
Recognition of fold and sugar linkage for glycosyltransferases by multivariate sequence analysis
42
Recognition of fold and sugar linkage for glycosyltransferases by multivariate sequence analysis Maria L. Rosén 1 , Maria Edman 1,2 , Michael Sjöström 2 , and Åke Wieslander 1 * From the 1 Department of Biochemistry & Biophysics, Stockholm University, SE 106 91 Stockholm, Sweden, and the 2 Department of Chemistry, Organic Chemistry, Research Group for Chemometrics, Umeå University, SE 901 87 Umeå, Sweden Running title: Fold classification by multivariate sequence analysis *Corresponding author. Phone: +46-8-16 24 63 Fax: +46-8-15 36 79 E-mail: [email protected] Full address: Department of Biochemistry & Biophysics, Stockholm University, SE 106 91 Stockholm, Sweden JBC Papers in Press. Published on May 17, 2004 as Manuscript M402925200 Copyright 2004 by The American Society for Biochemistry and Molecular Biology, Inc. by guest on January 10, 2019 http://www.jbc.org/ Downloaded from
Transcript of Recognition of fold and sugar linkage for glycosyltransferases by multivariate sequence analysis

Recognition of fold and sugar linkage for glycosyltransferases by multivariate sequence
analysis
Maria L. Rosén1, Maria Edman1,2, Michael Sjöström2, and Åke Wieslander1*
From the 1 Department of Biochemistry & Biophysics, Stockholm University,
SE 106 91 Stockholm, Sweden, and the 2 Department of Chemistry, Organic Chemistry,
Research Group for Chemometrics, Umeå University, SE 901 87 Umeå, Sweden
Running title: Fold classification by multivariate sequence analysis
*Corresponding author. Phone: +46-8-16 24 63 Fax: +46-8-15 36 79
E-mail: [email protected]
Full address:
Department of Biochemistry & Biophysics, Stockholm University,
SE 106 91 Stockholm, Sweden
JBC Papers in Press. Published on May 17, 2004 as Manuscript M402925200
Copyright 2004 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

2
SUMMARY
Glycosyltransferases (GTs) are among the largest groups of enzymes found and are usually
classified on basis of sequence comparisons into many families of varying similarity (CAZy
systematics). Only two different Rossman-like folds have been detected (GT-A and GT-B)
within the small number of established crystal structures. A third uncharacterized fold has
been indicated with transmembrane organisation (GT-C). We here use a method based on
multivariate data analyses (MVDA) of property patterns in amino acid sequences, and can
with high accuracy recognise the correct fold in a large data set of GTs. Likewise, a retaining
or inverting enzymatic mechanism for attachment of the donor sugar could be properly
revealed in the GT-A and GT-B fold group sequences by such analyses. Sequence alignments
could be correlated to important variables in MVDA, and the separating amino acid positions
could be mapped over the active sites. These seem to be localised to similar positions in space
for the α/β/α binding motifs in the GT-B fold group structures. Analogous, active-site
sequence positions were found for the GT-A fold group. Multivariate property patterns could
also easily group most GTs annotated in the genomes of Escherichia coli and Synechocystis to
proper fold or organisation group, according to benchmarking comparisons at the MetaServer.
We conclude that the sequence property patterns revealed by the multivariate analyses seem
more conserved than amino acid types for these GT groups, and these patterns are also
conserved in the structures. Such patterns may also potentially define substrate preferences.
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

3
INTRODUCTION
Glycosyltransferases (GTs) are one of the largest and most diverse enzyme groups in all
living cells. This enzyme group performs many critical functions such as the synthesis of
glycogen, and carbohydrate-polymers, they act on proteins that mediate cell-cell interactions
and glycosylate transcription regulators (1). Hence, the variety of acceptors that GTs act on is
highly diverse, with saccharides, lipids, proteins, and nucleic acid as the most common. To
reflect the structural variation of the acceptors and donators that glycosyltransferases can use,
and the fact that the sequence similarity within the GTases is low, a large diversity of folds of
these enzymes have been expected (2). Glycosylhydrolases, enzymes performing the reverse
reaction have been found to have many different fold types (2), but so far only two different
folds have been discovered within the solved crystal structures for the GTs, named GT-A and
GT-B respectively (3). A third glycosyltransferase group (GT-C) has been discovered by
iterative BLAST searches and by structural comparisons (4). These proteins are integral
membrane proteins with the active site in the long loop, and with the transmembrane helix
number varying between 8 to 13. The GT-C family can also be found with Hidden Markov
method searches within the GT families. This method has also identified a fourth family,
unique for eukaryotes, named GT-D (5).
The GT-A fold consists of two tightly associated β/α/β domains, of varying sizes, with
separated nucleotide (SGC) and acceptor binding domains (6). The majority of the proteins in
this fold group have a short N-terminal cytoplasmic domain followed by a transmembrane
(TM) segment, a stem region to reach out from the membrane, and finally the large globular
enzyme part (3). The GT-B group has two similar, but less tightly associated Rossman-like
β/α/β fold domains, and are frequently membrane associated (7). However, only a very
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

4
limited number of proteins from the GT-B group seem to have TM segments. The GT-A
group presently consists of nine solved crystal structures from sequence families GT-2, GT-6,
GT-7, GT-8, GT-13, and GT-43, and the GT-B group of seven structures from GT-1, GT-4,
GT-20, GT-28, GT-35, GT-63, and GT-64, respectively, in the CAZy systematics sequence
database (http://afmb.cnrs-mrs.fr/~cazy/CAZY/index.html). Reaction mechanisms of both
retaining and inverting types are included in both A and B fold groups (denoted clans). Within
the GT-A fold family with the retaining mechanism, the stereochemistry of the C1 position of
the donor sugar substrate is conserved and well studied (2,8). The retaining mechanism is
poorly understood and may consist of a few steps involving stable intermediates. Hence, there
is no coupling between reaction mechanism and fold.
Most comparative studies of glycosyltransferases have been based on amino acid sequence
comparisons using BLAST and similar methods, which mainly will account for amino acid
similarities and identities at specific positions. In the CAZy database, glycosyltransferases
have been divided into about 70 families based on such sequence similarities (9), over a
comparatively short stretch of the protein. To be classified within the same family an E value
of less then E-3 over at least 100 amino acids is needed (9). The database consists of both
predicted ORFs and fully functionally determined proteins. GTs within the same sequence
family can have highly diverse substrate specificities, e.g. like members of family 2, but still
share substantial sequence similarities (10). The opposite has also been recorded, enzymes
having very low sequence similarity can utilise the same substrates (7) and have the same fold
(11). There is often a low sequence similarity between different families, and the fold within a
family is expected to be conserved for its members (9,11,12). The catalytic mechanisms are
also expected to be conserved in each CAZy family (13). However, in general very few amino
acid positions are conserved among the GTs, and the sequence similarities for the structures
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

5
within the two established fold groups are surprisingly low. Furthermore, the number of
predicted glycosyltransferases (from translated gene sequences) within an organism does not
reflect the size of the genome. For example Escherichia coli (K12) with a genome size of
4639 kb has 34 predicted glycosyltransferases according to CAZy, Bacillus subtilis 4215 kb
has 28 GTs, Synechocystis 3573 kb has 61 GTs, and finally Mycoplasma pneumoniae of 816
kb has three predicted GTs. To predict the function of new or encoded glycosyltransferases,
the ORF of interest needs to have a close sequence similarity with an enzyme of known
function. Likewise, to predict the function of new enzyme groups by sequence similarity
methods is almost impossible.
A new approach is needed. One possible way used here to search for new GTs, would be to
look for physico-chemical properties patterns along the whole sequence. Protein sequences
behave in manners far from random and the amino acid sequence is organised to reflect the
structure (14). Hence proteins with the same (or similar) structure or function are expected to
have similar property patterns in the sequence, even at low sequence similarities, and over
their full lengths or only partially (e.g. for certain domains). This is very evident for proteins
with repeated motifs, e.g. TIM barrels (14,15), and must be valid for others as well.
Furthermore, for proteins with the same function but different folds, certain local patterns in
the sequence determining the function can still be the same. GTs of a given family performing
the same reaction would be expected to have similar properties. With the multivariate
sequence analysis methods, based on amino acid properties, proteins with no sequence
similarity can be grouped together, visualising conserved property patterns within the
proteins. The sequence is translated into values describing different amino acid properties, i.e.
hydrophobicity, bulk volume, polarisability and charge. The periodicity of properties along a
sequence is calculated followed by a multivariate analysis (16). Furthermore, sequence length
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

6
variations are of less importance. This method has been used to predict location of cellular
proteins in Synechocystis 2(Rajalahti et. al., in preparation), to characterise E.coli
compartment proteins (17), and to classify signal peptides (18).
In the present study, as a first step, a reference set of glycosyltransferases from families with
known structure members was compiled from enzymes classified by CAZy. The multivariate
analysis method could conveniently separate the three different fold types from each other and
furthermore divide the two different reaction mechanisms within the A and B structural fold
groups using sequence information. The ability to predict the fold and the sugar orientation
properly was also established. Furthermore, ORFs of unknown function and that share no
sequence homology with known glycosyltransferases could potentially be identified with this
method to be glycosyltransferases.
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

7
EXPERIMENTAL PROCEDURES
Data Set─In this study 141 selected glycosyltransferases from 13 different CAZy families
(see supplementary material) were used as reference sets. The data set mainly includes
sequences with known function or with high sequence similarity with proteins with known
function, where the probability for the same function is high. The references set include both
prokaryotic and eukaryotic enzymes, using various substrates and acceptors. Full-length
amino acid sequences were used, i.e. no signal peptide or anchor domains found especially in
the GT-A group have been removed. The number of amino acids in the protein sequences
varies between 229 and 994. This data set is non-redundant and contains no pair of sequences
that share more than 55% identity.
Multivariate Sequence Analysis─The aim of this study is to search and use periodic physical
features in the proteins that separate GTs according to structure and reaction mechanism.
Amino acids can be described and characterised in a number of ways. Parameters such as
retention-times in different chromatographic systems, electric properties, molecular mass etc
are commonly used, as described by Wold et al., 1993 (16). To decrease the number of
parameters that describes each amino acid, and at the same time keep the information in a few
variables, so-called z-scales are used. These z-scales are derived from 29 physical-chemical
experimental parameters for the amino acids with principal component analysis (19). They can
approximately be translated as z1 “hydrophobicity/hydrophilicity”, z2 “bulk of side-chain”
and z3 as “polarisability/charge”, see Table I. To describe the periodicities in a protein, auto-
cross-covariance in the z-values, are used (ACC) (16). The ACC-program multiplies the first
z-value for the first amino acid with the second, followed with the first and third up to the
highest lag. The same procedure is performed with the second and the third z-scales and all
combinations of them, see Fig. 1. The ACC-terms are the average value for every interaction-
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

8
term at different lags, hence z(1)1 x z (1)2, z(1)1 x z (2)2, z(1)1 x z(3)2 etc. For short peptides such
as signal peptides, the size of the maximal lag is dependent on the shortest peptide in a set, but
for large polypeptides such as glycosyltransferases, the optimal lag seems to be between 15-
25 aa. After optimisation, a window size of 19 was chosen in this study, which gives 171
variables (19x3x3) for each sequence. Auto covariances with lag= 1, 2, 3...L were calculated
by the equation:
Index j is used for the z-scales (j=1,2,3), index i is the aa position (i=1,2...n) and n is the
number of amino acids in the sequence (cf. Fig 1). The crossed covariances between the two
different scales j and k are given by (note the difference between ACCjk and ACCkj):
Partial least squares projections to latent structures discriminant analysis (PLS-DA) finds
the relationships between a X-matrix and a Y-matrix, i.e. find relationship between sequence
properties of the X-matrix, and a Y-matrix defining the group membership (20). In this study
the Y-matrix is composed of dummy variables, hence a value of 1 is given to members of the
same group and 0 for non-members. The method is using class membership in the Y-matrix,
that in PLS is composed of features that are responses to the variables in the X matrix, here
fold group or reaction mechanism. PLS-DA is using the assumption that sequences belonging
to the same class have common features and therefore will behave similarly in the analysis, as
visualised in the score plot. This method can also be used to predict relationships for new
unclassified sequences. In PLS-DA a multidimensional space is formed where every variable
(e.g. z(1)1z(1)2, z(1)1z(2)2, z(1)1z(3)2) represents one dimension and every object (data from one
sequence) is a point in this space. For the reference set, this means 141 points in a 171
∑−
+
−
×=
lagn
i
lagijijlagj lagn
zzACC ,,
,
∑−
+≠ −
×=
lagn
i
lagikijlagkj lagn
zzACC ,,
,
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

9
dimensional space. To get the first PLS component a line that best approximates the data (best
separates the objects according to group classification) is fitted to the multidimensional space
and the data points are projected onto this line in order to get coordinate values or scores. To
get the second component, a new line is fitted to the data space that describes the second-most
of the variation. Only two components are plotted at one time, hence the first, best separating
dimension and the second in this work. The PLS model contains information both regarding
the relationship among the objects (scores) and the contribution of the variables to the model
(PLS weights). A weight plot shows the contribution of the variables for the separation, hence
what periodic features that are responsible for the separation between the fold structures or
reaction mechanisms. These features can then be searched for on the sequence level. The
objects best separated, hence localised far away from the centre of the plot, are the ones best
described by the variables with the largest weight. Such variables are therefore most likely to
be identified at the sequence level in these proteins.
To evaluate the complexity i.e. the numbers of PLS components to use in the model cross
validation is preformed, where all objects are withdrawn, here 1/10 at a time, and their y-
values are predicted from an updated model based on 9/10 of the objects. This procedure is
repeated ten times. A Q (cum)2 value is calculated which describes how much of the variance
in the Y matrix can be predicted by the model. To obtain a perfect score of 1, all objects
should be predicted back to the exact position given by the the Y matrix. A Q(cum)2 larger
then 0.1 corresponds to a 95% significance of the model (21).
Sequence alignment methods─Lalign was used to make pair-wise sequence alignments
(http://www.ch.embnet.org/software/LALIGN_form.html). To combine the pair-wise
alignments ClustalW was used (22). The ACC variables were searched for in the sequences
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

10
with the use of the established alignments from the above method. The sequences were
translated to the z-values found to be important followed by sliding along the translated
sequences according to the ACC variable, e.g. aligning the first amino acid with the 18th
according to variable z(1)1z(1)18 for the GT-A group, see results and Fig. 4 below. The
products (according to e.g. z(1)1z(1)18) between the two aligned amino acids at the positions
indicated were then calculated, and the values compared for the aligned positions. Product
values with equal signs (cf. z-values in Table I), hence positive or negative values, were
marked. These positions were indicated on the sequence alignment (cf. Fig. 4 below), and in
selected protein structures using Swiss-PdbViewer (23). The prediction of transmembrane
segments was done using TMHMM at http://www.cbs.dtu.dk/services/TMHMM-2.0/ (24).
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

11
RESULTS
Fold and reaction mechanism revealed by multivariate analysis─Do glycosyltransferases
contain sequence property periodicities related to protein architecture and enzyme function?
GTs from prokaryotes and eukaryotes using many different substrates were used in the
reference sets selected from the CAZy classification, and mainly from families where
members have at least one established crystal structure. A few related families were also
included when needed. Proteins belonging to the same CAZy family are believed to have the
same fold. MVDA, here PLS-DA, could divide the glycosyltransferase sequences according
to the established (or predicted) fold (Fig. 2). In the structure division between GT-A, GT-B
and the third newly discovered GT-C group, with transmembrane topology, a Q(cum)2 value
of 0.730 was obtained (Fig. 2 A). This is a high Q(cum)2 value (“prediction ability” of the
model, maximum is 1.0; cf. experimental procedures), but the separation between GT-A and
GT-B was less pronounced and partially overlapping. Note that the number of transmembrane
segments seems to have less impact on the distribution of the GT-C sequences, i.e. no
grouping. To further investigate the differences between the different fold groups GT-A and
GT-B were each analysed separately with GT-C, yielding Q(cum)2 values of 0.868 (Fig. 2 B)
and 0.91 (data not shown), respectively. A division between the GT- A and GT-B yielded a
Q(cum)2 of 0.655 (Fig. 2 C). A further separation within the GT-A and GT-B groups was
achieved on basis of reaction mechanism (Fig. 3 A & B), and Q(cum)2 values of 0.702 and
0.562 respectively, were obtained for the inverting and retaining clans within these two fold
groups. Hence, multivariate ACC analyses of potential sequence property profiles for
glycosyltransferases revealed clear groupings for fold and reaction mechanism by robust PLS
models with high cross validation values.
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

12
Separating sequence features─A PLS weight plot showing the distribution of each variable
for the reaction mechanism division for each score plot can be drawn (cf. experimental
procedures). Here only the corresponding weight plot for the division between retaining and
inverting GTs for the GT-B group are shown (Fig. 3). Variables that are on the same side as
the sequence class of interest in the score plot are positively correlated, and the variables on
the diagonal side of the plot are negatively or oppositely correlated. The variable information
was used to investigate if properties responsible for the separation of reaction mechanism
could be outlined in the sequences. Sequences from proteins with known structure and the
same reaction mechanism and fold were aligned to search for the variables in the sequences.
The proteins used to represent the retaining enzymes of the GT-A fold group were: α3GalT
(PDB, 1FG5) (25), GTA (PDB code, 1LZ0) (26), GTB (PDB, 1LZ7) (26) from CAZy
family GT6, LgtC (PDB, 1G9R) (27) from family GT-8, and the inverting β4Gal-T1 (1FGX)
(28) GT7; GlcaT-I (PDB, 1FGG) (29) family GT43; SpsA, (PDB, 1QGQ) (30) GT2 and
GnT1 (1FO8) (8) from family GT13. The chosen sequences were aligned by various methods,
both multiple (ClustalW), and pair-wise (LALIGN) alignments, and for both full length and
partial protein sequences. This was preformed to establish if there are any conserved regions
between the different GT families achieving the same stereochemistry for the sugar linkage. A
stretch of 84 amino acids in the retaining group (cf. above) could successfully be aligned. The
members of family 6 and 8 have no significant sequence similarity when comparing the whole
proteins. However, no long gap-free sequence alignment between all the different proteins
belonging to the inverting group (cf. sequence Table in supplementary material) could be
found using the above methods. A selection of variables with high weights found to be
important for the reaction mechanism separation within fold group GT-A (as illustrated in Fig.
3) were (in rank) z(2)1z(3)13, z(2)1z(2)5, z(1)1z(1)18, z(2)1z(2)10, z(2)1z(1)4, and z(1)1z(1)14.
Important variables at shorter distances were size (z(2)) and hydrophilicity/hydrophobicity
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

13
(z(1)) patterns, e.g. positions 1 to 4 and 1 to 5, corresponding to 3 and 4 amino acids apart,
such as amino acids on the same side of a helix. Autocorrelation analyses have shown that
proteins with several helices have a strong periodicity of hydrophobicity, of approximately
3.7 (31). This is close to the 1 to 5 and 1 to 4 positions in this work (cf. above). The longer
correlation distances (1 to 13, 1 to 18 above) are longer than the average lengths of alpha
helices and beta strands in proteins, but in Rossman-like folds important functional residues
are frequently found in the connecting loops (32,33). The established alignments between the
selected retaining enzymes were used to search for the latter variables at the sequence level,
but shorter distances, hence z(2)1z(2)5, z(2)1z(1)4, may be more difficult to visualise due to the
number of helices in the structures. The variable z(2)1z(3)13 was also used in the search for
property patterns in the alignment, but the pattern was not as clear as the z(1)1z(1)18 variable
and could be of importance in other regions of the proteins. Parts of this alignment
coincidentally overlap with the UDP binding site of the donor substrates (34). Using the z-
values from Table I the correlation for the product of the variable z(1)1z(1)18 along the
sequences in the alignment was tentatively identified (Fig. 4). Within the established
alignment, 12 variables (i.e. products) were negative and 5 positive for the retaining GT-A
group (Fig. 4, top section). This variable should according to the PLS weight plot be
negatively correlated to this group. Positions in the alignment that are members of such
variable pairs can be superimposed onto each other in the corresponding crystal structures
indicating similar position in space, as illustrated for the GT-B fold group below (Fig. 5).
However, no comparison could be made between the retaining and inverting GT-A groups
(clans), since a useable comparative alignment for the inverting mechanism sequences could
not be established for the families involved.
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

14
In the GT-B fold group the most important variables were (in rank) z(2)1z(2)5, z(1)1z(1)16
z(2)1z(2)8, z(1)1z(3)14, z(3)1z(1)11, and z(2)1z(2)2 for the separation of the retaining and inverting
sequences (Fig. 3 B), according to the PLS weight plot (Fig. 3 C). More of the major variables
here seemed to involve correlations over shorter distances than for the GT-A group; e.g.
z(2)1z(2)2 indicates side-chain volumes for residues on opposite sides of β-strands, and
z(2)1z(2)5 volumes for residues next to each other on the face of an α helix, respectively. The
alignment method described above was also applied here, and established between sequences
from the GT1 and GT28 families, with no or low sequence homology over the whole proteins.
GtfB (PDB, 1IIR)(35) and MurG (PDB, 1NLM) (36) were chosen in order to correlate the
alignment to the structures. Two more sequences from each family were chosen to get a more
stable alignment. Furthermore, to compare the inverting and retaining mechanisms, four
sequences were chosen from family GT4, a retaining family, but without solved crystal
structures. Two of these have a well established (validated) fold model, the alDGS and
spMGS lipid glycosyltransferases (37). An alignment could be made for both reaction
mechanisms for these proteins over the same region, which contains the UDP-binding sites
(Fig. 4, middle/bottom sections). The variable z(1)1z(3)16 (cf. above) was traced at the
sequence level by its z products, and should be positively correlated to the inverting enzyme
group according to Fig. 3. In the alignment 18 positive and 7 negative pairs was found within
the inverting group, and 3 positive and 7 negative pairs were found for the retaining families
(Fig. 4), confirming the importance of the z(1)1z(3)16 variable. A visualisation of the donor
substrate binding regions in the MurG and GtfB structures (both inverting) showed that these
positions seem to occupy similar positions in the structure space (Fig. 5 A-C), potentially so
also in the model for a retaining enzyme (Fig. 5 D). The difference between inverting and
retaining reaction mechanism within the GT-B fold group sequences was the amino acid
hydrophobicity. In the inverting group, where positive values are important, the property
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

15
pattern pairs consist of one hydrophobic and one hydrophilic amino acid (see Table I). The
retaining group should have equal sign hydrophobicity values, hence two hydrophobic or two
hydrophilic amino acids at positions 1 and 16. The pattern was similar for the GT-A fold
group, where the variable is z(1)1z(1)18, however the pattern has not yet been identified in the
inverting enzyme clan.
Predictions from Genomes─The three GT fold groups could be well separated from each
other, where the division between GT-C and each of the two others separately were stronger
(Fig. 2). All proposed GTs from E.coli annotated in the CAZy database were used to evaluate
the prediction power of the ACC/PLS model. The results from the fold predictions are shown
in Figure 2 D and Table II. Many family members of the GT-A fold group are known to have
an N-terminal helix that anchors the protein to the membrane, TM segments in the GT-B
group are however rare. A pair-wise division between the GT-A and GT-B groups’
individually with the GT-C group was also made and the E.coli proteins were predicted into
these models (data not shown), revealing the same results as with the three fold groups
together. Evaluation of Table II shows that seven E.coli glycosyltransferases were predicted
to belong to the GT-C fold group. All these have one or more hydrophobic TM segments
each. No proteins without TM segments were incorrectly grouped to this family for the E.coli
set. E.coli has four other proteins that have one proposed TM segment each, but they were not
grouped to the GT-C fold. The latter ones only come from two families, GT8 and GT51. GT8
has a member with a solved crystal structure, LgtC of the GT-A fold group (27). The C-
terminal part of LgtC consists of a domain very rich in basic residues and several hydrophobic
and aromatic residues in an amphiphatic organisation, but no TM segments. Hence,
presence/absence of amphiphatic segments seemed of minor importance (data not shown).
This domain is believed to bind to the membrane and is relatively conserved within the family
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

16
(38). The E.coli proteins described above from GT8 have the conserved amino acids (data not
shown) indicating that these protein are correctly grouped; hence they do not have any TM
segments. Multiple fold predictions, made by the superior MetaServer (39) (at
http://bioinfo.pl/meta), were used to evaluate the ACC/PLS predictions here. The family
GT51 had neither of the established fold type according to the MetaServer, supporting the
ACC/PLS results. The E.coli glycosyltransferases with predicted transmembrane helices from
GT2 all have the GT-A fold as a separate domain in between or after the integral membrane
domains. Generally, the results from the MetaServer coincided with the results obtained with
the multivariate method used here (see Table III).
The 61 annotated, potential glycosyltransferases from Synechocystis found in the CAZy
database (data not shown), were also analysed by the same method. Here, 10 GTs were
predicted to belong to the GT-C group. The separation between the GT-A and GT-B fold, and
versus the GT-C group (like in Fig. 2 C & D), was also preformed for the Synechocystis GTs.
The results were again very similar between the different classification methods (Table III).
The proteins grouped to the GT-C fold type all have a high hydrophobic TM segment content.
There were however a few exceptions; one protein from family 19 was grouped to GT-C
group but has no predicted hydrophobic TM segment, and there were eight GTs that have
predicted TM segments, but were not recognised by the ACC method. Within the latter, three
are from CAZy GT51 family. This group was not grouped to the GT-C fold type (cf. above),
even though they all have one predicted TM segment (but not experimentally verified). A
total evaluation of the prediction results using the MetaServer for “benchmarking” revealed
that only that 10 out of 54 or 19% were “incorrectly” grouped (including TM ones). These
results include classification of proteins with TM segments as a correct result. Within these
ten, two contain both the GT-A and GT-B fold types (gene number sll1528 and slr1063) and
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

17
had intermediate PLS results, grouping to either of the groups with a prediction value close to
0.5. These enzyme sequences were spliced (in silico) according to fold type. This division
distributed the new sequence segments to the “correct” fold types (data not shown). Two other
proteins (slr1816 and slr0626) also consisted of multiple domains with different functions,
like a known GT fold linked to a Trp domain involved in protein interactions (40). The
domain with a known GT fold fell into the correct fold type group when spliced (data not
shown).
In summary, glycosyltransferases can be classified and analysed by multivariate analysis
methods on basis of sequence property patterns. The method could successfully predict the
fold of GTs and also the orientation of the catalyzed bond between the donor sugar and the
acceptor molecule, i.e. retaining or inversion mechanisms. Potentially important amino acid
positions in the donor sites were also suggested. From genomic analyses new fold types,
where the two major folds GT-A and GT-B were found within the same protein, could also be
recognised.
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

18
DISCUSSION
Fold predictions of glycosyltransferases have recently been discussed by many (4,41,42).
This is an important enzyme group, since the enzymes utilise many different sugar substrates
and also acts on an extensive number of acceptors, with only a few fold groups described. The
sequence similarity varies greatly between enzymes performing the same enzymatic reaction
but the fold can still be the same. GTs have also been used to evaluate different fold
recognitions methods, and the fold knowledge has been used in attempts to identify new GTs
in organisms, e.g. Mycobacterium tuberculosis (42). In the latter study, multivariate methods
were used to analyse a large data set, including fold prediction results, as well as molecular
mass and theoretical isoelectric points (42). In the present study a different approach was
developed. Glycosyltransferases of known structure were analysed by translating the amino
acid sequence into z-scores describing their physico-chemical properties (19), followed by
comparing property patterns between the sequences. Of course, some other supervised
classification methods that are applicable for problems with more variables than objects could
have been used here. For example support vector machines, have successfully been applied by
Chou and co-workers (43,44) to predict structural domains in whole proteins. However, since
at present, we are unaware of a more general comparison between the PLS-DA and the
support vector machine classifier we will not speculate of the outcome of a change of
classification method, but most advanced classification methods if correctly trained usually
give rather similar results.
Conservation of properties─ The bearing idea was here that properties are more conserved
than amino acid types, leading to conservation of structures (cf. Mirny & Shakhnovich 1999)
(33). Describing and comparing the GTs based on property patterns worked well both for fold
type classification and stereochemistry of the enzyme product sugar linkage (Fig. 2 & 3). The
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

19
division between different fold types was better resolved when only analysing two fold types
at the time, especially when membrane-spanning group GT-C was included. When all three
fold types were separated in the same model, a partial overlap between GT-A and GT-B was
seen. Both GT-A and GT-B consist of Rossman-like folds, with alternating alpha helices and
beta strands. This was recognised by the method, and the difference between these two fold
types was smaller than the difference between globular proteins and transmembrane ones with
a high TM helix content (i.e. GT-A plus GT-B versus GT-C) (Fig. 2 A). Amphipathic helices
were not interfering, hence property patterns are very different for this transmembrane group
compared to the cytosolic and surface-bound proteins.
Evaluation of genome data─The multivariate method was also used to predict the fold for all
GTs in E.coli and Synechocystis included in CAZy (Table II & III). In E.coli the proportion of
GTs with established functions is high, and Synechocystis is an organism with very high GT
content. The data set consists of GTs belonging to the same GT families as the reference set,
but also from other families. This method works best with proteins from families included in
the reference set; it becomes easier for the program to predict the membership probability if
the predicted protein share some homology with the proteins in the reference set, most evident
for the GT-A and GT-B fold groups.
The MVDA method used here recognised the hydrophobic helices and grouped these
proteins according to the TM helix content to the GT-C family, even when the dominating
part of the protein had the GT-A or GT-B fold. In the E.coli set, seven GTs with high TM
content were predicted to belong to the GT-C group (Table II). These proteins are annotated
in CAZy family 2 (GT-A fold) and 51 (indifferent), and are not considered part of the GT-C
group (4). Five other E.coli proteins containing transmembrane segments were not grouped to
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

20
this latter fold group. Note that the TM content within the same GT family seems to vary
greatly, and TM segments are not accounted for in CAZy. Despite this, fold and reaction
mechanism could properly be predicted for proteins grouped to other GT families than the
major ones.
The predictions were consistent, independent of the fold families included in the separation
analysis, and independent on if two or three fold classes were used. The results obtained from
the MetaServer were used as a benchmark comparison. When predicting the fold, with all
three fold groups in the reference set, 82% of the E.coli GTs were predicted to have the right
fold, and excluding the GT-C group increased the prediction correctness to 86% (Table III).
In the first fold prediction, recognition of TM segments was regarded as the correct result.
The scores were improved when the GT-C group was removed from the prediction, hence the
TM content is no longer accounted for (Table III). Predicted glycosyltransferases from
Synechocystis, was also analysed by the same method, but even though the fraction of GTs
belonging to the major families GT2 and GT4 are larger, the prediction ability was somewhat
lower (Table III). However, most of these are not as well studied as the E.coli enzymes.
Retaining and inverting mechanisms─The ability to predict retaining and inversion
mechanisms was even higher than the structural predictions, 100% for the E.coli and 86% for
the Synechocystis set within the GT-A fold group, and 60% versus 80% for the GT-B group.
The same comparison for the GT-C group cannot be made, since it seems to consist only of
inverting enzymes. Little is understood about the differences at the sequence level between
inversion and retention. The importance of acidic residues in the active site has been
suggested, but exact conserved positions have not been established (41). Extensive sequence
alignments revealed here that there are charge differences in the UDP-binding motif within
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

21
the GT-B fold family (GT4, GT20 and GT1, GT28, respectively), data not showed. The
conserved Ex7E motif has an Asp or Glu in the first position in GT4 and GT-20 (retaining),
but the inverting enzymes in GT-1 and GT-28 have a His, Lys or Arg. The Lys261 in MurG is
known to bind to a phosphate in the UDP-GalNAc donor (36), His293 in GtfA is known to
have the same function (45), hence there is a charge difference between the two reaction
mechanism within this motif.
The established alignment over the UDP-binding region was further analysed, to search for
differences between the inverting/retaining clans in the GT-B family. The differences between
the inversion enzyme clan within the GT-A group were too high and no alignment could be
established, and choosing only a few families could give incorrect results. Within the GT-B
group there are three solved crystal structures for the inversion group. There is one solved
structure (OtsA) within the retaining enzymes, and three fold models within the retaining GT4
family. The OtsA could not be aligned over the active site with the members of GT4 due to
longer loops and helixes in OtsA than the other enzymes (46). However, a comparison
between the two different reaction mechanisms could be achieved at the sequence level and
even on the structure level. The conserved positive variable patterns within the inverting clan
(Fig. 4) were found to be superimposed when comparing the structures within the GT-B fold
group (Fig. 5). The best comparison could be made for the inverting enzyme group where the
three different crystal structures have been solved, GftA(45), GtfB (35), and MurG (36,47).
GtfA was not used in the property alignment due to a different donor sugar nucleotide (45). A
preliminary comparison could also be made between inverting and retaining mechanism
within the GT-B group. When superimposing a fold model of alDGS (retaining) on to GtfB
and MurG the corresponding negative variable pairs in the retaining group (Fig. 4) were found
to be located in the same area (Fig. 5). The positions in MurG (inverting, GT-B fold) that are
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

22
known to bind UDP-GlcNAc, A264, L265, T266, E269, Q288, and Q289 (36), are located
within the alignment (Fig. 4), hence the property pattern positions might be involved in
guiding the donor substrate to the right orientation. They can also be important for the right
structure of the α/β/α motif of the active site (47). The positions in the GT-A retaining group
were also found around the nucleotide binding area (Fig. 4) and may have similar functions
(34). The property pattern positions from this alignment could also be superimposed at the
structure level, indicating a functional importance (data not shown). These GT-B and GT-A
sites are good targets for functional (mutational) analyses.
Conclusions─The ACC/PLS (multivariate) method that we describe here structurally
classifies and identifies glycosyltransferases with high accuracy, on basis of amino acid
sequence information. The method can even be used for predicting the stereochemistry of the
reaction mechanism. Potential separating sequence parameters between the inverting and
retaining mechanism have also been suggested. The positions found to be conserved within
fold groups performing the same stereo chemical reaction are good candidates for
mutagenesis, to better understand the differences between the two reaction types. This study
has also identified four proteins containing more than one fold type, i.e. Synechocystis
slr1816, sll1528, slr0626, slr1063. Multivariate analysis of all GTs annotated in the CAZy
database may find new fold groups. Detailed analyses of large GT sequence families, such as
GT-2 and GT-4, could potentially also find subgroups related to specific substrates, products
and small structure differences such as high TM content.
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

23
Acknowledgements
We thank Dr. Anders Öhman, Umeå University, for his help with the fold analysis, and the
Swedish Natural Science Research Council for financial support.
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

24
References:
1. Boix, E., Swaminathan, G. J., Zhang, Y., Natesh, R., Brew, K., and Acharya, K. R.
(2001) J. Biol. Chem. 276, 48608-48614
2. Bourne, Y., and Henrissat, B. (2001) Curr. Opin. Struct. Biol. 11, 593-600
3. Breton, C., and Imberty, A. (1999) Curr. Opin. Struct. Biol. 9, 563-571
4. Liu, J., and Mushegian, A. (2003) Protein Sci. 12, 1418-1431
5. Kikuchi, N., Kwon, Y. D., Gotoh, M., and Narimatsu, H. (2003) Biochem. Biophys.
Res. Commun. 310, 574-579
6. Unligil, U. M., and Rini, J. M. (2000) Curr. Opin. Struct. Biol. 10, 510-517
7. Breton, C., Mucha, J., and Jeanneau, C. (2001) Biochimie 83, 713-718
8. Unligil, U. M., Zhou, S., Yuwaraj, S., Sarkar, M., Schachter, H., and Rini, J. M.
(2000) EMBO J. 19, 5269-5280
9. Campbell, J. A., Davies, G. J., Bulone, V., and Henrissat, B. (1997) Biochem. J. 326,
929-939
10. Henrissat, B., and Davies, G. J. (2000) Plant Physiol. 124, 1515-1519
11. Davies, G., and Henrissat, B. (1995) Structure 3, 853-859
12. Henrissat, B., and Davies, G. (1997) Curr. Opin. Struct. Biol. 7, 637-644
13. Gebler, J., Gilkes, N. R., Claeyssens, M., Wilson, D. B., Beguin, P., Wakarchuk, W.
W., Kilburn, D. G., Miller, R. C., Jr., Warren, R. A., and Withers, S. G. (1992) J. Biol.
Chem. 267, 12559-12561
14. Rackovsky, S. (1998) Proc. Natl. Acad. Sci. U S A 95, 8580-8584
15. Wold, S., and Sjöström, M. (1998) Acta Chemica Scandinavica 52, 517-523
16. Wold, S., Jonsson, M., Sjöström, M., Sandberg, M., and Rännar, S. (1993) Anal.
Chim. Acta. 277, 239-253
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

25
17. Sjötröm, M., Rännar, S., and Wieslander, Å. (1995) Chemometrics and Intelligent
Laboratory Systems 29, 295-305
18. Edman, M., Jarhede, T., Sjöström, M., and Wieslander, A. (1999) Proteins 35, 195-
205
19. Hellberg, S., Sjöström, M., Skagerberg, B., and Wold, S. (1987) J. Med. Chem. 30,
1126-1135
20. Wold, S., Eriksson, L., and Sjöström, M. (1998) in Encyclopedia of Computational
Chemistry (Schleyer, v. R., ed), pp. 2006-2022, John Wiley & Sons, New York
21. Eriksson, L., Johansson, E., Kettaneh-Wold, N., and Wold, S. (2001) Multi- and
Megavariate Data Analysis Principles and Applications, Umetrics, Umeå
22. Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994) Nucleic. Acids. Res.22,
4673-4680
23. Guex, N., and Peitsch, M. C. (1997) Electrophoresis 18, 2714-2723
24. Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. (2001) J. Mol. Biol.
305, 567-580
25. Gastinel, L. N., Bignon, C., Misra, A. K., Hindsgaul, O., Shaper, J. H., and Joziasse,
D. H. (2001) EMBO J. 20, 638-649
26. Patenaude, S. I., Seto, N. O., Borisova, S. N., Szpacenko, A., Marcus, S. L., Palcic, M.
M., and Evans, S. V. (2002) Nat. Struct. Biol. 9, 685-690
27. Persson, K., Ly, H. D., Dieckelmann, M., Wakarchuk, W. W., Withers, S. G., and
Strynadka, N. C. (2001) Nat. Struct. Biol. 8, 166-175
28. Gastinel, L. N., Cambillau, C., and Bourne, Y. (1999) EMBO J. 18, 3546-3557
29. Pedersen, L. C., Tsuchida, K., Kitagawa, H., Sugahara, K., Darden, T. A., and
Negishi, M. (2000) J. Biol. Chem. 275, 34580-34585
30. Tarbouriech, N., Charnock, S. J., and Davies, G. J. (2001) J. Mol. Biol. 314, 655-661
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

26
31. Horne, D. S. (1988) Biopolymers 27, 451-477
32. Brändén, C.-I., and Tooze, J. (1999) Introduction to protein structure, 2. Ed., Garland,
New York
33. Mirny, L. A., and Shakhnovich, E. I. (1999) J. Mol. Biol. 291, 177-196
34. Heissigerova, H., Breton, C., Moravcova, J., and Imberty, A. (2003) Glycobiology 13,
377-386
35. Mulichak, A. M., Losey, H. C., Walsh, C. T., and Garavito, R. M. (2001) Structure 9,
547-557
36. Hu, Y., Chen, L., Ha, S., Gross, B., Falcone, B., Walker, D., Mokhtarzadeh, M., and
Walker, S. (2003) Proc. Natl. Acad. Sci. U S A 100, 845-849
37. Edman, M., Berg, S., Storm, P., Wikström, M., Vikström, S., Öhman, A., and
Wieslander, Å. (2003) J. Biol. Chem. 278, 8420–8428,
38. Wakarchuk, W. W., Cunningham, A., Watson, D. C., and Young, N. M. (1998)
Protein Engineering 11, 295-302
39. Ginalski, K., Elofsson, A., Fischer, D., and Rychlewski, L. (2003) Bioinformatics 19,
1015-1018
40. Blatch, G. L., and Lassle, M. (1999) Bioessays 21, 932-939
41. Franco, O. L., and Rigden, D. J. (2003) Glycobiology 13, 707R-712R
42. Wimmerova, M., Engelsen, S. B., Bettler, E., Breton, C., and Imberty, A. (2003)
Biochimie 85, 691-700
43. Chou, K. C., and Cai, Y. D. (2002) J. Biol. Chem. 277, 45765-45769
44. Cai, Y. D., Lin, S. L., and Chou, K. C. (2003) Peptides 24, 159-161
45. Mulichak, A. M., Losey, H. C., Lu, W., Wawrzak, Z., Walsh, C. T., and Garavito, R.
M. (2003) Proc. Natl. Acad. Sci. U S A 100, 9238-9243
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

27
46. Gibson, R. P., Tarling, C. A., Roberts, S., Withers, S. G., and Davies, G. J. (2003) J.
Biol. Chem. 279, 1950-1955
47. Ha, S., Walker, D., Shi, Y., and Walker, S. (2000) Protein Sci. 9, 1045-1052
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

28
Footnotes
1 The abbreviations used are: CAZy, carbohydrate active enzymes; MVDA, multivariate data
analyses; GT, Glycosyltransferase; BLAST, basic local alignment search tool ; SGC, SpsA
GnT I core; UDP, uridine diphosphate; GlcNAc, N-acetyl-d-glucosamine; TM,
transmembrane; PDB, protein data bank; ORF, open reading frame; ACC, auto cross
covariance; PLS-DA, Partial least squares projections to latent structures discriminant
analysis;
2 Unpublished cited work:
Rajalahti, T., Huang, F., Sjöström, M., Norling, B., and Wieslander, Å., in preparation
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

29
FIGURE LEGENDS
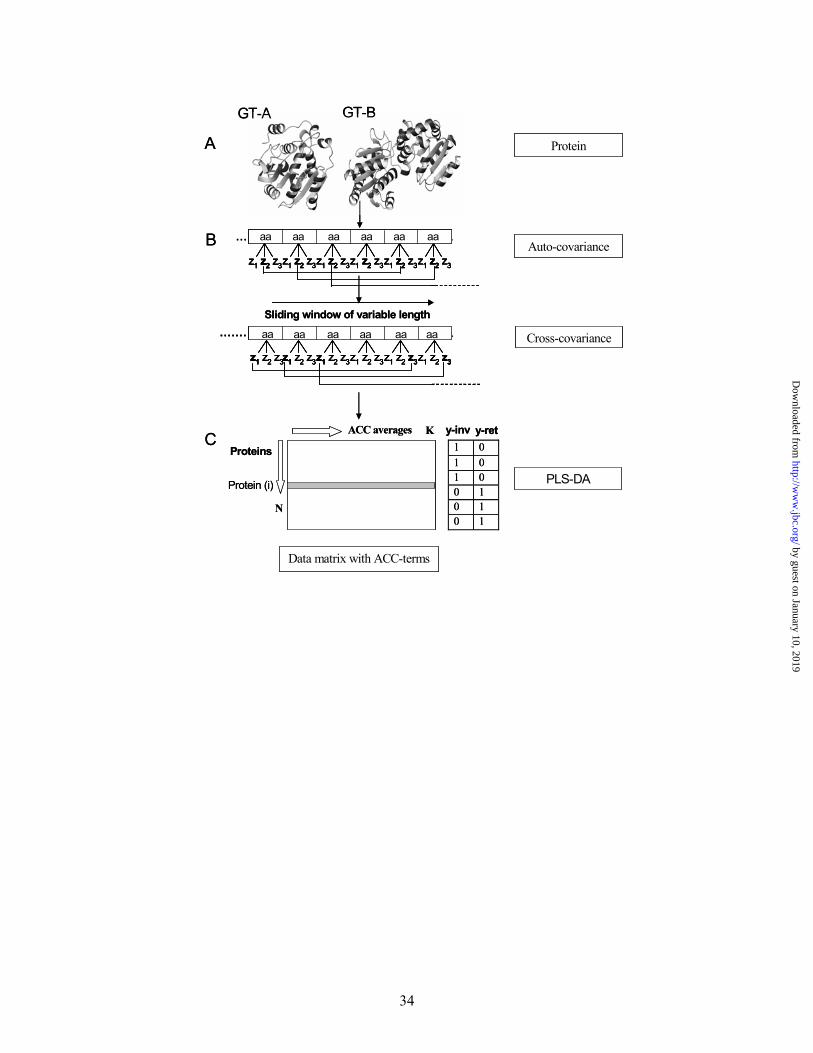
FIG 1. The steps used in analysing protein sequences with an ACC description of the
periodicity of the protein sequence followed by PLS-DA analysis. (A) Proteins are
collected from CAZy with a known fold type, (B) the amino acids are translated into the three
z-scales (from Table I) and are used for (C) calculation of auto covariance and cross
covariance variables, and (D) the data is collected in a data matrix. A discriminant vector for
each class is constructed and used as Y in the PLS-DA (e.g. y (GT-A) and y (GT-B)).
FIG. 2. Fold division of the glycosyltransferase reference sets based on sequence
property patterns. Q (cum)2 values (“prediction ability” of the model) for each comparison
are given in the plots, showing the two first score vectors t1 and t2 from the PLS-DA. The
predicted transmembrane segment content within each enzyme is indicated with numbers and
the fold group with colours. GT-A, red; GT-B, blue; GT-C green; and predicted E.coli GTs in
black. A, division between the GT-A, GT-B, and GT-C fold types; B, division between GT-A
and GT-C; C, GT-A and GT-B division; D, GT-A, GT-B, and GT-C fold division with the
E.coli GTs predicted. PS stands for prediction set.
FIG. 3. Division between the reaction stereochemistry within fold types. Panel (A) and
(B) showing the two first score vectors t1 and t2 from the PLS-DA. Based on sequence
property patterns; retaining (□) or inversion (▼) of the sugar linkage within the GT-A (A) and
GT-B (B) fold types. The Q (cum)2 values are given for each division. C, The corresponding
PLS-weight plot (w1*c1 / w2*c2) for the GT-B analysis in the B-panel, showing the x-variable
and y variable weights (w* and c respectively), where variables to the fare left and right in
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

30
the plot have the most separating information. A variable found on the same side as the
inversion group is positively correlated to this group and negatively correlated to the retaining
group. A weight plot for GT-A is not shown.
FIG. 4. Sequence alignment of selected GTs within the reference sets. The variable pairs
indicated were aligned with ClustalW. p, first position in positive variable pairs, e.g.
z(1)1z(1)18 (indicated from the PLS weight plots), (+) the second position (e.g. 18); n, first
position in a negative variable pair, (-) the second position. Conserved amino acids are
marked with (*), (:) amino acids with the same properties, and (.) similar properties as marked
in ClustalW.
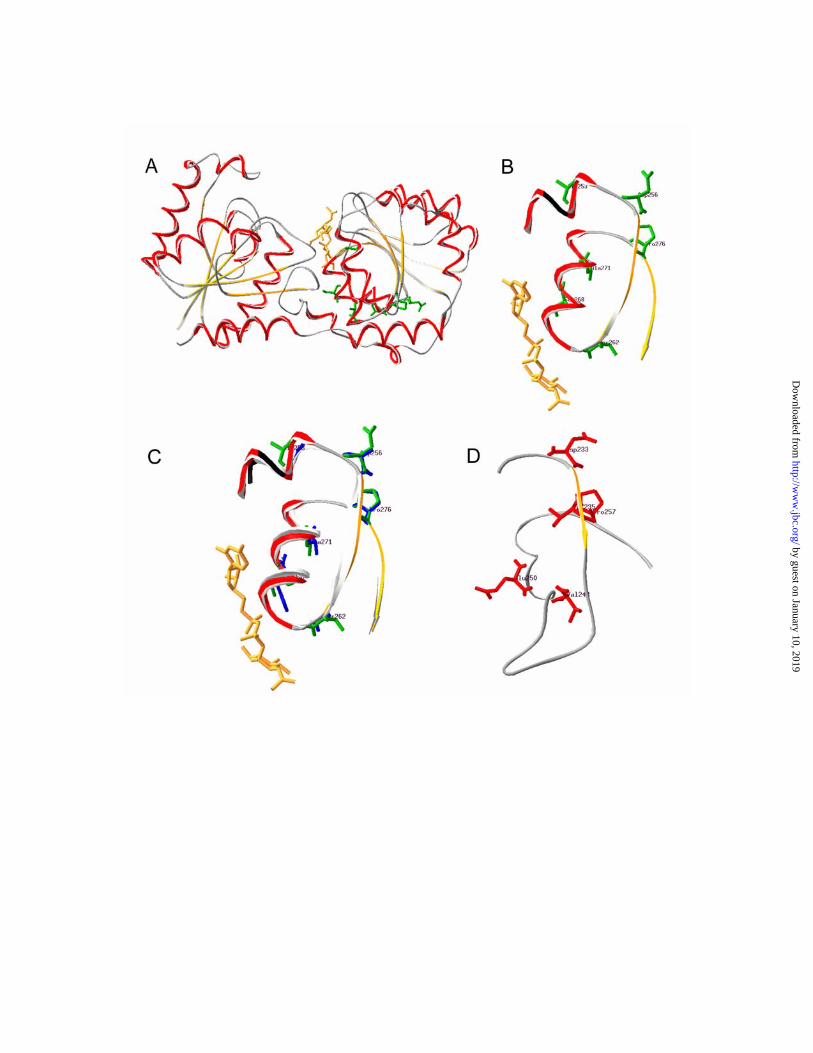
FIG. 5. Important sequence pattern positions in active site structures. A, Ribbon
structure of MurG (PDB code) from E.coli with the UDP-GlcNAc donor substrate shown in
yellow. Amino acids conserved in the variable sequence pairs and found around the active site
are marked in green. B, The UDP-GlcNAc binding region with the variable sequence pairs
indicated: Ala253, Asp256, Ser262, Ser268, Ala271, and Pro276. C, MurG and GtfB (PDB)
ribbon structures of the UDP-binding area with the amino acids Gly300, Ala303 Gly309,
His315, Ala318, and Pro323 in GtfB superimposed onto the MurG structure. D, The
corresponding region in alDGS lipid GT from Acholeplasma laidlawii. The positions marked,
Asp233, Val235, Glu242, Val248, Glu250, and Pro257, are members of negative variables
pairs.
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

31
TABLE I Descriptor Scales for the Coded Amino Acidsa
Amino acid z1 z2 z3 Phe (F) -4.92 1.30 0.45Trp (W) -4.75 3.65 0.85
Ile (I) -4.44 -1.68 -1.03Leu (L) -4.19 -1.03 -0.98Val (V) -2.69 -2.53 -1.29Met (M) -2.49 -0.27 -0.41Tyr (Y) -1.39 2.32 0.01Pro (P) -1.22 0.88 2.23Ala (A) 0.07 -1.73 0.09Cys (C) 0.71 -0.97 4.13Thr (T) 0.92 -2.09 -1.40Ser (S) 1.96 -1.63 0.57Gln (Q) 2.19 0.53 -1.14Gly (G) 2.23 -5.36 0.30His (H) 2.41 1.74 1.11Lys (K) 2.84 1.41 -3.14Arg (R) 2.88 2.52 -3.44Glu (E) 3.08 0.039 -0.07Asn (N) 3.22 1.45 0.84Asp (D) 3.64 1.13 2.36
aDerived by a principal component analysis of 29 physico-chemical properties for the amino
acids (19). z1 reflects ‘‘hydrophobicity/hydrophilicity’’, z2 side-chain ‘‘bulk volume’’, and
z3‘‘polarizability and charge’’, respectively.
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from
32
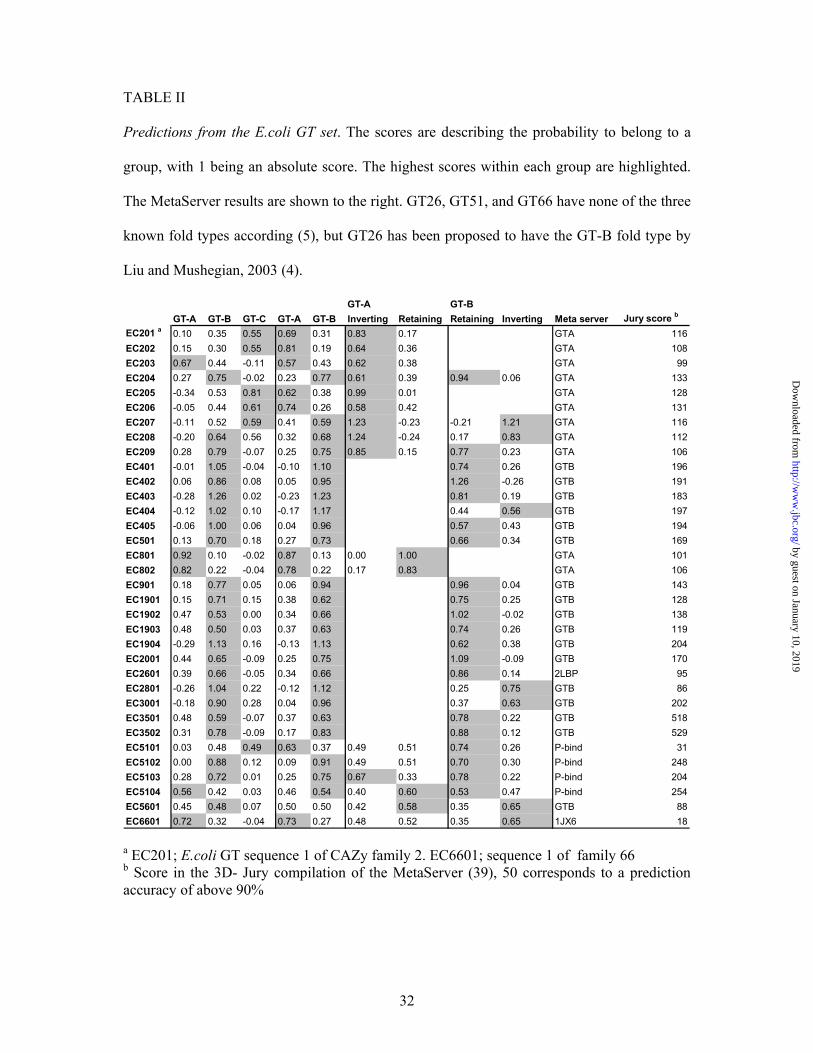
TABLE II
Predictions from the E.coli GT set. The scores are describing the probability to belong to a
group, with 1 being an absolute score. The highest scores within each group are highlighted.
The MetaServer results are shown to the right. GT26, GT51, and GT66 have none of the three
known fold types according (5), but GT26 has been proposed to have the GT-B fold type by
Liu and Mushegian, 2003 (4).
GT-A GT-B GT-A GT-B GT-C GT-A GT-B Inverting Retaining Retaining Inverting Meta server Jury score b
EC201 a 0.10 0.35 0.55 0.69 0.31 0.83 0.17 GTA 116EC202 0.15 0.30 0.55 0.81 0.19 0.64 0.36 GTA 108EC203 0.67 0.44 -0.11 0.57 0.43 0.62 0.38 GTA 99EC204 0.27 0.75 -0.02 0.23 0.77 0.61 0.39 0.94 0.06 GTA 133EC205 -0.34 0.53 0.81 0.62 0.38 0.99 0.01 GTA 128EC206 -0.05 0.44 0.61 0.74 0.26 0.58 0.42 GTA 131EC207 -0.11 0.52 0.59 0.41 0.59 1.23 -0.23 -0.21 1.21 GTA 116EC208 -0.20 0.64 0.56 0.32 0.68 1.24 -0.24 0.17 0.83 GTA 112EC209 0.28 0.79 -0.07 0.25 0.75 0.85 0.15 0.77 0.23 GTA 106EC401 -0.01 1.05 -0.04 -0.10 1.10 0.74 0.26 GTB 196EC402 0.06 0.86 0.08 0.05 0.95 1.26 -0.26 GTB 191EC403 -0.28 1.26 0.02 -0.23 1.23 0.81 0.19 GTB 183EC404 -0.12 1.02 0.10 -0.17 1.17 0.44 0.56 GTB 197EC405 -0.06 1.00 0.06 0.04 0.96 0.57 0.43 GTB 194EC501 0.13 0.70 0.18 0.27 0.73 0.66 0.34 GTB 169EC801 0.92 0.10 -0.02 0.87 0.13 0.00 1.00 GTA 101EC802 0.82 0.22 -0.04 0.78 0.22 0.17 0.83 GTA 106EC901 0.18 0.77 0.05 0.06 0.94 0.96 0.04 GTB 143EC1901 0.15 0.71 0.15 0.38 0.62 0.75 0.25 GTB 128EC1902 0.47 0.53 0.00 0.34 0.66 1.02 -0.02 GTB 138EC1903 0.48 0.50 0.03 0.37 0.63 0.74 0.26 GTB 119EC1904 -0.29 1.13 0.16 -0.13 1.13 0.62 0.38 GTB 204EC2001 0.44 0.65 -0.09 0.25 0.75 1.09 -0.09 GTB 170EC2601 0.39 0.66 -0.05 0.34 0.66 0.86 0.14 2LBP 95EC2801 -0.26 1.04 0.22 -0.12 1.12 0.25 0.75 GTB 86EC3001 -0.18 0.90 0.28 0.04 0.96 0.37 0.63 GTB 202EC3501 0.48 0.59 -0.07 0.37 0.63 0.78 0.22 GTB 518EC3502 0.31 0.78 -0.09 0.17 0.83 0.88 0.12 GTB 529EC5101 0.03 0.48 0.49 0.63 0.37 0.49 0.51 0.74 0.26 P-bind 31EC5102 0.00 0.88 0.12 0.09 0.91 0.49 0.51 0.70 0.30 P-bind 248EC5103 0.28 0.72 0.01 0.25 0.75 0.67 0.33 0.78 0.22 P-bind 204EC5104 0.56 0.42 0.03 0.46 0.54 0.40 0.60 0.53 0.47 P-bind 254EC5601 0.45 0.48 0.07 0.50 0.50 0.42 0.58 0.35 0.65 GTB 88EC6601 0.72 0.32 -0.04 0.73 0.27 0.48 0.52 0.35 0.65 1JX6 18
a EC201; E.coli GT sequence 1 of CAZy family 2. EC6601; sequence 1 of family 66 b Score in the 3D- Jury compilation of the MetaServer (39), 50 corresponds to a prediction accuracy of above 90%
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

33
Table III
Prediction yields from different fold and reaction mechanisms for the E.coli and
Synechocystis genome sets a.
Division E.coli (%) Synechosystis (%)
Allb Fold 82 67GTA/ GTB b Fold 86 80GTA Inv / Ret d 100 86GTB Inv / Ret d 60 80
a Intermediate prediction results, hence results close to 0.5 have been removed, see Table II. b GT-A, GT-B, GT-C fold types included. All sequences are included in the calculations,
however sequences from families with unknown fold type neither taken as correct nor wrong. c GTA/GTB; No true GT-C families are seen in E. coli and only one in Synechocystis. The
GT-C group have then been removed and the results recalculated based on the division
between only the two remaining fold groups. d Inv/Ret; Results from inverting (inv) and retaining (ret) mechanisms.
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from
34
Protein
PLS-DA
Auto-covariance
Cross-covariance
.......
.......Sliding window of variable length
Data matrix with ACC-terms
Proteins
ACC averages K
N
Protein (i)
.......
.......z1 z2 z3 z1 z2 z3z1 z2 z3 z1 z2 z3z1 z2 z3z1 z2 z3
aa aa aa aa aa aa
z1 z2 z3z1 z2 z3 z1 z2 z3z1 z2 z3z1 z2 z3 z1 z2 z3
aa aa aa aa aa aa
A
B
C
GT-A GT-B
y-inv y-ret1 0 1 0 1 0 0 1 0 1 0 1
Protein
PLS-DA
Auto-covariance
Cross-covariance
.......
.......Sliding window of variable length
Data matrix with ACC-terms
Proteins
ACC averages K
N
Protein (i)
.......
.......z1 z2 z3 z1 z2 z3z1 z2 z3 z1 z2 z3z1 z2 z3z1 z2 z3
aa aa aa aa aa aa
z1 z2 z3z1 z2 z3 z1 z2 z3z1 z2 z3z1 z2 z3 z1 z2 z3
aa aa aa aa aa aa
.......z1 z2 z3z1 z2 z3 z1 z2 z3z1 z2 z3z1 z2 z3z1 z2 z3 z1 z2 z3z1 z2 z3z1 z2 z3z1 z2 z3z1 z2 z3z1 z2 z3
aa aa aa aa aa aa
z1 z2 z3z1 z2 z3z1 z2 z3z1 z2 z3 z1 z2 z3z1 z2 z3z1 z2 z3z1 z2 z3z1 z2 z3z1 z2 z3 z1 z2 z3z1 z2 z3
aa aa aa aa aa aa
A
B
C
GT-A GT-B
y-inv y-ret1 0 1 0 1 0 0 1 0 1 0 1 by guest on January 10, 2019
http://ww
w.jbc.org/
Dow
nloaded from

35
-5
0
5
-1 0 -5 0 5
t[2]
t[1 ]
0
0
0
0
0
00
0
0
0
0
0
11
11 1
1 11
1
1
1
1
10
000
0
011
1111
33
4
3
1
1
1 1
1
1
1
1
111 1
1
1
00 0
0
0
0
0
00
0
0
0
00
0
0
00 0
0
00
0
0
0
0
0
000 00
00
0
000
0
20
0
00
1
0
0
01
Q2=0.655C
-5
0
5
-1 0 -5 0 5 1 0
t[2]
t [1 ]
000
0
0
0
0
0
00
00
11 11
1
1
1
111
1
11
000
0
0
0
1111
11
3
3
4
3
1
111
1
1
111
111
1
1
6777
5
910
1198
12
10
1010 11
1312
10
13 1312
1011 912
10
7
15887
9
8910
-1 0
-5
0
5
-1 0 -5 0 5 1 0
t[2]
t [1 ]
0
0
0
00
00 0
0
0
001
1
111 11
11
1
1
11 00
00
00
1111
11
33 431
111
11
1
11
111
1
1
00
000
0
0 00
0
0
000000
0
0
0
00
00
0
0
0000
00
00
0 0
0
0
02
0
0
0
0
1 0
0
0
1
6
7
7
75
9
10
11
9
8
12
10
1010
11
13 121013 1312
10
11912
10
7
15887 98
9
10
A
Q2=0.868B
-1 0
-5
0
5
-1 0 -5 0 5 1 0
tPS
[2]
tP S [1 ]
10
6
0
0
4
5
2 2
0
00
0
0
00
10
0
000
0
0
0
0
0
00
1
11
10
0
00
0
00
00 0
0
0
001
1
111 11
11
1
1
11 00
00
00
1111
11
33 431
111 1
1
1
11
111
1
1
000000
0 00
0
0
000000
0
0
0
00
00
0
0
0000
00
00
0 0
0
0
02
0
0
0
0
1 0
0
0
1
6
7
7
75
9
10
11
9
8
12
10
1010
11
13 121013 1312
10
11912
10
7
15887 98
9
10
D
Q2=0.730
Q2=0.730
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

36
-5
0
5
-5 0 5
t[2]
t[1 ]
-0
-0
0
0
0
-0 -0 0 0 0
w*c
[2]
w*c[1]
z11z12
z11z22
z11z32
z11z13
z11z23
z11z33
z11z14
z11z24z11z34z11z15
z11z25
z11z35
z11z16
z11z26 z11z36
z11z17
z11z27
z11z37
z11z18
z11z28
z11z38
z11z19
z11z29
z11z39
z11z110
z11z210
z11z310
z11z111
z11z211z11z311z11z112z11z212
z11z312
z11z113z11z213
z11z313
z11z114
z11z214
z11z314
z11z115
z11z215
z11z315
z11z116
z11z216
z11z316z11z117
z11z217
z11z317z11z118
z11z218
z11z318
z11z119
z11z219
z11z319
z11z120z11z220
z11z320
z21z12
z21z22 z21z32
z21z13
z21z23
z21z33
z21z14
z21z24z21z34
z21z15z21z25z21z35
z21z16
z21z26
z21z36
z21z17z21z27
z21z37
z21z18z21z28z21z38
z21z19
z21z29
z21z39z21z110
z21z210 z21z310z21z111
z21z211
z21z311z21z112z21z212
z21z312z21z113
z21z213
z21z313
z21z114
z21z214
z21z314
z21z115
z21z215
z21z315
z21z116
z21z216
z21z316
z21z117
z21z217
z21z317
z21z118
z21z218
z21z318
z21z119
z21z219z21z319z21z120
z21z220
z21z320
z31z12z31z22
z31z32z31z13
z31z23
z31z33
z31z14
z31z24
z31z34
z31z15
z31z25
z31z35
z31z16
z31z26
z31z36
z31z17
z31z27
z31z37z31z18
z31z28
z31z38
z31z19
z31z29
z31z39
z31z110
z31z210z31z310
z31z111
z31z211
z31z311
z31z112z31z212z31z312z31z113
z31z213
z31z313
z31z114z31z214
z31z314z31z115
z31z215
z31z315
z31z116z31z216
z31z316
z31z117
z31z217
z31z317
z31z118
z31z218
z31z318
z31z119
z31z219
z31z319z31z120z31z220
z31z320
$M1
$M1.DA2
-5
0
5
-5 0 5
t[2]
t[1 ]
A
B
C
Q2=0.702
Q2=0.562
• y-inv
•y-ret
-5
0
5
-5 0 5
t[2]
t[1 ]
-0
-0
0
0
0
-0 -0 0 0 0
w*c
[2]
w*c[1]
z11z12
z11z22
z11z32
z11z13
z11z23
z11z33
z11z14
z11z24z11z34z11z15
z11z25
z11z35
z11z16
z11z26 z11z36
z11z17
z11z27
z11z37
z11z18
z11z28
z11z38
z11z19
z11z29
z11z39
z11z110
z11z210
z11z310
z11z111
z11z211z11z311z11z112z11z212
z11z312
z11z113z11z213
z11z313
z11z114
z11z214
z11z314
z11z115
z11z215
z11z315
z11z116
z11z216
z11z316z11z117
z11z217
z11z317z11z118
z11z218
z11z318
z11z119
z11z219
z11z319
z11z120z11z220
z11z320
z21z12
z21z22 z21z32
z21z13
z21z23
z21z33
z21z14
z21z24z21z34
z21z15z21z25z21z35
z21z16
z21z26
z21z36
z21z17z21z27
z21z37
z21z18z21z28z21z38
z21z19
z21z29
z21z39z21z110
z21z210 z21z310z21z111
z21z211
z21z311z21z112z21z212
z21z312z21z113
z21z213
z21z313
z21z114
z21z214
z21z314
z21z115
z21z215
z21z315
z21z116
z21z216
z21z316
z21z117
z21z217
z21z317
z21z118
z21z218
z21z318
z21z119
z21z219z21z319z21z120
z21z220
z21z320
z31z12z31z22
z31z32z31z13
z31z23
z31z33
z31z14
z31z24
z31z34
z31z15
z31z25
z31z35
z31z16
z31z26
z31z36
z31z17
z31z27
z31z37z31z18
z31z28
z31z38
z31z19
z31z29
z31z39
z31z110
z31z210z31z310
z31z111
z31z211
z31z311
z31z112z31z212z31z312z31z113
z31z213
z31z313
z31z114z31z214
z31z314z31z115
z31z215
z31z315
z31z116z31z216
z31z316
z31z117
z31z217
z31z317
z31z118
z31z218
z31z318
z31z119
z31z219
z31z319z31z120z31z220
z31z320
$M1
$M1.DA2
-5
0
5
-5 0 5
t[2]
t[1 ]
A
B
C
Q2=0.702
Q2=0.562
• y-inv
•y-ret
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

37
GT-A Z(1)1z(1)18 Retaining linkage nn p n n pn p n- p+nn nnp+n n - + -- --+ - - HsBgGtB (O14758) EILTPLFG-TLHPSFYGSSREAFTYERRPQSQAYIPKDEGDFYYMGAFFGGSVQEVQRLTRACHQAMMVDQANGIEAVWHDESHL- HsBgGtA (P16442) EILTPLFG-TLHPGFYGSSREAFTYERRPQSQAYIPKDEGDFYYLGGFFGGSVQEVQRLTRACHQAMMVDQANGIEAVWHDESHL- BsGalT (P14769) ETLGESVA-QLQAWWYKADPNDFTYERRKESAAYIPFGEGDFYYHAAIFGGTPTQVLNITQECFKGILKDKKNDIEAQWHDESHL- NmLgtC (P96945) -SLTPLWDTDLGDNWLGACIDLF-VERQEGYKQKIGMADGEYYFNAGVLLINLKKWRRHDIFKMSCEWVEQYKDVM-QYQDQDIL- * * : : : * **: * :*::*: ...: . . . . :: :.: ::*:. * GT-B Z(1)1z(1)16 Inverting linkage - + + + ++ p p nn pnn n + + -- p-- p p n p n p - p+ p pp pp p p +pp ++ + + ++ AoGtfD (gi13591783) QALFRRVAAVIHHGSAGTEHVATRAGVPQLVIPRNTD----QPYFAGRVAALGIGVAHDGPTPTFESLSAALTTVLAPETRARAEAVAGMVLTDGAAAAADLVLAAVGREKPAVPA 112 AoGtfB (P96559) QVLFGRVAAVIHHGGAGTTHVAARAGAPQILLPQMAD----QPYYAGRVAELGVGVAHDGPIPTFDSLSAALATALTPETHARATAVAGTIRTDGAAVAARLLLDAVS-------- 104 NmMurG (gi7380690) VSAYRDADLVICRAGALTIAELTAAGLGALLVPYPHAVDDHQTANARFMVQAEAGLLLPQTQLTAEKLAEILGGLNREKCLKWAENARTLALPHSADDVAEAAIACAA-------- 108 EcMurG (P17443) AAAYAWADVVVCRSGALTVSEIAAAGLPALFVPFQHK+DRQQYWNALPLEKAGAAKIIEQPQLSVDAVANTLAGWSRETLLTMAERARAASIPDATERVANEVSRVAR-------- 107 : . *: :..* * : ** :.:* * * : . . : : :: * * . ...: .* . Retaining linkage + + - p n n p n - -n pnn n - +-- - alDGS (gi22641488) VDGAVIKGAFSGADCVFFPSYEETEGIVVLEGLASKTPVVLRDIPVYYDWLFHK spMGS (gi15958596) IAPSETALYYKAADFFISASTSETQGLTYLESLASGTPVIAHGNPYLNNLISDKMFG bbMGS (gi2681362) IPWEEIYYYYKISDIFASLSKSEVYPMTVIEALTAGIPAILINDYIYKDVIKEGIN EcMtfB (P47594) VPDEDLPYLYAAARTFVYPSFYEGFGLPILEAMSCGVPVVCSNVTSLPEVVGDAG : : . * * : :*.::. .: . :
by guest on January 10, 2019 http://www.jbc.org/ Downloaded from

39
SUPPLEMENTARY MATERIAL Rosén et al., 2004 Recognition of fold and sugar linkage for glycosyltransferases by multivariate sequence
analysis
Glycosyltransferase sequences used as references for training of the MVDA method. Number Organsim Gi number Function TM
helixes CAZy
Famliy 1 Amycolatopsis orientalis 5971640 Tdp-Epi-Vancosaminyltransferase 0 1 2 Homo sapiens 1407590 ceramide galactosyl transferase 2 1 3 Nicotiana tabacum 20146091 glucosyltransferase NTGT3 0 1 4 Amycolatopsis orientalis 13591783 glycosyltransferase 0 1 5 Arabidopsis thaliana 9392679 glucosyltransferase 0 1 6 Brassica napus 9794913 glucosyltransferase 0 1 7 Felis catus 2842546 UDP-glucuronosyltransferase 1 1 8 Stevia rebaudiana 21435782 UDP-glucosyltransferase 0 1 9 Basillus subtilis 580877 glycosyl transferase 0 2
10 Haemophilus ducreyi 8118046 beta 1-4 glucosyltransferase 0 2 11 Sinorhizobium meliloti 605654 exoM 0 2 12 Streptococcus agalactiae 13022167 beta-1,3-glucosyltransferase 0 2 13 Klebsiella pneumoniae 5006991 glucosyl transferase 0 2 14 Bradyrhizobium sp 12642177 nodulation N-acetylglucosaminyltransferase 0 2 15 Neisseria gonorrhoeae 595813 glycosyl transferase 0 2 16 Streptococcus agalactiae 3721919 N-acetylglucosaminyltransferase 0 2 17 Streptococcus pneumoniae 2198542 ss-1,3-N-acetylglucosaminyltransferase 0 2 18 Campylobacter jejuni 12004281 beta-1,3-galactosyltransferase 0 2 19 Acetobacter xylinus 3298349 beta-D-1,6 Glucosyl transferase 0 2 20 Streptococcus agalactiae 13022168 beta-1,3-galactosyltransferase 0 2 21 Acholeplasma laidlawii 22651488 alpha1,2-glucosyltransferase 0 4 22 Lactococcus lactis 15674119 glycosyltransferase 0 4 23 Streptococcus pneumoniae 15900944 glycosyltransferase 0 4 24 Streptococcus pneumoniae 15900945 glycosyltransferase 0 4 25 Acholeplasma laidlawii 14043013 1,2-diacylglycerol 3-glucosyltransferase 0 4 26 Borrelia burgdorferi 15594799 glycosyltransferase 0 4 27 Klebsiella pneumoniae 557195 galactosyl transferase 0 4 28 Streptococcus thermophilus 1276879 glycosyltransferase 0 4 29 Arabidopsis thaliana 11357895 sulfogalactosetransferase 0 4 30 Synechocystis sp. PCC 6803 1001478 sulfogalactosetransferase 0 4 31 Gluconacetobacter xylinus 1054906 alpha-mannosyltransferse 0 4 32 Mycobacterium tuberculosis 3719234 alpha-mannosyltransferse 0 4 33 Campylobacter coli 1486283 galactosyltransferase 0 4 34 Escherichia coli 3142172 mannosyl transferase 0 4 35 Escherichia coli 598471 mannosyltransferase B 0 4 36 Pseudomonas aeruginosa 3249545 glycosyltransferase WbpX 0 4 37 Pseudomonas aeruginosa 3249551 glycosyltransferase WbpY 0 4 38 Pseudomonas aeruginosa 3249553 glycosyltransferase WbpZ 0 4 39 Salmonella typhimurium 3132887 alpha1,3-glucosyltransferase 0 4 40 Synechococcus sp 14595230 tetrahydrobiopterin glucosyltransferase 0 4
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from
40
41 Callithrix sp. 1086056 alpha 1,3 galactosyltransferase 1 6 42 Homo sapiens 4590454 alpha 1,3 galactosyltransferase 1 6 43 Mus musculus 15419872 alpha-1,3-galactosyltransferase 1 6 44 Sus scrofa 609567 alpha-1,3-galactosyltransferase 1 6 45 Bos taurus 163124 alpha 1-3 galactosyltransferase 1 6 46 Gallus gallus 1469908 beta-1,4-galactosyltransferase 1 7 47 Gallus gallus 1469906 beta-1,4-galactosyltransferase 1 7 48 Homo sapiens 3132898 beta-1,4-galactosyltransferase 1 7 49 Bos taurus 127820 Beta-1,4-galactosyltransferase 1 1 7 50 Ciona intestinalis 9229932 beta 4 galactosyltransferase 1 7 51 Homo sapiens 3132896 beta-1,4-galactosyltransferase 1 7 52 Mus musulus 6651182 beta-1,4-galactosyltransferase III 1 7 53 Rattus norvegicus 3258653 beta-1,4-galactosyltransferase 1 7 54 Escherichia coli 26110705 Lipopolysaccharide 1,3-galactosyltransferase 0 8 55 Salmonella typhimurium 3132884 alpha1,3-galactosyltransferase 0 8 56 Escherichia coli 26110704 Lipopolysaccharide 1,2-glucosyltransferase 0 8 57 Salmonella typhimurium 16422285 alpha1,2-glucosyltransferase 0 8 58 Escherichia coli 3821841 alpha1,3-glucosyltransferase 0 8 59 Neisseria meningitidis 21654776 alpha 1,4 galactosyltransferase 0 8 60 Oryctolagus cuniculus 165782 acetylglucosaminyltransferase I 1 13 61 Cricetulus griseus 14388961 N-acetylglucoaminyltransferase I 1 13 62 Drosophila melanogaster 7804912 N-acetylglucoaminyltransferase I 1 13 63 Mus musculus 6754684 N-acetylglucoaminyltransferase I 1 13 64 Solanum tuberosum 6779206 N-acetylglucoaminyltransferase I 1 13 65 Xenopus laevis 15211610 N-acetylglucoaminyltransferase I 1 13 66 Arabidopsis thaliana 1865677 trehalose-6-phosphate synthase 0 20 67 Aspergillus niger 551471 trehalose-6-phosphate synthase 0 20 68 Candida albicans 1488038 trehalose-6-phosphate synthase 0 20 69 Methanothermobacter
thermautotrophicus 2104413 trehalose-6-phosphate synthase 0 20
70 Zygosaccharomyces rouxii 8886767 trehalose-6-phosphate phosphatase 0 20 71 Escherichia coli 862973 trehalose-6-phosphate synthase 0 20 72 Pichia pastoris 14718993 ceramide glucosyltransferase 3 21 73 Rattus norvegicus 4105567 ceramide glycosyltransferase 3 21 74 Gossypium arboreum 14718995 ceramide glucosyltransferase 4 21 75 Homo sapiens 1325917 ceramide glucosyltransferase 3 21 76 Magnaporthe grisea 14718991 ceramide glucosyltransferase 1 21 77 Mus musculus 9256626 alpha-mannosyltransferase 6 22 78 Oryza sativa 13161358 alpha-mannosyltransferase 7 22 79 Caenorhabditis elegans 19069522 alpha-mannosyltransferase 7 22 80 Drosophila melanogaster 23092941 alpha-mannosyltransferase 7 22 81 Schizosaccharomyces pombe 19075493 alpha-mannosyltransferase 5 22 82 Saccharomyces cerevisiae 6321296 alpha-mannosyltransferase 9 22 83 Trypanosoma brucei 7657993 alpha-mannosyltransferase 10 22 84 Caenorhabditis elegans 17566740 alpha-mannosyltransferase 10 22 85 Drosophila melanogaster 14549429 polypeptide N-acetylgalactosaminyltransferase 1 27 86 Drosophila melanogaster 7303062 polypeptide N-acetylgalactosaminyltransferase 1 27 87 Homo sapiens 1136285 polypeptide N-acetylgalactosaminyltransferase 1 27 88 Homo sapiens 971461 polypeptide N-acetylgalactosaminyl transferase 1 27 89 Homo sapiens 6318186 polypeptide N-acetylgalactosaminyltransferase 7 1 27 90 Macaca fascicularis 11041469 polypeptide N-acetylgalactosaminyltransferase 1 27 91 Mus musculus 1575723 polypeptide N-acetylgalactosaminyltransferase-T3 1 27 92 Mus musculus 13650039 polypeptide GalNAc transferase-T2 1 27 93 Mus musculus 13650041 polypeptide N-acetylgalactosaminyltransferase 7 1 27 94 Rattus norvegicus 1141792 polypeptide GalNAc transferase 1 27
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from
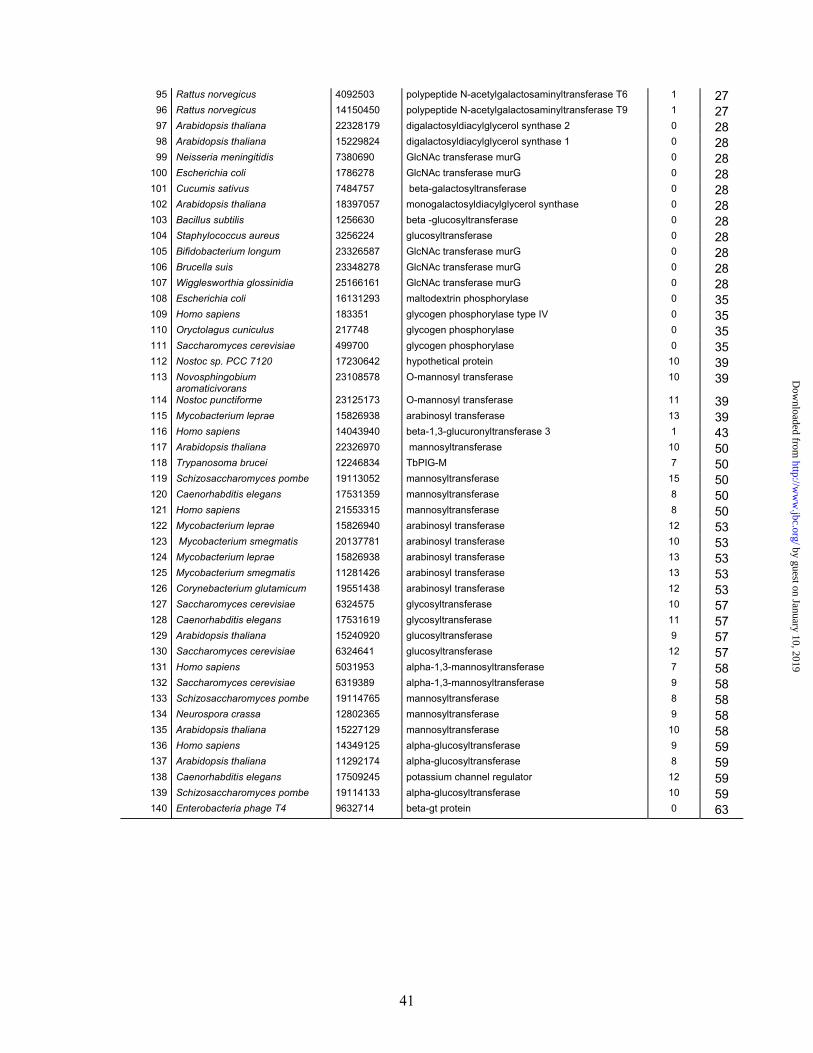
41
95 Rattus norvegicus 4092503 polypeptide N-acetylgalactosaminyltransferase T6 1 27 96 Rattus norvegicus 14150450 polypeptide N-acetylgalactosaminyltransferase T9 1 27 97 Arabidopsis thaliana 22328179 digalactosyldiacylglycerol synthase 2 0 28 98 Arabidopsis thaliana 15229824 digalactosyldiacylglycerol synthase 1 0 28 99 Neisseria meningitidis 7380690 GlcNAc transferase murG 0 28
100 Escherichia coli 1786278 GlcNAc transferase murG 0 28 101 Cucumis sativus 7484757 beta-galactosyltransferase 0 28 102 Arabidopsis thaliana 18397057 monogalactosyldiacylglycerol synthase 0 28 103 Bacillus subtilis 1256630 beta -glucosyltransferase 0 28 104 Staphylococcus aureus 3256224 glucosyltransferase 0 28 105 Bifidobacterium longum 23326587 GlcNAc transferase murG 0 28 106 Brucella suis 23348278 GlcNAc transferase murG 0 28 107 Wigglesworthia glossinidia 25166161 GlcNAc transferase murG 0 28 108 Escherichia coli 16131293 maltodextrin phosphorylase 0 35 109 Homo sapiens 183351 glycogen phosphorylase type IV 0 35 110 Oryctolagus cuniculus 217748 glycogen phosphorylase 0 35 111 Saccharomyces cerevisiae 499700 glycogen phosphorylase 0 35 112 Nostoc sp. PCC 7120 17230642 hypothetical protein 10 39 113 Novosphingobium
aromaticivorans 23108578 O-mannosyl transferase 10 39
114 Nostoc punctiforme 23125173 O-mannosyl transferase 11 39 115 Mycobacterium leprae 15826938 arabinosyl transferase 13 39 116 Homo sapiens 14043940 beta-1,3-glucuronyltransferase 3 1 43 117 Arabidopsis thaliana 22326970 mannosyltransferase 10 50 118 Trypanosoma brucei 12246834 TbPIG-M 7 50 119 Schizosaccharomyces pombe 19113052 mannosyltransferase 15 50 120 Caenorhabditis elegans 17531359 mannosyltransferase 8 50 121 Homo sapiens 21553315 mannosyltransferase 8 50 122 Mycobacterium leprae 15826940 arabinosyl transferase 12 53 123 Mycobacterium smegmatis 20137781 arabinosyl transferase 10 53 124 Mycobacterium leprae 15826938 arabinosyl transferase 13 53 125 Mycobacterium smegmatis 11281426 arabinosyl transferase 13 53 126 Corynebacterium glutamicum 19551438 arabinosyl transferase 12 53 127 Saccharomyces cerevisiae 6324575 glycosyltransferase 10 57 128 Caenorhabditis elegans 17531619 glycosyltransferase 11 57 129 Arabidopsis thaliana 15240920 glucosyltransferase 9 57 130 Saccharomyces cerevisiae 6324641 glucosyltransferase 12 57 131 Homo sapiens 5031953 alpha-1,3-mannosyltransferase 7 58 132 Saccharomyces cerevisiae 6319389 alpha-1,3-mannosyltransferase 9 58 133 Schizosaccharomyces pombe 19114765 mannosyltransferase 8 58 134 Neurospora crassa 12802365 mannosyltransferase 9 58 135 Arabidopsis thaliana 15227129 mannosyltransferase 10 58 136 Homo sapiens 14349125 alpha-glucosyltransferase 9 59 137 Arabidopsis thaliana 11292174 alpha-glucosyltransferase 8 59 138 Caenorhabditis elegans 17509245 potassium channel regulator 12 59 139 Schizosaccharomyces pombe 19114133 alpha-glucosyltransferase 10 59 140 Enterobacteria phage T4 9632714 beta-gt protein 0 63
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from

Maria L. Rosén, Maria Edman, Michael Sjöström and Åke Wieslandersequence analysis
Recognition of fold and sugar linkage for glycosyltransferases by multivariate
published online May 17, 2004J. Biol. Chem.
10.1074/jbc.M402925200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2004/06/10/M402925200.DC1
by guest on January 10, 2019http://w
ww
.jbc.org/D
ownloaded from









![Putative Glycosyltransferases and Other Plant Golgi ... · Putative Glycosyltransferases and Other Plant Golgi Apparatus Proteins Are Revealed by LOPIT Proteomics1[W] Nino Nikolovski,](https://static.fdocuments.in/doc/165x107/5beabde209d3f2ff498bfa69/putative-glycosyltransferases-and-other-plant-golgi-putative-glycosyltransferases.jpg)








