
RECOGNIZING METHYL-CPG SITES INVESTIGATNG … 4-Macromolecular Structure and... ·...
17
RECOGNIZING METHYL-CPG SITES: INVESTIGATNG ZBTB PROTEIN REGULATION OF EPIGENTICALLY MODIFIED DNA Albert M. Lund, 1 Tommy W. Terooatea, 1 Kuei-Ho Chen 1 , and Bethany A. Buck-Koehntop 1* 1 Department of Chemistry, University of Utah, 315 S. 1400 E., Rm. 2020, Salt Lake City, UT, USA * Correspondence to: [email protected] ABSTRACT Methylation of cytosine in CpG dinucleotides leads to subsequent gene suppression and can result in cancer progression if misregulated. These epigenetic DNA mod- ifications and the factors that regulate them are attrac- tive cancer therapeutic targets since the damage is re- versible unlike in genomic mutations. Here we utilize a multi-disciplinary approach to investigate the role that the Zbtb family of methyl-CpG binding proteins (MBPs) plays in modulating these epigenetic signals in cancer. INTRODUCTION, RESULTS AND DISCUSSION, CONCLUSION In eukaryotes, methylation of CpG dinucleotides is es- sential for genomic stability, control of gene expression and the regulation of chromatin structure in normal cells. Aber- rant alterations in these genomic methylation patterns have a direct link to a variety of diseases, including cancer. The Zbtb family is a set of specialized transcription factors, which exhibit bimodal DNA recognition by specifically targeting both methylated DNA signals as well as sequence- specific non-methylated sites [1-3] through a set of three conserved Cys 2 His 2 zinc fingers. DNA recognition subse- quently recruits chromatin remodelling machinery to the target site resulting in chromatin compaction and gene si- lencing. While there are sufficient findings to suggest that the Zbtb family of MBPs participates in cancer progression, an extensive analysis of the gene targets and subsequent signal- ing pathways regulated by each protein has yet to be inves- tigated. Here we utilize an interdisciplinary approach com- bining structural biology, molecular biology and cellular biology to determine the epigenetic signaling pathways reg- ulated by the Zbtb family of MBPs and to identify the pro- tein region(s) necessary for high-affinity DNA recognition. We have designed multiple protein constructs around the conserved Cys 2 His 2 zinc finger DNA binding region as well as additional zinc fingers located in the C-terminal regions of Zbtb4 and Zbtb38 as the functions of these additional zinc finger regions have not yet been identified. It is possi- ble that these regions also participate in DNA recognition and may be partly responsible for direct targeting of these proteins to differential gene targets. The constructs have been screened by HSQC analysis to determine whether they were amenable for further structural characterization and whether they exhibit DNA binding properties. Additionally, using western blot analysis (Figure 1), we have determined that the three Zbtb proteins exhibit differ- ential endogenous expression levels in a variety of cancer cell lines. Of particular interest, all of these proteins were down-regulated in the indolent form of prostate cancer (LNCaP) but up-regulated in the aggressive form (PC3). Interestingly, the MBD family of MBPs exhibits the oppo- site trend between the indolent and aggressive forms [4]. Through ChIP-Seq and qPCR analysis, we have begun to catalog the gene occupancy of these proteins in prostate cancer cells. In conclusion, our preliminary results indicate that the preferential gene targets in the PC3 cancer cells may define a new set of potential biomarkers that can be utilized to dis- tinguish aggressive from indolent forms of prostate cancer. Further, our initial structural analysis of the Zbtb proteins and their DNA interactions will provide the basis for a larg- er program in which atomic-level structural knowledge will be utilized for the design of novel and highly selective epi- genetic-based cancer therapeutics directed against the Zbtb family of MBPs. REFERENCES 1. Daniel, J.M., Spring, C.M., Crawford, H.C., Reynolds, A.B., Baig, A. Nuc. Acid Res., 2002, 30, 2911-2919. 2. Fillion, G.J.P., Zhenilo, S., Salozhin, S. Yamada, D., Prokhortchouk, E., Defossez, P.-A. Mol. Cell. Biol., 2006, 26, 169-181. 3. Buck-Koehntop, B.A., Martinez-Yamout, M.A., Dyson, H.J., Wright, P.E. FEBS Lett., 2012, 586, 734-739. 4. Pulukuri, S.M., Rao, J.S. Oncogene, 2006, 25, 4559- 4572. Figure 1. Western blot indicating the endogenous protein expres- sion levels for the three Zbtb family MBPs in various cancer cells. 154
Transcript of RECOGNIZING METHYL-CPG SITES INVESTIGATNG … 4-Macromolecular Structure and... ·...

RECOGNIZING METHYL-CPG SITES: INVESTIGATNG ZBTB PROTEIN REGULATION OF EPIGENTICALLY MODIFIED DNA
Albert M. Lund,1 Tommy W. Terooatea,1 Kuei-Ho Chen1, and Bethany A. Buck-Koehntop1*
1Department of Chemistry, University of Utah, 315 S. 1400 E., Rm. 2020, Salt Lake City, UT, USA
* Correspondence to: [email protected]
ABSTRACT
Methylation of cytosine in CpG dinucleotides leads to subsequent gene suppression and can result in cancer progression if misregulated. These epigenetic DNA mod-ifications and the factors that regulate them are attrac-tive cancer therapeutic targets since the damage is re-versible unlike in genomic mutations. Here we utilize a multi-disciplinary approach to investigate the role that the Zbtb family of methyl-CpG binding proteins (MBPs) plays in modulating these epigenetic signals in cancer.
INTRODUCTION, RESULTS AND DISCUSSION, CONCLUSION
In eukaryotes, methylation of CpG dinucleotides is es-sential for genomic stability, control of gene expression and the regulation of chromatin structure in normal cells. Aber-rant alterations in these genomic methylation patterns have a direct link to a variety of diseases, including cancer. The Zbtb family is a set of specialized transcription factors, which exhibit bimodal DNA recognition by specifically targeting both methylated DNA signals as well as sequence-specific non-methylated sites [1-3] through a set of three conserved Cys2His2 zinc fingers. DNA recognition subse-quently recruits chromatin remodelling machinery to the target site resulting in chromatin compaction and gene si-lencing.
While there are sufficient findings to suggest that the Zbtb family of MBPs participates in cancer progression, an extensive analysis of the gene targets and subsequent signal-ing pathways regulated by each protein has yet to be inves-tigated. Here we utilize an interdisciplinary approach com-bining structural biology, molecular biology and cellular
biology to determine the epigenetic signaling pathways reg-ulated by the Zbtb family of MBPs and to identify the pro-tein region(s) necessary for high-affinity DNA recognition.
We have designed multiple protein constructs around the conserved Cys2His2 zinc finger DNA binding region as well as additional zinc fingers located in the C-terminal regions of Zbtb4 and Zbtb38 as the functions of these additional zinc finger regions have not yet been identified. It is possi-ble that these regions also participate in DNA recognition and may be partly responsible for direct targeting of these proteins to differential gene targets. The constructs have been screened by HSQC analysis to determine whether they were amenable for further structural characterization and whether they exhibit DNA binding properties.
Additionally, using western blot analysis (Figure 1), we have determined that the three Zbtb proteins exhibit differ-ential endogenous expression levels in a variety of cancer cell lines. Of particular interest, all of these proteins were down-regulated in the indolent form of prostate cancer (LNCaP) but up-regulated in the aggressive form (PC3). Interestingly, the MBD family of MBPs exhibits the oppo-site trend between the indolent and aggressive forms [4]. Through ChIP-Seq and qPCR analysis, we have begun to catalog the gene occupancy of these proteins in prostate cancer cells.
In conclusion, our preliminary results indicate that the preferential gene targets in the PC3 cancer cells may define a new set of potential biomarkers that can be utilized to dis-tinguish aggressive from indolent forms of prostate cancer. Further, our initial structural analysis of the Zbtb proteins and their DNA interactions will provide the basis for a larg-er program in which atomic-level structural knowledge will be utilized for the design of novel and highly selective epi-genetic-based cancer therapeutics directed against the Zbtb family of MBPs.
REFERENCES
1. Daniel, J.M., Spring, C.M., Crawford, H.C., Reynolds, A.B., Baig, A. Nuc. Acid Res., 2002, 30, 2911-2919.
2. Fillion, G.J.P., Zhenilo, S., Salozhin, S. Yamada, D., Prokhortchouk, E., Defossez, P.-A. Mol. Cell. Biol., 2006, 26, 169-181.
3. Buck-Koehntop, B.A., Martinez-Yamout, M.A., Dyson, H.J., Wright, P.E. FEBS Lett., 2012, 586, 734-739.
4. Pulukuri, S.M., Rao, J.S. Oncogene, 2006, 25, 4559-4572.
Figure 1. Western blot indicating the endogenous protein expres-sion levels for the three Zbtb family MBPs in various cancer cells.
154

ONE-THIRD-OF-THE-SITES BINDING OF TRANSITION-STATE ANALOGUES BY TRIMERIC
PURINE NUCLEOSIDE PHOSPHPORYLASES IN THE LIGHT OF NEW FINDINGS
Marta Narczyk,1 Beata Wielgus-Kutrowska,
1 Katarzyna Breer,
1 Mariko Hashimoto,
2
Sadao Hikishima,2 Tsutomu Yokomatsu
2 and Agnieszka Bzowska
1*
1Division of Biophysics, Institute of Experimental Physics, University of Warsaw, Żwirki i Wigury 93, 02-089 Warsaw,
Poland and 2School of Pharmacy, Tokyo University of Pharmacy and Life Sciences, 1432-1 Horinouchi, Hachioji, To-
kyo 192-0392, Japan * Correspondence to: [email protected]
ABSTRACT
One-third-of-the-sites binding of transition-state ana-
logues, immucillins was postulated to occur in the case of
trimeric purine nucleoside phosphorylases. We have
synthesized a new analogue with the PNP transition-
state features, 9-{[N-[3′′,3′′-difluoro-3′′-(diethylphospho-
no)propyl]amino]methyl}-9-deazaguanine, and showed
by calorimetric and fluorimetric titrations that it binds
similarly to all three sites of the enzyme. We give possi-
ble explanation why experiments with immucillin were
interpreted in terms of one-third-of-the-sites binding.
INTRODUCTION,
Purine nucleoside phosphorylases (PNP, E.C. 2.4.2.1)
from mammalian sources are homotrimeric enzymes, and
important drug targets for potential immunosuppressive and
anticancer agents. It was postulated that PNP exhibits one-
third-of-the-sites binding, it means one molecule of transi-
tion-state (TS) analogue inhibitors, immucillins, binds to
each PNP trimer and blocks binding to the remaining two
sites [1]. Later, on the basis of isothermal titration calorime-
try (ITC) it was suggested that binding to subsequent sites
occurs with negative cooperativity, and dissociation con-
stants (Kd) were estimated: 56 pM, 12 nM and 15 M [2].
RESULTS AND DISCUSSION
Immucillins have two features of the PNP TS: proton at
position N(7) of the purine base and positive charge on the
pentose ring imitating the oxocarbenium ion character of the
nucleoside in TS (Scheme, upper left panel). We have pre-
viously synthesized analogue with one TS feature (DFPP-
DG, Scheme, lower left panel), and reported that it binds as
tightly as immucillin (Kd = 85 pM) but shows no one-third-
of-the-sites binding [3]. However, when the recombinant
PNP is used in binding studies, the apparent cooperativity
may appear because some PNP active sites may be blocked
by hypoxanthine (PNP product), moped from the host E.
coli cells [3]. Here we present synthesis (Scheme, right pan-
el) of 9-{[N-[3′′,3′′-difluoro-3′′-(diethylphosphono)propyl]-
amino]methyl}-9-deazaguanine (aza-DFPP-DG) having
both features of the TS of PNP (Scheme, compound 6). By
ITC and fluorimetric titrations, spanning broad enzyme con-
centration range, we demonstrate (Figure 1) that binding of
aza-DFPP-DG with trimeric PNP is characterized by one
dissociation constant, and stoichiometry of the complex is
three ligand molecules per enzyme trimer. Hence, no one-
third-of-the-sites binding is observed for this TS analogue.
Supported by the Polish Ministry of Science and Higher
Education (grants N N301 044939 and BW-1724/BF).
REFERENCES 1. Miles, R.W., Tyler, P.C., Furneaux,R.H., Bagdassarian,
C.K., Schramm, V.L. Biochem., 1998, 37, 8615-8621.
2. Edwards, A. A., Mason, J. M., Clinch, K., Tyler, P. C.,
Evans, G. B., Schramm, V. L. Biochem., 2009, 48,
5226-5238.
3. Breer, K., Wielgus-Kutrowska, B., Girstun, A., Staroń,
K., Hashimoto, M., Hikishima, S., Yokomatsu, T.,
Bzowska, A. Biochem. Biophys. Res. Commun., 2010,
391, 1203-1209.
Figure 1. ITC and fluorimetric titrations of calf PNP by aza-DFPP-
DG at pH 7.0, 25oC. Concentration of PNP subunits is shown.
N
N
N
O
H
NH2
H
CF2PO3H2
HN
N
N
OPOM
N CHNMe2R
HN
N
N
OPOM
N CHNMe2
NH
CF2PO3Et2
5
HN
N
N
OH
NH2
NH
CF2PO3H2
6
12
R=IR=CHO
a
c
d
+ XCF2PO3Et2
3
4
X=N3X=NH2
bNH
OH OH
OH
N
N
N
O
H
H
1
23
4
56
78
9
(H+)
7.6 M
0.05 M
0.5 M
155

SYNTHESIS OF BRANCHED RNAS, LARIAT MIRTRONS AND RNA MINI-LARIATS AND THEIR
USE IN DEBRANCHING-DEPENDENT RNA INTERFERENCE
Subha R. Das,* Eduardo Paredes and Debasish Grahacharya
Department of Chemistry and Center for Nucleic Acids Science and Technology, Carnegie Mellon University, Pittsburgh, PA 15213, USA. * Correspondence to: [email protected]
ABSTRACT
The synthesis of branched RNAs as well as lariat
RNAs that include native phosphodiester or trizole
backbone linkages is described. These RNAs are sub-
strates for debranching enzyme and could find use in
RNA silencing by a non-canonical pathway.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
Backbone branched RNAs (bRNAs) are involved in the
distinct but related processes of splicing, debranching and
retrotransposition that are key to cellular regulation by RNA.
The first step of splicing – either within the spliceosomal
apparatus or in the group II intron – generates an RNA in
which an invariant adenosine branch-point residue is linked
at the 2'-position to the 5'-end of an RNA sequence that cor-
responds to the conserved residues of the intron. This bRNA
occurs as a ‘Y’-shaped structure in trans-splicing or as a
lariat structure, in which the sequence through the 2'-5'-
linkage loops around to include the branch-point adenosine,
in cis- or pre-mRNA splicing. Subsequently, the lariat or
bRNA is the substrate for lariat debranching enzyme (Dbr1),
a manganese dependent phosphodiesterase that is related to
well-described metallophosphatases such as calcineurin, λ
protein phosphatase and Mre11, a nuclease involved in
DNA repair[1,2]. Debranching of the 2',5'-phosphodiester
linkage in bRNA from splicing is also required for the pro-
cessing and biogenesis of box C/D snoRNAs7 and intronic
microRNAs (mirtrons) that bypass Drosha[3,4]. Additional-
ly, a lariat RNA form for Ty1 RNA has been proposed,
though the specific role of Dbr that is required for re-
trotransposition from Ty1 elements in yeast to HIV remains
unclear.
The significance of natural bRNAs and lariat RNAs in
biology has prompted significant efforts toward their syn-
thesis – since the first solution phase syntheses of tetranu-
cleotides by Caruthers and coworkers in 1986 and Chatto-
padhyaya and coworkers shortly after, to the pioneering and
seminal efforts of Damha and coworkers in solid-phase syn-
thesis, and more recent approaches of Wang and Silverman
using deoxyribozymes[5-8]. Although these methods have
yielded branched oligomers and lariat RNAs, the ability to
readily access natural and modified sequences that include
biochemically useful probes or functional group substitu-
tions, has remained elusive.
Here we describe our successful efforts in the solid-phase
synthesis of branched RNA. The use of an orthogonal 2'-O-
protecting group in an internal residue permits facile branch
synthesis. The synthesis protocol is robust and issues of
migration of the phospho-diester or -triester are addressed.
The method is versatile, overcoming previous limitations to
sequences or modifications. All four natural nucleobases
could be installed as the branch-point residue. Several
bRNAs that include alternate branch points or single atom
substitutions were synthesized for biochemical interrogation
of Dbr1. While Dbr1 shares high active site homology with
Mre1, a metallophosphatase that acts on DNA, our analyses
with the bRNAs reveal a Dbr1 substrate feature that distin-
guishes it for its specific role.
With the branching RNA synthesis developed, we could
enhance it with the use azide-alkyne click-chemistry – both
in solid phase and in solution on naïve RNA (with free 2'-
hydroxyls) using a pseudo-ligandless protocol. These fur-
nishe branched RNA in which the loop is closed to complete
the lariat structure. This chemical ligation yields a triazole
linked lariat RNA with the location of the triazole linkage
either at the branch-point 2'-position or downstream.
These synthetic lariats or mini-lariats are substrates for
debranching in vitro. We therefore tested them in a dual
luciferase reporter assay for in vivo RNA silencing and
compare their effectiveness compared to single stranded
RNA as well as duplex siRNA. These results will be pre-
sented.
REFERENCES
1. Chapman, K. B. & Boeke, J. D. Cell 1991, 65, 483-492.
2. Khalid, M. F., Damha, M. J., Shuman, S. & Schwer, B.
Nucleic Acids Res 2005, 33, 6349-6360.
3. Okamura, K., Hagen, J.W., Duan, H., Tyler, D.M., Lai,
E.C., Cell, 2007, 130, 89–100.
4. Ruby, J.G., Jan, C.H., Bartel, D.P. Nature 2007, 448,
83-86.
5. Kierzek, R., Kopp, D. W., Edmonds, M. & Caruthers,
M. H. Nucleic Acids Res. 1986, 14, 4751-4764.
6. Zhou, X.X., Nyilas, A., Chattopadhyaya, J. Nucleic
Acids Symp Ser. 1987, 18, 93-96.
7. Carriero, S. & Damha, M. J. Solid-Phase Synthesis of
Branched Oligonucleotides 2001 in Current Protocols
in Nucleic Acid Chemistry; John Wiley & Sons, Inc.
8. Wang, Y. M. & Silverman, S. K. J Am Chem Soc 2003,
125, 6880-6881.
156

Detection of DNA Modifications and Enzymatic Repair Activities Based on a Multiple Spin-Labelling Strategy coupled with PELDOR Analysis
Mélanie Flaender 1, Giuseppe Sicoli 1, Samia Aci-Seche 2, Thomas Reignier 1, Vincent Maurel 1, Christine Saint-Pierre 1, Yves Boulard 2, Serge Gambarelli 1, Didier Gasparutto 1,*
1 SCIB - UMR E3 CEA / UJF Grenoble 1, INAC, CEA Grenoble, 38054 Grenoble Cedex 9, France 2 SBIGEM - IBITEC, CEA Saclay, 91191 Gif-sur-Yvette Cedex, France
* Correspondence to: [email protected]
ABSTRACT
Spin-labeled oligonucleotides produced by click chemis-try can be studied by EPR, by using a DEER sequence. This was used to test a complex triple-labelling strategy with damaged DNA. Extensive and accurate analysis of DNA structure and enzymatic repair processes were per-formed after digestion by an AP-endonuclease.
INTRODUCTION
Pulsed ELDOR (pulsed electron–electron double reso-nance) is a method of choice to measure distances in various double spin-labelled biological systems [1, 2]. Recently, this method was used to study the structure of normal and modi-fied DNA.[3-9]. By developing a “click chemistry” ap-proach to efficiently labels oligonucleotides with high yields, it is therefore possible to develop more complex la-belling scenarii involving at least three spin-label positions. Through triangulation measurements, these could provide more information on the DNA structure and can be used to study DNA-protein interactions. RESULTS AND DISCUSSION
In the present work we demonstrate that it is possible to efficiently synthesize and study such a sophisticated and informative structural probe, to study specific DNA damage and its enzymatic repair [10]. The results obtained confirm that it is possible to prepare duplex DNA constructs contain-ing three spin labels and one abasic site type modification with close to normal secondary structure. The nitroxide la-bels introduced by a “click-chemistry”-based strategy do not prevent recognition of the abasic site analogue by DNA re-pair systems, such as the enzyme Endo IV, and have no ef-fect on its cleavage activity (Scheme). In addition, we were able to effectively measure individual distances and the pre-cise widths of the distributions in the triangle formed by the three spin-labels. The current spin-labelling approach is easy to implement and provides nitroxide-containing mole-cules with relatively narrow distance distributions, making it possible to develop complex labelling strategies for the de-tection of precise biological events. This demonstrates the potential of our approach for the study of modified DNA structures and DNA-protein interactions. Acknowledgements: This work was funded by the ANR (Blanc-0064-01).
REFERENCES
1. Schiemann, O., Prisner, T.F. Q. Rev. Biophys. 2007.
2. Jeschke, G., Polyhach, Y. Phys. Chem. Chem. Phys. 2007.
3. Schiemann, O., Piton, N., Mu, Y., Stock, G., Engels, J.W., Prisner, T.F. J. Am. Chem. Soc. 2004.
4. Sicoli, G., Mathis, G., Aci-Seche, S., Saint-Pierre, C., Boulard, Y., Gasparutto, D., Gambarelli, S. Nucleic Ac-ids Res. 2009.
5. Sicoli, G., Mathis, G., Delalande, O., Boulard, Y., Gas-parutto, D., Gambarelli, S. Angew. Chem. Int. Ed. 2008.
6. Schiemann, O., Cekan, P., Margraf, D., Prisner, T.F., Sigurdsson, S.T. Angew. Chem. Int. Ed. 2009.
7. Marko, A., Margraf, D., Cekan, P., Sigurdsson, S.T., Schiemann, O., Prisner, T.F. Phys. Rev. 2010.
8. Singh, V., Azarkh, M., Exner, T.E., Hartig, J.S., Drescher, M. Angew. Chem. Int. Ed. 2009.
9. Romainczyk, O., Endeward, B., Prisner, T.F., Engels, J.W. Mol. Biosyst. 2011.
10. Flaender, M., Sicoli, G., Aci-Seche, S., Reignier, T., Maurel, V., Saint-Pierre, C., Boulard, Y., Gambarelli, S., Gasparutto, D. ChemBioChem 2011.
Scheme. PELDOR analysis of DNA lesion excision using an original triple-spin labeled probe
157
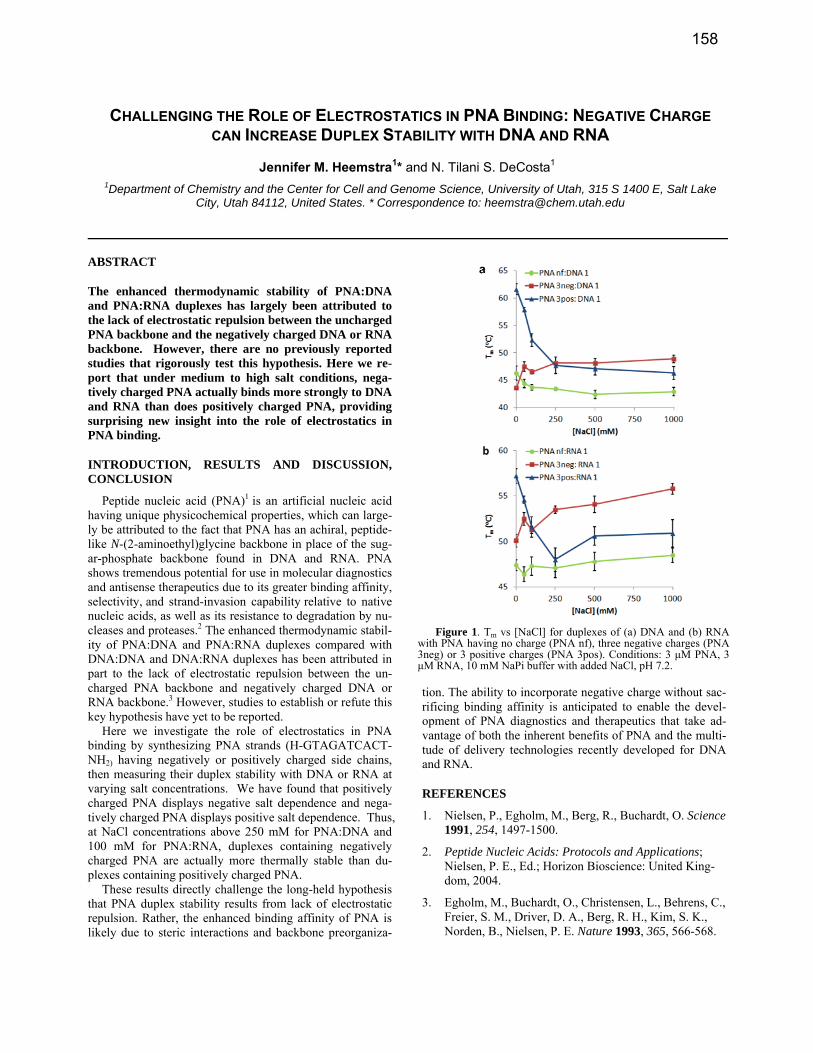
CHALLENGING THE ROLE OF ELECTROSTATICS IN PNA BINDING: NEGATIVE CHARGE CAN INCREASE DUPLEX STABILITY WITH DNA AND RNA
Jennifer M. Heemstra1* and N. Tilani S. DeCosta1
1Department of Chemistry and the Center for Cell and Genome Science, University of Utah, 315 S 1400 E, Salt Lake City, Utah 84112, United States. * Correspondence to: [email protected]
ABSTRACT
The enhanced thermodynamic stability of PNA:DNA and PNA:RNA duplexes has largely been attributed to the lack of electrostatic repulsion between the uncharged PNA backbone and the negatively charged DNA or RNA backbone. However, there are no previously reported studies that rigorously test this hypothesis. Here we re-port that under medium to high salt conditions, nega-tively charged PNA actually binds more strongly to DNA and RNA than does positively charged PNA, providing surprising new insight into the role of electrostatics in PNA binding.
INTRODUCTION, RESULTS AND DISCUSSION, CONCLUSION
Peptide nucleic acid (PNA)1 is an artificial nucleic acid having unique physicochemical properties, which can large-ly be attributed to the fact that PNA has an achiral, peptide-like N-(2-aminoethyl)glycine backbone in place of the sug-ar-phosphate backbone found in DNA and RNA. PNA shows tremendous potential for use in molecular diagnostics and antisense therapeutics due to its greater binding affinity, selectivity, and strand-invasion capability relative to native nucleic acids, as well as its resistance to degradation by nu-cleases and proteases.2 The enhanced thermodynamic stabil-ity of PNA:DNA and PNA:RNA duplexes compared with DNA:DNA and DNA:RNA duplexes has been attributed in part to the lack of electrostatic repulsion between the un-charged PNA backbone and negatively charged DNA or RNA backbone.3 However, studies to establish or refute this key hypothesis have yet to be reported.
Here we investigate the role of electrostatics in PNA binding by synthesizing PNA strands (H-GTAGATCACT-NH2) having negatively or positively charged side chains, then measuring their duplex stability with DNA or RNA at varying salt concentrations. We have found that positively charged PNA displays negative salt dependence and nega-tively charged PNA displays positive salt dependence. Thus, at NaCl concentrations above 250 mM for PNA:DNA and 100 mM for PNA:RNA, duplexes containing negatively charged PNA are actually more thermally stable than du-plexes containing positively charged PNA.
These results directly challenge the long-held hypothesis that PNA duplex stability results from lack of electrostatic repulsion. Rather, the enhanced binding affinity of PNA is likely due to steric interactions and backbone preorganiza-
tion. The ability to incorporate negative charge without sac-rificing binding affinity is anticipated to enable the devel-opment of PNA diagnostics and therapeutics that take ad-vantage of both the inherent benefits of PNA and the multi-tude of delivery technologies recently developed for DNA and RNA.
REFERENCES
1. Nielsen, P., Egholm, M., Berg, R., Buchardt, O. Science 1991, 254, 1497-1500.
2. Peptide Nucleic Acids: Protocols and Applications; Nielsen, P. E., Ed.; Horizon Bioscience: United King-dom, 2004.
3. Egholm, M., Buchardt, O., Christensen, L., Behrens, C., Freier, S. M., Driver, D. A., Berg, R. H., Kim, S. K., Norden, B., Nielsen, P. E. Nature 1993, 365, 566-568.
Figure 1. Tm vs [NaCl] for duplexes of (a) DNA and (b) RNA with PNA having no charge (PNA nf), three negative charges (PNA 3neg) or 3 positive charges (PNA 3pos). Conditions: 3 μM PNA, 3 μM RNA, 10 mM NaPi buffer with added NaCl, pH 7.2.
158

SYNTHESIS AND CHARACTERIZATION OF BRANCHED RNA THAT MIMIC LARIAT RNA
SPLICING INTERMEDIATES
Adam Katolik,1 Richard Johnsson,
1 David Sabatino,
1 Jeremy G. Lackey,
1 Eric Montemayor,
2
P. John Hart2 and Masad J. Damha
1*
1McGill University, 801 Sherbrooke St. W. Montréal, Québec H3A OB8,
2University of Texas Health Science Center,
7703 Floyd Curl Dr., San Antonio TX 78229. * Correspondence to: Email address [email protected]
ABSTRACT
We have developed new methods for the synthesis of
branched RNAs that mimic intronic lariat RNA inter-
mediates. These methods produce branched oligoribo-
nucleotide sequences of arbitrary length, base composi-
tion, and regiochemistry at the branch-point junction.
Lariat RNAs are intronic ‘lasso’-like structures whose 5′
ends are connected to an internal adenosine unit via a 2′,5′-
phosphodiester linkage. This linkage must be hydrolyzed by
the intron debranching enzyme (Dbr1) before a spliced in-
tron can be metabolized or processed into essential cellular
factors such as snoRNA and miRNA. Over the past several
years, our group has been synthesizing branched RNA
(bRNA) fragments as model systems for studying lariat
RNA land the structural basis of their recognition by dDbr1.
Much effort has been put forth to find a practical ways to
chemically synthesize bRNA via the solid-phase method.
Our first such approach utilized a 2′,3′-bis-O-
phosphoramidite to couple to two support-bound RNA
chains.[1] This method generated bRNAs with identical
sequences at the 2′ and 3′ positions of the branchpoint. Fol-
low up work focused on divergent methods that produced
branched RNA-DNA sequences of arbitrary base composi-
tion, length, and orientation around the branch-point junc-
tion.[2] This was accomplished by orthogonal deprotection
at an internal TBDMS group and growing the DNA se-
quence in the 5′-to-3′ direction using commercially available
reverse DNA amidites.[2a]
The present work is an extension of our ‘divergent’
method and produces branched oligonucleotides consisting
entirely of RNA. The major features of this strategy are the
use of (a) branching monomers (1-3) bearing 2′-ALE, 2′-DMTr, and 2′-Lev orthogonal protecting groups and b) ‘re-
verse’ RNA 2′-TBDMS 5′-phosphoramidites 4
(ChemGenes). The divergent, regiospecific methods for
constructing bRNA oligomers on controlled pore glass
(LCAA-CPG) are illustrated in Figure 1. Best results were
obtained when Method A and B were used in conjunction
with monomers 1 and 2, respectively. For example, when
Method A is used, the first arm (N4N3XN2N1) is constructed
in the 3′-to-5′ direction using commercially available 2′-TBDMS 3′-amidite monomers and branching synthon 1.
The cyanoethyl phosphate groups and 2′-ALE are removed
by treatment with (a) NEt3/MeCN at r.t. for 90 min; (b)
washing the support; and (c) treatment with 0.5 M hydrazine
hydrate, py/AcOH, r.t., 30 min. The second arm of the Y-
shape RNA is grown by adding a concentrated (0.3M) solu-
tion of 4 (N5) and continuing growing in the reverse 5’-to-3’
direction using 0.15M solutions of monomers 4.
Method C is analogous to that introduced in 1997 by Sproat
and co-workers [3], except that instead of their 2′-Lev, 3′-Pixyl 5′-amidite monomer [3], we employed the analogous 3
(i.e., 3′-DMTr instead of 3′-Pixyl). In our hands, removal
of the 3′-DMTr group causes 2-5% migration of the 3′-Lev
group to the vicinal 2′-OH position, resulting in the isolation
of the bRNA as regioisomeric mixtures. All bRNA se-
quences were characterized by MS and debranching assays
with Dbr1.
REFERENCES 1. Damha, M. J., Ganeshan, K., Hudson, R. H. Zabarylo S. V.
Nucleic Acids Research, 1992, 20(24), 6565-73.
2. (a) Braich, R.S., Damha, M.J. Bioconjugate Chemistry, 1997,
8(3), 370-377; (b) Damha, M.J. and Braich, R.S. Tetrahedron
Letters, 1998, 39, 3907-3910.
3. Grøtli, M., Eritja, R., Sproat, B, Tetrahedron, 1997, 53(33),
11317-11346.
159

Computing DNA oligonucleotides hybridization enthalpy within molecular dynamics modeling
A.A. Lomzov*, Y.N. Vorobjev, D.V. Pyshnyi
Institute of Chemical Biology and Fundamental Medicine SB RAS, Novosibirsk, Russia; *Correspondence to: [email protected]
Abstract: Development of new derivatives and analogues of nucleic acids (NA) and
reliable prediction of their physico-chemical properties is important both in practice and
basic research. Significant progress in development of software and hardware has made the
in silico research widely used. The goal of this work is to analyze an applicability of the
molecular dynamics (MD) modeling for calculating oligonucleotide hybridization enthalpy.
Methods and Algorithms: The enthalpies of DNA duplex formation were determined as a
difference of the internal energy of double- and single-stranded states which were
calculated from MD trajectory computed with Amber 11 software (UCSF, USA).
Computations were performed on NVIDIA GTX580 and Intel i7-2600 hardware.
Results: To determine optimal parameters of modeling we have used Dickerson-Drew
dodecamer (DDD) with well characterized secondary structure and thermal stability. We
have varied temperature, heating protocol, and ion concentration in implicit and explicit
solvent and compared averaged structures with those experimentally obtained. Using
optimal parameters of modeling we have shown that hybridization enthalpy of DDD
correlates well with experimental and calculated one via nearest neighbor model enthalpies.
The use of GPU has speeded up the modeling of DDD in implicit solvent up to 60 times
and up to 30 in explicit solvent in comparison with the single node CPU.
To verify the MD predictive ability we have collected database of experimentally
determined thermodynamic parameters (enthalpy, entropy and Gibbs energy) of
hybridization of 272 oligodeoxyribonucleotides. The total energy of oligonucleotide and its
complex were calculated from 2 ns trajectories simulated with optimal parameters. The
RMSD of calculated and experimental enthalpies was 15%.
Conclusion: The results obtained show that MD modeling allows one to calculate enthalpy
of matched DNA duplexes with high accuracy.
Availability: An extension of this work is retrieval of parameters of MD modeling for
more accurate prediction DNA duplex thermal stability, including complexes with
perturbation of the regular structure, that could be used instead experimental research.
This research has been supported by Integration grant SB RAS (86), RFBR (10-04-01492-
а) and by MCB programs of RAS.
160

RNA X AND THE IMPACT OF SECONDARY STRUCTURE ON THE STABILITY OF RNA
PHOSPHOTRIESTER LINKAGES
Tuomas Lönnberg1*
1Department of Chemistry, University of Turku, FIN-20014, Turku, Finland * Correspondence to: [email protected]
ABSTRACT
An oligonucleotide incorporating a branching RNA
phosphotriester linkage has been synthesized and its
hydrolysis studied in the presence of various partly
complementary oligonucleotides. Certain structural mo-
tifs stabilize the reactive phosphotriester by more than
two orders of magnitude, suggesting a half-life of several
hours for the so-called RNA X structure, proposed to be
formed during the splicing of mRNA.
INTRODUCTION
It has been suggested that two small nuclear RNAs, viz.
U2 and U6 snRNA of the human spliceosome, form a phos-
photriester structure (the so-called RNA X) upon attack of
the 2´-OH function of adenosine 21 of U2 snRNA on the
A53pG54 phosphodiester bond of U6 snRNA [1]. Given the
facile nucleophilic attack of the neighboring 2’-OH function
on the phosphotriester group of ribonucleoside 3´-
dialkylphosphates [2], this finding is rather unexpected. In
the proposed RNA X structure, however, the putative phos-
photriester linkage is part of a four-way junction motif that
might offer stabilization by orienting the flanking 2´-OH
unfavorably for nucleophilic attack (Fig. 1A). The extent of
such stabilization could be determined by studying the hy-
drolysis of a phosphate-branched RNA oligonucleotide
model in the presence of appropriately designed partly com-
plementary oligonucleotides (Fig. 1B-E).
RESULTS AND DISCUSSION
The oligonucleotide model incorporating a branching
3´,3´,5´-phosphotriester linkage was synthesized by manual
5-benzylthiotetrazole-promoted coupling of an appropriately
protected dimeric phosphoramidite building block to the
terminal 5´-OH of a CPG-bound oligonucleotide. Orthogo-
nal protections (DMTr and levulinoyl) were used in the 5´-
OH groups of the dimeric building block to allow synthesis
of the two branches independently. One of the 2´-OH
groups flanking the scissile phosphotriester linkage was
protected with a tert-butyldithiomethyl group, the other res-
idues being 2´-O-methylated.
Hydrolysis of the phosphate-branched oligonucleotide
model (Fig. 1B) was carried out at 25 °C in 50 mM tris(2-
carboxyethyl)phosphine buffer. Under these reducing condi-
tions, the disulphide bond of the tert-butyldithiomethyl pro-
tection is rapidly cleaved. Subsequent hydrolysis of the re-
sulting thiohemiacetal yields a free 2´-OH group, which
then attacks the phosphorus atom of the phosphotriester
linkage. Decomposition of the pentacoordinate phophorane
intermediate thus formed takes place by fission of the P-O5´
or one of the P-O3´ bonds and an order of magnitude slower
than previously reported for the respective trinucleoside
phosphotriester lacking oligonucleotide arms. Complete
hybridization of the main chain of the model with another
oligonucleotide (Fig. 1C) further stabilizes the phos-
photriester linkage by an order of magnitude and structures
placing this scissile linkage in a kinked position (Fig. 1D
and E) are even more stable, albeit only moderately.
CONCLUSION
Secondary structure can considerably stabilize an RNA
phosphotriester linkage, suggesting that the proposed RNA
X species could have a half-life of at least several hours
under physiological conditions.
REFERENCES
1. a) Valadkhan, S., Manley, J. L. Nature, 2001, 413, 701-
707; b) Valadkhan, S., Manley, J. L. RNA, 2003, 9, 892-
904.
2. a) Kosonen, M., Seppänen, R., Wichmann, O.,
Lönnberg, H. J. Chem. Soc., Perkin Trans. 2, 1999,
2433-2439; b) Mikkola, S., Kosonen, M., Lönnberg, H.
Curr. Org. Chem., 2002, 6, 523-538; c) Lönnberg, T.,
Kiiski, J., Mikkola, S. Org. Biomol. Chem., 2005, 3,
1089-1096.
Figure 1. A phosphotriester linkage within RNA X (A) and var-ious oligonucleotide model assemblies (B – E).
161

STRUCTURAL STUDIES ON FLUORINE MODIFIED NUCLEIC ACIDS
Nerea Martín-Pintado1, Maryam Yahyaee
2, Glen Deleavey
2, Anna Aviñó
3, Jonathan K. Watts
2, Ramón
Campos4, Guillem Portella
5, Modesto Orozco
5, Ramon Eritja
3, Masad Damha
2, and Carlos González
1*
1Instituto de Química Física Rocasolano, CSIC, C/ Serrano, 119, 28006 Madrid, Spain
2Department of Chemistry, McGill University, Montreal, QC, H3A 0B8, Canada
3Institute for Research in Biomedicine, IQAC-CSIC, CIBER-BBN Networking Centre on Bioengineering, Biomaterials
and Nanomedicine, E-08028 Barcelona, Spain 4Spectroscopy and NMR Unit, Structural and Computational Biology Programme, Spanish National Cancer Center
(CNIO), C. Melchor Fernández Almagro, 3, 28029, Madrid, Spain 5Joint IRB-BSC program on Computational Biology. Institute for Research in Biomedicine, Baldiri Reixac 10-12, E-
08028 Barcelona, Spain and Barcelona Supercomputing Center, Jordi Girona 29, 08034 Barcelona, Spain Department of Biochemistry. University of Barcelona. Diagonal 647, 08028 Barcelona, Spain
E-mail: [email protected]
ABSTRACT
In this communication we will present some of our
studies on fluorine-modified nucleic acids.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
Nucleic acids analogs containing 2’-fluoro-arabino (2’F-
ANA) and 2’-fluoro-ribose (2’F-RNA) are interesting
compounds for its potential applications in antisense and
interference RNA therapy. When 2’F-ANA hybridizes to
its complementary RNA, the resulting complex is a
substrate of the enzyme RNase H, which cleaves the
RNA strand of DNA/RNA hybrids, but is not active
against pure RNA duplexes. On the other hand, siRNAs
composed of combinations of 2’F-ANA and 2’F-RNA
can activate the RISC complex and elicit potent gene
silencing [1]. The favored conformations of these two
analogs are different, and combining 2’F-ANA and 2’F-
RNA led to reduce affinity relative to an RNA/RNA
duplex. Thus, the binding affinity at key regions of the
siRNA duplex could be tuned by changing the pattern of
incorporation of DNA-like and RNA-like nucleotides. By
combining 1H and
19F NMR spectroscopy, we have
determined the solution structure of several quimeric and
hybrid duplexes, which sequences combine different
patterns of 2’F-ANA and 2’F-RNA nucleotides [2]. In
this communication, we discuss the three-dimensional
structure of these duplexes, and compare them with the
structure of 2’F-ANA/RNA hybrids determined in a
previous study. We have also determined the structure of
a modified DNA/RNA hybrid duplex containing a 2’,2’-
difluoro-deoxycytidine nucleotide.
On the other hand, quadruplex structures have attracted
considerable attention during the last years. Chemical
modifications in this kind of structures help understand
their stability and structural properties and also have
potential applications in Biology and in Supramolecular
Chemistry. We will present here some of our recent NMR
studies on fluorine modified G-quadruplexes based on
telomeric sequences. Fluorine substitutions in the 2’-
position of the sugar (2’F-ANA and 2’F-RNA) stabilize
parallel propeller quadruplex conformations.
2′F-ANA 2′F-RNA
REFERENCES
1. Deleavey G. Watts J. Alain T. Robert F. Kalota A.
Aishwarya A. Pelletier J. Gewirt A. Sonenberg N.
Damha M. Nucleic Acids Research, 2010, 38, 4547-
4557
2. Watts JK, Martín-Pintado N, Gómez-Pinto I,
Schwartzentruber J, Portella G, Orozco M, Gonzalez
C and Damha MJ. Nucleic Acids Res, 2010, 38,
2498-2511.
162

SYNTHESIS OF MONO AND MULTIPLE CONJUGATED OLIGONUCLEOTIDES BY "CLICK
THIOL" CHEMISTRY AND COMBINATION WITH CUAAC "CLICK CHEMISTRY"
Albert Meyer, Jean-Jacques Vasseur and François Morvan*
Institut des Biomolécules Max Mousseron, UMR 5247 CNRS UM1 UM2, Université Montpellier 2, Place E. Bataillon, 34095 Montpellier cedex 5, France. *Correspondence to: [email protected]
ABSTRACT
Oligonucleotide conjugates were efficiently synthe-
sized by thiol click chemistry starting from a mono- or
poly-thiol oligonucleotides and different acrylamide de-
rivatives. This strategy was applied to form glycoclusters
and was also combined with CuAAC for bis-conjugation
through a sequential protocol.
INTRODUCTION
Oligonucleotide conjugates are widely used for various ap-
plications in biology, biotechnology, and medicine. Most
applications require labelling with dyes, redox tag or other
biomolecules such as biotin or carbohydrates.
We explored the use of thiol Michael-type addition (TMTA)
to prepare oligonucleotide conjugates. This addition corre-
sponds to the reaction of a thiol on an electron-deficient ene
forming a thioether linkage. This reaction is usually restrict-
ed to the reaction of thiol-oligonucleotides with maleimide
derivatives [1]. Herein we present the TMTA using acryla-
mide derivatives. Finally, TMTA was combined sequential-
ly with the Copper (I) catalyzed Azide Alkyne Cycloaddi-
tion (CuAAC) to synthesized bis conjugates.
RESULTS AND DISCUSSION
A 5'-S-acetyl-thiohexyl oligonucleotide was synthesized
using commercially available amidites on a DNA synthesiz-
er according to the phosphoramidite chemistry. Different
acrylamide derivatives exhibiting a phenyl, mannose, ferro-
cene, dansyl, biotin or deoxycholic moiety were prepared.
After the removal of cyanoethyl group by piperidine treat-
ment, the solid-supported thio-oligonucleotide was treated
with an acrylamide derivative in presence of TCEP and
K2CO3 methanol leading to the deprotection, release from
solid support and TMTA (Figure 1). Hence the monoconju-
gate oligonucleotide was obtained with a quantitative con-
version of the thiol-oligonucleotide. A size exclusion chro-
matography on cartridge allowed the isolation of almost
pure conjugate. The same strategy was applied to synthesize
a mannose-centered tetramannose oligonucleotide allowing
the formation of a glycocluster oligonucleotide conjugate
with high efficiency starting from a tetra-thiolhexyl oligo-
nucleotide.
Finally the TMTA was combined with the CuAAC ac-
cording to a sequentially protocol to synthesize bis-
conjugated oligonucleotides exhibiting biotin and carbohy-
drate, dansyl and mannose or mannose and galactose.
R
NH
NCH3H3C
SO
O
HN NH
O
S CO
HN
NHFe
HN
H3C H
CH3HO
HO
CH3
H
H
O
H
OHOHO
OH
O
HO
DNAOPO
OO
SNH
OR
DNAOPO
OO
AcSNH
OR
Thiol Click
OH
Figure 1. Structure of oligonucleotide conjugates synthesized
by "click thiol" chemistry
CONCLUSION
The TMTA is a very efficient click reaction to synthesize
oligonucleotides conjugated with various molecules. The
great advantages of this reaction are that it does not require
radical initiator and uv irradiation, and it occurs during the
deprotection and release of the oligonucleotide from solid
support affording the expected conjugates as the unique
molecule. TMTA could be applied to the synthesis of multi-
labelled oligonucleotides. Eventually, the sequential combi-
nation of the TMTA with the very popular CuAAC allowed
the synthesis of bis-conjugated oligonucleotides starting
either from the CuAAC and then TMTA or reverse.
REFERENCES 1. Singh, Y.; Murat, P.; Defrancq, E. Chem. Soc. Rev.
2010, 39, 2054-2070.
163

SYNTHESIS OF THE YPAA APTAMER BY CLICK LIGATION AND CONFORMATIONAL SWITCHING BY LIGAND SHAPE CONTROL
Maria Jenckel, Jennifer Frommer, Tamil Selvi Arunachalam, Bettina Appel and Sabine Müller*
Ernst Moritz Arndt Universität Greifswald, Institut für Biochemie, Felix-Hausdorff-Str. 4, 17487 Greifswald, Germany. *Correspondence to: [email protected]
ABSTRACT
We have investigated conformational switching in two
RNA systems: a small engineered hairpin ribozyme var-
iant and the ypaA aptamer of B. subtilis. Both RNAs are
responsive to flavine mononucleotide (FMN) and can be
conformationally controlled in dependence on the oxida-
tion state of FMN.
INTRODUCTION
Riboswitches play a very essential role in the regulation of gene expression. The FMN responsive riboswitch ypaA of B.
subtilis regulates expression of a gene encoding a riboflavin transport protein by effecting translation initiation. To study binding and folding of the ypaA aptamer domain, natural and specifically modified variants of the 129nts RNA were prepared in preparative scale using a combination of chemi-cal and enzymatic synthesis. A variety of methods was then used for binding and conformational analysis. Previously, we have developed a hairpin ribozyme variant responding to flavine mononucleotide (FMN) and several of its analogs [1-3]. FMN binds to the aptamer domain of the hairpin ribo-zyme derived aptazyme and stabilizes the catalytically com-petent conformation of the catalytic domain. Upon reduction, the molecular shape of FMN changes from planar to roof-like, leading to loss of its binding capacity, and subsequent-ly to down regulation of activity. Iterative cycles of oxida-tion/reduction allow for reversible switching of activity [2]. We have now used the established principle of FMN in-duced switching between two alternative RNA confor-mations with the ypaA aptamer.
RESULTS AND DISCUSSION
The aptamer domain of the ypaA riboswitch is a highly structured RNA, making its chemical preparation rather challenging. We started with the synthesis of aptamer frag-ments to be joined by enzymatic ligation. Screening several protocols, we succeeded with T4 RNA-Ligase splint ligation. Alternatively, ypaA aptamer variants were prepared by Click ligation using specifically functionalized RNA frag-ments (Figure 1). Folding of the ypaA variants was investi-gated in comparison to the natural aptamer by a number of methods. In particular, we used gelelectrophoretic gel mo-bility assays, CD and fluorescence spectroscopy and te-rahertz spectroscopy. Initial results indicate that the confor-mation of the ypaA riboswitch can be controlled by the oxi-
dation state of its ligand, analogous to the small engineered hairpin ribozyme variant described previously [1].
Figure 1. Click ligation used for generation of the ypaA ap-tamer.
Terahertz spectroscopy was carried out in collaboration
with the laboratory of Martina Havenith at Ruhr University Bochum, and showed a clear conformational transition of the RNA upon addition of FMN, associated with a change of water network dynamics. CD spectra of the aptamer rec-orded in the presence and absence of FMN as well as gel mobility analyses also clearly indicated FMN responsive conformational transitions. Furthermore, based on our pre-vious work on electrochemical reduction/oxidation of FMN, we have set up a new electrochemical assay involving im-mobilisation of the ligand on the electrode, to follow bind-ing and FMN dependent conformational switching.
CONCLUSION
Click-ligation was shown being an appropriate method for generation of functional ypaA aptamer variants. This allowed us to study FMN binding and conformational switching by a variety of methods, amongst them a novel electrochemical assay. The conformation of the aptamer can be controlled by changing the FMN oxidation state.
REFERENCES
1. Strohbach, D., Novak, N., Müller, S., Angew. Chem. Int.
Ed., 2006, 45, 2127-2129.
2. Strohbach, D., Turku, F., Schuhmann, W., Müller, S., Electroanalysis, 2008, 20, 935-940.
3. Müller, S., Appel, B., Krellenberg, T., Petkovic, S., IUBMB Life, 2012, 64, 34-47.
164

SYNTHESIS OF THE LIGHT-DRIVEN MOLECULAR MOTORS CONJUGATED WITH PEPTIDE
AND EVALUATION OF THE DNA BINDING PROPERTIES
Fumi Nagatsugi1,* Yusuke Takahashi
1, Maiko Kobayashi
1 and Shunsuke Kuwahara
2
1Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai-shi, Miyagi, 980-8577, Japan.
2Graduate School of Science Toho University, 2-2-1 Miyama ,Funabashi-shi ,Chiba, 274-
8510 * Correspondence to: Email address for [email protected]
ABSTRACT
Synthetic light-driven molecular motors are molecular
machine capable of rotation by photo-irradiation. In this
presentation, we wish to report the synthesis of the mo-
lecular motors conjugated with peptide and the evalua-
tion of the DNA binding properties.
INTRODUCTION
Synthetic molecular motors are one of an attractive mo-
lecular machine, which is capable of rotation under energy.
A variety of molecular motor systems have been designed in
past 10 years in which changes in shape, switching process-
es, or movements occur in response to external chemical,
electrochemical, or photochemical stimuli. But the applica-
tion of molecular motor in the movement of larger-scale
objects is limited and the useful functions of the molecular
motor have not been established. In this presentation, we
wish to report the development of the molecular motor,
which has the possibility to bind DNA duplex. DNA bind-
ing molecules have significant potential applications in ge-
nomic studies. Recently, the dynamic higher ordered DNA
structural change can play important roles in many cellular
processes. The small molecules inducing the structural
change of DNA have the potential to control the gene ex-
pression. We consider that the photo-driven molecular mo-
tor to bind DNA duplex has the potential to lead the struc-
tural change by photo irradiation. We have designed the
peptide-conjugated molecular motor, developed by Feringa 1and Harda
2, which can rotate in one rotational direction
through conversion of light energy into motor rotation
RESULTS AND DISCUSSION, CONCLUSION
The peptide conjugated molecular motors were synthesized
by using click chemistry between the molecular motor (1)
and two kinds of peptide derivatives (2) in good yields
(Scheme 1).
Trans-molecular motor (3a) and (4a) were rotated to cis-
olefin (3b) and (4b) by irradiated with UV light at 375 nm
for 15 s and heated at 37 ºC for overnight. Cis-molecular
motor (3b) and (4b) were also rotated to trans-olefin (3a)
and (3b) by irradiated with UV light at 415 nm for 15 s and
heated at 37 ºC for overnight. Binding affinity of the molec-
ular motors with duplex DNA was estimated by the ethidi-
um bromide displacement assay. Table 1 summarizes the
binding affinity of molecular motors (3) and (4) to duplex
DNA.
Peptide-conjugated trans-molecular motors (3a) and (4a)
showed lower C50 values toward all sequences of DNA than
those of cis-molecular motors (3b) and (4b). These results
have indicated that the binding affinity of trans-molecular
motor might be higher than that of cis-molecular motor.
In conclusion, we have successfully developed the molec-
ular motors to bind duplex DNA. We would like to present
these results in detail.
REFERENCES 1. ter Wiel, M. K. J.; van Delden, R. A.; Meetsma, A.; Feringa,
B. L., J. Am. Chem. Soc. 2003, 125, 15076-15086.
2. Fujita, T.; Kuwahara, S.; Harada, N., Eur. J. Org. Chem.,
2005, 4544-4556.
hn (312 nm)
hn (430 nm)
hn (330 nm)
D
hn (430 nm)
D
Fig.1. Light powered molecular motor
Scheme 1. Synthesis of the peptide conjugated molecular
motor
N3
N3
sodium ascorbate,
aq. CuSO4
DMF / H2O
N
N
N
N
N
N
(H2C)5
(CH2 )5
R
O
R
O
R (CH2)n
O
3a: trans R = KAKAK3b: cis R = KAKAK4a: trans R = PRGRP4b: cis R = PRGRP
1
2N
N
NN
N
N(CH2)3(H2C)3
RO
RO
hv (375 nm)and
hv (415 nm)and2a: R=KAKAK
2b: R=PRGRP
C50 (mM)3 (KAKAK) 4 (PRGEP)
PRGRP KAKAKtrans cis trans cis
DNA1 2.9 12.9 4.1 19.7 65.6 >100DNA2 2.3 5.6 1.7 10 >100 >100DNA3 3.1 4.9 2.7 8.2 >100 >100
1 mM DNA and 6 mM ethidium bromide in 5 mM cacodylate buffer containing 100 mM NaCl at pH 7.0
DNA3:5’CGCGCGCGCGCG3’
3’GCGCGCGCGCGC5’DNA1:5’CGCGAATTCGCG3’
3’GCGCTTAAGCGC5’
DNA2:5’CGATCGCGATCG3’
3’GCTAGCGCTAGC5’
Table 1. Binding affinity of molecular motors (C50, mM)
165

STUDY TOWARDS SYNTHESIS OF OLIGONUCLEOSIDES CONTAINING M1AA-DA, THE CONJUGATE MALONALDEHYDE-ACETALDEHYDE ADDUCT OF 2'-DEOXYADENOSINE
Donata Pluskota-Karwatka,* Kinga Salus and Henryk Koroniak
Adam Mickiewicz University, Faculty of Chemistry, Grunwaldzka 6, 60-780 Poznań, Poland. * Correspondence to: [email protected]
ABSTRACT
The methodology for preparation of oligonucleosides
containing M1AA-dA, the conjugate malonaldehyde-
-acetaldehyde adduct of 2'-deoxyadenosine has been
investigated. The synthetic strategy includes assembly
of oligonucleotide from 2-deoxyadenosine modified
by the malonaldehyde-acetaldehyde conjugate. The key
features for preparation of this derivative include
synthesis of the adenine adduct and then
transribosylation by using nucleoside 2-
deoxyribotransferase.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
Endogenous, metabolic processes are sources of highly
reactive chemicals that are responsible for DNA damage.
Lipid peroxidation induced by so-called reactive oxygen
species (ROS), results in generating bifunctional
electrophiles, mainly carbonyl compounds and epoxides that
exhibit a wide range of biological activities. These
electrophiles can damage DNA by the reaction at electron
rich sites of nucleobases leading to the formation
of exocyclic adducts. Beside transformations caused directly
by ROS, exocyclic adducts are the most frequent reason
of DNA damage. For understanding of these biological
implications it is essential to synthesise oligonucleotides
containing the modified nucleosides placed in a structurally
specific manner.
One of the major carbonyl products of lipid peroxidation
is malonaldehyde. This aldehyde is known to form adducts
with the DNA bases. Two strategies have been reported for
preparation of oligonucleotides containing
the malonaldehyde adducts: assembly of oligonucleotides
from modified nucleosides and postoligomerisation
(postsynthetic modification strategy) [1, 2].
Our previous studies resulted in the identification
of a series of DNA adducts of malonaldehyde formed in the
presence of acetaldehyde [3]. These adducts consist of units
derived from malonaldehyde-acetaldehyde conjugates
and are formed in the reactions of DNA bases with
the appropriate malonaldehyde-acetaldehyde condensation
products (Scheme 1). The deoxyadenosine adduct M1AA-
dA, is one of the main products formed when calf thymus
DNA is allowed to react simultaneously with
malonaldehyde and acetaldehyde under physiological pH
and temperature [3].
This study deals with the developing of the strategy
leading to the synthesis of oligonucleotides containing
the propenoformyl adduct M1AA-dA.
O
O O
O
CH3
M1AA
M2AA
N
N
N
H3C
O
RO
N
N N
N
N
CH3
OO
dR
N
N N
N
N
dR
H3C
O
M1AA-dA M1AA-Cyd
N
N
N
CH3
O O
OR
N
N
N N
NO
O
CH3
dR
O
M2AA-dGuo I
HN
N N
N
N
O
O
H3C
O
dR
M2AA-dGuo II
M2AA-dA M2AA-Cyd
dG
O O
H3C H
dA, C
dA, C
O O
O O
H3C H
O+
Scheme 1. Formation of malonaldehyde-acetaldehyde
conjugate adducts
This study has been financially supported by National Centre
of Science, projects No N N204 433340.
REFERENCES
1. Schnetz-Boutaud, N. C., Mao, H., Stone, M. P., Marnett
L. J. Chem. Res. Toxicol. 2000, 13, 90-95.
2. Wang, H., Kozekov, I. D., Kozekova, A., Tamura, P. J.,
Marnett, L. J., Harris, T. M., Rizzo, C. J. Chem. Res.
Toxicol. 2006, 19, 1467-1474.
3. Pluskota-Karwatka, D., Pawłowicz, A. J., Kronberg, L.
Chem. Res. Toxicol. 2006, 19, 921-926.
166

SYNTHESIS AND PROPERTIES OF MODIFIED DNA OLIGONUCLEOTIDES WITH A
ZWITTERIONIC BACKBONE STRUCTURE
Boris Schmidtgall,1* Claudia Höbartner
2 and Christian Ducho
1
1University of Paderborn, Department of Chemistry, Warburger Str. 100, 33 098 Paderborn, Germany and
2MPI for
Biophysical Chemistry, Am Faßberg 11, 37 077 Göttingen, Germany. *Correspondence to: [email protected]
ABSTRACT
The site-specific replacement of the natural phospho-
diester linkage by the positively charged NAA (nucleosyl
amino acid) motif within oligonucleotides has been
performed, thus yielding DNA analogues with a
(partially) zwitterionic backbone structure. The
synthesis of such NAA-modified oligonucleotides and
studies on their properties are presented.
INTRODUCTION
Despite extensive studies on the structure and function of
nucleic acids there are still many open questions to consider.
Interestingly, it has been proposed that only uniformly
charged polymers are suitable for the formation of self-
sustaining systems [1,2]. However, there is only limited
experimental proof for this hypothesis. We have therefore
designed oligonucleotides (ON) with zwitterionic backbone
structures (Figure 1) which were inspired by the nucleoside
core motif of naturally occurring muraymycin antibiotics [3]
and termed as NAA (nucleosyl amino acid)-modified ON.
RESULTS AND DISCUSSION, CONCLUSION
For the solid phase-supported synthesis of ON bearing
the NAA-modification, dimeric phosphoramidite building
blocks (6'S)-1 and (6'R)-1 (Figure 2) were used. These
compounds were synthesised via peptide coupling of
3'-aminothymidine derivative 2 with diastereomerically pure
(6’S)- or (6’R)-nucleosyl amino acids (NAA) 3 and
subsequent manipulations of the protecting group pattern.
The highly diastereoselective synthesis of NAA structures
of type 3 has been developed and reported by us [4,5].
Using the phosphoramidite reagents (6’S)-1 and (6’R)-1,
several ON (20 to 22-mer) with mixed sequences were
synthesized with varying positions and numbers of
modifications within the backbone. Melting experiments
were perfomed to assess the thermal stability of DNA/DNA-
and DNA/RNA-duplices having one NAA-modified DNA
strand. Mismatch recognition of the NAA-modified ON was
also surveyed.
The melting point experiments indicated only a slight
decrease in thermal stability (ca. 0 to max. -2.2°C per NAA-
modification for DNA/DNA systems) and no significant
harmful influence on mismatch recognition. No structural
distortion of NAA-modified duplices was observed by CD-
spectroscopy. Consequently, our results do not support the
assumption of the uniform charge pattern to be necessary for
replicable biopolymers as genetic molecules and thus
challenge this hypothesis.
REFERENCES
1. Westheimer, F. Science, 1987, 235, 1173-1178.
2. Benner, S. A., Hutter, D. Bioorg. Chem., 2002, 30, 62-
80.
3. McDonald, L. A., Barbieri, L. R., Carter, G. T., Lenoy,
E., Lotvin, J., Petersen, P. J., Siegel, M. M., Singh, G.,
Williamson, R. T. J. Am. Chem. Soc., 2002, 124,
10260-10261.
4. Spork, A. P., Ducho, C. Org. Biomol. Chem., 2010, 8,
2323-2326.
5. Spork, A. P., Wiegmann, D., Granitzka, M., Stalke, D.,
Ducho, C. J. Org. Chem., 2011, 76, 10083-10098.
O
O
OB
O
O
BOP
OO
O
HN
OB
O
O
BO
H3N6'
* 5'5'
B = nucleobase
NAA-modified oligonucleotide
(6'S) or (6'R)
DNA
Figure 1. Native DNA and NAA-modified oligonucleotides.
Figure 2. Synthesis of dimeric phosphoramidites 1 for the stereocontrolled preparation of NAA-modified oligonucleotides.
167

BENZOPHENONE MODIFIED DNA FOR PHOTOCATALYSIS
Michael J. Weinberger and Hans-Achim Wagenknecht*
Karlsruhe Institute of Technology (KIT), Institute for Organic Chemistry, Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany * Correspondence to: [email protected]
ABSTRACT
A Benzophenone – C – Nucleoside was synthesized
and incorporated into DNA to serve as a photosensitizer.
This modification allows the usage of light for chemical
reactions in structurally defined chiral vicinity.
INTRODUCTION
Using light and direct it in a certain way to a chemical
reaction via a photocatalyst combines demands of sustaina-
ble chemistry as well of modern catalysis. Moreover by add-
ing chiral information to the catalyst high enantioselectivi-
ties can be achieved. Benzophenone has already been ap-
plied successfully as a sensitizer for organocatalytic photo-
reactions by Bach and co-workers [1]. On the other hand
Feringa et al. [2] showed that DNA is a suitable source of
chiral information. We are now combining those two con-
cepts to build up DNA architectures with sensitizing abili-
ties. Therefore we synthesized the benzophenone C-
Nucleoside 1 and incorporated it into synthetic DNA. After
incorporation of 1, ss- and ds-DNA showed the benzophe-
none phosphorescence pattern [3]. Potentially we could use
this triplett energy to photocatalyse organic reactions, e.g.
[2+2]-cycloadditions or electron transfer induced radical
reactions.
RESULTS AND DISCUSSION
The key step during the synthetic route to 1 is the C-C-
coupling of a benzophenone derivative and Hoffer’s
Chlorosugar, which was performed in a Grignard reaction.
After deprotection to obtain 1 the following steps to the
DNA building block were standard phosphoramidite chem-
istry.
DNA modified with 1 clearly showed the benzophenone
phosphorescence pattern. These measurements gave the
evidence for the benzophenone triplet state in 1 and even in
DNA. To check for catalytic activity we chose the well-
known intramolecular [2+2]-cycloaddition of 4-(3-
butenyloxy)quinolone-2(1H)-one 2 [1,4-5].
The performed experiments using benzophenone or 1 in
- or -configuration as a catalyst worked efficient and
identical. This proofs that the modification of benzophenone
doesn’t affect the photophysical properties too much. But
before the modified Oligonucleotide itself can be used as an
efficient catalyst, it should also have the ability to bind the
substrate, thus to act as an aptamer.
To find a suitable aptamer sequence we are currently per-
forming SELEX on a random sequence 30-mer. We used
the quinolone derivative 5, where the carboxylic acid is
used as an anchor on oxirane modified polyacrylamide
beads.
After finding some aptamers targeting the quinolone
moiety, these sequences have to be modified by 1 and
checked for their ability to catalyse the benchmark reaction
of 2.
REFERENCES
1. Bauer, A., Wetkämper, F., Grimme, S., Bach, T. Nature,
2005, 436, 1139-1140.
2. Roelfes, G., Feringa, B. L. Angew. Chem. Int. Ed., 2005,
44, 3230-3232.
3. Weinberger, M., Wagenknecht, H.-A. Synthesis, 2012,
44, 648-652.
4. Kaneko, C., Suzuki, T., Sato, M., Naito, T. Chem.
Pharm. Bull., 1987, 35(1), 112-123.
5. Cauble, D., Lynch, V., Krische, M. JOC, 2003, 68, 15-
21.
168

COMPUTER MODELING OF THE CONFORMATIONAL PREFERENCES OF DAMAGED DNA
Stacey D. Wetmore*
Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive W, Lethbridge, Alberta, Canada *Correspondence to: [email protected]
ABSTRACT
Techniques in computational chemistry and models
ranging from damaged nucleobases to DNA helices pro-
vide an understanding of the conformational preferences
of C-linked deoxyguanosine phenolic adducts in duplex
environments. Through close collaborations with exper-
iments, conclusions can be drawn about the potential
biological effects of these lesions.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
The integrity of the genetic code is constantly threatened
by endogenous and exogenous sources that alter the chemi-
cal structure of DNA. Although many environmental con-
taminants have been identified as possible genotoxic agents
that covalently attach aryl groups to the DNA nucleobases,
little is known about the so-called bulky DNA “adducts”
(addition products). Furthermore, it appears to be difficult to
determine the exact mechanism of toxicity of these chemi-
cals, which is at least in part because of proposals that mul-
tiple mutations are induced when a single adduct adopts a
variety of conformations. In addition, the preferred confor-
mation of bulky adducts is dependent on a number of factors,
including adduct type, site of attachment, flanking bases,
complementary base, and environment (replication fork,
polymerase active site, etc.).
Without sufficient evidence that an environmental con-
taminant is a genotoxin, such as proof of a defined DNA
adduct structure, legislations that limit the extent of expo-
sure to humans via dietary intake, water, personal products,
etc., are significantly relaxed. Thus, studies of the exact
structure of DNA damage products and the possible muta-
genic mechanisms of compounds of potential risk are im-
portant for influencing legislative attitudes, as well as de-
veloping preventive and therapeutic measures for combating
potential carcinogenicity. As a result, we developed a sys-
tematic and graded computational approach to predict the
conformational and base-pairing preferences of the C-linked
phenolic 2-deoxyguanosine adducts (Figure 1). Specifically,
the nucleobase and nucleoside adducts are initially consid-
ered [1], and other factors are gradually incorporated into
the model including the backbone [2], complementary base
[3], flanking bases [4], and duplex environment [5].
Initial results from highly-accurate quantum chemical
calculations on small (nucleobase, nucleoside) models [1]
not only match the experimentally-predicted structure of the
adducts in a variety of solvents, but also explain observa-
tions regarding differences in orientations of the glycosidic
bond depending on the adduct formed. Although these stud-
ies predict a preference for the syn orientation about the
glycosidic bond, subsequent computational work considered
small models that are more relevant to B-DNA. For example,
calculations including the 5-monophosphate predict a very
small anti/syn energy difference [2], which suggests that a
complex conformational heterogeneity likely occurs in
DNA helices.
Using information gained by studying the preferred hy-
drogen-bonding patterns [3] and the effects of stacking with
the flanking nucleobases on the conformational preference
[4], the phenolic adducts were incoroprated into two strand
sequences, which differ in the nature of the bases flanking
the adduct. These sequences were studied both experimen-
tally and with molecular dynamics (MD) simulations [5].
Although experiments alone could not determine the con-
formation adopted by the adducts in DNA, the measured
melting temperatures combined with the calculated stabili-
ties of MD structures support a syn preference regardless of
the sequence or opposing base. This story highlights the
unique role computer calculations can play in elucidating
the structural preference of DNA damage when implement-
ed in conjunction with experiments, as well as unveiling the
biological implications of these lesions.
REFERENCES
1. Millen, A.L., Mclaughlin, C.K., Sun, K.M., Mander-
ville, R.A., Wetmore, S.D. J. Phys. Chem. A, 2008 112,
3742-3753 (2008).
2. Millen, A.L., Manderville, R.A., Wetmore, S.D. J. Phys.
Chem. B 2010, 114, 4373-4382.
3. Millen, A.L., Churchill, C.D.M., Manderville, R.A.,
Wetmore, S.D. J. Phys. Chem. B 2010, 114, 12995-
13004.
4. Millen, A.L., Kamenz, B.L., Leavens, F.M.V., Mander-
ville, R.A., Wetmore, S.D. J. Phys. Chem. B 2011, 115,
12993-13002.
5. Omumi, A., Millen, A.L., Wetmore, S.D., Manderville,
R.A. Chem. Res. Toxicol. 2011, 24, 1694-1709.
Figure 1. C-linked deoxyguanosine phenolic adducts.
169

COMPUTER MODELING OF DNA REPAIR ENZYMES: APPLICATIONS TO THE DNA GLYCOSYLASES
Stacey D. Wetmore*
Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive W, Lethbridge, Alberta, Canada *Correspondence to: [email protected]
ABSTRACT
A combined computational approach applied to DNA–
protein models can provide a greater understanding of
the mechanism of action of DNA repair enzymes. The
present contribution emphasizes the importance of such
applications by considering the DNA glycosylases, the
first enzymes involved in the base excision repair process.
INTRODUCTION, RESULTS AND DISCUSSION,
CONCLUSION
Although DNA can be damaged by a variety of sources,
intricate repair processes combat detrimental nucleobase
lesions. For example, the base excision repair (BER) pro-
cess uses several enzymes to identify and remove damaged
nucleobases from the DNA helix [1]. The first BER en-
zymes, the DNA glycosylases, are particularly intriguing
since they catalyse the cleavage of the inherently stable
base–sugar (glycosidic) bond, and some glycosylases are
bifunctional (exhibit -lyase activity).
Despite common themes in the proposed catalytic path-
ways for all glycosylases, conflicting conclusions and many
questions remain about the mechanism of events. Calcula-
tions can significantly contribute to this field since most
information has been conjectured from mutagenesis experi-
ments or crystal structures. The present contribution high-
lights recent work in the Wetmore group that combines
highly accurate quantum mechanical (QM) technqiues,
which describe reactions occuring in the active site, and
computationally efficient methodologies (MM, SE), which
treat the remainder of the DNA–enzyme complex.
Initial work investigated the effects of different factors
(nucleophile activation, substrate interactions) on the overall
barrier to glycosidic bond cleavage catalyzed by hUNG2 [2],
the glycosylase responsible for removing uracil (deaminated
cytosine) from DNA. A truncated active-site model (all res-
idues within approximately 10 Å of the substrate, Figure 1)
was developed for this most widely studied glycosylase,
which is justified by an abundance of reliable, high-
resolution crystal structures representing several steps of the
proposed reaction scheme. In this model, the substrate and
most important active-site residues were treated with DFT,
while the remaining atoms were treated with a lower-level
semi-empirical (SE) method. Using this ONIOM approach,
the relative energy was plotted as a function of the base–
sugar and nucleophile–sugar distances to identify the most
feasible reaction pathway. For the first time, a catalytic
pathway consistent with all current experimental data was
characterized. Furthermore, a modified mechanism was
proposed that explains a previously unresolved experimental
observation, and clarifies the identity and role of important
active site amino acid residues (including His268).
In contrast to hUNG2, the active site of the human en-
zyme responsible for repairing alkylation damage (AAG) is
lined solely with aromatic amino acids. Due to the low spec-
ificity of this enzyme and the lack of experimental crystal
structures that span the entire reaction and diversity of sub-
strates, the AAG catalytic mechanism was studied using a
computational model of the entire enzyme–DNA complex
(Figure 1) with neutral (ethanoadenine) and cationic (3-
methyladenine) lesions, as well as the natural A base [3].
The large size of this model requires an ONIOM approach
that uses DFT to treat the substrate and active site residues
and MM to treat the remainder of the system. This approach
accurately reproduces a recent experimental activation bar-
rier for the chemical step, and reveals important mechanistic
information including the (concerted) pathway used to ex-
cise both neutral and cationic lesions, and the dependence of
the role (catalytic bond cleavage or enhanced binding)
played by active-site aromatic amino acids on the lesion
being repaired. A novel explanation for why the natural A
base is not excised (poor nucleophilic activation) was also
developed, which suggests this enzyme may use the chemi-
cal step as a checkpoint to discriminate against natural pu-
rines.
REFERENCES 1. Berti, P. J., McCann, J. A. B. Chem. Rev. 2006, 106,
506-555.
2. Przybylski, J. L., Wetmore, S. D. Biochemistry, 2011,
50, 4218-4227.
3. Rutledge, L. R., Wetmore, S.D. J. Am. Chem. Soc. 2011,
133, 16258-16269.
Figure 1. ONIOM model for hUNG2 (left, DFT region in tube and SE region in wire frame) and AAG (right, DFT region dark blue and MM region in orange (mobile) and light green (fixed)).
170


















