
Reconstructing Kinship Relationships in Wild Populations I do not believe that the accident of birth...
24
Reconstructing Kinship Relationships in Wild Populations I do not believe that the accident of birth makes people sisters and brothers. It makes them siblings. Gives them mutuality of parentage. Maya Angelou Isabel Caballero UIC Priya Govindan Rutgers Chun-An (Joe) Chou Rutgers Saad Sheikh Ecole Polytechniq ue Alan Perez- Rathkeo UIC Mary Ashley UIC W. Art Chaovalitwongs e Rutgers Ashfaq Khokhar UIC Bhaskar DasGupta UIC Tanya Berger-Wolf UIC
-
Upload
shawn-ross -
Category
Documents
-
view
215 -
download
1
Transcript of Reconstructing Kinship Relationships in Wild Populations I do not believe that the accident of birth...

Reconstructing Kinship Relationshipsin Wild Populations
I do not believe that the accident of birth makes people sisters and brothers. It makes them siblings. Gives them mutuality of
parentage. Maya Angelou
Isabel CaballeroUIC
Priya GovindanRutgers
Chun-An (Joe) Chou
Rutgers
Saad SheikhEcole
Polytechnique
Alan Perez-Rathkeo
UIC
Mary AshleyUIC
W. Art Chaovalitwong
seRutgers
Ashfaq Khokhar
UIC
Bhaskar DasGupta
UIC
TanyaBerger-Wolf
UIC

Microsatellites (STR)
Advantages: Codominant (easy
inference of genotypes and allele frequencies)
Many heterozygous alleles per locus
Possible to estimate other population parameters
Cheaper than SNPs
But: Few loci
And: Large families Self-mating …
CACACACA5’
AllelesCACACACA
CACACACACACA
CACACACACACACA
#1
#2
#3
Genotypes
1/1 2/2 3/3 1/2 1/3 2/3

Siblings: two children with the same parentsQuestion: given a set of children, find the
sibling groups
Diploid Siblings
locusallele
father (.../...),(a /b ),(.../...),(.../...) (.../...),(c /d ),(.../...),(.../...) mother
(.../...),(e /f ),(.../...),(.../...) child
one from fatherone from mother

Why Reconstruct Sibling Relationships?
Used in: conservation biology, animal management, molecular ecology, genetic epidemiology
Necessary for: estimating heritability of quantitative characters, characterizing mating systems and fitness.
• But: hard to sample parent/offspring pairs. Sampling cohorts of juveniles is easier

The Problem
Ind Locus 1
Locus 2
allele 1/allele 2
1 1/2 1/2
2 1/3 3/4
3 1/4 3/5
4 3/3 7/6
5 1/3 3/4
6 1/3 3/7
7 1/5 8/2
8 1/6 2/2
Sibling Groups:
2, 4, 5, 6
1, 3
7, 8

Existing MethodsMethod Approach Error-
DetectionAssumptions
Almudevar & Field (1999,2003)
Minimal Sibling groups under likelihood
No Minimal sibgroups, representative allele frequencies
KinGroup (2004)
Markov Chain Monte Carlo/ML
No Allele Frequencies etc. are representative
Family Finder(2003)
Partition population using likelihood graphs
No Allele Frequencies etc. are representative
Pedigree (2001)
Markov Chain Monte Carlo/ML
No Allele Frequencies etc are representative
COLONY (2004)
Simulated Annealing/ ML
Yes Monogamy for one sex
Fernandez & Toro (2006)
Simulated Annealing/ ML
No Co-ancestry matrix is a good measure, parents can be reconstructed or are available

Inheritance Rulesfather (.../...),(a /b ),(.../...),(.../...) (.../...),(c /d ),(.../...),(.../...) mother
child 1 (.../...),(e1 /f1 ),(.../...),(.../...)
child 2 (.../...),(e2 /f2 ),(.../...),(.../...)child 3 (.../...),(e3 /f3 ),(.../...),(.../...)
child n (.../...),(en/fn ),(.../...),(.../...)
…4-allele rule: siblings have at most 4 distinct alleles in a locus
2-allele rule: In a locus in a sibling group: a + R ≤ 4
Num distinct alleles
Num alleles that appear with 3 others or are
homozygot

Our Approach: Mendelian Constrains
4-allele rule:siblings have at most 4 different alleles in a locus
Yes: 3/3, 1/3, 1/5, 1/6No: 3/3, 1/3, 1/5, 1/6, 3/2
2-allele rule: In a locus in a sibling group:a + R ≤ 4
Yes: 3/3, 1/3, 1/5No: 3/3, 1/3, 1/5, 1/6
Num distinct alleles
Num alleles that appear with 3 others or are
homozygot

Our Approach: Sibling Reconstruction
Given:n diploid individuals sampled at l loci
Find: Minimum number of 2-allele sets that contain all individuals
NP-complete even when we know sibsets are at most 31.0065 approximation gap Ashley et al ’09
ILP formulation Chaovalitwongse et al. ’07, ’10
Minimum Set Cover based algorithm with optimal solution (using CPLEX) Berger-Wolf et al. ’07
Parallel implementation Sheikh, Khokhar, BW ‘10

ID alleles1 1/2
2 2/3
3 2/1
4 1/3
5 3/2
6 1/4
Canonical families
1/1 1/2 1/3 1/4 2/2 2/3 2/4 3/4 3/3 4/4
1/1 1/1
1/2
2/1
2/21/3
1/4
2/3
2/4
3/1
4/1
3/2
4/2
1/1
1/2
2/1
1/1
1/3
2/1
2/3
3/1
2/1
3/2
1/2
1/3
2/1
3/1
ID alleles
1 55/43
2 43/114
3 43/55
4 55/114
5 114/43
6 55/78
1/3
2/1
2/3
2/1
3/2

Aside: Minimum Set Cover
Given: universe U = {1, 2, …, n} collection of sets S = {S1, S2,…,Sm}
where Si subset of U
Find: the smallest number of sets in Swhose union is the universe U
€
minI⊆[m ]
| I | such that Ui∈ISi =U
Minimal Set Cover is NP-hard
(1+ln n)-approximable (sharp)

Are we done?Challenges No ground truth available Growing number of methods Biologists need (one) reliable
reconstruction Genotyping errors
Answer: Consensus
Consensus is what many people say in chorus but do not believe as individuals
Abba Eban (1915 - 2002), Israeli diplomat In "The New Yorker," 23 Apr 1990

Consensus MethodsCombine multiple solutions to a problem to
generate one unified solution C: S*→ S Based on Social Choice Theory Commonly used where the real solution
is not known e.g. Phylogenetic Trees
Consensus...
S1 S2 Sk S

Error-Tolerant ApproachSheikh et al. 08
Locu
s
1
Locu
s
2
Locu
s
3
Locu
s
l
Sibling Reconstruc
tion Algorithm
...
Consensus...
S1 S2 Sk S

Distance-based Consensus
Consensus...S1 S2 Sk Ss
S
Search
f q
fq fd
Algorithm– Compute a consensus solution
S={g1,...,gk }– Search for a good solution near S
fd
NP-hard for any fd, fq or an arbitrary linear combination Sheikh et al. ‘08

A Greedy Approach - Algorithm Compute a strict consensus While total distance is not too large
Merge two sibgroups with minimal (total) distance
Quality: fq=n-|C| Distance function from solution C to C’
fd(C,C’) =sum of costs of merging groups in C to obtain C’
=sum of costs of assigning individuals to groups
Cost of assigning individual to a group:Benefit: Alleles and allele pairs sharedCost: Minimum Edit Distance

Change costs to average per locus costsCompare max group error on per locus basisTreat cost and benefit independentlyIn order to qualify a merge
Cost <= maxcostBenefit >= minbenefitBenefit = max benefit among possible merges
Auto Greedy Consensus

A Greedy Approach
{1,2} {3} {4} {5} {6,7}
{1,2} 3.5 1.1 2.5 5.1
{3} 0.5 0.3 0.5 0.1
{4} 1.0 3.0 0.6 1.1
{5} 2.0 1.2 3.5 4.9
{6,7} 0.6 0.9 1.2 4.1
S1 = { {1,2,3},{4,5},{6,7} }
S2 = { {1,2,3},{4}, {5,6,7} }
S3 = { {1,2},{3,4,5},{6,7} }
Strict Consensus
S = { {1,2}, {3}, {4}, {5}, {6,7} }
{1,2} {3,6,7} {4} {5} {6,7}
{1,2} 3.5 1.1 2.5 5.1
{3,6,7} 1.7 3.1 2.2 6.1
{4} 1.0 3.0 0.6 1.1
{5} 2.0 1.2 3.5 4.9
{6,7} 0.6 0.9 1.2 4.1
S = { {1,2}, {3}, {4}, {5}, {6,7} }
S={ {1,2}, {3,6,7}, {4}, {5} }

Testing and Validation: Protocol
1. Get a dataset with known sibgroups(real or simulated)
2. Find sibgroups using our alg3. Compare the solutions
Partition distrance, Gusfield ’03 = assignment problem
Compare to other sibship methods Family Finder, COLONY

Salmon (Salmo salar) - Herbinger et al., 1999 351 individuals, 6 families, 4 loci. No missing alleles
Shrimp (Penaeus monodon) - Jerry et al., 200659 individuals,13 families, 7 loci. Some missing alleles
Ants (Leptothorax acervorum )- Hammond et al., 2001Ants are haplodiploid species. The data consists of 377 worker diploid ants
Test Data
Simulated populations of juveniles for a range of values of number of parents, offspring per parent, alleles, per locus, number of loci, and the distributions of those.
Experimental Protocol
Generate F females and M males (F=M=5, 10, 20)
Each with l loci (l=2, 4, 6,8,10)Each locus with a alleles (a=10, 15)
Generate f families (f=5,10,20)For each family select female+male
uniformly at random
For each parent pair generate o offspring(o=5,10)
For each offspring for each locus choose allele outcome uniformly at random
Introduce random errors

Results
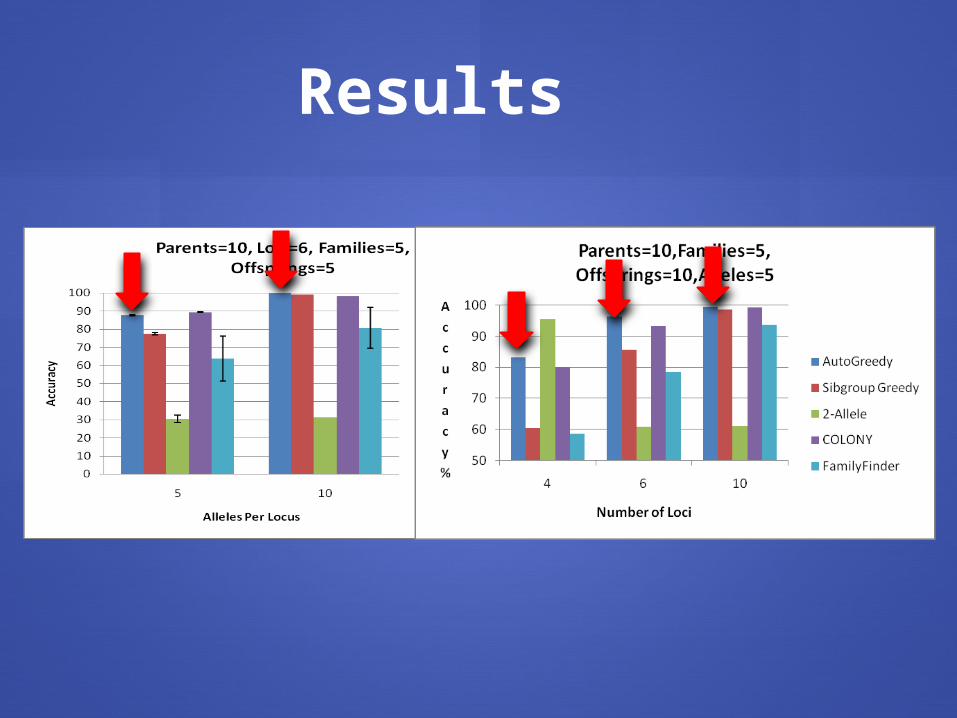
Results

ConclusionsCombinatorial algorithms with minimal
assumptionsBehaves well on real and simulated data Better than others with few loci, few large
familiesError tolerantUseful, high demand
New and improved: Efficient implementation Perez-Rathlke et al. (in submission)
Other objectives (bio vs math) Ashley et al. ‘10
Other genealogical relationships Sheikh et al. ‘09, ’10
Different combinatorial approach Brown & B-W, ‘10
Pedigree amalgamation


















