
Real-Time Degradation Dynamics of Hydrated Perfluoroalkyl ...
16
doi.org/10.26434/chemrxiv.11634348.v1 Real-Time Degradation Dynamics of Hydrated Perfluoroalkyl Substances (PFASs) in the Presence of Excess Electrons Sharma Yamijala, Ravindra Shinde, Bryan Wong Submitted date: 17/01/2020 • Posted date: 20/01/2020 Licence: CC BY-NC-ND 4.0 Citation information: Yamijala, Sharma; Shinde, Ravindra; Wong, Bryan (2020): Real-Time Degradation Dynamics of Hydrated Perfluoroalkyl Substances (PFASs) in the Presence of Excess Electrons. ChemRxiv. Preprint. https://doi.org/10.26434/chemrxiv.11634348.v1 Perfluoroalkyl substances (PFASs) are synthetic chemicals that are harmful to both the environment and human health. Using self-interaction-corrected Born-Oppenheimer molecular dynamics simulations, we provide the first real‐time assessment of PFAS degradation in the presence of excess electrons. In particular, we show that the initial phase of the degradation involves the transformation of an alkane-type C-C bond into an alkene-type C=C bond in the PFAS molecule, which is initiated by the trans elimination of fluorine atoms bonded to these adjacent carbon atoms. File list (2) download file view on ChemRxiv main_text_in_template.pdf (1.24 MiB) download file view on ChemRxiv supplementary_info.pdf (2.86 MiB)
Transcript of Real-Time Degradation Dynamics of Hydrated Perfluoroalkyl ...

doi.org/10.26434/chemrxiv.11634348.v1
Real-Time Degradation Dynamics of Hydrated Perfluoroalkyl Substances(PFASs) in the Presence of Excess ElectronsSharma Yamijala, Ravindra Shinde, Bryan Wong
Submitted date: 17/01/2020 • Posted date: 20/01/2020Licence: CC BY-NC-ND 4.0Citation information: Yamijala, Sharma; Shinde, Ravindra; Wong, Bryan (2020): Real-Time DegradationDynamics of Hydrated Perfluoroalkyl Substances (PFASs) in the Presence of Excess Electrons. ChemRxiv.Preprint. https://doi.org/10.26434/chemrxiv.11634348.v1
Perfluoroalkyl substances (PFASs) are synthetic chemicals that are harmful to both the environment andhuman health. Using self-interaction-corrected Born-Oppenheimer molecular dynamics simulations, weprovide the first real‐time assessment of PFAS degradation in the presence of excess electrons. In particular,we show that the initial phase of the degradation involves the transformation of an alkane-type C-C bond intoan alkene-type C=C bond in the PFAS molecule, which is initiated by the trans elimination of fluorine atomsbonded to these adjacent carbon atoms.
File list (2)
download fileview on ChemRxivmain_text_in_template.pdf (1.24 MiB)
download fileview on ChemRxivsupplementary_info.pdf (2.86 MiB)

COMMUNICATION
Please do not adjust margins
Please do not adjust margins
Received 17th December 2019,
Accepted 00th January 2020
DOI: 10.1039/x0xx00000x
Real-Time Degradation Dynamics of Hydrated Perfluoroalkyl Substances (PFASs) in the Presence of Excess Electrons
Sharma S.R.K.C. Yamijala, Ravindra Shinde, and Bryan M. Wong*
Perfluoroalkyl substances (PFASs) are synthetic chemicals that are
harmful to both the environment and human health. Using self-
interaction-corrected Born-Oppenheimer molecular dynamics
simulations, we provide the first real‐time assessment of PFAS
degradation in the presence of excess electrons. In particular, we
show that the initial phase of the degradation involves the
transformation of an alkane-type C-C bond into an alkene-type C=C
bond in the PFAS molecule, which is initiated by the trans
elimination of fluorine atoms bonded to these adjacent carbon
atoms.
The strongest and most stable bond in organic chemistry is
the carbon-fluorine (C-F) bond.1 The high stability of this bond,
which has enabled several technological advancements in the
past century, now poses a serious health hazard.2–5 In the last
century, the industrial sector has harnessed the intrinsic
strength of the C-F bond to incorporate perfluoroalkyl
substances (PFASs) in a wide variety of consumer and industrial
goods.6–8 Specifically, PFASs are regularly used in the
electronics, automotive, and aviation industries;6,7 several
consumer goods, including non-stick cookware, clothing,
carpets, shampoos, cleaning agents, and adhesives contain
these persistent pollutants.8 As such, the wide-spread usage of
these anthropogenic compounds has contaminated water
resources in the US and many other countries.9–11 Most
importantly, the subsequent consumption of this contaminated
water leads to the bio-accumulation of these compounds in
various organisms, including humans.9,10,12–14 In all of these
examples, the intrinsic strength of the C-F bond in PFASs
prevents most organisms from dissociating these compounds
through natural means,15 which facilitates their bio-
accumulation and toxicity. Thus, the intrinsic strength of the C-
F bond poses a severe threat to many forms of life, making the
treatment of these contaminated water sources essential.
An efficient treatment often includes either direct in-situ
destruction or removal of PFASs from contaminated water (for
example, using sorption or nanofiltration) and their subsequent
destruction in the wastewater concentrate.16,17 Since
perfluoroalkyl compounds primarily contain C-C and C-F bonds,
the degradation of PFASs requires the dissociation of both of
these bonds. However, breaking a C-C bond is less effective than
C-F bond cleavage,16,17 since the former generates short-chain
perfluoroalkyl substances that are still found to be toxic.18,19
Thus, the primary challenge in the complete treatment of PFAS-
contaminated water is the efficient dissociation of the strong C-
F bonds in these pollutants.
In the scientific literature, a few studies have suggested
possible avenues for C-F bond dissociation in the presence of
additional electrons.20–22 However, the majority of these studies
carried out their analysis after the degradation has occurred,
and specific mechanistic details of PFAS degradation are less
understood. Although a few computational studies have
addressed possible degradation of PFASs with excess
electrons,20 these studies were limited to static (i.e., non-
dynamic or stationary) electronic structure methods. As such,
these prior approaches restrict our understanding of PFAS
degradation dynamics. Also, to the best of our knowledge, there
is little or no information on defluorination timescales in PFASs.
To bridge this significant knowledge gap, we carried out
extensive first-principles Born-Oppenheimer Molecular
Dynamics (BOMD) simulations on both perfluorooctanoic acid
(PFOA) and perfluorooctanesulfonic acid (PFOS) molecules (the
most common PFAS contaminants in the environment) in the
presence of explicit water molecules. While we find these
pollutants to be stable in neutral aqueous environments, our
results indicate that both of these molecules readily
defluorinate at ultrafast timescales (< 100 fs) in the presence of
excess electrons. By considering a variety of different initial
conditions, we unambiguously show that defluorination in
PFASs often leads to the formation of an intermediate with an
alkene type C=C bond, which is crucial in the degradation of
PFASs. In addition, we also show that the formation of an HF
Department of Chemical & Environmental Engineering, Materials Science & Engineering Program, and Department of Physics & Astronomy. University of California, Riverside, Riverside, CA 92521, United States. E-mail:
[email protected]; Web: http://www.bmwong-group.com † Electronic Supplementary Information (ESI) available: Computational details and supporting data. See DOI: 10.1039/x0xx00000x Self-interaction-corrected density functional theory calculations were supported by the U.S. Department of Energy, Office of Science, Early Career Research Program under Award No. DE-SC0016269. Born-Oppenheimer Molecular Dynamics simulations of PFAS contaminants were supported by the National Science Foundation under Grant No. CHE-1808242.

COMMUNICATION PCCP
2 | Phys. Chem. Chem. Phys., 2019, 00, 1-4 This journal is © The Royal Society of Chemistry 2019
Please do not adjust margins
Please do not adjust margins
molecule is a possible outcome of the BOMD simulations. As
such, this work provides detailed mechanistic insight and
presents the first real-time picture of degradation dynamics of
PFASs in the presence of excess electrons.
Inspired by recent works demonstrating that reduction
processes (i.e., the addition of electrons) can degrade PFASs,20–
22 we studied the degradation dynamics of both PFOA and PFOS
in the presence of excess electrons. To avoid any spurious self-
interaction effects, which are usually observed with semi-local
functionals such as PBE, we have included self-interaction
corrections in all our MD simulations (further details are given
in the Supplementary Information). To mimic the water
environment, we solvated each of these PFASs with 43 explicit
water molecules. Due to this explicit solvent environment, both
PFOA and PFOS lose their acidic proton to water molecules (as
they should) during the NVT equilibration. In figure 1, we
present the geometries of PFOA and PFOS molecules (top and
bottom panels, respectively) after a 1-ps-long NVE simulation
carried out with a different number of excess electrons. In the
left panel, we show our results with no additional charge. As
expected, both PFOA and PFOS were stable (i.e., no bond
dissociation) during the entire simulation time. In contrast, for
simulations containing excess electrons, we observed an
apparent degradation of these molecules. In the middle and
right panels of figure 1, we show our results with -1 and -2
electronic charges, respectively. Both PFOA and PFOS were
defluorinated in the presence of excess electrons, and we find
that the number of dissociated C-F bonds is proportional to the
number of additional electrons in the simulation box. We
verified the robustness of these results by simulating six
different initial conditions (i.e., positions and velocities) for both
PFOA and PFOS (i.e., an additional 12 NVE simulations), and
these results are shown in figures S1 and S2 in the
Supplementary Information. The initial conditions for all these
additional runs were obtained from a 10-ps-long NVT
equilibration run, and a C-F bond dissociation was observed in
all these simulations. Collectively, these results demonstrate
that “excess electrons” can dissociate the crucial C-F bonds in
perfluoroalkyl substances to facilitate their degradation.
Next, we examined the defluorination timescales in both
PFOA and PFOS molecules with excess electrons. The initial
conditions for all our NVE runs were taken from NVT
calculations of neutral systems, with excess electrons
subsequently added to simulate the dynamics of
electrostatically charged environments. That is, the first frame
of all the NVE runs correspond to the neutral (i.e., uncharged)
geometries. For each of the dissociated C-F bonds shown in
figure 1, we studied the dissociation dynamics as a function of
time, which are presented in figure 2. Clearly, at the beginning
of all these simulations, the C-F bond distance is slightly above
1.3 Å, which is approximately the equilibrium C-F bond distance
in neutral PFASs. At the end of these simulations, the bond
distance increased to 3 Å, suggesting a complete dissociation of
these C-F bonds in the presence of excess electrons. However,
interestingly, in all these simulations, the C-F bond dissociation
occurred within the first 100 fs (see the insets of different
panels in figure 2). Here, it is essential to note that the
dissociated C-F bonds in the PFOA and PFOS molecules are
different, and even the dissociated C-F bonds in PFOA for the -1
and -2 electronic charges also differ (note the legends and top
panel of figure 2 for the various dissociated C-F bonds). Despite
these variations, we consistently observed an ultrafast
Figure 1: Geometries of hydrated PFOA (top panel) and PFOS (bottom panel) molecules after a 1 ps long NVE run with 0 (left), 1 (middle), and 2 (right) excess electrons. For both molecules, we observe defluorination in the presence of excess electrons. We verified the robustness of these results by simulating six different initial conditions for both PFOA and PFOS (i.e., an additional 12 NVE simulations), which are shown in figures S1 and S2 in the Supplementary Information.
Figure 2: Bond distance profiles of the dissociated C-F bonds for the structures shown in Figure 1. Results with -1 (middle panel) and -2 (bottom panel) electronic charges are presented for both PFOA (top left) and PFOS (top right) molecules. The insets in each panel show the variation in the C-F bond distances during the first 100 fs (= 0.1 ps) of the simulations. In all our simulations, we observed the dissociation of C-F bonds in less than 100 fs (i.e., an ultrafast dissociation). The legends in the figure designate the dissociated C-F bonds, and the corresponding atom numbering scheme is shown in the top panel. The structures in this panel were obtained after a 1 ps NVE simulation containing a -2 electronic charge (water molecules were omitted for clarity).

PCCP COMMUNICATION
This journal is © The Royal Society of Chemistry 2019 Phys. Chem. Chem. Phys ., 2019, 00, 1-4 | 3
Please do not adjust margins
Please do not adjust margins
degradation (i.e., < 100 fs) of PFASs in all these simulations.
Furthermore, we verified this ultrafast dissociation of C-F bonds
in all the other 12 NVE simulations (see the Supplementary
Information). Thus, we note that excess electrons not only
facilitate C-F bond dissociation in PFASs, but also initiate this
process on an ultrafast timescale.
In figure 3, we show the evolution of the spin-density as a
function of time for both PFOA and PFOS with -1 and -2
electronic charges. We note that the LUMO of neutral hydrated
PFOA and PFOS is localized entirely on the PFAS molecule (i.e.,
not on water); as such, the addition of an electron to this
composite system is expected to fill this unoccupied orbital on
PFAS. Indeed, as shown in the top panel of figure 3, the
additional charge (more precisely, spin) initially delocalizes on
the entire PFOA molecule; however, as the simulation proceeds,
the spin slowly accumulates at the site of dissociation. We also
obtained similar results with the PFOS molecule (see figure S3)
as well as in four additional NVE calculations performed with a
-1 electronic charge (not shown). These four calculations are
different from the 12 NVE calculations presented in the
Supplementary Information.
In a previous study on excess electrons,20 Su and co-workers
were unable to assign the initial site of the degradation process
through their electronic structure calculations. In all our
simulations, we also find that the site of dissociation is arbitrary.
However, our spin-density results provide additional reasoning
for Su and co-workers’ observation. First, since the excess
electron initially delocalizes over the entire PFAS molecule, all
the C-F bonds are weakened and have an almost equal
probability for dissociation. The specific C-F bond that
dissociates during the simulation is thus arbitrary and is most
possibly triggered by interactions between the contaminant
and the solvent (here, PFAS and water). Next, although we
mentioned that the site of dissociation is random, we never
observed the dissociation of fluorine atoms attached to the
terminal (primary) carbon atoms in any of our sixteen NVE
simulations. This facile dissociation of secondary C-F bonds is
due to their lower bond dissociation energy compared to the
primary C-F bonds, as noted in a few earlier studies.23,24
Contrary to the addition of a single electron, we find that the
spin-density can evolve in two different ways with the addition
of multiple electrons, as shown in the middle and bottom panels
of figure 3. We refer to these different regimes as a trans-type
(middle panel) or cis-type elimination (bottom panel) based on
how the fluorine atoms have dissociated from the PFAS
backbone structure. In both cases, initially, the spin-density of
each additional electron is localized at different parts of the
PFAS (i.e., either towards the head or tail group), introducing a
net spin-polarization in these compounds. However, as the
simulation proceeds, this spin-polarization is either retained
(cis-type) or completely suppressed (trans-type) depending on
whether the dissociated fluorine atoms were bonded to
adjacent carbon atoms or otherwise. As further demonstrated
below, the spin-polarization quickly (< 100 fs) decays when
fluorine atoms dissociate from adjacent carbon atoms, due to
the formation of a double bond between these carbon atoms
(in other cases, the spin polarization is retained).
In figure 4, we present dynamical plots of atoms exhibiting
maximum changes in the Mulliken charge and spin during the
initial stages of the simulations (i.e., during the C-F bond
dissociation). Primarily, the charge profiles of both PFOA and
PFOS with 2 excess electrons (figures 4a and 4c) show that the
fluorine atoms captured these excess charges during the
Figure 3: Evolution of spin-density as a function of time for both PFOA and PFOS with excess electrons. PFOA with a -1 (-2) electronic charge is shown in the top (middle) panel, and PFOS with a -2 charge is shown in the bottom panel. The spin density of PFOS with a -1 electronic charge (not shown for brevity) is similar to the top PFOA panel (see figure S3). An iso-value of 0.001, 0.00001, and 0.001 eÅ-3 was used for the top, middle, and bottom panels, respectively.
Figure 4: Atoms exhibiting the largest changes in Mulliken charges (A, C) and spin (B, D) during the initial stages of the simulation (i.e., during C-F bond dissociation). The left (right) panel depicts results for a PFOA (PFOS) molecule with two excess electrons. In both cases, after the C-F bond dissociation, negative charges were localized on the fluorine atoms. For only the cis-type elimination (see text for details), spins were localized on the carbon atoms (panel D). In the inset of panel A (B), the charge (spin) on the dissociated fluorine atoms are shown until 500 fs. In the inset of panel D, changes in the spin during the first 10 fs of the simulation are depicted. The atom numbering scheme is presented in the top panel.

COMMUNICATION PCCP
4 | Phys. Chem. Chem. Phys., 2019, 00, 1-4 This journal is © The Royal Society of Chemistry 2019
Please do not adjust margins
Please do not adjust margins
simulation. As such, there are no significant differences in the
charge profiles for both cis and trans-type eliminations. Even in
the simulations with a -1 electronic charge, we find that the
excess charge is primarily localized on the fluorine atoms (see
figure S4). We note that in simulations with a -1 electronic
charge (see figure S4), the carbon atoms retain the electronic
spin. In contrast to the charge profiles, the difference between
the cis and trans-type eliminations manifests strongly in the
spin profiles. In the cis-type elimination (figure 4d), opposite
spins were localized on different carbon atoms (C1 and C18); in
contrast, for the trans-type elimination, no atom is spin-
polarized at the end of the simulation, which leads to the
formation of a C=C bond along the PFAS backbone. Although the
total residual spin is zero in both cis and trans-type eliminations,
the cis-type elimination produces an antiferromagnetic
intermediate, and a trans-type elimination results in a non-
magnetic intermediate. This behaviour was confirmed in all the
16 NVE simulations performed with 2 excess electrons.
Since we observed a trans-type elimination in most of our
simulations (12 out of 16), we suggest that this intermediate
could be central to the degradation of perfluoroalkyl
substances. A few recent experiments further support this
finding. For example, Liu et al. have shown that fluorine atoms
attached to a C=C bond (i.e., sp2 carbon atoms) dissociate more
readily compared to fluorine atoms attached to a C-C bond (sp3
carbon atoms).23 Since most of our simulations with excess
electrons resulted in a trans-type elimination with the
formation of a C=C bond (which is known to be more susceptible
to defluorination), we infer that this trans-type intermediate
plays a crucial role in PFAS defluorination, which could explain
the PFAS degradation (with excess electrons) observed by Su et
al.20 Apart from the trans-type intermediate, we also found that
an HF molecule was formed during our simulations (see figure
S5 and the Supplementary Information for further details).
In conclusion, we have presented the first real-time,
dynamical study of PFAS degradation with excess electrons to
elucidate defluorination timescales and mechanisms. Using
several large-scale ab initio trajectory calculations (16 different
NVE simulations), we show that excess electrons enable the
dissociation of the strongest C-F bonds in these contaminants,
which occurs on an ultrafast timescale (< 100 fs). Moreover,
these large-scale simulations provide new mechanistic insight
into PFAS degradation processes that manifest themselves
strongly as spin and charge fluctuations as a function of time. In
particular, we have shown that a trans-type C-F elimination
(which leads to the formation of a C=C bond along the PFAS
backbone) is more probable compared to a cis-type elimination.
Since fluorine atoms attached to a C=C bond are more readily
dissociated, this trans-type intermediate could play a central
role in accelerating further PFAS defluorination processes. In
addition, our dynamics simulations also show the spontaneous
formation of an HF molecule, which could be another significant
intermediate/product of the PFAS degradation process. Taken
together, these first-principles-based molecular dynamics show
that (1) the use of excess electrons can be a viable approach for
breaking the strongest C-F bonds in PFAS contaminants, and (2)
the application of dynamical computational techniques can
shed essential mechanistic insight (such as degradation
timescales and short-lived intermediates) that cannot be readily
gleaned from static electronic structure studies of PFAS or other
aqueous contaminants.
Conflicts of interest
There are no conflicts to declare.
Notes and references
1 D. O’Hagan, Chem. Soc. Rev., 2008, 37, 308–319.
2 E. M. Sunderland, X. C. Hu, C. Dassuncao, A. K. Tokranov, C. C. Wagner and J. G. Allen,
J. Expo. Sci. Environ. Epidemiol., 2019, 29, 131–147.
3 G. Goldenman, M. Fernandes, M. Holland, T. Tugran, A. Nordin, C. Schoumacher and
A. McNeill, The cost of inaction: A socioeconomic analysis of environmental and health
impacts linked to exposure to PFAS, Nordic Council of Ministers, 2019.
4 https://pfas-1.itrcweb.org.
5 P. Grandjean and R. Clapp, New Solut., 2015, 25, 147–163.
6 E. Kissa, Fluorinated Surfactants and Repellents, Second Edition, CRC Press, 2001.
7 B. Ameduri and H. Sawada, Fluorinated Polymers: Volume 2: Applications, Royal
Society of Chemistry, 2016.
8 M. Kotthoff, J. Müller, H. Jürling, M. Schlummer and D. Fiedler, Environ. Sci. Pollut.
Res. Int., 2015, 22, 14546–14559.
9 R. Bossi, M. Dam and F. F. Rigét, Chemosphere, 2015, 129, 164–169.
10 A. Cordner, V. Y. De La Rosa, L. A. Schaider, R. A. Rudel, L. Richter and P. Brown, J.
Expo. Sci. Environ. Epidemiol., 2019, 29, 157–171.
11 J. S. Boone, C. Vigo, T. Boone, C. Byrne, J. Ferrario, R. Benson, J. Donohue, J. E.
Simmons, D. W. Kolpin, E. T. Furlong and S. T. Glassmeyer, Sci. Total Environ., 2019,
653, 359–369.
12 K. Kannan, Environ. Chem., 2011, 8, 333.
13 K. Kannan, S. Corsolini, J. Falandysz, G. Fillmann, K. S. Kumar, B. G. Loganathan, M. A.
Mohd, J. Olivero, N. Van Wouwe, J. H. Yang and K. M. Aldoust, Environ. Sci. Technol.,
2004, 38, 4489–4495.
14 L. W. Y. Yeung, M. K. So, G. Jiang, S. Taniyasu, N. Yamashita, M. Song, Y. Wu, J. Li, J. P.
Giesy, K. S. Guruge and P. K. S. Lam, Environ. Sci. Technol., 2006, 40, 715–720.
15 J. S.-C. Liou, B. Szostek, C. M. DeRito and E. L. Madsen, Chemosphere, 2010, 80, 176–
183.
16 N. B. Saleh, A. Khalid, Y. Tian, C. Ayres, I. V Sabaraya, J. Pietari, D. Hanigan, I.
Chowdhury and O. G. Apul, Environ. Sci. Water Res. Technol., 2019, 5, 198–208.
17 K. H. Kucharzyk, R. Darlington, M. Benotti, R. Deeb and E. Hawley, Novel treatment
technologies for PFAS compounds: A critical review, Academic Press, 2017, vol. 204.
18 National Toxicology Program, , DOI:10.22427/ntp-tox-97.
19 National Toxicology Program, , DOI:10.22427/ntp-tox-96.
20 Y. Su, U. Rao, C. M. Khor, M. G. Jensen, L. M. Teesch, B. M. Wong, D. M. Cwiertny and
D. Jassby, ACS Appl. Mater. Interfaces, 2019, 11, 33913–33922.
21 S. Huang and P. R. Jaffé, Environ. Sci. Technol., 2019, 53, 11410–11419.
22 R. K. Singh, S. Fernando, S. F. Baygi, N. Multari, S. M. Thagard and T. M. Holsen,
Environ. Sci. Technol., 2019, 53, 2731–2738.
23 J. Liu, D. J. Van Hoomissen, T. Liu, A. Maizel, X. Huo, S. R. Fernández, C. Ren, X. Xiao,
Y. Fang, C. E. Schaefer, C. P. Higgins, S. Vyas and T. J. Strathmann, Environ. Sci. Technol.
Lett., 2018, 5, 289–294.
24 A. Raza, S. Bardhan, L. Xu, S. S. R. K. C. Yamijala, C. Lian, H. Kwon and B. M. Wong,
Environ. Sci. Technol. Lett., 2019, 6, 624–629.

download fileview on ChemRxivmain_text_in_template.pdf (1.24 MiB)

S1
Supporting Information
Real-Time Degradation Dynamics of Hydrated
Perfluoroalkyl Substances (PFASs) in the Presence of
Excess Electrons
Sharma S.R.K.C. Yamijala, Ravindra Shinde, and Bryan M. Wong*
Department of Chemical & Environmental Engineering, Materials Science & Engineering Program, and
Department of Physics & Astronomy
University of California, Riverside, Riverside, CA 92521, United States
*Corresponding author. E-mail: [email protected]. Web: http://www.bmwong-group.com
Contents
p. S2: Figure S1: Final geometries of hydrated PFOA molecules obtained with six different initial BOMD conditions.
p. S3: Figure S2: Final geometries of hydrated PFOS molecules obtained with six different initial BOMD conditions.
p. S4: Figure S3: Evolution of the spin-density in a PFOS molecule with one excess electron during an NVE simulation.
p. S5: Figure S4: Atoms exhibiting the largest changes in Mulliken charges and spin during the initial stages of the simulation (i.e., during C-F bond dissociation).
p. S6: Figure S5: Formation of an HF molecule in the NVE simulations of (a) PFOA and (b) PFOS with a -2 electronic charge.
p. S7: Computational details.

S2
Figure S1. Final geometries of hydrated PFOA molecules obtained with six different initial BOMD conditions (positions and velocities). All NVE simulations were performed with 2 excess electrons, and all simulations were propagated for 150 fs or longer. For each of the different initial conditions, we observe a defluorination in the presence of excess electrons. All of these six initial conditions were obtained from an NVT simulation with a separation of 500-1000 fs between each initial condition.

S3
Figure S2. Final geometries of hydrated PFOS molecules obtained with six different initial BOMD conditions (positions and velocities). All NVE simulations were performed with 2 excess electrons, and all simulations were propagated for 150 fs or longer. For each of the different initial conditions, we observe a defluorination in the presence of excess electrons. All of these six initial conditions were obtained from an NVT simulation with a separation of 500-1000 fs between each initial condition.

S4
Figure S3. Evolution of the spin-density in a hydrated PFOS molecule with one excess electron during an NVE simulation.
S5
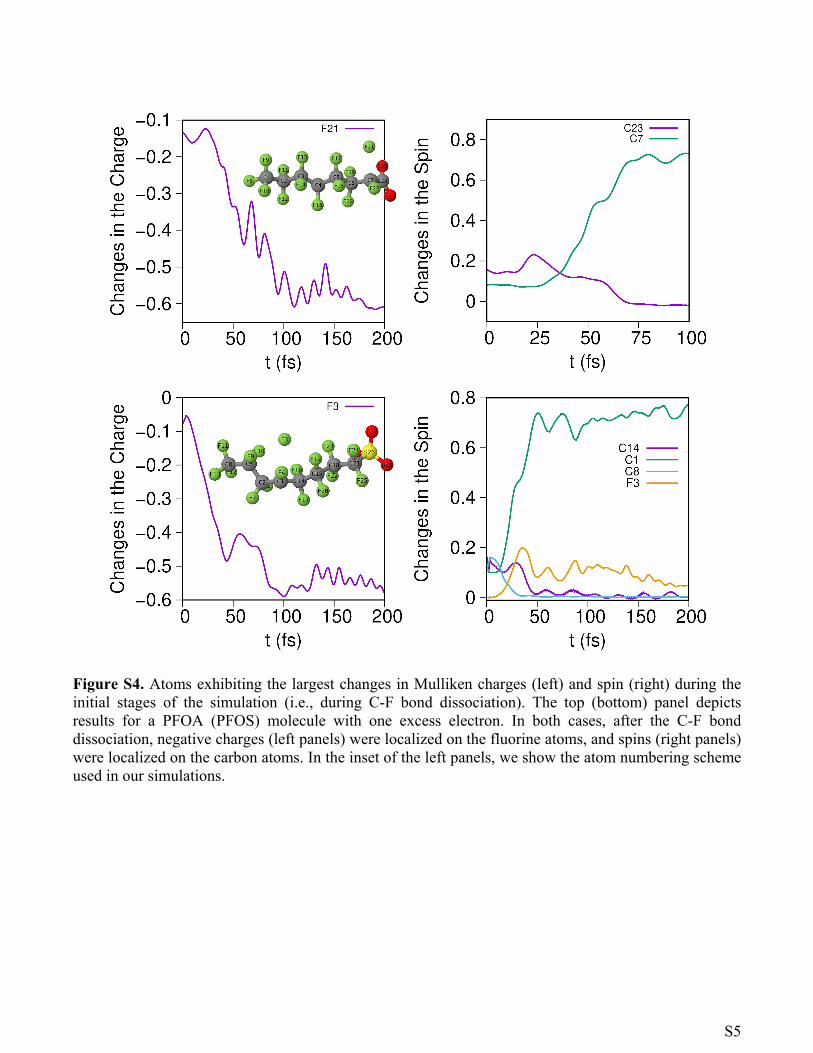
Figure S4. Atoms exhibiting the largest changes in Mulliken charges (left) and spin (right) during the initial stages of the simulation (i.e., during C-F bond dissociation). The top (bottom) panel depicts results for a PFOA (PFOS) molecule with one excess electron. In both cases, after the C-F bond dissociation, negative charges (left panels) were localized on the fluorine atoms, and spins (right panels) were localized on the carbon atoms. In the inset of the left panels, we show the atom numbering scheme used in our simulations.

S6
Figure S5. Formation of an HF molecule (bond length ~0.99 Å, circled in pink) in the NVE simulations of (a) PFOA and (b) PFOS with a -2 electronic charge. In both simulations, an H-F bond is formed after 2 ps. In panel (a), the formation of a C=C bond (~1.29 Å, circled in black) can also be seen.
Apart from the trans-type intermediate discussed in the main text, we also found that an HF
molecule was formed during our simulations (see Figure S5). Specifically, one of the dissociated
fluorine atoms from the PFAS molecule combines with a proton from the solvent (which originates
from PFOA/PFOS releasing its carboxylic/sulfonic acid proton into the solvent) to form an HF
molecule. In addition, we observed the formation of an HF molecule in our other NVE simulations as
well; as such, we suggest that an HF molecule could also be a significant intermediate or end product of
the PFAS degradation process.

S7
Computational Details
All of the Born-Oppenheimer Molecular Dynamics (BOMD) simulations were performed in a
microcanonical (NVE) ensemble using the Quick-Step method as implemented in the CP2K software
package.1 The BOMD equations of motion were integrated with a 0.5 fs time step, and initial velocities
and coordinates for all NVE runs were obtained by first running an NVT simulation at 300 K. For the
NVT simulations, we used the Nosé−Hoover thermostat of the chain length three. In all the NVT and
NVE runs, Grimme’s D3 dispersion2 correction was employed. To calculate the electronic energies and
gradients at each nuclear step, we used density functional theory with a self-interaction corrected PBE3
exchange-correlation functional. Specifically, we have included a self-interaction correction (SIC) for
all orbitals in our BOMD simulations using the average density SIC, as implemented in the CP2K
package.4 Following earlier work,5 we tuned the scaling parameters ‘a’ (=0.2) and ‘b’ (=0.25) to
reproduce the vertical electron affinities of PFOA and PFOS obtained with a non-empirically tuned
range-separated LC-BLYP functional (which typically gives energies that match experiment or high-
level wavefunction-based benchmarks6,7). To solve the self-consistent Kohn-Sham equations, we used
the molecularly optimized double-zeta quality (DZVP) basis-sets,8 which are compatible with the
employed Goedecker−Teter−Hutter (GTH) pseudopotentials.9,10 For the auxiliary plane-wave (PW)
basis used in the Gaussian-and-Plane-Waves method of CP2K,11 we used 600 Ry for the PW energy
cutoff and 60 Ry for the reference grid cutoff.
Calculations with Other Exchange-Correlation Functionals
Since prior studies on hydrated electrons have shown that the results are not sensitive to the
choice of semi-local functional (particularly PBE vs. BLYP),12,13 we omitted additional calculations
with the BLYP functional. However, to verify the robustness of our results, we performed additional
calculations with the B3LYP hybrid functional and found qualitatively similar results for some of our
initial studies. Due to the immense computational cost of hybrid functionals, we did not pursue

S8
additional BOMD calculations with the B3LYP functional, but we anticipate the results to be
qualitatively similar to our self-interaction corrected PBE calculations.
Tuning the Scaling Parameters
Following earlier studies,5 we tuned the scaling parameters ‘a’ and ‘b’ to reproduce the vertical
electron affinities of PFOA and PFOS, which we obtained with the non-empirically tuned range-
separated LC-BLYP functional. We found that the values of a = 0.2 and b = 0.25 reproduce these
benchmark electron affinities, which we subsequently used in our calculations. We also performed
additional calculations with a = 0.2 and b = 0.0, as used in prior studies;4 however, even with these
different parameters, we observed PFAS defluorination in the presence of excess charges, indicating the
robustness of our results.
Number of Explicit Water Molecules Considered in the Present Study
To mimic the surrounding water environment, we solvated each of these PFAS species with 43
explicit water molecules that were treated quantum mechanically. It is important to note that earlier
studies have already demonstrated that 31 water molecules are sufficient to reproduce the quantum
mechanical behavior of a hydrated electron.4 Considering the number of MD simulations performed in
this work and the substantial computational demand for each of these ab initio MD calculations, the
present choice of 43 water molecules was optimal for capturing all the necessary PFAS degradation
dynamics without sacrificing accuracy.
References
1 J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comp. Phys. Comm., 2005, 167, 103–128.
2 S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465.
3 J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868.
4 J. VandeVondele and M. Sprik, Phys. Chem. Chem. Phys., 2005, 7, 1363–1367.

S9
5 J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao, Q. Dong, C. Zhao, M. Leng, F. Ma, W. Liang, L. Wang, S. Jin, J. Han, L. Zhang, J. Etheridge, J. Wang, Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541–545.
6 L. N. Anderson, M. B. Oviedo and B. M. Wong, J. Chem. Theory Comput., 2017, 13, 1656–1666.
7 M. E. Foster and B. M. Wong, J. Chem. Theory Comput., 2012, 8, 2682–2687.
8 J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105.
9 S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B Condens. Matter, 1996, 54, 1703–1710.
10 M. Krack, Theor. Chem. Acc., 2005, 114, 145–152.
11 J. VandeVondele, M. Iannuzzi and J. Hutter, Springer, Berlin, Heidelberg, 2006, pp. 287–314.
12 T. Frigato, J. VandeVondele, B. Schmidt, C. Schütte and P. Jungwirth, J. Phys. Chem. A, 2008, 112, 6125–6133.
13 F. Uhlig, O. Marsalek and P. Jungwirth, J. Phys. Chem. Lett., 2012, 3, 3071–3075.

download fileview on ChemRxivsupplementary_info.pdf (2.86 MiB)


















