 60 ± 3 b
LVdp/dt (mm Hg/s) Control 3319 ± 115 2412 ± 250 2472 ± 235 1881 ± 403 900 ± 87Angiotensin II 3332 ± 153 2505 ± 123 2487 ± 118 1996 ± 64 1139 ± 93 a
PD 098,059 3365 ± 82 2470 ± 68 2600 ± 159 1853 ± 100 843 ± 78SB 202190 3253 ± 116 2544 ± 130 2527 ± 177 1717 ± 217 946 ± 213SP 600125 3221 ± 120 2301 ± 126 2225 ± 182 1773 ± 161 878 ± 108NAC 3176 ± 130 2387 ± 172 2509 ± 155 1850 ± 131 988 ± 67Ang II + PD 3578 ± 115 2781 ± 108 2786 ± 132 2115 ± 119 828 ± 138 b
Ang II + SB 3632 ± 164 2804 ± 101 2763 ± 169 2218 ± 218 1234 ± 113Ang II + SP 3441 ± 92 2826 ± 356 2725 ± 322 2200 ± 186 786 ± 55.8 b
Ang II + NAC 3065 ± 154 2392 ± 105 2508 ± 35 1798 ± 58 909 ± 66 b
Aortic flow (ml/min) Control 72 ± 5.2 43 ± 12.9 36 ± 12.7 19 ± 6.5 4.3 ± 1.4Angiotensin II 74 ± 4 47 ± 3.1 50 ± 3 37 ± 4.1 a 12.5 ± 2.8 a
PD 098,059 71 ± 3.4 46 ± 3.8 41 ± 4.8 20 ± 3 3.7 ± 1SB 202190 64 ± 5.1 34.5 ± 7.8 32.2 ± 5.1 12.6 ± 2.7 2.1 ± 1SP 600125 69 ± 6 32.4 ± 9.8 30 ± 4.4 12.2 ± 2 3.3 ± 0.8NAC 73 ± 4 44 ± 7 41 ± 3 23 ± 6 9 ± 2Ang II + PD 73 ± 2.8 49.3 ± 8 44 ± 7.4 18.5 ± 5.7 b 3.2 ± 1.8 b
Ang II + SB 73 ± 5.2 40 ± 7.5 39 ± 7.1 26.1 ± 7.2 9 ± 3.5Ang II + SP 71 ± 4.5 50 ± 6.1 51 ± 4.7 18.5 ± 4.9 b 3.4 ± 1.4 b
Ang II + NAC 68 ± 3 51 ± 3 47 ± 3 18 ± 3 b 5 ± 1 b
Coronary flow (ml/min) Control 24 ± 1.4 22 ± 1 23 ± 1.9 23 ± 1.3 22 ± 1.2Angiotensin II 26 ± 1.9 23 ± 1.5 24 ± 1.5 22 ± 1.1 20 ± 0.8PD 098,059 21 ± 1.4 20 ± 1.2 21 ± 1 21 ± 1.5 20 ± 1.8SB 202190 20.4 ± 1.5 19.5 ± 1 20.1 ± 1.1 20.3 ± 1 21 ± 0.9SP 600125 21 ± 1.8 20 ± 1 21 ± 1.2 23 ± 1.3 20 ± 0.8NAC 28 ± 1 26 ± 0.6 26 ± 0.9 25 ± 1 22 ± 0.9Ang II + PD 24.1 ± 1.8 23 ± 2 22 ± 1.8 22 ± 1.1 21 ± 1.6Ang II + SB 25 ± 1.7 22.1 ± 1.1 22.8 ± 1.6 23 ± 1.8 22 ± 2Ang II + SP 23 ± 1.5 23 ± 1.5 24 ± 1.5 24 ± 1.7 23 ± 1.7Ang II + NAC 28 ± 1 26 ± 1 26 ± 1 24 ± 0.9 22 ± 0.7
LVDP: Left ventricular developed pressure; LVdp/dt: Maximum first derivatives of developed pressure; R: Reperfusion.Results are expressed as mean ± SEM of 6 animals as groups.a P < 0.05 Angiotensin II vs control.b P < 0.05 (Ang II + PD) or (Ang II + SB) or (Ang II + SP) or (Ang II + NAC) vs Ang II.
251S. Das et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 248–255
RETRACTED
respective baseline values. Group II (Ang II) displayedsignificant recovery of postischemic myocardial function. Thecardioprotective effects of Ang II were evidenced by differencesin the LVDP from R-30 onwards (Table 1), the difference isespecially apparent at R-120 (81 ± 3.4 mm Hg vs. 53 ± 4.6 mmHg) and also in the LVdp/dt at R-120 (1139 ± 93 mm Hg/s vs.900 ± 87 mm Hg/s). Aortic flow was markedly higher in the
Ang II group throughout the reperfusion, but at R-60(37 ± 4.1 ml/min vs. 19 ± 6.5 ml/min) and also at R-120(12.5 ± 2.8 ml/min vs. 4.3 ± 1.4 ml/min) the increase wassignificant compared to the vehicle-treated group. With the useof ERK (1/2) inhibitor (PD 098,059), JNK inhibitor (SP600125) and free radical scavenger (NAC), Ang II lost itscardioprotective effects which were evidenced by significant

Fig. 2. Representative hearts showing cardiomyocyte apoptosis and myocardialinfarct size.
Fig. 3. Top. Effects of Ang II, PD 098,059, SB 202190, SP 600125 and NAC onmyocardial infarct size. The isolated hearts from vehicle control (n = 6), Ang IIpreperfused in the absence or presence of either PD 098,059 or SB 202190 or SP600125 or NAC (n = 6) rats were subjected to 30 min global ischemia followedby 2 h of reperfusion in working mode. Infarct size was measured by TTC dyemethod. Results are expressed as means ± SEM. *P < 0.05 vs. control and†P < 0.05 vs. Ang II. Bottom. Effects of Ang II, PD 098,059, SB 202190, SP600125 and NAC on cardiomyocyte apoptosis. The isolated hearts from vehiclecontrol (n = 6), Ang II preperfused in the absence or presence of either PD098,059 or SB 202190 or SP 600125 or NAC (n = 6) rats were subjected to30 min global ischemia followed by 2 h of reperfusion in working mode.Cardiomyocyte apoptosis was evaluated by Tunnel method in conjunction withantibody against α-myosin heavy chain. Results are expressed as means ± SEM.*P < 0.05 vs. control and †P < 0.05 vs. Ang II.
252 S. Das et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 248–255
ACT
differences in the post ischemic period of LVDP at R-120(65 ± 5 mm Hg, 63 ± 4.4 mm Hg and 60 ± 3 mm Hg vs.81 ± 3.4 mm Hg) and also from the significant decrease ofLVdp/dt at R-120 (828 ± 138 mm Hg/s, 786 ± 55.8 mm Hg/sand 909 ± 66 mm Hg/s vs. 1139 ± 93 mm Hg/s). This is alsoconfirmed from the Aortic flow value; which is markedly lowerthroughout the whole reperfusion period, especially at R-60(18.5 ± 5.7 ml/min, 18.5 ± 4.9 ml/min and 18 ± 3 ml/min vs.37 ± 4.1 ml/min) and also R-120 (3.2 ± 1.8 ml/min,3.4 ± 1.4 ml/min and 5 ± 1 ml/min vs. 12.5 ± 2.8 ml/min).In contrast, SB 202190 when used in conjunction with AngII, the cardioprotective effect of Ang II was almost unchanged,which was evidenced from all the three parameters, LVDP,LVdp/dt and Aortic flow (Table 1).
3.2. Effects of PD 098,059, SB 202190, SP 600125 and NACon infarct size-lowering ability of Ang II
Infarct size (percent of infarct vs. total area at risk) wasnoticeably reduced in Ang II group as compared to the control(25.6 ± 2.6% vs. 34.6 ± 2.1%) (Fig. 2). The infarct zone alsoreduced significantly when NAC was used as compared tocontrol (22.2 ± 3.9% vs. 34.6 ± 2.1%). The infarct zone wasincreased significantly when Ang II was used along with PD098,059 (34.6 ± 2.6% vs. 25.6 ± 2.6%) or SP 600125 (34.5 ± 2%vs. 25.6 ± 2.6%) or NAC (30.6 ± 1.2% vs. 25.6 ± 2.6%), but notwith SB 202190 (28.2 ± 2.3% vs. 25.6 ± 2.6%) as compared tothe Ang II group, as shown in the Fig. 3 (top).
3.3. Effects of PD 098,059, SB 202190, SP 600125 and NACon cardiomyocyte apoptotic cell death-lowering ability ofAng II
The percent of apoptotic cardiomyocytes was significantlyreduced in Ang II group as compared to the control(9.8 ± 2.7% vs. 22.7 ± 2.5%) (Fig. 2). NAC also significantly
RETR
reduced the apoptotic cell death (14.2 ± 2.3%). The apoptoticcell death was increased significantly when Ang II was usedalong with PD 098,059 or SP 600125, but not with SB202190. Thus, the apoptosis was significantly higher in AngII + PD 098,059 (21.9 ± 1.6%), Ang II + SP 600125(20.7 ± 2.0%) and Ang II + NAC (22.5 ± 2.2%) groups ascompared to the Ang II group Fig. 3 (bottom).
3.4. Effects of Ang II, and the inhibitors of Erk (1/2), p38MAPKand JNK as well as NAC on the expression of Akt, Bad andBcl-2
Ang II significantly enhanced the phosphorylation of Akt,Bcl-2 and Bad (Fig. 4, left). The phosphorylation of Akt wasincreased by 8-fold, Bcl-2 by 9-fold and Bad by 6-fold. Therewas no induction of Akt or Bad by Ang II. The Ang II-mediated phosphorylation of Bcl-2, Akt and Bad was reducedsignificantly by SP 600125, PD 098,059 and NAC, but notwith SB202190 (Fig. 4). The results of phosphorylatedproducts were normalized against their corresponding non-phosphorylated proteins, while the results of Bcl-2 were
ED
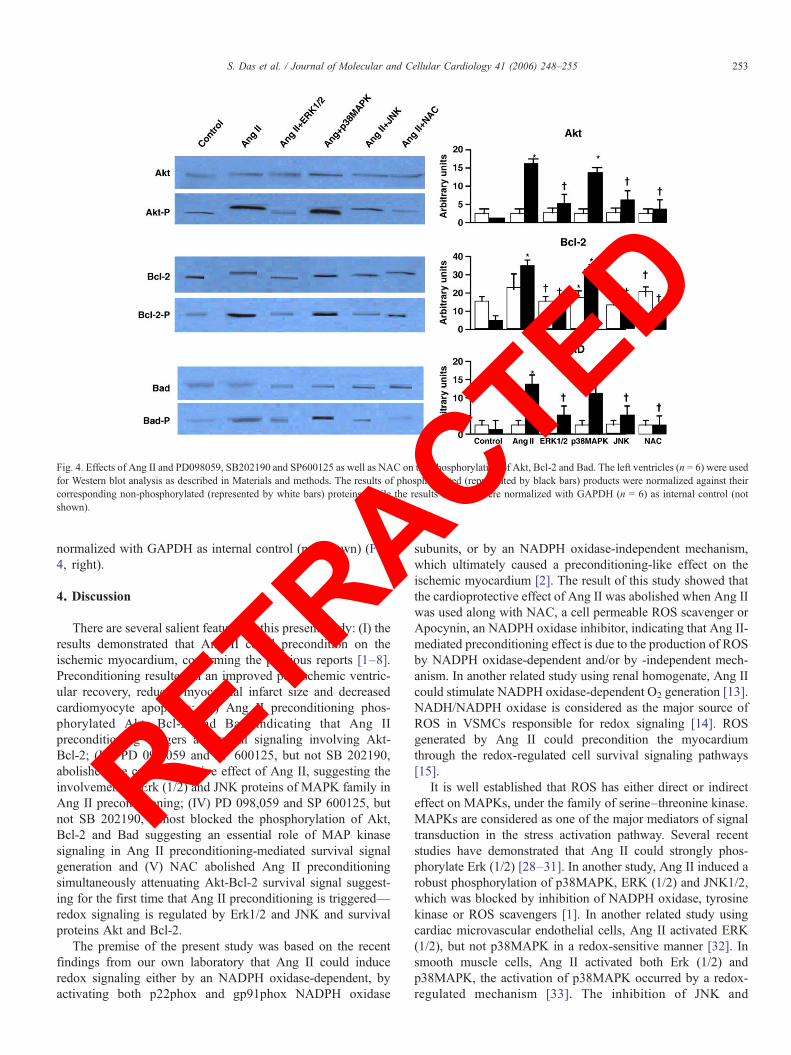
Fig. 4. Effects of Ang II and PD098059, SB202190 and SP600125 as well as NAC on the phosphorylation of Akt, Bcl-2 and Bad. The left ventricles (n = 6) were usedfor Western blot analysis as described in Materials and methods. The results of phosphorylated (represented by black bars) products were normalized against theircorresponding non-phosphorylated (represented by white bars) proteins, while the results of Bcl-2 were normalized with GAPDH (n = 6) as internal control (notshown).
253S. Das et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 248–255
ACTED
normalized with GAPDH as internal control (not shown) (Fig.4, right).4. Discussion
There are several salient features in this present study: (I) theresults demonstrated that Ang II could precondition on theischemic myocardium, confirming the previous reports [1–8].Preconditioning resulted in an improved postischemic ventric-ular recovery, reduced myocardial infarct size and decreasedcardiomyocyte apoptosis; (II) Ang II preconditioning phos-phorylated Akt, Bcl-2 and Bad indicating that Ang IIpreconditioning triggers a survival signaling involving Akt-Bcl-2; (III) PD 098,059 and SP 600125, but not SB 202190,abolished the cardioprotective effect of Ang II, suggesting theinvolvement of Erk (1/2) and JNK proteins of MAPK family inAng II preconditioning; (IV) PD 098,059 and SP 600125, butnot SB 202190, almost blocked the phosphorylation of Akt,Bcl-2 and Bad suggesting an essential role of MAP kinasesignaling in Ang II preconditioning-mediated survival signalgeneration and (V) NAC abolished Ang II preconditioningsimultaneously attenuating Akt-Bcl-2 survival signal suggest-ing for the first time that Ang II preconditioning is triggered—redox signaling is regulated by Erk1/2 and JNK and survivalproteins Akt and Bcl-2.
The premise of the present study was based on the recentfindings from our own laboratory that Ang II could induceredox signaling either by an NADPH oxidase-dependent, byactivating both p22phox and gp91phox NADPH oxidase
RETR
subunits, or by an NADPH oxidase-independent mechanism,which ultimately caused a preconditioning-like effect on theischemic myocardium [2]. The result of this study showed thatthe cardioprotective effect of Ang II was abolished when Ang IIwas used along with NAC, a cell permeable ROS scavenger orApocynin, an NADPH oxidase inhibitor, indicating that Ang II-mediated preconditioning effect is due to the production of ROSby NADPH oxidase-dependent and/or by -independent mech-anism. In another related study using renal homogenate, Ang IIcould stimulate NADPH oxidase-dependent O2 generation [13].NADH/NADPH oxidase is considered as the major source ofROS in VSMCs responsible for redox signaling [14]. ROSgenerated by Ang II could precondition the myocardiumthrough the redox-regulated cell survival signaling pathways[15].It is well established that ROS has either direct or indirecteffect on MAPKs, under the family of serine–threonine kinase.MAPKs are considered as one of the major mediators of signaltransduction in the stress activation pathway. Several recentstudies have demonstrated that Ang II could strongly phos-phorylate Erk (1/2) [28–31]. In another study, Ang II induced arobust phosphorylation of p38MAPK, ERK (1/2) and JNK1/2,which was blocked by inhibition of NADPH oxidase, tyrosinekinase or ROS scavengers [1]. In another related study usingcardiac microvascular endothelial cells, Ang II activated ERK(1/2), but not p38MAPK in a redox-sensitive manner [32]. Insmooth muscle cells, Ang II activated both Erk (1/2) andp38MAPK, the activation of p38MAPK occurred by a redox-regulated mechanism [33]. The inhibition of JNK and

254 S. Das et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 248–255
A
p38MAPK activation was shown by using an antioxidant in therat aortic smooth muscle cells, but interestingly the antioxidantdid not change the activation of ERK (1/2) [34]. In anotherstudy with adult rat cardiac fibroblasts, Ang II-stimulated ROSinduced osteoponin gene by the activation of ERK (1/2) andJNKs, but not p38 kinases [35].
The present study demonstrates that Ang II phosphorylatesAkt significantly. The phosphorylation of Akt was throughactivation of both ERK (1/2) and JNK pathways. However,Ang II failed to activate p38MAPK, as the phosphorylation ofAkt was still observed when the inhibitor of p38MAPK wasused in conjunction with Ang II. The observed differentialactivation of different MAPKs in myocardium is notsurprising since similar differential activation was observedin human vascular smooth muscle cells; IL-1β activated onlyp38MAPK and not JNK or ERK (1/2) [36]. This kind ofbehavior has been proven by other various studies in adultcardiac fibroblasts ROS-stimulated Erk (1/2) and JNK, andnot p38MAPK [35].
In summary, the results of the present study when viewed inthe light of previous studies [1–8], appear to suggest that Ang IIstimulates ROS production through NADPH-dependent and-independent pathways potentiating redox signaling. This leadsto the activation of ERK1/2 and JNK, but not p38MAPK, whichexert preconditioning-like effects on the ischemic myocardium.These results would tend to indicate that Ang II preconditioningdiffers from ischemic preconditioning, where p38MAPK playsa crucial role [16]. Both ischemic preconditioning and Ang IIpreconditioning appear to be regulated by ERK1/2 and JNKsignaling. Similar to ischemic preconditioning, Ang II pre-conditioning is also mediated through Akt-Bcl-2 survivalsignaling pathway.
Acknowledgments
This study was supported in part by NIH grants HL 34360,HL 22559, HL 33889 and HL 56803.
References
[1] Booz GW, Day JN, Baker KM. Interplay between the cardiac reninangiotensin system and JAK-STAT signaling: role in cardiac hypertrophy,ischemia/reperfusion dysfunction, and heart failure. J Mol Cell Cardiol2002;34:1443–53.
[2] Das S, Engelman RM, Maulik N, Das DK. Angiotensin preconditioning ofthe heart. Cell Biochem Biophys 2006;44:103–10.
[3] Diaz RJ, Wilson GJ. Selective blockade of AT1 angiotensin II receptorsabolishes ischemic preconditioning in isolated rabbit hearts. J Mol CellCardiol 1997;29:129–39.
[4] Ferreira AJ, Santos RA, Almeida AP. Angiotensin-(1–7): cardioprotectiveeffect in myocardial ischemia/reperfusion. Hypertension 2001;38:665–8.
[5] Nakano A, Miura AT, Ura N, Suzuki NK, Shimamoto K. Role of theangiotensin II type 1 receptor in preconditioning against infarction. CoronArtery Dis 1997;8:343–50.
[6] Sharma A, Singh M. Effect of ethylisopropyl amiloride, a Na+–H+exchange inhibitor, on cardioprotective effect of ischaemic and angiotensinpreconditioning. Mol Cell Biochem 2000;214:31–8.
[7] Sharma A, Singh M. Possible mechanism of cardioprotective effect ofangiotensin preconditioning in isolated rat heart. Eur J Pharmacol2000;406:85–92.
RETR
[8] Xiao XH, Allen DG. The role of endogenous angiotensin II in ischaemia,reperfusion and preconditioning of the isolated rat heart. Pflugers Arch2003;445:643–50.
[9] Liu Y, Tsuchida YA, Cohen MV, Downey JM. Pretreatment withangiotensin II activates protein kinase C and limits myocardialinfarction in isolated rabbit hearts. J Mol Cell Cardiol 1995;27:883–92.
[10] Vegh A, Papp JG, Parratt J. Attenuation of the antiarrhythmic effects ofischemic preconditioning by blockade of bradykinin B2 receptors. Br JPharmacol 1994;113:1167–72.
[11] Miki T, Miura T, Ura N, Ogawa T, Suzuki K, Shimamoto K, et al. Captoprilpotentiates the myocardial infarct size-limiting effect of ischemicpreconditioning through bradykinin B2 receptor activation. J Am CollCardiol 1996;28:1616–22.
[12] Davis RJ. MAPKs: new JNK expands the group. Trends Biochem Sci1994;19:470–3.
[13] Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. AngiotensinII stimulates NADH and NADPH oxidase activity in cultured vascularsmooth muscle cells. Circ Res 1994;74:1141–8.
[14] Pagano PJ, Ito Y, Tornheim K, Gallop PM, Tauber AI, Cohen RA. ANADPHoxidase superoxide-generating system in the rabbit aorta. Am JPhysiol 1995;268:H2274–80.
[15] Das DK, Maulik N, Engelman RM. Redox regulation of angiotensin IIsignaling in the heart. J Cell Mol Med 2000;214:31–8.
[16] Maulik N, Yoshida T, Zu YL, Sato M, Banerjee A, Das DK. Ischemicpreconditioning triggers tyrosine kinase signaling: a potential role forMAPKAP kinase 2. Am J Physiol 1998;275:H1857–64.
[17] Egan SE, Weinberg RA. The pathway to signal achievement. Nature1993;365:781–3.
[18] Das S, Tosaki A, Bagchi D, Maulik N, Das DK. Resveratrol-mediatedactivation of cAMP response element-binding protein through adenosineA3 receptor by Akt-dependent and-independent pathways. J PharmacolExp Ther 2005;314:762–9.
[19] Gauthier R, Laprise PE, Cardin C, Harnoi A, Plourde JC, Reed A, et al.Differential sensitivity to apoptosis between the human small and largeintestinal mucosae: linkage with segment-specific regulation of BCL-2homologs and involvement of signaling pathways. J Cell Biochem 2001;82:339–55.
[20] O'Gorman DM, McKenna SL, McGahon AJ, Knox KA, Cotter TG.Sensitisation of HL60 human leukaemic cells to cytotoxic drug-inducedapoptosis by inhibition of PI3-kinase survival signals. Leukemia2000;14:602–11.
[21] Hokenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer XY. Bcl-2functions in an antioxidant pathway to prevent apoptosis. Cell 1993;75:241–51.
[22] Maulik N, Engelman RM, Rousou JA, Flack JE, Deaton DW, Das DK.Ischemic preconditioning suppresses apoptosis by upregulating the anti-death gene, Bcl-2. Surg Forum 1998;49:209–11.
[23] Das S, Cordis GA, Maulik N, Das DK. Pharmacological preconditioningwith resveratrol: role of CREB-dependent Bcl-2 signaling via adenosineA3 receptor activation. Am J Physiol: Heart Circ Physiol 2005;288:H328–35.
[24] Hattori R, Hernandez TE, Zhu L, Maulik N, Otani H, Kaneda Y, et al.Essential role of the antioxidant gene Bcl-2 in myocardial adaptation toischemia: an insight with antisense Bcl-2 therapy. Antioxid Redox Signal2001;3:403–13.
[25] Maulik N, Engelman RM, Rousou JA, Flack III JE, Deaton D, Das DK.Ischemic preconditioning reduces apoptosis by upregulating anti-deathgene Bcl-2. Circulation 1999;100:II369–75.
[26] Engelman DT, Watanabe M, Engelman RM, Rousou JA, Kisin E, KaganVE, et al. Hypoxic preconditioning preserves antioxidant reserve in theworking rat heart. Cardiovasc Res 1995;29:133–40.
[27] Das S, Otani H, Maulik NN, Das DK. Lycopene, tomatoes, and coronaryheart disease. Free Radical Res 2005;39:449–55.
[28] Gorin Y, Ricono JM, Wagner B, Kim NH, Bhandari B, Choudhury GG,et al. Angiotensin II-induced ERK1/ERK2 activation and proteinsynthesis are redox-dependent in glomerular mesangial cells. Biochem J2004; 381:231–9.
CTED

255S. Das et al. / Journal of Molecular and Cellular Cardiology 41 (2006) 248–255
[29] Izawa Y, Yoshizumi M, Fujita Y, Ali N, Kanematsu Y, Ishizawa K,et al. ERK1/2 activation by angiotensin II inhibits insulin-inducedglucose uptake in vascular smooth muscle cells. Exp Cell Res 2005;308:291–9.
[30] Li JM, Wheatcroft S, Fan LM, Kearney MT, Shah AM. Opposing roles ofp47phox in basal versus angiotensin II-stimulated alterations in vascularO2-production, vascular tone, and mitogen-activated protein kinaseactivation. Circulation 2004;109:1307–13.
[31] Pinzar E, Wang T, Garrido MR, Xu W, Levy P, Bottari SP. Angiotensin IIinduces tyrosine nitration and activation of ERK1/2 in vascular smoothmuscle cells. FEBS Lett 2005;579:5100–4.
[32] Xie Z, Pimental DR, Lohan S, Vasertriger A, Pligavko C, Colucci WS, etal. Regulation of angiotensin II-stimulated osteopontin expression incardiac microvascular endothelial cells: role of p42/44 mitogen-activatedprotein kinase and reactive oxygen species. J Cell Physiol 2001;188:132–8.
RETRA
[33] Ushio-Fukai M, Zafari AM, Fukui T, Ishizaka N, Griendling KK. p22 phoxis a critical component of the superoxide-generating NADH/NADPHoxidase system and regulates angiotensin II-induced hypertrophy invascular smooth muscle cells. J Biol Chem 1996;271:23317–21.
[34] Kyaw M, Yoshizumi M, Tsuchiya K, Kirima K, Tamaki T. Antioxidantsinhibit JNK and p38 MAPK activation but not ERK 1/2 activation byangiotensin II in rat aortic smooth muscle cells. Hypertens Res 2001;24:251–61.
[35] Xie Z, Singh M, Singh K. ERK1/2 and JNKs, but not p38 kinase, areinvolved in reactive oxygen species-mediated induction of osteopontingene expression by angiotensin II and interleukin-1beta in adult rat cardiacfibroblasts. J Cell Physiol 2004;198:399–407.
[36] Jung YD, Liu W, Reinmuth N, Ahmad SA, Fan F, Gallick GE, et al.Vascular endothelial growth factor is upregulated by interleukin-1 beta inhuman vascular smooth muscle cells via the P38 mitogen-activated proteinkinase pathway. Angiogenesis 2001;4:155–62.
CTED


















