
Reata Pharmaceuticals: Creating New Opportunities for Patients
44
Creating New Opportunities for Patients
-
Upload
jon-le-culpepper -
Category
Health & Medicine
-
view
804 -
download
0
Transcript of Reata Pharmaceuticals: Creating New Opportunities for Patients

Creating New Opportunities for Patients

Forward-Looking Statements
This presentation contains certain “forward-looking” statements that are made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. All statements, other than statements of historical or present facts, are forward-looking statements, including statements regarding our future financial condition, business strategy, and plans and objectives of management for future operations. In some cases, you can identify forward-looking statements by terminology such as “believe,” “will,” “may,” “estimate,” “continue,” “aim,” “assume,” “anticipate,” “contemplate,” “intend,” “target,” “project,” “should,” “plan,” “expect,” “predict,” “could,” “possible,” “seek,” “goal”, “potential,” “hypothesize,” “likely” or the negative of these terms or other similar terms or expressions that concern our expectations, strategy, plans, or intentions. These statements are based on our intentions, beliefs, projections, outlook, analyses, or current expectations using currently available information, are not guarantees of future performance, and involve certain risks and uncertainties. Although we believe that the expectations reflected in these forward-looking statements are reasonable, we cannot assure you that our expectations will prove to be correct. Therefore, actual outcomes and results could materially differ from what is expressed, implied, or forecast in these statements. Any differences could be caused by a number of factors including but not limited to: the success, cost, and timing of our product development activities and clinical trials; our ability to advance our AIMs and other technologies; our ability to obtain and maintain regulatory approval of our product candidates, and limitations and warnings in the label of an approved product candidate; our ability to obtain funding for our operations, including funding necessary to complete further development and commercialization of our product candidates; our plans to research, develop, and commercialize our product candidates; the commercialization of our product candidates, if approved; the rate and degree of market acceptance of our product candidates; the size and growth potential of the markets for our product candidates, and our ability to identify target patient populations and serve those markets, especially for diseases with small patient populations; the success of competing therapies that are or may become available; our expectations regarding our ability to obtain and maintain intellectual property protection for our product candidates; the ability to license additional intellectual property relating to our product candidates and to comply with our existing license agreements; our ability to contract with third-party suppliers and manufacturers and their ability to perform adequately; our ability to attract collaborators with development, regulatory, and commercialization expertise;our ability to attract and retain key scientific or management personnel; our ability to grow our organization and increase the size of our facilities to meet our anticipated growth; the accuracy of our estimates regarding expenses, future revenue, capital requirements, and needs for additional financing; and regulatory developments in the United States and foreign countries.
Additional factors that could cause actual results to differ materially from our expectations can be found in our Securities and Exchange Commission filings. Moreover, we operate in a very competitive and rapidly changing environment. New risk factors emerge from time to time, and it is not possible for our management to predict all risk factors nor can we assess the effects of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in, or implied by, any forward-looking statements. All forward-looking statements included in this website are expressly qualified in their entirety by these cautionary statements. The forward-looking statements speak only as of the date made and, other than as required by law, we undertake no obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events, or otherwise.
2

Company Overview
Developing small-molecule drugs for orphan diseases where restoring mitochondrial function and reducing inflammation can produce meaningful clinical benefit
Lead programs: Nrf2-activators bardoxolone methyl (Bard) and omaveloxolone (Omav)– Mitochondrial dysfunction, chronic inflammation, and Nrf2 suppression are the pathogeneses
of the diseases we target– Nrf2 is a transcription factor that regulates over 200 genes, including many that restore energy
production, downregulate inflammation, and reduce fibrosis
Bard in Phase 3 for connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH) arising from scleroderma, lupus, and related CTD diseases – Phase 2 demonstrated significant improvement in six-minute walk distance (6MWD)– Phase 3 trial actively recruiting, with data expected H1 2018
Three orphan disease programs with potential to begin registrational portion of development during 2017– Friedreich’s Ataxia: Part 1 data 1H17, potential Part 2 pivotal program initiation 2H17– Alport Syndrome: Phase 2 data YE17, potential pivotal program initiation 2H17– Mitochondrial myopathy: Part 1 data 1H17, potential Part 2 pivotal program initiation 2H17
Additional Phase 2 programs ongoing for Bard in pulmonary hypertension caused by interstitial lung disease (PH-ILD) and Omav in immuno-oncology
3
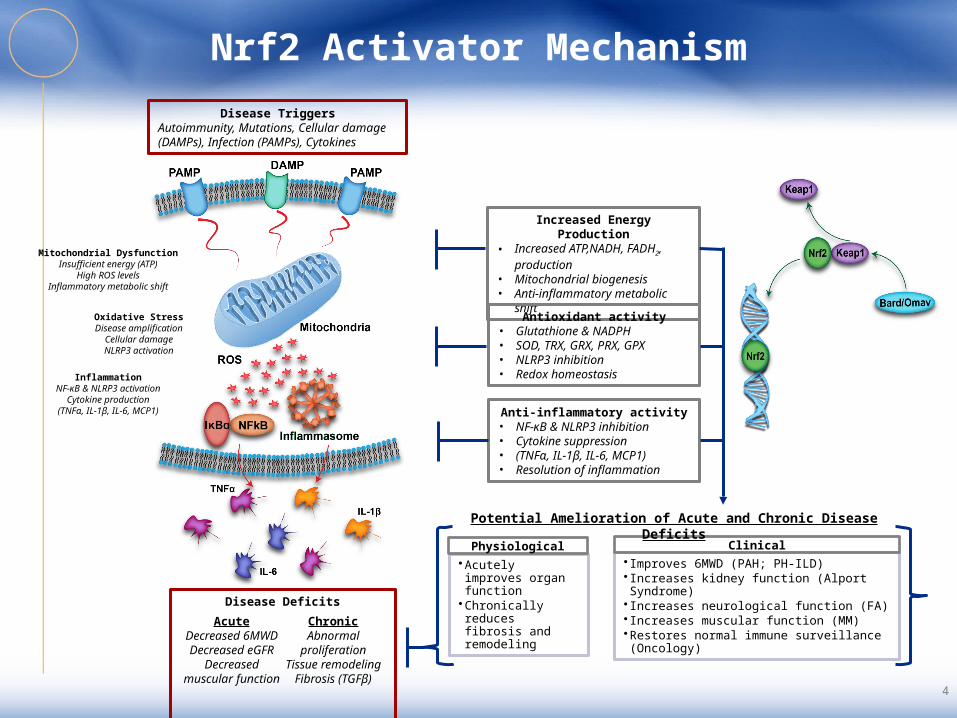
Nrf2 Activator Mechanism
Increased Energy Production• Increased ATP,NADH, FADH2,
production• Mitochondrial biogenesis• Anti-inflammatory metabolic shift
Antioxidant activity• Glutathione & NADPH • SOD, TRX, GRX, PRX, GPX• NLRP3 inhibition• Redox homeostasis
Anti-inflammatory activity• NF-κB & NLRP3 inhibition• Cytokine suppression• (TNFα, IL-1β, IL-6, MCP1)• Resolution of inflammation
Disease TriggersAutoimmunity, Mutations, Cellular damage (DAMPs), Infection (PAMPs), Cytokines
•Acutely improves organ function
•Chronically reduces fibrosis and remodeling
Physiological• Improves 6MWD (PAH; PH-ILD)• Increases kidney function (Alport Syndrome)• Increases neurological function (FA)• Increases muscular function (MM)•Restores normal immune surveillance (Oncology)
Clinical
Disease DeficitsAcute
Decreased 6MWDDecreased eGFR
Decreased muscular function
Chronic
Abnormal proliferation
Tissue remodelingFibrosis (TGFβ)
Potential Amelioration of Acute and Chronic Disease Deficits
4
Mitochondrial DysfunctionInsufficient energy (ATP)
High ROS levelsInflammatory metabolic shift
Oxidative StressDisease amplification
Cellular damageNLRP3 activation
InflammationNF-κB & NLRP3 activation
Cytokine production(TNFα, IL-1β, IL-6, MCP1)

Bardoxolone Methyl in CTD-PAH
5

CTD-PAH: A Brief Summary
CTD-PAH Pathophysiology– PAH that occurs in patients with connective tissue diseases such as scleroderma
and lupus erythematosus– Impaired bioenergetics, inflammation, fibrosis, and tissue remodeling implicated as
prime drivers of PAH
Patient Statistics– Among scleroderma or lupus patients, 10-15% have CTD-PAH– CTD-PAH is the leading cause of death for lupus and scleroderma patients– 5-year survival of scleroderma patients without PAH is 85% versus 10% with PAH– Prevalence of 12,000 patients in the US, 52,000 worldwide; 30% of all PAH
Disease Management– Vasodilators such as PDE-5 inhibitors, endothelin receptor antagonists (ERAs) and
prostacyclins are the only current treatment options– Vasodilators are significantly less potent in CTD-PAH versus idiopathic PAH (I-PAH)– Poor risk-benefit for vasodilators in CTD-PAH given minimal treatment effect and
adverse events resulting from systemic vasodilation
6

CTD-PAH is Distinct from Idiopathic PAH
Survival in CTD vs Idiopathic PAH Patients
CTD-PAH is a more severe disease than I-PAH– Median survival in U.S. is 4 years for CTD-PAH versus 7 years for I-PAH patients– Worse baseline six-minute walk distance (6MWD)
Compared to I-PAH, CTD-PAH is driven more by fibrosis than by impaired hemodynamics, reducing the impact of vasodilators– Vasodilators produce 3x the impact in 6MWD in I-PAH versus CTD-PAH– Fibrosis limits the ability of arteries to vasodilate, limiting vasodilator efficacy
*6MWD – Six-Minute Walk Distance
Rhee Meta-AnalysisHemodynamics and 6MWD Change for
CTD vs Idiopathic PAH Patients CTD-PAH I-PAHBaseline Characteristics RAP, mmHg 8 9.3
mPAP, mmHg 46 55
PVR, WU 10 13 6MWD (m) 351 365Response to Vasodilator Therapy 6MWD (m) 9.6 30.1
7

LARIAT: Phase 2 Trial of Bard Combined with Approved Therapies
US-only, Phase 2, double-blind, randomized, placebo-controlled trial– PAH patients required to be on 1-2 background therapies– Part 1: Dose ranging and titration cohorts
• Initially tested fixed doses of 2.5, 5, and 10 mg once daily (n=8 per dose group)• Subsequently tested titration design of 5 10 mg once daily
– Part 2: Open-label extension trial
Primary Objectives– Change in 6MWD from baseline through 16 weeks– Safety and tolerability through 16 weeks
6-Minute Walk TestE EchocardiogramT Telephone Contact
Screening Part 1
W2 Scr B W1 W8 W12Scr A
E
D1
E
D3
T
D10
T
W3
T
W4
E
W16
E
Bardoxolone Methyl
3:1
RPlacebo
Extension
Bardoxolone Methyl
8

Expanded CTD-PAH Data Demonstrate Robust Treatment Effect
Primary efficacy analysis of initial cohorts presented at CHEST 2015 showed a placebo-corrected 6MWD of 21 m (p=0.037) at doses of 2.5, 5, and 10 mg
Performed analysis of all CTD-PAH patients from LARIAT who were dosed up to 10 mg and completed Week 16– Included all patients and those who are CATALYST-eligible– Patients with moderate to severe anemia are excluded from CATALYST because
anemia treatments can affect 6MWD independent of study drug
Dataset Treatment NTime-Averaged Δ6MWD (m) Week 16 Δ6MWD (m)
Change from Baseline Placebo-corrected Change from
BaselinePlacebo-corrected
AllPlacebo 7 0.6
p=0.96 - 9.8p=0.44 -
BARD 15 26.7p<0.001
26.1P=0.06
38.2p<0.001
28.4p=0.07
CATALYST Eligible
Placebo 5 -10.1p=0.39 - -5.8
p=0.68 -
BARD 14 30.2p<0.001
40.3p=0.009
42.7p<0.001
48.5p=0.005
Time-averaged change in Bard patients consistent with earlier data– CATALYST-eligible patients showed larger change– Pooled standard deviation (SD) in LARIAT was 37 m, lower than estimated in trial
design for CATALYST (50 m)
9
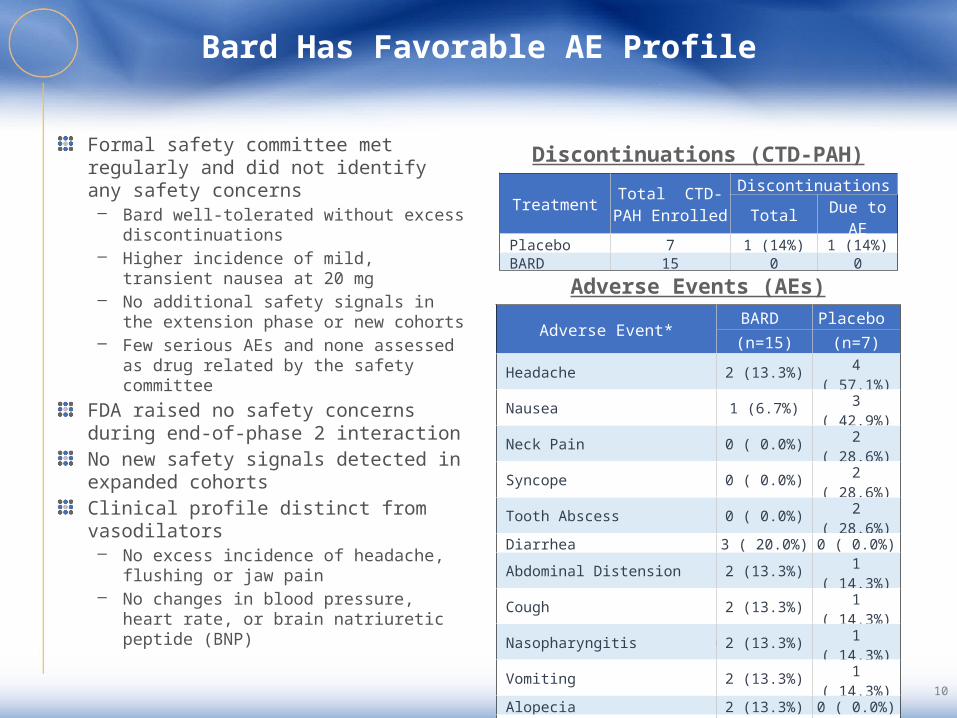
Bard Has Favorable AE Profile
Formal safety committee met regularly and did not identify any safety concerns– Bard well-tolerated without excess
discontinuations– Higher incidence of mild, transient
nausea at 20 mg– No additional safety signals in the
extension phase or new cohorts– Few serious AEs and none assessed as
drug related by the safety committeeFDA raised no safety concerns during end-of-phase 2 interactionNo new safety signals detected in expanded cohortsClinical profile distinct from vasodilators– No excess incidence of headache,
flushing or jaw pain– No changes in blood pressure, heart
rate, or brain natriuretic peptide (BNP)
*Listed are adverse events that occurred in ≥2 of placebo or BARD patients
Adverse Events (AEs)
Discontinuations (CTD-PAH)
Treatment Total CTD-PAH Enrolled
Discontinuations
Total Due to AE
Placebo 7 1 (14%) 1 (14%) BARD 15 0 0
Adverse Event*BARD Placebo (n=15) (n=7)
Headache 2 (13.3%) 4 ( 57.1%) Nausea 1 (6.7%) 3 ( 42.9%) Neck Pain 0 ( 0.0%) 2 ( 28.6%) Syncope 0 ( 0.0%) 2 ( 28.6%) Tooth Abscess 0 ( 0.0%) 2 ( 28.6%) Diarrhea 3 ( 20.0%) 0 ( 0.0%) Abdominal Distension 2 (13.3%) 1 ( 14.3%) Cough 2 (13.3%) 1 ( 14.3%) Nasopharyngitis 2 (13.3%) 1 ( 14.3%) Vomiting 2 (13.3%) 1 ( 14.3%) Alopecia 2 (13.3%) 0 ( 0.0%) Anemia 2 (13.3%) 0 ( 0.0%) Muscle Spasms 2 (13.3%) 0 ( 0.0%) Tachycardia 2 (13.3%) 0 ( 0.0%)
10

CATALYST: Phase 3 Trial in CTD-PAH Underway
FDA concurred with proposed Phase 3 trial in CTD-PAH– International, double-blind, randomized, placebo-controlled trial– Will enroll 130 to 220 patients on 0-2 background therapies, randomized 1:1 to
receive Bard (5 10 mg) or placebo for 24 weeks– Pre-specified adaptive component will assess baseline characteristics and
variability, as well as potentially modify the sample size to maintain power– Primary endpoint: 6MWD at week 24
6-Minute Walk TestT Telephone Contact
Screening Dose-Titration Period
W2W1 W8 W16Scr D1 D3
T
D10
T
W3
T
W4
Maintenance
W24
R
Placebo
1:1 Randomization
D31
T
W5
T
W6 W7
T
Bardoxolone Methyl (5 mg) Bardoxolone Methyl (10 mg)
6-Minute Walk TestT Telephone Contact
Screening Dose-Titration Period
W2W1 W8 W16Scr D1 D3
T
D10
T
W3
T
W4
Maintenance
W24
R
Placebo
1:1 Randomization
D31
T
W5
T
W6 W7
T
Bardoxolone Methyl (5 mg) Bardoxolone Methyl (10 mg)
Data confirm that power calculations for CATALYST are appropriate– LARIAT end-of-treatment analysis shows placebo-corrected change in 6MWD of
48.5 m (p=0.005) at week 16– CATALYST powered to detect 12.5 m change at 24 weeks with p<0.05
First patient enrolled, data expected H1 2018
11

Potential Bard PH Target Product Profile
Bard has potential to become the first treatment approved specifically for CTD-PAH– Bard may improve functional outcomes on top of optimized vasodilator therapy and can be
dosed in combination with approved PAH therapies due to a lack of systemic hemodynamic effects
– Bard may benefit patients who are underserved by vasodilator therapies, including patients with CTD-PAH, PH-ILD, and those with earlier stage disease
– Outcomes over time may improve due to Bard’s anti-inflammatory, anti-proliferative, and anti-fibrotic effects
– Bard’s toxicity profile does not compound that of vasodilators, e.g. jaw pain, dizziness– Convenient, once-daily, oral dosing
Large unmet need and market opportunity for CTD-PAH and PH-ILD– In the U.S., approximately 12,000 CTD-PAH patients and over 60,000 PH patients within the
subtypes being studied in LARIAT– Approximately 60,000 of the above patients have no approved therapy– Several vasodilators are already generic, and we believe more actively-marketed brands in
PAH will go generic prior to the launch of Bard
12

Omaveloxolone in Friedreich’s Ataxia
13

Friedreich’s Ataxia (FA): A Brief Summary
FA Pathophysiology– Caused by mutations that result in hypo-expression of frataxin– Frataxin is responsible for biosynthesis of iron-sulfur complexes in the mitochondria
used by OXPHOS complexes– Nrf2 is suppressed in FA due to frataxin deficiency, which leads to increased
oxidative stress and impaired energy production– Typically thought of as a neurodegenerative disease; however FA is a multi-system
disease that includes cardiomyopathy, diabetes, and fatigue
Patient Statistics– On average, patients are diagnosed in their early teens, wheelchair-bound in their
20s, and die in the 30s– Cardiomyopathy is the most common cause of death– Prevalence is approximately 6,000 in the U.S. and approximately 16,000 ex-U.S.
Disease Management– No treatments are currently approved – Roughly 50% of patients take vitamin cocktails and antioxidants which have shown
no reproducible activity
14

Nrf2 and Mitochondria are Dysregulated in FA
Nrf2 is suppressed and signaling is grossly impaired in FA and contributes to oxidative stress, mitochondrial dysfunction and reduced ATP production– Reduced nuclear expression of Nrf2– Reduced levels of glutathione and antioxidant Nrf2 target genes
In FA patients, mitochondrial function (oxygen consumption) is inversely correlated with neurologic function (FARS)Omav has the potential to address underlying pathophysiology of FA and MM by rescuing mitochondrial function and increasing ATP production
Shan Y et al., Antioxidant Redox Signal (2013); D’Oria V et al., Int J Mol Sci (2013); V Paupe et al, PLoS One (2009); Drinkard BE et al., Arch Phys Med Rehabil (2010)
Mito Function in FA Mito Function in FA BiopsiesNrf2 Activation Status
15

Friedreich’s Ataxia Clinical Program
Met with FDA in 2014 to discuss FA program – Recommended two-part design with a robust dose evaluation as “Part 1” and a randomized,
placebo controlled “Part 2” that could support registration – Recommended collection of multiple clinically-meaningful endpoints
Currently enrolling Part 1 of MOXIe, a randomized, placebo-controlled, international trial – MOXIe Part 1 is the only large, placebo-controlled, multi-center Phase 2 trial run in FA– Conducted in genetically-confirmed patients– Will enroll up to 72 patients, with a treatment duration of 12 weeks
Robust design includes relatively large number of patients with multiple clinically-meaningful endpoints and independent safety oversight– Neurological assessments: modified FARS (mFARS), 9-hole peg test, timed 25 foot walk test– Muscular and physiological assessments– Safety overseen by data safety monitoring board– Not powered for efficacy; assessing dose-dependent activity and separation from placebo
16

Friedreich’s Ataxia Clinical Program
Enrollment in Part 1 proceeding well with no safety or operational issues to date– Dose ranging study from 2.5 up to 300 mg given orally once daily– Cohorts of 8 patients randomized 3:1 to RTA 408 or placebo– 12 week treatment duration– Goal is to demonstrate pharmacodynamic changes associated with Nrf2 activation that
translate to improvements in clinical assessmentsOpportunity to amend Part 2 design based on Part 1 resultsMOXIe Part 2 will be a pivotal, randomized, international, placebo-controlled trial– Will select active dose from Part 1– 12 week or longer treatment duration– Assessing same endpoints as Part 1– Expected to support registration
Part 1 data available in H1 2017, can move to pivotal H2 2017
17

Potential Omav FA Target Product Profile
Omav has potential to become the first treatment approved for FA patients– FA is a deadly, genetically-linked disease impacting both sexes– FA is severely debilitating and ultimately fatal disease that predominately affects
children
Omav may directly improve neurological performance of FA patients and has the potential to prevent or delay disease progression– To date, Bard has demonstrated an acceptable safety profile in FA and other trials– Convenient, once-daily, oral dosing
Prevalence in U.S. is approximately 6,000 and approximately 16,000 outside the U.S.– Without existing treatment options, prevalence may be currently underestimated– With a therapy available, awareness of genetic testing may increase diagnosed
population
Omav pricing expected to be consistent with other disease-modifying therapies for orphan and ultra-orphan indications
18

Bardoxolone Methyl in Alport Syndrome
19

Alport Syndrome: A Brief Summary
Alport Syndrome Pathophysiology– Mutations in type IV collagen cause defects in the glomerular basement membrane– Patients usually present with hematuria, and then develop proteinuria and
deterioration of kidney function, leading to end-stage renal disease (ESRD)– Due to loss of type IV collagen, deafness and ocular abnormalities also common– Like diabetic chronic kidney disease (CKD), inflammation, fibrosis, and oxidative
stress are known to promote disease and drive progressive loss of kidney function
Patient Statistics– Median age at initiation of ESRD for males with most common form is 25– Median survival of approximately 55 years– Prevalence of approximately 12,000 patients in the US and over 100,000 globally
Current Disease Management– No approved therapies– ACE inhibitors and ARBs are commonly used off-label– Dialysis or renal transplant is needed in most patients
20

Pathogenesis of Alport Syndrome
Mutations in type IV collagen result in an abnormal composition of the glomerular basement membrane (GBM)Progressive damage to the GBM leads to:– Increased leakage of proteins – Increased activation of inflammatory mediators (NF-κB, IL-6, MCP-1, etc.) and
remodeling and fibrosis pathways (TGFβ and CTGF)– Inflammation, glomerular sclerosis, tubular atrophy, and interstitial fibrosis
Distinct insult from diabetic CKD (collagen mutation vs hyperglycemia) but similar mechanism of disease progression
Kowh et al., Annu Rev Pathol Mech Dis (2006); Gross et al., Matrix Biology (2010)21

Bard has Consistently Improved Renal Function in Clinical Trials
Bard significantly increased markers of renal function such as estimated glomerular filtration rate (eGFR), inulin clearance, and creatinine clearance across 10 clinical trials enrolling over 2,800 patients
StudyPhase/
CountryPatient
Population N∆eGFR
(mL/min/1.73m2)
402-C-0903 (BEACON) 3/Global CKD/Diabetes 2185 6.4 (p<0.001 vs PBO)
402-C-0804 (BEAM) 2/US CKD/Diabetes 227 8.6 (p<0.001 vs PBO)
RTA402-005 (TSUBAKI) 2/Japan CKD/Diabetes 40 Data not yet publicly disclosed
402-C-0902 2/US CKD/Diabetes 131 6.5 (p<0.001)
402-C-0801 (Stratum 1) 2a/US CKD/Diabetes 60 6.7 (p<0.001)
402-C-0801 (Stratum 2) 2b/US CKD/Diabetes 20 7.2 (p<0.001)
402-C-1102 1/US CKD/Diabetes 24 9.0 (p<0.05)
402-C-0501 1/US Cancer 47 18.2 (p<0.0001)
402-C-0702 1/2/US Cancer 34 32.2 (p=0.001)
402-C-1302 (LARIAT) 2/US Pulmonary Hypertension 54 14.7 (p<0.001 vs PBO)
22

Patients with CKD and Type 2 Diabetes Show Sustained Renal Function Improvement
Bard significantly increased eGFR (p<0.0001) in BEAM and BEACON
Change was durable through one year, the longest duration tested
Changes were associated with fewer renal SAEs and ESRD events
Pergola et al., NEJM (2011); De Zeeuw et al., NEJM (2013)
Number of PatientsPBO 1093 1023 885 726 547 402 281 125BARD 1092 958 795 628 461 345 241 103
0 8 16 24 32 40 48 5620.0
24.0
28.0
32.0
PBO BARD
Treatment WeekM
ean
eGFR
± S
E (m
l/min
/1.7
3 m
2)
-4
0
4
8
12
16
20
0 4 8 12 16 20 24 28 32 36 40 44 48 52
Mea
n eG
FR C
hang
e (m
l/min
/1.7
3 m
2 )
Week
150 mg (n = 14)
75 mg (n = 42)
25 mg (n = 69)
Placebo (n=51)
BEACONBEAM
23

Improvements in Renal Function are Retained After Withdrawal of Bard
In BEAM and BEACON, a significant, placebo-corrected improvement in eGFR was retained after withdrawal of drug for four weeks– eGFR assessed after sub-therapeutic concentrations reached– End-of-treatment and withdrawal eGFR changes correlate– The larger the effect on-treatment, the larger the retained benefit– Data suggest Bard affects chronic remodeling and fibrosis
Withdrawal Analysis
Baseline eGFR
Placebo-Corrected
∆eGFR Post-Withdrawal
P-value
BEAM (n=172)
Low Dose 33 0.6 p>0.05
Mid Dose 32 4.7 p<0.05
High Dose 32 5.0 p<0.05
BEACON (n=498)
20 mg 23 1.8 p<0.001
24

Development Path for Bard in Alport Syndrome
August 2016, Reata sought guidance from the FDA on a Phase 2 program in CKD caused by Alport syndrome
In October 2016, the FDA proposed a more efficient path to approval, recommending Reata run a single, pivotal trial
FDA provided guidance that change in eGFR would support registration – Did not require a time to dialysis or transplant endpoint (ESRD)– Accelerated approval would be supported by a retained improvement in eGFR following
48 weeks of treatment and 4 weeks off drug– Full approval would be supported by a retained improvement in eGFR following 2 years
of treatment and 4 weeks off drug
Enrollment of open-label Phase 2 portion of Phase 2/3 trial begins 1H17
Data from Phase 2 and go-forward decision on Phase 3 expected YE 2017
Year-one data from Phase 3 expected H1 2019
25

Alport Syndrome Phase 2/3 Trial Design
Trial designed with input from international key opinion leaders and the Alport Syndrome Foundation– Will enroll patients from 12 to 60 years old– Will enroll a broad range of kidney function (eGFR between 30-90 mL/min/1.73 m2)– Patients with stage 4 CKD, cardiovascular disease, or fluid retention will be excluded
Phase 2 Cohort– Open-label cohort enrolling a total of 30 patients
• 15 with microalbuminuria• 15 with macroalbuminuria
– Will measure change in eGFR following 12 weeks of treatment– Data not included in analysis of primary and secondary endpoints for Phase 3– Enrollment expected to begin 1H17
Phase 3 Cohort– Will enroll up to 180 patients with two years of follow-up– Placebo-controlled, double-blind trial with 1:1 randomization
26

Alport Syndrome Phase 2/3 Trial Design
Titration to be used to reach goal dose of 20 or 30 mg given orally, once daily
Endpoints for Accelerated and Full Approval– Primary endpoint of on-treatment eGFR change at Week 48– Key Secondary endpoint of eGFR change after 4 weeks of drug withdrawal (Week 52)– Additional secondary endpoints of eGFR change at Week 100 and after 4 weeks of drug
withdrawal (Week 104)– Significant and robust effect on primary and key secondary endpoint (Week 52) could serve
as the basis for accelerated approval– Continued effect at Week 104 could serve as the basis for full approval
Screening Dose-Titration Period
W2W1 W8 W12ScrA D1 D3
T
D10
T
W3
T
W4
Maintenance
W24
R
Placebo*
1:1 Randomization
D31
T
W5
T
W6 W7
T
BARD (5 mg)* BARD (20 mg)
W36 W48 W52
W/D
Off Study Drug
BARD (10 mg)
Primary efficacy analysisO eGFR determinationT Telephone Contact
W64 W76 W88 W100
BARD (20 mg)
ScrB
Maintenance
Placebo
BARD (5 mg)* BARD (20 mg)BARD (10 mg) BARD (30 mg) BARD (30 mg)
Baseline ACR ≤ 300 mg/g
Baseline ACR > 300 mg/g
W104
F/U
Off Study Drug
* Patients under the age of 18 will receive study drug (BARD or placebo) every other day during Week 1
Phase 3 Cohort
27

Design Implications From Previous Trials
Design leverages data from 2,600 CKD patients and 400 treated for 48 weeks– Withdrawal change predicted by on-treatment change – On-treatment change is predicted by baseline eGFR, baseline ACR, and drug exposure
Monitoring and dose titration to minimize risk of adverse events– Both approaches have worked well in partner’s ongoing diabetic CKD trial– Titration has minimized adverse events and drop-outs in our trials (PAH/PH-ILD)– Careful monitoring after randomization allows for early intervention
99% and 90% power to achieve primary and secondary endpoints– Goal is to replicate BEAM (4.7 mL/min/1.73 m2 difference) with twice as many patients– Study could be successful with effect that is approximately 50% less than BEAM
NBL
eGFR
Placebo-Corrected ∆eGFR
Week 12 One Year Withdrawal
BEAM: Mid Dose 90 32.3 12 16 (p<0.001) 4.7 (p=0.02)
CARDINAL Modeling
Replicate BEAM with more patients 180 60 12 16 (p<0.001) 4.7 (p<0.001)
Minimal detectable difference 2.5 (p<0.05)
28

Potential Bard Alport Syndrome Target Product Profile
Bard has potential to become the first treatment approved for Alport syndrome– No therapies are approved for this deadly, chronic, orphan disease– Off-label use of RAAS agents has minimal impact of slowing disease progression
Bard has a novel, potent mechanism that affects inflammation, fibrosis and GFR– Bard targets the underlying renal pathophysiology driving disease– Bard inhibits the acute and chronic inflammation-mediated pathways, and exerts anti-
proliferative and anti-fibrotic effects that affect tissue remodeling and GFR loss– Bard is an easy-to-administer, oral, once-daily capsule– Bard has the potential to be combined with any of class of RAAS compounds
Meeting approval endpoint of retained benefit is strong evidence of disease-modifying activity and can drive adoption and reimbursement
Approximately 12,000 patients in the U.S. and over 100,000 outside the U.S.
Bard pricing expected to be consistent with other disease modifying therapies for orphan and ultra-orphan indications
29

Omaveloxolone in Mitochondrial Myopathies
30

Mitochondrial Myopathies (MM): A Brief Summary
MM Pathophysiology – MM refers to an array of diseases caused by mutations in either mitochondrial
DNA (mtDNA) or nuclear genes encoding mitochondrial proteins used in OXPHOS – Severe reductions in ATP production affect muscle and neurons disproportionately
given the high energy needs of these cell types
Patient Characteristics– MM patients share the phenotype of reduced ATP production, but experience this
deficit in different ways, manifesting as diseases with distinct characteristics. Included in MM are:
Disease Management – No treatments are currently approved for any MM– Supplements such as creatine and antioxidants are often used but have shown no
reproducible activity
• Leigh syndrome and maternally inherited Leigh syndrome (MILS)
• Kearns-Sayre syndrome (KSS) Mitochondrial DNA depletion syndrome (MDS)
• Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS)
• Progressive external ophthalmoplegia (PEO)
• Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)
• Myoclonus epilepsy with ragged red fibers (MERRF)
• Neuropathy, ataxia and retinitis pigmentosa (NARP)
• Pearson syndrome
31

Omav Addresses Mitochondrial Deficits of MM
Mitochondrial dysfunction and reduced ATP production are the cause of MM– Muscle biopsies show impaired bioenergetics– Causes reduced exercise capacity and fatigue
Exercise capacity is routinely assessed clinically – MM patients have capacity to increase physical
activity with exercise training– Exercise training increases density of
mitochondria
Omav could improve exercise capacity by:– Increasing mitochondria in normal muscle – Rescuing ATP production in dysfunctional
myocytes
Taivassalo et al., Brain (2003); Jeppesen et al., Brain (2006); Tuppen HAL et al., Biochimica Biophysica Acta (2009); Murphy JL et al., Neuromuscular Dis (2012) 32

Mitochondrial Myopathies Clinical Program
Met with FDA in 2014 to discuss MM program – Recommended two-part design with a robust dose evaluation as “Part 1” and a
randomized, placebo controlled “Part 2” that could support registration – Recommended collection of multiple clinically meaningful endpoints
Currently enrolling Part 1 of MOTOR, a randomized, placebo-controlled, international trial – Conducted in genetically-confirmed patients– Will enroll up to 72 patients, with a treatment duration of 12 weeks
Robust design includes relatively large number of patients with multiple clinically-meaningful endpoints and independent safety oversight– Muscular and physiological assessments: exercise testing using metabolic cart– Functional capacity as assessed by 6MWD– Quality of life instruments to assess fatigue and activities of daily living– Safety overseen by data safety monitoring board– Not powered for efficacy; assessing dose-dependent activity and separation from
placebo
33

Mitochondrial Myopathies Clinical Program
Enrollment in Part 1 proceeding well with no safety or operational issues to date– Dose ranging study from 2.5 up to 300 mg given orally once daily– Cohorts of 8 patients randomized 3:1 to RTA 408 or placebo– 12 week treatment duration– Goal is to demonstrate pharmacodynamic changes associated with Nrf2 activation
that translate to improvements in clinical assessmentsOpportunity to amend Part 2 design based on Part 1 resultsMOTOR Part 2 will be a pivotal, randomized, international, placebo-controlled trial– Will select active dose from Part 1– 12 week or longer treatment duration– Assessing same endpoints as Part 1– Expected to support registration
Part 1 data available in H1 2017, can move to pivotal H2 2017
34

Potential Bard MM Target Product Profile
Omav has potential to become the first treatment approved for MM patients– MM is a deadly, genetically-linked group of diseases impacting both sexes– MM is severely debilitating group of diseases that affects both children and adults
Omav may directly improve functional capacity of MM patients and has the potential to prevent or delay disease progression– To date, Omav has demonstrated an acceptable safety profile in FA and other trials– Convenient, once-daily, oral dosing
Prevalence in U.S. is approximately 20,000 and approximately 60,000 outside U.S.– Without existing treatment options, prevalence may be currently underestimated– With a therapy available, awareness of genetic testing may increase diagnosed population
Omav pricing expected to be consistent with other disease modifying therapies for orphan and ultra-orphan indications
35

Additional Programs
36

Bard in PH-ILD Patients
LARIAT protocol expanded to include 4 randomized, placebo-controlled trials for Bard on top of optimized background therapy in ILD subtypes– Enrollment underway– Expecting 12-week data on 6MWD for all trials in 2H17
PH from interstitial lung disease is similar to PAH from connective tissue disease– Similar etiology: whereas CTD-PAH is driven by fibrosis in the pulmonary artery, PH-ILD
is driven by fibrosis in the lungs and often involves pulmonary vasculature– Current therapies address disease aspects other than pulmonary hypertension– Similar clinical symptoms, progression, and endpoints for approval
ILD Subtype Estimated US Prevalence Estimated WW Prevalence
Sarcoidosis (SAPH) 31,500 50,050
Idiopathic Pulmonary Fibrosis (IPF) 23,500 64,500
Connective Tissue Disease (CTD-ILD) 3,900 17,800
Idiopathic Interstitial Pneumonia (IIP) 4,300 34,550
37

Omav in Immuno-Oncology
Omav addresses a major mechanism of immune avoidance by tumors– Myeloid-derived suppressor cells (MDSCs) “cloak” tumors– Killer T cells become unable to recognize and attack tumors– Explains why most patients do not respond to checkpoint
inhibitors
Omav suppresses MDSCs and augments activity of immune therapies in preclinical models
REVEAL is a 1b/2 trial of Omav in combination with nivolumab in patients with iNOS+ (MDSC marker) unresectable or metastatic melanoma– iNOS is associated with poor survival and is present in
~75% of patients– Multiple endpoints assessing clinical, biological, and
pharmacodynamic activity
Phase 1b portion of trial ongoing with no safety issues identified to date
Phase 1b data expected in H2 2017
Omav Enhances Anti-PD-1 Activity and Induces Immune Rejection of Tumors
Gabrilovich et al., Nature Reviews Immunology (2012); Lu T et al., JCI (2011); Nagaraj et al., Clin Cancer Res, (2010)
0
2000
4000
6000
0 7 14 21 28Days Post Tumor Injection
Vehicle + IsoVehicle + PD-11 mg/kg Omav + PD-1
Omav Treatment Daily
Tum
or S
ize
(mm
3 )
38

Preclinical Development Programs
RTA 901 – A neuroprotective Hsp90 inhibitor– RTA 901 is a novel C-terminal Hsp90 inhibitor under development for neurologic conditions
– Hsp90/Hsp70 are key cellular stress management pathways with critical roles in mitochondrial protein import and protein chaperone activity
– Profound preclinical activity in a wide range of models of neurologic disease, including diabetic neuropathy and demyelinating nerve injury
– IND-enabling studies ongoing• GLP studies to date have demonstrated good tolerability and wide margins• PK/PD indicates likely once-daily oral dosing in humans
– Key clinical indications under consideration include diabetic neuropathy, ALS, SCA3, and SBMA
RORɣT - inflammation and immune modulating nuclear receptor– RORɣT has been recently recognized as a critical target for modulating the pro-
inflammatory and immune activity of Th17 cells– Appreciated as a central regulator of autoimmunity, with relevance in many autoimmune
conditions, such as MS, RA, and IBD– Through focused screening, Reata has identified potent and selective modulators of
RORɣT
39

Upcoming Key Events and Timing
(1) Bard is currently being developed unilaterally by Reata, and AbbVie retains the right to opt back into development. Reata retains commercial rights in US market; KHK has rights in certain Asian markets; AbbVie has rights in non-KHK markets outside the US.
(2) IPF means idiopathic pulmonary fibrosis, IIP means idiopathic interstitial pneumonia, and Sarc means sarcoidosis.(3) Reata’s next milestone is data from the Phase 3 trial in CTD-PAH. Reata began enrolling patients in October 2016.(4) Reata retains US commercialization; Omaveloxolone is currently being developed unilaterally by Reata, and AbbVie retains the
right to opt back into development and commercialization.
40
Stage Disease Drug Event Timing
Late Stage
Friedreich's Ataxia(4)
OmavP2 data 1H17
Pivotal trial initiation 2H17
Alport Syndrome(1)
Bard Initiate P2 1H17
Pivotal trial initiation 2H17
Mitochondrial Myopathies(4)
OmavP2 data 2H17
Pivotal trial initiation 1H18
CTD-PAH(1,3) Bard P3 data 1H18
Early Stage Pulmonary Hypertension (ILD)(1,2) Bard P2 data 2H17
Melanoma(4) Omav P1b/2 data 2H17

Appendix

CONFIDENTIAL
Appendix A - Reata Patent Portfolio
Bardoxolone methyl- Five major patent families– Morphic form patent- e.g., US 8,088,824
• Claims amorphous (commercial) and crystalline forms- only known forms of pure Bard• Statutory expiration May 2029 US, Aug. 2028 ROW• Likely term (with extensions) 13 to 14 years post-approval (varies with approval date)• Amorphous form claims granted in US, EU, Japan, Canada, China, Mexico, Eurasia, 6 other
territories– Still pending in 8 additional countries– Crystalline form claims granted/allowed in US, EU, Eurasia, Mexico, Singapore, Taiwan
– Formulation patent- e.g., US 8,747,901; claims cover commercial, alternative formulations • Statutory expiration Nov. 2030 US, Feb. 2030 ROW• Granted/allowed in 10 countries including US, Japan, Canada, Mexico; pending in 11
territories• Would be difficult for a competitor to achieve bioequivalence without infringing
– Methods of use in treating cardiovascular, renal, metabolic disease• Statutory expiration Jan. 2029• Pursuing claims for treatment of endothelial dysfunction (essential component of PAH)
– Methods of use in treating PAH, other forms of PH, and CV disease generally• Statutory expiration Aug. 2034• Claims use of Bard in patients who lack problem characteristics identified in BEACON
– Original patents claiming Bard expire Apr. 2022 US, June 2019 ROW

CONFIDENTIAL
Appendix A - Reata Patent Portfolio
Omav covered by two families of patents– Genus claims in US 8,124,799, related foreign cases- statutory expiration Dec. 2029
US, Apr. 2029 ROW• Granted in 11 foreign territories, pending in 7 others
– Molecule-specific patent (8,993,640) granted in US, pending in 30 other territories• Statutory expiration Apr. 2033• Potential post-approval term 14 years US (maximum allowed), 15 years EU/JP
RTA 901 specifically claimed in PCT/US2013/025387 and related applications– Cases filed in US, Europe, China, Japan, Canada, Mexico, 7 other countries– Statutory expiration Feb. 2033- term extensions likely to be available– Manufacturing process research has identified novel form that will be newly
patentable• Likely to have statutory expiration falling in 4Q 2036

Appendix B – BEACON Analysis and Findings
Conducted phase 3 trial of Bard in diabetic stage 4 CKD patients Bard increased eGFR and reduced renal SAEs and ESRD events as expectedTrial stopped for imbalance in cardiac events with unknown etiology at time– Post termination analysis identified fluid overload hospitalizations, during first three weeks
only, as source of increased cardiac events– 8.8% Bard versus 5.0% placebo and IV diuretics resolved symptoms– No other cardiac or major safety findings
Susceptible patients were in heart failure (HF) at baseline and identifiable– Analyses identified elevated BNP and history of HF as risk factors– Events balanced in patients without risk factors
Increased CHF events are observed with ERAs and prostacyclinsBard safety analysis reviewed by FDA and Japanese PMDA– FDA reviewed safety data in PAH patients to date and concurred with sponsor plan to
conduct Phase 3 trial in CTD-PAH– KHK initiated Phase 2 CKD trial in Japanese patients
BNP ≤ 200, No Prior HF
PBO BARD
Primary Composite 25 (5%) 15 (3%)
ESRD 20 (4%) 8 (2%)
CV Death 6 (1%) 7 (1%)
Heart Failure 10 (2%) 12 (2%)
BEACON Renal Data
PBO BARDWK48 ∆eGFR ≤ -3 67% 20%Renal SAEs 71 52ESRD Events 51 43


















