
Rational combination therapy with PARP and MEK inhibitors ...Rational combination therapy with PARP...
19
CANCER 2017 © The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers Chaoyang Sun, 1,2 * Yong Fang, 1 Jun Yin, 1 Jian Chen, 1,3 Zhenlin Ju, 4 Dong Zhang, 1 Xiaohua Chen, 1 Christopher P. Vellano, 1 Kang Jin Jeong, 1 Patrick Kwok-Shing Ng, 5 Agda Karina B. Eterovic, 1 Neil H. Bhola, 6 Yiling Lu, 1 Shannon N. Westin, 7 Jennifer R. Grandis, 6 Shiaw-Yih Lin, 1 Kenneth L. Scott, 8 Guang Peng, 9 Joan Brugge, 10 Gordon B. Mills 1 Mutant RAS has remained recalcitrant to targeted therapy efforts. We demonstrate that combined treatment with poly(adenosine diphosphate–ribose) polymerase (PARP) inhibitors and mitogen-activated protein kinase (MAPK) kinase (MEK) inhibitors evokes unanticipated, synergistic cytotoxic effects in vitro and in vivo in multiple RAS mutant tumor models across tumor lineages where RAS mutations are prevalent. The effects of PARP and MEK inhibitor combinations are independent of BRCA1/2 and p53 mutation status, suggesting that the synergistic activity is likely to be generalizable. Synergistic activity of PARP and MEK inhibitor combinations in RAS mutant tumors is associated with (i) induction of BIM-mediated apoptosis, (ii) decrease in expression of components of the homologous recombination DNA repair pathway, (iii) decrease in homologous recombination DNA damage re- pair capacity, (iv) decrease in DNA damage checkpoint activity, (v) increase in PARP inhibitor–induced DNA damage, (vi) decrease in vascularity that could increase PARP inhibitor efficacy by inducing hypoxia, and (vii) elevated PARP1 protein, which increases trapping activity of PARP inhibitors. Mechanistically, enforced expression of FOXO3a, which is a target of the RAS/MAPK pathway, was sufficient to recapitulate the functional consequences of MEK inhibitors including synergy with PARP inhibitors. Thus, the ability of mutant RAS to suppress FOXO3a and its reversal by MEK inhibitors accounts, at least in part, for the synergy of PARP and MEK inhibitors in RAS mutant tumors. The rational combination of PARP and MEK inhibitors warrants clinical investigation in patients with RAS mutant tumors where there are few effective therapeutic options. INTRODUCTION Although some patients with cancer initially respond to targeted therapy, clinical responses are usually short-lived, thus warranting development and implementation of effective combination therapies to increase patient benefit. The ability of tumor cells to adapt to stress engendered by targeted therapies represents a key mechanism of re- sistance that, if effectively targeted, could lead to tumor cell death and improved patient outcomes. Blocking adaptive responses to targeted therapies represents an attractive means toward development of rational combination therapies ( 1–3). Nowhere is the need for targeted therapies greater than for cancers driven by oncogenic RAS, which represents one of the most common types of mutations in cancer. KRAS is mutationally activated in >90% of pancreatic ductal adenocarcinomas, half of colorectal cancers, and about 30% of lung cancers (4). Similarly, about 30% of melanomas are driven by oncogenic NRAS, whereas HRAS is commonly mutated in squamous cell carcinomas (4). Despite the dominant oncogenic role of mutant RAS in these and other cancer types, activated RAS isoforms remain undruggable by current therapeutic modalities. This has led to wide research interest including establishment of the National Cancer Institute RAS Initiative (www.cancer.gov/research/key-initiatives/ras). Half of type 1 ovarian cancers are driven by oncogenic KRAS. This subtype consists primarily of low-grade tumors that are notoriously in- sensitive to chemotherapy and are almost uniformly lethal once spread to the peritoneal cavity (5). In contrast, more common type 2 ovarian cancers, which predominantly consist of high-grade serous ovarian cancers (HGSOCs), rarely contain RAS mutations but still exhibit RAS pathway activation in ~25% of tumors, thus demonstrating the im- portance of the RAS pathway in both ovarian cancer subtypes (6–8). About half of all HGSOC tumors exhibit aberrations in components of the homologous recombination (HR) DNA repair pathway (6) that likely contribute to efficacy of platinum drugs and of poly(adenosine diphosphate–ribose) polymerase (PARP) inhibitors (PARPis). PARP, a critical component of the single-strand break (SSB) repair (SSBR) pathway, came into focus as a target when SSBR was identified as a syn- thetic lethal partner with defects in the HR pathway induced by BRCA1/2 mutations (9, 10). Mechanistically, blocking PARP1 enzymatic activ- ity compromises base excision repair, resulting in conversion of SSBs to double-strand breaks (DSBs) during DNA replication, thus inducing synthetic lethality in cancer cells with HR defects. Normal cells, in con- trast, retain the ability to repair DSBs through HR and are therefore re- sistant to PARPi (11). Because PARP1 participates in additional DNA repair processes including inhibition of nonhomologous end joining 1 Department of Systems Biology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA. 2 Department of Obstetrics and Gynecology, Tongji Hos- pital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China. 3 Department of General Surgery, Second Affiliated Hospi- tal, Zhejiang University School of Medicine, Zhejiang Province, Hangzhou 310000, China. 4 Department of Bioinformatics and Computational Biology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA. 5 Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA. 6 Provost Office, Uni- versity of California, San Francisco, San Francisco, CA 94158, USA. 7 Department of Gynecologic Oncology and Reproductive Medicine, University of Texas MD An- derson Cancer Center, Houston, TX 77030, USA. 8 Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA. 9 Depart- ment of Clinical Cancer Prevention, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA. 10 Department of Cell Biology, Ludwig Center at Harvard, Harvard Medical School, Boston, MA 02115, USA. *Corresponding author. Email: [email protected] SCIENCE TRANSLATIONAL MEDICINE | RESEARCH ARTICLE Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017 1 of 18 by guest on May 22, 2020 http://stm.sciencemag.org/ Downloaded from
Transcript of Rational combination therapy with PARP and MEK inhibitors ...Rational combination therapy with PARP...

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
CANCER
1Department of Systems Biology, University of Texas MD Anderson Cancer Center,Houston, TX 77030, USA. 2Department of Obstetrics and Gynecology, Tongji Hos-pital, Tongji Medical College, Huazhong University of Science and Technology,Wuhan 430030, China. 3Department of General Surgery, Second Affiliated Hospi-tal, Zhejiang University School of Medicine, Zhejiang Province, Hangzhou 310000,China. 4Department of Bioinformatics and Computational Biology, University ofTexas MD Anderson Cancer Center, Houston, TX 77030, USA. 5Sheikh KhalifaBin Zayed Al Nahyan Institute for Personalized Cancer Therapy, University ofTexas MD Anderson Cancer Center, Houston, TX 77030, USA. 6Provost Office, Uni-versity of California, San Francisco, San Francisco, CA 94158, USA. 7Department ofGynecologic Oncology and Reproductive Medicine, University of Texas MD An-derson Cancer Center, Houston, TX 77030, USA. 8Department of Molecular andHuman Genetics, Baylor College of Medicine, Houston, TX 77030, USA. 9Depart-ment of Clinical Cancer Prevention, University of Texas MD Anderson CancerCenter, Houston, TX 77030, USA. 10Department of Cell Biology, Ludwig Centerat Harvard, Harvard Medical School, Boston, MA 02115, USA.*Corresponding author. Email: [email protected]
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
2017 © The Authors,
some rights reserved;
exclusive licensee
American Association
for the Advancement
of Science.
http://stm.scienc
Dow
nloaded from
Rational combination therapy with PARP and MEKinhibitors capitalizes on therapeutic liabilities in RASmutant cancersChaoyang Sun,1,2* Yong Fang,1 Jun Yin,1 Jian Chen,1,3 Zhenlin Ju,4 Dong Zhang,1 Xiaohua Chen,1
Christopher P. Vellano,1 Kang Jin Jeong,1 Patrick Kwok-Shing Ng,5 Agda Karina B. Eterovic,1
Neil H. Bhola,6 Yiling Lu,1 Shannon N. Westin,7 Jennifer R. Grandis,6 Shiaw-Yih Lin,1
Kenneth L. Scott,8 Guang Peng,9 Joan Brugge,10 Gordon B. Mills1
Mutant RAS has remained recalcitrant to targeted therapy efforts. We demonstrate that combined treatmentwith poly(adenosine diphosphate–ribose) polymerase (PARP) inhibitors and mitogen-activated protein kinase(MAPK) kinase (MEK) inhibitors evokes unanticipated, synergistic cytotoxic effects in vitro and in vivo in multipleRAS mutant tumor models across tumor lineages where RAS mutations are prevalent. The effects of PARP andMEK inhibitor combinations are independent of BRCA1/2 and p53 mutation status, suggesting that the synergisticactivity is likely to be generalizable. Synergistic activity of PARP and MEK inhibitor combinations in RAS mutanttumors is associated with (i) induction of BIM-mediated apoptosis, (ii) decrease in expression of components ofthe homologous recombination DNA repair pathway, (iii) decrease in homologous recombination DNA damage re-pair capacity, (iv) decrease in DNA damage checkpoint activity, (v) increase in PARP inhibitor–induced DNA damage,(vi) decrease in vascularity that could increase PARP inhibitor efficacy by inducing hypoxia, and (vii) elevated PARP1protein, which increases trapping activity of PARP inhibitors. Mechanistically, enforced expression of FOXO3a, whichis a target of the RAS/MAPK pathway, was sufficient to recapitulate the functional consequences of MEK inhibitorsincluding synergy with PARP inhibitors. Thus, the ability of mutant RAS to suppress FOXO3a and its reversal by MEKinhibitors accounts, at least in part, for the synergy of PARP and MEK inhibitors in RAS mutant tumors. The rationalcombination of PARP and MEK inhibitors warrants clinical investigation in patients with RAS mutant tumors wherethere are few effective therapeutic options.
ema
by guest on May 22, 2020
g.org/
INTRODUCTIONAlthough some patients with cancer initially respond to targetedtherapy, clinical responses are usually short-lived, thus warrantingdevelopment and implementation of effective combination therapiesto increase patient benefit. The ability of tumor cells to adapt to stressengendered by targeted therapies represents a key mechanism of re-sistance that, if effectively targeted, could lead to tumor cell death andimproved patient outcomes. Blocking adaptive responses to targetedtherapies represents an attractive means toward development of rationalcombination therapies (1–3).
Nowhere is the need for targeted therapies greater than for cancersdriven by oncogenic RAS, which represents one of the most commontypes ofmutations in cancer.KRAS ismutationally activated in >90% ofpancreatic ductal adenocarcinomas, half of colorectal cancers, and
about 30% of lung cancers (4). Similarly, about 30% of melanomasare driven by oncogenic NRAS, whereas HRAS is commonly mutatedin squamous cell carcinomas (4). Despite the dominant oncogenic roleof mutant RAS in these and other cancer types, activated RAS isoformsremain undruggable by current therapeutic modalities. This has led towide research interest including establishment of the National CancerInstitute RAS Initiative (www.cancer.gov/research/key-initiatives/ras).
Half of type 1 ovarian cancers are driven by oncogenic KRAS. Thissubtype consists primarily of low-grade tumors that are notoriously in-sensitive to chemotherapy and are almost uniformly lethal once spreadto the peritoneal cavity (5). In contrast, more common type 2 ovariancancers, which predominantly consist of high-grade serous ovariancancers (HGSOCs), rarely contain RAS mutations but still exhibitRAS pathway activation in ~25%of tumors, thus demonstrating the im-portance of the RAS pathway in both ovarian cancer subtypes (6–8).
About half of all HGSOC tumors exhibit aberrations in componentsof the homologous recombination (HR) DNA repair pathway (6) thatlikely contribute to efficacy of platinum drugs and of poly(adenosinediphosphate–ribose) polymerase (PARP) inhibitors (PARPis). PARP,a critical component of the single-strand break (SSB) repair (SSBR)pathway, came into focus as a target when SSBRwas identified as a syn-thetic lethal partnerwith defects in theHRpathway induced byBRCA1/2mutations (9, 10).Mechanistically, blocking PARP1 enzymatic activ-ity compromises base excision repair, resulting in conversion of SSBs todouble-strand breaks (DSBs) during DNA replication, thus inducingsynthetic lethality in cancer cells with HR defects. Normal cells, in con-trast, retain the ability to repair DSBs through HR and are therefore re-sistant to PARPi (11). Because PARP1 participates in additional DNArepair processes including inhibition of nonhomologous end joining
1 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2
http://stm.sciencem
ag.org/D
ownloaded from
(NHEJ) and alternative NHEJ and in recruitment of DNA repair pro-teins (12), patients whose tumors are HR-proficient may also benefitfrom PARPi. Furthermore, several PARPis “trap” PARP proteins atsites of DNA damage, with trapped PARPs being more toxic than SSBsor DSBs (13, 14).
Recently, a number of potent trapping PARPis including olaparib,niraparib, and rucaparib have been approved for ovarian cancer ther-apy. However, like most other targeted therapies, responses to PARPiare all too frequently transient. A number of combination therapieshave been implemented with PARPi to attempt to induce HR defectsin tumors with intact HR and thus engender PARP sensitivity or to in-crease efficacy of PARPi by blocking DNA repair either by inducinghypoxia or by blocking DNA damage cell cycle checkpoints. These in-clude inhibitors of signaling through the phosphatidylinositol 3-kinase(PI3K) pathway (NCT01623349 and NCT02208375), vascular endo-thelial growth factor receptor (VEGFR) (NCT02345265), and cell cyclecheckpoints including WEE1 (NCT02576444 and NCT02511795)(ClinicalTrials.gov).
Here, we evaluated adaptive responses that could mediate resistanceto PARPi through reverse phase protein arrays (RPPAs), which quan-titate hundreds of critical signalingmolecules in terms of both total pro-tein and posttranslational modification analysis. Transient treatmentwith PARPi induced a marked increase in RAS/mitogen-activated pro-tein kinase (MAPK) pathway activation including down-regulation ofthe key RAS/MAPK targets: FOXO3a and BIM1, which was recapitu-lated in PARPi-resistant cell lines. On the basis of this observation, weassessed the activity of PARPi in combination with MAPK kinase(MEK) or extracellular signal–regulated kinase (ERK) inhibition anddemonstrated that the combination was synergistic in a subset of ovar-ian cancer cell lines. These studies led to the serendipitous observationthatRASmutant cell lines acrossmultiple lineages are resistant to PAR-Pi and that this resistance could be reversed byMEK or ERK inhibition.We subsequently demonstrated that MEK inhibitors (MEKis) sensitizeRASmutantmodels to PARPi primarily through the induction of FOX-O3a, at least in part through increasing apoptotic sensitivity, altering theexpression of PARP1, decreasingDNAdamage sensing, and decreasingHR DNA repair capacity. Consistent with these observations, PARPiand MEKi/ERKi demonstrated synergistic activity in vitro and in vivoin tumors with mutant RAS.
020
RESULTSPARPi and MEKi induce inverse adaptive responsesTo identify adaptive responses that mediate resistance to PARPi, weused RPPA to assess signaling pathway perturbations in response totreatment with BMN673 (talazoparib), a potent trapping PARPi in 10breast, ovarian, and endometrial cancer cell lines (fig. S1A). Replicatesfor each treatment condition [monolayer two-dimensional (2D),spheroid 3D, and two time points] were averaged for each line (Fig. 1A).We next rank-ordered changes by summing median-centered proteinamounts normalized to control [dimethyl sulfoxide (DMSO)–treatedcells at the same time point and culture condition] and visualized datato provide an assessment of decreases or increases (represented by greenand red, respectively) in expression or phosphorylation of proteins (Fig.1 and fig. S1). Proteins exhibiting increased expression or phosphoryl-ation (indicated by “p” before the protein name) in response to PARPitreatment include those involved in DNA damage repair or DNA dam-age checkpoints, such as FOXM1, CHK1/pCHK1, CHK2/pCHK2,ATM/pATM, pWEE1, RAD51, and RAD50. In addition, we observed
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
marked changes in cell cycle regulators including pRB, CDK1, and cy-clin B1, as well as activation ofmultiple components of the RAS/MAPKsignaling pathway including pMEK, pMAPK, pPKC, pYB1, pBAD, andpS6 (15–19). PARPi treatment decreased FOXO3a, P27, and BIM,which is consistent with PARPi-induced activation of the RAS/MAPKpathway, because FOXO3a is down-regulated by RAS/MAPK pathwayactivation, P27 and BIM are downstream of FOXO3a (20), and BIM istargeted for proteolysis by direct ERKphosphorylation (21). This obser-vation is also consistent with RPPA analysis of The Cancer GenomeAtlas (TCGA) samples (Pan-Can set in TCPAportal.org v4.0), indicat-ing that pMEK and BIM were inversely correlated (P = 8.7 × 10−53)across multiple tumor lineages. PARPi-induced decreases in BIM ex-pression and increased phosphorylation of BAD would be expectedto decrease propensity of cells to undergo apoptosis and thus contributeto adaptation of cells to stress induced by treatmentwithPARPi. Finally,we observed that PARPi treatment decreased PARP1, consistent withchanges in PARP1 expression contributing to adaptation to PARPi, giv-en that PARP1 expression correlates with sensitivity to PARPi (22).
Because PARPi altered expression of multiple proteins in the RAS/MAPKpathway,we also assessed adaptive responses to theGSK1120212B(MEKi) by treating five breast and ovarian cancer cell lines (Fig. 1B andfig. S1B). This analysis revealed marked GSK1120212B-mediated de-creases in phosphorylation of proteins downstream of MEK (pERK,pYB1, pPKC, pBAD, and pS6; Fig. 1B), although pMEK was increasedlikely due to a previously described positive feedback loop (23).GSK1120212B treatment also decreased the amounts or phosphoryl-ation of proteins involved in DNA damage repair and DNA damagecheckpoints including pATM, ATR, BRCA2, CHK1/pCHK1, CHK2/pCHK2, FOXM1, MRE11, RAD50, RAD51, and WEE1 (Fig. 1B). Asexpected, MEKi increased P27, FOXO3a, and BIM and modestlyincreased 53BP1 and PARP1. PARPi and MEKi induced reciprocaleffects on key proteins involved in DNA damage repair, DNA damagecheckpoints, and cell viability (P < 0.001; Fig. 1, C and D, and fig. S1, Cand D). Together, the data raised the possibility that PARPi and MEKicould each block adaptive responses induced by the other drug and thusinduce a synthetic lethal interaction.
PARPi and MEKi demonstrate synergy in a subset of ovariancancer cell linesTo explore potential synergy between PARPi and MEKi, we assessedeffects ofmonotherapy and combination therapy on 24well-characterizedovarian cancer cell lines selected to represent different subtypes of ovar-ian cancer (Fig. 2A) (24). For 8 of the 24 cell lines, the PARPi/MEKicombinationwas synergistic, with a combination index (CI) of less than0.5 (Fig. 2A, red). Some of the other cell lines exhibited marked sensi-tivity to BMN673 without any significant interaction with MEKi (Fig.2A, blue), and some cell lines exhibited modest to no response to eithersingle or combination treatment (Fig. 2A, black). Given that the effectsof PARPi most clearly manifest after multiple cell divisions, we also as-sessed the effects of the inhibitors in long-term assays. PARPi andMEKiat low concentrations resulted in synergistic inhibition of cell viability(fig. S2A) and colony formation of OVCAR8 in 3D (fig. S2B) and 2D(fig. S2C) compared to treatment with either inhibitor alone.
Mutant KRAS mediates resistance to PARPi and sensitivity toPARPi and MEKiWe next sought to identify molecular underpinnings responsible forsynergy by the PARPi and MEKi combination. In ovarian cancer celllines examined, KRAS mutations were unexpectedly associated with
2 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
Fig. 1. PARPi and MEKi induce inverse adaptive responses. (A) Ten breast, ovarian, and endometrial cell lines [BT474 (PIK3CA_Mut, HER2_Amp), HCC1954 (PIK3CA_Mutand HER2_Amp), HCC1937 (BRCA1_Mut), MDA-MB-468 (EGFR_Overexpression and PTEN_Mut), IGROV-1 (BRCA1_Mut), TOV21G (PIK3CA_Mut and KRAS_Mut), KLE (TP53_Mut),ETN-1 (PTEN_Mut), SKBr3 (HER2_Amp), SKOV3 (PIK3CA_Mut and HER2_Amp)] were cultured in Matrigel (3D) or monolayer (2D) and treated for 7 or 3 days, respectively, withDMSO or BMN673 at a median inhibitory concentration (IC50) determined experimentally for each line for 2D and 3D conditions. Protein lysates were analyzed for 220 totaland phosphoproteins by RPPA. For visualization, 2D and 3D conditions and time were averaged (see fig. S1 for all samples). Heat map represents “rank-ordered” changesinduced by BMN673 treatment, calculated by summing median-centered protein amount normalized to control. Proteins with consistent decreases are at the top (green) andincreases are at the bottom (red) of the heat map. (B) Five cell lines from (A) (MDA-MB-468, HCC1954, BT474, SKOV3, and SKBr3) were cultured in Matrigel (3D) or monolayer(2D) and treated with MEKi (GSK1120212B) at two concentrations (2 and 20 nM) or DMSO for 24 or 48 hours, after which protein lysates were analyzed for 220 total andphosphoproteins by RPPA. (C) The left column of the heat map shows protein changes ordered by the effects of BMN673 treatment from (A), sorted in increasing orderfrom top to bottom (green indicates the most consistently decreased proteins, and red indicates the most consistently increased proteins after treatment). The rightcolumn in the heat map shows protein order after GSK1120212B treatment from (B). (D) Comparison (2 × 2) of selected proteins [marked in (A) and (B)] after BMN673and GSK1120212B treatment.
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017 3 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
resistance to BMN673 (P = 4.7 × 10−5) (Fig. 2B). AlthoughKRASmuta-tions are rare in HGSOCs, the indicated ovarian cancer cell lines haveclassical activating KRAS mutations, with the exception of OVCAR8,whichhas a rareKRASP121Hmutation of unknown significance.OVCAR8
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
has a RAS/MAPK activation score (24) similar to that of the ovariancancer cell lines with classicalKRASmutations. As expected,KRASmu-tations were associated with increased sensitivity to MEKi (P = 0.026;Fig. 2C). Consistent with our observation of reciprocal effects of MEKi
Fig. 2. Mutant KRAS mediates resistance to PARPi and sensitivity to PARPi and MEKi (A) Drug response curves for BMN673 combined with AZD6244 in 24 well-characterized ovarian cancer cell lines treated with varying concentrations of the two compounds for 96 hours. CI was calculated using CalcuSyn software with theChou-Talalay equation. CI values reflect the sign and magnitude of drug-drug interaction: synergy, <0.5; additivity, 0.5 to 1; antagonism, >1. Red cell line name: synergy(CI, <0.5); blue cell line name: cells intrinsically sensitive to PARPi; black cell line name: cells exhibited modest to no response to either single or combination treatment.(B) Top: Percentage of cell viability at 10 mM BMN673 from (A). Bottom: Selected mutations in cell lines. KRAS and BRCA2 correlations with BMN673 response weresignificant (P = 4.7 × 10−5 and P = 0.023, respectively). (C) Top: Percentage of cell growth inhibition by AZD6244 from (A). Bottom: Selected mutations in cell lines. KRAScorrelation with AZD6244 response was significant (P = 0.026). (D) Correlation between IC50 of BMN673 and AZD6244 (Pearson’s r = −0.412; P = 0.049). (E) Top: CI values;cells were arranged by CI into synergistic (green) and nonsynergistic (red) (cutoff, 0.5) based on (A). Bottom: Selected mutations in cell lines. KRASmutation is correlatedwith synergistic effect between PARPi and MEKi (P = 3 × 10−6). (F) Interactions between BMN673 and AZD6244 (MEKi), GSK1120212B (MEKi), SCH772984 (ERKi),GSK2118436A (BRAFi), SP600125 (JNKi), PD0332991 (CDK4/6 inhibitor), and BKM120 [PI3K inhibitor (PI3Ki)] were examined across a panel of 10 cell lines including8 RASmutant lines and 2 RAS wild-type lines (cell line data in fig. S3, A and B). (G) Response of Ba/F3 cells rendered interleukin-3–independent with KRAS_WT, KRASG12D,KRASG12V, or PIK3CAH1047R to BMN673 and AZD6244/SCH772984. (H) Heat map of CI of combinations of BMN673 with AZD6244 and SCH772984 from (G). Combinationswith BMN673 and GSK2118436A (BRAFi) and SP600125 (JNKi) are also presented.
4 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
and PARPi on adaptive responses (Fig. 1), we detected a significantinverse correlation between sensitivity of cell lines to PARPi andMEKi(P = 0.049; Fig. 2D). Strikingly, KRAS mutations emerged as the mostsignificant predictor of synergistic effects of PARPi and MEKi (P <0.001; Fig. 2E). This synergy between PARPi and MEKi did not associ-ate with mutation status of BRCA1 or BRCA2 (Fig. 2E), suggesting thatthe observed drug synergy is dependent on amechanism(s) independentof HR deficiency induced by BRCA1/2mutations. Furthermore, the syn-ergistic response to the MEKi and PARPi combination was independentof p53mutation status (Fig. 2E), indicating its potential efficacy on bothp53 wild-type and mutant tumors.
To determine which targets within the RAS/MAPK pathway medi-ate synergy with PARPi, we assessed the efficacy of targeting differentnodes in the RAS/MAPK pathway. The eight KRAS mutant ovariancancer cell lines that demonstrated synergy between BMN673 andAZD6244 were included along with two RAS wild-type lines, HOC8and ES2, which were unresponsive to the MEKi/PARPi combination(Fig. 2A). CI in the rescreening studywas highly correlatedwith the resultsfrom our initial screen (r = 0.93; fig. S3A). Two MEKis (GSK1120212Band AZD6244) and an ERKi (SCH772984) demonstrated similarpatterns of synergy with PARPi (Fig. 2F and fig. S3B). In contrast, theBRAF inhibitor (BRAFi) (GSK2118436A) only demonstrated synergyin TOV21G and modest additivity with PARPi in other lines. The fail-ure of BRAFis to demonstrate consistent synergy with PARPi is likelydue to the ability of other RAF homologs to bypass the effect of BRAFinhibition (25). Three unrelated inhibitors targeting c-Jun N-terminalkinase (JNK) (SP600125), CDK4/6 (PD0332991), or PI3K (BKM120)did not demonstrate synergy with PARPi in any of the lines, which isconsistent with selectivity of interactions between MEKi/ERKi andPARPi in RAS mutant cell lines (Fig. 2F). Thus, both MEKi and ERKishow marked synergy with PARPi in KRAS mutant ovarian cancercell lines.
To directly demonstrate that mutant KRAS mediates resistanceto PARPi and sensitizes to PARPi and MEKi combinations, we devel-oped Ba/F3 lines (26, 27) stably expressing wild-type or activatedforms of KRAS (G12V and G12C). Cells expressing wild-type KRASor expressing activated PIK3CAH1047R were sensitive to PARPi andmoderately sensitive to MEKi but did not demonstrate synergy withthe PARPi and MEKi combination. This was, in part, due to themarked sensitivity to PARPi (Fig. 2, G and H). Strikingly, both KRASmutant expressing cell lines exhibited marked resistance to PARPi butwere sensitive to combinations of PARPi and MEKi or ERKi (Fig. 2G).Similar to effects in naturally occurring lines, synergistic interactionswere observed with PARPi and MEKi or ERKi but not with BRAFiand JNKi (JNK inhibitor) (Fig. 2H).
We further explored the role of mutant KRAS in resistance toPARPi and in synergy between MEKi and PARPi using doxycycline(Dox)–inducible HPDE-iKRASG12D pancreatic cancer cells (28) andcell lines from a KRAS-inducible mouse pancreatic cancer model[p48 Cre_tetO_LKrasG12D ROSA_rtTAL+ p53L+ mice (LKrasG12D)(29)]. In HPDE-iKRASG12D, induction of mutant KRAS induced RAS/MAPK pathway activation and decreased BIM (fig. S3C), as expected.Induction of mutant KRAS decreased a second BH3-only protein, BID(BID was not on the arrays in Fig. 1), which could also contribute todecreased sensitivity to cell death. Dox-induced mutant KRAS expressiondecreased sensitivity to BMN673 in both inducible KRASmodels (fig. S3,D and E). The MEKi and PARPi combination was not synergistic innoninduced HPDE but was synergistic in HPDE where mutant KRASwas induced by Dox (fig. S3D). Further, in two different lines from the
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
LKrasG12D model, the combination of BMN673 with MEKi or ERKi wassynergistic (fig. S3F).
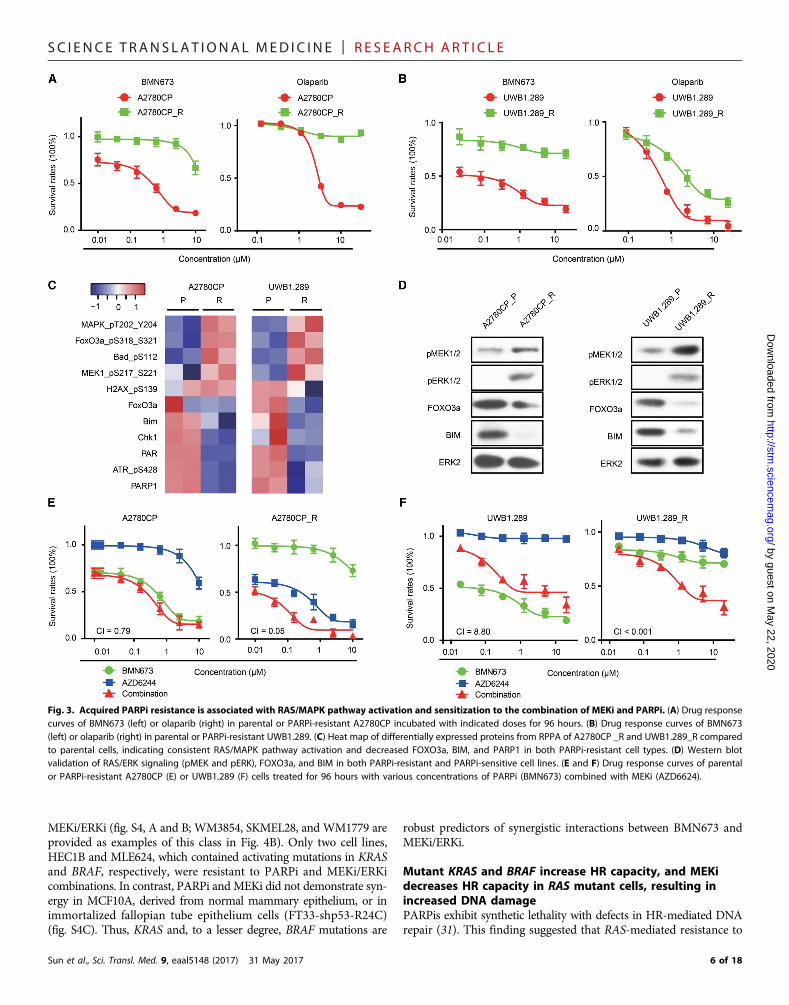
Acquired PARPi resistance is associated with RAS/MAPKpathway activation and sensitization to the combination ofMEKi and PARPiTo explore whetherMEKi could resensitize PARPi-resistant cells to theeffects of PARPi, we developedPARPi-resistant cells by culturing highlyPARPi-sensitive cells (UWB1.289 and A27980CP, both RAS wild type;see Fig. 2) in the continued presence of BMN673 for 3 to 4 months, atwhich time drug-resistant clones emerged. A2780CP PARPi-resistant(A2780CP_R) and UWB1.289 PARPi-resistant (UWB1.289_R) cloneswere highly resistant to BMN673 and cross-resistant to olaparib (Fig. 3,A and B). RPPA analysis demonstrated that RAS/MAPK pathway ac-tivity [increased pMEK, pBAD, and pFOXO3a (inactive form)] was up-regulated in PARPi-resistant clones (Fig. 3C).Moreover, resistant clonesshowed lower total FOXO3a and BIM, as expected from increased RAS/MAPK pathway activity. The decreased PAR and PARP1 expression inthe resistant cells could also contribute to PARPi resistance becausePARP1 expression is associated with PARPi sensitivity (22). Westernblotting confirmed increased RAS/MEK pathway activity with concom-itant decreases in FOXO3a and BIM in resistant cells (Fig. 3D). Overall,the signaling changes in long-termPARPi-resistant cells exhibitedmanysimilarities to adaptive responses to short-term PARPi treatment (seeFig. 1). Despite increased RAS/MEKpathway activity,KRAS sequencingdemonstrated that the resistant lines did not acquire classical activatingKRAS mutations. However, deep next-generation sequencing (NGS)and Sanger sequencing of individual PARPi-resistant clones fromA2780CP_R demonstrated the presence of KRASA146T, KRASA59T, andMAP2K1A283T in 19, 11, and 6%of cells, respectively, but not inA2780CPparental cells. Prolonged culture of the lines without PARPi resulted inloss of the mutant KRAS and MAP2K1 clones. The KRASA146T mutanthas been demonstrated to be modestly activating (30). The selection ofKRAS mutations in a PARPi-resistant line supports the concept thatRAS mutations and RAS/MAPK pathway activation are key mediatorsof PARP resistance. As expected from increases in RAS/MAPK activityin PARPi-resistant cell lines and KRAS and MAPK1 mutations,A2780CP_R clones were markedly more sensitive and UWB1.289_Rclones were modestly more sensitive to MEKi (Fig. 3, E and F). MEKiresensitized bothPARPi-resistant clones to PARPi (Fig. 3, E and F). Thus,MEKis have the potential to resensitize PARPi-resistant human tumorsto PARPi.
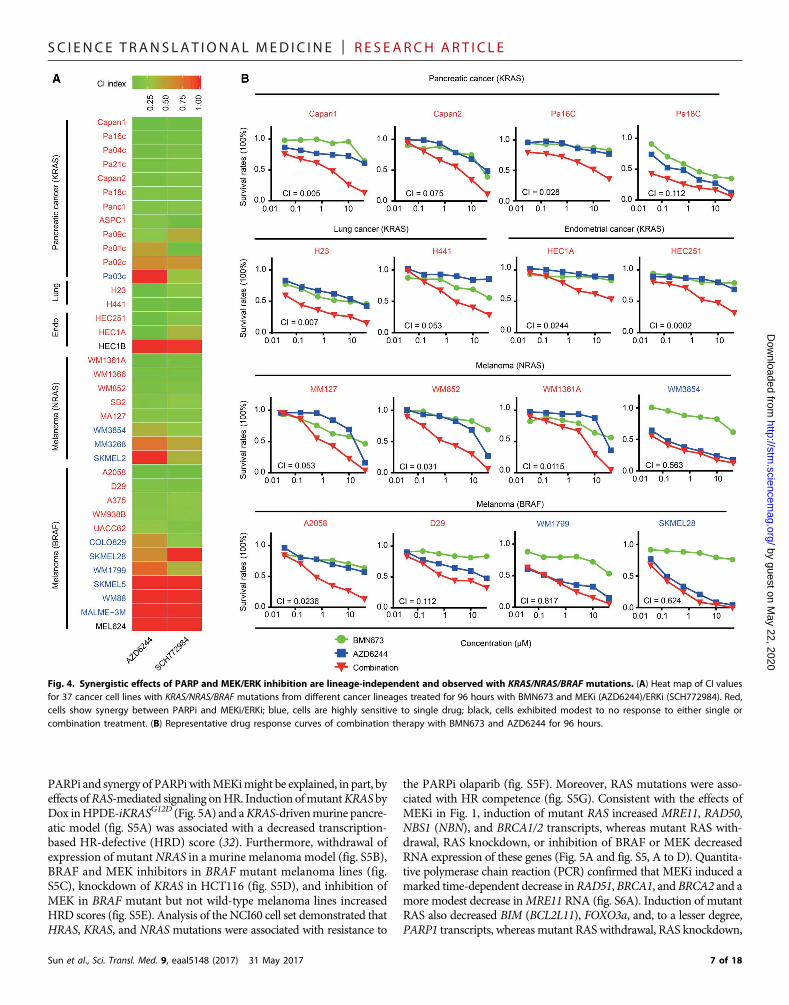
Synergistic effects of PARP and MEK/ERK inhibition arelineage-independent and observed with KRAS/NRAS/BRAF mutationsTo determine whether synergistic effects of combined PARPi andMEKi/ERKi can be generalized across different lineages and differentRAS/ERK pathway aberrations, we next performed drug response as-says using a panel of 17 KRAS mutant cancer lines (12 pancreatic, 2lung, and 3 endometrial), 8 NRAS mutant melanoma lines, and 12BRAF mutant melanoma lines (Fig. 4, A and B, and fig. S4, A andB). All RASmutant cell lines were resistant to BMN673 monotherapy,including Capan1, which has a BRCA2 mutation. However, markedsynergism (CI, <0.5) between BMN673 and MEKi/ERKi was observedin 25 of 37 of KRAS/NRAS and BRAF mutant cell lines. In a subset ofcell lines, interactions between PARPi and MEKi/ERKi were modestdespite a CI indicating synergism. Most (10 of 12) lines that did notdemonstrate synergy were highly sensitive to monotherapy with
5 of 18
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
MEKi/ERKi (fig. S4, A and B; WM3854, SKMEL28, and WM1779 areprovided as examples of this class in Fig. 4B). Only two cell lines,HEC1B and MLE624, which contained activating mutations in KRASand BRAF, respectively, were resistant to PARPi and MEKi/ERKicombinations. In contrast, PARPi and MEKi did not demonstrate syn-ergy in MCF10A, derived from normal mammary epithelium, or inimmortalized fallopian tube epithelium cells (FT33-shp53-R24C)(fig. S4C). Thus, KRAS and, to a lesser degree, BRAF mutations are
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
robust predictors of synergistic interactions between BMN673 andMEKi/ERKi.
Mutant KRAS and BRAF increase HR capacity, and MEKidecreases HR capacity in RAS mutant cells, resulting inincreased DNA damagePARPis exhibit synthetic lethality with defects in HR-mediated DNArepair (31). This finding suggested that RAS-mediated resistance to
Fig. 3. Acquired PARPi resistance is associated with RAS/MAPK pathway activation and sensitization to the combination of MEKi and PARPi. (A) Drug responsecurves of BMN673 (left) or olaparib (right) in parental or PARPi-resistant A2780CP incubated with indicated doses for 96 hours. (B) Drug response curves of BMN673(left) or olaparib (right) in parental or PARPi-resistant UWB1.289. (C) Heat map of differentially expressed proteins from RPPA of A2780CP _R and UWB1.289_R comparedto parental cells, indicating consistent RAS/MAPK pathway activation and decreased FOXO3a, BIM, and PARP1 in both PARPi-resistant cell types. (D) Western blotvalidation of RAS/ERK signaling (pMEK and pERK), FOXO3a, and BIM in both PARPi-resistant and PARPi-sensitive cell lines. (E and F) Drug response curves of parentalor PARPi-resistant A2780CP (E) or UWB1.289 (F) cells treated for 96 hours with various concentrations of PARPi (BMN673) combined with MEKi (AZD6624).
6 of 18
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
PARPi and synergy of PARPiwithMEKimight be explained, in part, byeffects ofRAS-mediated signaling onHR. Induction ofmutantKRAS byDox inHPDE-iKRASG12D (Fig. 5A) and aKRAS-drivenmurine pancre-atic model (fig. S5A) was associated with a decreased transcription-based HR-defective (HRD) score (32). Furthermore, withdrawal ofexpression of mutantNRAS in a murine melanomamodel (fig. S5B),BRAF and MEK inhibitors in BRAF mutant melanoma lines (fig.S5C), knockdown of KRAS in HCT116 (fig. S5D), and inhibition ofMEK in BRAF mutant but not wild-type melanoma lines increasedHRD scores (fig. S5E). Analysis of the NCI60 cell set demonstrated thatHRAS, KRAS, and NRAS mutations were associated with resistance to
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
the PARPi olaparib (fig. S5F). Moreover, RAS mutations were asso-ciated with HR competence (fig. S5G). Consistent with the effects ofMEKi in Fig. 1, induction of mutant RAS increased MRE11, RAD50,NBS1 (NBN), and BRCA1/2 transcripts, whereas mutant RAS with-drawal, RAS knockdown, or inhibition of BRAF or MEK decreasedRNA expression of these genes (Fig. 5A and fig. S5, A to D). Quantita-tive polymerase chain reaction (PCR) confirmed that MEKi induced amarked time-dependent decrease in RAD51, BRCA1, and BRCA2 and amore modest decrease inMRE11 RNA (fig. S6A). Induction of mutantRAS also decreased BIM (BCL2L11), FOXO3a, and, to a lesser degree,PARP1 transcripts, whereas mutant RAS withdrawal, RAS knockdown,
Fig. 4. Synergistic effects of PARP and MEK/ERK inhibition are lineage-independent and observed with KRAS/NRAS/BRAF mutations. (A) Heat map of CI valuesfor 37 cancer cell lines with KRAS/NRAS/BRAF mutations from different cancer lineages treated for 96 hours with BMN673 and MEKi (AZD6244)/ERKi (SCH772984). Red,cells show synergy between PARPi and MEKi/ERKi; blue, cells are highly sensitive to single drug; black, cells exhibited modest to no response to either single orcombination treatment. (B) Representative drug response curves of combination therapy with BMN673 and AZD6244 for 96 hours.
7 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
Fig. 5. Mutant KRAS increases HR capacity and MEKi decreases HR capacity in RAS mutant cells, causing increased DNA damage. (A) Microarray data fromHPDE-iKRASG12D cell lines with or without Dox induction were analyzed by unsupervised clustering for HRD gene signatures. Heat maps of clusters indicate that cellswith KRASG12D induction are more likely HR-intact (left). Quantification of HRD scores of HPDE-iKRASG12D with or without Dox induction was calculated on the basis ofcorrelation to HRD gene signatures (right; higher scores are more likely to have HR defects). (B) Western blot showing dose-dependent protein changes in OVCAR8 after24 hours of treatment with the indicated concentrations of AZD6244. (C) Immunofluorescence staining of FOXO3a/BRCA1 and RAD51/g-H2AX after treatment withBMN673 (1 mM)/AZD6244 (5 mM)/combination therapy in OVCAR8 for 48 hours. Scale bars, 20 mm. (D) Comet assay in HPDE-iKRASG12D cell lines after treatment withBMN673 (200 nM)/AZD6244 (200 nM)/combination therapy for 72 hours with or without Dox induction. DNA damage was quantified via % DNA in tails. Each data pointrepresents at least 50 cells. (E) Comet assay in OVCAR8 cell lines after treatment with BMN673 (1 mM)/AZD6244 (5 mM)/combination therapy for 72 hours. DNA damagewas quantified via % DNA in tails. Each data point represents at least 50 cells. (F) Direct repeat (DR)–GFP assay to measure HR-mediated DNA DSB repair in DR-GFPU2OS. Frequency of GFP+ cells by flow cytometry after infection with the I-Sce1 endonuclease and incubation for 48 hours with or without 100 nM AZD6244 (top).NHEJ-mediated DNA DSB repair in EJ5-GFP U2OS. Frequency of GFP+ cells by flow cytometry after infection with the I-Sce1 endonuclease and incubation for 48 hourswith or without 100 nM AZD6244 (bottom). (G and H) Immunofluorescence staining for RAD51, g-H2AX, and MRE11 before and after IR in inducible HPDE-iKRASG12D (G)and OVCAR8 (H). Right: Images for MRE11 foci staining and quantification of MRE11 foci-positive cells at 2 hours after IR. Left: Because of relatively late recruitment ofRAD51, quantification of RAD51 staining was performed at different times (0, 2, 4, 6, and 8 hours after IR), and representative images are shown. Scale bars, 20 mm. DAPI,4′,6-diamidino-2-phenylindole. Error bars represent SEM of three independent experiments. Student’s t test: **P < 0.01 and ***P < 0.001.
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017 8 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
or inhibition of BRAF or MEK increased mRNA for these genes (Fig.5A and fig. S5, A to D). Western blot analysis confirmed that MEKiincreased BIM, FOXO3a, and g-H2AX while down-regulating pERKas well as RAD51 and BRCA1 (Fig. 5B and fig. S6B). The effects ofMEKi on MRE11 protein at early time points were modest, consistentwith the limited decrease inMRE11mRNA (Fig. 5B and fig. S6B).How-ever, at later time points, MEKis were sufficient to induce marked de-creases in BRCA1, Rad51, and MRE11 (fig. S6B). MEKi failed to alter53BP1 in OVCAR8 (Fig. 5B), consistent with themajor effects ofMEKibeing on HR competence rather than NHEJ. Immunofluorescenceanalysis demonstrated low FOXO3a, BRCA1, and g-H2AX withreadily detectable diffuse RAD51 cytosolic and nuclear expressionin resting OVCAR8 and HOC1 (Fig. 5C and fig. S6C). Consistentwith the Western blotting data, RAD51 was markedly decreasedby MEKi. Strikingly, the PARPi BMN673 induced a marked increasein BRCA1 nuclear staining, which was abrogated by the addition ofMEKi (Fig. 5C). The decrease in BRCA1 protein induced by MEKiand maintained in the combination of MEKi and PARPi (Fig. 5B andfig. S6, B and D) may contribute, in part, to the decrease in BRCA1 nu-clear localization. However, the more marked decrease in nuclearBRCA1 staining compared to total protein raises the possibility that de-creased expression of multiple components of the HR pathway such asRAD51 combined to result in the marked decrease in BRCA1 nuclearlocalization. g-H2AX was increased by the combination of PARPi andMEKi in multiple cell lines studied (fig. S6D). Together, this argues thatinhibition of the RAS/ERK pathway in RASmutant cells decreases HRcapacity, rendering cells HRD, and increases accumulation of DNAdamage in the presence of PARPi.
RAS mutants can activate both the MAPK pathway and the PI3Kpathway. Furthermore, PI3K pathway inhibitors have demonstratedsynergy with PARPi. However, as indicated in Fig. 2F, PI3Kis did notdemonstrate synergy with PARPi in RAS mutant cells. As indicated infig. S6 (E and F), althoughMEKi inducedmarked nuclear translocationof FOXO3a in the presence and absence of PARPi, the pan-PI3KiBKM120 had modest effects on FOXO3a nuclear translocation. Con-sistent with this observation, PI3Kis hadmodest effects on BIM expres-sion in RAS mutant cells (fig. S6E).
We used a comet DNA damage detection assay to directly examinewhether activation of KRAS would result in decreased DNA damagethat could be reversed by MEKi. In noninduced HPDE-iKRASG12D,BMN673 treatment increased the amount of DNA in tails, consistentwith accumulation of DNA damage (Fig. 5D). However, induction ofmutant KRAS rendered cells resistant to BMN673-induced DNAdamage, as indicated by decreased accumulation of DNA in tails.Combined treatment with MEKi and PARPi reversed the effects ofKRAS, as evidenced by an increase in comet formation (Fig. 5D).We observed similar effects in OVCAR8, where BMN673 but notMEKi increased DNA damage and the combination increased cometformation further (Fig. 5E). To directly assess the effects of MEKi onHR capacity, we used U2OS and an HR–green fluorescent protein(GFP) reporter assay. MEKi markedly decreased the ability of cellsto reconstitute GFP in the test plasmid (Fig. 5E). In contrast, MEKihad modest effects on NHEJ (Fig. 5F), consistent with modest effectson 53BP1 (Fig. 5B).
DSBs recruit the MRN complex including MRE11, NBS1, andRAD50, which is required for resection of DNA ends at DSBs as wellas for the initiation of DNA repair and DNA damage checkpointpathways. Increased DNA damage concurrent with decreased recruit-ment of BRCA1 to the nucleus suggested that MRN function might be
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
defective in cells treated with the PARPi and MEKi combination. Inboth OVCAR8 and HPDE-iKRASG12D, MEKi decreased ionizing ra-diation (IR)–induced MRE11 and RAD51 foci (Fig. 5, G and H). Be-cause efficient recruitment of RAD51 to DSBs requires MRN function,this provides independent support for MEKi compromising MRNfunction. The marked decrease in both MRE11 and RAD51 foci aswell as the decrease in BRCA1 nuclear localization and increased ac-cumulation of DNA damage suggests that MEKi interferes with theDNA damage repair pathway at multiple sites.
A FOXO3a-BIM cascade mediates sensitivity to PARP andMEK inhibitionAs indicated in Fig. 1, PARPi and MEKi demonstrated opposite effectson BIM, P27, and FOXO3a. The amounts of basal BIM, a BH3-onlyprotein that is a key mediator of apoptotic balance (33), in cell linesin Fig. 2 were strongly correlated with MEKi and PARPi synergism(P = 0.001). Decreased FOXO3a and BIM in PARPi-resistant cells(Fig. 3, C and D) and KRAS-induced cells (fig. S5) are consistent withFOXO3a and BIM contributing to resistance of RAS mutant cells toPARPi. Thus, low basal BIM or treatment-induced increases in BIMcould potentially identify tumors likely to respond to PARPi andMEKi combinations.
Low basal expression of BIM in KRAS mutant cell lines or KRAS-induced lines and the ability of PARPi to decrease BIM suggest thatFOXO3a- and BIM-induced apoptosis may contribute to synergy be-tween MEKi and PARPi. Down-regulation of BIM or FOXO3a withsmall interfering RNA (siRNA) rendered OVCAR8 more resistant toBMN673 and abrogated synergistic effects of PARPi and MEKi (Fig. 6A;see Fig. 6B for BIMand FOXO3a protein content). In contrast, enforcedexpression of FOXO3a or BIM was sufficient to sensitize OVCAR8 toBMN673monotherapy (Fig. 6C, left; see Fig. 6D for BIM and FOXO3aprotein content) and to PARPi andMEKi combinations (Fig. 6C, right).These data indicate that expression of FOXO3a or BIM is sufficient topartially mimic the effects of MEKi, suggesting that BIM is both neces-sary and sufficient to sensitize cells to PARPi.
PARPi and MEKi monotherapy each induced a modest increasein apoptosis, as assessed by an annexin V apoptosis assay (Fig. 6E).However, treatment with both PARPi andMEKi led to a robust increasein apoptosis (Fig. 6E). Consistent with a role for apoptosis in the effectsof PARPi and MEKi combinations, Z-VAD-FMK (a pan-caspase inhib-itor) markedly inhibited the effects of PARPi and MEKi combina-tions (Fig. 6F).
FOXO3a knockdown decreased BIM as expected, yet depletion ofBIM had no effect on FOXO3a (Fig. 6G). MEKi efficiently decreasedpERK and increased pMEK, likely due to a feedback loop. Changesin cleaved caspase-3 and cleaved PARP were consistent with annexinV results (Fig. 6F) and demonstrated that PARPi, MEKi, and particu-larly the combination induced apoptosis that was decreased in cellswith knockdown of BIM or FOXO3a (Fig. 6G). Similarly, overex-pression of BIM or FOXO3a increased apoptosis induced by PARPi,MEKi, and the combination of both agents (Fig. 6G). The effects ofBIM and FOXO3a on pERK likely reflect a feedback loop from thesedownstream mediators. Both MEKi and the combination of MEKiand PARPi increased apoptosis, as indicated by increased cleavedPARP and cleaved caspase in HOC1 and HOC7 KRAS mutant cells(Fig. 6H), suggesting generalizability.
Consistent with effects of PARPi and MEKi on FOXO3a protein(Fig. 1), PARPi andMEKi decreased and increased, respectively, FOXO3aactivity, as assessed by a FOXO reporter assay (34) in OVCAR8 (Fig. 6I).
9 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
The addition ofMEKi to PARPi reversed the effects of PARPi onFOXO3atranscription in OVCAR8 (Fig. 6I). In a chromatin immunoprecipitation(ChIP) assay, PARPi decreased the association of FOXO with the BIMpromoter (Fig. 6J), consistent with decreases in BIM in response to PARPi(Fig. 1).
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
The ability of FOXO3a expression to mimic the effects of MEKi incombination with PARPi (Fig. 6, A andC) and of siRNA to FOXO3a toreverse the effects of theMEKi and PARPi combination on cell viabilityprompted us to explore whether FOXO3a contributed to the effectsof mutant RAS and MEKi on DNA damage repair. On the basis of
Fig. 6. A FOXO3a-BIMcascade mediates sensi-tivity to PARP and MEK in-hibition. (A) OVCAR8 cellswere treated with 10 nM con-trol siRNA, BIM siRNA, orFOXO3a siRNA. The next day,cells were treated with in-creasing doses of BMN673(left) or combination withincreasing doses of BMN673and 5 mM MEKi (AZD6244)(right). Cell viability was as-sessed with PrestoBlue af-ter 96 hours. (B) OVCAR8cells were transfected withBIM siRNA or control siRNA.Western blotting for BIMdemonstrated effective BIMknockdown by siRNA (top).OVCAR8 cells were trans-fected with FOXO3a siRNAor control siRNA. Westernblotting for FOXO3a dem-onstrated effective FOXO3aknockdown by siRNA. BIM isdown-regulated by FOXO3aknockdown (bottom). ERK2was used as loading con-trol. (C) BIM and FOXO3awere expressed in OVCAR8and then treated as indi-cated. Cell viability was as-sessed with PrestoBlue after96 hours. In the combina-tion, cells were treated withincreasing doses of BMN673and a constant 5 mM MEKi(AZD6244). (D) OVCAR8 cellswere transfected with BIMor control plasmid. Westernblotting for BIM demon-strated effective BIM overex-pression (OE; top). OVCAR8
cells were transfected with FOXO3a or control plasmid. Western blotting for FOXO3a demonstrated effective FOXO3a overexpression. BIM is up-regulated by FOXO3a over-expression (bottom). ERK2 was used as loading control. (E) OVCAR8 cells were treated with BMN673 (1 mM), AZD6244 (5 mM), or combination therapy with BMN673 (1 mM) andAZD6244 (5 mM) for 96 hours and subjected to annexin V–fluorescein isothiocyanate/propidium iodide apoptosis analysis. Each value represents mean ± SEM from threeindependent experiments. (F) A pan-caspase inhibitor (Z-VAD-FMK) (50 mM) was added to OVCAR8. Cells were treated as indicated. PrestoBlue assay was carried out after 96 hoursof combination therapy with BMN673 dosed as indicated and a constant dose of 5 mMMEKi (AZD6244). (G) Left: BIM/FOXO3a proteins were knocked down by siRNA in OVCAR8.Forty hours later, cells were treated with BMN673 (1 mM), AZD6244 (5 mM), or combination therapy with BMN673 (1 mM) and AZD6244 (5 mM) for 96 hours. Western blot detectionof cleaved PARP1 (Cl-PARP1) and cleaved caspase-3 (Cl-Caspase3) was used to measure apoptotic cell death (left). Right: BIM/FOXO3a cells were overexpressed in OVCAR8 for48 hours. Cells were then treated with BMN673 (1 mM), AZD6244 (5 mM), or combination therapy with BMN673 (1 mM) and AZD6244 (5 mM) for 96 hours. Western blot detection ofcleaved PARP and cleaved caspase-3 was used to measure apoptotic cell death (right). ERK2 was used as loading control. (H) Two RAS mutant cell lines (HOC1 and HOC7) weretreated with BMN673 (1 mM), AZD6244 (1 mM), or combination therapy with BMN673 (1 mM) and AZD6244 (1 mM) for 96 hours. Western blot detection of cleaved PARP andcleaved caspase-3 was used to measure apoptotic cell death. ERK2 was used as loading control. (I) OVCAR8 cells were transfected with FOXO reporter for 24 hours and then treatedas indicated. FOXO activity was assayed 48 hours after treatments (n = 3). In all reporter assays, the luciferase-based reporter signal was normalized to the expression of a co-transfected Renilla luciferase control plasmid. (J) OVCAR8 cells were treated with BMN673 for 48 hours. ChIP-PCR was used to determine percentage input of the BIM promoterprecipitated with endogenous FOXO3a. Results are means ± SEM of three independent experiments. Student’s t test: ***P < 0.001.10 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
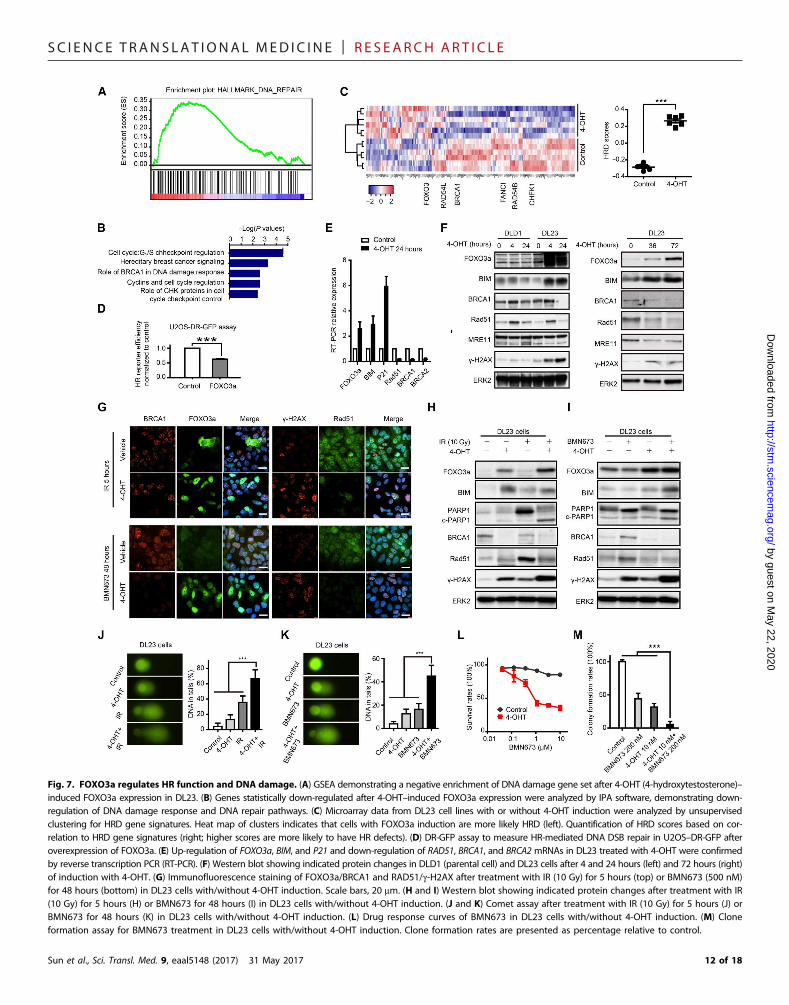
published transcriptional profiles, induction of FOXO3a in RASmu-tant DL23 colon cancer cells induced amarked decrease in hallmarksassociated with DNA damage repair based on Gene Set EnrichmentAnalysis (GSEA) and Ingenuity Pathway Analysis (IPA) analysis(Fig. 7, A and B) (35). Furthermore, induction of FOXO3a markedlyincreased HRD transcriptional scores (Fig. 7C) (33). In addition, ex-pression of FOXO3a in U2OS decreased the ability of cells to repair adefective plasmid by HR (Fig. 7D). Consistent with the alterations inHR, FOXO3a expression decreased RAD51, BRCA1, and BRCA2RNA as well as BRCA1 and RAD51 protein, including those inducedby either IR or BMN673 (Fig. 7, E and F). This was associated withincreased DNA damage, as reflected in g-H2AX accumulation. Asexpected, expression of FOXO3a increased BIM and P21 RNA aswell as BIM protein and induced apoptosis, as indicated by accumu-lation of cleaved PARP. Although FOXO3a induction had modesteffects at early time points, prolonged induction of FOXO3a was suf-ficient to decrease MRE11 protein (Fig. 7F). This was similar to theeffects of MEKi on MRE11 that were only observed at late timepoints (fig. S6B).
As assessed by immunofluorescence, induction of FOXO3amarked-ly decreased nuclear localization of BRCA1 induced by IR (Fig. 7G).Similarly, induction of FOXO3a markedly decreased RAD51 andincreased g-H2AX foci in cells treated with IR and PARPi (Fig. 7G).Further, induction of FOXO3a was sufficient to decrease BRCA1 pro-tein (Fig. 7, H and I) and to inhibit the increase inRAD51 induced by IRand the increase in BRCA1 and RAD51 induced by PARPi (Fig. 7, Hand I). In addition, expression of FOXO3a was sufficient to modestlyincrease DNA damage, as indicated by g-H2AX accumulation (Fig. 7,F to I) and comet formation (Fig. 7, J and K) and to markedly increaseDNA damage induced by either IR or PARPi. Expression of FOXO3awas sufficient tomarkedly decrease cell number, as assessed by dye con-version (Fig. 7L) and colony formation (Fig. 7M). Together, these dataindicate that FOXO3a induction is sufficient to mimic many of theeffects of MEKi in RAS mutant cells and to explain, at least in part,the synergy between PARPi and MEKi.
PARP1 expression contributes to sensitivity to PARPi andMEKi combinationsPARP1 expression is decreased by PARPi, increased by MEKi (Fig. 1),and decreased in PARPi-resistant cells (Fig. 3), suggesting that PARP1could contribute to synergistic activity of PARPi and MEKi, becausePARP1 is required for the optimal activity of PARPi. PARP1 is inverselycorrelated with RAS/MAPK pathway activation, including a direct cor-relation with BIM acrossmore than 700 cell lines (http://tcpaportal.org/mclp/#/; fig. S7A). Furthermore, PARP1 is directly correlated with ex-pression of multiple members of DNA damage and DNA checkpointpathways, consistent with the role of PARP1 in DNA damage repair(fig. S7A). We thus determined the effects of knockdown of PARP1on sensitivity of OVCAR8 to PARPi andMEKi. Knockdown of PARP1markedly decreased sensitivity to PARPi and PARPi andMEKi combi-nations (fig. S7B), consistent with the effects of PARPi andMEKi com-binations being due, at least in part, to PARP trapping.
MRE11 contributes to sensitivity to PARPi andMEKi combinationTheMRN complex that includesMRE11, RAD50, andNBS1 is requiredforDNAdamage sensing and for excision of overhangs in double-strandDNA breaks as well as for activity of PARPi (36, 37). On the basis ofRPPA analysis of more than 700 cell lines (http://tcpaportal.org/
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
mclp/#/), MRE11 protein correlates with RAS/MAPK pathway activ-ity, as evidenced by a direct correlation between MRE11 and pMEK aswell as an inverse correlation between MRE11 and FOXO3a and BIMprotein (fig. S7C). Furthermore, MRE11 protein correlated with mul-tiple DNA damage repair and DNA damage checkpoint mediators(fig. S7C). Similarly, MRE11A RNAwas significantly elevated in KRASmutant tumors within the Cancer Cell Line Encyclopedia (cBioPortal.org; P = 4.3 × 10−4).
Our data indicate thatMEKi leads to decreasedMRE11 and RAD50protein (NBS1 is not in the protein array) (Fig. 1) and RNA (fig. S5),concomitant with decreased phosphorylation ofmultiple DNA damagerepair and DNA damage checkpoint mediators. MRE11 foci were alsodecreased by MEKi (Fig. 5, G and H). These data suggest thatknockdown of MRE11 might mimic the effects of MEKi and increasethe activity of PARPi in RAS mutant tumors. MRE11 siRNA increasedsensitivity of OVCAR8 to PARPi (fig. S7D). Knockdown ofMRE11 didnot further increase the activity of PARPi and MEKi combinations,consistent with the marked decrease in MRE11 induced by MEKi.
FOXM1 does not mediate sensitivity to the PARPi andMEKi combinationPARPi and MEKi treatment also led to increases and decreases, re-spectively, in another forkhead factor, FOXM1, which has been im-plicated in tumorigenesis (38). However, FOXM1 siRNA did not alteractivity of PARPi in RAS mutant OVCAR8 or MCAS (fig. S7E). Fur-thermore, the FOXM1 inhibitor sinomycin or FDI-6 did not demon-strate synergism with BMN673 (fig. S7F). Thus, FOXM1 does not appearto be a rate-limiting mediator of synergy between PARPi and MEKi.
MEKi and PARPi are synergistic in KRAS mutant tumorsin vivoOn the basis of synergy of MEKi and PARPi in RAS mutant cell linesin vitro, we explored the effects of MEKi and PARPi in KRAS mutanttumors in vivo. In established OVCAR8 tumors, combinations ofMEKi and PARPi induced tumor regression followed by prolongeddecreases in tumor volume (Fig. 8A). In contrast, OVCAR8 tumorswere completely resistant to BMN673 at the dose used and demon-strated only a modest response to MEKi.
We also assessed the efficacy of PARPi and MEKi combinations inHPDE-iKRASG12D tumors. Mutant KRAS was induced with Dox, andtumors were allowed to establish in vivo. In this model, PARPi orMEKi alone were without activity (Fig. 8B). Once again, MEKi andPARPi combinations markedly decreased tumor growth. As demon-strated in previous studies (28), removal of Dox and thus KRAS ex-pression resulted in complete tumor regression.
We also assessed the effects of PARPi and MEKi combinations inan MMTV-LPA receptor transgene–induced transplantable tumorthat had acquired a spontaneous KRASQ61H mutation (LPA1-T127)(39). Like human PDX, the LPA1-T127 tumor has never been culturedon plastic and thus may be more representative of human tumors.Furthermore, LPA receptor transgene–induced tumors are late onset,heterogeneous, and associated with an inflammatory response similarto human tumors (38). In this syngeneic tumor model, PARPi andMEKi combinations induced tumor regressions that were maintained(Fig. 8C). Complete tumor regressions extended beyond the need toterminate mice due to tumor volume in other treatment groups. Therewas no evidence of toxicity, as indicated by weight loss in mice witheither monotherapy or combination therapy with PARPi and MEKi(fig. S8A).
11 of 18
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
Fig. 7. FOXO3a regulates HR function and DNA damage. (A) GSEA demonstrating a negative enrichment of DNA damage gene set after 4-OHT (4-hydroxytestosterone)–induced FOXO3a expression in DL23. (B) Genes statistically down-regulated after 4-OHT–induced FOXO3a expression were analyzed by IPA software, demonstrating down-regulation of DNA damage response and DNA repair pathways. (C) Microarray data from DL23 cell lines with or without 4-OHT induction were analyzed by unsupervisedclustering for HRD gene signatures. Heat map of clusters indicates that cells with FOXO3a induction are more likely HRD (left). Quantification of HRD scores based on cor-relation to HRD gene signatures (right; higher scores are more likely to have HR defects). (D) DR-GFP assay to measure HR-mediated DNA DSB repair in U2OS–DR-GFP afteroverexpression of FOXO3a. (E) Up-regulation of FOXO3a, BIM, and P21 and down-regulation of RAD51, BRCA1, and BRCA2 mRNAs in DL23 treated with 4-OHT were confirmedby reverse transcription PCR (RT-PCR). (F) Western blot showing indicated protein changes in DLD1 (parental cell) and DL23 cells after 4 and 24 hours (left) and 72 hours (right)of induction with 4-OHT. (G) Immunofluorescence staining of FOXO3a/BRCA1 and RAD51/g-H2AX after treatment with IR (10 Gy) for 5 hours (top) or BMN673 (500 nM)for 48 hours (bottom) in DL23 cells with/without 4-OHT induction. Scale bars, 20 mm. (H and I) Western blot showing indicated protein changes after treatment with IR(10 Gy) for 5 hours (H) or BMN673 for 48 hours (I) in DL23 cells with/without 4-OHT induction. (J and K) Comet assay after treatment with IR (10 Gy) for 5 hours (J) orBMN673 for 48 hours (K) in DL23 cells with/without 4-OHT induction. (L) Drug response curves of BMN673 in DL23 cells with/without 4-OHT induction. (M) Cloneformation assay for BMN673 treatment in DL23 cells with/without 4-OHT induction. Clone formation rates are presented as percentage relative to control.
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017 12 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
Immunohistochemical analysis of OVCAR8 andHPDE-iKRASG12D
tumors at the termination of the study demonstrated that pERK wasdecreased in MEKi-treated tumors, consistent with MEKi fullyinhibiting its target at doses used throughout the duration of the study(Fig. 8, D and E). As expected, FOXO3a and BIM were decreased inBMN673-treated tumors, increased in MEKi-treated tumors, andincreased compared to PARPi-treated tumors in mice that receivedMEKi/PARPi combinations. This was associated with the inductionof activated caspase-3 indicative of apoptosis and of g-H2AX indicativeof DNA damage, particularly in MEKi- and PARPi-treated cells (Fig. 8,D and E).
CD31, which is expressed by endothelial cells, was markedly de-creased in MEKi/PARPi-treated tumors (Fig. 8, D and E). This wasconsistent with a blanched appearance of tumors excised from themice(fig. S8B). The observations thatmutantRAS inducesVEGFproductionand that VEGFR signals through the RAS/MAPK pathway (40, 41)could contribute to decreases in vascularity observed with PARPi andMEKi combinations. VEGF expression is dependent on RAS pathwayactivity inmultipleRASmutant cell linemodels (fig. S5, A toD). PARP1was decreased in PARPi-treated cells but increased inMEKi-treated tu-mors, with PARP1 being higher in PARPi plus MEKi-treated tumorsthan in PARPi monotherapy–treated tumors (Fig. 8, D and E).
MRE11 was markedly decreased in MEKi-treated tumors and re-mained low in tumors treated with both PARPi and MEKi (Fig. 8, Dand E). In data from TCGA samples (TCPAportal.com), MRE11 pro-tein was highly correlated with pCHK2 (r = 0.5; P = 3 × 10−287) andCD31 (r = 0.47; P = 4.9 × 10−255), suggesting that the relationship be-tweenMRE11 and CD31 inMEKi- and PARPi-treated tumors may re-flect concurrent effects of RAS/MAPK activity on both pathways.Because MRE11 and the MRN complex are required for optimal acti-vation of ATM (42), parallel effects on MRE11 and pATM support theimportance of decreased MRE11 and MRN complex function in theactivity of PARPi andMEKi combinations. 53BP1 was not significantlyaltered in tumors treated with PARPi orMEKi or the combination (Fig.8, D and E). This is consistent with the major effects of the MEKi onsensitivity to PARPi being mediated by the effects on HR rather thanNHEJ (Fig. 5). Western blotting data of tumors confirmed that MEKidecreased the expression of BRCA1 andRAD51while increasing that ofBIM and FOXO3a (Fig. 8, F and G).
Our in vitro studies provided evidence that PARPi-resistant cellswould be sensitive to the effects of PARPi and MEKi combinations(Fig. 3). To test this further, we recultured cells from residual OVCAR8and HPDE-iKRASG12D tumors after treatment and determined theirsensitivity to the drugs. As noted in Fig. 8 (H and I), responses of recul-tured cells from vehicle-treated tumors recapitulated responses ofparental cells to PARPi and MEKi. Although OVCAR8 from BMN673-treated tumors did not demonstrate differential responses to the inhibi-tors, HPDE-iKRASG12D fromBMN673-treated tumors were resistant toPARPi but not to MEKi or PARPi and MEKi combinations. Similarly,HPDE-iKRASG12D fromMEKi-treated tumors demonstrated decreasedsensitivity toMEKi (OVCAR8 cells are constitutively resistant toMEKi,so a change could not be assessed) but were sensitive to both PARPi andthe combination. Thus, both OVCAR8 and HPDE-iKRASG12D fromPARPi- or MEKi-treated tumors retained sensitivity to PARPi andMEKi combinations, suggesting that MEKi- or PARPi-treated patientscould still benefit from PARPi plus MEKi combinations. In contrast,OVCAR8 and HPDE-iKRASG12D from tumors treated with PARPiand MEKi combinations were modestly resistant to individual drugsand more resistant to combinations than DMSO-treated cells, suggest-
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
ing that patients who fail the combination are likely to need alternativetherapies.
DISCUSSIONWe demonstrated thatKRAS andNRASmutant tumors and, to a lesserextent, BRAFmutant tumors are highly sensitive to PARPi and MEKi/ERKi combinations in vitro and, for KRAS mutant models and thePARPi andMEKi combination, in vivo. The combination was well tol-erated at active doses in mice, and further doses used in mice weresimilar to those used in previous studies and represent achievablemonotherapy doses in humans (43, 44). Synergistic activity was ob-served with two structurally distinct PARPis, indicating that the ac-tivity is likely on target and a class effect. Similarly, two differentMEKisand an ERKi demonstrated synergy with PARPi, again consistent withthe RAS/MAPK pathway being a direct mediator. However, BRAFiswere less effective in combination with PARPi, suggesting that targetingspecific nodes in the RAS/MAPK pathwaymight demonstrate differen-tial efficacy potentially due to feedback or bypass mechanisms activatedby inhibition of different nodes (25, 45). The sensitivity of RASmutantcells to the combination appears to be independent of intrinsic gene ex-pression patterns, as observed across multiple different lineages. Be-cause synergistic responses to MEKi and PARPi combinations wereindependent of p53 mutation status, the approach should be effectivein both p53 wild-type and mutant tumors. Together, the in vitro andin vivo data argue that a MEKi and PARPi combination has thepotential to induce cell death and increase the magnitude, duration,and spectrum of PARPi activity.
Many patients who initially respond to PARPi rapidly developresistance and progress (46). Cell lines rendered resistant to PARPihave elevated RAS/MAPK signaling independent of mutations in KRAS.Resistance to PARPi in cell lines selected for resistance in vitro as well asin cells recultured from PARPi-treated tumors in vivo was reversed, atleast in part, by MEKi, suggesting that PARPi and MEKi combinationmay be active in patients who have failed PARPi. Because many breastand ovarian cancer patients are receiving PARPi, and patients rapidlybecome resistant to PARPi, PARPi-resistant patients will soon representa large population of breast and ovarian cancer patients who requiretherapeutic alternatives.
PARPi and MEKi combinations were effective in cells that did nothave aberrations in BRCA1/2, suggesting that the combination couldexpand the spectrum of patients likely to benefit from PARPi beyondthose with tumors with intrinsic HR defects. PARPi and MEKi com-binations were highly effective in cell lines with high HR competencedue to RAS or BRAFmutations. Although RAS and, to a lesser degree,BRAF mutations are robust predictors of synergistic interactions be-tween PARPi and MEKi/ERKi, not all RAS or BRAF mutant linesdemonstrated synergy with the combination. The combination dem-onstrated marked activity in 95% of the RAS and BRAF mutant linestested based on synergy in 8 of 8 RASmutant ovarian lines and 25 of37 RAS and BRAF mutant lines from other lineages combined withmonotherapy activity of PARPi or MEKi in 10 of the 12 RAS orBRAF mutant lines that did not demonstrate synergism. Thus, if ef-fective biomarkers of response with greater predictive value thanRAS/BRAF mutations or BIM protein cannot be identified, and ifthe combination has acceptable tolerability in humans as it does inmice, the combination would be expected to demonstrate activity inmost of the patients with RAS or BRAF mutant tumors (defined asresponse to the monotherapy or the combination therapy).
13 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
Fig. 8. PARPi and MEKi are synergistic in OVCAR8 xenograft, HPDE-iKRASG12D xenograft, and LPA1-T127 syngeneic breast cancer models. (A) Human OVCAR8cells were grown as xenograft tumors in athymic nude mice. When tumors were 50 to 200 mm3, mice were randomized into treatment cohorts: vehicle (0.5% hydro-xypropylmethylcellulose and 0.2% Tween 80), BMN673 (0.333 mg/kg per day), GSK1120212B (2 mg/kg per day), or the combination of BMN673 and GSK1120212B (n =8 for each group). Tumor measurements were performed two times per week by calipers, and average tumor volume ± SEM for each cohort is displayed. P values weredetermined by one-way analysis of variance (ANOVA) test. (B) HPDE-iKRASG12D were injected into athymic nude mice subcutaneously. Seven days after tumor injection,mice were randomly assigned into “Dox off” and four treatment groups with “Dox on” [via Dox diet (200 mg/kg; BioServ)]: vehicle, BMN673 (0.333 mg/kg per day),GSK1120212B (2 mg/kg per day), or the combination of BMN673 and GSK1120212B (n = 8 per group)]. (C) LPA1-T127 tumor tissues were transplanted into the mam-mary fat pads of FVB mice. Eight days later, mice were randomly assigned and treated accordingly. Mice were followed until death or sacrificed when tumor sizereached 2 cm in diameter. (D and E) Tumor tissues from OVCAR8 (D) and HPDE-iKRASG12D (E) xenografts were subjected to immunohistochemical analyses and probedwith indicated antibodies. Representative images of immunohistochemistry are shown with treatment indicated. (F and G) Western blots showing indicated proteinchanges in tumor tissues after treatment with MEKi or vehicle in OVCAR8 xenografts (F) and HPDE-iKRASG12D xenografts (G). (H and I) Cells from OVCAR8 (H) and HPDE-iKRASG12D (I) tumor-bearing mice were recultured in the absence of drug. Drug response curves are shown for individual and combination therapy with BMN673 andGSK1120212B for 96 hours in recultured cells from OVCAR8 and HPDE-iKRASG12D xenografts. Three recultured cell lines from each group were assessed, and repre-sentative results are presented as means ± SEM of three independent experiments.
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017 14 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
Given that only a subset of tumors with BRCA1/2mutants respondsto PARPi, it is reasonable to hypothesize thatRASmutations or pathwayactivation plays a role in resistance to PARPi in some BRCA1/2mutanttumors such as those represented by Capan1, a BRCA2/KRAS mutantcell line resistant to PARPi.RASmutations are rare inBRCA1/2mutatedovarian and breast cancers, but physiological pathway activation (6, 7)could mediate PARPi resistance, which could potentially be reversed byMEKi. In contrast, in other lineages such as pancreas and stomach,BRCA1/2 and KRAS mutations do co-occur (cBioPortal.org).
The adaptive responses induced by PARPi and MEKi suggestedmultiple potential mechanisms underlying synergistic activity of thecombination that were further validated in vitro and in vivo. The syn-ergistic activity of PARPi and MEKi combinations is associated with(i) altered RAS/MAPK signaling; (ii) decreased HR repair capacity in-cluding decreased BRCA1/2 and DNA checkpoint activity, leading toincreased DNA damage; (iii) decreased expression of MRN compo-nents, in particular MRE11; (iv) FOXO3a- and BIM-mediated apopto-sis; (v) increased PARP1, which is required for optimal activity oftrapping PARPi; and (vi) decreased vascularity, which could increasehypoxia and resultant PARPi efficacy. Most of the changes in totalprotein amounts could be detected at an mRNA level, suggesting thatmany of the effects are transcriptional. However, several were best re-flected by changes in phosphorylation, consistent with posttransla-tional effects.
The ability ofMEKi to increase FOXO3a appears to be central to theeffects of MEKi in RASmutant cells and to explain, to a major degree,the synergy between PARPi andMEKi. Enforced expression of FOXO3awas sufficient to mimic many of the effects of MEKi. Although the in-duction of apoptosis due to FOXO3a induction of BIM was expected,surprisingly, FOXO3a was sufficient to decrease MRE11, BRCA1, andRAD51, consistent with decreased ability to repair DNA damage in un-treated cells or in the presence of either IR or PARPi. Although PI3K islocated downstream of mutant RAS, and PI3K regulates FOXO3a (47),PI3Kis were not synergistic with PARPi in RAS mutant cells. Further-more, the effects of PI3Kis on BIM and FOXO3a were modest com-pared to those of MEKis in RAS mutant tumors. This may be, inpart, due to RAS/MAPK regulating BIM stability as well astranscription. PARPi andMEKi combinations were associated with de-creased vascularity and CD31 expression in treated tumors. The obser-vations that mutant RAS induces VEGF production and that VEGFRsignals through the RAS/MAPK pathway (40, 41) could contribute todecreases in vascularity observed with MEKi. Our data indicate thatVEGF expression is RAS-dependent in multiple systems. Furthermore,FOXO3a regulates VEGF production through FOXM1 (48). Becausehypoxia increases the activity of PARPi, and the combination of a PARPiand cediranib (a VEGFR inhibitor) is highly active in patients (49),the effect of MEKi and PARPi on vascularity could contribute to theactivity of the combination in vivo. The correlation between MRE11and CD31 in TCGA patient samples before treatment suggests thatthere may be an additional unexpected relationship between DNAdamage and tumor vascularity.
MRE11 and decreased activity of theMRN complex and subsequentactivation ofDNAdamage repair andDNAdamage checkpoint activityinduced by MEKi appear to contribute to the efficacy of PARPi andMEKi combinations. Down-regulation of MRE11 was sufficient to in-crease the activity of PARPi. MEKi and FOXO3a induction induced adelayed decrease in MRE11 as monotherapy. However, FOXO3a andMEKiwere not sufficient tomarkedly decrease RAD50orNBS1 proteinin vitro. In vivo, MEKi monotherapy markedly decreased MRE11
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
protein, likely due to the prolonged in vivo treatment. Thus, down-regulation ofMRE11 appears to require prolonged exposure toMEKi.MRE11 inhibitors have been identified (36) and may represent analternative approach to MEKi to increase the activity of PARPi. Theseinhibitors only block the enzyme activity of MRE11 and not the scaf-folding activity and thus may not fully recapitulate loss of MRE11.Further, MRE11 inhibition may not be sufficient to mimic the fullspectrum of activities of MEKi in RAS mutant tumors.
The ability of MEKi to increase PARP1 could not be explained bythe actions of FOXO3a alone. The ability of PARPi and MEKi com-binations to optimally decrease cell viability is dependent on the amountof PARP1. This is compatible with the contention that PARP1 is nec-essary for toxicity mediated by trapping PARPi (13, 22). This suggeststhat nontrapping PARPis may not demonstrate the same degree of syn-ergy with MEKi as do trapping PARPis. FOXO3a induction was insuf-ficient to increase PARP.
The studies herein add to a number of previous studies that suggestthat adaptive responses to targeted therapies provide an opportunityto rapidly and efficiently identify potential therapeutic interventionsthat would convert cytostatic responses observed with many targetedtherapies into cytotoxicity with a greater benefit (1–3). Although anal-ysis of RNA or other omics approaches such as metabolomics orlipidomics provides an opportunity to identify adaptive responses, mosttargeted therapies that are being developed target proteins, and changesin protein function mediate resistance to targeted therapies. RNA andprotein expression and particularly the amounts of posttranslationallymodified and functional proteins are frequently weakly correlated (50).Although it may be possible to impute protein function by quantifyingdownstream RNA master regulators (51), analysis of proteins and pro-tein function may provide a more direct assessment. Thus, analysis ofprotein function may provide a more efficient approach to identifyadaptive responses. Although emerging mass spectrometry approacheshave the ability to identify adaptive responses, RPPA provides a cost-effective, efficient approach for sparse unbiased analysis of pathwaysinvolved in adaptive responses. In particular, RPPA has increased sen-sitivity for a number of phosphoproteins over mass spectrometry, andmass spectrometry is “blind” to a number of phosphoproteins due to alack of convenient cleavage sites (52).
This study has a number of limitations. Although we have demon-strated synergy between PARP and MEK in KRAS and NRAS and, toa lesser degree, BRAF mutant lines in vitro, we have not assessedHRAS mutant models and have only assessed KRAS mutant modelsin vivo. Thus, we do not know whether the combination would beeffective when the RAS/MAPK pathway is activated by other mecha-nisms such as loss of NF1 or activation of cell surface receptors. Al-though OVCAR8 used in these studies has a KRAS mutation andactivation of the RAS/MAPK pathway similar to cells with KRAS mu-tations, it is not clear whether the KRAS mutation in OVCAR8 is adriver or passenger. At most, it is modestly activating. We have de-monstrated that PARPi-resistant A2780CP clones acquire KRASA146T
or KRASA59T and MAP2K1A283T mutations and that these are modu-lated by incubation with PARPi, but we have not demonstrated thatthese mutations drive resistance to PARPi; further, we have not iden-tified a mechanism explaining the PARPi resistance in the UWB1.289clone. This is particularly important because the parental UWB1.289clone has a deleterious BRCA1 mutation and is PARPi-sensitive. Al-though we are able to explain almost all of the effects of inhibition ofMEKi on the sensitization of cells to PARPi by an induction of FOXO3afunction, we have not elucidated the mechanism underlying increases
15 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
Dow
n
in PARP1, and although there is literature support for a FOXO3apathway to CD31, we have not demonstrated this functionally inthe models tested. Finally, of course, although the preclinical dataare strongly supportive of both efficacy and acceptable toxicity, wehave not yet demonstrated the activity and tolerability of the combi-nation in human patients.
In summary, MEKi and ERKi are synergistic with PARPi inmost oftheRASmutant lines evaluated aswell as in PARPi-resistant cells.MEKialter apoptotic balance, induce HR deficiency, and decrease DNA dam-age checkpoint activity in RAS mutant cells. These act together to in-crease DNA damage induced by PARPi and to increase apoptosis inresponse to PARPi. The activity of the combination is independentof BRCA1/2 and p53mutation status. Together, the data indicate thatRAS mutant tumors as well as tumors that have become resistant toPARPi are likely to benefit from the combination. These data indi-cate that exciting clinical activity of PARPi in BRCA1/2 mutant tu-mors could be extended to RAS and BRAF mutant tumors. Thus,PARPi and MEKi/ERKi combination therapy warrants exploration inhuman trials.
by guest on May 22, 2020
http://stm.sciencem
ag.org/loaded from
MATERIALS AND METHODSStudy designThe objective of this study was to evaluate the adaptive resistance toPARPi through high-throughput pathway surveillance tools (RPPA)and to develop a rational combinational therapy that would lead to syn-thetic lethality. We demonstrate that PARPi and MEKi demonstratesynergy in vitro and in vivo in a wide spectrum of RASmutant tumormodels. Sample size in experiments was selected on the basis of pre-vious experience and was sufficient to detect statistically significantdifferences between treatments. Mice were randomized to differenttreatment groups, without blinding. Study groups were followed untilindividual tumor or entire cohort measurements reached 2 cm in di-ameter, at which point sacrifice was indicated in accordance with In-stitutional Animal Care and Use Committee protocols. All in vitroexperiments were run in biological triplicates, except where specifiedotherwise
Statistical analysisTwo-sided Student’s t test was used to compare differences between twogroups of cells in vitro.Data are presented asmeans± SEM, andP<0.05is considered significant. Analysis of variance was used to compare dif-ferences among multiple groups. Differences in survival of mice wereexamined by Kaplan-Meier curves with the log-rank test. Two-sidedP less than 0.05 was considered significant. All statistical analyses weredone using SPSS 17.0 (SPSS Inc.).
SUPPLEMENTARY MATERIALSwww.sciencetranslationalmedicine.org/cgi/content/full/9/392/eaal5148/DC1Materials and MethodsFig. S1. PARPi and MEKi induce inverse adaptive responses.Fig. S2. Long-term assay of combination therapy with PARPi and MEKi demonstrates that lowdoses of drugs show synergy in OVCAR8 cells.
Fig. S3. PARPi plus MEKi/ERKi demonstrate synergy in cells with RAS mutations.
Fig. S4. Synergistic effects of PARP and MEK/ERK inhibition are lineage-independent andobserved with KRAS/NRAS/BRAF mutations.
Fig. S5. RAS/ERK pathway activity is associated with HR competence, DNA damagecheckpoints, and apoptotic sensitivity.
Fig. S6. MEKis decrease HR competence and enhance DNA damage induced byIR or PARPi.
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
Fig. S7. PARP and MRE11, but not FOXM1, contribute to the synergistic effects of PARPiand MEKi.Fig. S8. PARPi, MEKi, and combination therapy have no effect on the body weight of mice.References (53, 54)
REFERENCES AND NOTES1. T. Muranen, L. M. Selfors, D. T. Worster, M. P. Iwanicki, L. Song, F. C. Morales, S. Gao,
G. B. Mills, J. S. Brugge, Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 21, 227–239 (2012).
2. C. H. Kugel III, E. J. Hartsough, M. A. Davies, Y. Y. Setiady, A. E. Aplin, Function-blockingERBB3 antibody inhibits the adaptive response to RAF inhibitor. Cancer Res. 74,4122–4132 (2014).
3. L. N. Kwong, G. M. Boland, D. T. Frederick, T. L. Helms, A. T. Akid, J. P. Miller, S. Jiang,Z. A. Cooper, X. Song, S. Seth, J. Kamara, A. Protopopov, G. B. Mills, K. T. Flaherty,J. A. Wargo, L. Chin, Co-clinical assessment identifies patterns of BRAF inhibitor resistancein melanoma. J. Clin. Invest. 125, 1459–1470 (2015).
4. J. Gao, B. A. Aksoy, U. Dogrusoz, G. Dresdner, B. Gross, S. O. Sumer, Y. Sun, A. Jacobsen,R. Sinha, E. Larsson, E. Cerami, C. Sander, N. Schultz, Integrative analysis of complexcancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
5. I. Diaz-Padilla, A. L. Malpica, L. Minig, L. M. Chiva, D. M. Gershenson, A. Gonzalez-Martin,Ovarian low-grade serous carcinoma: A comprehensive update. Gynecol. Oncol. 126,279–285 (2012).
6. The Cancer Genome Atlas Research Network, Integrated genomic analyses of ovariancarcinoma. Nature 474, 609–615 (2011).
7. K. E. Hew, P. C. Miller, D. El-Ashry, J. Sun, A. H. Besser, T. A. Ince, M. Gu, Z. Wei, G. Zhang,P. Brafford, W. Gao, Y. Lu, G. B. Mills, J. M. Slingerland, F. Simpkins, MAPK activationpredicts poor outcome and the MEK inhibitor, selumetinib, reverses antiestrogenresistance in ER-positive high-grade serous ovarian cancer. Clin. Cancer Res. 22, 935–947(2016).
8. S. E. Patton, M. L. Martin, L. L. Nelsen, X. Fang, G. B. Mills, R. C. Bast Jr., M. C. Ostrowski,Activation of the ras-mitogen-activated protein kinase pathway and phosphorylationof ets-2 at position threonine 72 in human ovarian cancer cell lines. Cancer Res. 58,2253–2259 (1998).
9. A. Ashworth, A synthetic lethal therapeutic approach: Poly(ADP) ribose polymeraseinhibitors for the treatment of cancers deficient in DNA double-strand break repair.J. Clin. Oncol. 26, 3785–3790 (2008).
10. N. Schultz, E. Lopez, N. Saleh-Gohari, T. Helleday, Poly(ADP-ribose) polymerase (PARP-1)has a controlling role in homologous recombination. Nucleic Acids Res. 31, 4959–4964(2003).
11. J. D. Iglehart, D. P. Silver, Synthetic lethality—A new direction in cancer-drugdevelopment. N. Engl. J. Med. 361, 189–191 (2009).
12. M. Wang, W. Wu, W. Wu, B. Rosidi, L. Zhang, H. Wang, G. Iliakis, PARP-1 and Kucompete for repair of DNA double strand breaks by distinct NHEJ pathways.Nucleic Acids Res. 34, 6170–6182 (2006).
13. J. Murai, S.-y. Huang, B. B. Das, A. Renaud, Y. Zhang, J. H. Doroshow, J. Ji, S. Takeda,Y. Pommier, Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 72,5588–5599 (2012).
14. J. Murai, S.-Y. Huang, A. Renaud, Y. Zhang, J. Ji, S. Takeda, J. Morris, B. Teicher,J. H. Doroshow, Y. Pommier, Stereospecific PARP trapping by BMN 673 and comparisonwith olaparib and rucaparib. Mol. Cancer Ther. 13, 433–443 (2014).
15. R. Akbani, P. K. Ng, H. M. J. Werner, M. Shahmoradgoli, F. Zhang, Z. Ju, W. Liu,J.-Y. Yang, K. Yoshihara, J. Li, S. Ling, E. G. Seviour, P. T. Ram, J. D. Minna, L. Diao,P. Tong, J. V. Heymach, S. M. Hill, F. Dondelinger, N. Städler, L. A. Byers,F. Meric-Bernstam, J. N. Weinstein, B. M. Broom, R. G. W. Verhaak, H. Liang,S. Mukherjee, Y. Lu, G. B. Mills, A pan-cancer proteomic perspective on The CancerGenome Atlas. Nat. Commun. 5, 3887 (2014).
16. M. Toulany, T.-A. Schickfluss, W. Eicheler, R. Kehlbach, B. Schittek, H. P. Rodemann,Impact of oncogenic K-RAS on YB-1 phosphorylation induced by ionizing radiation.Breast Cancer Res. 13, R28 (2011).
17. X. Fang, S. Yu, A. Eder, M. Mao, R. C. Bast Jr., D. Boyd, G. B. Mills, Regulation of BADphosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway.Oncogene 18, 6635–6640 (1999).
18. P. P. Roux, D. Shahbazian, H. Vu, M. K. Holz, M. S. Cohen, J. Taunton, N. Sonenberg,J. Blenis, RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylationvia RSK and stimulates cap-dependent translation. J. Biol. Chem. 282, 14056–14064(2007).
19. N. Panupinthu, S. Yu, D. Zhang, F. Zhang, M. Gagea, Y. Lu, J. R. Grandis, S. E. Dunn,H. Y. Lee, G. B. Mills, Self-reinforcing loop of amphiregulin and Y-box bindingprotein-1 contributes to poor outcomes in ovarian cancer. Oncogene 33, 2846–2856(2014).
16 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from
20. M. Stahl, P. F. Dijkers, G. J. P. L. Kops, S. M. A. Lens, P. J. Coffer, B. M. T. Burgering,R. H. Medema, The forkhead transcription factor FoxO regulates transcription of p27Kip1
and Bim in response to IL-2. J. Immunol. 168, 5024–5031 (2002).21. F. Luciano, A. Jacquel, P. Colosetti, M. Herrant, S. Cagnol, G. Pages, P. Auberger,
Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via theproteasome pathway and regulates its proapoptotic function. Oncogene 22, 6785–6793(2003).
22. L. A. Byers, J. Wang, M. B. Nilsson, J. Fujimoto, P. Saintigny, J. Yordy, U. Giri, M. Peyton,Y. H. Fan, L. Diao, F. Masrorpour, L. Shen, W. Liu, B. Duchemann, P. Tumula,V. Bhardwaj, J. Welsh, S. Weber, B. S. Glisson, N. Kalhor, I. I. Wistuba, L. Girard,S. M. Lippman, G. B. Mills, K. R. Coombes, J. N. Weinstein, J. D. Minna, J. V. Heymach,Proteomic profiling identifies dysregulated pathways in small cell lung cancer andnovel therapeutic targets including PARP1. Cancer Discov. 2, 798–811 (2012).
23. C. J. Caunt, M. J. Sale, P. D. Smith, S. J. Cook, MEK1 and MEK2 inhibitors and cancertherapy: The long and winding road. Nat. Rev. Cancer 15, 577–592 (2015).
24. S. Domcke, R. Sinha, D. A. Levine, C. Sander, N. Schultz, Evaluating cell lines as tumourmodels by comparison of genomic profiles. Nat. Commun. 4, 2126 (2013).
25. W. Kolch, M. Halasz, M. Granovskaya, B. N. Kholodenko, The dynamic control of signaltransduction networks in cancer cells. Nat. Rev. Cancer 15, 515–527 (2015).
26. L. W. T. Cheung, S. Yu, D. Zhang, J. Li, P. K. S. Ng, N. Panupinthu, S. Mitra, Z. Ju, Q. Yu, H. Liang,D. H. Hawke, Y. Lu, R. R. Broaddus, G. B. Mills, Naturally occurring neomorphic PIK3R1mutations activate the MAPK pathway, dictating therapeutic response to MAPK pathwayinhibitors. Cancer Cell 26, 479–494 (2014).
27. H. Liang, L. W. T. Cheung, J. Li, Z. Ju, S. Yu, K. Stemke-Hale, T. Dogruluk, Y. Lu, X. Liu,C. Gu, W. Guo, S. E. Scherer, H. Carter, S. N. Westin, M. D. Dyer, R. G. W. Verhaak,F. Zhang, R. Karchin, C.-G. Liu, K. H. Lu, R. R. Broaddus, K. L. Scott, B. T. Hennessy,G. B. Mills, Whole-exome sequencing combined with functional genomics revealsnovel candidate driver cancer genes in endometrial cancer. Genome Res. 22,2120–2129 (2012).
28. Y. H. Tsang, T. Dogruluk, P. M. Tedeschi, J. Wardwell-Ozgo, H. Lu, M. Espitia, N. Nair,R. Minelli, Z. Chong, F. Chen, Q. E. Chang, J. B. Dennison, A. Dogruluk, M. Li, H. Ying,J. R. Bertino, M.-C. Gingras, M. Ittmann, J. Kerrigan, K. Chen, C. J. Creighton, K. Eterovic,G. B. Mills, K. L. Scott, Functional annotation of rare gene aberration drivers of pancreaticcancer. Nat. Commun. 7, 10500 (2016).
29. H. Ying, A. C. Kimmelman, C. A. Lyssiotis, S. Hua, G. C. Chu, E. Fletcher-Sananikone,J. W. Locasale, J. Son, H. Zhang, J. L. Coloff, H. Yan, W. Wang, S. Chen, A. Viale, H. Zheng,J.-h. Paik, C. Lim, A. R. Guimaraes, E. S. Martin, J. Chang, A. F. Hezel, S. R. Perry, J. Hu,B. Gan, Y. Xiao, J. M. Asara, R. Weissleder, Y. A. Wang, L. Chin, L. C. Cantley, R. A. DePinho,Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucosemetabolism. Cell 149, 656–670 (2012).
30. J. W. Tyner, H. Erickson, M. W. N. Deininger, S. G. Willis, C. A. Eide, R. L. Levine,M. C. Heinrich, N. Gattermann, D. G. Gilliland, B. J. Druker, M. M. Loriaux, High–throughputsequencing screen reveals novel, transforming RAS mutations in myeloid leukemiapatients. Blood 113, 1749–1755 (2009).
31. H. E. Bryant, N. Schultz, H. D. Thomas, K. M. Parker, D. Flower, E. Lopez, S. Kyle, M. Meuth,N. J. Curtin, T. Helleday, Specific killing of BRCA2-deficient tumours with inhibitors ofpoly(ADP-ribose) polymerase. Nature 434, 913–917 (2005).
32. G. Peng, C. Chun-Jen Lin, W. Mo, H. Dai, Y.-Y. Park, S. M. Kim, Y. Peng, Q. Mo, S. Siwko,R. Hu, J.-S. Lee, B. Hennessy, S. Hanash, G. B. Mills, S.-Y. Lin, Genome-wide transcriptomeprofiling of homologous recombination DNA repair. Nat. Commun. 5, 3361 (2014).
33. L. O’Connor, A. Strasser, L. A. O’Reilly, G. Hausmann, J. M. Adams, S. Cory,D. C. S. Huang, Bim: A novel member of the Bcl-2 family that promotes apoptosis.EMBO J. 17, 384–395 (1998).
34. K. Kannike, M. Sepp, C. Zuccato, E. Cattaneo, T. Timmusk, Forkhead transcription factorFOXO3a levels are increased in Huntington disease because of overactivated positiveautofeedback loop. J. Biol. Chem. 289, 32845–32857 (2014).
35. S. P. Tenbaum, P. Ordóñez-Morán, I. Puig, I. Chicote, O. Arqués, S. Landolfi,Y. Fernández, J. R. Herance, J. D. Gispert, L. Mendizabal, S. Aguilar, S. R. Cajal,S. Schwartz Jr., A. Vivancos, E. Espin, S. Rojas, J. Baselga, J. Tabernero, A. Muñoz,H. G. Palmer, b-Catenin confers resistance to PI3K and AKT inhibitors and subvertsFOXO3a to promote metastasis in colon cancer. Nat. Med. 18, 892–901 (2012).
36. A. Shibata, D. Moiani, A. S. Arvai, J. Perry, S. M. Harding, M.-M. Genois, R. Maity,S. van Rossum-Fikkert, A. Kertokalio, F. Romoli, A. Ismail, E. Ismalaj, E. Petricci,M. J. Neale, R. G. Bristow, J.-Y. Masson, C. Wyman, P. A. Jeggo, J. A. Tainer, DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities.Mol. Cell 53, 7–18 (2014).
37. E. Vilar, C. M. Bartnik, S. L. Stenzel, L. Raskin, J. Ahn, V. Moreno, B. Mukherjee,M. D. Iniesta, M. A. Morgan, G. Rennert, S. B. Gruber, MRE11 deficiency increasessensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstablecolorectal cancers. Cancer Res. 71, 2632–2642 (2011).
38. S. Zona, L. Bella, M. J. Burton, G. Nestal de Moraes, E. W.-F. Lam, FOXM1: An emergingmaster regulator of DNA damage response and genotoxic agent resistance.Biochim. Biophys. Acta 1839, 1316–1322 (2014).
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
39. S. Liu, M. Umezu-Goto, M. Murph, Y. Lu, W. Liu, F. Zhang, S. Yu, L. C. Stephens,X. Cui, G. Murrow, K. Coombes, W. Muller, M.-C. Hung, C. M. Perou, A. V. Lee,X. Fang, G. B. Mills, Expression of autotaxin and lysophosphatidic acid receptorsincreases mammary tumorigenesis, invasion, and metastases. Cancer Cell 15, 539–550(2009).
40. O. Takahashi, R. Komaki, P. D. Smith, J. M. Jürgensmeier, A. Ryan, B. N. Bekele,I. I. Wistuba, J. J. Jacoby, M. V. Korshunova, A. Biernacka, B. Erez, K. Hosho, R. S. Herbst,M. S. O’Reilly, Combined MEK and VEGFR inhibition in orthotopic human lungcancer models results in enhanced inhibition of tumor angiogenesis, growth, andmetastasis. Clin. Cancer Res. 18, 1641–1654 (2012).
41. J. Rak, Y. Mitsuhashi, L. Bayko, J. Filmus, S. Shirasawa, T. Sasazuki, R. S. Kerbel, Mutantras oncogenes upregulate VEGF/VPF expression: Implications for induction andinhibition of tumor angiogenesis. Cancer Res. 55, 4575–4580 (1995).
42. J.-H. Lee, T. T. Paull, Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1complex. Science 304, 93–96 (2004).
43. Y. Shen, F. L. Rehman, Y. Feng, J. Boshuizen, I. Bajrami, R. Elliott, B. Wang, C. J. Lord,L. E. Post, A. Ashworth, BMN 673, a novel and highly potent PARP1/2 inhibitor for thetreatment of human cancers with DNA repair deficiency. Clin. Cancer Res. 19, 5003–5015(2013).
44. A. G. Gilmartin, M. R. Bleam, A. Groy, K. G. Moss, E. A. Minthorn, S. G. Kulkarni,C. M. Rominger, S. Erskine, K. E. Fisher, J. Yang, F. Zappacosta, R. Annan, D. Sutton,S. G. Laquerre, GSK1120212 (JTP-74057) is an inhibitor of MEK activity andactivation with favorable pharmacokinetic properties for sustained in vivo pathwayinhibition. Clin. Cancer Res. 17, 989–1000 (2011).
45. J. Villanueva, A. Vultur, J. T. Lee, R. Somasundaram, M. Fukunaga-Kalabis, A. K. Cipolla,B. Wubbenhorst, X. Xu, P. A. Gimotty, D. Kee, A. E. Santiago-Walker, R. Letrero, K. D’Andrea,A. Pushparajan, J. E. Hayden, K. D. Brown, S. Laquerre, G. A. McArthur, J. A. Sosman,K. L. Nathanson, M. Herlyn, Acquired resistance to BRAF inhibitors mediated by a RAFkinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K.Cancer Cell 18, 683–695 (2010).
46. J. Ledermann, P. Harter, C. Gourley, M. Friedlander, I. Vergote, G. Rustin, C. Scott, W. Meier,R. Shapira-Frommer, T. Safra, D. Matei, E. Macpherson, C. Watkins, J. Carmichael,U. Matulonis, Olaparib maintenance therapy in platinum-sensitive relapsed ovariancancer. N. Engl. J. Med. 366, 1382–1392 (2012).
47. A. Brunet, A. Bonni, M. J. Zigmond, M. Z. Lin, P. Juo, L. S. Hu, M. J. Anderson,K. C. Arden, J. Blenis, M. E. Greenberg, Akt promotes cell survival by phosphorylatingand inhibiting a Forkhead transcription factor. Cell 96, 857–868 (1999).
48. C. T. Karadedou, A. R. Gomes, J. Chen, M. Petkovic, K.-K. Ho, A. K. Zwolinska, A. Feltes,S. Y. Wong, K. Y. K. Chan, Y.-N. Cheung, J. W. H. Tsang, J. J. Brosens, U.-S. Khoo,E. W.-F. Lam, FOXO3a represses VEGF expression through FOXM1-dependentand -independent mechanisms in breast cancer. Oncogene 31, 1845–1858 (2012).
49. J. F. Liu, W. T. Barry, M. Birrer, J.-M. Lee, R. J. Buckanovich, G. F. Fleming, B. J. Rimel,M. K. Buss, S. Nattam, J. Hurteau, W. Luo, P. Quy, C. Whalen, L. Obermayer, H. Lee,E. P. Winer, E. C. Kohn, S. P. Ivy, U. A. Matulonis, Combination cediranib andolaparib versus olaparib alone for women with recurrent platinum-sensitive ovariancancer: A randomised phase 2 study. Lancet Oncol. 15, 1207–1214 (2014).
50. C. Cenik, E. S. Cenik, G. W. Byeon, F. Grubert, S. I. Candille, D. Spacek, B. Alsallakh,H. Tilgner, C. L. Araya, H. Tang, E. Ricci, M. P. Snyder, Integrative analysis ofRNA, translation, and protein levels reveals distinct regulatory variation acrosshumans. Genome Res. 25, 1610–1621 (2015).
51. M. J. Alvarez, Y. Shen, F. M. Giorgi, A. Lachmann, B. B. Ding, B. H. Ye, A. Califano, Functionalcharacterization of somatic mutations in cancer using network-based inference ofprotein activity. Nat. Genet. 48, 838–847 (2016).
52. P. Mertins, D. R. Mani, K. V. Ruggles, M. A. Gillette, K. R. Clauser, P. Wang, X. Wang,J. W. Qiao, S. Cao, F. Petralia, E. Kawaler, F. Mundt, K. Krug, Z. Tu, J. T. Lei, M. L. Gatza,M. Wilkerson, C. M. Perou, V. Yellapantula, K.-l. Huang, C. Lin, M. D. McLellan, P. Yan,S. R. Davies, R. R. Townsend, S. J. Skates, J. Wang, B. Zhang, C. R. Kinsinger, M. Mesri,H. Rodriguez, L. Ding, A. G. Paulovich, D. Fenyö, M. J. Ellis, S. A. Carr; NCI CPTAC,Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534,55–62 (2016).
53. S. Jones, X. Zhang, D. W. Parsons, J. C.-H. Lin, R. J. Leary, P. Angenendt, P. Mankoo,H. Carter, H. Kamiyama, A. Jimeno, S.-M. Hong, B. Fu, M.-T. Lin, E. S. Calhoun,M. Kamiyama, K. Walter, T. Nikolskaya, Y. Nikolsky, J. Hartigan, D. R. Smith, M. Hidalgo,S. D. Leach, A. P. Klein, E. M. Jaffee, M. Goggins, A. Maitra, C. Iacobuzio-Donahue,J. R. Eshleman, S. E. Kern, R. H. Hruban, R. Karchin, N. Papadopoulos, G. Parmigiani,B. Vogelstein, V. E. Velculescu, K. W. Kinzler, Core signaling pathways in humanpancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806(2008).
54. A. Eijkelenboom, M. Mokry, E. de Wit, L. M. Smits, P. E. Polderman, M. H. van Triest,R. van Boxtel, A. Schulze, W. de Laat, E. Cuppen, B. M. T. Burgering, Genome-wide analysisof FOXO3 mediated transcription regulation through RNA polymerase II profiling.Mol. Syst. Biol. 9, 638 (2013).
17 of 18

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
Acknowledgments: We thank A. Maitra (Division of Pathology/Lab Medicine, University ofTexas MD Anderson Cancer Center) for iKRAS1, iKRAS2, Pa01c, Pa02c, Pa03C, Pa04c,Pa09c, Pa16c, Pa18c, and Pa21c and B. M. T. Burgering (Department of MolecularCancer Research, University Medical Centre, Utrecht, Netherlands) for DLD1 and DL23.Funding: This study is supported by Susan G. Komen grant SAC110052 (to G.B.M.); StandUp to Cancer Dream Team Translational Research grant (SU2C-AACR-DT0209 to G.B.M.).NIH grants 5U01CA168394 (to G.B.M. and K.L.S.), 5P50CA098258 (to G.B.M. and S.N.W.),and 5P50CA083639 (to G.B.M. and S.N.W.); Andrew Sabin Family Fellowship Program(to S.N.W.); Cancer Center Support grant CA016672; and the Dr. Miriam and SheldonG. Adelson Medical Research Foundation (to G.B.M. and J.B.). Author contributions: C.S., Y.F.,J.Y., and J.C. conceived and performed experiments. Z.J. analyzed RPPA data. X.C., C.P.V.,and Y.L. performed RPPA. D.Z. conducted some immunohistochemical evaluation. K.J.J. andA.K.B.E. performed deep NGS and Sanger sequencing of PARPi-resistant clones. P.K.-S.N.established wild-type KRAS or mutant KRAS and PIK3CA Ba/F3 cell lines. N.H.B., S.N.W., J.R.G.,S.-Y.L., K.L.S., G.P., J.B., and G.B.M. provided expertise and feedback. G.B.M. conceived andcoordinated the project and wrote the manuscript. Competing interests: G.B.M. has licensedan HRD assay to Myriad Genetics; is a SAB member/consultant with AstraZeneca, CatenaPharmaceuticals, Critical Outcome Technologies, ImmunoMET, Ionis, MedImmune,
Sun et al., Sci. Transl. Med. 9, eaal5148 (2017) 31 May 2017
Nuevolution, Pfizer, Precision Medicine, SignalChem Lifesciences, Symphogen, Takeda/Millennium Pharmaceuticals, and Tarveda; and has stock options with Catena Pharmaceuticals,ImmunoMet, Spindle Top Ventures, and Tarveda. S.N.W. is a consultant with AstraZeneca,Clovis Oncology, Genentech, Medivation, Casdin Capital, and Vermillion. G.B.M. receivesresearch support from AbbVie, Adelson Medical Research Foundation, AstraZeneca, BreastCancer Research Foundation, Critical Outcomes Technology, Horizon Diagnostics, Illumina,Ionis, Karus Therapeutics, Komen Research Foundation, NanoString, Pfizer, Takeda/MillenniumPharmaceuticals, and Tesaro.
Submitted 1 December 2016Accepted 29 March 2017Published 31 May 201710.1126/scitranslmed.aal5148
Citation: C. Sun, Y. Fang, J. Yin, J. Chen, Z. Ju, D. Zhang, X. Chen, C. P. Vellano, K. J. Jeong,P. K.-S. Ng, A. K. B. Eterovic, N. H. Bhola, Y. Lu, S. N. Westin, J. R. Grandis, S.-Y. Lin, K. L. Scott,G. Peng, J. Brugge, G. B. Mills, Rational combination therapy with PARP and MEK inhibitorscapitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med. 9, eaal5148 (2017).
18 of 18
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from

mutant cancersRASliabilities in Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic
Grandis, Shiaw-Yih Lin, Kenneth L. Scott, Guang Peng, Joan Brugge and Gordon B. MillsJin Jeong, Patrick Kwok-Shing Ng, Agda Karina B. Eterovic, Neil H. Bhola, Yiling Lu, Shannon N. Westin, Jennifer R. Chaoyang Sun, Yong Fang, Jun Yin, Jian Chen, Zhenlin Ju, Dong Zhang, Xiaohua Chen, Christopher P. Vellano, Kang
DOI: 10.1126/scitranslmed.aal5148, eaal5148.9Sci Transl Med
tumors such as ovarian and pancreatic cancer.therapeutic combination, and the authors have demonstrated its effectiveness in mouse models of aggressive KRAS mutant tumors. MEK and PARP inhibitors are clinically approved drugs that provide a readily translatablediscovered that inhibition of MEK or ERK (proteins in the RAS pathway) can reverse PARP inhibitor resistance in
. haveet alnewest classes of anticancer therapeutics, and many other chemotherapy types. However, Sun and also among the most difficult to treat. RAS mutant tumors are usually resistant to PARP inhibitors, one of the
Alterations in one of the RAS proteins, such as KRAS, are among the most common oncogenic mutationsNo escape for KRAS mutant tumors
ARTICLE TOOLS http://stm.sciencemag.org/content/9/392/eaal5148
MATERIALSSUPPLEMENTARY http://stm.sciencemag.org/content/suppl/2017/05/26/9.392.eaal5148.DC1
CONTENTRELATED
http://science.sciencemag.org/content/sci/368/6486/eaax6367.fullhttp://science.sciencemag.org/content/sci/363/6433/1280.fullhttp://science.sciencemag.org/content/sci/362/6419/1171.fullhttp://stm.sciencemag.org/content/scitransmed/10/453/eaan0941.fullhttp://stke.sciencemag.org/content/sigtrans/11/540/eaat0229.fullhttp://stm.sciencemag.org/content/scitransmed/10/450/eaaq1093.fullhttp://science.sciencemag.org/content/sci/359/6381/1217.fullhttp://stm.sciencemag.org/content/scitransmed/10/436/eaao5931.fullhttp://stm.sciencemag.org/content/scitransmed/9/400/eaal1645.fullhttp://science.sciencemag.org/content/sci/356/6343/1111.fullhttp://stm.sciencemag.org/content/scitransmed/6/224/224ra26.fullhttp://stm.sciencemag.org/content/scitransmed/2/53/53ra75.fullhttp://stm.sciencemag.org/content/scitransmed/8/362/362ps17.fullhttp://stm.sciencemag.org/content/scitransmed/9/375/eaal2463.full
REFERENCES
http://stm.sciencemag.org/content/9/392/eaal5148#BIBLThis article cites 54 articles, 23 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
registered trademark of AAAS. is aScience Translational MedicineScience, 1200 New York Avenue NW, Washington, DC 20005. The title
(ISSN 1946-6242) is published by the American Association for the Advancement ofScience Translational Medicine
Copyright © 2017, American Association for the Advancement of Science
by guest on May 22, 2020
http://stm.sciencem
ag.org/D
ownloaded from


















