
Rajagopal, Mogana Sundari (2014) Canarium patentinervium miq....
424
CANARIUM PATENTINERVIUM MIQ. (BURSERACEAE KUNTH.): A PHYTOCHEMICAL AND PHARMACOLOGICAL STUDY MOGANA SUNDARI RAJAGOPAL B. Pharm. (Hons) THESIS SUBMITTED TO THE UNIVERSITY OF NOTTINGHAM (MALAYSIA CAMPUS) FOR THE DEGREE OF DOCTOR OF PHILOSOPHY AUGUST 2014
Transcript of Rajagopal, Mogana Sundari (2014) Canarium patentinervium miq....

CANARIUM PATENTINERVIUM MIQ.
(BURSERACEAE KUNTH.):
A PHYTOCHEMICAL AND
PHARMACOLOGICAL STUDY
MOGANA SUNDARI RAJAGOPAL
B. Pharm. (Hons)
THESIS SUBMITTED TO THE UNIVERSITY OF
NOTTINGHAM (MALAYSIA CAMPUS) FOR THE
DEGREE OF DOCTOR OF PHILOSOPHY
AUGUST 2014

ii
DECLARATION
I, R.Mogana, declare that this thesis is my own work. It is being submitted for the
Degree of Doctor of Philosophy, at the School of Pharmacy, Faculty of Sciences,
University of Nottingham, Malaysia. It has not been submitted before for any
degree or examination at this or any other University.
«««««
Signature
«««««
Date

iii
DEDICATION
To my beloved Sri Sri Radha Krsna, my beloved parents, G. Rajagopal
and P. Muthuletchumy and my beloved husband P. Ramesh Kumar

iv
ACKNOWLEDGEMENTS
.
I offer my greatest thanks to my dear husband and beloved parents for their love,
ongoing support and motivation.
I am grateful to my supervisor Associate Professor Dr.Christophe Wiart for his
constant enthusiasm and commitment, as well as his support all through the course of
this research. Thank you for inspiring me and for the steadfast encouragement in the
field of phytochemistry and emotional support. You have been a great inspiration.
Thank you!
I offer thanks to my co-supervisor Dr. Khoo Teng Jin for his kind attention and
guidance. I would also like to thank Dr Achyut Adhikari for his valuable expertise in
NMR. In addition I would also like to thank Dr.Tracey Bradshaw on her assistance
with the anticancer work, Dr Banasri Hazra on her assistance with the antileishmanial
and Assoc. Prof. Nongyao Sawangjaroen for the assistance on the parasitic work.

v
ABSTRACT
Canarium patentinervium Miq. belongs to the family of Burseraceae best known for
producing resins of economic, medicinal, and cultural values such as frankincense,
myrrh, and copal. This family consists of 18 genera and 700 species of trees. In the
Asia-Pacific region, about 20 species of Burseraceae are used to treat haemorrhoids,
heal wounds and to treat skin infections. This plant has been used to heal wounds
amongst the indigenous people of Malaysia. Furthermore no pharmacological and
phytochemical studies have been reported on this species. This study was undertaken
to screen the phytochemical and pharmacological aspects of this plant. Qualitative
phytochemical properties of the crude extract was determined for the presence of
tannins, flavonoids, alkaloids, saponins or sterols. Phytochemical analysis of
Canarium patentinervium Miq. revealed presence of tannins and flavonoids in the
ethanol extract of leaves and barks. Bioassay guided fractionation led to isolation of
eight secondary metabolites by chromatography which were identified by NMR
techniques. Two coumarins (scopoletin and scoparone), five phenols (cynaroside,
hyperin, (+)-catechin, lioxin and syringic acid) and a norsesquiterpene with
cyclohexenone ring (vomifoliol). The latter three compounds were isolated for the
first time from the genus Canarium. The plant and the isolated compounds were then
subjected to six biological assays comprising of antibacterial, antioxidant, anticancer,
anti-inflammatory, anti-acetylcholinesterase and anti-parasitic activities. Infectious
diseases remain the leading cause of death worldwide and bacteria have become more
resistant to conventional antibiotic and the search for novel antimicrobial agents from
medicinal plants has become crucial. Antibacterial test was done using disc diffusion
method, minimum inhibitory concentration (MIC), minimum bactericidal

vi
concentration (MBC) and death kinetic assay with ampicillin as the positive control.
The ethanol extract of leaves and the hexane extract of bark displayed remarkable
antibacterial activity against both Gram-positive bacteria and Gram-negative bacteria.
All isolated compounds tested against S.aureus ATCC 11632 showed bacterial
growth inhibition. Scopoletin, scoparone, hyperin, cynaroside and syringic acid had
bactericidal effect <100 µg/ml. Only scopoletin had bactericidal effect and complete
kill at MBC 50.00 µg/ml. Bacterial infections have been known to generate extensive
formation of free radicals which is becoming increasingly recognized in the
pathogenesis of the many human diseases. The role of free radicals and active oxygen
is becoming increasingly recognized in the pathogenesis of the many human diseases,
including cancer, neurodegenerative diseases, ageing, and atherosclerosis. Five
various antioxidant assays with different mode of action [2, 2-diphenyl-1-
picrylhydrazyl (DPPH) and 2, 2'-azinobis (3-ethylbenzothiazoline-6-sulfonic acid)
(ABTS), ferric reducing antioxidant power (FRAP), ȕ-carotene bleaching assay and
superoxide dismutase (SOD) assay] were used to test the antioxidant scavenging
abilities of this plant. Vitamin C (L-ascorbic acid), quercetin and trolox were used as
positive controls. The ethanol extract of leaves and barks displayed superior
antioxidant capacities. The EC50 values of the samples were consistently low in SET
methods (ABTS, DPPH and FRAP) superior to standard as opposed to HAT method
ȕ-carotene bleaching assay). Hyperin and (+)-catechin exhibited the most consistent
free radical scavenging capability across the five antioxidant assay. Hyperin and (+)-
catechin have significantly lower IC50 (0.75±0.03 µg/ml and 0.94±0.27 µg/ml
respectively) compared to SOD enzyme (IC50 1.59±04 µg/ml). Scopoletin exhibited
potent antioxidant activity compared to scoparone with significantly lower EC50
values in ABTS (IC50 1.08±0.03 µg/ml) compared to ascorbic acid (EC50 1.54±0.03

vii
µg/ml) and lower values in FRAP assay (FRAP value 49.00±0.64 µg/ml) than
quercetin (FRAP value 86.00±0.24 µg/ml) and ascorbic acid (FRAP value
347.00±0.23 µg/ml). 9RPLIROLRO KDG SRWHQW ȕ-carotene bleaching activity with IC50
6.85±0.37 µg/ml. Infections and free radical generation are recognized in the
pathogenesis of cancer. The ethanol and chloroform extract of barks showed
significant anticancer activities with GI50 values of 34.40µg/ml and 23.44µg/ml. The
most susceptible cell lines were found to be the breast cancer cell line, MDA 468.
Scopoletin displayed potent anticancer effect against breast cancer cell line MDA 468
(GI50 0.09±0.25 µg/ml) and colorectal cancer cell line HT-29 (GI50 0.17±0.05 µg/ml),
the latter being more significant that positive control doxorubicin (GI50 0.66±0.60
µg/ml). In recent years, roles have been identified for several inflammatory cells and
for a large number of inflammatory mediators in important pathologies not previously
linked to inflammation, such DV $O]KHLPHU¶V GLVHDVH DQG FDUGLRYDVFXODU GLVRUGHUV
including atherosclerosis, as well as cancer. Recently, reports have appeared
regarding so-FDOOHG ³GXDO LQKLELWRUV´ DJHQWV WKDW LQKLELW QRW RQO\ cyclooxygenase-1
(COX-1) and cyclooxygenase-2 (COX-2), but also 5-lipoxygenase (5-LOX).
Chloroform extract of the barks had the lowest 5-LOX inhibition (IC50=29.53±0.0
ȝJPO ZKHQ FRPSDUHG WR 1'*$ (IC50= ȝJPO Ethanol extract of
leaves had superior COX-1 inhibition (IC50 = 0.60±0.01µg/ml) compared to COX-2
inhibition (IC50 = 1.07±0.01 µg/ml), whereas the barks had superior COX-2 inhibition
(IC50 = 9.39±0.03 µg/ml) as opposed to COX-1 (IC50 = 11.41±0.03 µg/ml). All
isolated compounds exhibited significantly lower 5-LOX inhibition than NDGA.
Scopoletin and scoparone were potent inhibitors of 5-LOX recording lowest IC50
values (IC50 0.34±0.01 µg/ml and 0.20±0.01 µg/ml respectively). However (+)-
catechin had a more comprehensive anti-inflammatory activity with dual inhibition of

viii
5-LOX (IC50 16.10±0.03 µg/ml) and COX (COX-1; IC50 12.08±0.02 µg/ml, COX-2;
IC50 83.89±0.03 µg/ml). Syringic acid exhibited potent 5-LOX inhibition (IC50
1.38±0.03 µg/ml) and moderate COX-1 inhibition (IC50 34.89±0.02 µg/ml).
Furthermore, oxidative and inflammatory processes are among the pathological
features associated with the central QHUYRXVV\VWHPLQ$O]KHLPHU¶VGLVHDVH7KHUH LV
evidence that acetyl cholinesterase (AChE) inhibitors have an anti-inflammatory role
through action against free radicals and amyloid toxicity, as well as through
decreasing release of cytokines from activated microglia in the brain and blood.
Chloroform extract of the barks displayed the best activity (IC50 ȝJPO
as opposed to positive control, galanthamine (IC50 = 0.74±0.ȝJPO7KHHWKDQRO
extract of barks and leaves follow through with IC50 ȝJPODQG,&50 =
ȝJPOUHVSHFWLYHO\. Only scopoletin, scoparone, vomifoliol and syringic
acid showed AChE inhibition at IC50 <100 µg/ml. Syringic acid exhibited good AChE
inhibition (IC50 29.53±0.19 µg/ml), lowest of all compounds tested. Choline is the
precursor of phosphatidylcholine (PC), a main component of Leishmania
promastigote membranes. Therefore, inhibition of choline formation may decrease
Leishmania survival. This hypothesis can be tested by using inhibitors of the
acetylcholinesterase enzyme (AChE), which catalyzes the hydrolysis of acetylcholine
to choline and acetic acid, as leishmanicidal compounds. The hexane extract of leaves
showed moderate antileshmanial activity with IC50 YDOXHVRIȝJPO2QO\
ethanol extracts showed activity against Giardia intestinalis and Entamoeba
histolytica DW FRQFHQWUDWLRQ RI ȝJPO 6copoletin was tested against all three
parasite amd it was more potent against Leishmania donovani (IC50 163.30±0.32
µg/ml) and MIC of >200 µg/ml for both Giardia intestinalis and Entamoeba
histolytica. Six kinds of major biological effects were evident in the crude and

ix
compounds namely, antioxidant, antibacterial, anti-inflammatory, anti-AChE, anti-
parasitic, and anticancer all of which were reported for the first time from this plant.
Given the aforementioned evidence it is tempting to speculate that Canarium
patentinervium Miq. represents an exciting scaffold from which to develop leads for
treatment of inflammatory and oxidative stress related diseases.

x
PUBLICATIONS ARISING FROM THIS STUDY*
x R. Mogana, K. Teng--LQDQG&:LDUW³,Q9LWUR$QWLPLFURELDO$QWLR[LGDQW
Activities and Phytochemical Analysis of Canarium patentinervium Miq. from
0DOD\VLD´ %LRWHFKQRO 5HV ,QW YRO S (abstract pg
377).
x R. Mogana, T. D. Bradshaw, T. J. KhRRDQG&:LDUW³,Q9LWUR$QWLWXPRU
potential of Canarium patentinervium 0LT´$FDG-&DQFHU5HVYROQR
1, pp. 1±4, 2011 (abstract pg 378).
x R. Mogana DQG & :LDUW ³Canarium / ௗ $ 3K\WRFKHPLFDO DQG
3KDUPDFRORJLFDO5HYLHZ´-3KDUP5HVYRO. 4, no. 8, pp. 2482±2489, 2011
(abstract pg 379).
x A Nematollahi , N Aminimoghadamfarouj , M Rajagopal , TJ Khoo, C.Wiart,
³7KH ILUVW DQWLEDFWHULDO DFWLYLW\ UHSRUW RI WKUHH VHOHFWHG0DOD\VLDQ UDLQIRUHVW
PHGLFLQDOSODQWV´3ODQWD0HGYRO±PL9, 2011 (abstract pg 380).
x R. Mogana DQG & :LDUW ³$QWL-Inflammatory, Anticholinesterase, and
Antioxidant Potential of Scopoletin Isolated from Canarium patentinervium
0LT%XUVHUDFHDH.XQWK´(YLGHQFH-Based Complement. Altern. Med., vol.
2013, p. 734824, 7 pages, 2013 (abstract pg 381).
x R. Mogana, K. Teng--LQ DQG &:LDUW ³7KH0HGLFLQDO 7LPEHUCanarium
patentinervium Miq. (Burseraceae Kunth.) Is an Anti-Inflammatory
Bioresource of Dual Inhibitors of Cyclooxygenase (COX) and 5-Lipoxygenase
(5-/2;´,651%LRWHFKQRl., vol. 2013, pp. 1±8, 2013 (abstract pg 382).
*Appendix C

xi
CONFERENCE PROCEEDINGS AND TALKS RELATED TO
THIS STUDY
x Oral presentation at the 25th Scientific Meeting of the Malaysian Society of
Pharmacology & Physiology, 25th-26th of May 2011 at Faculty of Medicine &
Medical Sciences, Universiti Putra Malaysia. ± ³Canarium- a source of new
DQWLELRWLFVDQGDQWLR[LGDQWV"´
x Poster presentation at the 59th International Congress and Annual Meeting
of the Society for Medicinal Plant and Natural Product Research, 4 ± 9th
September 2011, Maritim Pine Beach Resort Hotel Antalya, Turkey. ³The
first antibacterial activity report of three selected Malaysian rainforest
PHGLFLQDOSODQWV´
x Poster presentation at the International Conference on Natural Products, 13-
16th November 2011, Palm Garden IOI Resort, Putrajaya, 0DOD\VLD³In vitro
antimicrobial, antioxidant, anticancer activities and phytochemical
analysis of Canarium patentinervium Miq. From Malaysia ³
x Oral presentation at the International Conference on Natural Products, 4-6th
March 2013, Shah Alam Convention Centre0DOD\VLD ³Antiinflammatory,
anticholinesterase and antioxidant activities of scopoletin isolated from
Canarium VS%XUVHUDFHDH.XQWK´
x Oral presentation at the Graduate School Talk, 27th June 2012, University of
Nottingham (Malaysia Campus). ³Canarium patentinervium Miq.: The
SK\WRFKHPLFDODQGSKDUPDFRORJLFDOVWXG\´.

xii
LIST OF FIGURES
Page
Figure 1.1: Chemical structures of secondary metabolites from Canarium
L.
24
Figure 1.2: A diagrammatic summary of steps in the study of Canarium
patentinervium Miq. evaluating the phytochemistry and
pharmacological activities.
46
Figure 2.1: Fresh and dried herbarium sample of Canarium
patentinervium Miq.
48
Figure 3.1: Positive flavonoid result for leaf ethanol extract (left) against
blank and and bark ethanol extract (right) against blank.
55
Figure 3.2: Positive tannin result for leaf ethanol extract marked as test
WXEHµ/(¶OHIWDQGEDUNHWKDQROH[WUDFWPDUNHGDVWHVW
WXEHµ6¶ULJKWDJDLQVWEODQN-), positive standard tannic
acid(+) and ferric chloride.
56
Figure 3.3: Positive sterol test for leaf ethanol extract (left) and bark
ethanol extract (right) against blank.
57
Figure 3.4: Positive sterol test for bark hexane extract (left) and bark
chloroform extract (right) against blank.
58
Figure 4.1: Diagrammatic representation of chemical reaction of the
reaction of DPPH in the presence of an electron donating
antioxidant.
67
Figure 4.2: Diagrammatic representation of the formation of the ABTS
radical after the addition of potassium persulphate.
70

xiii
Figure 4.3: Formation of (Fe2+-TPTZ) complex from (Fe3+-TPTZ)
complex by antioxidant
73
Figure 4.4: )RUPDWLRQRIDGGXFWVIURPE\ȕ-carotene and antioxidant
with a lipid peroxide radical.
75
Figure 4.5: DPPH test result with 96 well microtiter plate results for bark
(left) and leaf (right) ethanol extract.
81
Figure 4.6: DPPH scavenging activity (%) of Canarium patentinervium
Miq.
83
Figure 4.7: ABTS scavenging activity (%) of Canarium patentinervium
Miq.
84
Figure 4.8: Fe (II) calibration curve in FRAP assay. 87
Figure 4.9: Leaf hexane (LH) extract calibration curve in FRAP assay. 88
Figure 4.10: Leaf chloroform (LC) extract calibration curve in FRAP
assay.
89
Figure 4.11: Leaf ethanol (LE) extract calibration curve in FRAP assay. 90
Figure 4.12: Bark hexane (BH) extract calibration curve in FRAP assay. 91
Figure 4.13: Bark chloroform (BC) extract calibration curve in FRAP
assay.
92
Figure 4.14: Bark ethanol (BE) extract calibration curve in FRAP assay. 93
Figure 4.15: Ascorbic acid (AA) calibration curve in FRAP assay. 94
Figure 4.16: Trolox (TRO) calibration curve in FRAP assay. 95
Figure 4.17: Quercetin (QC) calibration curve in FRAP assay. 96
Figure 4.18: ȕ-carotene decolourisation (%) of Canarium patentinervium
Miq.
97
Figure 4.19: Gallic acid calibration curve in Folin-Ciocalteu assay. 98

xiv
Figure 4.20: Quercetin calibration curve in total flavonoid content
determination.
99
Figure 5.1: Arachidonic acid metabolites and inflammation. 108
Figure 5.2: The translocation of 5-lipoxygenase and cytosolic
phospholipase A2, upon cellular stimulation, to the nuclear
membrane, followed by substantial generation of
leukotrienes.
112
Figure 5.3: Cyclooxygenase and arachidonic acid metabolism. 114
Figure 5.4: COX inhibition by BE. 122
Figure 5.5: COX inhibition by LE. 123
Figure 6.1: Cholinergic neurotransmission. 132
Figure 6.2: AChE inhibition by BC. 139
Figure 6.3: AChE inhibition by galanthamine. 140
Figure 7.1: Zone of inhibition against Staphylococcus aureus (left plate)
and methicillin resistant Staphylococcus aureus (right plate)
for leaf ethanol extract at 1 mg/disc.
165
Figure 7.2: Zone of inhibition against Bacillus cereus (left plate) and
Pseudomonas aeruginosa (right plate) for leaf ethanol extract
at 1 mg/disc.
166
Figure 7.3: Time-kill plot for MSSA ATCC 11632 in presence of LE. 171
Figure 7.4: Time-kill plot for MRSA ATCC 43300 in presence of LE. 172
Figure 7.5: Time-kill plot for MSSA (clinical strain) in presence of LE. 173
Figure 7.6: Time-kill plot for MRSA (clinical strain) in presence of LE. 174
Figure 7.7: Time-kill plot for MSSA (clinical strain) in presence of BE. 175
Figure 7.8: Time-kill plot for MRSA (clinical strain) in presence of BE. 176

xv
Figure 7.9: Time-kill plot for Bacillus cereus ATCC 10876 in presence
of LE.
177
Figure 7.10: Time-kill plot for Bacillus cereus ATCC 10876 in presence
of BE.
178
Figure 7.11: Time-kill plot for Escherichia coli (clinical strain) in
presence of BE.
179
Figure 7.12: Time-kill plot for coagulase-negative Staphylococcus aureus:
O (R) (clinical strain) in presence of LE.
180
Figure 7.13: Time-kill curve for coagulase-negative Staphylococcus
aureus: O (R) (clinical strain) in presence of BH.
181
Figure 7.14: Time-kill plot for coagulase-negative Staphylococcus aureus:
O (R) (clinical strain) in presence of BE.
182
Figure 7.15: Time-kill plot for coagulase-negative Staphylococcus.aureus:
O (S) (clinical strain) in presence of LE.
183
Figure 7.16: Time-kill plot for coagulase-negative Staphylococcus aureus:
O (S) (clinical strain) in presence of BE.
184
Figure 7.17: Time-kill plot for Enterococcus faecalis (clinical strain) in
presence of LE.
185
Figure 7.18: Time-kill plot for Enterococcus faecalis (clinical strain) in
presence of BE.
186
Figure 7.19: Time-kill plot for Klebsiella sp. (clinical strain) in presence
of LE.
187
Figure 7.20: Time-kill plot for Klebsiella sp. (clinical strain) in presence
of BE.
188

xvi
Figure 7.21: Time-kill plot for Klebsiella pneumonia ESBL (clinical
strain) in presence of LE.
189
Figure 7.22: Time-kill plot for Klebsiella pneumonia ESBL (clinical
strain) in presence of BE.
190
Figure 8.1: Appearance of trophozoites under microscope in negative
control, positive control and with LE.
208
Figure 8.2: MTT assay of Canarium patentinervium Miq. against
Leishmania donovani.
210
Figure 9.1a: Effects of extract of leaves against MDA 468 growth. 222
Figure 9.1b: Effects of extract of barks against MDA 468 growth. 223
Figure 9.2a: Effects of extract of leaves against MCF-7 growth. 224
Figure 9.2b: Effects of extract of barks against MCF-7 growth. 225
Figure 9.3a: Effects of extract of leaves against HCT 116 growth. 226
Figure 9.3b: Effects of extract of barks against HCT 116 growth. 227
Figure 10.1: Schematic representation of the isolation and purification of
compound 1 (scopoletin), compound 2 (scoparone),
compound 3 (+-catechin), compound 4 (hyperin) and
compound 5 (cynaroside) isolated from Canarium
patentinervium Miq.
246
Figure 10.2: Schematic representation of the isolation and purification of
compound 6 (vomifoliol), compound 7 (lioxin) and
compound 8 syringic acid isolated from Canarium
patentinervium Miq.
247
Figure 10.3: The PTLC of chloroform extract of barks. 248
Figure 10.4: The chemical structure of compound 1 (scopoletin). 250

xvii
Figure 10.5: The chemical structure of compound 2 (scoparone). 251
Figure 10.6: The chemical structure of compound 3 [(+)-catechin]. 253
Figure 10.7: The chemical structure of compound 4 (hyperin). 253
Figure 10.8: The chemical structure of compound 5 (cynaroside). 255
Figure 10.9: The chemical structure of compound 6 (vomifoliol). 256
Figure 10.10: The chemical structure of compound 7 (lioxin). 257
Figure 10.11: The chemical structure of compound 8 (syringic acid). 258
Figure 11.1: Schematic of the major branch pathways of (poly) phenol
biosynthesis.
268
Figure 11.2: Main classes of phenolics. 269
Figure 11.3: 5-LOX inhibition by scopoletin and scoparone isolated from
Canarium patentinervium Miq.
284
Figure 11.4: 5-LOX inhibition by compounds isolated from Canarium
patentinervium Miq.
285
Figure 11.5: COX inhibition by (+)-catechin isolated from Canarium
patentinervium Miq.
286
Figure 11.6: AChE inhibition by isolated compounds from Canarium
patentinervium Miq.
289
Figure 11.7a: Effect of scopoletin isolated compounds from Canarium
patentinervium Miq. against growth of breast cancer cell line
MDA 468 (GI50 = 0.09µg/ml).
291
Figure 11.7b: Effect of scopoletin isolated compounds from Canarium
patentinervium Miq against growth of colon carcinoma cell
line HT-29 (GI50 = 0.17 µg/ml).
292

xviii
Figure 11.8: Time-kill plot for MSSA ATCC 11632 in presence of
scopoletin isolated from Canarium patentinervium Miq.
295
Figure 11.9: Structural features of flavonoids with high antioxidant
activity.
299
Figure 11.10: (+)-catechin and the corresponding fully oxidized ortho-
quinone product.
300
Figure A1: 1H NMR spectrum of compound 1 356
Figure A2: 13C NMR spectrum of compound 1 357
Figure A3: 1H NMR spectrum of compound 2 358
Figure A4: 13C NMR spectrum of compound 2 359
Figure A5: 1H NMR spectrum of compound 3 360
Figure A6: 13C NMR spectrum of compound 3 361
Figure A7: 1H NMR spectrum of compound 4 362
Figure A8: 13C NMR spectrum of compound 4 363
Figure A9: 1H NMR spectrum of compound 5 364
Figure A10: 13C NMR spectrum of compound 5 365
Figure A11: 1H NMR spectrum of compound 6 366
Figure A12: 13C NMR spectrum of compound 6 367
Figure A13: HMBC spectrum of compound 6 368
Figure A14: COSY spectrum of compound 6 369
Figure A15: 1H NMR spectrum of compound 7 370
Figure A16: 13C NMR spectrum of compound 7 371
Figure A17: 1H NMR spectrum of compound 8 372
Figure A18: 13C NMR spectrum of compound 8 373
Figure B1: HPLC chromatogram for compound 6 374

xix
Figure B2: HPLC chromatogram for compound 7 375
Figure B3: HPLC chromatogram for compound 8 376

xx
LIST OF TABLES
Page
Table 1.1: Drugs derived from plants, their clinical uses, sources and
recent plant derived drugs in global market till 2004.
4
Table 1.2: List of genera in the Burseraceae Kunth., approximate number
of species, tribal and subtribal placement, and geographic
range.
6
Table 1.3: Canarium L. species and their geographic distribution. 7
Table 1.4: Biological and pharmacological activities (in vitro) of
Canarium L. extracts and pure constituents.
14
Table 1.5: Biological and pharmacological activities (in vivo) of
Canarium L. extracts and pure constituents.
15
Table 1.6: Secondary metabolites of Canarium L. 18
Table 3.1: Preliminary phytochemical analysis of the crude extract of
Canarium patentinervium Miq.
54
Table 4.1: DPPH radical scavenging activity at 10 µg/ml of extract. 82
Table 4.2: Antioxidant values of Canarium patentinervium Miq. 100
Table 4.3: The mean r2 values for correlation between total phenolics
contents, total flavonoid contents and antioxidant activities
(EC50).
101
Table 5.1: Anti-inflammatory values of Canarium patentinervium Miq. 124
Table 6.1: Anti-acetylcholinesterase values of Canarium patentinervium
Miq.
138

xxi
Table 7.1: Antibacterial activity of Canarium patentinervium Miq.
(1 mg/disc) vs. ampicillin against 4 ATCC bacteria and 10
clinical bacterial strains and 3 clinical yeast strains tested by
disc diffusion assay.
167
Table 7.2: MIC values for Canarium patentinervium Miq. extracts
against 4 ATCC bacteria and 9 clinical bacterial strains and 1
clinical yeast strain.
168
Table 7.3: MBC values for Canarium patentinervium Miq. extracts
against 4 ATCC bacterias and 9 clinical bacterial strains and 1
clinical yeast strain.
169
Table 7.4: Bacteriostatic (-) and Bactericidal (+) effects of Canarium
patentinervium Miq. extracts.
170
Table 8.1: Human endoparasites in study. 199
Table 8.2: MIC values of Canarium patentinervium Miq. incubated for
24 hrs with Giardia intestinalis and Entamoeba histolytica
growing in vitro.
207
Table 8.3: Antiparasitic values of Canarium patentinervium Miq. against
Leishmania donovani (AG 83) promastigotes.
209
Table 9.1: Cytotoxic drugs developed from plant sources. 217
Table 9.2: Growth inhibition of human cancer cell lines by crude plant
extracts of Canarium patentinervium Miq.
221
Table 10.1: 1H and 13C NMR spectral data (CDCL3, 500 MHz) of
scopoletin.
250
Table 10.2: 1H and 13C NMR spectral data (CDCL3, 500 MHz) of
scoparone.
251

xxii
Table 10.3: 1H and 13C NMR spectral data (CD3OD, 500 MHz) of (+)-
catechin.
252
Table 10.4: 1H and 13C NMR spectral data (CD3OD, 500 MHz) of hyperin. 254
Table 10.5: 1H and 13C NMR spectral data (CD3OD, 500 MHz) of
cynaroside.
255
Table 10.6: 1H and 13C NMR spectral data (CD3OD, 500 MHz) of
vomifoliol.
256
Table 10.7: 1H and 13C NMR spectral data (CD3OD, 500 MHz) of lioxin. 257
Table 10.8: 1H and 13C NMR spectral data (D2O, 500 MHz) of syringic
acid.
258
Table 11.1: Antioxidant values of isolated compounds from Canarium
patentinervium Miq.
282
Table 11.2: Anti-inflammatory values of the isolated compound from
Canarium patentinervium Miq.
283
Table 11.3: Anti- acetylcholinesterase values for isolated compounds from
Canarium patentinervium Miq.
288
Table 11.4: Growth inhibition of human cancer cell lines by isolated
compounds from Canarium patentinervium Miq.
290
Table 11.5: MIC, MBC and MBC/MIC ratio for isolated compounds from
Canarium patentinervium Miq. against Staphylococcus aureus
ATCC 11632.
294
Table 11.6: Antiparasitic values of isolated compounds from Canarium
patentinervium Miq. against Leishmania donovani (AG 83)
promastigotes and MIC values against Giardia intestinalis and
Entamoeba histolytica growing in vitro.
296

xxiii
ABBREVIATIONS
1H NMR- Proton nuclear magnetic resonance
13C NMR- Carbon nuclear magnetic resonance
5-LOX- 5-lipoxygenase
5-HPETE- 5(S)- hydroxyperoxyeicosatetraenoic acid
5-S-HETE- 5-hydroxy- 6,8,11,1-eicosatetraenoic acid
µg- Microgram
µl- Microliter
µM- Micromolar
oC- Celcius
AAE- Ascorbic acid equivalent
AChE- Acetylcholinesterase
ACh- Acetylcholine
AD- $O]KHLPHU¶VGLVHDVH
AIDS- Acquired immunodeficiency syndrome
amu- Atomic mass unit
AST- Antimicrobial susceptibility testing
ATCC- American Type Culture Collection
BChE- Butyrylcholinesterase
BDE- Bond dissociation energy
CAT- Choline acetyltransferase
CC- Column chromatrography
CCl4- Carbon tetrachloride

xxiv
cfu- Colony forming units
cm- Centimeter
CNS- central nervous system
COSY- correlation spectroscopy
COX-1- Cyclooxygenase-1
COX-2- Cyclooxygenase-2
COX-3- Cyclooxygenase-3
cPLA2- Cytosolic phospholipase A2
d- Doublet
dd- Doublet of doublet
D-GaIN- D-galactosamine
DMAPP- Dimethyl allyl diphosphate
DMSO- Dimethyl sulfoxide
DPPH- 1,1,-diphenyl-2-2-picrylhydrazyl
DTNB- 5,5´-Dithio-bis(2-nitrobenzoic) acid
EC50- Effective concentration at 50 % activity
EDTA- Ethylene diamine tetra-acetic acid
FC- Folin-Ciocalteu
FLAP- 5-lipoxygenase activating protein
FRAP - Ferric reducing antioxidant power
g- Gram
GAE- Gallic acid equivalent
GC- Gas chromatography
GOT- Glutamic oxaloacetic transaminase
GPT- Glutamic pyruvic transaminase

xxv
HAT- Hydrogen atom transfer reaction
HMBC- heteronuclear multiple-bond correlation spectroscopy
HPLC- High performance liquid chromatography
hr- Hour
HO2- Hydroperoxyl
IC50- Inhibitory concentration at 50 % activity
iNOS- Inducible NO (nitric oxide) synthase
INT- p-iodonitrotetrazolium
IP- Ionization potential
IPP- Isopentyl diphosphate
IR- Infrared spectroscopy
i.p- Intra peritoneal
kg- Kilogram
L- Litre
lb- Pound
LOO- Peroxy radical
LTA4- Leukotriene A4
LTB4- Leukotriene B4
M- Molar
m- Multiplet
mM- Milimolar
MBC- Minimum bactericidal assay
mg- Miligram
MHB- Mueller Hilton broth
MHA- Mueller Hilton agar

xxvi
ml- Mililitre
MIC- Minimum inhibitory concentration
Min- Minutes
MS- Mass spectroscopy
MTT- Methylthiazol tetrazolium
NCE- New chemical entities
NDGA- Nordihydroguairetic acid
NCCLS- National Committee for Clinical Laboratory Standards
nm- Nanometer
NMR- Nuclear magnetic resonance
NSAID- Non-steroidal anti-inflammatory drugs
NO2- Nitrogen dioxide
N2O3- Dinitrogen trioxide
O2- - Superoxide
ONOO- Peroxy nitrate
ORAC- Oxygen radical absorbance capacity
PC- Phospholipase C
PGs- Prostaglandins
PGG2- Prostaglandin G2
PGI2- Prostacyclin
PLA2- Phospholipase A2
PNS- Peripheral nervous system
ppm- Parts per million
PTLC- Preparative thin layer chromatography
RA- Rheumatoid arthritis

xxvii
ROS- Reactive oxygen species
RNS- Reactive nitrogen species
Rt- Retention time
ROO- Peroxyl
s- Singlet
SET- Single electron transfer
SD- Standard deviation
SOD- Superoxide dismutase
t- Triplet
TEAC- Trolox equivalent antioxidant capacity
TLC- Thin layer chromatography
TMPD- N,N,N¶,N¶-tetramethyl-p-phenylenediamine
TPTZ- 2,4,6-tripyridyl-s-triazine
TSA- Tryptic soya agar
TSB- Tryptic soya broth
TxA2- Thromboxane A2
UV- Ultra-violet spectroscopy
WST-1- (2-(4-Iodophenyl)- 3-(4- nitrophenyl)-5-(2,4-disulfophenyl)- 2H-
tetrazolium, monosodium salt)
XO- Xanthine oxidase

xxviii
TABLE OF CONTENTS
Page Declaration
ii
Dedication
iii
Acknowledgements
iv
Abstract
v
Publications arising from this study
x
Conference proceedings and talks related to this study
xi
List of figures
xii
List of tables
xx
Abbreviations
xxiii
Table of contents
xxviii
CHAPTER 1: GENERAL INTRODUCTION
1
1.1 Introduction 1
1.2 An ethnopharmacological research 2
1.3 An introduction to the family Burseraceae Kunth. and genus
Canarium L.
3
1.3.1 The family Burseraceae Kunth 3
1.3.2 The genus Canarium L. 5
1.3.3 Botanical features of Canarium L. 8
1.4 Traditional and medicinal uses of Canarium L. 9

xxix
1.4.1 In vitro and in vivo pharmacological activities of Canarium L. 10
1.5 The phytochemistry of Canarium L. 13
1.6 The commercial uses of Canarium L. produces 17
1.7 Selection of plant material
42
1.7.1 The selection of Canarium patentinervium Miq.
43
1.7.2 The selection of bioassays performed
43
1.8 Aims of study
44
1.9 Objectives of study
44
CHAPTER 2: PLANT COLLECTION AND EXTRACTION
47
2.1 Brief introduction to Canarium patentinervium Miq. 47
2.2 Plant collection and extraction 47
CHAPTER 3: PHYTOCHEMICAL METHODS
50
3.1 Introduction
50
3.2 Phytochemical assay protocol
50
3.3 Results
52
3.4 Discussion
53
3.5 Conclusion
53
CHAPTER 4: THE ANTIOXIDANT ACTIVITY
59
4.1 Introduction 59

xxx
4.1.1 Free radicals and their mechanism of action
59
4.1.2 Natural antioxidants
61
4.1.3 Therapeutic potential of phenolic substances
62
4.1.4 Antioxidant potential of Canarium L.
63
4.2 Materials and methods
64
4.2.1 Antioxidant determination assays
64
4.2.2 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay 66
4.2.2.1 Principle of the assay
66
4.2.2.2 Colorimetric spectrophotometric assay
68
4.2.3 ¶-Azino-bis(3-ethyl-benzthiazoline-6-sulfonic acid)
(ABTS) assay
69
4.2.3.1 Principle of the assay
69
4.2.3.2 Colorimetric spectrophotometric assay
71
4.2.4 Ferric reducing ability of plasma (FRAP) assay
72
4.2.4.1 Principle of assay 72
4.2.4.2 Colorimetric spectrophotometric assay 74
4.2.5 ȕ-carotene bleaching assay 75
4.2.5.1 Principle of assay 75
4.2.5.2 Colorimetric spectrophotometric assay 76
4.2.6 Superoxide dismutase assay 77
4.2.6.1 Principle of assay 77
4.2.6.2 Colorimetric spectrophotometric assay 77
4.2.7 Total phenolic content determination
78
4.2.7.1 Principle of assay 78

xxxi
4.2.7.2 Colorimetric spectrophotometric assay 78
4.2.8 Total flavonoid content determination 79
4.2.8.1 Principle of assay
79
4.2.8.2 Colorimetric spectrophotometric assay 79
4.2.9 Statistical analysis 79
4.3 Results 80
4.3.1 DPPH assay 81
4.3.2 ABTS assay 81
4.3.3 FRAP assay 81
4.3.4 ȕ-carotene bleaching assay 85
4.3.5 Superoxide dismutase assay 85
4.3.6 Total phenolic content determination 85
4.3.7 Total flavonoid content determination 86
4.3.8 The antioxidant values, total phenolic and flavonoid content
and their correlations
86
4.4 Discussion 102
4.5 Conclusion 105
CHAPTER 5: THE ANTI-INFLAMMATORY ACTIIVTY 106
5.1 Introduction 106
5.1.1 Inflammation and arachidonic system 106
5.1.2 The lipoxygenase system 110
5.1.3 The cyclooxygenase system 111
5.1.4 Dual inhibitors 115

xxxii
5.1.5 Anti-inflammatory potential of Canarium L. 115
5.2 Materials and methods 116
5.2.1 Anti-inflammatory determination assays 116
5.2.2 The 5-LOX inhibition assay 117
5.2.2.1 Principle of assay 117
5.2.2.2 Protocol 118
5.2.3 The peroxidase endpoint assay for COX-1 and COX-2 118
5.2.3.1 Principle of assay 118
5.2.3.2 Protocol 119
5.2.4 Statistical analysis 119
5.3 Results 120
5.3.1 The 5-LOX inhibition assay 120
5.3.2 The peroxidase endpoint assay for COX-1 and COX-2 121
5.4 Discussion 121
5.5 Conclusion 127
CHAPTER 6: ANTI-ACETYLCHOLINESTERASE ACTIVITY 128
6.1 Introduction 128
6.1.1 3DWKRSK\VLRORJ\RI$O]KHLPHU¶VGLVHDVH 128
6.1.2 Acetylcholine and its functions 130
6.1.3 Distribution and function of cholinesterase 130
6.1.4 Cholinesterase inhibitors 131
6.1.5 Plants as potential anticholinesterase inhibitors 134

xxxiii
6.2 Materials and methods 135
6.2.1 Anti-acetylcholinesterase determination assays 135
6.2.2.1 Principle of assay 135
6.2.2.2 Protocol 136
6.2.2 Statistical analysis 137
6.3 Results 137
6.4 Discussion 141
6.5 Conclusion 142
CHAPTER 7: ANTIMICROBIAL ACTIVITY 144
7.1 Introduction 144
7.1.1 Infectious disease 144
7.1.2 Chemotherapeutic agents: Factors affecting their effectiveness 144
7.1.3 Drug resistance 145
7.1.4 Natural products and their role in drug discovery 146
7.1.5 Canarium species and their known antimicrobial activity 147
7.2 Materials and methods 148
7.2.1 Disc diffusion assay 148
7.2.1.1 Principle of method 148
7.2.1.2 Protocol 150
7.2.2 Minimum inhibitory concentration assay 153
7.2.2.1 Principle of method 153
7.2.2.2 Protocol 153
7.2.3 Minimum bactericidal concentration (MBC) 157

xxxiv
7.2.3.1 Principle of method 157
7.2.3.2 Protocol 157
7.2.4 Time-kill assay 157
7.2.4.1 Principle of method 157
7.2.4.2 Protocol 158
7.2.5 Statistical analysis 159
7.3 Results 159
7.3.1 Disc diffusion assay 159
7.3.2 MIC assay 160
7.3.3 MBC assay and MBC/MIC ratio 161
7.3.4 Time-kill assay 162
7.4 Discussion 163
7.4.1 Disc diffusion assay 163
7.4.2 MIC, MBC and MIC/MBC ratio 191
7.4.3 Time-kill assay 196
7.5 Conclusion 196
CHAPTER 8: ANTIPARASITIC ACTIVITY 198
8.1 Introduction 198
8.1.1 Parasitic infection: causative factor and treatment 198
8.1.2 Natural products and parasitic defence 201
8.2 Materials and methods 202
8.2.1 Principle of method 202
8.2.2 Protocol 201

xxxv
8.2.2.1 Antiparasitic assay against Giardia intestinalis
and Entamoeba histolytica
201
8.2.2.1.1 Test organisms 202
8.2.2.1.2 Cultivation of organisms 203
8.2.2.1.3 Antiprotozoal assay 203
8.2.2.2 Antiparasitic activity against Leishmania
donovani promastigotes
204
8.2.3 Statistical analysis 205
8.3 Results 205
8.4 Discussion 206
8.5 Conclusion 212
CHAPTER 9: ANTICANCER ACTIVITY 213
9.1 Introduction 213
9.1.1 Carcinogenesis 213
9.1.2 Natural products and carcinogenesis defence 214
9.2 Materials and methods 218
9.2.1 Principle of method 218
9.2.2 Protocol 218
9.2.2.1 Cell lines and cell culture 218
9.2.2.2 Preparation of plant samples 219
9.2.2.3 The MTT assay 219
9.2.3 Statistical analysis 219

xxxvi
9.3 Results 220
9.4 Discussion 220
9.5 Conclusion 228
CHAPTER 10: ISOLATION AND IDENTIFICATION OF
COMPOUNDS
229
10.1 Introduction 229
10.1.1 Secondary metabolites 229
10.1.2 Isolation methods 232
10.1.3 Crude fractionation 233
10.1.4 Chromatography 233
10.1.4.1 Thin layer chromatography (TLC) 234
10.1.4.2 Liquid chromatography (LC) 236
10.1.4.3 Size exclusion chromatography 237
10.1.4.4 High performance liquid chromatography system 239
10.1.5 Structure elucidation 240
10.1.5.1 Nuclear magnetic resonance (NMR) 241
10.2 Materials and methods 242
10.2.1 Isolation of compound 1-5 (crude fractionation) 242
10.2.2 Isolation of compound 1 & 2 242
10.2.2.1 Silica gel column chromatography 242
10.2.2.2 Preparative thin layer chromatography (PTLC) 243
10.2.3 Isolation of compound 3,4 & 5 243
10.2.3.1 Size-exclusion column chromatography 243

xxxvii
10.2.4 Isolation of compound 6-8 244
10.2.4.1 Isolation of compound 6-8 (PTLC and solvent
partioning
244
10.2.4.2 Isolation of compound 6-8 (HPLC) 245
10.2.5 Nuclear magnetic resonance (NMR) 249
10.3 Results 250
10.4 Discussion 259
10.5 Conclusion 263
CHAPTER 11: BIOACTIVITY OF ISOLATED SECONDARY
METABOLITES
264
11.1 Introduction 264
11.1.1 Terpenes and phenols 264
11.1.2 General antioxidant mechanism of phenolics 266
11.1.3 Flavonoids 270
11.1.4 Phenolic acids 272
11.1.5 Coumarins 273
11.2 Materials and methods 275
11.2.1 Antioxidant capacity assays 275
11.2.2 Anti-inflammatory determination assays 276
11.2.3 Anti-acetylcholinesterase determination assay 276
11.2.4 Anticancer assay 277
11.2.5 Antibacterial assays 277
11.2.6 Antiparasitic assays 278

xxxviii
11.2.7 Formulation of scopoletin 278
11.2.8 Statistical analysis 278
11.3 Results 279
11.3.1 Antioxidant capacity assays 279
11.3.2 Anti-inflammatory determination assays 281
11.3.3 Anti-acetylcholinesterase detmination assay 281
11.3.4 Anticancer assay 287
11.3.5 Antibacterial assays 287
11.3.6 Antiparasitic assays 293
11.4 Discussions 293
11.4.1 Antioxidant activity 298
11.4.2 Anti-inflammatory activity 303
11.4.3 Anti-acetylcholinesterase activity 305
11.4.4 Antiparasitic activity 306
11.4.5 Anticancer activity 307
11.4.6 Antibacterial activity 308
11.4.7 Formulation of scopoletin as a gel 308
11.5 Conclusion 310
CHAPTER 12: GENERAL CONCLUSIONS 312
CHAPTER 13: RECOMMENDATIONS FOR FUTURE WORK 321
REFERENCES 324
APPENDIX A: NMR DATA 356

xxxix
APPENDIX B: HPLC CHROMATOGRAMS 374
APPENDIX C: PUBLICATIONS 377
APPENDIX D: CONTINUOUS EDUCATION 383

1
CHAPTER 1
GENERAL INTRODUCTION
1.1 Introduction
Pharmacognosy is not a familiar term, even to many scientists which is defined as the
study of crude drugs of plant and animal origin. The scope of pharmacognosy is also
defined as the study of physical, chemical, biochemical and biological properties of
drugs, drug substances, or potential drugs or drug substances of natural origin as well
as the search for new drugs from natural origin as well as the search for new drugs
from natural sources [1]. Natural products offer unmatched chemical diversity with
structural complexity and biological potency. It is estimated about 100-fold higher hit
rate for natural products over synthetic compounds. Natural products occupy different
chemical space that is sometimes difficult to access compared with synthetic
compounds. Not only do the natural product databases contain many more scaffolds,
but an important proportion of the ring systems are not found at all in other drug
databases. Such unexploited scaffolds represent promising new starting points in drug
discovery.
Natural products compounds not only serve as drugs or templates for drugs but lead to
a better understanding of targets and pathways involved in disease process. They also
create opportunities for additional drug targets to be identified and exploited in these
pathways. The elucidation of the anti-inflammatory mechanism of action of aspirin
led to the discovery of the cyclooxygenase isozymes COX-1 and COX-2, which were
used in the development of novel anti-inflammatory drugs [2]. Over half of the

2
ZRUOG¶VWRSEHVW-selling pharmaceuticals drugs in 1991 owed their origin to natural
products. Higher plant-derived products represent around 25% of the total number of
clinically used drugs and include the classical drugs atropine, codeine, digoxin,
morphine and quinine [1].
Number of drug molecules obtained and developed industrially from plants and used
in modern medicines increased drastically from 121 in 1995, 130 in 1997, 143 in 2000
and 166 in 2006. From 1981 and 2000, 61% of the small molecule- new chemical
entities (NCE) that were introduced as drugs worldwide can be traced to or were
inspired by natural products. Between 2001 and 2005, 23 new drugs derived from
natural products were introduced for the treatment of bacterial and fungal infection,
FDQFHU GLDEHWHV G\VOLSLGHPLD DWRSLF GHUPDWLWLV $O]KHLPHU¶V GLVHDVH DQG JHQHWLF
disease such as tyrosinaemia and Gaucher disease [3]. Table 1.1 lists the drugs
derived from plants, their clinical uses and sources as well as recent plant-derived
drugs in the global market.
1.2 An ethnopharmacological research
The study of plants used in traditional medicine requires the effective integration of
information on chemical composition of extracts, pharmacological activities of
isolated compounds, as well as indigenous knowledge of traditional healers. The
acquisition of ethnobotanical information remains an empirical aspect in any such
study [4]. There is a great need and ethical obligation to accurately document
investigative findings on plants used for health purposes. This will also aid in the

3
efficient preservation and conservation of traditional knowledge which may
potentially benefit society in general.
1.3 An introduction to the family Burseraceae Kunth. and genus Canarium L.
1.3.1 The family Burseraceae Kunth.
The Burseraceae Kunth is one of the nine flowering plant families belonging to the
order Sapindales Juss. ex Bercht. & J. Pearl that comprise the monophyletic group
(5700 species), whose first known fossils appear in Europe 65 million years ago (Ma).
The Burseraceae Kunth. consists of approximately 18 genera and 700 species. This
family is divided into 3 tribes namely Canariceae, Protieae which are determined to be
monophyletic and Burserae which was shown to be polyphyletic. The list of genera in
the Burseraceae Kunth., approximate number of species, tribal and subtribal in the
Burseraceae Kunth., approximate number of species, tribal and subtribal placement,
and geographic range of genera are shown in Table 1.2. The family is distributed
pantropically across a broad range of low-elevation, frost-free habitats including
rainforest, dry deciduous forest and deserts [5]. The Burseraceae Kunth. are best
known for producing resins of economic, medicinal, and cultural values such as
frankincense, myrrh and copal [6].

4
Table 1.1 : Drugs derived from plants, their clinical uses, sources and recent plant derived drugs in global market till 2004 [3][7].
Drug Clinical Use/Action Plant source
Atropine Anticholinergic Atropa belladonna Linn.
Colchicine Antitumor, antigout agent Colchicum autumnale L.
Digitoxin Cardiotonic Digitalis purpurea L.
Emetine Emetic, amoebicide Cephaelis ipecacuanha (Brot.) A. Rich.
Morphine Analgesic Papaver somniferum L.
Pilocarpine Parasympathomimetic Pilocarpus jaborandi (Holmes.)
Quinine Antimalarial Cinchona ledgeriana (Howard) Bern.
Apomorphine HCL Potent dopamine receptor agonist Papaver somniferum L.
3DUNLQVRQ¶VGLVHDVH
Tiotropium bromide Longer-acting antibronchospasm Atropa belladonna Linn.
Nitisinone For hereditary tyrosinaemia type-1 Callistemone citrinus (Curtis) Skeels.
Galanthamine HBr Selective acetylcholinesterase Galanthus nivalis L.
,QKLELWRU$O]KHLPHU¶VGLVHDVH
Arteether Antimalarial Artemisia annua L.

5
1.3.2 The genus Canarium L.
The genus Canarium L. probably originated from the North American continent, not
Gondwanaland [5]. This clade embraces 75 species of trees which are mainly found in
tropical Asia and the Pacific, and a few species in tropical Africa [8], about 9 species
were found in the Philippines [8]. The geographical centre of their genetic diversity is
the Molucca Islands of eastern Indonesia, but their centre of cultivated diversity is
undoubtedly western Melanesia. Twenty to 25 species are found in the South Pacific,
of which 21 are in Papua New Guinea [9], eight in the Solomon Islands [10] and 3 or
4 in Vanuatu [11]. The known Canarium species and their distribution are listed in
Table 1.3.
The word Canarium L. GHULYHV IURP WKH 0DOD\ QDPH µNDQDUL¶[8]. Canarium L.
species often produce edible kernels, called canarium nut of commercial interest:
Canarium indicum L., Canarium solomonense B.L.Burtt, Canarium harveyi Seem,
Canarium odontophyllum Miq. and Canarium album L. Another economical interest
of Canarium L. species, is the production of resins used in foods (Canarium
luzonicum Miq.), in the making of incense and varnishing. In spite of these
commercial potentials of Canarium L., little attention has been given to the collection
and conservation of Canarium L. species [12]. The genetic diversity thus derogates at
an alarming rate.

6
Table 1.2: List of genera in the Burseraceae Kunth., approximate number of species, tribal
and subtribal placement, and geographic range [5].
Bursereae Burserinae
Bursera Jacq. ca. 100 spp. Caribbean, Mexico, Central, and S. America Commiphora Jacq. ca. 190 spp. Africa, India, S. America
Boswelliinae Aucoumea Pierre 1 sp. W. Africa Beiselia Forman 1sp. SW Mexico Boswellia Roxb. ca. 30 spp. NE Africa, Arabia, India Triomma Hook. f. 1sp. W Malesian region Unnamed subtribe Garuga Roxb. 4 spp. India, SE Asia
Canarieae Canarium L. ca. 105 spp. SE Asia, Malaysia, Africa Dacryodes Vahl 66 spp. Caribbean, Mexico, C. and S. America, SE Asia, Africa Haplolobus H.J. Lam 22 spp. E. Malaysia Pseudodacryodes R. Pierlot 1 sp. Central Africa Rosselia Forman 1 sp. Rossel Island, New Guinea Santiria Blume 24 spp. W. Malesian region, Philipines, Moluccas, New Guinea, Africa Scutinanthe Thwaites 2 spp. Sri Lanka, S. Myanmar, Celebes, Sumatra, Malay Peninsula,Borneo Trattinnickia Willd. 13 spp. C. and S. America
Protieae Crepidospermum Hook. f. 6 spp. S. America Protium Burm. f. 150 spp. Mexico, C. and S. America, Africa, SE Asia Tetragastris Gaertn. 9 spp. Central and S. America
Garuga was placed informally within the Bursereae by Harley and Daly (1995).

7
Table 1.3: Canarium L. species and their geographic distribution [13]. Species Distribution
C. acutifolium (DC.) Merr. Moluccas, New Guinea, Central Celebes C. album (Lour.) Raeusch. Annam, Tonkin, S. China, Hainan C. apertum H.J. Lam Sumatra, Malay Peninsula, Borneo C. asperum Benth. Solomon Is., Bawean and Kangean Is subsp. asperum Lesser Sunda Is., Borneo, Philippines, subsp. papuanum (H.J. Lam) Leenh. Celebes, Moluccas, New Guinea C. australianum F.v.M N. Australia, SE New Guinea C. baileyanum Leenh. Queensland, NS Wales C. balansae Engl. Loyalty Is. C. balsamiferum Willd. Celebes, Moluccas, New Guinea C. luzonicum (Bl.) A. Gray Philippines C. macadamii Leenh. New Guinea C. madagascariense Engl. Madagascar, Tanganyika, subsp. Madagascariense Mozambique subsp. Obtusifolium (S.Elliot) Leenh. C. maluense Laut. Central Celebes, Moluccas, New Guinea subsp. maluense subsp. borneense Leenh. C. manii King Middle and S. Andaman C. megacarpum Leenh. W. New Guinea C. megalanthum Merr. Sumatra, Malay Peninsula, Borneo C. merrillii H. J. Lam Borneo C. muelleri F.M. Bailey Australia (Queensland) C. odontophyllum Miq. Sumatra, Borneo, Philippines C. oleiferum Baill. New Caledonia C. oleosum (Lamk) Engl. New Britain, Lesser Sunda Island.,
N. Celebes, Moluccas, New Guinea C. ovatum Engl. Philippines C. paniculatum (Lamk) Benth. ex Engl. Mauritius C. parvum Leenh. Tonkin, N. Annam C. patentinervium Miq. Sumatra, Malay Peninsula, Banka,
Anambas Island., Borneo C. perlisanum Leenh. Malay Peninsula C. piloso-sylvestre Leenh. W. New Guinea C. pilosum Benn. Sumatra, Malay Peninsula, Borneo subsp. pilosum subsp. borneensis Leenh. C. pimela Leenh. S. China, Hainan, Tonkin, Laos,
Annam, Cambodia C. polyphyllum K. Sch. New Guinea C. pseudodecumanum Hochr. Sumatra, Malay Peninsula, Borneo C. pseudopatentinervium S. Sumatra, Banka, Borneo H.J. Lam C. pseudosumatranum Leenh. Malay Peninsula C. rigidum (Bl.) Zipp. ex Miq. New Guinea C. salomonense B.L. Burtt Solomon Island., E. New Guinea subsp. salomonense subsp. papuanum C. samoense Engl. Samoa C. schlecteri Laut. New Britain, E. New Guinea

8
Table 1.3: Canarium L. species and their geographic distribution [13]. (continuation) Species Distribution
C. schweinfurthii Engl. Trop. W and Central Africa C. smithii Leenh. Fiji C. strictum Roxb. SW Deccan, Sikkim, Assam, Upper Burm C. subulatum Guill. S. China, Hainan, Tonkin, Laos, Annam, Cambodia C. sumatranum Boerl. Sumatra, Malay Peninsula and Koord. C. sylvestre Gaertn. Moluccas, New Guinea C. trifoliatum Engl. New Caledonia C. trigonum H.J. Lam Central Celebes C. vanikoroense Leenh. New Hebrides, Fiji C. vitiense A. Gray Fiji C. vrieseanum Engl. Philippines, Central and N. Celebes C. vulgare Leenh. Kangean and Bawean Island., Lesser Sunda Island., Celebes, Moluccas C. whitei Guill. New Caledonia C. zeylanicum (Retz.) Bl. Sri Lanka
1.3.3. Botanical features of Canarium L.
The members of the genus Canarium L. consist of medium to large buttressed trees
up to 40-50m tall, or rarely a shrub. The barks are greenish grey, fawn or light yellow
brown that are usually smooth, scaly or dippled with many small lenticels. Outer bark
are thin while the inner barks are pinkish brown or reddish brown, laminated, soft and
aromatic with a clear sticky or rarely oily exudate. The stems are usually terete. The
leaves are pinnate, spiral and stipulated. The rachis is terete flattened to channeled
swollen at base, and bears 5-21 folioles. The folioles are oblique at base, entire,
dentate or serrate at margin, often thick and acuminiate at apex. The secondary nerves
are arching and joined near margin. The tertiary nerves are reticulate. The
infloresence is an axillary or terminal panicle. The calyx is cupular. The corolla
includes 3 creamy petals. The androecium comprises a whorl of 6 stamens. The disc
within the stamens is 6 lobed. The gynaecium consist of 3 carpels united into a 3

9
lobular ovary. The drupes are seated on a persistent enlarged calyx and enclose a
woody stone [14].
1.4 Traditional and medicinal uses of Canarium L.
Elemi (British Pharmaceutical Codex, 1934) is an oleoresin exuded through the bark
of Canarium luzonicum Miq. or Canarium commune L. which has been used in the
form of an ointment as a stomach stimulant and as an expectorant [15]. The barks of
Canarium indicum L. has been used for chest pains where else the oil has been
patented for treatment of arthritis pain and the oleoresin of the tree is applied as a
poultice for ulcerated wounds. The resin of Canarium tonkinense Engl. has been used
as a stimulant, rubefacient and anti-rheumatic when applied externally. The oleoresin
has been applied as ointment for ulcers [16]. The dried fruit of chinese olive or
Canarium album (Lour.) Raeusch. is used in China and used to treat bacterial and
viral infections, inflammation, poisoning and for detoxification [17].
In Chinese folk medicine, the dried fruits of Canarium album (Lour.) Raeusch have
been used for treatment of angina, dysentery, snake bites, cough-hematemesis,
enteritis, diarrhoea, toxicosis from swellfish and alcohol [18]. Canarium
schweinfurthii Engl. is used by traditional healers as a remedy for diabetes mellitus in
southern Senegal [19] while in Congo and Central African Republic the plant is used
in fever, as stimulant, emollient, in post-partum pain, constipation, malaria, diarrhoea,
sexual infections and rheumatism [20]. In Indonesia, the bark of Canarium littorale
Bl. is used to make a decoction taken to heal haemorrhoids [21].

10
1.4.1 In vitro and in vivo pharmacological activities of Canarium L.
Only 12 % of 75 species have been studied for their pharmacological activities.
Extracts and pure compounds derived from Canarium L. were reported to have a
variety of pharmacological activities of which antioxidant, antibacterial, antifungal,
antitumor, anti-inflammatory, hepatoprotective, analgesic and anti-diabetic (Table
1.4).
i. Antioxidant
Antioxidant activities were reported in Canarium album (Lour.) Raeusch, Canarium
odontophyllum Miq. and Canarium schweinfurthii Engl. A tonic soup made of
Canarium album (Lour.) Raeusch used mainly in China displayed significant
antioxidant activity by 1,1,-diphenyl-2-2-picrylhydrazyl (DPPH) and ferric reducing
antioxidant power (FRAP) assay [22]. Tannins extracted from the leaves, twigs and
stem barks of Canarium album (Lour.) Raeusch showed potent antioxidant activity in
the DPPH radical scavenging activity with IC50 values of 56.86 µg/ml, 62.31 µg/ml
and 54.80 µg/ml respectively and ferric reducing power of 4.28, 3.74 and 4.49 mmol
AAE/g equivalent of dried tannin [23]. Pure compounds from this species includes
brevifolin, hyperin and ellagic acid which showed free radical scavenging activity in
DPPH assay [24].
The essential oil of Canarium schweinfurthii Engl. was tested for the antioxidant
DFWLYLW\ZLWK WKH'33+DVVD\DQGE\ȕ-carotene bleaching test. It exhibited highest
antioxidant activity at 150 µg/ml activity in both assay [25]. The ethyl acetate fraction

11
of peel of Canarium odontophyllum Miq. exhibited 95±1.00% scavenging activity at
the concentration of 40 µg/ml [26][27].
ii. Antibacterial and antifungal activities
Antibacterial activities were reported in Canarium schweinfurthii Engl. and Canarium
patentinervium Miq. Dichloromethane extract of Canarium schweinfurthii Engl. had
bactericidal activity against Gram-negative Vibrio cholerae with minimum inhibitory
concentration (MIC) of 0.62 mg/ml while the ethylacetate extract was active against
Gram-positive and Gram-negative bacteria namely Staphylococcus aureus and
Proteus vulgaris with MIC values of 10 mg/ml and 5 mg/ml respectively. Ethanol
extract was active against Gram-negative Vibrio cholerae and Proteus vulgaris with
MIC values of 0.62 mg/ml and 10 mg/ml respectively [28]. In a separate disc
diffusion assay, the essential oil of Canarium schweinfurthii Engl. abrogated the
survival of Gram-negative Salmonella enterica, Gram-positive Streptococcus pyogens
and Staphylococcus aureus with an inhibition zone of 27 mm, 25 mm and 18 mm
respectively. However the author did not inform on the concentration of the extract
per disc. The oil was also fungicidal against Candida albicans with an inhibition
zone of 23 mm [25].
iii. Hepatoprotective activities
Hepatoprotective activity was exhibited in Canarium manii King. and Canarium
album (Lour.) Raeusch. The biflavanoid agathisflavone from Canarium manii King.
preserved the integrity of the liver cells membrane of rodents as evidenced by the

12
decrease in the CCl4-induced rise of glutamic oxaloacetic transaminase (GOT) and
glutamic pyruvic transaminase (GPT) levels. GPT which is predominantly found in
the liver showed a dose-dependent and significant reduction [29]. The triterpenes urs-
12-ene-Įȕ-diol, olean-12-ene-Įȕ-diol and urs-12-ene-ȕȕ-diol from from
Canarium album (Lour.) Raeusch markedly reduced the amount of alanine
aminotransferase leakage from the primary cultured hepatocytes intoxicated with 0.2
mM of D-galactosamine (D-GaIN) [30]. The phenols brevifolin and ellagic acid
protected rat hepatocytes against D-GaIN-induced insults [24].
iv. Other Biological Activities
The essential oil of Canarium schweinfurthii Engl. collected from the region of
Cameroon inhibited the enzymatic activity of lipoxygenase with an IC50 value of 62.6
ppm [31]. However in a separate study of the same species collected from central
African region did not show any activity in the cotton pellet induced granuloma
formation experiment [20]. This may suggest possible evidence of variety between
secondary metabolites constituents according to regions. The essential oil of
Canarium schweinfurthii Engl. at a dose of 1, 2 and 3 mg/kg i.p. displayed potent
analgesic effect in the acetic acid-induced writhing and hot plate experiments [20].
Anti-diabetic activity was reported in the methanol/methylene chloride extract of stem
bark of Canarium schweinfurthii Engl. At a dose of 300 mg/kg there was 67.1 %
reduction in blood glucose levels after a once daily subcutaneous injection on
streptozotocin-induced diabetic male rats over 14 days, versus insulin that had 76.8 %
reduction. Weight gain was only 6.6 % as opposed to untreated rats that had lost 14.1

13
% of body weight. There was also significant reduction in food and fluid consumption
by 68.5 % and 79.7 %. These results showed the extract could reverse hyperglycemia,
polyphagia and polydipsia provoked by streptozotocin, thus having anti-diabetic
activity [19].
1.5 The phytochemistry of Canarium L.
Isolation and structure elucidations of secondary metabolites in Canarium L. has been
carried out since the 50¶s [32]. Majority of investigations include the resin and the
fruit of the species. To date about 96 compounds have been isolated from 9 species,
which are Canarium schweinfurthii Engl., Canarium boivinii Engl., Canarium
odontophllum Miq., Canarium manii King., Canarium album (Lour) Raeusch,
Canarium zeylanicum (Retz.) Blume, Canarium commune L., Canarium muelleri
F.M. Bailey and Canarium bengalense Roxb. The extensively researched species are
Canarium schweinfurthii Engl. and Canarium album (Lour.) Raeusch
[17][18][23][33][34].

14
Table 1.4: Biological and pharmacological activities (in vitro) of Canarium L. extracts and pure
constituents
Extract/Compound Species Pharmacological activity References
Polymeric
procyanidins(tannins)
from leaves, twigs and
stem bark
Canarium album
(Lour.) Raeusch
Significant DPPH radical
scavenging activity,
Ferric reducing antioxidant
activity
[23]
Ethyl acetate fraction
of the fruit peel
Canarium
odontophyllum
Miq.
Antioxidant activity with
DPPH assay, FRAP assay and
hemoglobin oxidation assay
[26]
Carotenoids from
peel,pulp and seed
extracts
Canarium
odontophyllum
Miq.
Significant antioxidant activity
with beta-carotene bleaching
assay, ABTS assay, DPPH
assay and hemoglobin
oxidation assay
[27]
Essential oil of resins Canarium
schweinfurthii Engl.
Bactericidal for Enterococcus
faecalis, Listeria innocua,
Salmonella enterica,
Staphylococcus aureus,
Staphylococcus camorum
Fungicidal for Candida
albicans
$QWLR[LGDQWIRU'33+DQGȕ-
carotene bleaching test
[25]
Essential oil of
resins(monoterpenes
hydrocarbon)
Canarium
schweinfurthii Engl.
Significant anti-inflammatory
activity via lipooxygenase
method with IC50 of 62.6 ppm
[31]
Brevifolin, ellagic
acid and hyperin
Canarium album
(Lour.) Raeusch
Significant antioxidant activity
and inhibitory effect on lipid
peroxidation assay
[24]
Dichloromethane
extract of barks
Canarium
schweinfurthii Engl.
Antimicrobial activity against
V.cholerae
[28]
Extract of whole plant Canarium album
(Lour.) Raeusch
Antioxidant activities in DPPH
and FRAP assay
[22]

15
Table 1.5: Biological and pharmacological activities (in vivo) of Canarium L. extracts and pure
constituents
Extract/Compound Species Pharmacological activity References
Agathisflavone
(biflavanoid)
Canarium manii
King.
Hepatoprotective activity
against experimentally-induced
carbon tetrachloride-
hepatotoxicity in rats and mice
[29]
urs-12-ene-Įȕ-
diol, olean-12-
ene-ĮȕGLRO(triterpene)
Canarium album
(Lour.) Raeusch
Hepatoprotective activity in
primary cultured rat
hepatocytes intoxicated with D-
galactosamine
[30]
brevifolin, ellagic acid Canarium album
(Lour.) Raeusch
Reduction of carbon
tetrachloride induced liver
damage in mice. Reduction in
elevated GPT and GOT levels
after intraperitoneal
administration
[24]
Essential oil of
resins(composed
mainly of nerolidol
and octylacetate)
Canarium
schweinfurthii Engl.
Significant analgesic effect
using acetic acid-induced
writhing and hot plate methods
with swiss mice
[20]
Methanol/methylene
chloride extract of
stem barks
Canarium
schweinfurthii Engl.
Anti-diabetic activity that
reverses hyperglycemia,
polyphagia and polydipsia in
streptozotocin-induced diabetic
rats. Significant reduction 69.9
% reduction in blood glucose
level after 14 days at 300
mg/kg
[19]

16
The isolation and separation technique is very much dependent on the type of
fractions. Essential oils are analysed with gas chromatography (GC) and mass
spectroscopy (MS) [20][31][35]. Other substances are separated with liquid
chromatography using different solvent mixtures with silica gel [29][36], charcoal
[37], sephadex [18] and multiple column packing such as AB-8 adsorption resin,
polyamide, and TSK Toyopearl HW-40(S). Other types of analytical techniques
include thin layer chromatography (TLC) and high performance liquid
chromatography (HPLC) [17][27][34][38][39].
The structures are mainly established by mass spectroscopy (MS), ultra-violet
spectroscopy (UV), infrared spectroscopy (IR) and 1H and/or 13C nuclear magnetic
resonance (NMR). 1H and/or 13C spectroscopy is probably the most useful method in
structure elucidation [29]. Among the secondary metabolites isolated from members
of the genus Canarium L. are terpenes (monoterpenes, triterpenes, tetraterpenes like
carotenoids, sesquiterpenes, cyclohexane and sterols), carboxylic acids, coumarins,
furans, lipids and phenols (flavonoids, tannins, phenolic acids). The main secondary
.metabolites isolated so far from the genus Canarium L. consists of terpenes with 58
compounds and flavonoids with 11 compounds. The profile of all known secondary
metabolites of Canarium L. as found in literature are included in Table 1.6 and their
structures are included in Figure 1.1.

17
1.6 The commercial uses of Canarium L. produces
(OHPL GHULYHG IURP WKH $UDELF µ$O-ODPL¶ LV D FROOHFWLYH WHUP DSSOLHG WR VHYHUDO
oleoresins obtained from different plants of the family Burseraceae Kunth. The most
important and widely known of these oleoresins is Manila Gum Elemi which exudates
from the trunk of Canarium luzonicum Miq. or Canarium commune L. The natural
constituent of elemi oil is elemicin. Variety of foodstuffs are flavoured with elemi oil
and in Europe its used in spices and seasonings. In US elemi oil is also used in
fragrances to approximately 1000 lb/year [40]. At present, at least 4 species of
Canarium L. nuts are of economic importance. Canarium ovatum Engl. (known
ORFDOO\ DV µSLOL¶ DQG µSLODXL¶ LV WKH PRVW LPSRUWDQW QXW-producing species in the
Philippines. Canarium luzonicum Miq. most commonly known in the Philippines as
µSLVD¶ DQG µEDVLDG¶ LV LPSRUWDQW QRW DV DQ HGLEOH QXW EXW IRU LWV RLO\ UHVLQ NQRZQ
ORFDOO\DVµVDKLQJ¶ZKLFKLVWDSSHGIURPWKHWUXQN:KHQSURFHVVHGLWLVFDOOHGµEUHD
EODQFD¶ZKLWH Sitch) and is exported as Manila elemi Canarium indicum L. is an
important nut-SURGXFLQJVSHFLHVLQWKH6RORPRQ,VODQGVORFDOO\FDOOHGµQJDOL¶3DSXD
1HZ*XLQHDORFDOO\FDOOHGµJDOLS¶DQG9DQXDWXZKHUHLWLVNQRZQDVµQDQJDL¶ [10].
Canarium album (Lour) Raeusch.NQRZQLQ(QJOLVKDV&KLQHVHROLYHµVDPRFKHHQ¶
LQ7KDLODQGDQGµWUDPWUDQJ¶LQ9LHWQDPLVLPSRUWDQWLQWKHVHFRXQWULHVIRULWVHGLEOH
pulp and kernel [13].

18
Table 1.6 Secondary metabolites of Canarium L.
Compound name Species Chemical
formula
Structure
number
Plant part References
CARBOXYLIC ACIDS Octyl acetate
Canarium schweinfurthii Engl.
C10H20O2
1
resin,oil
[20]
2,5-Dimethoxytoluene Canarium schweinfurthii Engl.
C9H12O2 2 resin,oil [20]
COUMARINS Scoparone
Canarium album (Lour.) Raeusch
C11H10O4
3
fruit
[41]
Scopoletin Canarium album (Lour.) Raeusch
C10H8O4 4 fruit [41]
HETEROCYCLIC COMPOUNDS-FURANS 2-acetylfuran
Canarium schweinfurthii Engl.
C6H6O2
5
resin,oil
[20]
n-octanol Canarium schweinfurthii Engl.
C8H18O 6 resin,oil [20]
LIPIDS Hexadecanoic acid
Canarium schweinfurthii Engl.
C16H32O2
7
fruit,oil
[42]
9-octadecenoic acid Canarium schweinfurthii Engl.
C18H34O2 8 fruit,oil [42]
6,9-octadecadienoic acid
Canarium schweinfurthii Engl.
C18H32O2 9 fruit,oil [42]
9,12,15-octadecatrienoic acid
Canarium schweinfurthii Engl.
C18H32O4 10 fruit,oil [42]
Oleic acid Canarium schweinfurthii Engl.
C18H34O2 11 fruit,oil [43]
Linoleic acid Canarium schweinfurthii Engl.
C18H32O2 12 fruit,oil [43]
n-decanol Canarium schweinfurthii Engl.
C10H22O 13 resin, oil [20]
n-dodecanol Canarium schweinfurthii Engl.
C12H26O 14 resin,oil [20]
PHENOLS- FLAVONOIDS Luteolin
Canarium album (Lour.) Raeusch
C15H10O6
15
fruit
[18]
Luteolin-7-O-ȕ-D-glucoside Canarium album (Lour.) Raeusch
C21H20O11 16 fruit [18]
Quercetin Canarium album C15H10O7 17 fruit [18]

19
Table 1.6 Secondary metabolites of Canarium L. (continuation)
Compound name Species Chemical
formula
Structure
number
Plant part References
Quercetin-3-O-ȕ-D-glucoside Kaempferol ¶¶-tetrahydroxyflavanone
Canarium album (Lour.) Raeusch Canarium album (Lour.) Raeusch Canarium album (Lour.) Raeusch
C21H19O12 C15H10O6
C15H10O6
18 19 20
fruit fruit fruit
[18] [18] [18]
3,5,7,3'-tetrahydroxy-4'-methoxyflavanonol
Canarium album (Lour.) Raeusch
C15H10O6 21 fruit [18]
Hyperin/ Quercetin-3-galactoside
Canarium album (Lour.) Raeusch
C21H20O12 22 dried stem, leaf,fruit
[24] [17]
Kaempferol-3-glucoside Canarium album (Lour.) Raeusch
C21H20O11 23 fruit [17]
Amentoflavone Canarium album (Lour.) Raeusch
C30H18O10 24 fruit [17]
Agathisflavone Canarium manii King. C30H18O10 25 stem,bark [29] PHENOLIC ACIDS Sinapic acid
Canarium album (Lour.) Raeusch
C11H12O5
26
fruit
[34]
Corilagin Gallic acid
Canarium album (Lour.) Raeusch Canarium album (Lour.) Raeusch
C27H22O18 C7H6O5
27 28
fruit fruit
[41] [17]
Ellagic acid Canarium album (Lour.) Raeusch
C14H6O8 29 dried stem,leaf
[24]
Brevifolin carboxylic acid Canarium album (Lour.) Raeusch
C13H8O8 30 fruit [34]
3-O-galloyl quinic acid butyl ester
Canarium album (Lour.) Raeusch
C18H24O10 31 fruit [33]
3,4-dihydroxybenzoic acid ethyl ether
Canarium album (Lour.) Raeusch
C9H10O4 32 fruit [18]
2-hydroxybenzoic acid Canarium album (Lour.) Raeusch
C7H6O3 33 fruit [18]
TANNINS Ethyl gallate
Canarium album (Lour.) Raeusch
C9H10O5
34
fruit
[18] [17]
Methyl gallate Canarium album (Lour.) Raeusch
C8H8O5 35 fruit [17]
Elemicin Canarium commune L. C12H16O3 36 fruit
[40]

20
Table 1.6 Secondary metabolites of Canarium L. (continuation)
Compound name Species Chemical
formula
Structure
number
Plant part References
SAPONINS- HYDROXY ACIDS Elemadienonic acid
Canarium boivinii Engl. Canarium schweinfurthii Engl.
C30H46O3
C30H46O3
37 37
resin resin
[44] [32]
TERPENES- CYCLOHEXANE Limonene Furfuryl butanoate
Canarium schweinfurthii Engl. Canarium boivinii Engl. Canarium zeylanicum (Retz.) Blume . Canarium schweinfurthii Engl.
C9H12O3
C9H12O3
38 39
resin,oil resin oleoresin resin, oil
[20] [36] [44] [20]
MONOTERPENES Į-pinene
Canarium album (Lour.) Raeusch
C10H16
40
resin
[35]
ȕ-Pinene Canarium album (Lour.) Raeusch
C10H16 41 resin [35]
Myrcene Canarium album (Lour.) Raeusch
C10H16 42 resin [35]
Į-Fenchene Canarium album (Lour.) Raeusch
C10H16 43 resin [35]
p-1-Menthene Canarium album (Lour.) Raeusch
C10H18 44 resin [35]
¨-3-Carene Canarium album (Lour.) Raeusch
C10H16 45 resin [35]
Į-Terpinene Canarium album (Lour.) Raeusch
C10H16 46 resin [35]
cis-Sabinene hydrate Canarium album (Lour.) Raeusch
C10H18O 47 resin [35]
Terpinolene Canarium album (Lour.) Raeusch
C10H16 48 resin [35]
Linalool Canarium album (Lour.) Raeusch Canarium schweinfurthii Engl.
C10H18O 49 resin resin,oil
[35] [20]

21
Table 1.6 Secondary metabolites of Canarium L. (continuation)
Compound name Species Chemical
formula
Structure
number
Plant part References
cis-p-Menth-2-en-1-ol
Canarium album (Lour.) Raeusch
C10H18O
50
resin
[35]
trans-p-Menth-2-en-1-ol Canarium album (Lour.) Raeusch
C10H18O 51 resin [35]
Terpinen-4-ol Canarium album (Lour.) Raeusch
C10H18O 52 resin [35]
Į-Terpineol Canarium album (Lour.) Raeusch Canarium schweinfurthii Engl.
C10H18O 53 resin resin,oil
[35] [20]
cis-Piperitone Canarium album (Lour.) Raeusch
C10H16O2 54 resin [35]
Isobornyl acetate Canarium album (Lour.) Raeusch
C12H20O2 55 resin [35]
1,8-cineole Canarium schweinfurthii Engl.
C10H18O 56 reisn,oil [20]
Citronellyl acetate Canarium schweinfurthii Engl.
C12H22O2 57 resin,oil [20]
Neryl acetate Canarium schweinfurthii Engl.
C12H20O2 58 resin,oil [20]
Decyl Acetate Canarium schweinfurthii Engl.
C12H24O2 59 resin,oil [20]
a-pinene/ Į-pinene
Canarium boivinii Engl. Canarium zeylanicum (Retz.) Blume
C10H16 40 Resin Oleoresin, timber
[36] [44]
Carvone Canarium zeylanicum (Retz.) Blume
C10H14O 60 oleoresin [36]
Į-phellandrene Canarium zeylanicum (Retz.) Blume
C10H16 61 oleoresin, bark,timber
[36]
ȕ-phellandrene Canarium zeylanicum (Retz.) Blume
C10H16 62 oleoresin, bark,timber
[36]
Terpineol Canarium zeylanicum (Retz.) Blume
C10H18O 63 oleoresin, bark
[36]
TRITERPENES Į-amyrin/ a-amyrin/ (urs-12-en-ȕ-ol)
Canarium album (Lour.) Raeusch Canarium boivinii Engl. Canarium zeylanicum (Retz.) Blume
C30H50O
64
dried stem, leaf Resin, oleoresin, bark,timber
[30] [36] [44]

22
Table 1.6 Secondary metabolites of Canarium L. (continuation)
Compound name Species Chemical
formula
Structure
number
Plant part References
ȕ-amyrin(olean-12-en-ȕ-ol)
Canarium album (Lour.) Raeusch Canarium boivinii Engl. Canarium zeylanicum (Retz.) Blume
C30H50O
65
dried stem, leaf Resin oleoresin, bark,timber
[30] [36] [44]
3-epi-Į-amyrin Canarium album (Lour.) Raeusch
C30H50O 66 dried stem, leaf
[30]
3-epi-ȕ-amyrin Canarium album (Lour.) Raeusch
C30H50O 67 dried stem, leaf
[30]
Į-Amyrenone (urs-12-en-3-one)
Canarium zeylanicum (Retz.) Blume
C30H48O 68 oleoresin [36]
ȕ-Amyrenone (olean-12-en-3-one)
Canarium zeylanicum (Retz.) Blume
C30H48O 69 oleoresin [36]
Taraxerol Canarium zeylanicum (Retz.) Blume
C30H50O 70 bark [36]
urs-12-ene-Įȕ-diol Canarium album (Lour.) Raeusch
C30H50O2 71 dried stem,leaf
[30]
olean-12-ene-ĮȕGLRO Canarium album (Lour.) Raeusch
C30H50O2 72 dried stem, leaf
[30]
TETRATERPENES CAROTENOIDS All-trans-lutein
Canarium odontophyllum Miq.
C40H56O2
73
peel,pulp, seed
[26]
9-cis-lutein Canarium odontophyllum Miq.
C40H56O2 74 peel,pulp, seed
[26]
13-cis-lutein Canarium odontophyllum Miq.
C40H56O2 75 peel,pulp, seed
[26]
15-cis-ȕ-carotene Canarium odontophyllum Miq.
C40H56 76 peel,pulp, seed
[26]
9-cis-ȕ-carotene Canarium odontophyllum Miq.
C40H56 77 peel,pulp, seed
[26]
All-trans-ȕ-carotene Canarium odontophyllum Miq.
C40H56 78 peel,pulp, seed
[26]
SESQUITERPENES Į-Cubenene
Canarium album (Lour.) Raeusch
C15H24
79
resin
[35]
Į-Copaene Canarium album (Lour.) Raeusch
C15H24 80 resin [35]

23
Table 1.6 Secondary metabolites of Canarium L. (continuation)
Compound name Species Chemical
formula
Structure
number
Plant part References
ȕ-Cubebene
Canarium album (Lour.) Raeusch
C15H24
81
resin
[35]
Canaric acid Canarium muelleri F.M. Bailey Canarium zeylanicum (Retz.) Blume
C30H48O2 82 oleoresin [36] [37]
(E)-ȕ-Caryophyllene Canarium album (Lour.) Raeusch
C15H24 83 resin [35]
Į-Humelene Canarium album (Lour.) Raeusch
C15H24 84 resin [35]
Germacrene D Canarium album (Lour.) Raeusch
C15H24 85 resin [35]
Spathulenol Canarium album (Lour.) Raeusch Canarium schweinfurthii Engl.
C15H24O 86 Resin resin,oil
[20] [35]
Viridiflorol Canarium schweinfurthii Engl.
C15H26O 87 resin, oil [20]
Caryophyllene epoxide Canarium album (Lour.) Raeusch
C15H24O 88 resin [35]
(E)- Nerolidol Canarium schweinfurthii Engl.
C15H26O 89 resin,oil [20]
Elemene Canarium zeylanicum (Retz.) Blume
C15H24 90 oleoresin, bark
[36]
Elemol Canarium zeylanicum (Retz.) Blume
C15H26O 91 oleoresin, bark
[36]
Brevifolin Canarium album (Lour.) Raeusch
C10H12O4 92 dried stem, leaf
[24]
į-Elemene Canarium album (Lour.) Raeusch
C15H24 93 resin [35]
STEROLS Sitosterol
Canarium zeylanicum (Retz.) Blume
C29H50O
94
oleoresin, bark,timber
[36]
ȕ-sitosterol Canarium benglense Roxb.
C29H50O 95 bark [45]
1HRLOH[RQROȕ-Hydroxyurs-12-en-11-one)
Canarium zeylanicum (Retz.) Blume
C30H48O2 96 oleoresin, bark
[36]

24
1.
2.
3.
4.
5.
6.
7.
8.
9.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

25
10.
11.
12.
13.
14.
15.
16.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

26
17.
18.
19.
20.
21.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

27
22.
23.
24.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

28
25.
26.
27.
28.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

29
29.
30.
31.
32.
33.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

30
34.
35.
36.
37.
38.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

31
39.
40.
41.
42.
43.
44.
45.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

32
46.
47.
48.
49.
50.
51.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

33
52.
53.
54.
55.
56.
57.
58.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

34
59.
60.
61.
62.
63.
64.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

35
65.
66.
67.
68.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

36
69.
70.
71.
72.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

37
73.
74.
75.
76.
77.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

38
78.
79.
80.
81.
82.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

39
83.
84.
85.
86.
87.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

40
88.
89.
90.
91.
92.
93.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

41
94.
95.
96.
Figure 1.1: Chemical structures of secondary metabolites from Canarium L.

42
In Melanesia, marketing operations for Canarium indicum L. and Canarium harveyi
Seem. range from private and community based production, processing, and
marketing of kernel oil for cosmetic and medicinal use, to government-backed
nationwide purchasing of kernels for sale as confections and oil. Canarium
solomonense B.L. Burtt in the Solomons islands are used as general-purpose timber
[46]. Because of the potential of Canarium L. as a high-value export crop for nuts (for
confections) and/or oil extraction, some research is being done on its taxonomy,
production and marketing. Additional research needs include selection, evaluation,
and improvement of promising varieties for timber production, investigation of
cultural aspects, phenological studies, and vegetative propagation [47].
1.7 Selection of plant material
The selection of plant material for the screening of biological activity can be based on
a random selection or based on ethnopharmacology, where existing knowledge of the
particular healing properties have been handed down from generation to generation
especially amongst traditional healers. An additional mechanism for the identification
of the plants for the study of its chemical constituents is based on chemotaxonomy.
Chemotaxonomy is a science focusing on the correlation between related plant
species and the occurrence of similar secondary metabolites.

43
1.7.1. The selection of Canarium patentinervium Miq.
Despite reasonable traditional use of Canarium L. species, as well as extreme
botanical diversity, the Canarium species in the Malaysian region have neither been
the subject of pharmacological nor extensive phytochemical studies. Claims of the
traditional usage to heal wounds [12] therefore requires validation and accurate
documentation. For this purpose, this study was initiated as a basis for scientific
verification regarding the traditional use of Canarium patentinervium Miq. . The
leaves and barks of Canarium patentinervium Miq. were collected from one
individual tree in April 2010 from Bukit Putih, Selangor, Malaysia (3°5'24 "N
101°46'0"E). The plant was identified by Mr. Kamaruddin (Forest Research Institute
of Malaysia). A herbarium sample (PID 251210-12) has been deposited in the Forest
Research Institute of Malaysia.
1.7.2 The selection of bioassays performed
The origin and design of a screening process incorporates knowledge attained in
ethnomedicine, traditional uses of the plant species, phytochemical evaluation and
correlation to specific biological targets as well as the use of natural product libraries
and general or targeted literature review. Stable standardized crude extracts are
prepared and assayed for the claimed activities for which the particular plant species
is traditionally used. The present study is focused on antioxidant, anti-inflammatory,
anti-acetylcholinesterase, antimicrobial, antiparasitic and anticancer activities.

44
1.8 Aims of study
This study is undertaken to extract and to isolate and purify active constituents from
plants by phytochemical techniques and to characterize, elucidate and identify the
structure of the pure compound and to assay isolated plant active constituents for
antibacterial, antiparasitic, antioxidant, anti-inflammatory, anti-acetylcholinesterase
and anticancer activities (Figure 1.2).
1.9 Objectives of the study
i. Extraction of leaves and barks of plant with solvents of different polarity to
obtain crude extracts.
ii. Investigation of the secondary metabolites present in the crude extracts of
leaves and barks by qualitative phytochemical analysis.
iii. Investigation of the antioxidant activity of the crude extracts of both leaves
and barks using the 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging
assay, ferric reducing ability of plasma (FRAP) assay, ¶-Azino-bis (3-ethyl-
benzthiazoline-6-sulfonic acid) (ABTS) assay, ȕ-carotene bleaching assay and
superoxide dismutase assay (SOD).
iv. Investigation of the anti-inflammatory potential of the crude extracts against 5-
lipoxygenase (5-LOX) and cycloxygenase (COX) systems in vitro.
v. Investigation of the anti-acetylcholinesterase (AChE) potential of the crude
extracts in vitro.

45
vi. Investigation of the in vitro antibacterial activity of the crude extracts of leaves
and barks using the disc diffusion assay and determination of minimum
inhibitory concentration, minimum bactericidal concentration and death kill
rate against four ATCC bacterial strains namely Gram-positive bacteria
Staphylococcus aureus, Bacillus cereus, methicillin-resistant Staphylococcus
aureus and Gram-negative Pseudomonas aeruginosa and nine clinical strain
bacteria.
vii. Investigation of the in vitro antifungal activity of the crude extracts of leaves
and barks using the disc diffusion assay against three clinical strain fungi
namely, Candida parapsilosis, Candida glabrata and Candida albicans.
viii. Investigation of the anticancer activity of the crude extracts of the leaves and
barks against human derived cell lines [HCT 116,colon cancer cell line and
MCF-7 (ER+), MDA 468 (ER-) breast carcinoma] using MTT assay.
ix. Investigation of the in vitro anti-parasitic activity of the crude extracts of
leaves and barks against Leishmania donovani promastigotes, Entamoeba
histolytica and Giardia intestinalis.
x. Isolation and identification of the chemical compounds responsible for the
antibacterial and antioxidant activity in the most active extract through
bioassay-guided fractionation using liquid-liquid extraction or solvent
partioning, column chromatography, thin layer chromatography (TLC) and
nuclear magnetic resonance (NMR).
xi. Bioactivity screening of all isolated compounds using the same bioassay as
mentioned in iii-viii.

46
Leaves and barks of Canarium patentinervium
Miq.
Succesive maceration extraction with solvent hexane, chloroform and
ethanol
Hexane extract
Chloroform extract
Ethanol extract
Bioassay ( in vitro)
Most active extract
Preliminary fractionation with solvent partioning and
column chromatography
Bioassay ( in vitro)
Active fraction(s)
Different chromatographic
techniques for isolation and purification of
compound(s) guided by in vitro bioassay
Active compound (s)
Structure determination by NMR
Bioassay of pure compound (s) using different concentrations
to determine potency
Figure 1.2: A diagrammatic summary of steps in the study of Canarium patentinervium Miq. evaluating the phytochemistry and pharmacological activities

47
CHAPTER 2
PLANT COLLECTION AND EXTRACTION
2.1 Brief introduction to Canarium patentinervium Miq.
Canarium patentinervium Miq. is fairly widespread species which occurs in lowland
primary and secondary mixed dipterocarp forest and kerangas forest. It is recorded as
a common understorey tree,W¶VDVPDOOWRPHGLXP-sized tree reaching a height of 50-
60 feet. The bark is grey brown that is smooth to cracking with many small lenticels
and often inconspicuous hoop marks. The inner bark is reddish brown and loosely
fibrous with clear resinous exudate. The sapwood is whitish with strong resinous
smell from slash. Stipules are caduceus, inserted at the junction of twigs and rachis.
Leaves are about 6-15 inches long and thickened at both ends, blade ovate to oblong
lanceolate. Apex is gradually to rather shortly and bluntly acuminate. Fruits are
ellipsoid to obovoid, rounded with a stone triangular in cross section. Its¶ strongly
glaucous and strongly aromatic (Figure 2.1). Tree flowers mainly from November to
May and fruits from September to April [14].
2.2 Plant collection and extraction
The leaves and barks of Canarium patentinervium Miq. were collected from one
individual tree in April 2010 from Bukit Putih, Selangor, Malaysia (3°5'24 "N
101°46'0"E). The plant was identified by Mr. Kamaruddin (Forest Research Institute

48
Figure 2.1: Fresh and dried herbarium sample of Canarium patentinervium Miq.

49
of Malaysia). A herbarium sample (PID 251210-12) has been deposited in the Forest
Research Institute of Malaysia. The leaves and barks were air dried at ambient
temperature (25 oC) in the laboratory for about 15 days. Thereafter the leaves were
separated from the barks and any fruit present were removed and stored separately.
The plant material was crushed to a fine powder using an industrial grinder.
Extraction was performed with maceration that involves leaving the pulverized plant
to soak in a suitable solvent in a closed container at room temperature [48]. Dried and
grinded sample of leaves (2.8 kg) and barks (1.7 kg) were soaked in hexane with the
ratio of 1:3 parts of sample to solvent for 2 hr in a 60 oC water bath, then filtered and
concentrated with a rotary evaporator (Buchi, R-200 Switzerland). This was repeated
3 times. Thereafter the leaves and barks were left to air dry completely for 3 days
before repeating the whole process with chloroform and then ethanol respectively.
The yield for the hexane, chloroform and ethanol extract of leaves were 1.25 %, 1.11
% and 6.45 % respectively. The yield for the hexane, chloroform and ethanol extract
of barks were 1.04 %, 0.40 % and 2.61 %. Crude extracts were kept at -20 oC until
further use.

50
CHAPTER 3
PHYTOCHEMICAL METHODS
3.1 Introduction
Phytochemical screening is basically done to detect the types of secondary
metabolites that may be present in the plant extract. A positive reaction should not be
taken as proof of presence of a certain type of secondary metabolite because other
compound types may give false-positive reactions. Despite this caveat, these detection
methods are often effective for generating hyphotheses about what types of secondary
PHWDEROLWHV PD\ EH SUHVHQW LQ D PL[WXUH RI ³XQNQRZQV´ DQG RI PRQLWRULQJ WKH
presence of compounds of interest [48]. Qualitative phytochemical analysis of the
crude extract was determined as outlined below.
3.2 Phytochemical assay protocol
Qualitative phytochemical analysis of the crude extract was determined as outlined
below [48][49].
i. Alkaloids test
200 mg of the extract was dissolved in 10 ml of methanol and heated on a
boiling water bath with 2M HCl (5 ml). After cooling, the mixture was
filtered and the filtrate was divided into two equal portions. One portion
was treated with a few drops of Mayer's reagent and the other with equal

51
amounts of Wagner's reagent. The samples were then observed for the
presence of turbidity or precipitation. A (+) score was recorded if the
reagent produced only a slight opaqueness; a (++) score was recorded if a
definite turbidity, but no flocculation was observed and a (+++) score was
recorded if a definite heavy precipitate or flocculation was produced.
0D\HU¶VUHDJHQWFRQVLVWVRIVROXWLRQVGHVFULEHGKHUHDVVROXWLRQ,DQGVROXWLRQ,,
Solution I: 1.36 g HgCl2 was dissolved in 60 ml water. Solution II: 5 g KI was
dissolved in 10 ml water. Both solutions are combined and diluted with water to 100
ml. If alkaloids are present a white to yellowish precipitate will appear. Wagner
reagent consists of 1.27 g I2 (sublimed) and 2 g KI was dissolved in 20 ml water and
water is made up to 100 ml. A brown precipitate indicates the presence of alkaloids.
ii. Flavonoids test
40 mg plant material was dissolved in 2 ml ethanol and filtered. The
filtrate was treated with a few drops of concentrated HCl and magnesium
turnings (0.5 g). The presence of flavonoids was indicative if pink or
magenta-red color developed within 3 min.
iii. Saponins test
About 2.5 g of the plant material was extracted with boiling water. After
cooling, the extract was shaken vigorously to froth and was then allowed
to stand for 15-20 min and classified for saponin content as follows: (no
froth = negative; froth less than 1 cm = weakly positive; froth 1.2 cm high
= positive; and froth greater than 2 cm high = strongly positive).

52
iv. Tannins test
About 10 mg of extract was dissolved in 6 ml of hot distilled water and
filtered. The solution is divided in three test tubes. To the first 0.9 %
sodium chloride solution was added, to the second 0.9 % sodium chloride
and 1 % gelatine solution was added and to the third ferric chloride (FeCl3)
was added. Formation of a precipitate in the second treatment suggests the
presence of tannins, and a positive response after addition of FeCl3 to the
third portion which will result in a characteristic blue, blue-black, green or
blue-green color supports this inference.
v. Sterols (Salkowski reaction) test.
40mg of extract was dissolved in 2 ml of chloroform and filtered. The
filtrate was then added to 1 ml of concentrated H2SO4. The presence of
sterols was indicated by the 2 phase formation with a red color in the
chloroform phase.
3.3 Results
Phytochemical analysis results exhibits the presence of flavonoid, tannin and sterols
in the respective leaves and barks extracts as shown in Table 3.1. Presence of
flavonoid and tannin was shown in the leaf and bark extract of ethanol (Figure 3.1 and
3.2). Steroids were present in all extracts except leaf extract of hexane and chloroform
(Figure 3.3 and Figure 3.4).

53
3.4 Discussion
Phytochemical analysis of Canarium patentinervium Miq.(Table 3.1) revealed
presence of tannins and flavonoids in the ethanol extract of leaves and barks.
Triterpenoids can be divided into at least four groups of compounds namely true
triterpenes, steroids, saponins and cardiac glycosides [50]. Steroids were detected in
the ethanol extract of leaves and all tested bark extracts.
3.5 Conclusion
Results from our phytochemical analysis revealed that the ethanol extract of leaves
and barks of Canarium patentinervium Miq. accumulate substantial amounts of
flavonoids and tannins which could be possibly result in antioxidant or antibacterial
activity [51].

54
Table 3.1: Preliminary phytochemical analysis of the crude extract of Canarium patentinervium Miq
Sample Alkaloids Flavonoids Saponins Tannins Steroids
LH - - - - -
LC - - - - -
LE - +++ - +++ +++
BH - - - - +++
BC - - - - ++
BE - +++ - +++ +++
LH-leaf hexane extract, LC- leaf chloroform extract, LE-leaf ethanol extract, BH- bark hexane extract, BC- bark chloroform extract, BE- bark ethanol extract -:negative, +:trace, ++:positive, +++: strongly positive

55
Figure 3.1: Positive flavonoid result for leaf ethanol extract (left) against blank and and bark ethanol extract (right) against blank.

56
Figure 3.2: Positive tannin result for OHDIHWKDQROH[WUDFWPDUNHGDVWHVWWXEHµ56LE¶OHIW DQGEDUNHWKDQROH[WUDFWPDUNHGDVWHVWWXEHµ6¶ULJKW against
blank (-), positive standard tannic acid(+) and ferric chloride .

57
Figure 3.3: Positive sterol test for leaf ethanol extract (left) and bark ethanol extract (right) against blank.

58
Figure 3.4: Positive sterol test for bark hexane extract (left) and bark chloroforml extract (right) against blank.

59
CHAPTER 4
THE ANTIOXIDANT ACTIVITY
4.1 Introduction
4.1.1 Free radicals and their mechanism of action
Oxygen is important for life processes to occur, however, an excess of oxygen could
result in oxidative damage, which may even lead to death. The damage is not due to
the presence of oxygen, but rather due to its role in the reduction of certain products
to toxic free radicals as natural by products of cell metabolism [52]. These free
UDGLFDOVDUHSURGXFHGZLWKLQ OLYLQJFHOOVDQGDUHSDUWRI WKHFHOO¶V normal metabolic
processes, including detoxification processes and immune system defences. Free
radicals may either be oxygen derived (ROS, reactive oxygen species) or nitrogen
derived (RNS, reactive nitrogen species) [53]. The oxygen derived molecules are O2-
(superoxide), HO (hydroxyl), HO2 (hydroperoxyl), ROO (peroxyl), RO (alkoxyl) as
free radical and H2O2 oxygen as non-radical. Nitrogen derived oxidant species are
mainly NO (nitric oxide), ONOO (peroxy nitrate), NO2 (nitrogen dioxide) and N2O3
(dinitrogen trioxide) [54].
In normal cell, there are appropriate oxidant and antioxidant balance. However this
balance can be shifted, when the production species is increased or when levels of
antioxidants are diminished. This stage is called oxidative stress [53]. Oxidative stress
results in the damage of biopolymers including nucleic acids, proteins,
polyunsaturated fatty acids and carbohydrates. Oxidative stress causes serious cell

60
GDPDJHOHDGLQJWRDYDULHW\RIKXPDQGLVHDVHVOLNH$O]KHLPHU¶VGLVHDVH3DUNLQVRQV¶V
disease, atherosclerosis, cancer, liver disease, inflammation, diabetes, AIDS, arthritis,
immunological incompetence, neurodegenerative disorders and etc [55][56].
In aerobic organisms, the defence system against these free radicals is provided by
free radical scavengers which act as anti-oxidants. Antioxidants are defined as a
molecule capable of slowing or preventing the oxidation of other molecules, whereas
DELRORJLFDODQWLR[LGDQWKDVEHHQGHILQHGDV³DQ\VXEVWDQFHWKDWZKHQSUHVHQWDWORZ
concentrations compared to those of an oxidizable substrate, significantly delays or
SUHYHQWVR[LGDWLRQRIWKDWVXEVWUDWH´[56] . There are at least four general sources of
antioxidants: (1) enzymes, for example super oxide dismutase, gluthathione
peroxidase and catalse; (2) large molecules (albumin, ceruloplasmin, ferritin, other
proteins); (3) small molecules [ascorbic acid, glutathione, uric acid, tocopherol,
carotenoids, (poly)phenols]; and (4) some hormones (estrogen, angiotensin,
melatonin, etc) [54][56].
Mechanisms of antioxidant action include serving as (1) physical barriers to prevent
ROS generation or ROS access to important biological sites, eg UV filters, cell
PHPEUDQHV FKHPLFDO WUDSVVLQNV WKDW ³DEVRUE´ HQHUJ\ DQG HOHFWURQV TXHQFKLQJ
ROS, eg carotenoids and anthocyanidins; (3) catalytic systems that neutralize or divert
ROS, eg SOD (superoxide dismutase), catalse and gluthathione peroxidase; (4)
binding/inactivation of metal ions to prevent generation of ROS, eg ferritin,
ceruloplasmin, catechins; and (5) chain breaking antioxidants which scavenge and
destroy ROS, eg ascorbic acid (vitamin C), tocopherols (vitamin E), uric acid,
gluthathione, flavonoids [57].

61
4.1.2 Natural antioxidants
The term, antioxidant is used to describe a component that can function to decrease
tissue damage by reactive oxygen. Defence provided for by the anti-oxidant systems
is crucial to survival and can operate at different levels within the cells through the
prevention of radical formation, intercepting formed radicals, repairing oxidative
damage, increasing the elimination of damaged molecules, and recognition of
excessively damaged molecules, which are not being repaired but rather eliminated to
prevent mutations from occurring during replication [58]. Non-enzymatic anti-
oxidants are classified as being either water-soluble or lipid-soluble, depending on
whether they act primarily in the aqueous phase or in the lipophilic region of the cell
membranes [51].
The hydrophilic anti-oxidants include vitamin C (ascorbic acid) and certain
polyphenol flavonoid groups, while the lipophilic anti-oxidants include ubiquinols,
retinoids, carotenoids, apocynin, procyanidins, certain polyphenol flavonoid groups
and tocopherols [59]. Other non-enzymatic anti-oxidants include antioxidant enzyme
cofactors, oxidative enzyme inhibitors and transition metal chelators such as ethylene
diamine tetra-acetic acid (EDTA) [58].

62
4.1.3 Therapeutic potential of phenolic substances
The establishment of an inverse correlation between the intake of fruits and
vegetables and the occurrence of diseases such as inflammation, age-related disorders,
cancer and cardiovascular disease is derived from clinical trials and epidemiological
studies [58][59]. Phenolic substances, which are known to possess high antioxidative
activity, are actually common phytochemicals in fruits and leafy vegetables. Plants
containing phenolic compounds have been reported to possess strong antioxidant
properties. Most of these phenolics are classified into two principal groups of phenol;
carboxylic acids and flavonoids, the latter being the most significant [58]. Phenolic
compounds are found abundantly in all parts of the plant, such as wood, bark, stems,
leaves, fruit, root, flowers, pollen and seeds. Antioxidative activity of phenolic
compounds is based on their ability to donate hydrogen atoms to free radicals. Many
phenolic compounds, particularly flavonoids, exhibit a wide range of biological
effects, including antioxidant activity, antibacterial, antiviral, anti-inflammatory,
antiallergic, anticancer, anti-thrombotic, vasodilatory actions and the ability to lower
the risk of coronary heart diseases [58].
Knowledge of the potential antioxidant compounds present within a plant species
does not necessarily indicate its antioxidant capacity, as the total anti-oxidant effect
may be greater than the individual antioxidant activity of one compound, due to
synergism between different anti-oxidant compounds. Methods to measure the anti-
oxidant activity in plant material generally involve both the generation of radicals
(and their related compounds), and the addition of anti-oxidants, the latter resulting in
the reduction of the radical and its consequent disappearance [60][61]. The role of

63
free radicals and active oxygen is becoming increasingly recognized in the
pathogenesis of the many human diseases, including cancer, neurodegenerative
diseases, ageing and atherosclerosis [62].
Free radicals can also cause lipid peroxidation in foods that leads to their
deterioration. Synthetic antioxidants, such as butylated hydroxyanisole (BHA) and
butylated hydroxytoluene (BHT), have restricted use in food as they are suspected of
carcinogenicity and of hormonal perturbation [63][64]. In search for sources of novel
anti-oxidants with low toxicity, medicinal plants have over the past few years been
studied extensively for their radical scavenging activity [65]. As plants produce a
large number of anti-oxidants to control the oxidative stress caused by sunbeams and
oxygen, it is clear that plants may represent a source of new compounds with
antioxidant activity [66].
4.1.4 Antioxidant potential of Canarium L.
Antioxidant activities were reported in Canarium album (Lour.) Raeusch, Canarium
odontophyllum Miq. and Canarium schweinfurthii Engl. A tonic soup made of
Canarium album (Lour.) Raeusch used mainly in China displayed significant
antioxidant activity by 1,1,-diphenyl-2-2-picrylhydrazyl (DPPH) and ferric reducing
antioxidant power (FRAP) assay [22]. Tannins extracted from the leaves, twigs and
stem barks of Canarium album (Lour.) Raeusch showed potent antioxidant activity in
the DPPH radical scavenging activity with IC50 values of 56.86 µg/ml, 62.31 µg/ml
and 54.80 µg/ml respectively and ferric reducing power of 4.28, 3.74 and 4.49 mmol
AAE/g equivalent of dried tannin [67]. Pure compounds from this species includes

64
brevifolin, hyperin and ellagic acid which showed free radical scavenging activity in
DPPH assay [24]. The essential oil of Canarium schweinfurthii Engl. was tested for
WKH DQWLR[LGDQW DFWLYLW\ ZLWK WKH '33+ DVVD\ DQG E\ ȕ-carotene bleaching test. It
exhibited highest antioxidant activity at 150 µg/ml activity in both assay [25]. The
ethyl acetate fraction of peel of Canarium odontophyllum Miq. exhibited 95±1.00%
scavenging activity at the concentration of 40 µg/ml [26][68].
4.2 Materials and methods
4.2.1 Antioxidant determination assays
Major antioxidant capacity methods have been generally divided into two categories:
(1) hydrogen atom transfer (HAT) reaction and (2) single electron transfer (SET)
reaction-based method. SET and HAT almost always occur together with the balance
determined by antioxidant structure and pH. Bond dissociation energy (BDE) and
ionization potential (IP) are two major factors that determine the mechanism and the
efficacy of the antioxidants. HAT-based methods measure the classical ability of an
antioxidant to quench free radicals by hydrogen donation. Relative reactivity in HAT
methods are determined by the BDE of the H-donating group in the potential
antioxidant and IP. Most HAT-based methods monitor competitive reaction kinetics
and are rapid.

65
;$+ ;+$
SET-based methods detect the ability of a potential antioxidant to transfer one
electron to reduce any compound, including metals, carbonyls and radicals.
;$+ X- $+
Relative reactivity is based on deprotonation and IP of the reactive functional group
so SET is pH dependent. In general, IP decrease with increasing pH, reflecting
increased electron-donating capacity with deprotonation. At acidic conditions, the
reducing capacity may be restrained due to protonation on antioxidant compounds,
whereas in basic conditions, proton dissociation of phenolic compounds would
increase samples reducing capacity. SET methods can be relatively slow and involve
two components, antioxidant and oxidant (probe).
Probe (oxidant) + e (from antioxidant) reduced probe + oxidised antioxidant
Huang et al. [69] broadly classified antioxidant capacity assays based on the reaction
mechanisms as follows : SET [Folin-Ciocalteu (Folin-C), 1,1-diphenyl-2-
picrylhydrazine (DPPH) radical scavenging, trolox equivalent antioxidant capacity
(TEAC), ferric reducing ability of plasma (FRAP)]; and HAT [oxygen radical
absorbance capacity (ORAC), ȕ-carotene/linoleic acid model system and inhibition of
phospholipid peroxidation]. Prior et al. [70] classified DPPH, TEAC and Folin-C
antioxidant assays into a category that utilises both SET and HAT reaction
mechanisms, though predominantly based on electron transfer [70][53].

66
4.2.2 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay
4.2.2.1 Principle of the assay
The antioxidant activity of each of the plant extracts was determined using the
colorimetric DPPH assay, as described by Juan Badaturuge [71], was employed to
determine the radical scavenging activity of the plant extracts. In DPPH radical-
scavenging assay, antioxidants react with DPPH, and convert it to yellow coloured
ĮĮ-diphenyl-ȕ-picryl hydrazine. The degree of discolouration indicates the radical-
scavenging potential of the antioxidant activities [72] (Figure 4.1). The hydrogen
donating capacity of test samples is quantified in terms of their ability to scavenge the
relatively stable, organic free radical DPPH and by consequent reduction thereof. The
absorption of the deep violet DPPH solution is measured at 550 nm, after which
absorption decreases due to decolourisation to a yellow-white colour, in the event of
reduction [73]. This decrease in absorption is stoichiometric according to the degree
of reduction. The remaining DPPH is measured at a time interval of 30 min after the
addition of the DPPH, which corresponds inversely to the radical scavenging activity
of the sample extract or anti-oxidant.

67
FIOH ± flavonoid compound, FIO. ± flavonoid having donated a hydrogen
Figure 4.1: Diagrammatic representation of chemical reaction of the reaction of DPPH in the presence of an electron donating antioxidant [74].
DPPH relatively stable
organic radical ± deep violet colour
DPPH reduced in presence of anti-
oxidant ± yellow-white colour

68
4.2.2.2 Colorimetric spectrophotometric assay
Aliquots of plant extract dissolved in dimethyl sulfoxide (DMSO, R&M) were plated
out in triplicate in a 96-well microtiter plate at different concentrations, prepared as
serial dilutions ranging from 10 µg/ml to 0.3125 µg/ml. The 0.1 mM DPPH solution
(AldrichR) was added to alternating columns of the test samples and methanol for
control of test samples, in the remaining columns. The plate was shaken for 2 min and
incubated for 30 min in the dark. The percentage decolourisation was obtained
spectrophotometrically at 550 nm using the Thermo Scientific Varioskan Flash
microtiter plate reader, linked to a computer equipped with (SkanIt Software 2.4.3).
Percentage decolourisation was plotted against the concentration of the sample and
the EC50 values were determined using Prism 5.00 software. Vitamin C (l-ascorbic
acid), quercetin and trolox was used as positive control. At least three independent
tests were performed for each sample. The DPPH absorbance decreases with an
increase in DPPH radical scavenging activity. This activity is given as percent DPPH
radical scavenging, which is calculated with the equation:
DPPH radical scavenging activity (%) = [(Abscontrol - Abssample)]/ (Abscontrol)] x
100
Abscontrol is the absorbance of DPPH radical + methanol;
Abssample is the absorbance of DPPH radical + sample extract /standard.

69
4.2.3 ¶-Azino-bis(3-ethyl-benzthiazoline-6-sulfonic acid) (ABTS) assay
4.2.3.1 Principle of the assay
The antioxidant activity of each of the plant extracts was determined using the
colorimetric ABTS assay, as described by Miller et al. [75], Rice-Evans [76] and
Roberta et al. [77] was employed to determine the radical scavenging activity of the
plant extracts. The ABTS anti-oxidant assay, also known as the Trolox equivalent
anti-oxidant capacity (TEAC) assay, assesses the total radical scavenging capacity of
the plant extracts. This is determined through the ability of these extracts to scavenge
the long-lived specific ABTS radical cation chromophore in relation to that of Trolox,
the water-soluble analogue of vitamin E [78]. The generation of the ABTS+
blue/green chromophore occurs through the oxidation of ABTS diammonium salt in
the presence of potassium persulfate (Figure 3.4), with the absorption maxima
occurring at wavelengths 645 nm, 734 nm and 815 nm. Anti-oxidants will reduce the
pre-formed radical cation to ABTS, and in so doing bring about the decolourisation of
ABTS+ to a colourless product [76]. The extent of this decolourisation is a measure of
the ABTS+ radical cation that has been scavenged, after a fixed time period, and is
relative to the Trolox standard.

70
Figure 4.2: Diagrammatic representation of the formation of the ABTS radical after the addition of potassium persulphate.
ABTS radical
relatively stable
organic radical- deep
turquoise green
ABTS- transparent

71
4.2.3.2 Colorimetric spectrophotometric assay
Aliquots of plant extract dissolved in dimethyl sulfoxide (DMSO, R&M) were plated
out in triplicate in a 96-well microtiter plate at different concentrations. At least three
independent tests were performed for each sample. Trolox (6-hydroxy-2,5,7,8-
tetramethylchromon-2-carboxylic acid, Sigma-Aldrich) Vitamin C (l-ascorbic acid)
and quercetin was used as positive control and prepared in ethanol and serial
dilutions of this positive control were also prepared. Ethanol was used as the negative
control. The stock solution included 7 mM ABTS solution and 2.4 mM potassium
persulfate solution. The working solution was then prepared by mixing the 2 stock
solutions in equal quantities. This solution was then stored in the dark for 12 - 16
hours in order to stabilise it before use. It remains stable for 2 - 3 days in the dark.
The concentrated ABTS+ solution was diluted with cold ethanol shortly before
conducting the assay, to a final absorbance of 0.70 ± 0.01 at 734 nm at 37 °C, in a 3
cm cuvette. The total scavenging capacity of the extracts was quantified through the
addition of 100 ȝO$%76to 100 ȝORISODQWH[WUDFW7KHVROutions were heated to 37
°C for 7 min, after which the absorbance was read at 734 nm on a spectrophotometer
(Thermo Scientific Varioskan Flash microtiter plate reader, linked to a computer
equipped with SkanIt Software 2.4.3). All experiments were done in triplicate. The
percentage decolourisation was calculated using equation below and the extent of
inhibition of the absorbance of the ABTS+ was plotted as a function of the
concentration.

72
This activity is given as percent ABTS radical scavenging, which is calculated with
the equation:
ABTS radical scavenging capacity (%) = [(Abscontrol - Abssample)]/ (Abscontrol)] x
100
Abscontrol is the absorbance of ABTS radical +ethanol;
Abssample is the absorbance of ABTS radical + sample extract /standard.
4.2.4. Ferric reducing ability of plasma (FRAP) assay
.
4.2.4.1 Principle of assay
The antioxidant activity of each of the plant extracts was determined using the
colorimetric FRAP assay, as described by Benzie & Strain [79] with slight
modifications. The mechanism associated with this method is shown in Figure 4.3.
This method was first developed to quantitate ascorbic acid in serum or plasma. When
a Fe3+-TPTZ complex is reduced to the Fe2+ form by an antioxidant under acidic
conditions, an intense blue color with maximum absorption develops at 593nm.
Therefore, the antioxidant effect (reducing ability) can be evaluated by monitoring the
formation of a Fe2+-TPTZ complex with a spectrophotometer. The FRAP assay gives
fast and reproducible results.

73
Figure 4.3: Formation of (Fe2+-TPTZ) complex from (Fe3+-TPTZ) complex by antioxidant.

74
4.2.4.2 Colorimetric spectrophotometric assay
Aliquots of plant extract dissolved in dimethyl sulfoxide (DMSO, R&M) were plated
out in triplicate in a 96-well microtiter plate at different concentrations. Vitamin C (l-
ascorbic acid), quercetin and trolox were used as positive control. At least three
independent tests were performed for each sample. The working FRAP reagent was
prepared just before assay by mixing 300 mM of acetate buffer (pH 3.6), 10 mM of
2,4,6-tripyridyl-s-triazine (TPTZ) and 20 mM of FeCl3.6H2O in ratio of 10:1:1.
Briefly 180 µl of the FRAP reagent was mixed with 20 µl of the test sample so that
the final dilution of the test sample in the reaction mixture was 1/10. After 30
minutes, the absorbance of the coloured product (ferrous triphridyltriazine complex)
was recorded using a spectrophotometer (Thermo Scientific Varioskan Flash
microtiter plate reader, linked to a computer equipped with SkanIt Software 2.4.3). Fe
(II) concentrations in the range of 1 µM-100 µM (FeSO4.7H2O) was used as standard
for calibration curve and equation of linearity is determined (y=ax+b). From the
linearity equation, concentration of sample that produced same absorbance as 1mM of
Fe (II) was determined (y of sample filled in equation to obtain x). The antioxidant
activity was calculated as Ferrous Equivalents, the concentration of samples which
produced an absorbance value equal to that of 1 mM FeSO4.

75
4.2.5 ȕ-carotene bleaching assay
4.2.5.1 Principle of assay
7KHȕ-carotene bleaching assay was conducted according to the method described by
Habtemariam et al. [80] with some modifications. ,W KDV ORQJ EHHQ NQRZQ WKDW ȕ-
FDURWHQHUHDFWVZLWKWKHSHUR[\UDGLFDOWRSURGXFHWKDWȕ-carotene epoxides. Therefore
that ȕ-carotene has received attention as a radical scavenger or antioxidant. Later an
DQWLR[LGDQW DVVD\ XVLQJ WKDW ȕ-carotene combined with lipids such as linolenic acid
was established [80]. As shown in Figure 4.4, lipids such as linolenic acid forms as
SHUR[\UDGLFDO/22LQSUHVence of the ROS and O2. This peroxy radical reacts with
ȕ-carotene to form a stable ȕ-FDURWHQHUDGLFDOVXEVHTXHQWO\WKHDPRXQWRIȕ-carotene
reduces in the testing solution. If an antioxidant is present in the testing solution, it
reacts competitively with the peroxy radical. Therefore, antioxidant effects are easily
monitored by the bleaching of the colour of a test solution with a spectrophotometer at
470 QPZKLFKLVWKHW\SLFDODEVRUEDQFHE\ȕ-carotene [56].
ȕ-carotene /22ȕ-carotene adduct
/22
Antioxidant /22ȕ-carotene adduct
Figure 4.4: )RUPDWLRQRIDGGXFWVIURPE\ȕ-carotene and antioxidant with lipid
peroxide radical.

76
4.2.5.2 Colorimetric spectrophotometric assay
Aliquots of plant extract dissolved in dimethyl sulfoxide (DMSO, R&M) were plated
out in triplicate in a 96-well microtiter plate at different concentrations. Vitamin C (l-
ascorbic acid), quercetin and trolox were used as positive control. %ULHIO\PORIDȕ-
carotene solution in chloroform (2 mg in 10 ml) was pipetted into a round bottom
flask containing 40 µl of linoleic acid and 500 µl of Tween 20. After the removal of
chloroform using a rotary vacuum evaporator at 45 oC, 100 ml of deionised water
were added with vigorous agitation. 180 µl of the emulsion was added to 20 µl of test
samples at varying concentrations in 96-well microtitre plate. The absorbance was
measured at 470 nm immediately against a blank consisting of the emulsion without
ȕ-carotene and after 3 hr of incubation at 50 oC using a spectrophotometer (Thermo
Scientific Varioskan Flash microtiter plate reader, linked to a computer equipped with
SkanIt Software 2.4.3). All determinations were carried out in triplicates.
The antioxidant activity of test agents was evaluated in terms of bleaching RI ȕ-
carotene using the following formula:
Antioxidant activity AA (%) = [1- (A0-At)/(Aoƍ-Atƍ@[
A0 and Aoƍ DUHabsorbances measured at zero time of incubation for the test sample
and control, respectively; At and AtƍDUHthe absorbances measured in the test sample
and control, respectively, after incubation for 3 hr.

77
4.2.6 Superoxide dismutase assay
4.2.6.1 Principle of assay
This assay was conducted with Sigma SOD assay kit [81]. SOD Assay Kit-WST
DOORZV YHU\ FRQYHQLHQW 62' DVVD\LQJ E\ XWLOL]LQJ 'RMLQGR¶V KLJKO\ ZDWHU-soluble
tetrazolium salt, WST-1 (2-(4-Iodophenyl)- 3-(4- nitrophenyl)-5-(2,4-disulfophenyl)-
2H-tetrazolium, monosodium salt) that produces a water-soluble formazan dye upon
reduction with a superoxide anion. The rate of reduction with oxygen is linearly
related to the xanthine oxidase (XO) activity, and is inhibited by SOD. Therefore, the
IC50 (50% inhibition activity of SOD or SOD-like materials) can be determined by a
colorimetric method. Since the absorbance at 450 nm is proportional to the amount of
superoxide anion, the SOD activity as an inhibition activity can be quantified by
measuring the decrease in the colour development at 450 nm.
4.2.6.2 Colorimetric spectrophotometric assay
Twenty microliters of sample was plated at different concentrations in a 96-well
microtiter plate. Then, 200 µl of WST solution and 20 µl of enzyme was added and
incubated for 20 mins at 37 oC. Absorbance was then recorded at 450nm using
Thermo Scientific Varioskan Flash microtiter plate reader, linked to a computer
equipped with (SkanIt Software 2.4.3) and SOD inhibition rate was determined. SOD
enzyme (Nacalai Tesque) was used as standard control.

78
4.2.7 Total phenolic content determination
4.2.7.1 Principle of assay
The total phenolic content of the crude extract and fractions was determined with the
Folin±Ciocalteu reagent, following the modified method of Singleton and Rossi [82].
The Folin-Ciocalteu reagent is sensitive to reducing compounds including
polyphenols, thereby producing a blue colour upon reaction. This blue colour is
measured spectrophotometrically, thus determining the total phenolic content.
4.2.7.2 Colorimetric spectrophotometric assay
Briefly, 20 µl of test samples dissolved in methanol (1 mg/ ml) were added to 100 µl
of Folin±Ciocalteu reagent and 1.58 ml of deionised water. After allowing the mixture
to stand at room temperature for 5 min, 300 µl of 20% (w/v) sodium carbonate were
added. Reaction mixtures were further incubated at room temperature for 30 mins,
following which absorbance at 765 nm was read against a blank, using a Jenway 6305
UV±Vis spectrophotometer (Jenway Ltd., Essex, UK). The standard calibration curve
was plotted using gallic acid (50-250 µg/ml), from which total phenolic content was
expressed as gallic acid equivalents (mg/g extract).

79
4.2.8 Total flavonoid content determination
4.2.8.1 Principle of assay
The total flavonoid content of the crude extract and fractions was determined with the
aluminium chloride colorimetric method of Froehlicher et al. [83]. The principle of
this assay is that aluminium chloride forms acid stable complexes with the C-4 keto
group and either the C-3 or C-5 hydroxyl group of flavones and flavonols. In addition,
aluminium chloride forms acid labile complexes with the orthodihydroxyl groups in
the A- or B- ring of flavonoids [84].
4.2.8.2 Colorimetric spectrophotometric assay
Briefly, 0.5 ml of test samples dissolved in methanol (1 mg/ ml) were added to 1.5 ml
of 2% methanolic solution of aluminium chloride in sealed tubes and kept in dark for
15 mins. Absorbance was then read at 430 nm using UV-vis spectrophotometer
against blank of methanolic aluminium chloride solution. The standard calibration
curve was plotted using quercetin (50-250 µg/ml), from which total flavonoid content
was expressed as quercetin equivalents (mg/g extract).
4.2.9 Statistical analysis
Concentration±response curves were calculated using the Prism software package
5.00 for Windows, GraphPad Software, San Diego California USA,
www.graphpad.com (GraphPad, San Diego, USA) and data were reported as mean

80
and SD values obtained from a minimum of three determinations. Non linear best fit
was plotted with SD and 95% confidence interval. All data were expressed as mean ±
standard deviation. SD was computed but values were not shown in the graphs via
this software. Data were analyzed using one way Anova followed by Tukey test using
GraphPad Prism5 software. A significant difference was considered at the level of P <
0.01.
4.3 Results
4.3.1 DPPH assay
In this test the ethanol extract of leaves and barks exhibited profound antioxidant
activities (Figure 4.5). The antioxidant activities were highest for ethanol extract of
barks (77.80±0.01 %) followed by the ethanol extract of leaves (65.80±0.01 %), the
hexane extract of leaves (38.90±0.02 %), the chloroform extract of barks (8.00±0.02
%), the hexane extract of barks (5.00±0.01 %) and the chloroform extract of leaves
(3.00±0.01 %) against standard ascorbic acid which was 97.90±0.00 % at the dose of
10 µg/ml (Table 4.1). The EC50 (µg/ml) for the ethanol extract of barks (2.33 µg/ml)
and leaves (2.93 µg/ml) were as good as ascorbic acid (1.88 µg/ml) as shown in
Figure 4.6 and Table 4.2.

81
Figure 4.5: DPPH test result with 96 well microtiter plate results for bark (left) and leaf
(right) ethanol extract.
4.3.2 ABTS assay
In this test the ethanol extract of leaves and barks exhibited good antioxidant activities
(Figure 4.7). The EC50 (µg/ml) for the ethanol extract of barks (0.93±0.01 µg/ml) and
leaves (2.28±0.01 µg/ml. The EC50 for ethanol extract of bark was significantly lower
than ascorbic acid (1.54±0.06 µg/ml) (Table 4.2).
4.3.3 FRAP assay
The FRAP values are represented in terms of concentration of sample that produced
same absorbance as 1 mM of Fe (II) was determined (y of sample filled in equation to
obtain x). The antioxidant activity was calculated as Ferrous Equivalents, the
concentration of samples which produced an absorbance value equal to that of 1 mM
FeSO4.

82
Table 4.1: DPPH radical scavenging activity at 10 µg/ml of extract
Extracts % DPPH radical scavenging
LH 38.90±0.02
LC 3.00±0.01
LE 65.80±0.01
BH 5.00±0.01
BC 8.00±0.02
BE 77.80±0.01
AA 97.90±0.00
LH-leaf hexane extract, LC- leaf chloroform extract, LE-leaf ethanol extract
BH- bark hexane extract, BC- bark chloroform extract, BE- bark ethanol extract
AA- ascorbic acid

83
0 2 4 6 8 10
0
20
40
60
80
100
AA
LH
LC
LE
BH
BC
BE
Quercetin
Trolox
concentration(Pg/ml)
% D
PP
H
Figure 4.6: DPPH scavenging activity (%) of Canarium patentinervium Miq.

84
0 20 40 60
0
20
40
60
80
100
LH
LC
LE
BH
BC
BE
Trolox
QuercetinAscorbic acid
concentration(Pg/ml)
% A
BT
S d
ecol
ouri
satio
n
Figure 4.7: ABTS scavenging activity (%) of Canarium patentinervium Miq.

85
Linear regression curve for all samples and positive controls are shown in Figure 4.9-
4.17. From the standard graph of ferrous sulphate (Figure 4.8), the values for six
extracts were determined using the standard formula (Table 4.2). The FRAP activity
was calculated from regression equation of ferrous sulphate calibration curve
(y=0.0105x+0.0136, r2- 0.9817). The ethanol extract of bark showed significant
FRAP activity of 67.00±0.32 µg/ml compared to quercetin and ascorbic acid. The
ethanol extract of the leaves showed significant FRAP activity of 200.00±0.07 µg/ml
compared to ascorbic acid. The poorest activity was shown by hexane extract of barks
that correlates with the low r2 in its regression equation (Figure 4.12).
4.3.4 ȕ-carotene bleaching assay
The ȕ-carotene decolourisation (Figure 4.18) was most prominent in ethanol extract
of leaves and barks with EC50 (µg/ml) of 6.04±0.02 µg/ml and 7.04±0.04 µg/ml
respectively (Table 4.2)
4.3.5 Superoxide dismutase assay
The ethanol extract of the leaves shows highest SOD-like activity with IC50 of
ȝJPO7DEOH.
4.3.6 Total phenolic content determination
The total phenolics in the extracts was determined with the Folin-Ciocalteu assay,
calculated from regression equation of calibration curve of gallic acid

86
(y=0.1762x+0.0047, r2- 0.9994) (Figure 4.19) and is expressed as gallic acid
equivalents, GAE (mg/g). The highest phenolic content was in ethanol extract of
leaves followed by barks (Table 4.2).
4.3.7 Total flavonoid content determination
The total flavonoids in the extracts was determined with the aluminium chloride
assay, calculated from regression equation of calibration curve (y=0.09330x+0.0023,
r2-0.9992) (Figure 4.20) of quercetin and is expressed as quercetin equivalent, (mg/g).
The highest flavonoid content was in ethanol extract of leaves followed by barks
(Table 4.2).
4.3.8 The antioxidant values, total phenolic and flavonoid content and their
correlations
The total antioxidant activity investigated with the relevant total phenolic and
flavonoid content are summarized in table 4.3.

87
Figure 4.8: Fe (II) calibration curve in FRAP assay.
y = 0.0105x + 0.0136 r² = 0.9817
0
0.1
0.2
0.3
0.4
0.5
0.6
0 10 20 30 40 50 60
Ab
sorb
ance
, 59
3n
m
Concentration µM
FRAP: Fe(II) calibration curve

88
Figure 4.9: Leaf hexane (LH) extract calibration curve in FRAP assay.
y = 0.0036x + 0.2681 r² = 0.9576
0.0
0.1
0.2
0.3
0.4
0.5
0 10 20 30 40 50 60
Ab
sorb
ance
, 59
3 n
m
Concentration µg/ml
Frap LH

89
Figure 4.10: Leaf chloroform (LC) extract calibration curve in FRAP assay.
y = 0.0077x + 0.2794 r² = 0.9857
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0 10 20 30 40 50 60
Ab
sorb
ance
,59
3n
m
Concentration µg/ml
FRAP: LC calibration curve

90
Figure 4.11: Leaf ethanol (LE) extract calibration curve in FRAP assay.
y = 0.0525x + 0.0136 r² = 0.9817
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0 2 4 6 8 10 12 14
Ab
sorb
ance
,59
3n
m
Concentration µg/ml
FRAP: LE calibration curve

91
Figure 4.12: Bark hexane (BH) extract calibration curve in FRAP assay.
y = 0.0029x + 0.2733 r² = 0.6982
0
0.1
0.2
0.3
0.4
0.5
0 10 20 30 40 50 60
Ab
sorb
ance
,59
3n
m
Concentration µg/ml
FRAP: BH calibration curve

92
Figure 4.13: Bark chloroform (BC) extract calibration curve in FRAP assay.
y = 0.0082x + 0.3288 r² = 0.9894
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0 10 20 30 40 50 60
Ab
sorb
ance
,59
3n
m
Concentration µg/ml
FRAP: BC calibration curve

93
Figure 4.14: Bark ethanol (BE) extract calibration curve in FRAP assay.
y = 0.1652x + 0.3254 r² = 0.9989
0.0
0.5
1.0
1.5
2.0
2.5
0 2 4 6 8 10 12 14
Ab
sorb
ance
,59
3n
m
Concentration µg/ml
FRAP: BE calibration curve

94
Figure 4.15: Ascorbic acid (AA) calibration curve in FRAP assay.
y = 0.0301x + 0.0831 r² = 0.9799
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
0 10 20 30 40 50 60
Ab
sorb
ance
,59
3n
m
Concentration µg/ml
FRAP: AA calibration curve

95
Figure 4.16: Trolox (TRO) calibration curve in FRAP assay.
y = 0.1193x + 0.2815 r² = 0.9904
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
0 2 4 6 8 10 12 14
Ab
sorb
ance
,59
3n
m
Concentration µg/ml
FRAP: TRO calibration curve

96
Figure 4.17: Quercetin (QC) calibration curve in FRAP assay.
y = 0.3053x + 0.4857 r² = 0.9646
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
0 2 4 6 8 10 12 14
Ab
sorb
ance
,59
3n
m
Concentration µg/ml
FRAP: QC calibration curve

97
0 20 40 60
0
20
40
60
80
100
LH
LC
LE
BH
BC
BE
TROLOX
QUERCETIN
concentration(Pg/ml)
%E -
caro
tene
dec
olou
risat
ion
Figure 4.18ȕ-carotene decolourisation (%) of Canarium patentinervium Miq.

98
Gallic Acid calibration curve
y=0.1762x+0.0047, r2=0.9994
0 2 4 6
0.0
0.2
0.4
0.6
0.8
1.0
concentration(Pg/ml)
Abso
rban
ce (O
765
nm)
Figure 4.19: Gallic acid calibration curve in Folin-Ciocalteu assay.

99
Quercetin calibration curve
y=0.009330x-0.002314, r2=0.9992
20 40 60
-0.1
0.0
0.1
0.2
0.3
0.4
0.5
Concentration (Pg/ml)
Absorb
ance (O
430 n
m)
Figure 4.20: Quercetin calibration curve in total flavonoid content determination.

100
Table 4.2: Antioxidant values of Canarium patentinervium Miq.
Extracts
Total phenolic content, Gallic acid equivalent
(mg/g)
Total flavonoid content, Quercetin equivalent (mg/g)
ABTS assay, EC50 ȝJPO
DPPH assay, EC50 ȝJPO
FRAP assay, FRAP value
ȝJPO
ȕ-carotene bleaching
assay, EC50
ȝJPO
Superoxide dismutase
assay IC50ȝJPO
LH 6.61±0.01 18.99±0.02 27.30±0.01 17.45±0.03 2845.00±0.15 9.39±0.05 80.54±0.03
LC 14.28±0.03 12.57±0.04 31.10±0.02 134.00±0.04 1329.00±0.18 21.81±0.04 26.84±0.03
LE 204.97±0.05 125.32±0.03 2.28±0.01 2.93±0.00B 200.00±0.07 6.04±0.02 3.05±0.01
BH 5.19±0.01 1.55±0.02 521.00±0.01 1857.00±0.01 3531.00±0.14 125.00±0.08 238.00±0.04
BC 13.99±0.03 12.14±0.02 34.90±0.02 87.80±0.05 1242.00±0.19 50.40±0.03 20.13±0.02
BE 100.26±0.01 24.57±0.02 0.93±0.01A 2.33±0.02 67.00±0.32 7.04±0.04 8.51±0.07
AA NA NA 1.54±0.06 1.88±0.01 347.00±0.23 NA NA
QC NA NA 0.88±0.03A 2.88±0.02B 86.00±0.24 1.64±0.04C NA
TRO NA NA 0.68±0.02 4.77±0.04 33.00±0.54 1.65±0.03C NA
SOD NA NA NA NA NA NA 1.59±0.04
LH: leaf hexane extract, LC: leaf chloroform extract, LE: leaf ethanol extract, BH: bark hexane extract, BC: bark chloroform extract, BE: bark ethanol extract, AA: ascorbic acid, QC:quercetin, TRO: trolox and SOD: superoxide dismutase Data were obtained from three independent experiments, each performed in triplicates (n=9) and represented as mean ± SD. Values with the same capital letter are not significantly different (P<0.01) according to Tukey multiple comparison test.

101
Table 4.3: The mean r2 values for correlation between total phenolics contents, total flavonoid contents and antioxidant activities (EC50)
ABTS DPPH FRAP BC
Total phenolic contents 0.1410 0.1285 0.5246 0.2287
Total flavonoid contents 0.1388 0.1319 0.3179 0.2146
ABTS na 0.9977*** 0.5398 0.9008**
DPPH 0.9977*** na 0.5029 0.8977**
FRAP 0.5398 0.5029 na 0.4770
BC 0.9008** 0.8977** 0.4770 na
** significant with p<0.01
*** significant with p<0.0001, na- not applicable

102
4.4 Discussion
In vitro antioxidant capacity can be determined by hydrogen atom transfer (HAT)
method and single electron transfer (SET) method [56]. HAT based methods measure
the ability of an antioxidant to scavenge free radical by hydrogen donation to form a
stable compound. SET based methods detect the ability of the antioxidant to transfer
one electron to reduce compound including metals, carbonyls and radicals [70][85]. ȕ-
carotene bleaching assay involves HAT method, FRAP assay involves SET method
while DPPH and ABTS assay involves both method predominantly via SET method
[55][57].
FRAP is the ferric reducing power of antioxidants by the reduction of the ferric ions
to the ferrous ions, which form a blue colored ferrous-tripyridyltriazine complex
(ferric TPTZ) which is detected at 593 nm. Deeper blue colour indicates higher
antioxidant potential [86]. The samples absorbance equivalent to 1 mM FeSO4 was
calculated from equation of linearity of individual sample. Total FRAP value was
determined from the absorbance obtained from samples above using the standard Fe
(II) calibration curve equation (y=0.0105x+0.0136, r2=0.9817). Ethanol extract of
barks displayed significant (p< 0.01) FRAP value ( ȝJPO compared to
ascorbic acid (ȝJPODQGTXHUFHWLQȝJPO
,Q WKH ȕ-carotene/linoleic model, linoleic acid reacts with ROS and O2 to form an
XQVWDEOHSHUR[\UDGLFDOȕ-carotene being an antioxidant will react with this radical to
form stable epoxide causing the bleaching of yellow solution. Competition reaction
occurs with the presence of another antioxidant (sample) to react with the peroxy

103
radical resulting in slower bleaching of solution detected at 470 nm
spectrophotometrically [87]. Ethanol extract of leaves and barks displayed best EC50
value of ȝJPO DQG ȝJPO UHVSHFWLYHO\ Our results are in
agreement to the previous reports which pointed out that ascorbic acid did not show
its antioxidant activity under similar assay [88] Such phenomenon was termed as
³SRODU SDUDGR[´ZKHUHE\ SRODU DQWLR[LGDQWV UHPDLQLQJ LQ WKH DTXHRXV SKDVH RI WKH
emulsion are more diluted in lipid phase and thus, they are less effective [89].
ABTS assay involves the reduction of the blue-green 2,2-azino-bis(3-
thylbenzothiazoline-6- VXOIRQDWH UDGLFDO FDWLRQ $%76ǜ E\ DQWLR[LGDQWV WR LWV
original colourless ABTS form. Greater discolourisation results in lower absorbance
at 734 nm indicating higher antioxidant capacity [75][76]. Ethanol extract of the bark
displayed significant (p<0.01) EC50 ( ȝJPO) compared to ascorbic acid
(ȝJPO).
The DPPH radical has a deep purple colour and absorbs strongly at a wavelength of
550 nm, whereas the yellowish reduction product DPPH2 does not. In DPPH assay
(another SET method), ethanol extract of leaves and barks displayed significantly
lower EC50 value of ȝJPODQGȝJPOUHVSHFWLYHO\FRPSDUHGWR
trolox (EC50 = ȝJPO[90]. The radical scavenging potential against DPPH
organic radical directly depends on the number of hydroxyl groups present in ring B
of flavonoids, with an increase in the number of hydroxyl groups resulting in an
increase in radical scavenging activity [91]. Phenols, amino and thiophenol groups are
commonly known to be the active groups for DPPH scavenging. The mechanism by
which DPPH is scavenged, aids in elucidating the structure-activity relationship

104
(SAR) of the antioxidant and in doing so, may be beneficial in the rational design of
novel flavonoid-derived antioxidants with improved pharmacological profiles [92].
The DPPH and ABTS assays have the same mechanism of action, but, in most cases,
the results obtained from the ABTS assay are higher than those from DPPH assay. It
has been documented that results reported for the ABTS assay do not only take into
account the activity of the parent compound, but also the contribution of reaction
products and other individual compounds on the activity, which is not the case in the
DPPH assay [93][94].
SOD which catalyses the dismutation of the superoxide anion (O2-) into hydrogen
peroxide and molecular oxygen, is one of the most important anti-oxidative enzymes.
Preventive antioxidants, such as SOD are described either as preventing introduction
of initiating radicals or as inhibiting the rate which new chains are set up. The
analytical methods for determination of O2- scavenging capacity employ enzymatic or
nonenzymatic O2- generation systems. In this study the enzymatic system that uses
xanthine oxidase (XOD) to generate O2- radicals was used [57]. XOD was used for
formation of O2- that is the reduced to oxygen by SOD-like activities in samples. The
WST-1 is used as a probe that undergoes competitive reaction with SOD-like samples
to reduce O2- to oxygen forming a formazan coloured dye. Thus higher rate of SOD
activity results in less dye formation and less absorbance. The ethanol extract of the
leaves shows highest SOD-like activity with IC50 RIȝJPO
In the above experiments, the ethanol extract of leaves and barks displayed superior
antioxidant capacities. The EC50 values of the samples were consistently low in SET

105
methods (ABTS, DPPH and FRAP) superior to standard as opposed to HAT method
ȕ-carotene bleaching) assay. The present study demonstrates that Canarium
patentinervium Miq. is a potent source of antioxidants that exhibits its antioxidant
activity predominantly via the SET method. Several reports emphasized on the fact
that there is a positive relationship between total phenolic contents and antioxidant
activity [95][96]. Our study demonstrated the existence of a low positive correlation
between the total phenolic contents and the FRAP values (y = -0.0125x + 2.2564, r2 =
0.5246) and total phenolic contents and ȕ-carotene bleaching values (y = -0.2747x +
52.42, r2 = 0.2287), respectively. Therefore, one could draw an inference that non
phenolic by itself or in synergy with the phenolics and flavonoids impart Canarium
patentinervium Miq its antioxidant properties.
4.5 Conclusion
The present study demonstrates that Canarium patentinervium Miq. is a potent
source of antioxidants that exhibits its antioxidant activity predominantly via the SET
method. Pronounced radical scavenging activity has been reported in plants with
phenolic moieties, the presence of which is common in natural antioxidants. These
phenolic moieties include substances such as tannins, flavonoids, phenolic acids,
tocopherol, and cathecheses. Tannins are, at least in part, responsible for the free
radical scavenging activities working synergistically with other antioxidant substances
[86].

106
CHAPTER 5
ANTI-INFLAMMATORY ACTIVITY
5.1 Introduction
5.1.1 Inflammation and arachidonic system
Over the last 10 years, a significant body of evidence has emerged indicating that
chemically diverse classes of naturally-occurring substances derived from higher
plants are of potential interest for therapeutic interventions in several inflammatory
diseases [97]. Inflammation response is a critical protective reaction to irritation,
injury, or infection, characterised by redness, heat, swelling, loss of function and pain.
Redness and heat result from an increase in blood flow, swelling is associated with
increased vascular permeability, and pain is the consequence of activation and
sensitisation of primary afferent nerve fibres. In normal conditions, these changes in
inflamed tissue serve to isolate the effects of the insult and thereby limit the threat to
the organism [97].
In recent years, roles have been identified for several inflammatory cells and for a
large number of inflammatory mediators in important pathologies not previously
OLQNHG WR LQIODPPDWLRQ VXFK DV $O]KHLPHU¶V GLVHDVH DQG FDUGLRYDVFXODU GLVRUGHUV
including atherosclerosis, as well as cancer [98]. These findings have significantly
increased the importance of understanding the cellular and molecular mechanisms
forming the basis of the inflammatory process and in identifying new targets for the

107
development of innovative and safe therapeutic strategies for the management of
inflammatory diseases. Most plant-derived secondary metabolites are known to
interfere directly or indirectly with the following molecules and/or mechanisms
[98][99]:
1. various inflammatory mediators (eg arachidonic acid metabolites, peptides,
cytokines, excitatory amino acids etc).
2. the production and/or action of second messengers ( such as cGMP, cAMP,
various protein kinases, and calcium among others).
3. the expression of transcription factors such as AP-1, NF-Ƹ% DQG SURWR-
oncogenes (c-jun, c-fos, and c-myc),
4. the expression of key pro-inflammatory molecules such as inducible NO
synthase (iNOS), cyclooxygenase (COX- F\WRNLQHV ,/ ȕ 71)-Į HWF
neuropeptidases and proteases.
In this study we reviewed and investigated the contribution of naturally occurring
substances from higher plants acting on the arachidonic acid pathway. Inflammation
in injured cells is both initiated and maintained by the overproduction of
prostaglandins and leukotrienes, which are produced by separate enzymatic pathways,
viz. the cyclo-oxygenase (COX) and lipoxygenase (LOX) pathways, respectively.
Both the prostaglandins as well as the leukotrienes are biosynthesised on demand
from arachidonic acid, which is a 20-carbon fatty acid, derived from the breakdown of
cell membrane phospholipids by any number of phospholipase A2 (PLA2) isoforms
[100] (Figure 5.1).

108
.
Other HPETEs
lipoxygenases
HETEs
Prostaglandin G2 (PGG2) 5-HPETE 5-HETE
Prostaglandin H2 (PGH2)
Leukotriene A4 (LTA4) LTB4
Prostacyclin Thromboxane A2
(PGI2) (TXA2) Leukotriene C4 (LTC4)
Leukotriene D4 (LTD4)
Leukotriene E4 (LTE4)
PGD2 PGE2 Lipoxin A4 Lipoxin B4
(LXA4) (LXB4)
Figure 5.1: Arachidonic acid metabolites and inflammation [101].
Cell membrane phospholipids
ARACHIDONIC ACID
Phospholipases
Cyclooxygenase 5-Lipoxygenase
Causes vasodilation,
inhibits platlet
aggregation
Causes vasocontriction,
promotes platlet
aggregation
Vasodilation Increased vascular
permeability
Chemotaxis
Vasoconstriction Bronchospasm Increrased vascular permeability Inhibit neutrophil
adhesion and chemotaxis
12-

109
Arachidonic acid is normally stored in membrane-bound phospholipids and released
by the action of phospholipases. Enzymatic conversion of released arachidonic acid
into biologically active derivatives proceeds through one of several routes [99].
Cyclooxygenase converts arachidonic acid to unstable cyclic endoperoxides from
which prostaglandins, prostacyclin and thromboxanes are derived. Formation of the
leukotrienes from arachidonic acid is initiated by the action of 5-lipoxygenase
producing leukotriene A4. Hydrolysis of leukotriene A4, or the incorporation of
glutathione results in the formation of leukotriene B4 and C4, respectively [102]. In
addition, 12- and 15-lipoxygenase can catalyse arachidonic acid conversion and
lipoxins A and B are amongst the possible products. Many of these metabolites of
arachidonic acid feature prominently in the development of inflammation.
Prostaglandin E2 and prostacyclin are potent vasodilators, while leukotriene D4 causes
cellular adhesion, chemotaxis of neutrophils and degranulation. Leukotrienes C4, D4
and E4 contribute to inflammation by increasing vascular permeability. Leukotrienes
are also believed to play an important pathophysiological role in allergic broncho-
constriction of asthma [103].
Through pharmacological intervention in the arachidonic acid cascade various anti-
inflammatory agents have been developed. These include aspirin-like drugs, which
inhibit cyclo-oxygenase. Corticosteroids appear to indirectly inhibit phospholipases
thus preventing release of arachidonic acid. Future progress in this field is likely to
produce drugs which antagonise arachidonic acid derivatives or inhibit the enzymes
involved in their synthesis with greater specificity [99][103].

110
5.1.2 The lipoxygenase system
5-LOX presents either in the cytosol or the nucleus of a resting cell (depending on the
cell), as a soluble enzyme. Upon cellular stimulation, 5-LOX and cytosolic
phospholipase A2 (cPLA2) co-migrate to the nucleus. It is here that cPLA2 liberates
arachidonic acid from the membrane phospholipids [104] (Figure 5.2). Arachadonic
acid is the main substrate for the lipoxygenase enzyme, and is presented to 5-LOX for
metabolism by the 5-lipoxygenase activating protein (FLAP). The metabolism of
arachidonic acid produces leukotrienes and lipoxins via the LOX pathway [105]. 5-
LOX, catalysing the oxidation of arachidonic acid, produces 5(S)-
hydroxyperoxyeicosatetraenoic acid (5-HPETE), a hydroperoxide intermediate, which
undergoes dehydration, resulting in the formation of leukotriene A4 (LTA4).
Enzymatic hydrolysis of LTA4, as well as conjugation with other substances, leads to
the formation of inflammatory mediators [106]. These inflammatory mediators are
responsible for the powerful chemo-attractive effects on the eosinophils, neutrophils
and macrophages, as well as the increased release of pro-inflammatory cytokines by
macrophages and lymphocytes.
Two other pathways in the LOX system, 15-LOX and 12-LOX, are responsible for
the production of lipoxins, the latter having the potential to counteract the pro-
inflammatory effects of the leukotrienes. 5-Lipoxygenase is inhibited by quinones,
hydroxyquinones, and a variety of phenolic compounds, including certain flavonoids
such as, quercetin, kaempferol, morin, myricetin and cirsiliol [107]. At the onset of
the inflammatory process, arachidonic acid is converted to eicosanoids and
leukotriene B4 (LTB4) by LOX [104], which is coupled with the production of

111
prostaglandins and thromboxanes by cyclooxygenase (COX). The leukotrienes
present at the onset of inflammation are also responsible for the maintenance thereof.
Leukotrienes have been identified as mediators of a number of inflammatory and
allergic reactions [103]..
These include rheumatoid arthritis, inflammatory bowel disease, atopic dermatitis,
psoriasis, chronic urticaria, asthma [108] and allergic rhinitis [109][103]. The 5-LOX
pathway has also recently been linked to the development of atherosclerosis,
osteoporosis and certain types of cancers [105]. As a result of the pathophysiological
implications of 5-LOX products and the potential benefits of anti-leukotriene therapy,
different strategies have been employed (targeting PLA2, 5-LOX, FLAP, LTA4
hydrolase and leukotriene C4 (LTC4) synthase with 5-LOX being the ideal and most
promising target [105].
5.1.3 The cyclooxygenase system
Cyclooxygenase (COX) is a key enzyme that catalyzes the transformation of
arachidonic acid to several biologically active eicosanoids, such as prostaglandins
(PGs), thromboxane (e.g., TxA2), and prostacyclin (e.g., PGI2). This enzyme actually
catalyzes two major reactions, as shown in Figure 5.3. Arachidonic acid, the most
important physiological precursor of eicosanoids biosynthesis, can be released from
the membrane phospholipid by activation of phospholipase A2 (PLA2) and/or
phospholipase C (PC). Cyclooxygenase then catalyzes the addition of molecular
oxygen at the C-11 of arachidonic acid to form the 11-peroxy intermediate, following

112
by rearrangement to a cyclic endoperoxide and introduction of a second molecule of
oxygen at C-15 to form prostaglandin G2 (PGG2).
Ligand
Receptor
Figure 5.2: The translocation of 5-lipoxygenase and cytosolic phospholipase A2, upon
cellular stimulation, to the nuclear membrane, followed by substantial generation of
leukotrienes [105]. 5-LOX = 5-lipoxygenase, cPLA2 = cytosolic phospholipase A2, FLAP =
5-lipoxygenase activating protein, PKA = protein kinase A, cAMP = cyclic adenosine
monophosphate, MK2 = kinases, ERK1,2 = extracellular signal-regulated kinase 1,2.
Cell Stimulation
5-LOX
?
Phospholipase C
5-LOX
MAP Kinases ERK 1,2
+ Ca 2+
+ DAG
?
FLAP
cPL
Nucleu
s
Leukotrienes
SH3- Interactions
Antiinflammatory Signal (cAMP)
CELL
p 38 MK2

113
The hydroperoxidase activity reduces PGG2 to generate PGH2 which is the immediate
precursor for production of eicosanoids, such as PGD2, PGE2, PGF2a, PGI2, and TxA2,
through corresponding synthase activities. These arachidonic acid metabolites derived
from PGH2 are important mediators of inflammatory responses, immunological
effects, and tumour development [100].
Cyclooxygenase (COX) exist as three isoforms: cyclooxygenase-1 (COX-1),
expressed in most cells constitutively, inducible cyclooxygenase-2 (COX-2), triggered
by pro-inflammatory stimuli, and COX-3, present mainly in the cerebral cortex and
human heart [110][111] . COX-1 is a constitutive form of the enzyme that has been
linked to the production of physiologically important prostaglandins that may play a
role in homeostasis (gastric, renal, etc). COX-2 exists in a constitutive form in kidney
and brain and is inducible by cytokines and growth factors. Induction of the COX-2 is
linked to inflammatory cell types and tissues and COX-2 is believed to be the target
for the anti-inflammatory actions of NSAIDs. Thus there is an on-going effort to
identify compounds that will inhibit COX-2 in preference to COX-1 since such an
agent may be safer and perhaps more efficacious [112].
Despite of the undesired side effects such as increased cardiovascular risk of COX-2-
selective inhibitors, much research spanning many years has been focused on finding
COX-2 selective inhibitors, owing to the remarkable reduction of adverse
gastrointestinal and renal effects associated with conventional non-steroidal anti-
inflammatory drugs (NSAIDs) as well as to their potential therapeutic benefits in
several diseases, including certain types of cancer [113]. Thus, it is of value to study
cyclooxygenase inhibitors.

114
Phospholipase A2
Arachidonic acid
Constitutive & Constitutive Inducible
PGG2
Hydroperoxidase
PGH2
Prostanoids
Receptors
SOME BIOLOGICAL EFFECTS
Figure 5.3: Cyclooxygenase and arachidonic acid metabolism.
Membrane phospholipids
COX-2 COX-1
Cell specific isomerases
PGI2 TXA2 PGE2 PGD2 PGFĮ
Prostacylin synthase PGE Synthase 3*)Į6\QWKDVH
Thromboxane Synthase
PGD Synthase
IP 73Į73ȕ EP1, EP2,
EP3, EP4
DP1, DP2 )3Į)3ȕ
Vasodilator Antiaggregant
Vasoconstrictor Proaggregant
Vasodilator Neuroprotective Neurodestructive
Vasodilator Anti- inflammatory Proapoptotic
Vasodilator Antiaggregant

115
5.1.4 Dual inhibitors
Conventional pharmacological management of inflammatory disease like
osteoarthritis involves treatment with non-steroidal anti-inflammatory drugs
(NSAIDs) or selective COX-2 inhibitors that block the formation of PGs without
modulating 5-LOX enzyme activity. Inhibition of one or both of the COX enzymes
PD\³VKXQW´$$PHWDEROLVPGRZQWKH-LOX pathway, which can aggravate toxicity
associated with the lack of PGs and excess production of LTs. For example, NSAID-
induced gastric ulcers have been shown to have high concentrations of LTB4 in their
walls, which attracts leukocytes to the stomach and may contribute to ulceration
[114][115].
Recently, reports have appeared regarding so-FDOOHG ³GXDO LQKLELWRUV´ DJHQWV WKDW
inhibit not only COX-1 and COX-2, but also 5- LOX [116][117][118][119]. These
agents with anti-oxidative properties may be particularly effective for managing the
metabolic processes underlying inflammatory conditions and reducing both gastric
and cardiovascular side effects by balancing AA metabolism in the body. This dual
inhibition activity will be investigated for this plant.
5.1.5 Anti-inflammatory potential of Canarium L.
The essential oil of Canarium schweinfurthii Engl. collected from the region of
Cameroon inhibited the enzymatic activity of lipoxygenase with an IC50 value of 62.6
ppm [19]. However in a separate study of the same species collected from central
African region did not show any activity in the cotton pellet induced granuloma

116
formation experiment [13]. This may suggest possible evidence of variety between
secondary metabolites constituents according to regions. The essential oil of
Canarium schweinfurthii Engl. at a dose of 1, 2 and 3 mg/ kg i.p. displayed potent
analgesic effect in the acetic acid-induced writhing and hot plate experiments [13].
5.2 Materials and methods
5.2.1 Anti-inflammatory determination assays
The anti-inflammatory activity of the plant extracts was determined using the in vitro
5- LOX assay. This assay measures the inhibitory activity against the 5-LOX enzyme,
which is a key enzyme in the metabolism of arachidonic acid that is responsible for
the formation of leukotrienes (which play a pivotal role in the pathophysiology of
chronic inflammatory and allergic diseases) as first determined by Sircar et al. [120]
and later modified by Evans [121].
There are three ways to measure COX activity, one is to measure the final product of
the reaction, PGE2; another is to measure directly the oxygen uptake that occurs
during the first step of the reaction using the oxygen sensor and third is to measure the
second reaction (peroxidase function) spectrophotometrically [112]. The peroxidase
co-substrate oxidation assay can detect cyclooxygenase inhibitors which may directly
interfere with enzyme activity or scavenge the free radical intermediates to block the
reaction process. However, the oxygen consumption assay is mainly reflective of
inhibitors which block the interaction of enzyme and arachidonic acid. We currently

117
describe a peroxidase co-substrate oxidation assay system adapted to a 96-well plate
format. This requires less enzyme and reaction material and yields savings in time and
money. The peroxidase co-substrate oxidation assay is useful as a pre-screening
method [100][112].
5.2.2 The 5-LOX inhibition assay
5.2.2.1 Principle of the assay
The 5-LOX assay was conducted according to the method described by Baylac &
Racine [122] with some modifications. Lipoxygenases (LOX) are dioxygenases that
catalyse the addition of molecular oxygen to polyunsaturated fatty acids containing a
1,4-pentadiene group. The 5-LOX enzyme thus converts its substrate, arachidonic
acid, to the conjugated diene product 5-hydroxy- 6,8,11,1-eicosatetraenoic acid (5-S-
HETE), which, in turn, is converted to LTA4, and then to LTB4, by LTA4 hydrolase.
For the purposes of this experiment, linoleic acid was used as the substrate, as it
shares a high degree of structural similarity to arachidonic acid (containing the 1,4-
pentadiene group in question), it is far easier to handle as well as having a stronger
affinity for the 5-LOX enzyme resulting in greater UV absorbance readings [123].
The experiment specifically determines increases in absorbance at 234 nm as a result
of the formation of conjugate double bonds in linoleic acid hydroperoxide (from a
1,4-diene to a 1,3-diene), as used in the biochemical evaluation of the LOX pathway
of soybean plants submitted to wounding [124].

118
5.2.2.2 Protocol
5-LOX enzyme (human recombinant from Calbiochem) was used. Ice-cold buffer
(potassium phosphate) at 4 oC was mixed with 100 U of the thawed enzyme. Twenty
microliters of samples dissolved in DMSO was plated out in triplicate in a 96-well
microtiter plate at different concentrations, followed by 160 µl of 0.1 M potassium
phosphate buffer (pH 6.3) maintained at 25 oC and 20 µl of enzyme solution. Mixture
was agitated and 10 µl of linoleic acid was added and incubated for 10 mins at 25 o C.
Absorbance was recorded at 234 nm using Thermo Scientific Varioskan Flash
microtiter plate reader, linked to a computer equipped with (SkanIt Software 2.4.3). 5-
Lipoxygenase is known to catalyse oxidation of unsaturated fatty acids containing 1-4
diene and the modification of linoleic acid (1-4-diene into 1- 3-diene) can be detected
at 234 nm. Percentage inhibition of enzyme was determined by comparison of rates of
reaction of samples relative to blank sample using the formula (E-S)/E x 100, where E
is the activity of enzyme without test sample and S is the activity of enzyme with test
sample. The experiments were done in triplicate. Nordihydroguaiaretic acid (NDGA)
was used as positive control.
5.2.3 The peroxidase endpoint assay for COX-1 and COX-2
5.2.3.1 Principle of the assay
The COX-1 and COX-2 peroxidase endpoint assay was conducted according to
method described by Gierse et al. [112]. The peroxidase endpoint assay system is
based on sequential reactions of the cyclooxygenase (generating PGG2) and the

119
peroxidase (converting PGG2 to PGH2) activities. Its success relies on the peroxidase
co-substrate being transformed to a comparatively stable chromophore in a
predictable stoichiometric relationship to the reduction of PGG2. N,N,N¶,N¶-
tetramethyl-p-phenylenediamine (TMPD) is a convenient co-substrate for use in the
assay.
5.2.3.2 Protocol
COX-1 and COX-2 enzyme from Calbiochem was used. Twenty microliters of
sample dissolved in DMSO was plated in triplicates at different concentrations in a
96-well microtiter plate followed by 20 µl of enzyme solution. Then, 160 µl of
endpoint assay mix was added and incubated for 10 mins at room temperature.
Absorbance was recorded at 611 nm using Thermo Scientific Varioskan Flash
microtiter plate reader, linked to a computer equipped with (SkanIt Software 2.4.3).
Percentage inhibition of enzyme was determined by comparison of rates of reaction of
samples relative to blank sample using the formula (E-S)/E x 100, where E is the
activity of enzyme without test sample and S is the activity of enzyme with test
sample. Indomethacin was used as positive control.
5.2.4 Statistical analysis
Concentration±response curves were calculated using the Prism software package
5.00 for Windows, GraphPad Software, San Diego California USA,
www.graphpad.com (GraphPad, San Diego, USA) and data were reported as mean
and SD values obtained from a minimum of three determinations. Non linear best fit

120
was plotted with SD and 95% confidence interval. All data were expressed as mean ±
standard deviation. SD was computed but values were not shown in the graphs via
this software. Data were analyzed using one way Anova followed by Tukey test
using GraphPad Prism5 software. A significant difference was considered at the level
of P < 0.01.
5.3 Results
5.3.1 The 5-LOX inhibition assay
In the 5-LOX assay, chloroform extract of the barks displayed potent enzyme
inhibition (IC50=29.53±0.03 µg/ml) which was close to nordihydroguaiaretic acid
(IC50=29.19±0.02 µg/ml). Ethanol extract of leaves and barks follow through with
IC50=49.66±0.02 µg/ml and IC50=59.06±0.07 µg/ml respectively. Thus the potency of
activity against 5-LOX was BC > LE > BE (Table 5.1.). According to the 5-LOX
enzyme inhibition activity measurement guide by Kamatou et al. [94], [IC50 <30
µg/ml: good activity; 30 < IC50 <80 µg/ml : moderate activity; IC50 >80 µg/ml : poor
activity] chloroform extract of barks of Canarium patentinervium Miq displays good
5-LOX enzyme inhibition activity followed by moderate activity by ethanol extract of
leaves and barks.

121
5.3.2 The peroxidase endpoint assay for COX-1 and COX-2
In the COX assay, only ethanol extract of leaves and barks exhibited IC50 below 100
µg/ml. Ethanol extract of leaves had superior COX-1 inhibition (IC50 = 0.60±0.01
µg/ml) compared to COX-2 inhibition (IC50 = 1.07±0.01 µg/ml) (Figure 5.5) whereas
the barks had superior COX-2 inhibition (IC50 = 9.39±0.03 µg/ml) as opposed to
COX-1 (IC50 = 11.41±0.03 µg/ml) (Figure 5.4) .
5.4 Discussion
These assay measures the inhibitory activity against 5-LOX and COX enzymes. At
the onset of the inflammatory process, arachidonic acid is converted to eicosanoids
and leukotriene B4 (LTB4) by LOX [104], which is coupled with the production of
prostaglandins and thromboxanes by cyclo-oxygenase (COX). 5-LOX is the key
enzyme in the metabolism of arachidonic acid that is responsible for the formation of
leukotrienes which play a pivotal role in the pathophysiology of chronic inflammatory
and allergic diseases. Lipoxygenase catalysing the oxidation of arachidonic acid,
produces 5(S) - hydroxyperoxyeicosatetraenoic acid (5-HPETE), a hydroperoxide
intermediate, which undergoes dehydration, resulting in the formation of leukotriene
A4 (LTA4). Enzymatic hydrolysis of LTA4, as well as conjugation with other
substances, leads to the formation of inflammatory mediators [106]. These
inflammatory mediators are responsible for the powerful chemo-attractive effects on

122
0 20 40 60 80 100
0
20
40
60
80
100
COX-1
COX-2
concentration(Pg/ml)
% inhib
itio
n o
f C
OX
Figure 5.4: COX inhibition by BE.

123
0 2 4 6
0
20
40
60
80
100
COX-1
COX-2
% in
hibi
tion
of C
OX
concentration(Pg/ml)
Figure 5.5: COX inhibition by LE.

124
Table 5.1.Anti-inflammatory values of Canarium patentinervium Miq.
Extracts Anti-inflammatory assay 5-LOX, IC50
ȝJPO
COX-1 inhibition assay IC50ȝJPO
COX-2 inhibition assay IC50ȝJPO
COX-1/COX-2 ratio
LH 206.00±0.02 >100 >100 NA
LC 104.69±0.04 >100 >100 NA
LE 49.66±0.02 0.60±0.01 1.07±0.01 0.56
BH 110.07±0.04 >100 >100 NA
BC 29.53±0.03 >100 >100 NA
BE 59.06±0.07 11.41±0.03 9.39±0.03 1.22
NDGA 29.19±0.02 NA NA NA
INDO NA 0.29±0.04 0.26±0.03 1.11
LH: leaf hexane extract, LC: leaf chloroform extract, LE: leaf ethanol extract, BH: bark hexane extract, BC: bark chloroform extract, BE: bark ethanol extract, AA: ascorbic acid, QC: quercetin, TRO: trolox and INDO: indomethacin. Data were obtained from three independent experiments, each performed in triplicates (n=9) and represented as mean ± SD. Values with the same capital letter are not significantly different (P<0.01) according to Tukey multiple comparison test.

125
the eosinophils, neutrophils and macrophages, as well as the increased release of pro-
inflammatory cytokines by macrophages and lymphocytes [104].
Other inflammatory mediators such as histamine and immunoglobulin E, cause
spasms in the smooth muscle of bronchi and blood vessels, playing an eminent role in
asthmatic attacks. As leukotrienes thus play a major role in the pathophysiology of
chronic inflammatory diseases, it has been suggested that 5-LOX inhibitors may thus
be useful in the treatment of various conditions [125]. The 5-LOX pathway has also
been implicated in cardiovascular disease, including atherosclerosis, stroke,
myocardial infarction and the weakening of large artery walls and the formation of
aneurysms [126][105]. The presence of the 5-LOX pathway, the production of
leukotrienes and presence of the enzymes concerned, as well as leukotriene receptors
is expressed in diseased tissue. Genetic studies have been carried out on mice linking
the 5-LOX pathway to atherosclerosis, and population genetic studies involving
humans, correlates genotypes of 5-LOX, FLAP and LTA4 hydrolase to cardiovascular
disease, as shown in studies by Dwyer et al. [127] and Helgadottir et al.
[128][129][130].
The results obtained from the leaves and barks of the plant indicate good anti-
inflammatory activity via 5-LOX inhibition cascade in the chloroform extract of the
barks (IC50=29.53±0.0 ȝJPOZKHQFRPSDUHG WR1'*$ (IC50=29.19±0.02 ȝJPO
Moderate activity was shown in BE and LE while poor activity was depicted in LH
and BH in accordance with measure of activity by Kamatou et al. [94], [IC50 <30
µg/ml: good activity; 30 < IC50 <80 µg/ml : moderate activity; IC50 >80 µg/ml : poor
activity].

126
COX (prostaglandin H2 synthase) inhibition is the mechanism of action of most
NSAIDS. COX-1 is a constitutive form of the enzyme that has been linked to the
production of physiologically important prostaglandins that may play a role in
homeostasis (gastric, renal, etc). COX-2 is a form of the enzyme that is inducible by
cytokines and growth factors. Induction of COX-2 is linked to inflammatory cell
types and tissues. There is an ongoing effort to identify compounds that might inhibit
COX-2 in preference to COX-1 since such an agent may be safer and perhaps more
efficacious. In this study, COX activity is measured by utilizing peroxidase activity
and the electron donor TMPD, which turns blue upon reduction as a co-substrate.
Arachidonic acid is used as a substrate which must first be converted to
hydroperoxide thus yielding an indirect measure of COX activity. This assay has been
noted to be a high-thorough put method [112].
In this study, only the ethanol extract of leaves and barks showed inhibition against
COX-1 and COX-2 enzymes with IC50 below 100 µg/ml. Ethanol extract of leaves
had superior COX-1 inhibition (IC50 = 0.60±0.01µg/ml) compared to COX-2
inhibition (IC50 = 1.07±0.01 µg/ml) whereas the barks had superior COX-2 inhibition
(IC50 = 9.39±0.03 µg/ml) as opposed to COX-1 (IC50 = 11.41±0.03 µg/ml).
In order to determine the inhibitory selectivity on COX-1 and COX-2 enzymes, the
COX-1/COX-2 ratio was applied. According to Burnett et al. [131], A COX-1
selective inhibitor will have ratio <1, whereas a COX-2 selective inhibitor will have
ratio of > 1. In this study ethanol extract of bark was COX-2 selective (ratio 1.22)
while the leaves were COX-1 selective (ratio 0.56). The COX-2 selective behaviour

127
of the ethanol extract of the barks was similar to positive control indomethacin (ratio
1.11). This indicates the presence of lead compound that would have a more potent
COX-2 selectivity than indomethacin in the ethanol extract of barks. In view of dual
inhibition activity, the ethanol extract of barks seemed to have moderate inhibition
against 5-LOX and good inhibition against COX enzymes (selective COX-2).
Chloroform extract of the bark though had good 5-LOX inhibition but did not have
activity against COX enzymes below 100 µg/ml.
5.5 Conclusion
A combination of anti-inflammatory and antioxidant activity constitutes a good
indication on potential anti-inflammatory activity of a drug [132][133]. Keeping this
in mind the ethanol fraction of the plant warranted further studies. The pathway
mechanism of the dual inhibition of the fraction against 5-LOX and COX needs to be
investigated. However it would be interesting to investigate the bioactive component
of the chloroform extract of barks as it exhibited an activity similar to our positive
control. The clinical significance of these data is quite clear that they support a role
for Canarium patentinervium Miq. (Burseraceae Kunth.) as a source of lead
compounds in the management of inflammatory diseases.

128
CHAPTER 6
ANTI-ACETYLCHOLINESTERASE ACTIVITY
6.1 Introduction
6.1.1 Pathophysiology of $O]KHLPHU¶VGLVHDVH
Neurodegenerative disease is a term applied to a variety of conditions arising from a
chronic breakdown and deterioration of the neurons, particularly those of the central
nervous system [134] $O]KHLPHU¶V GLVHDVH $' ZDV ILUVW GHVFULEHG LQ E\ D
Bavarian neuropsychiatrist Alois Alzheimer [135]. It is a complex, multifactoral,
progressive, neurodegenerative disease primarily affecting the elderly population and
is estimated to account for 50 ± 60 % of dementia cases in persons over 65 years of
age [136]. The pathophysiology of AD is complex and involves several different
biochemical pathways.
The first neurotransmitter defect discovered in AD involved acetylcholine (ACh),
which plays an important role in memory and learning. In the CNS, ACh stimulation
of the nicotinic receptors appears to be associated with cognitive function. Normally,
ACh is stored in the nerve terminals, in structures called vesicles and is released from
the nerve endings when the nerve terminal is depolarized, thereby entering the
synapse and binding to the receptor [137]. However, in patients with AD, the ACh
which is released has a very short half-life due to the presence of large amounts of the
enzymes; acetylcholinesterase (AChE) and butyrylcholinesterase (BChE), which are

129
both present in the brain and are detected among neurofibrillary tangles and neuritic
plaques [138][139]. These enzymes hydrolyse the ester bond in the ACh molecule,
leading to loss of stimulatory activity. Approaches to enhance cholinergic function in
AD have included stimulation of cholinergic receptors or prolonging the availability
of ACh released into the neuronal synaptic cleft by use of agents which restore the
level of acetylcholine through inhibition of both AChE and BChE.
Recently, Hodges [140] demonstrated that the inhibition of AChE holds a key role not
only to enhance cholinergic transmission in the brain but also to reduce the
DJJUHJDWLRQ RI ȕ-amyloid and the formation of the neurotoxic fibrils in AD.
Therefore, AChE and BChE inhibitors have become remarkable alternatives in
treatment of AD [139]. Existing anticholinesterase drugs (example, tacrine, donepezil,
physostigmine, galantamine and heptylphysostigmine) for the treatment of dementia
are reported to have several dangerous adverse effects such as hepatotoxicity, short
duration of biological action, low bioavailability, adverse cholinergic side effects in
the periphery and a narrow therapeutic window [141][142]. This necessitates the
interest in finding better AChE inhibitors from natural resources.
The history of drug discovery has shown that plants contain active compounds that
have become new sources to investigate for the pharmaceutical industry. Plant
constituents may not only act synergistically with other constituents from the same
plant but may also enhance the activity of compounds or counteract toxic effects of
compounds from other plant species [143]. In traditional practices, numerous plants
have been used to treat cognitive disorders, including neurodegenerative diseases and
different neuropharmacological disorders [144].

130
6.1.2 Acetylcholine and its functions
Acetylcholine (ACh), a neurotransmitter in the brain plays a critical role in the
function of learning and memory. ACh is synthesized from acetyl-CoA and choline
by choline acetyltransferase, and is released into the synaptic cleft which then is
hydrolyzed by AChE to become choline and acetic acid (Figure 6.1). Choline is taken
up again into the presynaptic neurons for use in ACh synthesis. Free choline within
the nerve terminal is acetylated by a cytosolic enzyme, choline acetyltransferase
(CAT), the source of the acetyl groups being acetyl-CoA [141].
The rate-limiting process in the Ach synthesis appears to be the choline transport, the
activity of which is regulated according to the rate at which ACh is released. ACh has
functions both in the peripheral nervous system (PNS) and in the central nervous
system (CNS) as a neuromodulator [141]. Its receptors have very high binding
constants. In the peripheral nervous system, acetylcholine activates muscles, and is a
major neurotransmitter in the autonomic nervous system. In the central nervous
system, ACh and the associated neurons form a neurotransmitter system, the
cholinergic system, which tends to cause anti-excitatory actions. Cholinesterase is
present in the presynaptic nerve terminals and Ach is continually being hydrolysed
and re-synthesised [145][146].
6.1.3 Distribution and function of cholinesterase
There are two distinct types of cholinesterase both belonging to the family of serine
hydrolases, namely acetylcholinesterase (AChE) and butyrylcholinesterase (BChE),

131
related closely in molecular structure but differing in their distribution, substrate
specificity and functions [145].
AChE is widely distributed in the central nervous system (CNS) and the peripheral
nervous system AChE is one of the most efficient enzymes of the nervous system
which is bound to the basement membrane in the synaptic cleft at cholinergic
synapses, where its function is to hydrolyse the released transmitter. AChE is quite
specific to acetylcholine (Ach). BChE has a widespread distribution, being found in
tissues such as liver, skin, brain and gastrointestinal smooth muscles, as well as
soluble form in the plasma. It is not particularly associated with cholinergic synapses
and has broader substrate specificity than AChE. It hydrolyses butyrylcholine more
rapidly than ACh [145][147].
6.1.4 Cholinesterase inhibitors
The most important drugs that enhance cholinergic transmission act either by
inhibiting cholinesterase or by increasing acetylcholine release by AChE. The choline
released in the process is reused in synthesizing new ACh. Inhibition of AChE increases the
amount of ACh available for neurotransmission.
There are three main effects of cholinesterase inhibition:
x effects on autonomic cholinergic synapses
x effects on neuromuscular junction
x effects on the central nervous system (CNS)

132
aptic cell
Na+ Choline
Transporter
f
AChE +
Figure 6.1: Cholinergic neurotransmission. The neurotransmitter Ach is synthesised in
presynaptic cholinergic neurons by choline acetyltransferase (CAT or ChAT). The process
entails transfer of an acetyl group from acetyl-coenzyme A to choline. Until needed, the ACh
molecules are stored in discrete vehicles at the ends of the presynaptic neurons. Arrival of a
nerve impulse triggers the release of Ca2+ ions, which activate actin microfilaments that in
turn pull the storage vesicles into position for ACh release. In a single event the vesicles
empty their contents into the synaptic cleft. Most of these molecules bind to cholinergic
receptors on adjacent postsynaptic neurons. Any that remain unbound are rapidly hydrolysed
by AChE. The choline released in the process is reused in synthesizing new ACh. Inhibition
of AChE increases the amount of ACh available for neurotransmission.
Presynaptic
cell
Choline
ACh
ACh
ChAT
ACh
Acetyl CoA
+
Choline
Acetic acid
ACh
Transporter
Postsynaptic cell
Ach receptors

133
Autonomic effects include bradycardia, hypotension, excessive secretions,
bronchoconstriction, gastrointestinal hypermotility and decrease of intraocular
pressure. This inhibition can be useful in the treatment of glaucoma that has increased
intraocular pressure. Drugs that have been used for this condition are physostigmine
and ecothiopate as eye drops. Neuromuscular action causes muscular fasciculation
and increased twitch tension and can produce depolarisation block [147] .
The clinical uses of this action would be in myasthenia gravis where transmission
fails due to too few ACh receptors due to immunological reasons, and cholinesterase
inhibition improves transmission. Drugs targeting this action would be neostigmine
and pyridostigmine taken orally. Cholinesterase inhibition at CNS has been used to
alleviate the cholinergic deficiency associated with Alzheimers disease (AD).
Although the overall AChE activity is reduced, it is increased in the neuritic plaque
and neurofibrillary tangles at the early stages of a AD patient brain [146]. It has been
VXJJHVWHG WKD\ $&K(PD\ SURPRWH DJJUHJDWLRQ RI $EHWD ȕ-amyloid) into a more
toxic amyloid form. Thus inhibiting AChE activity might increase ACh
neurotransmission in the synaptic cleft of the brain and diminish the Abeta burden
resulting in improved cognitive function and alleviating the process of amyloid
deposition. Among several existing hypotheses explaining the origin of AD, including
the cholinergic, tau and amyloid theories [148][149], the cholinergic one is most
studied. Majority of drugs on the market are thus AChE inhibitors like donepezil,
tacrine, rivstigmine and galathamine [147].

134
6.1.5 Plants as potential anticholinesterase inhibitors
In traditional practices, numerous plants have been used to treat cognitive disorders,
including neurodegenerative diseases and different neuropharmacological disorders.
Ethnopharmacological approach and bioassay-guided isolation have provided a lead
in identifying potential AChE inhibitors from plant sources, including those for
memory disorders. A variety of plants has been reported to show AChE inhibitory
activity and so may be relevant to the treatment of neurodegenerative disorders such
as AD [144].
Majority of studies have focused on the anticholinesterase alkaloids, such as physostigmine
and galantamine. So far, more than 35 alkaloids have been reported to have AChE inhibitory
activity [144]. The other major classes of compound reported to have such activity are the
terpenoids, glycosides and coumarins. Plants belonging to families Acanthaceae,
Apocynaceae, Amaryllidaceae, Angelicae, Araceae, Asclepiadaceae, Berberidaceae,
Buxaceae, Combretaceae, Compositae, Coniferae, Cyperaceae, Ebenaceae, Ericaceae,
Euphorbiaceae, Fumariaceae, Gentianaceae, Guttiferae, Lamiaceae, Leguminosae, Lilliaceae,
Lycopodiaceae, Malvaceae, Magnoliaceae, Menispermaceae, Molluginaceae, Moraceae,
Musaceae, Nelumbonaceae, Papaveraceae, Piperaceae, Rubiaceae, Rutaceae, Sapotaceae,
Solanaceae and Tamaricaceae have been reported to have AChE inhibitory potential [149].
The present study was undertaken to screen the anti-acetylcholinesterase potential of
Canarium patentinervium Miq. from family of Burseraceae. No anti-
acetylcholinesterase studies have been reported on this species to date.

135
6.2 Materials and methods
6.2.1 Anti-acetylcholinesterase determination assays
6.2.1.1 Principle of the assay
Acetylcholinesterase (AChE) inhibitory activity was measured by slightly modifying the
spectrophotometric method developed by Ellman et al. [150]. Acethylcholine iodide which
is the substrate is hydrolysed by acethylcholinesterase enzyme resulting in the
formation thiocholine that reacts with dithiobisnitrobenzoate ion (DTNB) resulting in
yellow 5-thio-2-nitrobenzoate anion observed at a wavelength of 412 nm.
acethylcholine thiocholine + acetate
Thiocholine + dithiobisnitrobenzoate yellow color
This is accomplished by the continuous reaction of the thiol ZLWK ¶ dithiobis-2-
nitrobenzoate ion (I) to produce the yellow anion of 5-thio-2-nitro-benzoic acid (II).
H2O + (CH3)3N+CH2CH2SCOCH3 (enzyme) (CH3)3N
+CH2CH2S-
+ CH3+COO- + 2H+
(CH3)3N+CH2CH2S
- + RSSR (I) (CH3)3N+CH2CH2SSR
+ RS- (II)
R =
The reaction with the thiol has been shown to be sufficiently rapid so as not to be rate limiting
in the measurement of the enzyme, and in the concentrations used does not inhibit the
enzymic hydrolysis.

136
6.2.1.2 Protocol
5,5´-Dithio-bis(2-nitrobenzoic) acid (DTNB), galanthamine, nordihydroguairetic acid
(NDGA) and electric eel acetylcholinesterase (AChE) (Type-VI-S, EC 3.1.1.7) purchased
from Sigma AldrichR was used. Samples were dissolved in dimethyl sulfoxide (DMSO,
R&M) prior to assay and serial dilutions were done. In brief, 130 µL of 0.1 mM sodium
phosphate buffer (pH 8.0), 20 µ/ RI '71% ȝ/ RI WHVt solution and 20 µL of AChE
solution were added by multichannel automatic pipette (Eppendorf, Germany) in a 96-well
microplate and incubated for 15 min at 25 °C. The reaction was then initiated with the
addition of 10 µL of acetylthiocholine iodide. The hydrolysis of acetylthiocholine iodide was
monitored by the formation of the yellow 5-thio-2-nitrobenzoate anion as a result of the
reaction of DTNB with thiocholines, catalyzed by enzymes at a wavelength of 412 nm
utilizing a 96-well microplate Thermo Scientific Varioskan Flash microtiter plate
reader, linked to a computer equipped with (SkanIt Software 2.4.3).
Percentage inhibition of AChE was determined by comparison of rates of reaction of
samples relative to blank sample (ethanol in phosphate buffer pH=8) using the
formula (E-S)/E x 100, where E is the activity of enzyme without test sample and S is
the activity of enzyme with test sample. The experiments were done in triplicate.
Galanthamine was purchased from Sigma (St. Louis, MO, USA) and used as
reference.

137
6.2.2 Statistical analysis
Concentration±response curves were calculated using the Prism software package
5.00 for Windows, GraphPad Software, San Diego California USA,
www.graphpad.com (GraphPad, San Diego, USA) and data were reported as mean
and SD values obtained from a minimum of three determinations. Non-linear best fit
was plotted with SD and 95% confidence interval. All data were expressed as mean ±
standard deviation. SD was computed but values were not shown in the graphs via
this software. Data were analyzed using one way Anova followed by Tukey test using
GraphPad Prism5 software. A significant difference was considered at the level of P <
0.01.
6.3 Results
In the anti-acethylcholinesterase assay, chloroform extract of the barks displayed the
best activity (IC50 = 88.59±0.14 ȝJPO as opposed to galanthamine (IC50 = 0.74±0.06
ȝJPO)LJXUH7KHHWKDQROH[WUDFWRIEDUNVDQGOHDYHVIROORZWKURXJKZLWKIC50
= 186.00±0.15 ȝJPODQGIC50 = 201.24±0.15 ȝJPOUHVSHFWLYHO\. Hexane extracts of
bark and leaves and the chloroform extract of leaves had the lowest enzyme inhibition
activity (IC50 = 570.00±0.08 ȝJPO IC50 = 842.00±0.25 ȝJPO DQG IC50 =
1780.00±0.24 ȝJPOUHVSHFWLYHO\. Thus the potency of activity against AChE was BC
> BE > LE > BH >LH > LC (Table 6.1).

138
Extracts Anti-acetylcholinesterase assay, IC50ȝJPOLH 842.00±0.25
LC 1780.00±0.24
LE 201.34±0.15
BH 570.00±0.08
BC 88.59±0.14
BE 186.00±0.15
Galantamine 0.74±0.06
LH: leaf hexane extract, LC: leaf chloroform extract, LE: leaf ethanol extract, BH: bark hexane extract, BC: bark
chloroform extract, BE: bark ethanol extract. Data were obtained from three independent experiments, each
performed in triplicates (n=9) and represented as mean ± SD.Values with the same capital letter are not
significantly different (P<0.01) according to Tukey multiple comparison test.
Table 6.1. Anti-acetylcholinesterase values of Canarium patentinervium Miq.
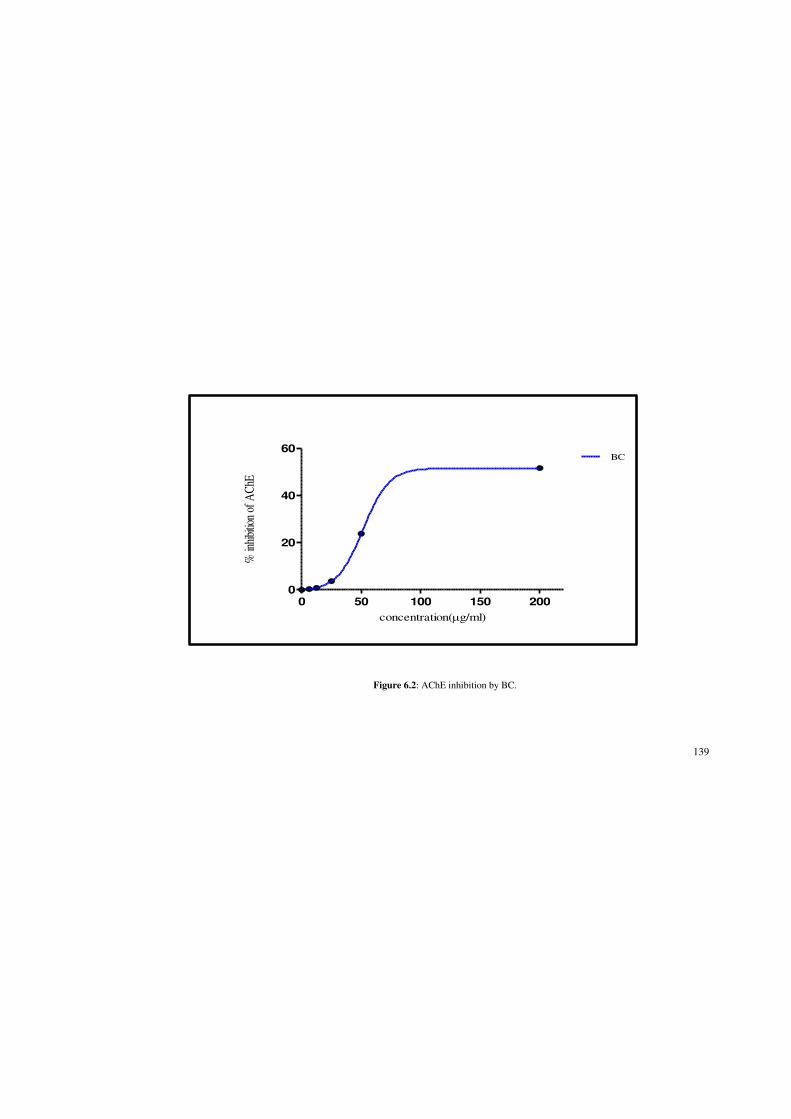
139
0 50 100 150 200
0
20
40
60BC
concentration(Pg/ml)
% in
hibi
tion
of A
ChE
Figure 6.2: AChE inhibition by BC.

140
0 2 4 6
0
20
40
60
80
100
GL
concentration(Pg/ml)
% in
hibi
tion
of A
ChE
Figure 6.3: AChE inhibition by galanthamine.

141
6.4. Discussion
This assay measures the inhibition properties against AChE, which is the key enzyme
in the hydrolysis of acetylcholine that is responsible for muscle and organ relaxations.
Acetylcholinesterase inhibitors are used medicinally to treat myasthenia gravis to
LQFUHDVHQHXURPXVFXODU WUDQVPLVVLRQDQG WR WUHDW$O]KHLPHU¶VGLVHDVH GHILFLHQF\ LQ
the production of acetylcholine). Furthermore, oxidative and inflammatory processes
are among the pathological features associated with the central nervous system in
AlzheiPHU¶VGLVHDVH [151]. The brain of patients suffering from AD is said to be under
oxidative stress as a result of perturbed ionic calcium balances within their neurons
and mitochondria [152][153]. Accumulating evidence suggests that oxidative damage
to neurons plays an important role in the AD pathogenesis [154].
Thus, efforts to reduce oxidative injury may prove beneficial in retarding or
preventing the onset and progression of AD in patients. In this study (Chapter 4), BC
had displayed good antioxidant potential via enzymatic and non-enzymatic assays.
This in addition to its anti-acetylcholinesterase activity exhibited in this chapter
suggest that chloroform extract of the barks hold lead compounds that inhibit
acetylcholinesterase activity as well as reduce the oxidative stress with possible
neuroprotective effects. Moreover, there is evidence that AChE inhibitors have an
anti-inflammatory role through action against free radicals and amyloid toxicity, as
well as through decreasing release of cytokines from activated microglia in the brain
and blood [153].
There is an established link between the cholinergic system and inflammation as
acetylcholine, the principle neurotransmitter, is reported to attenuate the release of

142
cytokines in the parasympathetic anti-inflammatory pathway by which the brain
modulates systemic inflammatory responses to endotoxin [155]. Destruction of
neurons due to inflammation around Abeta plaques is thought to be a major factor in
the pathogenesis of AD [2]. In Chapter 5 it has been found that BC had an anti-
inflammatory action against 5-LOX (IC50 = 29.53±0.03 µg/ml) against the positive
control NDGA ((IC50 = 29.19±0.02 µg/ml). This in relation to the anti-
acetylcholinesterase activity exhibited herein suggest that BC may also acts as an anti-
inflammatory agent related to AD pathogenesis.
Because of the unclear pathogenesis of AD, there have been several hypothesis
associated with the disease such as amyloid-ȕ peptide-containing plaque formation,
excess metal ions, oxidative stress, and reduced acetylcholine levels. As proposed by
Zhang et al. [156], finding more than one approach with multifunction for AD
treatment draws attention to researchers. Thus, BC that exhibits good antioxidant
potential, anti-inflammatory and anti-acetylcholinesterase action warrants further
study for isolation of its bioactive principle.
6.5 Conclusion
The creation of effective therapeutic agents for AD would be a major medical
milestone. Different causation targets are being targeted and medicinal,
pharmacological, clinical and pathological research is on-going around the world. The
anti-acetylcholinesterase activity of Canarium patentinervium Miq. in agreement with
its potent antioxidant and anti-inflammatory activity demonstrated in Chapter 4 and 5

143
respectively represents an exciting scaffold from which to develop leads for treatment
of neurodegenerative disease.

144
CHAPTER 7
ANTIMICROBIAL ACTIVITY
7.1 Introduction
7.1.1 Infectious disease
Since their introduction, antimicrobials (antibiotics) have played an essential role in
decreasing morbidity due to infectious diseases. Antimicrobials are used for treatment
of infections and for prophylaxis against infections in humans and animals, for growth
promotion in food animal rearing and in agriculture. However infectious diseases
remain the leading cause of death worldwide and bacteria have become more resistant
to conventional antibiotic in recent years. The widespread use of these compounds is
thought to further encourage the emergence of antimicrobial resistance [157]. The
number of resistant pathogenic bacteria grows at an alarming rate worldwide and the
search for novel antimicrobial agents from medicinal plants to combat such pathogens
has become crucial for avoiding the emergence of untreatable bacterial infections
[158][159].
7.1.2 Chemotherapeutic agents: Factors affecting their effectiveness
The ideal chemotherapeutic agent has a high therapeutic index with selective toxicity,
thereby resulting in lethal damage to pathogens through the inhibition of cell wall
synthesis, protein synthesis or nucleic acid synthesis, as well as through the disruption

145
of the cell membrane and the inhibition of certain essential enzymes. This results in
selective disruption of the specific structure and/or function essential to bacterial
growth and survival, without causing similar effects to its eukaryotic host [160].
The efficacy of antimicrobial agents is influenced by a number of factors. Firstly, it is
of obvious importance that the antimicrobial agent reaches the site of the infection.
This greatly depends on the stability of the drug, its lipophilic or hydrophilic nature,
its absorption from a specific site and the presence of blood clots or necrotic tissue,
the latter of which may protect the pathogen against the antibiotic. Secondly, the
susceptibility of the pathogen to the particular chemotherapeutic agent is of utmost
importance, as well as the specific growth phase in which the pathogen is in at that
particular stage [160].
7.1.3 Drug resistance
Bacteria have evolved many different mechanisms of resistance. These can be
classified as: a) alteration in, or addition of, the target site of antimicrobial binding; b)
alteration in access to the target site for instance the decreased permeability of cell
wall or efflux mechanisms; and c) inactivation of the antimicrobial binding.
Furthermore resistance may arise through mutation or by acquisition of resistance
genes by horizontal transmission from another bacterial species [157].
On the other hand, drug resistance may be brought about by the limited drug diffusion
into the biofilm matrix, enzyme-mediated resistance, genetic adaptation, efflux
pumps, as well as through the adaptation of the outer microbial membrane, the latter

146
occurring either through the lack of or through the overexpression of certain
membrane proteins [161].
This phenomenon of increased drug resistance, combined with the multiplicity of side
effects by existing agents and the emergence of diseases for which no treatment yet
exists, makes the search for the new antimicrobial agents a highly relevant and
important subject for research. For centuries, plants have been used in the traditional
treatment of microbial infections. This assembly of knowledge by indigenous peoples
about plants and their products continue to play an essential role in health care of a
great proportion of the population [162].
7.1.4 Natural products and their role in drug discovery
Natural products have played a pivotal role in the discovery of antimicrobial drugs,
with the drug either being completely derived from the natural product, or serving as a
lead for novel drug discovery. Most antimicrobials discovered during the past 6-7
decades have been discovered through screening of soil samples, of which the
antimicrobial efficacies were determined first in vivo and later in vitro [162].
Plants synthesize a diverse array of secondary metabolites, which play a key role in
the natural defence mechanisms employed by the plant against predation by
microorganisms and insects. It is thus no surprise that these aromatic compounds
have, in numerous instances, been found to be useful antimicrobial phytochemicals
and, as a result, these compounds are now divided into different chemical categories:

147
phenolics, terpenoids and essential oils, alkaloids, lectins and polypeptides, as well as
polyacetylenes [163]. An increase in the isolation and identification of such
compounds may thus contribute greatly to the success in antibiotic discovery.
7.1.5 Canarium species and their known antimicrobial activity
Studies conducted on Canarium species have suggested that these species may well
be active against micro-organisms. Antibacterial activities was previously reported in
Canarium schweinfurthii Engl. Dichloromethane extract of Canarium schweinfurthii
Engl. had bactericidal activity against Gram-negative Vibrio cholerae with minimum
inhibitory concentration (MIC) of 0.62 mg/ml while the ethylacetate extract was
active against Gram-positive and Gram-negative bacteria namely Staphylococcus
aureus and Proteus vulgaris with MIC values of 10 mg/ml and 5 mg/ml respectively.
Ethanol extract was active against Gram-negative Vibrio cholerae and Proteus
vulgaris with MIC values of 0.62 mg/ml and 10 mg/ml respectively. The oil was also
fungicidal against Candida albicans with an inhibition zone of 23 mm [28]. In a
separate disc diffusion assay, the essential oil of Canarium schweinfurthii Engl.
abrogated the survival of Gram-negative Salmonella enterica, Gram positive
Streptococcus pyogenes and Staphylococcus aureus with an inhibition zone of 27 mm,
25 mm and 18 mm respectively [25]. Canarium patentinervium Miq. have been used
traditionally in healing wounds [164], as such this study will attempt to validate these
ethnopharmacological claims.

148
7.2 Materials and Methods
The in vitro antimicrobial properties of Canarium patentinervium Miq. was evaluated
using the disc diffusion assay, minimum inhibitory concentration (MIC) assay,
minimum bactericidal assay (MBC) and death kinetic assay. The hexane, chloroform
and ethanol extracts of Canarium patentinervium Miq. were prepared (see Chapter 2
for the extraction process). The following bacterial strains were employed in the
screening:
i. Gram-positive bacteria such as Staphylococcus aureus (ATCC 11632),
Bacillus cereus (ATCC 10876), methicillin-resistant Staphylococcus
aureus (ATCC 43300) and Gram-negative bacteria such as Escherichia
coli (ATCC 10536) and Pseudomonas aeruginosa (ATCC 10145).
ii. Ten clinical bacterial strains and 3 clinical strain of yeasts obtained from
the Bacteriology Unit, Department of Microbiology and Immunology of
UKM Medical Centre. The assays involving these clinical strains were
also perfomed at HUKM.
7.2.1 Disc diffusion assay
7.2.1.1 Principle of method
The principles of determining the effectivity of a noxious agent to a bacterium were
well enumerated by Rideal, Walker and others at the turn of the century. The
discovery of antibiotics made these tests (or their modification) too cumbersome to be
put up as a routine due to the large number of tests it requires [165]. The ditch plate

149
method of agar diffusion used by Alexander Fleming was the forerunner of a variety
of agar diffusion methods devised by workers in this field. The Oxford group used
these methods initially to assay the antibiotic contained in blood by allowing the
antibiotics to diffuse out of reservoirs in the medium in containers placed on the
surface [165]. With the introduction of a variety of antimicrobials it became necessary
to perform the antimicrobial susceptibility test as a routine.
In this assay, the antimicrobial contained in a reservoir was allowed to diffuse out into
the medium and interact in a plate freshly seeded with the test organisms. Even now a
variety of antimicrobial containing reservoirs are used but the antimicrobial
impregnated absorbent paper disc is by far the commonest type used. The disc
diffusion method of antimicrobial susceptibility testing (AST) is the most practical
method and is still the method of choice for the average laboratory. Automation may
force the method out of the diagnostic laboratory but in this country as well as in the
smaller laboratories of many countries, it will certainly be the most commonly carried
out microbiological test for many years to come [165].
The Kirby-Bauer and Stokes' methods are usually used for antimicrobial susceptibility
testing, with the Kirby-Bauer method being recommended by the National Committee
for Clinical Laboratory Standards (NCCLS). The accuracy and reproducibility of this
test are dependent on maintaining a standard set of procedures. NCCLS is an
international, interdisciplinary, non-profit, non-governmental organization composed
of medical professionals, government, industry, healthcare providers, educators etc. It
promotes accurate antimicrobial susceptibility testing (AST) and appropriate reporting
by developing standard reference methods, interpretative criteria for the results of

150
standard AST methods, establishing quality control parameters for standard test
methods, provides testing and reporting strategies that are clinically relevant and cost-
effective [165]. Interpretative criteria of NCCLS are developed based on international
FROODERUDWLYHVWXGLHVDQGZHOOFRUUHODWHGZLWK0,&¶VDQGWKHUHVXOWVKDYHFRUURERUDWHG
with clinical data. Based on study results NCCLS interpretative criteria are revised
frequently. NCCLS is approved by FDA-USA and recommended by WHO
[165][166].
7.2.1.2 Protocol [166][167][168]
i. Preparation of Mueller Hilton Agar
Mueller Hilton Agar was prepared from a commercially available dehydrated base
DFFRUGLQJWRWKHPDQXIDFWXUHU¶VLQVWUXFWLRQV The agar solution was then autoclaved.
It was then allowed to cool in a 45-50 oC water bath. The agar was then poured into
plastic flat bottomed petri dishes on a level, horizontal surface to give a uniform depth
of approximately 4 mm. This corresponds to 25 ml for plates with a diameter of 100
mm. The agar medium was then allowed to cool to room temperature and unless plate
is used the same day, stored in a refrigerator (2 to 8 oC). A representative sample of
each batch of plates was examined for sterility by incubation at 30-35 oC for 24 hrs or
longer.
ii. Preparation of dried filter paper discs/ cotton swabs/ tryptic soy broth/
normal saline solution
Whatman filter paper no.1 was used to prepare discs approximately 6 mm in diameter.
They are then placed in a bijou bottle and autoclaved for sterility. Same was done for

151
cotton swabs. Tryptic soy broth (TSB) (20 ml) ZDVSUHSDUHG DV WKHPDQXIDFWXUHU¶V
instructions. About 5 ml was poured into a Bijou bottle and autoclaved. About 150
ml of sodium chloride solution of 0.9 % was prepared and autoclaved
iii. Preparation of sample extracts, negative control and standard antibiotic
solution.
Sample extracts were dissolved in DMSO at a concentration of 100 mg/ml and
filtered. Negative control was DMSO. Standards used were ampicillin and
streptomycin which were prepared at the concentration of 100 µg/ml.
iv. Preparation of fresh/ pure colonies of bacteria
Tryptic soy agar was prepared according to the manufacturers intructions. Agar was
autoclaved and poured on petri dishes to 4 mm, approximately 25 ml to a 100 mm
petri dish. Bacterias are streaked on the tryptic soya agar (TSA) plates in 4 density
level around the plates. Plates are then sealed with a parafilm and incubated for 18 hrs
to obtain single colonies.
v. Preparation of disc with extracts, control and standards
About 10 µl of sample extracts was pipetted onto the disc in triplicates. Same was
done for controls and standards. Impregnated discs are left to air dry overnight for 12
hrs.
vi. Inoculum Preparation
At least 3-5 well isolated colonies of the same morphological type are selected from
an agar plate culture. The top of each colony was touched with a sterilized loop and

152
the growth was transferred into the Bijou bottle containing TSB. This was repeated
for each bacteria into each Bijou bottle. The broth culture was incubated at 35 oC for
about 2 hrs till it achieved the turbidity of 0.5 Mcfarland standard/625 nm to yield 1 X
108 cfu/ml. Using optical density at 625 nm, the reading below must be obtained for
different bacteria, turbidity was adjusted with sterile normal saline.
vii. Inoculation of test plates
The bacterial broth was used within 15 mins after the turbidity of the inoculum
suspension was adjusted. Sterile cotton swab was dipped into the suspension and
rotated several times and pressed firmly on the inside wall of the tube above the fluid
level. This was to remove any excess inoculum from the swab. The dried surface of a
Mueller Hilton agar plate was inoculated by streaking the swab over the entire agar at
every 60 oC. As a final step, the rim of the agar was swabbed.
viii. Application of discs to inoculated agar plates
Each disc was pressed down on the agar to ensure complete contact according to
prepared template. Plates were placed in 35 oC incubator within 15 mins after discs
are applied.
viiii. Reading plates and interpretation of results
After 18 hrs of incubation, each plate was examined. Zone of inhibition was measure
using sliding calipers to the nearest millimeters. The inhibition zones would thus
include disc size of 6 mm. The experiment was done three times and the mean values
are presented.

153
7.2.2 Minimum inhibitory concentration assay
7.2.2.1 Principle of method
The MIC is the lowest concentration being beyond the concentration where no growth
inhibition of test organisms was observed. Minimum inhibitory concentration (MIC)
assays investigate the in vitro susceptibility of organisms to antimicrobial agents. This
method employs different dilutions of the antimicrobial agent and quantitatively
investigates the lowest concentration at which visible microbial growth inhibition is
achieved. Microbial growth is visualised through the addition of the tetrazolium salts,
specifically p-iodonitrotetrazolium (INT, Sigma-Aldrich). The assay is based on the
detection of dehydrogenase activity in living cells by being converted from a
colourless solution to an intensely coloured formazan (red) product [169]. This
method yields reproducible results within one doubling dilution of the end point of the
activity.
7.2.2.2 Protocol
The MIC of the plant extracts was determined by serial dilution, as described by Eloff
[169]. These dilution experiments were performed in sterile 96-well microtiter plates.
i. Preparation of sample
Stock solutions of the respective plant extracts were prepared in 1.5 ml
microcentrifuge tubes (Eppendorff) by dissolving dry plant extract in

154
dimethylsulphoxide (DMSO, Saarchem) to a final concentration of 64 mg/ml.
$OLTXRWVRIȝORIWKHVWRFNVRlution were transferred aseptically into the top row
RIPLFURWLWHUSODWHURZ$ZKLFKDOUHDG\FRQWDLQHGȝODOLTXRWVRIVWHULOHZDWHU
thereby resulting in a 50 % dilution of the stock solution to 32 mg/ml. After adequate
mixing of the contents of eacKZHOOȝODOLTXRWVRIURZ$ZHUHWUDQVIHUUHGIURP
URZ$WRWKHFRUUHVSRQGLQJZHOOVLQURZ%DOVRFRQWDLQLQJȝODOLTXRWVRIVWHULOH
water), followed by mixing and resulting in yet another 50% dilution of the plant
extract (to 16 mg/ml). This process was repeated IRU HYHU\ URZ UHVXOWLQJ LQȝO
aliquots ranging in concentration from 32 mg/ml (row A) to 0.25 mg/ml (row H).
ii. Preparation of inocolums
In order to determine the range of antimicrobial activity of Canarium patentinervium
Miq., four different microbial (reference) cultures and 10 clinical strains were used
and are listed below.
i. Gram-positive bacteria
x Staphylococcus aureus (ATCC 11632)
x Bacillus cereus (ATCC 10876)
x Methicillin-resistant Staphylococcus aureus (ATCC 43300)
x Staphylococcus aureus MSSA
x Staphylococcus aureus MRSA
x Coagulase negative Staph: Oxacillin (S)
x Coagulase negative Staph: Oxacillin (R)
x Enterococcus faecalis

155
ii. Gram-negative bacteria
x Pseudomonas aeruginosa (ATCC 10145)
x Escherichia coli
x Klebsiella sp
x Escherichia coli ESBL
x Klebsiella pneumoniae ESBL
iii. Yeast
x Candida parapsilosis
Bacterias are streaked on TSA plates in 4 density level around the plate. Plates are
then sealed with a parafilm and incubated for 18 hrs to obtain fresh single colonies. At
least 3-5 well isolated colonies of the same morphological type are selected from the
agar plate culture. The top of each colony was touched with a sterilized loop and the
growth was transferred into Bijou bottle containing TSB/MHB. This was repeated for
each bacteria placed into each Bijou bottle. Broth was incubated at 35 oC for about 2
hrs till it achieved 0.5 Mcfarland Standard/ 600 nm (OD-0.1 nm) to yield 1 x 108
cfu/ml [170]. Turbidity was adjusted with sterile normal saline. To obtain 10 x 105
cfu/ml, add 100 µl of adjusted broth to 9900 µl of TSB/MHB (1:100 dilution).
iii. MIC assay
7KLVZDVIROORZHGE\DGGLWLRQRIȝODWconcentration of 10 x 105 colony forming
units (cfu)/ml, of liquid microbial culture grown in Mueller Hilton Broth (MHB) to
HDFK ZHOO 7KLV \LHOGHG D ILQDO YROXPH RI ȝO LQ HDFK ZHOO DQG ILQDO H[WUDFW

156
concentrations ranging from 16 mg/ml in row A to 0.125 mg/ml in row H (Figure 4.1)
with bacteria 5 x 105 cfu/ml. The microtiter plates were incubated at 37 °C, overnight
for bacteria and 48 hours for yeasts (as yeasts require a greater time period for
growth). After incubation at 37 &ȝORIDPJPOVROXWLRQRI,17ZDVDGGHGWR
each well as an indicator of microbial growth. The plates were incubated at 37 oC for
30 min (bacteria) and 24 hr (yeast) and the MIC values visually determined. The
lowest concentration of each extract displaying no visible growth was recorded as the
minimum inhibitory concentration. The concentration that inhibited bacterial/yeast
growth completely (the first clear well) was taken as the MIC value. MIC values were
determined at least in duplicate and repeated to confirm activity
In order to determine the sensitivity of the microorganisms, positive control
experiments were conducted: (1) for bacterial strains, ampicillin (Sigma-Aldrich) at a
starting concentration of 0.10 mg/ml in sterile water, and (2) for yeast strains,
Amphotericin B (Sigma-Aldrich), at a starting concentration of 0.10 mg/ml in DMSO
and water (where 1.00 mg/ml was prepared in DMSO, and diluted to 0.10 mg/ml in
sterile water thereafter). The final concentrations for these experiments ranged from
25.00 µg/ml (row A) to 0.19 µg/ml (row H). A negative control experiment was
conducted using only DMSO.

157
7.2.3 Minimum bactericidal concentration (MBC)
7.2.3.1 Principle of method
Minimum bactericidal concentration (MBC) was recorded as a lowest extract
concentration killing 99.9 % of the bacterial inocula after 24 hr incubation at 37 °C.
Each experiment was repeated at least three times.
7.2.3.2 Protocol
The determination of MBC was performed using the method of Ozturk & Ercisli
[171] only for the susceptible bacterias/ fungi from the MIC assay (Section 7.2.2).
Ten microliters were taken from the well obtained from the MIC experiment (MIC
value) and two wells above the MIC value well and spread on MHA plates. The
number of colony was counted after 18-24 hrs of incubation at 37 °C. The
concentration of sample that produces < 10 colonies was considered as MBC value.
7.2.4 Time-kill assay
7.2.4.1 Principle of method
Death kinetic studies are often referred to as time-kill studies, and are used to
determine the rate at which the antimicrobial agent kills pathogens over time, as well
as the extent at which the activity occurs. Upon introduction of a micro-organism into
a new environment, its growth displays a lag phase during which no cell growth

158
occurs, after which its growth enters the exponential phase, where microbial cell
multiplication occurs at an exponential rate (doubling in number at regular intervals).
This exponential phase is followed by the stationary phase, the latter of which usually
results from the depletion of nutrients (carbon, nitrogen and/or oxygen sources) [160].
However, upon exposure to a constant concentration of an antimicrobial agent, the
organism will remain within the lag phase for a certain amount of time. This is
followed by a log-linear killing phase, during which the number of microbial colonies
are decreased until it enters into a second lag phase. Re-growth may occur after this
second lag phase, but documentation of this phenomenon rarely occurs as time-kill
studies are usually performed over a 24 hour period only [172][173].
7.2.4.2 Protocol
Only samples that have MIC values of lower or equivalent to 0.5 mg/ml (strong
inhibitors) are tested for the time-kill [174]. Concentration of samples to be tested is
determined base on the MIC values (up to 4 times the MIC value) [175]. Samples and
incolums are prepared as in section 7.2.2.2 (i-ii). This was followed by addition of
ȝORIPLFURELDOEURWKDWconcentration of 10 x 105 colony forming units (cfu)/ml,
of liquid microbial culture grown in Mueller Hilton Broth (MHB) to each well. This
\LHOGHG D ILQDO YROXPH RI ȝO LQ HDFK ZHOO of the extract. The plates are then
incubated at 37 °C and optical density was recorded at 1 hr intervals up to 18 hrs at
wavelength of 600 nm. Graphs were plotted on the basis of the turbidity varying over
a period of time. The growth rate thus obtained was studied for any signs of
bactericidal effects of the plant extract. Ampicillin was used as positive control. A

159
solution of the solvent in which dried extract was dissolved served as negative
control.
7.2.5 Statistical analysis
Growth curve of bacteria were analysed using MS-Excel and data were reported as
mean and SD values obtained from a minimum of three determinations. Bacterial
growth curve was plotted with SD and 95% confidence interval. All data were
expressed as mean ± standard deviation. Data were analyzed using one way Anova
followed by Tukey test. A significant difference was considered at the level of P <
0.01.
7.3 Results
7.3.1 Disc diffusion assay
The six extracts of Canarium patentinervium Miq. that were screened for
antimicrobial activity using the disc diffusion assay displayed good activity (Table
7.1). The extracts were screened for activity against Gram-positive bacteria
(Staphylococcus aureus ATCC 11632, MRSA ATCC 43300, Bacillus cereus ATCC
10876, clinical strains consisting of MSSA, MRSA, Coagulase-negative
Staphyloccocus: oxacillin sensitive/resistant, Enterococcus faecalis and Streptococcus
pneumoniae), Gram-negative bacteria (Escherichia coli ATCC 10536, Pseudomonas
aeruginosa ATCC 10145, clinical strains consisting of Esherichia coli, Klebsiella sp,

160
Esherichia coli ESBL and Klebsiella pneumoniae ESBL and yeasts (Candida
albicans, Candida glabrata and Candida parapsilosis). All the bacterias and yeast
were sensitive to the extracts except Streptococcus pneumoniae, Eschericia coli
ATCC 10536, Candida glabrata and Candida albicans. The ethanol extract of leaves
and the hexane extract of bark displayed good antibacterial activity against both
Gram-positive bacteria and Gram-negative bacteria. The ethanol extracts and hexane
extract of the bark had the best sensitivity to Candida parapsilopsis (inhibition zone
14 mm).
Both ATCC and clinical strains of MSSA and MRSA were sensitive to the ethanol
extracts (9-12 mm) (Figure 7.1) and hexane extract of barks (8-14 mm). Coagulase-
negative Staphylococcus aureus: oxacillin sensitive and resistant and Enterococcus
faecalis were most sensitive to the ethanol extracts (inhibition zone 8-12 mm). Gram-
negative bacteria Klebsiella pneumoniae ESBL (inhibition zone 9-14 mm) had
sensitivity to all the bark extracts while Klebsiella sp showed sensitivity only towards
the ethanol extracts (inhibition zone 11-13 mm). Pseudomonas aeruginosa ATCC
10145 was inhibited by the ethanol extract of leaves (inhibition zone 11 mm) (Figure
7.2) and hexane extract of barks (inhibition zone 12 mm) while poor activity was
seen with Esherichia coli with all extracts.
7.3.2 MIC assay
All extracts displayed some activity against Gram-positive and Gram-negative
bacteria and yeast (Table 7.2). The activity against MSSA reference and clinical
strains ranged from 0.50 mg/ml to 2.00 mg/ml with the strongest activity seen with

161
the ethanol extract of leaves. The activity against MRSA reference and clinical strains
ranged from 0.25 mg/ml to 2.00 mg/ml with the lowest MIC seen in the ethanol
extract of leaves. All the extracts had activity against coagulase-negative
Staphylococcus aureus: oxacillin resistant and sensitive with MIC ranging from as
low as 0.50 mg/ml (ethanol extract of leaves) to 16.00 mg/ml (hexane extract of
leaves). Enterococcus faecalis had activity only against the ethanol extracts with an
MIC of 0.50 mg/ml. Bacillus cereus ATCC 10876 showed sensitivity only against the
ethanol extracts (MIC 0.5 mg/ml) and hexane extract of barks (MIC 1.00 mg/ml).
All the bark extracts showed activity against Gram-negative Klebsiella sp. with an
MIC range of 0.50 mg/ml to 4.00 mg/ml while only the ethanol extracts of leaves and
barks showed activity against Klebsiella pneumoniae ESBL with MIC of 0.5 mg/ml.
Pseudomonas aeruginosa ATCC 10145 was sensitive towards ethanol extract of
leaves and hexane extract of barks with an MIC of 1.00 mg/ml and 4.00 mg/ml
respectively. Esherichia coli clinical strain was sensitive only towards ethanol extract
of barks with an MIC of 0.50 mg/ml while Escherichia coli ESBL was inhibited by
the chloroform extracts and hexane extract of barks with MIC of 8.00 mg/ml. The
ethanol extracts and hexane extract of bark both had activity against yeast Candida
parapsilopsis with the MIC of 2.00 mg/ml and 8.00 mg/ml respectively.
7.3.3 MBC assay and MBC/MIC ratio
The action of an antibacterial on the bacterial strains can be characterized with two
parameters such as minimum inhibitory concentration (MIC) and minimum
bactericidal concentration (MBC). According to the ratio MBC/MIC, we appreciated

162
antibacterial activity. If the ratio 0%&0,& , the effect was considered as
bactericidal but if the ratio MBC/MIC > 4 , the effect was defined as bacteriostatic
[176][177]. The MBC and MBC/MIC ratio are displayed in Table 7.3 and 7.4
respectively.
Both the ethanol extract of leaf and bark were bactericidal against Bacillus cereus
ATCC 10876 (MBC of 1.00 mg/ml), MSSA ATCC 11632 (MBC range of 1.00 to
2.00 mg/ml), Klebsiella sp and Klebsiella pneumonia ESBL (MBC of 2.00 mg/ml)
and yeast Candida parapsilopsis (MBC 8.00 mg/ml). The hexane extract of barks was
bactericidal against MSSA, MRSA (both strains) and Pseudomonas aeruginosa
ATCC 10145 with MBC range of 2.00 mg/ml to 8.00 mg/ml. Chloroform extract of
barks showed bactericidal activity against MSSA (ATCC 11632 and clinical strains)
with MBC of 1.00 mg/ml and 4.00 mg/ml respectively. Both hexane and chloroform
extract of barks were bactericidal against Klebsiella sp with MBC/MIC ratio of 4.
Rest of the MBC as displayed in Table 7.3 were all having bacteriostatic action with
an MBC/MIC ratio > 4.
7.3.4 Time-kill assay
Only samples that have MIC values of lower or equivalent to 0.5 mg/ml (strong
inhibitors) are tested for the time-kill [174]. The results obtained for the time-kill
study of Canarium patentinervium Miq. are shown in Figure 7.3-7.22. The MBC
values (Table 7.3) was consistent with the cidal concentration as displayed in the
growth curve of this time-kill study. Blank control (broth alone) was the curve marked
as 0 mg/ml of extract. The antibacterial activity was observed at different times upon

163
exposure of bacterias to the different concentration of extracts. This observation was
made by varying turbidity over time for a period of 18 hrs.
7.4 Discussion
7.4.1 Disc diffusion assay
According to Gislene et al. [178], chemicals that have antibacterial activity with zones
of inhibition of 7 mm and above can be considered as potential antimicrobial
candidates. In the present study, antibacterial screening of six extracts of Canarium
patentinervium Miq. showed varying degrees of antibacterial activity against human
pathogenic bacteria such as Staphyloccocus aureus, MRSA, Bacillus cereus (ATCC
strain respectively), coagulase-negative Staphylococcus aureus, Enterococcus
faecalis, Klebsiella pneumoniae ESBL and yeast Candida parapsilopsis (clinical
strain respectively) cultured in vitro. Out of six extracts, the ethanol extract of leaves
and hexane extract of barks exhibited significant antibacterial activity against both
Gram-positive and Gram-negative bacteria. The best sensitivity to the ethanol extract
of the leaves at 1 mg/disc was obtained against Staphyloccocus aureus (12 mm),
MRSA (10 mm), Bacillus cereus (12 mm) and Enterococcus faecalis (12 mm),
Klebsiella pneumoniae ESBL (13 mm) and yeast Candida parasilopsis (14 mm). The
hexane extract of the barks showed best activity against MSSA (14 mm), Klebsiella
sp (14 mm) and yeast Candida parapsilopsis (14 mm).

164
This is of special interest since most Gram-negative bacteria are more resistant to
plant extracts [179]. Results from our phytochemical analysis revealed that the
ethanol extract of leaves and barks of Canarium patentinervium Miq. accumulate
substantial amounts of flavonoids and tannins which could be well correlated with the
activities measured.

165
Figure 7.1: Zone of inhibition against Staphylococcus aureus (left plate) and methicillin resistant Staphylococcus aureus (right plate) for leaf ethanol extract
at 1 mg/disc.

166
Figure 7.2: Zone of inhibition against Bacillus cereus (left plate) and Pseudomonas aeruginosa (right plate) for leaf ethanol extract at 1 mg/disc.

167
Table 7.1: Antibacterial activity of Canarium patentinervium Miq. (1 mg/disc ) vs. ampicillin against 5 ATCC bacterias and 10 clinical bacterial strains and 3 clinical yeast strains tested by disc diffusion assay
Bacteria/ yeast Plant extract (Zone of inhibition) (mm) a
LH LC LE BH BC BE Ampicilinc Amphotericinc
MSSA Staphylococcus aureus ATCC 11632 - - 12.00±0.21 11.00±0.25 7.00±0.29 9.00±0.58 10.00±0.66 NA
MRSA Staphylococcus aureus ATCC 43300 - - 10.00±0.42 9.00±0.25A - 9.00±0.00A - NA
Bacillus cereus ATCC 10876 - - 12.00±0.25B 12.00±0.45B - 9.00±0.00 8.00±0.00 NA
Eschericia coli ATCC 10536 - - - - - - 7.00±0.00 NA
Pseudomonas aeruginosa ATCC 10145 - - 11.00±0.34 12.00±0.47 - - - NA
MSSA Staphylococcus aureus - - 11.00±0.23 14.00±0.43 13.00±0.17 10.00±0.45 7.00±0.26 NA
MRSA Staphylococcus aureus - - 10.00±0.37C 8.00±0.29 - 10.00±0.47C - NA
Eschericia coli - - - - - 8.00±0.57 - NA
Eschericia coli ESBL - 7.00±0.18 - 8.00±0.38D 8.00±0.26D - - NA
Coagulase-negative Staphylococcus aureus: O (R) - 7.00±0.59 10.00±0.00 8.00±0.34E 8.00±0.36E 9.00±0.34 - NA
Coagulase-negative Staphylococcus aureus: O (S) 7.00±0.37F 7.00±0.16F 12.00±0.29G - - 12.00±0.23G 10.00±0.27 NA
Enterococcus faecalis - - 12.00±0.46 - - 8.00±0.15H 8.00±0.28H NA
Streptococcus pneumoniae - - - - - - - NA
Klebsiella sp - - 11.00±0.38I 14.00±0.00 11.00±0.34I 9.00±0.42 10.00±0.14 NA
Klebsiella pneumoniae ESBL - - 13.00±0.25 - - 11.00±0.27 - NA
Candida parapsilosis - - 14.00±0.12K 14.00±0.24K - 14.00±0.10K NA 11.00±0.23
Candida glabrata - - - - - - NA 9.00±0.15
Candida albicans - - - - - - NA -
LH-leaf hexane extract, LC- leaf chloroform extract, LE-leaf ethanol extract, BH- bark hexane extract, BC- bark chloroform extract, BE- bark ethanol extract, NA-not applicable
a: inhibition zones are the mean including disc (6 mm) b: sample extract at the concentration of 100 mg/ml (1mg/disc), c: ampicillin/amphotericin at 100 µg/ml = 1 µg/disc, -: no activity noted
Data were obtained from three independent experiments, each performed in triplicates (n=9) and represented as mean ± SD.
Values with the same capital letter are not significantly different (P<0.01) according to Tukey multiple comparison test.

168
Table 7.2: MIC values for Canarium patentinervium Miq. extracts against 4 ATCC bacterias and 9 clinical bacterial strains and 1 clinical yeast strain
Bacteria/ yeast Plant extract minimum inhibitory concentration MIC (mg/ml) and positive controla MIC (µg/ml)
LH LC LE BH BC BE Ampicilina Amphotericina
MSSA Staphylococcus aureus ATCC 11632 - - 0.50±0.10 2.00±0.10 1.00±0.00A 1.00±0.00A 1.56±0.00 NA
MRSA Staphylococcus aureus ATCC 43300 - - 0.50±0.00 1.00±0.00B - 1.00±0.00B - NA
Bacillus cereus ATCC 10876 - - 0.50±0.15C 1.00±0.00 - 0.50±0.00C 6.25±0.00 NA
Pseudomonas aeruginosa ATCC 10145# - - 1.00±0.00 4.00±0.05 - - - NA
MSSA Staphylococcus aureus - - 0.50±0.10 2.00±0.00D 2.00±0.00D 0.50±0.00 >25.00±0.00 NA
MRSA Staphylococcus aureus - - 0.25±0.00E 2.00±0.10 - 0.25±0.00E - NA
Eschericia coli - - - - - 0.50±0.00 - NA
Eschericia coli ESBL - 8.00±0.00F - 8.00±0.00F 8.00±0.00F - - NA
Coagulase-negative Staphylococcus aureus: O (R) - 8.00±0.00 1.00±0.00 0.50±0.00 2.00±0.00 0.50±0.00 - NA
Coagulase-negative Staphylococcus aureus: O (S) 16.00±0.00 8.00±0.00 0.50±0.00G - - 0.50±0.10G 8.00±0.00 NA
Enterococcus faecalis - - 0.50±0.00I - - 0.50±0.00I 3.13±0.00 NA
Klebsiella sp - - 0.50±0.00J 4.00±0.00K 4.00±0.00K 0.50±0.00J >25.00±0.00 NA
Klebsiella pneumoniae ESBL# - - 0.50±0.00 - - 0.50±0.00 - NA
Candida parasilopsis - - 2.00±0.00K 8.00±0.00 - 2.00±0.00K NA 0.78±0.00
LH-leaf hexane extract, LC- leaf chloroform extract, LE-leaf ethanol extract, BH- bark hexane extract, BC- bark chloroform extract, BE- bark ethanol extract
Data were obtained from three independent experiments, each performed in duplicates (n=6) and represented as mean ± SD, -: no activity, NA-not applicable
Values with the same capital letter are not significantly different (P<0.01) according to Tukey multiple comparison test. #: statistical analysis not performed due to insufficient data

169
Table 7.3: MBC values for Canarium patentinervium Miq. extracts against 4 ATCC bacterias and 9 clinical bacterial strains and 1 clinical yeast strain
Bacteria/ yeast Plant extract minimum inhibitory concentration,MBC (mg/ml) and positive controla MBC (µg/ml)
LH LC LE BH BC BE Ampicilina Amphotericina
MSSA Staphylococcus aureus ATCC 11632 - - 2.00±0.00A 2.00±0.00A 1.00±0.00B 1.00±0.00B 6.25±0.00 NA
MRSA Staphylococcus aureus ATCC 43300 - - 2.00±0.00 4.00±0.00C - 4.00±0.00C - NA
Bacillus cereus ATCC 10876 - - 1.00±0.10D 8.00±0.00 - 1.00±0.00D >25.00±0.00 NA
Pseudomonas aeruginosa ATCC 10145 - - 4.00±0.00 8.00±0.05 - - - NA
MSSA Staphylococcus aureus - - 4.00±0.10E 4.00±0.00E 4.00±0.00E 2.00±0.00 >25.00±0.00 NA
MRSA Staphylococcus aureus - - 2.00±0.00 8.00±0.10 - 1.00±0.00 - NA
Eschericia coli# - - - - - 2.00±0.00 - NA
Eschericia coli ESBL# >16.00±0.00 - >16.00±0.00 >16.00±0.00 - - NA
Coagulase-negative Staphylococcus aureus: O (R) >16.00±0.00 4.00±0.00F 2.00±0.00 8.00±0.00 4.00±0.00F - NA
Coagulase-negative Staphylococcus aureus: O (S)# >16.00±0.00 >16.00±0.00 2.00±0.00 - - 2.00±0.10 12.50±0.00 NA
Enterococcus faecalis# - - 4.00±0.00 - - 4.00±0.00 6.25±0.00 NA
Klebsiella sp - - 2.00±0.00I 16.00±0.00J 16.00±0.00J 2.00±0.00I >25.00±0.00 NA
Klebsiella pneumoniae ESBL# - - 2.00±0.00 - - 2.00±0.00 - NA
Candida parapsilopsis# - - 8.00±0.00 >16.00±0.00 - 8.00±0.00 NA 3.13±0.00
LH-leaf hexane extract, LC- leaf chloroform extract, LE-leaf ethanol extract, BH- bark hexane extract, BC- bark chloroform extract, BE- bark ethanol extract
Data were obtained from three independent experiments, each performed in duplicates (n=6) and represented as mean ± SD, -: no activity, NA-not applicable
Values with the same capital letter are not significantly different (P<0.01) according to Tukey multiple comparison test. #: statistical analysis not performed due to insufficient data

170
Table 7.4: Bacteriostatic (-) and Bactericidal (+) effects of Canarium patentinervium Miq. extracts
Bacteria/ yeast MBC/MIC ratio (+/-)
LE BH BC BE
MSSA Staphylococcus aureus ATCC 11632 4 (+) 1 (+) 1 (+) 1 (+)
MRSA Staphylococcus aureus ATCC 43300 4 (+) 4 (+) - 4 (+)
Bacillus cereus ATCC 10876 2 (+) 8 (-) - 2 (+)
Pseudomonas aeruginosa ATCC 10145 4 (+) 2 (+) - -
MSSA Staphylococcus aureus 8 (-) 2 (+) 2 (+) 4 (+)
MRSA Staphylococcus aureus 8 (-) 4 (+) - 4 (+)
Eschericia coli - - - 4 (+)
Coagulase-negative Staphylococcus aureus: O (R) 4 (+) 4 (+) 4 (+) 8 (-)
Coagulase-negative Staphylococcus aureus: O (S) 4 (+) - - 4 (+)
Enterococcus faecalis 8 (-) - - 8 (-)
Klebsiella sp 4 (+) 4 (+) 4 (+) 4 (+)
Klebsiella pneumoniae ESBL 4 (+) - - 4 (+)
Candida parapsilopsis 4 (+) ND - 4 (+)
LE-leaf ethanol extract, BH- bark hexane extract, BC- bark chloroform extract, BE- bark ethanol extract
-: no activity, ND: not determined

171
Figure 7.3: Time-kill plot for MSSA ATCC 11632 in presence of LE
-0.1
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in
OD
)
Time (hr)
0 mg/ml 1mg/ml 0.25mg/ml 0.5mg/ml 2mg/ml

172
Figure 7.4: Time-kill plot for MRSA ATCC 43300 in presence of LE
-0.2-0.1
00.10.20.30.40.50.60.70.80.9
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in
OD
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml

173
Figure 7.5: Time-kill plot for MSSA (clinical strain) in presence of LE
-0.10
0.10.20.30.40.50.60.70.80.9
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in
OD
)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

174
Figure 7.6: Time-kill plot for MRSA (clinical strain) in presence of LE
-0.20.00.20.40.60.81.01.21.41.6
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in
OD
)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

175
Figure 7.7: Time-kill plot for MSSA (clinical strain) in presence of BE
-0.10
0.10.20.30.40.50.60.70.8
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in
OD
)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

176
Figure 7.8: Time-kill plot for MRSA (clinical strain) in presence of BE
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18Bac
teri
al g
row
th (
incr
ease
in
OD
)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

177
Figure 7.9: Time-kill plot for Bacillus cereus ATCC 10876 in presence of LE
-0.10.00.10.10.20.20.30.30.40.40.50.5
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in
OD
)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml

178
Figure 7.10: Time-kill plot for Bacillus cereus ATCC 10876 in presence of BE
-0.1
-0.1
0.0
0.1
0.1
0.2
0.2
0.3
0.3
0.4
0.4
0.5
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in
OD
)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml

179
Figure 7.11: Time-kill plot for for Escherichia coli (clinical strain) in presence of BE
-0.2
0
0.2
0.4
0.6
0.8
1
1.2
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in
OD
)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

180
Figure 7.12: Time-kill plot for coagulase-negative Staphylococcus aureus: O (R) (clinical strain) in presence of LE
-0.10
0.10.20.30.40.50.60.70.80.9
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in
OD
)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

181
Figure 7.13: Time-kill plot for coagulase-negative Staphylococcus aureus: O (R) (clinical strain) in presence of BH
-0.10
0.10.20.30.40.50.60.70.80.9
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in
OD
)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

182
Figure 7.14: Time-kill plot for coagulase-negative Staphylococcus aureus: O (R) (clinical strain) in presence of BE
-0.10
0.10.20.30.40.50.60.70.80.9
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in
OD
)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

183
Figure 7.15: Time-kill plot for coagulase-negative Staphylococcus.aureus: O (S) (clinical strain) in presence of LE
-0.2
0
0.2
0.4
0.6
0.8
1
1.2
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in O
D)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

184
Figure 7.16: Time-kill plot for coagulase-negative Staphylococcus aureus: O (S) (clinical strain) in presence of BE
-0.10
0.10.20.30.40.50.60.70.8
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in O
D)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

185
Figure 7.17: Time-kill plot for Enterococcus faecalis (clinical strain) in presence of LE
-0.1
0
0.1
0.2
0.3
0.4
0.5
0.6
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in O
D)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

186
Figure 7.18: Time-kill plot for Enterococcus faecalis (clinical strain) in presence of BE
-0.10
0.10.20.30.40.50.60.70.8
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in O
D)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

187
Figure 7.19: Time-kill plot for Klebsiella sp. (clinical strain) in presence of LE
0.00.10.20.30.40.50.60.70.80.9
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in O
D)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

188
Figure 7.20: Time-kill plot for Klebsiella sp. (clinical strain) in presence of BE
0.00.10.20.30.40.50.60.70.80.9
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18Bac
teri
al g
row
th (
incr
ease
in O
D)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

189
Figure 7.21: Time-kill plot for Klebsiella pneumonia ESBL (clinical strain) in presence of LE
0.0
0.1
0.2
0.3
0.4
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in O
D)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

190
Figure 7.22: Time-kill plot for Klebsiella pneumonia ESBL (clinical strain) in presence of BE
0.0
0.1
0.2
0.3
0.4
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in O
D)
Time (hr)
0 mg/ml 0.25mg/ml 0.5mg/ml 1mg/ml 2mg/ml 4mg/ml

191
7.4.2 MIC, MBC and MIC/MBC ratio
Upon identifying possible antimicrobial action with the disc diffusion assay, a more
comprehensive investigation was done to evaluate the total bactericidal/ bacteriostatic
activity of an extract against a microb using the MIC/ MBC and time-kill assay. All
plant species with MIC values of up to 8 mg/ml are considered to possess at least
some degree of inhibitory effect, and any concentration exceeding this should not be
considered effective, according to Fabry et al. [180]. The majority of the crude bark
and leaf extracts of the six Canarium patentinervium Miq. exhibited moderate to
relatively good activity against Gram-positive and Gram-negative pathogens with the
MIC values ranging from 0.25 mg/ml to 8.00 mg/ml. Only the hexane extract of
leaves exhibiting weak antimicrobial activity. All active extracts displayed
concentration-dependent antimicrobial activity. Aligiannis et al. [181] proposed a
classification system based on MIC results obtained for plant materials, which was
consequently described and implemented by Duarte et al. [182].
Strong microbial inhibitors possessed MIC values of equal or lower than 0.50 mg/ml;
a clear indication that the MIC value of 0.25 mg/ml obtained for ethanol extracts (leaf
and bark) against MRSA Staphylococcus aureus (clinical strain) indicates exceptional
antimicrobial activity. The ethanol extract of leaves also resulted with the MIC of
0.50 mg/ml against MSSA (both strains) and MRSA (ATCC 43300). Coagulase-
negative Staphylococcus aureus: oxacillin resistant and sensitive strains were both
sensitive against both ethanol extracts with an MIC of 0.5 mg/ml for the bark extracts
and MIC range of 0.5 mg/ml to 1.00 mg/ml for the leaves. Staphylococcus aureus is a
Gram-positive bacteria that can cause a range of illnesses, from minor skin infections,

192
such as pimples, impetigo, boils, celluliti, folliculitis, carbuncles, scalded skin
syndrome and abscesses to life threatening diseases such as pneumonia, meningitis,
osteomyelitis, endocarditis, toxic shock syndrome, bacteremia and sepsis. Its
incidence ranges from skin, soft tissue, respiratory, bone, joint, endovascular to
wound infections [183]. MRSA is one of the greatly feared strains of Staphylococcus
aureus ZKLFKKDYHEHFRPHUHVLVWDQWWRPRVWȕ-lactam antibiotics and mostly hospital-
accquired. Coagulase-negative Staphylococcus aureus are differentiated by their
inability to produce free coagulase resulting in their inability to clot blood plasma
[184].
The ethanol extracts (bark and leaves) were bactericidal against Bacillus cereus with a
MIC of 0.50 mg/ml and MBC of 1.00 mg/ml. Bacillus cereus is a causative agent of
both gastrointestinal infections (diarrheal and emetic type of food poisoning) and non-
gastrointestinal infections (post traumatic wound and burn infections. ophthalmic
infections, endocarditis, postoperative meningitis and urinary tract infections) [185].
The ethanol extracts were also bactericidal against Gram-negative Klebsiella sp and
Klebsiella pneumoniae ESBL with MIC of 0.50 mg/ml and MBC of 2.00 mg/ml.
Klebsiella is well known to most clinicians as a cause of community-acquired
bacterial pneumonia, occurring particularly in chronic alcoholics which have a high
fatality rate if untreated. Nosocomial Klebsiella infections are caused mainly by
Klebsiella pneumoniae, the medically most important species of the genus. Due to the
extensive spread of antibiotic-resistant strains, especially of extended-spectrum b-
lactamase (ESBL)-producing strains, there has been renewed interest in Klebsiella
infections. Since ESBL production frequently is accompanied by multiresistance to
antibiotics, therapeutic options become limited [186].

193
The points above will make the discovery of an antimicrobial agent as effective as
Canarium patentinervium Miq. against these pathogens highly significant. The
ethanol extract of leaves displayed good microbial inhibition action against all
microbs tested except for Pseudomonas aeruginosa and Candida parapsilopsis with
moderate inhibition but bactericidal action with the MIC of 1.00 mg/ml and 2.00
mg/ml respectively. Pseudomonas aeruginosa is a Gram-negative bacterium and an
opportunistic, nosocomial pathogen in immunocompromised individuals. It typically
infects the pulmonary tract, urinary tract, burns, wounds and blood infections [187].
Moderate microbial inhibitors are described by Aligiannis et al. [181] as those plant
extracts with MIC values ranging between 0.60 mg/ml and 1.50 mg/ml. Amongst the
extracts investigated in the present study, hexane extracts of the barks seemed to have
most of the moderate inhibitors with an interesting bactericidal action against MRSA
ATCC 43300 strain (MIC of 1.00 mg/ml and MBC of 4.00 mg/ml). Weak microbial
inhibitors are classified as those agents with MIC values of between 1.60 mg/ml and
8.00 mg/ml [181]. None of the extracts in the present study yielded MIC values in
excess of 8.00 mg/ml against Gram-positive bacteria except with hexane extract of
leaves. The results obtained against the Gram-positive and Gram-negative bacteria
thus support the traditional use of Canarium patentinervium Miq. in wound
healing[164] and hold potential in the treatment of colds, wound healing and as an
antiseptic.
In general, the Gram-negative bacteria displayed the least sensitivity towards the
extracts, and most of the plant extracts (except ethanol extract of barks) exhibited
poor and unvaried activity against Esherichia coli, indicating the resistance of this

194
bacterium to the plant extracts. This was to be expected, as Gram-negative bacteria
offer a much more complex barrier system against permeation of foreign substances
(in this case, the antimicrobial agent). This is attributed to the specialised cell wall
structure and especially the presence of the outer envelope resulting in the
impermeability of these micro-organisms to biocides and antibiotics, and at times,
resulting in regulation and prevention of their passage to the target region [188].
Resistance to the plant extracts is, thus, exhibited to a far greater extent by the Gram-
negative bacteria than by Gram-positive bacteria [188]. The lipophilic or hydrophilic
nature of compounds also plays a role in the activity, or lack thereof, against the
micro-organisms. Compounds considered to be more effective against Gram-negative
bacteria are considerably less lipophilic. This is as a result of the structure of the
Gram-negative cell wall which also has a higher lipid content [189]. This could
explain why in this study the ethanol extracts which contain more hydrophilic
compounds showed more inhibition against Gram-negative bacteria such as
Esherichia coli and Klebsiella species.
The ethanol extracts and hexane extract of barks also showed inhibition against
Candida parapsilopsis with a cidal and static action respectively. Both ethanol
extracts had MIC of 2.00 mg/ml (MBC of 8.00 mg/ml) and hexane extract of barks
had an MIC of 8.00 mg/ml (MBC >16.00 mg/ml). More than 80% of all fungal
bloodstream infections are a direct result from the Candida species and since the
eighties there has been an explosive rise in the rate of Candidal infections. Over the
past decade, the incidence of Candida parapsilosis has dramatically increased. In fact,
reports indicate that Candida parapsilosis is often the second most commonly isolated

195
Candida species from blood cultures and Candida parapsilosis even outranks
Candida albicans in some European, Asian, and South American hospitals [190].
The ethanol extracts have shown to have polyphenols such as tannins and flavonoids
from the phytochemical analysis (chapter 2). Polyphenols, such as tannins and
flavonoids, have important antibacterial activity [191]. The antimicrobial activity of
flavonoids is due to their ability to complex with extracellular and soluble protein and
to complex with bacterial cell wall while that of tannins may be related to their ability
to inactivate microbial adhesions, enzymes and cell envelop proteins [163]. Flavones
are phenolic structures containing one carbonyl group (as opposed to the two
carbonyls in quinones). The addition of a 3-hydroxyl group yields a flavonol.
Flavonoids are also hydroxylated phenolic substances but occur as a C6-C3 unit
linked to an aromatic ring. Their activity is probably due to their ability to complex
with extracellular and soluble proteins and to complex with bacterial cell walls. More
lipophilic flavonoids may also disrupt microbial membranes [192].
Tannin is a general descriptive name for a group of polymeric phenolic substances
capable of tanning leather or precipitating gelatin from solution, a property known as
astringency. They are found in almost every plant part: bark, wood, leaves, fruits and
roots. Tannins may be formed by condensations of flavan derivatives or by the
polymerization of quinine units [191]. Many human physiological activities, such as
stimulation of phagocytic cell, host mediated tumour activity and a wide range of anti-
infective actions, have been assigned to tannins. Their mode of antimicrobial action as
described for quinone, may be related to their ability to inactivate microbial
adhesions, enzymes, cell envelope, transport-proteins etc [191]. They also form a

196
complex with polysaccharide. According to a number of studies, tannins can also be
toxic to filamentous fungi, yeasts and bacteria [192]. However, it is important to note
that, if tannins were solely responsible for the activity presented by these results, this
activity would be observed against all organisms and would not be limited to Gram-
positive bacteria or yeasts. The current hypothesis is thus that tannins are at least
partially responsible for the antibiotic activity. Coumarins have also demonstrated
antimicrobial activity [163].
7.4.3 Time-kill assay
$OOH[WUDFWVZLWKJRRGLQKLELWRUV0,&YDOXHPJPOwas tested for the time-kill
efficacy. All extracts displayed killing (bactericidal) action except the ethanol extracts
(leaves and barks) against Enterococcus faecalis which was only bacteriostatic
(Figure 7.17 & 7.18) and ethanol extract of barks that was bacteriostatic against
coagulase-negative Staphylococcus aureus: oxacillin resistant (Figure 7.14). The other
extracts bactericidal effect was achieved in the concentration of 2.00 to 4.00 mg/ml.
Killing rate was observed at varying times from range of 1 hr to 10 hrs all resulting in
complete kill.
7.5 Conclusion
All crude extracts of Canarium patentinervium Miq. under investigation exhibited
exceptional concentration-dependent antimicrobial activity against both Gram-
positive and Gram-negative bacteria as well as against yeasts. The most promising

197
activity was displayed against Gram-positive bacteria Staphylococcus aureus, MRSA,
Bacillus cereus and Gram-negative bacteria and Pseudomonas aeruginosa, Klebsiella
sp, Klebsiella pneumoniae ESBL and yeast Candida parapsilopsis. This serves as a
clear indication of the potential of these extracts for further chemical studies as
antimicrobial agents. The antimicrobial activity exhibited by the bark extracts and
ethanol extract of leaves against MSSA and MRSA indicates exceptional
antimicrobial activity. All the tested bacterias and yeast were susceptible to either
bark or leaf extracts.
In general, the Gram-negative bacteria Escherichia coli displayed the least sensitivity
towards the extracts, being inhibited only by the ethanol extract of the bark indicating
the resistance of this bacterium to the rest of the plant extracts. The activity against
Klebsiella pneumonia ESBL and Klebsiella sp. was, however, far more promising
with variability amongst the plant extracts. It is at this stage important to note that the
failure of a plant extract to demonstrate in vitro activity during the general screening
process does not necessarily imply a total absence of inherent medicinal value. The
possible presence of synergistic interactions between the different plant constituents
in crude preparations may result in activities that are not exhibited by isolated
compounds, and should not be excluded.

198
CHAPTER 8
ANTIPARASITIC ACTIVITY
8.1 Introduction
8.1.1 Parasitic infection: causative factor and treatment
Parasitic infection is one of a major health problem in the developing world. Twenty-
five percent of the world's population could be suffering from parasitic infection
Highest prevalence is in underdeveloped agricultural and rural areas in the tropical
and subtropical regions. In some areas incidence may reach 90 % of the population.
During the evolution of humans a broad set of parasites have evolved, that use us as a
host organism [193]. Usually a parasite will not kill its host (at least not immediately),
as this would be an evolutionary dead end for a parasite. However, most parasites are
either unpleasant for us (think of lice and fleas) or weaken our health (most internal
parasites). However, some parasitic infections, such as malaria, trypanosomiasis or
Chagas can be deadly if the patients are not treated with adequate therapeutics.
Because humans usually live in close proximity and often without good hygienic
conditions the transmission of parasites within a human population is often facilitated
[194].
Many parasitic infections are the cause of tropical diseases, such as malaria,
trypanosomiasis, leishmaniasis, Chagas disease, schistosomiasis, onchocerciasis,
lymphatic filariasis, and helminthiases. Parasites are responsible for probably more
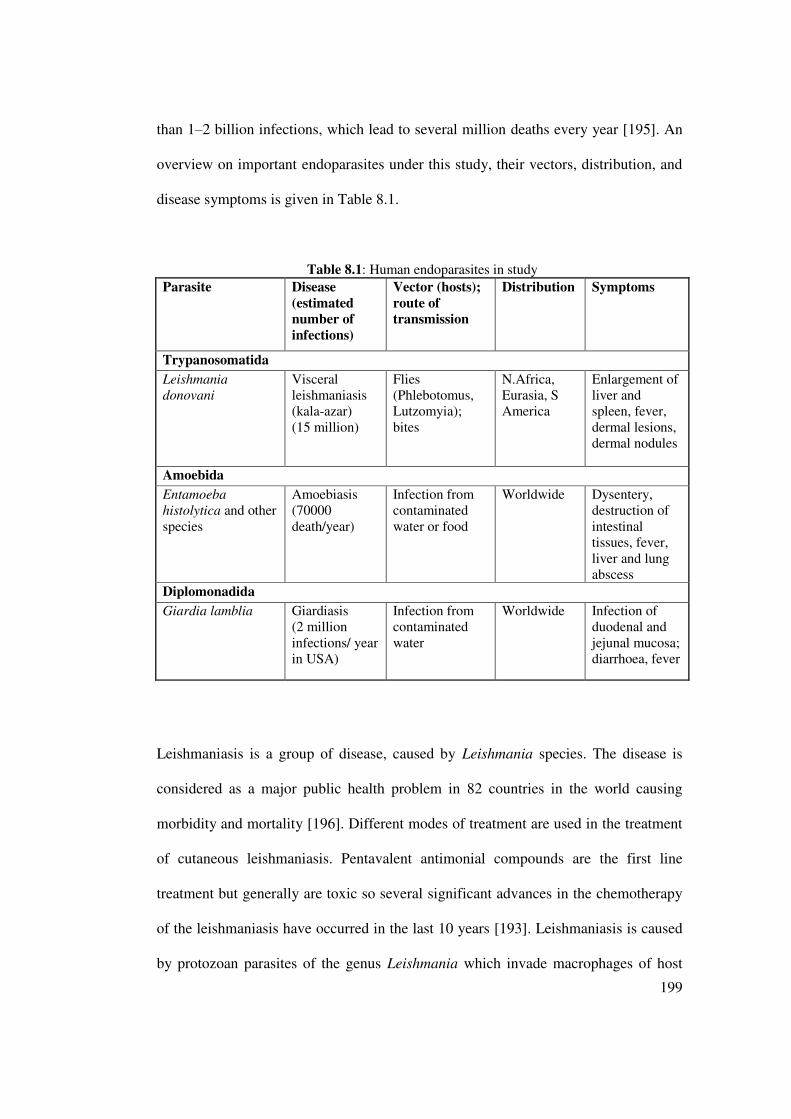
199
than 1±2 billion infections, which lead to several million deaths every year [195]. An
overview on important endoparasites under this study, their vectors, distribution, and
disease symptoms is given in Table 8.1.
Table 8.1: Human endoparasites in study
Parasite Disease (estimated number of infections)
Vector (hosts); route of transmission
Distribution Symptoms
Trypanosomatida
Leishmania donovani
Visceral leishmaniasis (kala-azar) (15 million)
Flies (Phlebotomus, Lutzomyia); bites
N.Africa, Eurasia, S America
Enlargement of liver and spleen, fever, dermal lesions, dermal nodules
Amoebida
Entamoeba histolytica and other species
Amoebiasis (70000 death/year)
Infection from contaminated water or food
Worldwide Dysentery, destruction of intestinal tissues, fever, liver and lung abscess
Diplomonadida
Giardia lamblia Giardiasis (2 million infections/ year in USA)
Infection from contaminated water
Worldwide Infection of duodenal and jejunal mucosa; diarrhoea, fever
Leishmaniasis is a group of disease, caused by Leishmania species. The disease is
considered as a major public health problem in 82 countries in the world causing
morbidity and mortality [196]. Different modes of treatment are used in the treatment
of cutaneous leishmaniasis. Pentavalent antimonial compounds are the first line
treatment but generally are toxic so several significant advances in the chemotherapy
of the leishmaniasis have occurred in the last 10 years [193]. Leishmaniasis is caused
by protozoan parasites of the genus Leishmania which invade macrophages of host

200
organisms. A distinction is made between cutaneous, mucocutaneous, visceral and
diffuse leishmaniasis, of which visceral leishmaniasis is a fatal disease causing
approximately 60,000 death per year [197]. Leishmaniasis and HIV infections often
co-occur and these patients usually have a poor prognosis. Patients are treated with
the synthetic drugs stibogluconate, meglumine and pentamidine (developed 70 years
ago), which have severe side effects and fail to work in North Bihar (India)
[198][199]. Also the macrolide antibiotic amphotericin B has been employed, which
can also be toxic for patients. New developments include the anticancer drug
miltefosine, the aminoglycoside antibiotic paronomycin, and the 8-aminoquinoline
sitamaquine [197]. Among natural products, berberine (which occurs in many TCM
plants) had promising anti-leishmanial activities [198][199].
Entamoeba histolytica and Giardia intestinalis are parasite of large and small
intestine of human, respectively. Although infection with E. histolytica is
asymptomatic in 90% of cases, about 50 million people are estimated to suffer from
the symptoms of amoebiasis such as haemorrhagic colitis and amoebic liver abscess.
These infections result in 50,000 to 100,000 deaths annually [200]. Giardia
intestinalis (Synonyms: Giardia duodenalis, Giardia lamblia) is one of the most
common universal pathogenic intestinal protozoan parasites of humans [201]. It is
also widely spread in various animals such as dogs [202], cats [203] and cattle [204].
It is frequently reported to be a significant cause of diarrhea in people traveling to
WURSLFDOFRXQWULHVWUDYHOHU¶VGLDUUKHD$OWKRXJKWKHPDMRULW\RILQIHFWHGKXPDQVVKHG
Giardia cysts without any symptoms, the major clinical signs of giardiasis in humans
include acute or chronic diarrhea, nutritional disorders and weight loss. Recent studies
have shown that Giardia genotypes may be shared between human and numerous

201
other species of animal [205][206] indicating transmission between humans and
animal vectors may occur, possibly through exposure to contaminated water.
Metronidazole, the current drug of choice, can cause mutagenicity in bacteria [207]
and is carcinogenic in rodents [208][209]. It also possesses undesirable side effects
and failures in treatment have been reported [210][211]. We therefore plan to evaluate
the in vitro activity of Canarium patentinervium Miq., against Giardia intestinalis in
the search for an alternative drug suitable for use in preventing and treating cases of
diarrhoea.
8.1.2 Natural products and parasitic defence
We know that humans have used medicinal plants for several thousands of years to
treat illness and health disorders [212]. Medicinal chemists have synthesized a
number of drugs which can be used against many but by far not all endoparasites. A
major problem is that many of these drugs were developed many years ago and some
parasitic strains have become resistant to them. The development of new antiparasitic
drugs has not been much of a priority for the pharmaceutical industry because many
of the parasitic diseases occur in poor countries where the populations cannot afford
to pay a high price for the drugs. Thus an investment in drug development against
parasitic diseases is a risky affair. An alternative to synthetic drugs is the search for
anti-parasitic plant extracts or secondary metabolites derived from them. Natural
products still play an important role in therapy: between 1981 and 2006, 1,184 new
drugs were registered of which 28% were natural products or their derivatives.
Another 24% of the new drugs had pharmacophores (i.e., functional groups with

202
pharmacological activity) derived from natural products [213]. A good starting point
to find antiparasitic natural products would be traditional medicinal plants, such as
those known from Asia, Africa or America [212] that have been employed to treat
infections.
8.2 Materials and Methods
8.2.1 Principle of method
The antiprotozoal activity of the compounds was perform according to the standard
methods as described by Sawangjaroen et al. [214][200]. The antileishmanial activity
of the compounds was perform according to the standard methods as described by
Mosmann [215].
8.2.2 Protocol
8.2.2.1 Antiparasitic assay against Giardia intestinalis and Entamoeba histolytica
8.2.2.1.1 Test organisms
Giardia intestinalis
A local Thai strain of Giardia intestinalis, originally described by Siripanth et al.
[216] was used throughout this experiment.

203
Entamoeba histolytica
Entamoeba histolytica strain HM1: IMSS, purchase from ATCC was used throughout
this experiment.
8.2.2.1.2 Cultivation of organisms
Both Giardia intestinalis and Entamoeba histolytica were cultured axenically in screw
capped tubes at 37 oC, under anaerobic condition, on YI medium [217] supplemented
with 10% heat-inactivated horse serum. Subculture was performed every 48 hr. For
assays, trophozoites were harvested by chilling the tube on ice for 15 min to detach
the monolayer and centrifuged at 300 g for 5 min. The supernatant was decanted and
cells were re-suspended in fresh medium. The numbers of viable cells were calculated
using a haemocytometer and 0.4 % (w/v) trypan blue. The criteria used for viability
were motility and dye exclusion.
8.2.2.1.3 Antiprotozoal assay
Briefly, trophozoites, 2x105 cells/ml, was incubated, in triplicate, in the presence of
serial two fold dilutions of plant extracts that ranged from 31.25 to 500 Pg/ml for
crude extract in 96 well tissue culture plates (200 Pl/well). Metronidazole and
complete medium with added DMSO will be used as negative and positive control,
respectively. After 24 hr of incubation at 37 oC under anaerobic conditions, the
trophozoites from each well will examine with an inverted microscope. The
appearance of trophozoites will score and presented as score values from 1 to 4 with 1
showing the most inhibition of growth and 4 showing no inhibition, according to

204
Upcroft and Upcroft [218]. The minimum inhibitory concentration (MIC) will be
recorded (the lowest concentration at which >90% of the trophozoites rounded up).
Each concentration was tested in triplicates and at least three experiments were
performed on separate occasions.
8.2.2.2 Antiparasitic activity against Leishmania donovani promastigotes
Antileishmanial activity of tested plant extract, against Leishmania donovani (strain
MHOM/IN/1983/AG83) promastigotes, was evaluated by a quantitative colorimetric
assay using MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide;
Sisco Research Laboratory, Mumbai, India]. Amphotericin B (Sigma-Aldrich Co.,
MO, USA; IC50 =0.4±0.1µM) was used as the positive control in all the experiments.
Promastigotes (5×105 cells/ml; 300 µl), were treated with and without tested samples
at concentrations of 100 and 500 µg/mL, and incubated at 22 ± 2 oC. After 72 hr, cells
were harvested, and re-suspended in PBS (500 µl) containing MTT (0.3 mg/ml).
Purple formazan crystals were dissolved in DMSO and the optical density (O.D.) was
measured at 570 nm in an ELISA reader (BIO-RAD; model 680, USA). The number
of viable cells was directly proportional to the amount of formazan produced through
the reduction of yellow MTT by the dehydrogenase enzymes present in the inner
mitochondrial membrane of the living cells.
The percentage of growth inhibition was calculated as follows:
% inhibition = [(O.D. of untreated control - O.D. of treated set)/ O.D. of untreated
control] x 100.

205
8.2.3 Statistical analysis
The IC50 values (concentration of drug which inhibited at least 50% cell growth) for
each compounds were determined from respective dose-responsive percentage
inhibition curves with the help of Microsoft Excel and data were reported as mean and
SD values obtained from a minimum of three determinations. Graph was plotted with
SD and 95% confidence interval.
8.3 Results
The result of the in vitro effect of extracts on Giardia intestinalis and Entamoeba
histolytica was summarized in Table 8.2. It was found that the ethanol extracts were
both active against growth of Giardia intestinalis in vitro with MIC 500 µg/ml (Table
8.2). The MIC of metronidazole was 2.5 µg/ml and the remaining extracts showed no
activity (MIC >500µg/ml for crude extract). The appearance of trophozoites in
negative, positive and with exposure to LE under microscope is shown in Figure 8.1.
The result of the in vitro effect of extracts on Leishmania donovani was summarized
in Table 8.3. The potency of extracts against Leishmania donovani was LH > BH >
BC > LC. Both ethanol extracts of leaves and barks had IC50 values of above 500
µg/ml. Hexane extract displayed lowest IC50 value of 257.40±0.30 µg/ml. Figure 8.2
shows the inhibition of the hexane and chloroform extracts.

206
8.4 Discussion
Parasites are eukaryotes and therefore share most molecular and biochemical
properties with their eukaryotic hosts, making it often difficult to find antiparasitic
drugs which are both effective and non-toxic for humans. This limitation always has
to be kept in mind when discussing the numerous findings that some drug or extract
from a medicinal plant is active against parasites in vitro. In order to be medicinally
useful, such a drug must have bioavailability and should not intoxicate the patient. A
first guidance is the determination of a selectivity index (SI) which compares the
cytotoxicity of a drug against a parasite and a library of human cells [194].
Most of the antiparasitic properties of extracts and isolated natural products have been
tested in vitro only. Translation of the in vitro research results into in vivo trials is
urgently required. Furthermore, even if animal experiments were successful, we
would need clinical trials of the new compounds alone or in combination with
established parasiticidal drugs to prove their efficacy and safety. These developments
are costly and it is presently difficult to attract the pharmaceutical industries into these
fields for various reasons. TCM and other traditional medicine systems employ
several thousand of medicinal plants; some of which have known antiparasitic
properties.

207
Table 8.2: MIC values of Canarium patentinervium Miq. incubated for 24 hr with Giardia intestinalis and Entamoeba histolytica growing in vitro.
Sample MIC (µg/ml)
Giardia intestinalis Entamoeba histolytica
LH >500 >500
LC >500 >500
LE 500 >500
BH >500 >500
BC >500 >500
BE 500 >500
Metronidazole 2.5 2.5
LH: leaf hexane extract, LC: leaf chloroform extract, LE: leaf ethanol extract, BH: bark hexane extract, BC: bark chloroform extract, BE: bark ethanol extract. Data were obtained from three independent experiments, each performed in triplicates (n=9) and represented as mean ± SD. Values with the same capital letter are not significantly different (P<0.01) according to Tukey multiple comparison test.

208
Figure 8.1: Appearance of trophozoites under microscope in negative control, positive control and with LE
Negative control of G. intestinalis
Negative control of E. histolytica
Positive control: 10 Pg/ml of metronidazole ;cell deathͿ
Positive control: 10 Pg/ml of metronidazole;cell deathͿ

209
Table 8.3: Antiparasitic values of Canarium patentinervium Miq. against Leishmania donovani (AG 83) promastigotes
Sample IC50 (µg/ml)
LH 257.40±0.30
LC 457.70±0.25
LE >500
BH 284.20±0.40
BC 359.90±0.20
BE >500
LH: leaf hexane extract, LC: leaf chloroform extract, LE: leaf ethanol extract, BH: bark hexane extract, BC: bark chloroform extract, BE: bark ethanol extract. Data were obtained from three independent experiments, each performed in triplicates (n=9) and represented as mean ± SD. Values with the same capital letter are not significantly different (P<0.01) according to Tukey multiple comparison test.

210
0 200 400 600
0
20
40
60
80BH
BC
LH
LC
% in
hibi
tion
agai
nst
L.
do
no
van
i
concentration(Pg/ml)
Figure 8.2: MTT assay of Canarium patentinervium Miq. against Leishmania donovani.

211
They offer a unique opportunity to identify natural products which could be
potentially used to treat parasitic infections. The success story of artemisinin from
Artemisia annua [213][219] can probably be repeated. A number of medicinal plants
and secondary metabolites isolated from them have been screened for anti-
trypanosomal activity [220]. Active natural products include several groups of
alkaloids, phenolics, saponins, cardiac glycosides, other terpenoids, and
polyacetylenes (common in Apiaceae, Asteraceae and Araliaceae). Although some
natural products are active in the submicromolar range and show good selectivity,
only few have been studied in vivo in an animal model.
In the present study, six different extracts of Canarium patentinervium Miq.
(Burseraceae) leaves and barks were screened for their in vitro antiparasitic activities,
among the different extracts tested, the hexane extract of leaves showed moderate
antileshmanial activity with IC50 values of 257.40±0.30 µg/ml. This could be due to
essential oils present in the hexane extracts as shown previously in the family of
Burseraceae [221]. Only ethanol extracts showed activity against Giardia intestinalis
and Entamoeba histolytica at concentration of 500 µg/ml. Other extracts were inactive
against these two intestinal parasites.
Small lipophilic secondary metabolites, such as terpenoids or phenylpropanoids as
found in the essential oil of many plants (especially in Lamiaceae, Myrtaceae,
Rubiaceae, Apiaceae, Asteraceae, Lauraceae, Rutaceae, Burseraceae, Verbenaceae,
Pinaceae, Cupressaceae), can dissolve in biomembranes and disturb their fluidity and
the function of membrane proteins [221]. Therefore, many of the lipophilic mono- and
sesquiterpenes, phenylpropanoids and isothiocyanates (as present in Brassicaceae,

212
Tropaeoleaceae) have a certain degree of antimicrobial and antiparasitic properties
[222].
8.5 Conclusion
Plant substances continue to serve as a wellspring of drugs for the world population
and several plant-based drugs are in extensive clinical use. Preliminary antiparasitic
screening of six extracts of Canarium patentinervium Miq. showed that the ethanol
extract had promising growth inhibition on Giardia intestinalis and Entamoeba
histolytica. The hexane extract of leaves was active against Leshmanial donovani. The
results obtained suggest that further isolation studies can be performed on Canarium
patentinervium Miq to identify the active constituents responsible for the antiparasitic
activities.

213
CHAPTER 9
ANTICANCER ACTIVITY
9.1 Introduction
9.1.1 Carcinogenesis
Cancer, a cellular malignancy that results in the loss of normal cell-cycle control, such
as unregulated growth and the lack of differentiation, can develop in any tissue of any
organ, and at any time [223] .
Carcinogenesis is the transformation of a normal cell to a cancerous cell through
many stages, which occur over a number of years or even decades. The first stage of
carcinogenesis is the initiation stage, which involves the reaction between the
carcinogens and the DNA of the cells. Inhibiting this early stage of cancer is an
important strategy in cancer prevention or treatment. Promotion is the second stage
and may occur slowly over an extended period of time, ranging from several months
to years. Beneficial effects may arise from a change in lifestyle and diet, which may
result in the individual not developing cancer during his or her lifetime. The third
stage is the progressive stage, involving the spread of the cancer. It is evident that,
upon entering into this stage, preventative factors such as diet have less of an impact.

214
Despite the therapeutic advances made in understanding the processes involved in
carcinogenesis, cancer has become one of the most serious medical problems today.
The worldwide mortality rate increases annually, with more than seven million deaths
occurring per year. For this reason, cancer chemotherapy has become a major focus
area of research. Different lifestyles, risk factors (such as age, gender, race, genetic
disposition) and the exposure to different environmental carcinogens, lead to the
varying patterns of cancer incidence [223]. At least 35% of all cancers worldwide
result from an incorrect diet, and in the case of colon cancer, diet may account for
80% of these cases [224]. One of the most important contributions to the development
of cancer is the oxidative damage to DNA [225]. Permanent genetic alterations may
occur in those cells where DNA is damaged and where division of this DNA occurs
before it can be repaired. These cells may begin to divide more rapidly and result in
carcinogenesis [224].
9.1.2 Natural products and carcinogenesis defence
³&KHPRSUHYHQWLRQ´ LV GHILQHG as a process to delay or prevent carcinogenesis in
humans through the ingestion of dietary or pharmaceutical agents. This also implies
the identification of chemical entities (specifically cytotoxic entities) that are effective
against a range of cancer cell lines, although less active or non-toxic against the
normal (healthy) cell population. The search for such anticancer agents from plant
VRXUFHV VWDUWHG LQ WKH ¶V DQG Slant products have proven to be an important
source of anticancer drugs [226]. This directly results from the biological and
chemical diversity of nature, which allows for the discovery of completely new
chemical classes of compounds.

215
The discovery and development of plant-derived compounds led to the first treatment
of cancer, specifically upon administration of these compounds in combination with
synthetic agents. At present more than 450 different compounds have been isolated
from active plants that have shown in vitro and/or in vivo antitumor activity. Virtually
every major class of natural chemical compound is represented in the list of active
constituents [227]. Of the 121 medications being prescribed for use in cancer
theraphy, 90 are sourced from plants. It was also determined that approximately 74%
of these discoveries were as a result of an investigation into the claims made by
folkloric tradition [228]. A number of natural products are used as chemoprotective
agents against commonly occurring cancers. Examples of these compounds used as
cytotoxic drugs are shown in Table 9.1.
A number of mechanisms exist by which phytochemicals aid in the prevention of
cancer. This preventative action most probably results from the additive or synergistic
effects of a number of phytochemicals, since cancer is a multi-step process [227].
Proposed mechanisms by which phytochemicals may prevent cancer include: (i) anti-
oxidant and free radical scavenging activity; (ii) antiproliferative activity; (iii) cell-
cycle arresting activity; (iv) induction of apoptosis; (v) activity as enzyme cofactors;
(vi) enzyme inhibition; (vii) gene regulation; (viii) activity as hepatic phase I enzyme
inducers, and (ix) activity as hepatic phase II enzyme inducers. Oxidative damage to
DNA, proteins and lipids, resulting from an increase in oxidative stress, is considered
to be one of the most important mechanisms contributing to the development of
cancer [227] .

216
The phytochemicals that most often appear to be protective against cancer are
curcumin, genistein, resveratrol, diallyl sulfide, (S)-allyl cystein, allicin, lycopene,
ellagic acid, ursolic acid, catechins, eugenol, isoeugenol, isoflavones, protease
inhibitors, saponins, phytosterols, vitamin C, lutein, folic acid, beta carotene, vitamin
E and flavonoids, to name but a few [224]. These phytochemicals suppress the
inflammatory processes that lead to transformation, hyperproliferation, and initiation
of carcinogenesis. The inhibitory influences of these phytochemicals may ultimately
suppress the final steps of carcinogenesis via angiogenesis and metastasis.
The present study was undertaken to screen the anticancer potential Canarium
patentinervium Miq. No anticancer studies have previously been reported on this
species to date.

217
Table 9.1: Cytotoxic drugs developed from plant sources.
Therapeutic agent Plant source Mechanism of action
Treatment of cancer type
Reference
Vincristine, vinblastine
Catharanthus roseus
Inhibition of tubulin polymerisation
+RGJNLQ¶VGLVHDVH [229]
Etoposide, teniposide Podophyllum peltatum
Inhibition of topoisomerase II
Testicular cancer, and small cell lung carcinoma, leukaemias, lymphomas
[229] [230] [231]
Paclitaxel, docetaxel Taxus brevifolia
Promotion of tubulin stabilization
Ovarian and breast carcinoma
[229]
Irinotecan, topotecan, 9-aminocampothecin, 9-nitrocamptothecin
Camptotheca acuminata
Inhibition of topoisomerase I
Advanced colorectal cancer, also active in lung, cervix and ovarian cancer
[230]
Homoharingtonine Harringtonia cephalotaxus
Inhibition of DNA polymerase
Various leukaemias [229]
4-Ipomeanol Ipomoea batatas
Cytochrome P-450- mediated conversion into DNA-binding metabolites
Lung cancer [229]
Elliptinium Bleekeria vitensis
Inhibition of topoisomerase II
Advanced breast cancer
[229]
Flavopiridol Amoora rohituka, Dysoxylum binectariferum
Inhibition of cyclin- dependent kinases
Encouraging results noted in a variety of solid and haematological malignancies, in patients with colorectal, prostate, lung, renal carcinoma, non-+RGJNLQ¶Vlymphoma and chronic lymphocytic leukaemia
[229]

218
9.2 Materials and Methods
9.2.1 Principle of the MTT assay
MTT assay is dependent on the reduction of the tetrazolium salt MTT (3-(4,5-
dimethylthazol-2-yl)-2,5-diphenyl tetrazolium bromide) by the mitochondrial
dehydrogenase of viable cells to form a blue formazan product. The assay measures
cell respiration and the amount of formazan produced is proportional to the number of
living cells present in culture which will result in lower optical density (OD) [232].
Biological in vitro assay was determined as follows [233].
9.2.2 Protocol
9.2.2.1 Cell lines and cell culture
Human derived cell lines [HCT 116,colon cancer cell line and MCF-7 (ER+), MDA
468 (ER-) breast carcinoma] were routinely cultivated at 37 °C in an atmosphere of 5
% CO2 in RPMI 1640 medium supplemented with 2 mM L-glutamine and 10 % fetal
calf serum and subcultured twice weekly to maintain continuous logarithmic growth.
Cells were seeded into 96-well microtiter plates at a density of 3-5 × 103 per well and
allowed 24 hr to adhere before extracts were introduced (final concentration 200
µg/ml to 1 µg/ml).

219
9.2.2.2 Preparation of plant samples
Extracts were prepared as 400 µg/ml top stock solutions, dissolved in DMSO, and
stored at 4 oC, protected from light for a maximum period of 4 weeks. Serial sample
dilutions were prepared in medium immediately prior to each assay.
9.2.2.3 The MTT assay
At the time of extract addition and following 72 hr exposure, MTT (3-(4,5-
dimethylthazol-2-yl)-2,5-diphenyl tetrazolium bromide) was added to each well.
Incubation at 37 °C for 4 hr allowed reduction of MTT by viable cells to an insoluble
formazan product. Well contents were aspirated and formazan solubilized by addition
of DMSO:glycine buffer (pH 10.5) (4:1). Absorbance was read on an Anthos Labtec
System plate reader at 550 nm as a measure of cell viability, thus cell growth or
extract toxicity was determined. Doxorubicin was used as positive control.
9.2.3 Statistical analysis
Concentration±response curves were calculated using Microsoft Excel and data were
reported as mean and SD values obtained from a minimum of three determinations.
Graph was plotted with SD and 95% confidence interval.

220
9.3 Results
A range of extracts were evaluated in MTT assays following a 3-day exposure against
a panel of two human breast cancer cell lines, MCF-7 (ER+) and MDA 468 (ER-),
and a colon cancer cell line, HCT 116. In general, the breast cancer cell lines were
more sensitive to the extracts, and the MDA 468 line was the most sensitive of the
three lines (Table 9.2) The best growth inhibition was observed with the chloroform
and ethanol extract of barks, GI50 values of 23.44 µg/ml and 34.40 µg/ml respectively
(Figure 9.1-9.3).
9.4 Discussion
In the present study, six different extracts of Canarium patentinervium Miq.
(Burseraceae) leaves and barks were screened for their in vitro antitumor activities.
Among the different extracts tested, the chloroform and ethanol extract of barks
showed good antitumor activities with GI50 values of 23.44±0.05 µg/ml and
34.40±0.21 µg/ml respectively. The most susceptible cell lines were found to be the
breast cancer cell line, MDA 468.
It is known that nature is able to produce a wide variety of chemical entities of novel
structure. Many of the new and novel compounds isolated from natural sources might
otherwise have never been discovered, especially those of considerable complexity
requiring the development of methods for the creation of new ring systems.

221
Table 9.2: Growth inhibition of human cancer cell lines by crude plant extracts of Canarium patentinervium Miq.
Extracts Cell line/ GI50 (µg/ml)
MDA 468 HCT 116 MCF-7
LH >100 >100 >100
BH >100 >100 >100
LC >100 >100 >100
BC 23.44±0.05 62.46±0.23 64.50±0.27
LE 61.48±0.23 97.18±0.08 85.40±0.18
BE 34.4±0.21 46.53±0.12 39.06±0.16
Doxorubicin 0.06±0.12 0.70±0.18 0.34±0.26
LH: leaf hexane extract, LC: leaf chloroform extract, LE: leaf ethanol extract, BH: bark hexane extract, BC: bark chloroform extract, BE: bark ethanol xtract Data were obtained from three independent experiments, each performed in triplicates (n=9) and represented as mean ± SD. Values with the same letter are not significantly different (P<0.01) according to Tukey multiple comparison test.

222
Figure 9.1a: Effects of extract of leaves against MDA 468 growth
0
0.2
0.4
0.6
0.8
1
1.2
control 0.5 µg/ml 1 5 10 50 100
OD
550 n
m
concentration (µg/ml)
chloroform
ethanol
hexane
T zero

223
Figure 9.1b: Effects of extract of barks against MDA 468 growth
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
control 0.5 µg/ml 1 5 10 50 100
OD
550 n
m
concentration (µg/ml)
chloroform
ethanol
hexane
T zero

224
Figure 9.2a: Effects of extract of leaves against MCF-7 growth
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
control 0.5 µg/ml 1 5 10 50 100
OD
550 n
m
concentration (µg/ml)
chloroform
ethanol
hexane
T zero

225
Figure 9.2b: Effects of extract of barks against MCF-7 growth
-0.2
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
control 0.5 µg/ml 1 5 10 50 100
OD
550 n
m
concentration (µg/ml)
chloroform
ethanol
hexane
T zero
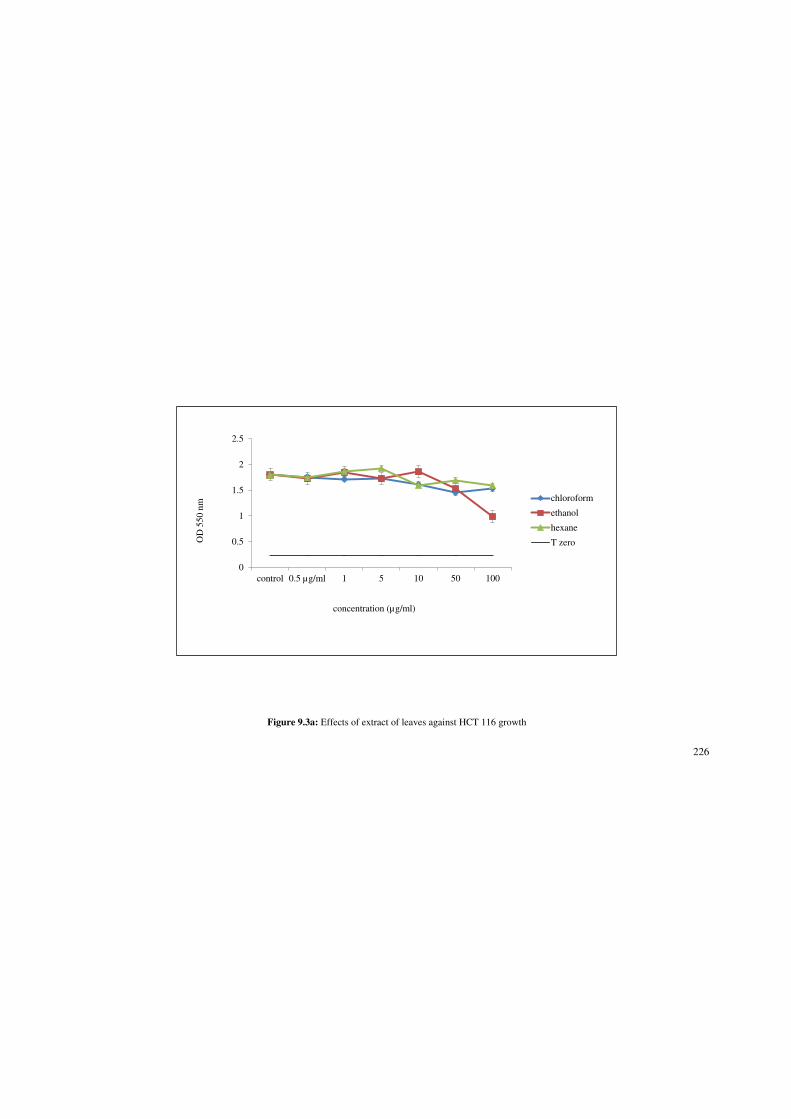
226
Figure 9.3a: Effects of extract of leaves against HCT 116 growth
0
0.5
1
1.5
2
2.5
control 0.5 µg/ml 1 5 10 50 100
OD
550 n
m
concentration (µg/ml)
chloroform
ethanol
hexane
T zero

227
Figure 9.3b: Effects of extract of barks against HCT 116 growth
-0.5
0
0.5
1
1.5
2
2.5
control 0.5 µg/ml 1 5 10 50 100
OD
550 n
m
concentration (µg/ml)
chloroform
ethanol
hexane
T zero

228
Natural products appeared to be a promising source for new types of compounds with
antitumor activity [234].
9.5 Conclusion
Plant substances continue to serve as a wellspring of drugs for the world population
and several plant-based drugs are in extensive clinical use [3]. Preliminary anticancer
screening of six extracts of Canarium patentinervium Miq. showed that the
chloroform and ethanol extract of the barks had promising effect on the cell viability
of breast cancer cell line MDA 468. The results obtained suggest that further isolation
studies can be performed on Canarium patentinervium Miq to identify the active
constituents responsible for the anticancer activities.

229
CHAPTER 10
ISOLATION AND IDENTIFICATION OF COMPOUNDS
10.1 Introduction
10.1.1 Secondary metabolites
Medicinal plants have formed the basis of health care since earliest times of humanity
and are still being widely used. The clinical, pharmaceutical and economic value
continues to grow, varying between countries. Chemodiversity in plants has proven to
be important in pharmacological research and drug development, not only for the
isolation of bio-active compounds used directly as therapeutic agents, but also as
leads to the synthesis of drugs or as models for pharmacologically active compounds
[235]. The rapid identification of these bio-active compounds, however, is critical if
this tool of drug discovery is to compete with developments in technology. Plant
preparations are distinguished from chemical drugs due by their complexity of
mixtures containing large numbers of bio-active compounds or secondary metabolites.
This brings about the challenge of drug discovery from natural sources [236].
Plants produce primary and secondary metabolites which encompass a wide array of
functions. Primary metabolites, which include amino acids, simple sugars, nucleic
acids, and lipids, are compounds that are necessary for cellular processes [237].
Secondary metabolites include compounds produced in response to stress, such as the
case when acting as a deterrent against herbivores. Plants can manufacture many

230
different types of secondary metabolites, which have been subsequently exploited by
humans for their beneficial role in a diverse array of applications [237]. Although
secondary metabolites from natural products have formed the basis of medicines, the
presence of these compounds in the biochemistry of the plant is very often difficult to
justify. It has been suggested that these compounds may have been synthesized by the
plant as part of the defense system of the plant, e.g. plants are known to produce
phytoalexins as a response to attack by bacteria and fungi [236]. The presence of
highly toxic natural products has also been highlighted in some animals namely the
Amazonian frogs so as to deter predation by other animals [236].
Secondary metabolites are organic molecules that are not involved in the normal
growth and development of an organism. While primary metabolites have a key role
in survival of the species, playing an active function in the photosynthesis and
respiration, the absence of secondary metabolites does not result in immediate death
but rather in long-term LPSDLUPHQWRI WKHRUJDQLVP¶VVXUYLYDELOLW\RIWHQSOD\LQJDQ
important role in plant defense [238]. Secondary metabolites are frequently produced
at highest levels during a transition from active growth to stationary phase [238]. The
producer organism can grow in the absence of their synthesis, suggesting that
secondary metabolism is not essential, at least for short term survival. A second view
proposes that the genes involved in secondary metabolism provide DµµJHQHWLFSOD\LQJ
ILHOG´ WKDW DOORZV PXWDWLRQ DQG QDWXUDO VHOHFWLRQ WR IL[ QHZ EHQHILFLDO traits via
evolution. A third view characterizes secondary metabolism as an integral part of
cellular metabolism and biology; it relies on primary metabolism to supply the
required enzymes, energy, substrates and cellular machinery and contributes to the
long term survival of the producer [238].

231
Different classes of these compounds are often associated to a narrow set of species
within a phylogenetic group and constitute the bioactive compound in several
medicinal, aromatic, colorant, and spice plants and/or functional foods. These
compounds are an extremely diverse group of natural products synthesized by plants,
fungi, bacteria, algae, and animals [239]. A simple classification of secondary
metabolites includes three main groups: terpenes (such as plant volatiles, cardiac
glycosides, carotenoids and sterols) constituting about 25,000 types, phenolics (such
as phenolic acids, coumarins, lignans, stilbenes, flavonoids, tannins and lignin)
constituting about 8,000 types and nitrogen containing compounds (such as alkaloids
and glucosinolates) constituting about 12,000 types [240]. Whatever the reasons for
the presence of these compounds in nature, they provide an invaluable resource that
have been used to find new drug molecules [241]. A number of traditional separation
techniques with various solvent systems and spray reagents, have been described as
having the ability to separate and identify secondary metabolites [48][238].
When an active extract has been identified, the first task is the identification of the
bio-active phytocompounds. The coupling of preliminary bioassay screening on crude
extracts and chromatographic methods such as column chromatography, preparative
thin layer chromatography (PTLC), high performance liquid chromatography
(HPLC), mass spectrometry (MS) and nuclear magnetic resonance (NMR)
spectroscopy, eventually leading to final bioassay screening, are important for the
acquisition of biologically active compounds.. The successful isolation of compounds
from plant materials largely depends on the type of solvent used for extraction [242].
The extract is separated into individual components and the biological activity of each
fraction is determined until a pure active compound is obtained. The pure compound

232
is then identified using methods such as mass spectrometry and nuclear magnetic
resonance spectroscopy.
Research has demonstrated that natural products represent an unparalleled reservoir of
molecular diversity. The isolation and identification of bio-active phytocompounds in
medicinal plant extracts generally used by local population to treat diseases would
prove an immeasurable contribution to drug discovery and development [235]. This
would not only validate the traditional use of herbal remedies but also provide leads in
the search for new active principles. Investigations surrounding the constituents of the
indigenous Canarium patentinervium Miq. in general, have not been the focus of any
phytochemical study.
10.1.2 Isolation methods
Once the extract has been obtained, the activity within can be demonstrated by
bioassay methods using both the crude extract or by using the fractionated extracts.
Fractionation has the added advantage of getting to the biologically active material
faster. One of the simplest separation methods is partitioning which is a widely used
method as an initial extract purification step. A combination of solvents²miscible
and immiscible ones are used to separate the phytochemicals making up the extract.
This method relies on the ability or the components to be either soluble in water or in
the organic phase [241]. This was then followed by various methods of
chromatography to isolate the bioactive molecules.

233
10.1.3 Crude Fractionation
The objectives at this stage are to simplify the extract composition by dividing it into
groups of compounds sharing similar physicochemical characteristics and/or to
remove the bulk of unwanted materials and thus enrich the extract with respect to the
target compounds. Procedures commonly employed involve solvent partitioning,
defatting, and desalting. The procedure used in this study is a modification of the
method developed by Kupchan [48]. It can be used for defatting and desalting as well.
Most of fats will go with the n-hexane fraction, while inorganic salts will go with the
aqueous one. The advantage of the method is total recovery of target compounds.
Drawbacks are the problems of emulsion formation, time ineffectiveness, and use of
large volumes of solvents.
10.1.4 Chromatography
µ&KURPDWRJUDSK\¶ LV WKH JHQHUDO WHUP IRU D YDULHW\ RI physico-chemical separation
techniques, all of which have in common the distribution of a component between a
mobile phase and a stationary phase. The stationary phase is fixed in the system and
the mobile phase which is a fluid, streams through the chromatographic system. The
various chromatographic techniques are sub-divided according to the physical state of
these two phases. The molecules of the analytes are distributed between the mobile
and the stationary phases. When present in the stationary phase, they are retained and
do not move through the system but when present in the mobile phase, they migrate
with a velocity equivalent to that of the mobile phase. Due to the different
distributions of the analytes, their mean residence time in the stationary phase differs,

234
resulting in a different 40 net migration velocity. This is the principle of the
chromatographic system [243]. Chromatograhic techniques have been instrumental in
the separation of natural products.
10.1.4.1 Thin Layer Chromatography (TLC)
One of the fastest and most widely used chromatographic techniques is Thin Layer
Chromatography. TLC is an analytical technique that can be used to monitor reactions
and for the qualitative analysis of complex mixtures and for the identification of
unknown compounds. It is also important for determining the correct solvent system
with which to run a column chromatography. TLC is composed of two phases, a
mobile and a solid phase. The solid phase is a thin solid support that usually consists
of alumina or silica [48].. The mobile phase is a solvent that moves through capillary
action right through the solid phase. In general, the solid phase is usually polar while
the mobile solvent is non polar relative to the solid phase. The choice of the solvent
depends on the properties of compounds that you want to separate and the nature of
the solid support you are using (ie, is it silica or alumina). If some compounds cannot
be visualized with eyes UV light can be used and a variety of visualisation reagents
can be used [48].
The behaviour of a compound on a TLC is usually described in terms of its relative
mobility or retention factor (Rf) value. Rf is a unique value for each compound under
the same conditions. The Rf for a compound is a constant from one experiment to the
next only if the chromatography conditions (solvent system, adsorbent, thickness of
adsorbent, amount of material spotted and temperature) are also constant [244]. Since

235
these factors are difficult to keep constant from experiment to experiment, relative Rf
YDOXHV DUH JHQHUDOO\ FRQVLGHUHG ³5HODWLYH 5I´ PHDQV WKDW WKH YDOXHV DUH UHSRUWHG
relative to a standard. Based on the Rf values, the identity and characteristics of the
different compounds can be determined. More polar compounds will have smaller Rf
values since they will have a stronger affinity for the polar solid phase. This would
result in the compound not being carried very far along the TLC plate. Compounds
with larger Rf values interact strongly with the less polar mobile phase and thus, tend
to be non polar themselves [238].
PTLC is used for analytical separations of larger quantities of materials. Because
sample mass loading capacity is proportional to the square root of the sorbent layer
thickness, thicker layers are used (>250 ȝPXSWRPP:KHQDELQGHULVUHTXLUHG
the softer inorganic binders (like calcium sulphate) are used so the sample bands can
be easily removed. The compounds to be separated are often applied as long streaks,
developed and then recovered by scraping the adsorbent from the plate and eluting
with a strong solvent [244].
TLC has the advantage of being a highly cost-effective qualitative technique in as
much as a large number or samples can be analysed or separated simultaneously.
Although it is a very simple and convenient technique, one of its limitations is that it
cannot tell the difference between enantiomers and some isomers. Another
disadvantage of TLC is that in order to identify specific compounds, the Rf values for
the compounds of interest must be known beforehand [245].

236
10.1.4.2 Liquid chromatography (column)
Column chromatography is the most common form of chromatography. Column
chromatography is suitable for the physical separation of microgram to kilogram
quantities of material. A solvent acts as the mobile phase while a finely divided solid
surface acts as the stationary phase. The stationary phase will adsorb the components
of the mixture to varying degrees. As the solution containing the mixture passes over
the adsorbent, the components are distributed between the solvent and adsorbent
surface. This process may be described by a three-way equilibrium between the
sample, the solvent and the adsorbent [48]. The solvent and sample compete for
positions on the solid adsorbent, the solvent displacing the sample reversibly and
continuously in the direction of the solvent flow. Consequently, a weakly adsorbed
compound will spend more time in the solvent, and will therefore be eluted first. The
stationary phase is solid and packed in a column made of glass, stainless steel, or
other inert material. The common solid adsorbents are alumina (aluminum oxide) and
silica gel (silicon dioxide). The sample mixture is applied to the top of the column
and the mobile phase passes through the column either by gravity, vacuum, or
pressure [238].
Liquid-solid column chromatography is an effective separation technique when all
appropriate parameters and equipment are used. This method is especially effective
when the compounds within the mixture are colored, as this gives the scientist the
ability to see the separation of the bands for the components in the sample solution.
Even if the bands are not visible, certain components can be observed by other
visualization methods. One method that may work for some compounds is irradiation

237
with ultraviolet light. This makes it relatively easy to collect samples one after
another. However, if the components within the solution are not visible by any of
these methods, it can be difficult to determine the efficacy of the separation that was
performed. In this case, separate collections from the column are taken at specified
time intervals. Since the human eye is the primary detector for this procedure, it is
most effective when the bands of the distinct compounds are visible [246].
10.1.4.3 Size Exclusion Chromatography
Size exclusion chromatography separates molecules by their size. This is done by
having the stationary phase be packed with small particles of silica or polymer to form
uniform pores. The smaller molecules will get trapped in the silica particles and will
elude from the column at a rate that is greater than that of larger molecules. Thus, the
retention time depends on the size of the molecules. Larger molecules will be swept
away in the mobile phase, therefore having a smaller retention time [245]. In this type
RI FKURPDWRJUDSK\ WKHUH LVQ¶W DQ\ LQWHUDFWLRQ EHLQJ SK\VLFDO RU FKHPLFDO EHWZHHQ
the analyte and the stationary phase. The most commonly used size inclusion sorbents
are the dextran gels, particularly the lipophilic versions such as Sephadex LH-20,
which are mostly used for the separation of small hydrophobic natural products from
WKHLUODUJHUµµFRQWDPLQDQWV¶¶XVXDOO\ chlorophylls, fatty acids, and glycerides [48]. In
organic solvents such as chloroform and methanol, these gels swell to form a matrix.
As compounds migrate with the solvent through the gel, small molecules become
included into the gel matrix, whereas larger ones are excluded and migrate at a greater
rate [238].

238
One of the most extensively used gels in natural product separation, especially
nonpolar or intermediate polarity compounds, is Sephadex LH-20, a
hydroxypropylated form of Sephadex G-25. The derivatization offers lipophilicity to
the gel, at the same time preserving its hydrophilicity. As a result of the added
lipophilicity, LH-20 gel swells sufficiently in organic solvents and allows handling of
natural products that are soluble in organic solvents [245]. The useful fractionation
range of LH-20 is approx. 100±4000 amu, and is particularly ideal for removal of
chlorophyll from plant extracts. It should be noted that separations on gels such as
Sephadex LH-20 also involve the mechanisms of adsorption, partition, possibly ion
exchange, and occasionally the trend of larger molecules eluting first and smaller ones
eluting last may be reversed [245]. The gel filtration mode is operational when a
single eluent is used. The partition mechanism comes into play when the eluent is a
mixture of solvents (usually a mixture of polar and nonpolar solvents); the more polar
of the solvents is taken up by the gel, resulting in a two-phase system with stationary
and mobile phases of different compositions [238] .
This form of chromatography has found considerable use in the removal of
µLQWHUIHULQJ¶SODQWSLJPHQWVVXFKDVWKHFKORURSK\OOV that tend to be larger and more
lipophilic than many plant natural products. Sephadex LH-20 has also been used for
the separation of various avermectins and is the most common method for the
preparative isolation of condensed and hydrolysable tannins [247]. This is a non-
destructive method for the recovery of a high quantity or extract [48].

239
10.1.4.4 High performance liquid chromatography system
HPLC instrumentation includes a pump, injector, column, detector and recorder or
data system. The heart of the system is the column where separation occurs. Since the
stationary phase is composed of micrometer size porous particles, a high pressure
pump is required to move the mobile phase through the column. The components (or
analytes) are first dissolved in a solvent, and then forced to flow through a
chromatographic column under a high pressure. The chromatographic process begins
by injecting the solute onto the top of the column where the mixture is resolved into
its components [243]. The amount of resolution is important and is dependant upon
the extent of the interaction between the solute components and the stationary phase.
The stationary phase is defined as the immobile packing material in the column.
Separation of components occurs as the analytes and mobile phase are pumped
through the column. Eventually each component elutes from the column as a narrow
band (or peak) on the recorder [245]. The interaction of the solute with the mobile and
stationary phases can be manipulated through different choices of both solvents and
stationary phases. As a result, HPLC acquires a high degree of versatility not found in
other chromatographic systems and it has the ability to easily separate a wide variety
of chemical mixtures [243].
Modern HPLC uses a non-polar solid phase, like C-18, and a polar liquid phase,
generally a mixture of water and another solvent. High pressures up to 400 bars are
required to elute the analytes through the column before they pass through a diode-
array detector. The detector measures the absorption spectra of the analytes to aid in
their identification. HPLC is useful for compounds that cannot be vaporized or that

240
decompose under high temperature [243]. Initially, pressure was selected as the
principle criterion of modern liquid FKURPDWRJUDSK\ DQG WKXV WKH QDPH ZDV µKLJK
SUHVVXUHOLTXLGFKURPDWRJUDSK\¶7KLVZDV however, an unfortunate term because it
seems to indicate that the improved performance is primarily due to the high pressure.
This is not true. In fact, high performance is the result of many factors: very small
particles of narrow distribution range and uniform pore size and distribution, high
pressure column slurry packing techniques, accurate low volume sample injectors,
sensitive low volume detectors and good pumping systems [243].
There is a growing interest in the chemical composition of plants and the use of
HPLC has several advantages over other methods. The technique is sensitive, rapid
and does not require the preparation of derivatives before separation. A further
advantage is that ultraviolet absorption detectors set in the aromatic region of the
spectrum (e.g. 275 nm) may also be used. Several reports have also been published of
the application of HPLC to various phenolic acids and aldehydes that occur in plants
[248].
10.1.5 Structure elucidation
In most cases of extraction and isolation of natural products, the end point is the
identification of the compound or the conclusive structure elucidation of the isolated
compound. However, structure elucidation of compounds isolated from plants, fungi,
bacteria, or other organisms is generally time consuming, and sometimes can be the
µµERWWOHQHFN¶¶ LQQDWXUDO SURGXFW UHVHDUFK [48]. There are many useful spectroscopic
methods of getting information about chemical structures, but the interpretation of

241
these spectra normally requires specialists with detailed spectroscopic knowledge and
wide experience in natural product chemistry. With the remarkable advances made in
the area of artificial intelligence and computing, there are a number of excellent
automated structure elucidation programs available that could be extremely useful
[238].
If the target compound is known, it is often easy to compare preliminary
spectroscopic data with literature data or to make direct comparison with the standard
sample. However, if the target compound is an unknown and complex natural
product, a comprehensive and systematic approach involving a variety of physical,
chemical, and spectroscopic techniques is required [48]. Information on the chemistry
of the genus or the family of plant or microbe under investigation could sometimes
provide additional hints regarding the possible chemical class of the unknown
compound [249]. Some of the spectroscopic methods used for structure elucidation
include ultraviolet-visible spectroscopy (UV-vis), infrared spectroscopy (IR), mass
spectrometry (MS) and nuclear magnetic resonance (NMR). In this study we have
used NMR.
10.1.5.1 Nuclear magnetic resonance (NMR)
Nuclear magnetic resonance (NMR) is a spectroscopic technique involving a
magnetic field in which a sample is placed. The sample is then subjected to
radiofrequency radiation at the appropriate frequency, allowing for the absorption of
energy depending on the type of nucleus, whether, for example, it is a 1H or 13C. The
frequency also depends on chemical environment of the nucleus, whether methyl or

242
hydroxyl protons are present, molecular conformations, and dynamic processes. Both
1H NMR and 13C NMR are used, with the latter providing information on the carbon
skeleton, and the former relating to the specifics of the hydrogen atoms, thus
complementing each other [245].
10.2 Materials and methods
10.2.1 Isolation of compound 1-5 (crude fractionation)
The isolation scheme for compound 1-5 is shown in Figure 10.1. The ethanol extract
of the leaves (LE-40 g) was dissolved in water. It is then partitioned with petroleum
ether, chloroform, and water to yield the respective solvent extracts (LE-PE, LE-CL
and LE-WA). Phytochemical analysis was repeated as in 3.2 (iv) to confirm the
absence of tannins in LE-PE and LE-CL and the presence of tannins in LE-WA. The
solvent partitioning thus concentrates all tannins in the water fraction.
10.2.2 Isolation of compound 1 & 2
10.2.2.1 Silica gel column chromatography
A glass column was clamped upright and packed with silica gel (size 0.063-0.200
mesh) mixed with the appropriate mobile phase and poured into the column (4 cm x
90 cm) as a compact even suspension. This constituted the stationary phase. The
extract (LE-CL weighing 6 g) was then mixed with a small amount of chloroform

243
and introduced as a thin band to the silica gel. Once the extract was loaded onto the
silica gel, the mobile phase was added at a constant flow rate. Gradient elution of
increasing polarity was initiated with chloroform/methanol gradient elution (the ratio
from 100:0 to 92:8). Thirteen column fractions were collected and analyzed by TLC
(chloroform/methanol). Fractions with similar TLC pattern were combined to total of
four fractions.
10.2.2.2 Preparative thin layer chromatography (PTLC)
Fraction 2 that was yielded from chloroform/ methanol ratio 96:4 was
rechromatographed on a preparative TLC (silica gel plate with 2 mm thickness) with
solvent system chloroform/ methanol (ratio of 1000:15) yielding total 7 bands. Band
five was collected and rechromatographed on preparative TLC (0.5 mm thickness)
with solvent system chloroform/ methanol (ratio of 89:11) to yield four bands, with
band two and band three sent for NMR characterisation.
10.2.3 Isolation of compound 3, 4 & 5
10.2.3.1 Size-exclusion column chromatography
The principle governing this method is based on the molecular size differences of the
compounds, which result in the separation of these compounds. The stationary phase
comprised of a porous three-dimensional polymeric matrix, namely SephadexTM LH-
20 (GE Healthcare). This matrix was initially saturated with ethanol in order to
facilitate swelling before use. SephadexTM LH-20 in ethanol adsorbs tannins while it

244
desorbs tannins in acetone [247]. This is then useful for the separation of tannin and
non-tannin containing fractions. Once swelling was achieved the matrix was then
introduced into a glass column and the fraction (LE-WA) which was dissolved in
ethanol was introduced as a thin band was then applied to the column. The mobile
phase consisted ethanol. Sixteen fractions were collected analyzed by TLC. Fractions
with similar TLC pattern were combined to total of four fractions. The adsorbed
tannins were then flushed with a change of mobile phase to acetone:water (70:30)
resulting in fraction WT. The phytochemical tannin test (section 3.2-iv) was again
performed to confirm the presence of tannins in WT and absence in the other
fractions. The matrix had irreversibly sorbed material that could be tannin [247].
Fraction WB was reintroduced to another column with SephadexTM LH-20 that was
swelled in methanol. Total 3 fractions were collected and fraction WB2 was sent for
nuclear magnetic resonance (NMR) characterisation. Fraction WD was also
introduced to SephadexTM LH-20 that was swelled in methanol. Total five fractions
were collected and fraction WD3 and WD4 were sent for nuclear magnetic resonance
(NMR) characterisation.
10.2.4 Isolation of compound 6-8
10.2.4.1 Isolation of compound 6-8 (PTLC and solvent partioning)
The isolation scheme for compound 6-8 is shown in Figure 10.2. The chloroform
extract of the barks were dissolved in dicloromethane:methanol (2:1). Mixture was
chromatographed on a PTLC (silica gel plate with 2 mm thickness) with solvent

245
system ethyl acetate: methanol (10:1) yielding total three bands (Figure 10.3). First
band (B1) was rechromatographed on PTLC (2 mm thickness) with solvent system
ethyl acetate: methanol (6:1) yielding four bands. The third band upon reconstitution
resulting in fraction B1-BA was subjected to HPLC (section 10.4.5). The second band
(B2) from Figure 10.3 was rechromatographed on PTLC (2 mm thickness) with
solvent system ethyl acetate: methanol (6:1) yielding three bands. The second band of
this PTLC plate upon reconstitution (B2-BA) was subjected to HPLC (section 10.4.5).
The third band (B3) from Fig 10.3 and all the other areas including origin was
combined and partitioned between water and ethyl acetate resulting in aqueous (B3-
AL) and organic layer (B3-OL). The B3-AL fraction was subjected to HPLC (section
10.4.5).
10.2.4.2 Isolation of compound 6-8 (HPLC)
Fraction B1-BA, B2-BA and B3-AL was concentrated in vacuum and then dissolved
in methanol. The solution was then subjected to HPLC individually. The samples
were then qualitatively analyzed by using Agilent 2690 semi-prep HPLC system
(Luna C-18 column, 100 mm x 30 mm) equipped with a 996 photodiode-array
detector (PDA). The injection of the sample was 5 µl and the flow rate was 2 ml/min.

246
Liquid- liquid partitioning
With petroleum ether with chloroform with water
Silica gel 60 SephadexTM LH-20 Final flushing Chloroform:methanol column with (100:0 92:8) Ethanol acetone:water
13 fractions recombined 16 fractions recombined (70:30) to 4 fractions to 4 fractions
PTLC SephadexTM-LH 20 Chloroform:methanol Methanol (1000:15) 7 fractions
PTLC Chloroform:methanol (89:11)
Figure 10.1: Schematic representation of the isolation and purification of compound 1 (scopoletin), compound 2 (scoparone), compound 3 (+-catechin), compound 4 (hyperin) and
compound 5 (cynaroside) isolated from Canarium patentinervium Miq.
Ethanol extract of leaves, LE Dissolved in water ± 40 g
Extraction of Canarium patentinervium Miq. bark and leaves (500 g)
Crude hexane extract Crude chloroform extract Crude ethanol extract
LE-PE ± 11 g LE-CL ± 6 g LE-WA ± 24 g
2% met
CA
4% met
CB
6% met
CC
8% met
CD
CB1 CB2 CB3 CB4 CB5 CB6 CB7
CB4-1 CB4-2 CB4-3 CB4-4
Compound 1 (scopoletin)
49 mg
Compound 2
(scoparone)
11 mg
WA WB WC WD
Final
fraction
WT
WB1 WB2 WB3
Compound 3
(+-catechin)
14 mg
WD1 WD2 WD3 WD4 WD5
Compound 4
(hyperin)
5.2 mg
Compound 5
(cynaroside)
4.8 mg

247
PTLC Ethyl acetate:methanol (10:1)
PTLC PTLC Combined and partitioned Ethyl acetate: methanol Ethyl acetate:methanol between water and (6:1) (6:1) ethyl acetate
Concentrated in vacuum Concentrated in vacuum Concentrated in vacuum Dissolved in methanol Dissolved in methanol Dissolved in methanol 5 ml of solution 5 ml of solution 5 ml of solution Semi-Prep HPLC Semi-Prep HPLC Semi-Prep HPLC Mobile phase Mobile phase Mobile phase (A: water, B: acetonitrile) (A:water, B:acetonitrile) (A:ammonium bicarbonate, B: acetonitrile)
Acetonitrile removed by Acetonitrile removed by Acetonitrile removed by Evaporation and evaporation and evaporation and freeze dried freeze dried freeze dried
Dissolved in methanol Dissolved in methanol Dried under nitrogen Dried under nitrogen
Figure 10.2: Schematic representation of the isolation and purification of compound 6 (vomifoliol), compound 7 (lioxin) and compound 8 syringic acid isolated from Canarium
patentinervium Miq.
Chloroform extract of barks (BC) Dissolved in dicloromethane:methanol
(2:1) -7.2g
Extraction of Canarium patentinervium Miq. bark and leaves (500 g)
Crude hexane extract Crude chloroform extract Crude ethanol extract
Band 1, B1 Band 2, B2 Band 3, B3 Others, BO
B1-BA Others
Fraction
(Rt= 14.1
min)
B1-14 (Solid)
Compound 6
(vomifoliol) -15 mg
B2-BA
Fraction
(Rt= 15.8
min)
B2-15 (Solid)
Compound 7
(lioxin) ± 3.4 mg
Others Aqueous layer
(B3-AL)
Organic
layer
Fraction (Rt=
3.3 min)
B3-3 (Solid)
Compound 8 (syringic
acid) ± 3.6 mg

248
Figure 10.3: The PTLC of chloroform extract of barks
Solution B1-BA: The mobile phase started off with 15 % acetonitrile and 85 % water.
The solvent ratio was changed through a linear gradient to 20 % acetonitrile and 80 %
water in 14 min. This ratio was maintained for 2 min and thereafter the solvent ratio
was changed back to the initial starting conditions. At Rt (retention time) = 14.1 min
the fraction eluted (B1-14) was collected and the acetonitrile was removed by
evaporation and freeze-drying. The solid in the vial was then redissolved in methanol
and dried under nitrogen. B1-14 was then sent for nuclear magnetic resonance (NMR)
characterisation.
Solution B2-BA: The mobile phase started off with 15 % acetonitrile and 85 % water.
The solvent ratio was changed through a linear gradient to 20 % acetonitrile and 80 %
water in 14 min. This ratio was maintained for 4 min and thereafter the solvent ratio
was changed back to the initial starting conditions. At Rt (retention time) = 15.8 min
the fraction eluted (B2-15) was collected and the acetonitrile was removed by
evaporation and freeze-drying. The solid in the vial was then redissolved in methanol
Diagram of TLC

249
and dried under nitrogen. B2-15 was then sent for nuclear magnetic resonance (NMR)
characterisation.
Solution B3-AL: The mobile phase started off with 5 % acetonitrile and 95 % water
(containing 10 mM ammonium bicarbonate). The solvent ratio was changed through a
linear gradient to 16% acetonitrile and 84 % water (containing 10 mM ammonium
bicarbonate) at 1 min. This ratio was maintained for 5 min and thereafter the solvent
ratio was changed back to the initial starting conditions. At Rt (retention time) = 3.3
min the fraction eluted (B3-3) was collected and the acetonitrile was removed by
evaporation and freeze-drying. The solid in the vial was then redissolved in methanol
and dried under nitrogen. B3-3 was then sent for nuclear magnetic resonance (NMR)
characterisation.
10.2.5 Nuclear magnetic resonance (NMR)
Final chemical characterisation of the 8 isolated compounds was achieved by NMR,
which was performed in collaboration with Assistant Professor Dr. Achyut Adhikari
(HEJ Research Institute of Chemistry, University of Karachi, Pakistan). Nuclear
magnetic resonance spectroscopy was performed on a Varian 500 MHz NMR
spectrometer. Samples CB4-2 and CB4-3 were recorded at room temperature in
deuterated chloroform (CDCL3), samples B1-14,B2-15, WB-2, WD-3 and WD-4
were recorded at room temperature in deuteromethanol (CD3OD) while sample B3-3
were recorded at room temperature in deuterated water (D2O).

250
10.3 Results
Compound 1 was isolated as a pale yellow powder. On the basis of the spectral data
obtained for 1H NMR and 13C NMR [Table 10.1 and Figure A1 & A2 (Appendix A )],
and compared to that of relative references [250][251][252], compound 1 was
identified as scopoletin (Figure 10.4). It has an empirical formula of C10H8O4 with
molecular weight of 192.16.
Table 10.1: 1H and 13C NMR spectral data (CDCL3, 500 MHz) of scopoletin
Position įC įH H's Type J (Hz)
2 161.6 - - - -
3 111.6 6.3 1H d 9.5
4 143.3 7.63 1H d 9.5
5 103.2 6.87 1H s -
6 144 - - - -
7 150.2 - - - -
8 107.4 6.95 1H s -
9 149.7 - - - -
10 113.5 - - - -
6- OCH3 56.4 3.98 3H s -
Figure 10.4: The chemical structure of compound 1 (scopoletin)

251
Compound 2 was isolated as a pale yellow powder. On the basis of the spectral data
obtained for 1H NMR and 13C NMR [Table 10.2 and Figure A3 & A4 (Appendix A)],
and compared to that of relative references [253][254], compound 2 was identified as
scoparone (Figure 10.5). It has an empirical formula of C11H10O4 with molecular
weight of 206.19.
Table 10.2: 1H and 13C NMR spectral data (CDCL3, 500 MHz) of scoparone
Position įC įH H's Type J (Hz)
2 161.41 - - - -
3 111.45 6.32 1H d 9.6
4 150.06 7.64 1H d 9.6
5 100.05 6.87 1H s
6 152.87 - - - -
7 146.37 - - - -
8 143.28 6.88 1H s
9 113.59 - - - -
10 107.98 - - - -
6-OCH3 56.38 3.95 3H s -
7-OCH3 56.4 3.98 3H s -
Figure 10.5: The chemical structure of compound 2 (scoparone)

252
Compound 3 was isolated as light yellow needles. On the basis of the spectral data
obtained for 1H NMR and 13C NMR [Table 10.3 and Figure A5 & A6 (Appendix A)],
and compared to that of relative references [255][256], compound 3 was identified as
catechin. Catechin exists in nature as (+) and (-) enantiomers. The optical rotation of
the compound in methanol was tested using Schmidt + Haensch Polartronic H532
Polarimeter. The experimental rotation was an average of +69.25 (n=4) concluding
that this compound is (+)-catechin (Figure 10.6). Previous published data on (+)-
catechin reports an optical rotation of +56.60 [257]. It has an empirical formula of
C15H14O6 with molecular weight of 290.26.
Table 10.3: 1H and 13C NMR spectral data (CD3OD, 500 MHz) of (+)-catechin
Position įC įH H's Type J (Hz)
2 81.46 4.01 1H d 7.5
3 67.41 3.98 1H m 5.5, 8.5
4 27.12
4a - 2.52 1H dd 8.2, 16.1
4b - 2.87 1H dd 5.5, 16.1
5 156.19 - - - -
6 94.86 5.94 1H d 2.3
7 156.45 - - - -
8 94.08 5.87 1H d 2.3
9 155.52 - - - -
10 99.40 2.52 1H dd -
OH - 4.58 1H d -
1' 130.82 - - - -
2' 113.84 6.85 1H d 1.8
3' 144.83 - - - -
4' 144.85 - - - -
5' 114.66 6.78 1H d 8.1
6' 118.62 6.74 1H dd 1.8, 8.1

253
Figure 10.6: The chemical structure of compound 3 [(+)-catechin]
Compound 4 was isolated as a pale yellow powder. On the basis of the spectral data
obtained for 1H NMR and 13C NMR [Table 10.4 and Figure A7 & A8 (Appendix A)],
and compared to that of relative references [258][259], compound 4 was identified as
hyperin (Figure 10.7). It has an empirical formula of C21H20O12 with molecular
weight of 460.37.
Figure 10.7: The chemical structure of compound 4 (hyperin)

254
Table 10.4: 1H and 13C NMR spectral data (CD3OD, 500 MHz) of hyperin
Position įC įH H's Type J (Hz)
2 157.37 - - - -
3 134.36 - - - -
4 178.13 - - - -
5 161.63 - - - -
6 98.55 6.23 1H d 2.10
7 164.90 - - - -
8 93.34 6.43 1H d 2.10
9 157.08 - - - -
10 104.17 - - - -
1' 121.51 - - - -
2' 114.67 7.86 1H d 2.60
3' 144.43 - - - -
4' 148.56 - - - -
5' 116.36 6.88 1H d 8.60
6' 121.51 7.60 1H dd 2.6, 8.6
1'' 103.99 5.19 1H d 8.20
2'' 71.77 3.84 1H dd 8.2, 8.0
3'' 75.80 3.60 1H dd 8.0, 2.1
4'' 68.62 3.87 1H dd 3.2, 2.1
5'' 77.88 3.45 1H m -
6'' 60.53 - - - -
6a'' - 3.67 1H dd 11.0, 3.0
6b'' - 3.56 1H dd 11.0, 5.8
Compound 5 was isolated as a light yellow powder. On the basis of the spectral data
obtained for 1H NMR and 13C NMR [Table 10.5 and Figure A9 & A10 (Appendix
A)], and compared to that of relative references [260][261], compound 5 was
identified as cynaroside (Figure 10.8). It has an empirical formula of C21H20O11 with
molecular weight of 448.38.

255
Table 10.5: 1H and 13C NMR spectral data (CD3OD, 500 MHz) of cynaroside
Position įC įH H's Type J (Hz)
2 165.52 - - - -
3 102.72 6.60 1H s
4 182.66 - - - -
5 161.51 - - - -
6 100.27 6.53 1H d 2.00
7 163.43 - - - -
8 94.65 6.83 1H d 2.00
9 157.58 - - - -
10 105.69 - - - -
1' 121.98 - - - -
2' 112.81 7.43 1H d 2.20
3' 145.77 - - - -
4' 149.80 - - - -
5' 115.40 6.93 1H d 8.40
6' 119.12 7.45 1H dd 8.40, 2.20
1'' 99.75 5.09 1H d 7.30
2'' 73.33 3.48 1H t 9.20
3'' 76.46 3.56 1H t 9.20
4'' 69.86 3.45 1H t 9.20
5'' 77.00 3.60 1H m -
6'' 61.06 - - - -
6a'' - 3.75 1H dd 12.40, 5.80
6b'' - 3.95 1H dd 12.40, 1.90
Figure 10.8: The chemical structure of compound 5 (cynaroside)

256
Compound 6 was isolated as white solid (HPLC chromatogram in Figure B1-
Appendix B). On the basis of the spectral data obtained for 1H NMR and 13C NMR
[Table 10.6 and Figure A11 & A12 (Appendix A)], and compared to that of relative
references [262][263], compound 5 was identified as vomifoliol (Figure 10.9). It has
an empirical formula of C13H20O3 with molecular weight of 224.30.
Table 10.6: 1H and 13C NMR spectral data (CD3OD, 500 MHz) of vomifoliol
Position įC įH H's Type J (Hz)
1 78.54 - - - -
2 41.03 - - - -
3a - 2.18 1H d 15.00
3b - 2.54 1H d 15.00
4 199.85 - - - -
5 125.69 5.90 1H m -
6 166.10 - - - -
7 128.71 5.81 1H m -
8 135.49 5.82 1H m -
9 67.34 4.34 1H m -
10 22.41 1.26 3H d 7.50
11 23.06 1.03 3H s -
12 22.05 1.06 3H s -
13 18.15 1.94 3H s -
Figure 10.9: The chemical structure of compound 6 (vomifoliol)

257
Compound 7 was isolated as white powder (HPLC chromatogram in Figure B2-
Appendix B). On the basis of the spectral data obtained for 1H NMR and 13C NMR
[Table 10.7 and Figure A13 & A14 (Appendix A)], and compared to that of relative
references [264][265], compound 7 was identified as lioxin (Figure 10.10). It has an
empirical formula of C8H8O3 with molecular weight of 152.15.
Table 10.7: 1H and 13C NMR spectral data (CD3OD, 500 MHz) of lioxin
Position įC įH H's Type
1 127.72 - - -
2 110.07 7.30 1H m
3 149.09 - - -
4 147.08 - - -
5 115.51 7.30 1H d
6 125.64 7.31 1H m
7 191.60 9.57 1H s
OH - 6.77 1H s
10 54.80 3.80 3H s
Figure 10.10: The chemical structure of compound 7 (lioxin)

258
Compound 8 was isolated as pale yellow powder (HPLC chromatogram in Figure B3-
Appendix B). On the basis of the spectral data obtained for 1H NMR and 13C NMR
[Table 10.8 and Figure A15 & A16 (Appendix A)], and compared to that of relative
references [266][267], compound 8 was identified as syringic acid (Figure 10.11). It
has an empirical formula of C9H10O5 with molecular weight of 198.17.
Table 10.8: 1H and 13C NMR spectral data (D2O, 500 MHz) of syringic acid
Position įC įH H's Type
1 121.08 - - -
2 106.92 7.11 1H s
3 147.08 - - -
4 148.41 - - -
5 147.08 - - -
6 106.92 7.11 1H s
2-OCH3, 6-OCH3 56.26 3.57 3H s
COOH 174.29 - - -
Figure 10.11: The chemical structure of compound 8 (syringic acid)

259
10.4 Discussion
All the compounds were isolated for the first time from Canarium patentinervium
Miq. Seven phenolic compounds (compounds 1-6 and 8) and one norsesquiterpene
with cyclohexenone ring (compound 7) was isolated.
The 1H-NMR spectrum of compound 1 (Table 10.1) revealed the presence of two
doublets at įH 7.63 (H-4, J = 9.5 Hz) and 6.30 (H-3, J = 9.5 Hz). The two singlet
signals appeared at įH 6.87 and 6.95 were assigned for the two protons H-5 and H-8,
respectively. The 1H NMR spectra of compound 2 identified as scoparone (Table
10.2) showed two doublets with coupling constant of 9.+]DWįDQGį 7.64,
which were assigned as H-3 and H-4, respectively, characteristic for coumarins [253].
The 1H NMR spectrum of scoparone was similar to that of scopoletin, except for an
additional siQJOHWDWįppm indicating its identity as scoparone [268].
Compound 3 were isolated as light yellow crystals. The structural identity of this
compound was determined through NMR spectral analysis (Table 10.3) that was
compared to literature [255][256]. Catechin possesses two benzene rings (called the
A- and B-rings) and a dihydropyran heterocycle (the C-ring) with a hydroxyl group
on carbon 3. The A ring is similar to a resorcinol moiety while the B ring is similar to
a catechol moiety. There are two chiral centers on the molecule on carbons 2 and 3
resulting in 4 optical isomers (+) catechin, (-) catechin, (+) epicatechin and (-)
epicatechin. Of these, (+) catechin and (-) epicatechin occur in nature [269]. From the
experimental data of optical rotation, this compound was confirmed to be (+)
catechin.

260
Compound 4 was isolated as pale yellow powders. In the 1H-NMR spectrum, the
typical proton signals of quercetin moiety, that is, those of an AMX system due to a
1',3',4'-WULVXEVWLWXWLRQRIULQJ%>į 7.86 (d, J = 2.+]GG- +]DQGį
6.88 (d, J = 8.6 Hz)], and a typical meta-coupled pattern for H-8 and H-SURWRQV>į
6.43 (d, J = 2.1 Hz) and 6.23 (d,J = 2.1 Hz)], were observed. The 13C-NMR of
compound 4 showed resonances for twenty one carbons and presence of a quercetin
moiety with the exception of monosaccharide moiety (Table 10.4). An anomeric
proton signals of compound 4 DSSHDUHGDWį 5.19 (d, J = 8.2 Hz) and other oxygenated
methines and methylene signals RIVXJDUPRLHW\DWį (dd, J = 3.2, 2.1 Hz, H-4''),
3.84 (dd, J = 8.2, 8.0 Hz, H-2''), 3.67 (dd, J = 11.0, 3.0, H-6''a), 3.56 (dd, J = 11.0, 5.8
Hz, H-6''b), 3.60 (dd, J = 8.0, 2.1 Hz, H-3'') and 3.45 (m, H-5''). Also, the carbon
VLJQDOVRIWKHVXJDUPRLHW\ZHUHREVHUYHGDWį 103.99 (C-1''), 77.88 (C-5''), 75.80 (C-
3''), 71.77 (C-2''), 68.62 (C-4'') and 60.53 (C- VXJJHVWLQJ WKH SUHVHQFH RI ȕ-
galactopyranoside units. Thus, the structure of compound 4 was identified as
quercetin-3-O-ȕ-D-galactopyranoside, hyperin [258].
The 1H NMR spectrum of compound 5, a light yellow powder, showed two meta±
coupled doublets (J=2.0 Hz) at į6.83 and 6.53, each integrating for one proton, and
were assigned to H-8 and H-6, respectively of ring A of 5,7-dihydroxyflavonoids
[270]. The presence of ABX system at į7.45 (dd, J = 8.4, 2.2 Hz, H-6'), 7.43 (d, J =
2.2 Hz, H-2') and 6.93 (d, J = 8.3 Hz, H-5'), characteristic of 1, 2, 4-trisubstituted
phenyl unit [270]. The only singlet at į6.60, integrating for one proton, was attributed
C-3 to proton of flavonoids (35). These spectral data revealed the presence of luteolin
skeleton [271]. In addition, the 1H NMR spectrum showed a series of signals between

261
įGG- +]+-6''b), 3.75 (dd, J = 12.40, 1.90 Hz, H-6''a), 3.60 (t,
H-5''), 3.56 (t, J = 9.2 Hz, H-3''), 3.48 (dd, J = 9.2, H-2'') and 3.45 (t, J = 9.2 Hz, H-4'')
attributable to a sugar moiety. The coupling constant (J = 7.3 Hz) of the anomeric
proton ORFDWHG DW į (d, J = 7.3 Hz, H-1'') and the 13C NMR (Figure 10.8)
chemical shifts of the sugar carbons į and 61.06)
UHYHDOHGWKHSUHVHQFHRIȕ-O-glucoside unit in luteolin- 7-O-glucoside. The 13C NMR
data showed the presence of a ketone carbonyl at C-į), two olefinic carbons
at C-2 and C-3 įDQG), and four hydroxyl carbons at C-7, C-5, C-4'
and C-į, 161.51, 149.80 and 145.77). On comparison with the literature on
flavonoids of luteolin, it was revealed that the physical and spectral data of luteolin-7-
O-glucoside isolated were in good agreement with those recorded for luteolin- 7-O-
glucoside or cynaroside [271][260].
The comparison of the corresponding NMR data of compound 6, a white solid (Table
10.6) with those reported in literature [262][263] led to its identification as vomifoliol.
The cyclohexenone structure was confirmed by the observation in the HMBC (Figure
A13-Appendix A) spectrum of correlations between the AB system H-3ab (įį
2.54, J = 15.00 Hz) and C-4 į ), C-1 į ) and C-5 į .69). The
location of the two high field methyl groups C-11 and C-12 at C-2 was ascertained by
the detection of the H3ab-C11 and H3ab-C12 correlations. The position of the side
chain on C-1 of the cyclohexenone ring is confirmed by the observation in the HMBC
spectrum of the H7-C1 correlation. The COSY (Figure A14-Appendix A) spectrum
revealing FRUUHODWLRQVEHWZHHQWKHROHILQLFVLJQDO+įZLWKWKHRWKHUROHILQLF
proton H8 į) which in turn had a cross peak with H9 į) permitted us to

262
confirm the structure of this side chain. All these NMR data together suggested the
structure of vomifoliol.
The 1H NMR spectrum of compound 7 which was isolated as white powder exhibited
the following signals (Table 10.7)į (CHO), multiplets at į 7.30, 7.31 (2H) and
į GRXEOHW DW į 7.04 (1H) (aromatic protons), and 3.80 ppm (CH3O). The 13C NMR
VSHFWUXPVKRZHGVLJQDOVDWį (CHO), 149.09, 147.08, 127.72, 125.64, 115.51,
and 110.07 (aromatic carbons) and 54.80 ppm (CH3O). The 1H and 13C spectra were
identical to those of 4-hydroxy-3-methoxybenzaldehyde (Figure 10.10) (also known
as vanillin or lioxin) [265].
Compound 8 eluted first to give a pale yellow powder on drying. The 13C NMR
showed two methoxyls, two olefinic methines and five quaternary carbons, including
a carboxy, and three oxygenated olefinic carbons. The only signals appearing in the
1H NMR were a 6H singlet for the two methoxyls (į) and a 2H singlet for two
aromatic protons (į 7.11) (Table 10.8). Further analysis of the NMR data indicated a
symmetric trisubstituted phenolic acid. Two of the substituents were methoxyl groups
and the third a hydroxyl group. The data was found to fit well with that of 4-hydroxy-
3,5-dimethoxybenzoic acid (syringic acid) [266][267]. Further evidence for the
structure of 8 was provided by a 3-bond correlation between H-2/6 (į7.11) and the
carboxy carbon (į 174.29) (Figure 10.11). The possibility of 4-hydroxy-2,6-
dimethoxybenzoic acid was thus eliminated.

263
10.5 Conclusion
Eight compounds were isolated for the first time from Canarium patentinervium Miq.
using various isolation techniques such as TLC, CC, HPLC and identified with NMR
method. Compounds isolated scopoletin, scoparone, (+)-catechin, hyperin,
cynaroside, lioxin and syringic acid were phenolics while vomifoliol was a
norsesquiterpene with a cyclohexenone ring. Lioxin, syringic acid and vomifoliol
were isolated from this genus for the first time. These findings agree with the
phytochemical analysis which was done in Chapter 2.

264
CHAPTER 11
BIOACTIVITY OF ISOLATED SECONDARY METABOLITES
11.1 Introduction
11.1.1 Terpenes and phenols
Terpenes are the most numerous and structurally diverse plant natural products. The
term terpene usually refers to a hydrocarbon molecule while terpenoid refers to a
terpene that has been modified, such as by the addition of oxygen. Terpenoids
(sometimes called as isoprenoids) are hydrocarbon natural products based on 5-
carbon (isoprene) units as their building blocks [237]. Terpenes are an enormous class
of natural products spanning well over 30,000 members having been discovered to-
date [272]. These compounds are classified as to the number of isoprene units, which
include the hemiterpenes (1), monoterpenes (2), sesquiterpenes (3), diterpenes (4),
sesterterpenes (5), tripterpenes (6), and polyterpenes (many units). Sometimes the
prefix nor- is added to name a structural analog that can be derived from a parent
compound by the removal of one carbon atom along with the accompanying hydrogen
atoms (norsesquiterpene) [237].
Of these different major classes of terpenoids, the diterpenes are structurally most
diversified, possessing at least 6 large structural groups. Biological active compounds
are found in each class of the terpenoids, particularly among the sesquiterpenes,
diterpenes and triterpenes [237]. Isopentyl diphosphate (IPP) and dimethyl allyl

265
diphosphate (DMAPP) are found to be the actual building blocks in terpene
biosynthesis [273]. There are 2 biosynthetic pathways for the production of IPP and
DMAPP, the mevalonate pathway and the more recently discovered deoxyxylulose
pathway. All terpenes are formed through the reactions of IPP and DMAPP [237].
Rare terpenoids such as norisoprenoids, like the C13-norisoprenoids 3-oxo-Į-ionol are
found to be present in Muscat of Alexandria leaves and 7,8-dihydroionone
derivatives, and megastigmane-3,9-diol and 3-oxo-7,8-dihydro-Į-ionol found in
Shiraz leaves (both grapes in the species Vitis vinifera) or wine (responsible for some
of the spice notes in Chardonnay) [274].
Polyphenols are the third most widespread class of metabolites in nature, and their
distribution is almost ubiquitous [241]. It is estimated that 100,000 to 200,000
secondary metabolites exist and some 20% of the carbon fixed by photosynthesis is
channeled into the phenylpropanoid pathway, thus generating the majority of the
natural-occurring phenolics, such as flavonoids and stilbenes [275].. Although a large
variety of plant phenols exists, most of these compounds arise from a common origin:
the amino acids phenylalanine or tyrosine. These aminoacids are deaminated to
cinnamic acids, which enter the phenylpropanoid pathway. As a general rule recently
proposed by Quideau et al. [276]WKHWHUPµSODQWSKHQROLFV¶VKRXOGEHVWULFWO\XVHGWR
refer to secondary natural metabolites arising biogenetically from the shikimate/
phenylpropanoid pathway, which directly provides phenylpropanoids (Figure 11.1),
RUWKHµSRO\NHWLGH¶acetate/malonate pathway, which can produce simple phenols, or
both of them [277]. A key step in this biosynthetic route is the introduction of one or
more hydroxyl groups into the phenyl ring. As result, these compounds are derived
from a common carbon skeleton building block: the C6-C3 phenylpropanoid unit [53].

266
Biosynthesis, according to this pathway, produces the large variety of plant phenols:
cinnamic acids (C6-C3), benzoic acids (C6- C1), flavonoids (C6-C3-C6),
proanthocyanidins [(C6-C3-C6)n], coumarins (C6-C3), stilbenes (C6-C2-C6), lignans
(C6-C3-C3-C6) and lignins [(C6-C3)n]. Main classes of phenolics include phenolic acid
(benzoic acid derivatives and cinnamic acid derivatives), flavonoids (flavones,
flavonols, flavanones, flavan-3-ols, anthocyanidins and isoflavones), coumarins,
stilbenes and lignans (Figure 11.2) [275]. These pathways produce a bewildering
array of monomeric and polymeric VWUXFWXUHVWKHWHUPµSRO\SKHQROV¶GHILQLQJWKRVH
with more than one phenolic ring) that fulfill a very broad range of physiological roles
in plants [277]. Although the bulk of these compounds play cell wall structural roles,
plant tissues synthesize a vast array of non-structural constituents that have various
roles in plant growth DQGVXUYLYDO7KXVWKHH[SUHVVLRQ³SODQWSKHQROLFV´HPEUDFHVD
highly diverse group with an extremely large structural diversity: tens of thousands of
diverse structures have been identified, with the number continually increasing
[276][278].
11.1.2 General antioxidant mechanisms of phenolics
Phenolics are able to act as antioxidants in a number of ways. Phenolic hydroxyl
groups are good hydrogen donors: hydrogen-donating antioxidants can react with
reactive oxygen and reactive nitrogen species in a termination reaction, which breaks
the cycle of generation of new radicals [279]. Following interaction with the initial
reactive species, a radical form of the antioxidant is produced, having a much greater
chemical stability than the initial radical. The interaction of the hydroxyl groups of
phenolics with the ʌ-electrons of the benzene ring gives the molecules special

267
properties, most notably the ability to generate free radicals where the radical is
stabilized by delocalization. The formation of these relatively long-lived radicals is
able to modify radical-mediated oxidation processes [280]. The antioxidant capacity
of phenolic compounds is also attributed to their ability to chelate metal ions involved
in the production of free radicals [281]. However, phenolics can act as pro-oxidants
by chelating metals in a manner that maintains or increases their catalytic activity or
by reducing metals, thus increasing their ability to form free radicals [282]. Phenolic
structures often have the potential to strongly interact with proteins, due to their
hydrophobic benzenoid rings and hydrogen-bonding potential of the phenolic
hydroxyl groups. This gives phenolics the ability to act as antioxidants also by virtue
of their capacity to inhibit some enzymes involved in radical generation, such as
various cytochrome P450 isoforms, lipoxygenases, cyclooxygenase and xanthine
oxidase [280][283].

268
Figure 11.1: Schematic of the major branch pathways of (poly) phenol biosynthesis. PAL: phenylalanine ammonia-lyase, C4H: cinnamate-4-hydrolase, 4CL: 4-coumaroyl:CoA-ligase, HCT:
hydroxycinnamoyl transferase, C3H: p-coumarate-3-hyroxylase, CHS: chalcone synthase, CHI: chalcone isomerase, ANS: anthocyanidinstnthase, DFR: dihydroflavonol reductase, FS: flavone
synthase, F3H: Flavanone 3-hydroxylase, IFS: isoflavone synthase, ANR: anthocyanidin reductase, LAR: leucoanthocyanidin reductase [277].

269
Figure 11.2: Main classes of phenolics [275]

270
11.1.3 Flavonoids
Approximately 9000 different flavonoids have been reported from plant sources, and
with almost certainty many more are still to be discovered, as they continue to capture
the interests of scientists from numerous disciplines. Based on the 10-carbon skeleton
of flavonoids, they can be substituted by a range of different groups, viz. hydroxyl,
methoxyl, methyl, isoprenyl and benzyl substituents [284]. Flavonoids are
characterized by a phenylbenzopyran chemical structure. The general structure
includes a C15 (C6-C3-C6) skeleton joined to a chroman ring (benzopyran moiety). The
heterocyclic benzopyran ring is known as the C ring, the fused aromatic ring as the A
ring, and the phenyl constituent as the B ring (Figure 11.2). Commonly flavonoids are
further modified by the addition of substituent groups such as methyl groups,
aromatic acyl groups, and/or sugar moieties [284]. Increasingly, flavonoids are
becoming the subject of medical research. Flavonoids play a role in disease
prevention in humans. They have been reported to possess many useful properties,
including anti-inflammatory, oestrogenic, enzyme inhibition, antimicrobial,
antiallergic, vascular and cytotoxic antitumour activity [285], but the antioxidant
activity is, without a doubt, the most studied one attributed to flavonoids. This well
established antioxidant activity of flavonoids is also responsible for other biological
activities in which the prevention of oxidative stress is beneficial [286].
The anti-oxidants activity of the flavonoids which neutralize damaging chemicals in
the body such as free radicals results in reduction of oxidative stress, which are
thought to contribute to a number of diseases, including atherosclerosis, degenerative
brain GLVHDVHVOLNH$O]KHLPHU¶VGLVHDVHDVZHOODVSUHPDWXUHDJHLQJ [52]. Flavonoids

271
have also been found to help the body fight viruses and decrease allergic reactions.
There is abundant evidence to support the role of flavonoids in disease prevention a
high dietary intake of quercetin is related to a reduced risk of heart disease [287].
Rutin has been found to help prevent stomach ulcers by protecting the stomach lining
and it also protects blood vessels. Flavonoids have also been found to reduce the
µVWLFNLQHVV¶ RI EORRG SODWHOHWV DQG thus reduce the formation of blood clots, which
reduce the risks of certain cardiovascular diseases [288]. Flavan-3-ols are thought to
interfere in the pathogenesis of cardiovascular disease via several mechanisms:
antioxidative, antithrombogenic, and antiinflammatory. In particular,
proanthocyanidins and flavan-3-ol monomers aid in lowering plasma cholesterol
levels, inhibit LDL oxidation, and activate endothelial nitric oxide synthase to prevent
platelet adhesion and aggregation that contribute to blood clot formation [289][290].
The anticancer activity of some flavonoid compounds is due to their ability to
scavenge free radicals, thus avoiding the early stages of cancer promotion. Besides
this mechanism, flavonoids have also been reported to act as anticancer agents via
regulation of signal transduction pathways of cell growth and proliferation,
suppression of oncogenes and tumor formation, induction of apoptosis, modulation of
enzyme activity related to detoxification, oxidation and reduction, stimulation of the
immune system and DNA repair, and regulation of hormone metabolism [289]. In the
past few years we have witnessed the establishment of other flavonoid classes as
potent molecules for the treatment of other pathologies that do not involve these
FRPSRXQGV¶ DQWLR[LGDQW properties. This is the case of some isoflavones, whose
estrogen-like capacity is now well established. The activity of these compounds is
related with their similarity to estradiol estrogen. Genistein and daidzein have

272
demonstrated to be promising molecules for the treatment of conditions in which the
agonist effect in estrogen receptors is beneficial, such as menopause conditions. In
fact, several preparations containing these compounds, mainly soya-derived, are now
used in therapeutics [291].
11.1.4 Phenolic acids
Phenolic acids have a carboxyl group attached or linked to benzene ring (Figure 11.2).
Two classes of phenolic acids can be distinguished depending on their structure:
benzoic acid derivatives (i.e. hydroxybenzoic acids, C6-C1) and cinnamic acid
derivatives (i.e. hydroxycinnamic acids, C6-C3) (Figure 11.2) [292]. Hydroxycinnamic
acid compounds (p-coumaric, caffeic acid, ferulic acid) occur most frequently as
simple esters with hydroxy carboxylic acids or glucose, while the hydroxybenzoic
acid compounds (p-hydroxybenzoic, gallic acid, ellagic acid) are present mainly in the
form of glucosides. Furthermore, phenolic acids may occur in food plants as esters or
glycosides conjugated with other natural compounds such as flavonoids, alcohols,
hydroxyfatty acids, sterols, and glucosides [293].
They have been the subject of a great number of chemical, biological, agricultural,
and medical studies. Among the wide diversity of naturally occurring phenolic acids,
at least 30 hydroxy- and polyhydroxybenzoic acids have been reported in the last 10
years to have biological activities [294]. They have antioxidant, antimutagenic and
even leaf movement regulating agents that protect the organism that produces them
from the oxidative stress created by metabolism and their physical environment [294].
They also have antiviral, antibacterial (bactericidal, bacteriostatic), algicidal, plant

273
growth regulating, phytotoxic, antifungal, antiprotozoal, nematicidal, insecticidal,
antifeedant, and mammalian estrogenic, keratolytic, platelet aggregation inhibiting,
hypoglycemic, cytotoxic, and neurotoxic activities that may serve to protect the
organism that biosynthesizes them from competing, pathogenic, and herbivorous
organisms in their biological environment [295][296]. The diverse biological
functions of these phenolic acids suggest potential pharmacological activities.
11.1.5 Coumarins
&RXPDULQV RZH WKHLU FODVV QDPH WR µFRXPDURX¶ WKH YHUQDFXODU QDPH RI WKH WRQND
bean (Dipteryx odorata Willd., Fabaceae), from which coumarin itself was isolated in
1820 [297]. The primary site of synthesis of coumarins is suggested to be the young,
actively growing leaves, with stems and roots playing a comparatively minor role.
Coumarins belong to a group of compounds known as the benzopyrones, all of which
consist of a benzene ring joined to a pyrone (Figure 11.2). To this days, around 1,300
coumarins are known, with all of them being derivatives of 5,6-benzo-2- pirone (Į-
chromone) (with OH, OCH3 or CH3 substituents on the benzoic ring.). Coumarins
may also be found in nature in combination with sugars, as glycosides. As derivatives
of simple coumarins, other compounds are known, such as furanocoumarins, which
include a furanic ring, linear pyranocoumarins, angular pyranocoumarins, dimeric
coumarins, of which dicoumarol is an example and also furanochromones [298].
Historically, the ability of dicoumarol to inhibit blood clotting, that later led to the
development of the anticoagulant drug warfarin, was the first call to this class of
FRPSRXQGV¶ ELRORJLFDO SURSHUWLHV Coumarin is the parent molecule of warfarin,

274
which acts as a vitamin K antagonist. Warfarin is a clinically useful anticoagulant and
widely employed rodenticide. Several biological activities have been reported in
natural-occurring coumarins, from photo sensitizers to vasodilatation. Recently, the
interest has been given to synthetic derivatives of coumarins, such as fluorinated and
1-azo coumarins, which displayed moderate analgesia properties, and excellent anti-
inflammatory and anti-microbial activities [299].
Coumarins have a variety of bioactivities including anticoagulant, estrogenic, dermal,
photosensitizing, antimicrobial, vasodilator, molluscidal, antihelmintic, sedative,
hypnotic, analgesic and hypothermic activity [300]. Various coumarins have been
reported to possess anti-inflammatory activity as shown in carrageenan-induced
inflammation and cotton pellet granuloma tests [301][302]. There have been reports
on efficacies of pure coumarins as well as extracts containing them against Gram-
positive and Gram-negative bacteria as well as fungi [303]. The free OH group at C-6
in the coumarins nucleus has been found to be important for antifungal activity, while
the free hydroxyl group at C-7 is important for antibacterial activity [304]. The
bioactivities of phototoxic psoralens and dicoumaroul derivatives are well known and
several of these compounds are used in antipsoriatic and anticoagulant therapy [305].
Other than psoriasis, skin disease like cutaneous T-cell lymphoma, atopic dermatitis,
urticaria pigmentosa and lichen planus [306] are treated with the photochemotherapy
of linear furanocoumarins (psoralens). The most widely used compound is
xanthotoxin [307]. Bergapten is considered a valuable alternative for chemotherapy of
psoriasis, since its clinical efficacy is comparable to that of xanthotoxin, although
bergapten requires higher cumulative UVA doses.

275
The inherent fluorescent properties of many coumarins are a key factor in many
applications. Areas where coumarins are widely used include estimation of enzymatic
activity [308], labeling of proteins, antibodies, DNA and lipids, derivatising agents in
chromatography, etc. Coumarins have a wide variety of uses in industry, mainly due
to their strong fragrant odour [308]. Uses include that of a sweetener and fixative of
perfumes (e.g. 3,4-dihydrocoumarin), an enhancer of natural oils such as lavender, a
food additive in combination with vanillin, a flavour/odour stabilizer in tobaccos and
an odour masker in paints and rubbers. 6-Methylcoumarin is mainly used as a flavour
enhancer, and 7- hydroxycoumarin is mainly used in sunscreens.
11.2 Material and methods
Eight compounds that have been isolated in Chapter 11 were subjected to various
biological assays.
11.2.1 Antioxidant capacity assays
Samples were dissolved in dimethyl sulfoxide (DMSO, R&M) prior to assay at a
stock concentration of 1 mg/ml, and serial dilution was done accordingly to obtain a
good EC50 curve. Trolox (6-hydroxy-2,5,7, 8-tetramethylchromon-2-carboxylic acid,
Sigma-Aldrich), vitamin C (l-ascorbic acid), and quercetin were used as positive
control at a stock concentration of 1 mg/ml. Assay was performed using Thermo
Scientific Varioskan Flash microtiter plate reader, linked to a computer equipped with
(SkanIt Software 2.4.3). EC50 values were determined using Prism 5.00 software. At

276
least three independent tests were performed for each sample. Method is as explained
in Chapter 4, DPPH assay (section 4.2.1.2), ABTS assay (section 4.2.2.2), FRAP
DVVD\ VHFWLRQ ȕ-carotene bleaching assay (section 4.2.4.2) and SOD assay
(section 4.2.5.2)
11.2.2 Anti-inflammatory determination assays
Test samples and positive controls were dissolved in dimethyl sulfoxide (DMSO,
R&M) prior to assay at a stock concentration of 1 mg/ml, and serial dilution was done
according to the assay to obtain a good EC50 curve. Absorbance was recorded using
Thermo Scientific Varioskan Flash microtiter plate reader, linked to a computer
equipped with (SkanIt Software 2.4.3). Percentage inhibition of enzyme was
determined by comparison of rates of reaction of samples relative to blank sample
using the formula (E ± S)/ E × 100, where E is the activity of enzyme without test
sample and S is the activity of enzyme with test sample. The experiments were done
in triplicate. Method has been explained in Chapter 5, 5-LOX inhibition assay (section
5.4.2.2) and the peroxidase endpoint assay for COX-1 and COX-2 (section 5.5.3.2)
11.2.3 Anti-acetylcholinesterase determination assays
Test samples and galanthamine which was used as positive control was dissolved in
dimethyl sulfoxide (DMSO, R&M) prior to assay at a stock concentration of 1 mg/ml,
and serial dilution was done accordingly to obtain a good EC50 curve. The reaction
was monitored utilizing a 96-well microplate Thermo Scientific Varioskan Flash
microtiter plate reader, linked to a computer equipped with (SkanIt Software 2.4.3).

277
Percentage inhibition of AChE was determined by comparison of rates of reaction of
samples relative to blank sample (ethanol in phosphate buffer pH = 8) using the
formula (E ± S)/ E × 100, where E is the activity of enzyme without test sample and S
is the activity of enzyme with test sample. The experiments were done in triplicate.
Method is explained in Chapter 6 (section 6.4.6.2).
11.2.4 Anticancer assay
All samples except syringic acid (insufficient yield) were tested for this activity
against human derived cell lines MDA 468 (ER-) breast carcinoma]. Samples were
prepared as 1 mg/ml stock solutions, dissolved in DMSO, and stored at 4 0C,
protected from light for a maximum period of 4 weeks. Serial drug dilutions were
prepared in medium immediately prior to each assay. Due to good activity exhibited,
scopoletin was also tested against HT-29, MCF-7 and HCT 116 cell lines. Method is
as explained in Chapter 9 section 9.3.2.
11.2.5 Antibacterial assay
Due to insufficient yield of compounds isolated, all samples were tested for the
antimicrobial effect only against S. aureus ATCC 11632. Samples was dissolved in
dimethyl sulfoxide (DMSO, R&M) prior to assay at a stock concentration of 1 mg/ml.
Method used is as explained previously, MIC assay (section 7.2.2.2), MBC assay
(section 7.2.3.2) and time-kill assay (section 7.2.4.2).

278
11.2.6 Antiparasitic assay
Due to insufficient yield of compounds isolated, scopoletin was tested for both assays
and only scoparone, (+)-catechin, lioxin and vomifoliol was tested for the
antileshmanial assay. Method is as explained in chapter 8, for antiparasitic assay
against Giardia intestinalis and Entamoeba histolytica (section 8.3.2.2.1) and
antileishmanial activity against Leishmania donovani promastigotes (section
8.3.2.2.2).
11.2.7 Formulation of scopoletin
Due to potent antioxidant, antibacterial and anti-inflammatory activity (to be
discussed in 11.8), scopoletin was formulated as a gel. Scopoletin (25 mg) was
prepared as a 0.5 % w/w preparation. Scopoletin was dissolved in few drops of carrier
solvent DMSO. Then orange oil is added to increase the penetration via skin. Pure
aloe vera gel was then added in sufficent amount to make up a total of 5 g of gel.
11.2.8 Statistical analysis
For section 11.2.1-11.2.3, concentration±response curves were calculated using the
Prism software package 5.00 for Windows, GraphPad Software, San Diego California
USA, www.graphpad.com (GraphPad, San Diego, USA) and data were reported as
mean and SD values obtained from a minimum of three determinations. Non linear
best fit was plotted with SD and 95% confidence interval. All data were expressed as

279
mean ± standard deviation. SD was computed but values were not shown in the
graphs via this software. For section 11.2.4-6 growth curve of bacteria/parasite/cell
were analysed using MS-Excel and data were reported as mean and SD values
obtained from a minimum of three determinations. All data were expressed as mean ±
standard deviation. Data were analyzed using one way Anova followed by Tukey test.
A significant difference was considered at the level of P < 0.01.
11.3 Results
11.3.1 Antioxidant capacity assays
The antioxidant capacity of the isolated compounds is shown in Table 11.1. In the
DPPH assay, the highest antioxidative action was exhibited by (+)-catechin (EC50
4.77±0.01 µg/ml) similar with positive control trolox (EC50 4.77±0.01 µg/ml),
followed by hyperin (EC50 4.89±0.02 µg/ml), cynaroside (EC50 7.89±0.03 µg/ml),
syringic acid (EC50 15.43±0.02 µg/ml) and scopoletin (EC50 36.80±0.01 µg/ml). Both
lioxin and vomifoliol had EC50 >100 µg/ml.
In the ABTS assay, the highest antioxidative action was exhibited by cynaroside
(EC50 0.26±0.02 µg/ml) followed by scopoletin (EC50 1.08±0.03 µg/ml), (+)-catechin
(EC50 5.03±0.06 µg/ml), syringic acid (EC50 7.38±0.02 µg/ml) hyperin (EC50
8.19±0.04 µg/ml), vomifoliol (EC50 34.89±0.04 µg/ml) and lioxin (EC50 83.22±0.03
µg/ml).

280
In the FRAP assay, antioxidant activity was calculated as Ferrous Equivalents, the
concentration of samples which produced an absorbance value equal to that of 1mM
FeSO4. The compounds absorbance equivalent to 1 mM FeSO4 was calculated from
equation of linearity of scopoletin (y=0.0203x+0.5259, r2=0.9837), scoparone
(y=0.0009x+0.1708, r2=0.5026), (+)-catechin (y=0.0141x+0.1442, r2=0.7415),
hyperin (y=0.0140x+0.2189, r2=0.8319), cynaroside (y=0.0156x+0.3277, r2=0.6009),
vomifoliol (y=0.0062x+0.0549, r2=0.6990), lioxin (y=0.0126x+0.0471, r2=0.8546),
syringic acid (y=0.0172x+0.2963, r2=0.6901). Total FRAP value was determined
from the absorbance value above using the standard Fe (II) calibration curve equation
(y=0.0105x+0.0136, r2=0.9817). Both scopoletin and (+)-catechin displayed
significantly lower FRAP value (49.00±0.64 µg/ml and 75.59±0.42 µg/ml
respectively) than positive control ascorbic acid and quercetin (347.00±0.23 µg/ml
and 86.00±0.24 µg/ml respectively).
In the ȕ-carotene decolourisation assay, the most potent activity was displayed by
vomifoliol (EC50 6.85±0.37 µg/ml), followed by lioxin (EC50 20.13±0.47 µg/ml),
hyperin (EC50 32.21±0.35 µg/ml), syringic acid (EC50 53.69±0.36 µg/ml), cynaroside
(EC50 56.38±0.38 µg/ml), (+)-catechin (EC50 83.22±0.56 µg/ml) and scopoletin (EC50
124.50±0.07 µg/ml). Only scoparone had EC50 of >100 µg/ml.
In the SOD assay, both hyperin and (+)-catechin has IC50 values significantly lower
(IC50 0.75±0.03 µg/ml and IC50 0.94±0.27 µg/ml respectively) than positive control
SOD enzyme (IC50 1.60±0.06 µg/ml). Cynaroside (IC50 1.87±0.03 µg/ml) had activity
similar to SOD, followed by scoparone (IC50 48.72±0.02 µg/ml) and lioxin (IC50
53.69±0.38 µg/ml). Scopoletin, vomifoliol and syringic acid had IC50 of >100 µg/ml.

281
11.3.2 Anti-inflammatory determination assays
The results are shown in Table 11.2 and Figure 11.3-11.5. In the 5-LOX inhibition
assay, all isolated compounds displayed significantly lower IC50 compared to positive
control NDGA. Both scoparone and scopoletin (IC50 0.20±0.01 µg/ml and 0.34±0.01
µg/ml respectively) had the most potent 5-LOX enzyme inhibition (Figure 11.3)
which was more than eightyfold compared to NDGA (IC50 29.21±0.05 µg/ml). This
was followed by syringic acid (IC50 1.38±0.03 µg/ml), hyperin (IC50 3.02±0.02
µg/ml), vomifoliol (IC50 8.72±0.02 µg/ml), (+)-catechin (IC50 16.10±0.03 µg/ml),
lioxin (IC50 17.79±0.03 µg/ml) and cynaroside (IC50 18.12±0.04 µg/ml) (Figure 11.4).
In the COX enzyme inhibition assay, only (+)-catechin and syringic acid showed
good enzyme inhibition while no enzyme inhibition was noticed with lioxin and all
other compounds had IC50 values >100 µg/ml. Compound (+)-catechin displayed
COX-1 selective activity with COX-1/COX-2 ratio of 0.14 with COX-1 enzyme IC50
of 12.08±0.02 µg/ml and COX-2 enzyme IC50 83.89±0.03 µg/ml (Figure 11.5).
Syringic acid had COX-1 enzyme inhibition at IC50 34.89±0.02 µg/ml with COX-2
inhibition at IC50 >100 µg/ml.
11.3.3 Anti-acetylcholinesterase determination assays
The anti-acetylcholinesterase values for isolated compounds are shown in Table 11.3
and Figure 11.6. Only 4 compounds showed moderate enzyme inhibition namely
syringic acid (IC50 29.53±0.19 µg/ml), scopoletin (IC50 51.00±0.02 µg/ml), scoparone
(IC50 86.58±0.05 µg/ml) and vomifoliol (IC50 96.64±0.09 µg/ml) as shown in Figure

282
Table 11.1: Antioxidant values of isolated compounds from Canarium patentinervium Miq.
Compounds ABTS assay, EC50 ȝJPO
DPPH assay, EC50 ȝJPO
FRAP assay, FRAP value
ȝJPO
ȕ-carotene bleaching assay,
EC50 ȝJPO
Superoxide dismutase assay, IC50
ȝJPO scopoletin 1.08±0.03 36.80±0.01. 49±0.64 124.5±0.07 >100
scoparone >100 >100 11492±0.16 >100 48.72±0.02
(+)-catechin 5.03±0.06 4.77±0.01A 75.59±0.42 83.22±0.56 0.94±0.27C
hyperin 8.19±0.04 4.89±0.02 791.96±0.52 32.21±0.35 1.87±0.03D
cynaroside 0.26±0.02 7.79±0.03 694.63±0.32 56.38±0.38 0.75±0.03C
vomifoliol 34.89±0.04 >100 1686.89±0.28 6.85±0.37 >100
lioxin 83.22±0.03 >100 830.67±0.53 20.13±0.47 53.69±0.38
syringic acid 7.38±0.02 15.43±0.02 594.03±0.41 53.69±0.36 >100
AA 1.57±0.06 1.83±0.01 345.15±0.23 NA NA
QC 0.98±0.03 2.78±0.02A 88.16±0.24 1.70±0.03B NA
TRO 0.65±0.02 4.77±0.01A 35.00±0.54 1.69±0.03B NA
SOD NA NA NA NA 1.60±0.06D
AA: ascorbic acid, QC:quercetin, TRO:trolox and SOD: superoxide dismutase Data were obtained from three independent experiments, each performed in triplicates (n=9) and represented as mean ± SD. Values with the same capital letter are not significantly different (P<0.05) according to Tukey multiple comparison test.

283
Table 11.2: Anti-inflammatory values of the isolated compound from Canarium patentinervium Miq.
Compounds Anti-inflammatory assay 5-LOX, IC50 ȝJPO
COX-1 inhibition assay IC50ȝJPO
COX-2 inhibition assay IC50ȝJPO
COX-1/COX-2 ratio
scopoletin 0.34±0.01 >100 >100
scoparone 0.20±0.01 >100 >100
(+)-catechin 16.1±0.03 12.08±0.02 83.89±0.03 0.14
hyperin 3.02±0.02 >100 >100
cynaroside 18.12±0.04 >100 >100
vomifoliol 8.72±0.02 >100 >100
lioxin 17.79±0.03 NO INH NO INH
syringic acid 1.38±0.03 34.89±0.02 >100
NDGA 29.21±0.05 NA NA NA
INDO NA 0.30±0.04 0.25±0.03 1.20
NDGA: nordihydroguairetic acid, INDO: indomethacin, NO INH: no inhibition noted Data were obtained from three independent experiments, each performed in triplicates (n=9) and represented as mean ± SD. Values with the same letter are not significantly different (P<0.01) according to Tukey multiple comparison test.

284
0 1 2 3 4 5
0
50
100scoparone
scopoletin
NDGA
concentration(Pg/ml)
% 5
-LO
X in
hibi
tion
Figure 11.3: 5-LOX inhibition by scopoletin and scoparone isolated from Canarium patentinervium Miq
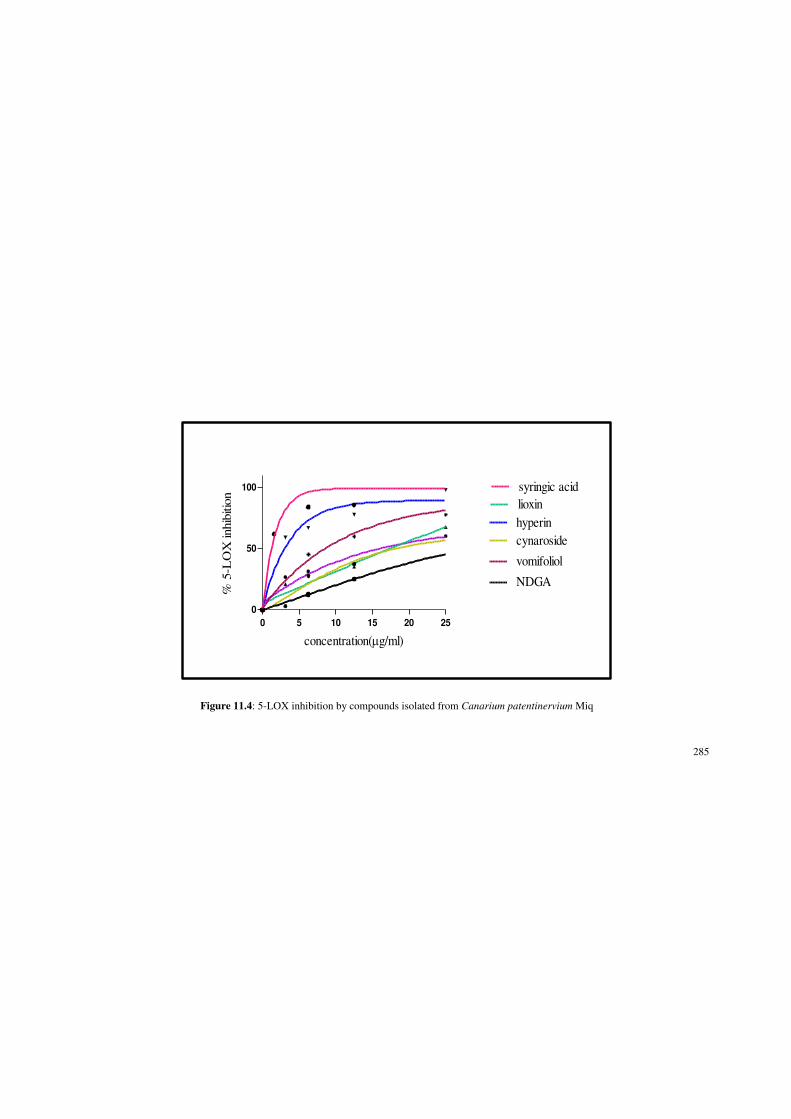
285
0 5 10 15 20 25
0
50
100
lioxin
cynaroside
vomifoliol
hyperin
syringic acid
NDGA
concentration(Pg/ml)
% 5
-LO
X inhib
itio
n
Figure 11.4: 5-LOX inhibition by compounds isolated from Canarium patentinervium Miq

286
0 20 40 60 80 100
0
20
40
60
80
COX-1
COX-2
concentration(ug/ml)
% I
nhib
ition
by
CO
X
Figure 11.5: COX inhibition by (+)-catechin isolated from Canarium patentinervium Miq.

287
11.6. Rest of the compounds showed poor enzyme inhibition with IC50 values >100
µg/ml.
11.3.4 Anticancer assay
The anticancer activity of the compounds that were tested are shown in Table 11.4
and Figure 11.7a and 11.7b. Isolated compound with the highest yield scopoletin was
tested against four cell lines, two human breast cancer cell lines, MCF-7 (ER+) and
MDA 468 (ER-), and two colon cancer cell line, HCT 116 and HT-29. Scopoletin
exhibited potent growth inhibition of MDA 468 (GI50 0.09±0.25 µg/ml) (Figure
11.7a) and HT-29 (GI50 0.17±0.05 µg/ml) (Figure 11.7b) with significantly lower GI50
compared to positive control doxorubicin (GI50 0.69±0.06 µg/ml) for the HT-29 cell
line. Cell line MCF-7 and HCT 116 were less sensitive to scopoletin with GI50 values
>2 µg/ml. Scoparone, (+)-catechin, hyperin, cynaroside, vomifoliol and lioxin were
tested only against MDA 468 cell line and all having GI50 values >2 µg/ml.
11.3.5 Antibacterial assay
The antibacterial assay results are shown in Table 11.5 and Figure 11.8.
Staphylococcus aureus ATCC 11632 was sensitive against all compounds tested.
Scopoletin had the best activity with MIC 25.00±0.00 µg/ml and MBC of 50.00±0.00
µg/ml with complete bacterial kill at 50.00±0.00 µg/ml (Figure 11.8). Scoparone,
Hyperin, cynaroside and syringic acid had bactericidal activity at 100.00±0.00 µg/ml.

288
Table 11.3: Anti- acetylcholinesterase values for isolated compounds from Canarium patentinervium Miq.
Compounds Anti-acetylcholinesterase assay, IC50ȝJPO scopoletin 51.00±0.02
scoparone 86.58±0.05
(+)-catechin >100
hyperin >100
cynaroside >100
vomifoliol 96.64±0.09
lioxin >100
syringic acid 29.53±0.19
galantamine 0.77±0.09
Data were obtained from three independent experiments, each performed in triplicates (n=9) and represented as mean ± SD.
Values with the same letter are not significantly different (P<0.01) according to Tukey multiple comparison test.

289
0 20 40 60 80 100
0
20
40
60
80
100syringic acid
vomifoliol
scoparone
scopoletin
concentration(Pg/ml)
% in
hibi
tion
of A
ChE
Figure 11.6: AChE inhibition by isolated compounds from Canarium patentinervium Miq.

290
Table 11.4: Growth inhibition of human cancer cell lines by isolated compounds from Canarium patentinervium Miq.
Compounds Cell line/ GI50 (µg/ml)
MDA 468 HT-29 MCF-7 HCT 116
scopoletin 0.09±0.25 0.17±0.05 >2 >2
scoparone >2 nt nt nt
(+)-catechin >2 nt nt nt
hyperin >2 nt nt nt
cynaroside >2 nt nt nt
vomifoliol >2 nt nt nt
lioxin >2 nt nt nt
doxorubicin 0.04±0.12 0.69±0.06 0.38±0.28 0.65±0.18
Nt = not tested, Data were obtained from three independent experiments, each performed in triplicates (n=9) and represented as mean ± SD. Values with the same letter are not significantly different (P<0.01) according to Tukey multiple comparison test.

291
Figure 11.7a: Effect of scopoletin isolated compounds from Canarium patentinervium Miq. against growth of breast cancer cell line MDA 468
(GI50 = 0.09 µg/ml)
0
0.5
1
1.5
2
2.5
control 0.01 0.02 0.1 0.2 1 2
OD
55
0 n
m
concentration (µg/ml)
scopoletin
T zero

292
Figure 11.7b: Effect of scopoletin isolated compounds from Canarium patentinervium Miq. against growth of colon carcinoma cell line HT-29
(GI50 = 0.17 µg/ml)
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
control 0.01 0.05 0.1 0.5 1 5
OD
55
0 n
m
concentration (µg/ml)
scopoletin
T zero

293
11.3.6 Antiparasitic assay
The antiparasitic activity of the compounds tested is shown in Table 11.6. Compound
scopoletin was tested against all three parasite amd it was more potent against
Leishmania donovani (IC50 163.30±0.32 µg/ml) and MIC of >200 µg/ml for both
Giardia intestinalis and Entamoeba histolytica. Sensitivity of Leishmania donovani is
then followed by with the presence of lioxin (IC50 211.48±0.32 µg/ml), vomifoliol
(IC50 302.80±0.33 µg/ml), scoparone (IC50 329.90±0.32 µg/ml) and (+)-catechin (IC50
478.93±0.28 µg/ml).
11.4 Discussion
Of the eight isolated compounds, five were phenolics (hyperin, cynaroside, (+)-
catechin, lioxin and syringic acid), two were coumarins (scopoletin and scoparone)
and one was a norsesquiterpene with a cyclohexenone ring (vomifoliol). Among the
phenolics, (+)-catechin is a flavanol, hyperin is a flavonol with a glucosidic moiety
and cynaroside is a flavone with a glycosidic moiety and two were benzoic acid
derivatives. The basic skeleton of these groups are shown in Figure 11.2.
Scopoletin possesses interesting activities, in particular, vasorelaxant [309],
antioxidant [310],antimicrobial [311], anti-inflammatory [312], anti-pyretic [313],
anti-platelet aggregation [314] and anti-diabetes mellitus properties [252]. In addition,

294
Table 11.5: MIC, MBC and MBC/MIC ratio for isolated compounds from Canarium patentinervium Miq. against Staphylococcus aureus ATCC 11632
Compounds Concentration (µg/ml)
MBC/MIC ratio MIC MBC
scopoletin 25.00±0.00 50.00±0.00 2 (+)
scoparone 50.00±0.00 100.00±0.00 2 (+)
(+)-catechin 50.00±0.00 >100 nd
hyperin 50.00±0.00 100.00±0.00 2 (+)
cynaroside 50.00±0.00 100.00±0.00 2 (+)
vomifoliol 100.00±0.00 >100 nd
lioxin 100.00±0.00 >100 nd
syringic acid 50.00±0.00 100.00±0.00 2 (+)
ND: not determined

295
Figure 11.8: Time-kill plot for MSSA ATCC 11632 in presence of scopoletin isolated from Canarium patentinervium Miq.
-0.1
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Bac
teri
al g
row
th (
incr
ease
in O
D)
Time (hr)
0 ug/ml 6.25ug/ml 12.5ug/ml 25ug/ml 50ug/ml

296
Table 11.6: Antiparasitic values of isolated compounds from Canarium patentinervium Miq. against
Leishmania donovani (AG 83) promastigotes and
MIC values against Giardia intestinalis and Entamoeba histolytica growing in vitro.
Compounds
IC50 ( (µg/ml) MIC (µg/ml)
Leishmania donovani Giardia intestinalis Entamoeba histolytica
scopoletin 163.30±0.32 >200 >200
scoparone 329.90±0.32 nt nt
(+)-catechin 478.93±0.28 nt nt
vomifoliol 302.80±0.33 nt nt
lioxin 211.48±0.32 nt nt
Data were obtained from three independent experiments, each performed in triplicates (n=9) and represented as mean ± SD. Values with the same letter are not significantly different (P<0.01) according to Tukey multiple comparison test. nt: not tested

297
it exerted neuroprotective [315] and hypotensive [316] activities in addition to
applications in cardiovascular disease [252], antitumor [317], anti-proliferation and
antithyroid [318] treatment. Scoparone has been studied for its potential
pharmacological properties including immunosuppression [319] and vasorelaxation
[320]. Compound (+)-catechin is reported to induce longevity in the nematode worm
Caenorhabditis elegans [321], reduces atherosclerotic lesion development in mice
[322], have opposite effects on glycogen metabolism in isolated rat hepatocytes [323],
inhibits intestinal tumor formation in mice [324], inhibits the oxidation of low density
lipoprotein [325] and as an antioxidant [326]. Cynaroside has been reported for
moderate inhibition of enzymes for the synthesis of thromboxane B2 and leukotriene
B4 as well as hydrogen peroxide scavenging activity, scavenge reactive oxygen and
nitrogen species [327] to chelate transition metals, antibacterial [328], antiviral [329]
and antifungal [330] activity.
Hyperin has been found to have antioxidant activity [331], inhibition of lipid
peroxidation in rat liver microsomes [332] and protective effects to PC12 cells against
cytotoxicity induced by hydrogen peroxide and tert-butyl hydroperoxide [333]. Lioxin
had been reported to exert strong antioxidant (oral protectant) [334], powerful wound
healing properties [335], protective effects against DNA damage [336] as well as
antimutagenic [337] and immunostimulating [338] properties. Syringic acid has
shown to have antioxidant, antibacterial [339] and hepatoprotective [340] activities.
Vomifoliol have been reported to have weak DPPH activity [341] and anti-
inflammatory activity by inhibition of TNF-Į [341] and plays an important role as an
endogenous regulator of the stomatal aperture [262].

298
11.4.1 Antioxidant activity
The antioxidant mechanisms of flavonoids comprise free radical scavenging, but also
enzyme inhibition/induction, metal chelation, interaction with receptors and
modulation of gene expression [342]. The direct scavenging of radicals is believed to
occur in cells, but, to avoid the interference of other mechanisms, this work was
developed using a cell-free system. The past few decades of structure-activity
relationships research have generated several consistent lines of evidence supporting
the role of specific structural components as requisites for antioxidant activity. The
antioxidant activity of flavonoids and their metabolites in vitro depends upon the
arrangement of functional groups about the nuclear structure, namely ring A, B and C
[343]. Consistent with most polyphenolic antioxidants, both the configuration and
total number of hydroxyl groups substantially influence several mechanisms of
antioxidant activity. The B-ring hydroxyl configuration is the most significant
determinant of scavenging of ROS and RNS. A-ring substitution correlates little with
antioxidant activity [344].
Some characteristics of flavonoids are important to the antioxidant capacity according
to the famous three Bors criteria [344] (Figure 11.9): (i) o-dihydroxy structure in the
B ring LHD¶¶-catechol structure, which confers higher stability to the radical form
and participates in electron delocalization ; (ii) C2-C3 double bond in conjugation with
a 4-oxo function in the C ring, responsible for electron delocalization of the aromatic
nucleus; (iii) 3- and 5-OH groups with 4-oxo function in the A and C rings, required
for maximum radical scavenging potential. Moreover, an additional criterion could be
added: (iv) In the absence of o-dihydroxy structure in the B ring, hydroxyl

299
substituents in a catechol structure on the A ring are able to compensate and become a
larger determinant of flavonoid antiradical activity.
Figure 11.9: Structural features of flavonoids with high antioxidant activity.
Of the three flavonoids, (+)-catechin and hyperin exhibited the most consistent free
radical scavenging capability across the five antioxidant assay (Table 11.1). Good
correlation was found between FRAP values and hydroxyl groups and especially the
catechol moiety and 3-OH [345], explaning the best activity shown by (+)-catechin
(FRAP value 75.59±0.42 µg/ml). Results in the ABTS and DPPH assay were
relatively similar as they have the same mechanism of action [53]. Sawai et al. [346]
have identified the antioxidant mechanism using NMR analytical approaches to
identify the antioxidative molecular mechanism in DPPH assay and found that the B
ring of (+)-catechin has been changed to a quinone structure as shown in Figure
11.10.

300
Figure 11.10: (+)-catechin and the corresponding fully oxidized ortho-quinone product
Flavanols, namely here (+)-catechin and flavonol (hyperin) are more effective free
radical scavengers than flavones (cynaroside) [343], which may be ascribed to the
greater number of hydroxyl groups and 3-OH to the former two. Plumb and co-
workers [347] reported that the antioxidant properties of flavonol glycosides from tea
decreased as the number of glycosidic moieties increased. In addition, glycosylation
of flavonoids mostly decreases their antioxidant activity. Multiple A-ring methoxy
groups as seen in cynaroside also reverse the positive effect of a B-ring catechol
[344].
+RZHYHU LQ WKH ȕ-carotene bleaching assay, which involves lipid peroxidation via
hydrogen atom transfer (HAT method- section 4.5), hyperin and cynaroside
outweighed activity of (+)-catechin. Flavonoids with a C2±C3 double bond in
conjugation with a 4-carbonyl group has been known to exhibit lower EC50 values
(indicative of stronger antioxidant activity) in a microsomal system compared to those
with saturated heterocycles [344] explaining the lower EC50 values of cynaroside
(56.38±0.38 µg/ml) and hyperin (32.21±0.35 µg/ml) LQ WKH ȕ-carotene bleaching
assay. However, hyperin has a lower EC50 than cynaroside in this assay as the C-3

301
position is an excellent choice for substitution to give the flavonoid an optimum
lipophilicity [348].
Between both glycosides, hyperin was superior to cynaroside in all five assays (Table
11.1) as C-glycosylation in the A-ring (seen in A ring of cynaroside) results in greater
diminution of activity than 3-glycosylation (seen in hyperin) in the heterocycle [349].
Studies have also shown that O-glycosylation at carbon 7, but not carbon 3, weakens
the antioxidant effect of flavonoids in rat mitochondria [349]. O-glycosylation
interferes with the coplanarity of the B-ring with the rest of the flavonoid and the
ability to delocalize electrons [350]. Though glycosides are usually weaker
antioxidants than aglycones, bioavailability is sometimes enhanced by a glucose
moiety [349]. The SOD assay involves flavonoids having SOD-like activity and
inhibition of XOD that produces the free radicals. Hyperin and (+)-catechin have
significantly lower IC50 (0.75±0.03 µg/ml and 0.94±0.27 µg/ml respectively)
compared to SOD enzyme. This is due to the structure arrangement of hyperin as the
planar flavonoid core (double bond between C-2 and C-3) and the presence of free
hydroxyl groups at C-5 and C-7 are important prerequisites for this activity [344]. (+)-
catechin has slightly less activity compared to hyperin as it lacks the double bond
between C-2 and C-3).
Phenolic acids have been investigated for the radical scavenging activity via ABTS
assay by Rice- Evans [351]. Structure-activity comparisons suggest that antioxidant
activity of phenolic acids depends on the number and orientation of hydroxyl groups
relative to the electron-withdrawing CO2H, CH2CO2H, or (CH)2CO2CH functional

302
group [39]. It can be seen that syringic acid has more electron withdrawing groups
compared to lioxin confirming the lower EC50 in ABTS assay (7.38±0.02 µg/ml).
Between both coumarins, scopoletin exhibited potent antioxidant activity compared to
scoparone. It had significantly lower EC50 values in ABTS (EC50 1.08±0.03 µg/ml)
compared to ascorbic acid (EC50 1.57±0.06 µg/ml) and lower values in FRAP assay
(FRAP value 49.00±0.64 µg/ml) than quercetin (FRAP value 88.16±0.24 µg/ml) and
ascorbic acid (FRAP value 345.00±0.23 µg/ml). The result is consistent with a
previous report that the scavenging capacity was higher for coumarin bearing OH
groups than the methoxy-substituted derivatives [352] as in scoparone. In the SOD
assay, the SOD-like activity and inhibition of XOD that produces the free radicals
seemed to be better with scoparone (IC50 48.72±0.02 µg/ml) than scopoletin (IC50
>100 µg/ml). Reports have stated that a methoxy substituted for 6- OH diminishes
inhibition potency against XOD [352] as in the result of both coumarins here. This
further enhances the hypothesis that the H atom of the 6-hydroxyl plays a more
significant role than the O atom. It also can be observed that perhaps the 7-methoxy
group in scoparone increases the inhibition activity.
Not much biological data is available on vomifoliol except weak DPPH activity and
anti-inflammatory activity by inhibition of TNF-Į [341]. However in this study,
though consistent low DPPH activity is seen (EC50 >100 µg/ml), moderate activity in
noted in ABTS (EC50 JPODQGSRWHQWȕ-carotene bleaching activity was
noted (EC50 6.85±0.37 µg/ml).

303
11.4.2 Anti-inflammatory activity
All isolated compounds exhibited significantly lower 5-LOX inhibition than positive
NDGA (IC50 29.21±0.05 µg/ml). Among the five phenolics, syringic acid and hyperin
had the most potent 5-LOX inhibitory activity (IC50 1.38±0.03 µg/ml and 3.02±0.02
µg/ml respectively). Hydroxylations at 5, 7, and 4' have also shown to enhance overall
anti-inflammatory activity in flavonoids [102]. Flavonols were found to be stronger
inhibitors of 5-LOX than flavones [353], as established in this study with hyperin
(IC50 3.02±0.02 µg/ml) being superior to cynaroside (IC50 18.12±0.04 µg/ml).
Although it is difficult to establish structural-activity relationships due to their
varieties of chemical structures, these inhibitory activities against 5-LOX could
explain, at least in part, the anti-inflammatory/ antiallergic activities of flavonoids. No
clinical data showing the relation of flavonoid intake and incidence (severity) of
inflammatory disorders such as RA and AD was available, although several studies
demonstrated some inverse correlation of flavonoid intake and incident rate of
cardiovascular failure [354].
Lipoxygenases (LOXs) also function as prooxidant enzymes that catalyze enzymatic
lipid peroxidation. Additionally, they are a source of free radicals initiating
nonenzymatic lipid peroxidation and other oxidative processes involved in the
formation of atherosclerotic lesions [354]. Consequently, inhibition of LOXs may
contribute to the universal antioxidant activities of flavonoids. Planar flavonoid
structure with delocalized electrons achieved by the C2-C3 double bond seems to be a
prerequisite for inhibitory activity. The following structural features were found to
enhance the inhibitory potency: (i) presence of a catechol moiety in the B or A ring,

304
(ii) 4-carbonyl group in the C ring [344], thus confirming the results of this study with
hyperin having more 5-LOX inhibition than (+)-catechin.
However (+)-catechin had a more comprehensive anti-inflammatory activity with dual
inhibition of 5-LOX (IC50 16.10±0.03 µg/ml) and COX (COX-1; IC50 12.08±0.02
µg/ml, COX-2; IC50 83.89±0.03 µg/ml). (+)-catechin displayed COX-1 selective
activity with the COX-1/COX-2 ratio <1 and this confirms previous findings [107].
Benzoic acid derivatives, syringic acid exhibited potent 5-LOX inhibition (IC50
1.38±0.03 µg/ml) and moderate COX-1 inhibition (IC50 34.89±0.02 µg/ml) compared
to lioxin that had good 5-LOX inhibition (IC50 17.79±0.03 µg/ml) but no COX
inhibition. Consideration of the structure-activity relationship of phenolic acids on the
anti-inflammatory action revealed that both C-4 hydroxy and C-3 methoxy radicals of
the benzoic acid derivatives play an important role in anti-inflammatory activities
[355]. However the presence of extra methoxy group at C-5 seemed to drastically
increase the activity as seen in syringic acid. One study reported potent COX-2
inhibition of syringic acid (IC50 0.40 µg/ml) using cell based study [356] but we
demonstrated poor COX-2 inhibition in this study.
Both coumarins scopoletin and scoparone were potent inhibitors of 5-LOX recording
lowest IC50 values (IC50 0.34±0.01 µg/ml and 0.20±0.01 µg/ml respectively).
Scoparone has slightly lower IC50 than scopoletin as the SAR findings has reported
that the chloro and methoxy substitution in coumarin ring showed increase anti 5-
LOX activity [357]. Cyclohexenone derivatives are well known lead molecules for the
treatment of inflammation and autoimmune diseases [358]. The anti-inflamatory
activity previously reported for vomifoliol were by inhibition of TNF-Į [341]. In this

305
study, vomifoliol has exhibited potent 5-LOX inhibition (IC50 8.72±0.02 µg/ml) and
poor COX inhibition.
11.4.3 Anti-acetylcholinesterase activity Only the scopoletin, scoparone, vomifoliol and syringic acid showed AChE inhibition
at IC50 <100 µg/ml. The studies have also shown that naturally occurring as well as
the chemically synthesised coumarin analogs exhibit potent AChE inhibitory activity
[359]. Coumarin ring seems to be essential for the optimal activity and its replacement
with related structural moiety such as chromone is associated with loss of AChE
inhibitory activity. The substituents at coumarin moiety particularly at 6th and 7th
position also influence the activity in a significant manner. The presence of electron-
donating groups such as ±OCH3, ± OH, and ±NH2 increase the activity and it has
been generally attributed to increase in lipophilicity of compounds [360]. This might
explain good activity of scopoletin (IC50 51.00±0.02 µg/ml) and scoparone (IC50
86.58±0.05 µg/ml) as both have electron donating groups at position 6th and 7th. The
presence of bulkier substituents at 6th and 7th positions of the coumarin is associated
with significant loss in AChE inhibitory activity indicating the critical role of
electronic as well as steric effects in influencing the AChE inhibitory activity [360],
this explains why scoparone has higher IC50 due to it having bulkier two methoxy
groups.
Up to date, quite a lot of studies have reported affirmative effects of phenolics in
neurodegenerative diseases depending upon their antioxidative properties [361].
However, there has been a small number of data on AChE inhibitory activities of

306
phenolic compounds. Among the phenolics isolated in this study only syringic acid
exhibited good AChE inhibition (IC50 29.53±0.19 µg/ml), lowest of all compounds
tested. The literature concerning the role of phenolic acids and their derivatives in the
neuroprotection of the CNS is, however, still incomplete. Thus the actual mechanism
of inhibition needs to be investigated. Cyclohexenone derivatives, vomifoliol had
moderate AChE inhibition (IC50 96.64±0.09 µg/ml), which is to the best of our
knowledge the first to be reported.
11.4.4 Antiparasitic activity
Only scopoletin exhibited moderate antileishmanial activity (IC50 163.30±0.32 µg/ml)
followed by lioxin, vomifoliol, scoparone and (+)-catechin. Poor activity of scopoletin
was noted against Giardia intestinalis and Entamoeba histolytica while the others
were not tested against them. Comparing the anti AChE and antiparasitic activity, it
can be deduced that same compounds seemed active in both assays. Choline is the
precursor of phosphatidylcholine (PC), a main component of Leishmania
promastigote membranes [362]. Therefore, inhibition of choline formation may de-
crease Leishmania survival. This hypothesis can be tested by using inhibitors of the
acetylcholinesterase enzyme (AChE), which catalyzes the hydrolysis of acetylcholine
to choline and acetic acid, as leishmanicidal compounds. This may identify another
mechanism of action for leishmanicidal activity [363].
The lactone groups present in coumarins are also present in the structures of
Annonaceous acetogenins that show leishmanicidal activity [364]. The AChE inhibi-
tory activity of a previous study on scoparone indicates a possible action mechanism

307
E\ GLVUXSWLQJ WKH YLDELOLW\ RI OHLVKPDQLD¶V FHOO PHPEUDQHV. There has been a
hypothesis that the coumarin and scoparone have mechanism of action acting on the
same pathway as above compounds, resulting in a net negative effects on choline
uptake by the parasite [363]. Only flavonoid (+)-catechin was tested in this study.
Flavonoids however, are able to inhibit acetylcholine hydrolysis and interfere in the
PC due to their low concentration of choline precursor from the host. Nevertheless,
the leishmanicidal activity of these compounds may be related to their ability to
chelate iron (Fe), depriving this essential nutrient from the intracellular forms [365].
11.4.5 Anticancer activity
In this study, scopoletin displayed potent anticancer effect against breast cancer cell
line MDA 468 (GI50 0.09±0.25 µg/ml) and colorectal cancer cell line HT-29 (GI50
0.17±0.05 µg/ml), the latter being more significant that positive control doxorubicin
(GI50 0.69±0.06 µg/ml). Previously studies on scopoletin have shown it to inhibit the
cell proliferation by inducing cell cycle arrest and the increase of apoptosis in human
prostate tumor cells [366], and it induced apoptosis of HL-60 cells [367]. The ability
of scopoletin have been said to induce programmed cell death in leukemic cells
representing a novel and important aspect of the discussion of the anti-tumoral effects
of various plants containing scopoletin [367]. In this study, scopoletin has shown
potent growth inhibition of these two cell lines warranting further study.

308
11.4.6 Antibacterial activity
All isolated compounds tested against Staphylococcus aureus ATCC 11632 showed
bacterial growth inhibition. Scopoletin, scoparone, hyperin, cynaroside and syringic
acid had bactericidal effect <100 µg/ml. Only scopoletin had bactericidal effect and
complete kill at MBC 50.00 µg/ml. The antibacterial properties of coumarins were
first recognised in 1945 when an investigation with dicoumarol was found to inhibit
the growth of several strains of bacteria. All the methods reported in the study of the
antibacterial activity of coumarins were disc diffusion methods [368]. It has then been
suggested that coumarins possess antibacterial activity act selectively against Gram-
positive microorganisms [368]. Free hydroxyl group at position 7 has been shown to
be important for antibacterial activity [298] as confirming the activity of scopoletin
that has free 7-OH.
11.4.7 Formulation of scopoletin as a gel
Previously study by Seon et al. [369] have demonstrated scopoletin to inhibit
tyrosinase kinase with an IC50 of 10.2 µg/ml suggecting its possible use in skin aging
and preventive activities. Biosynthesis of melanin in animals is normally initiated
through the oxidation of tyrosine into DOPA by tyrosinase which is the rate limiting
enzyme in this pathway. The production of abnormal pigmentation such as melasma,
freckles, senile lentigines, and other forms of melanin hyperpigmentation could be a
serious aesthetic problem [369]. In this study scopoletin was found to exhibit potent
antioxidant, anti-inflammatory and antibacterial properties. Scopoletin has been
studied to have good pharmacokinetics data with good absorption in the stomach and

309
colon of rats [370]. Reported physical properties of scopoletin which passes the
Lipinsky rule (mass < 500, log < 5, donor count < 5, and acceptor count < 10), for
possible lead compound in drug discovery [371] and, in agreement with its potent
antioxidant power, good anti-inflammatory and antibacterial activity enables it to be
considered for a formulation for further test and development.
Transdermal drug delivery offers the following advantages over oral administration:
(1) peak and valley levels in the serum are avoided; (2) first-pass metabolism is
avoided and the skin metabolism is relatively low; (3) less frequent dosing regimens
is needed due to the maintenance and longer sustainability of zero-order drug delivery
and (4) less inter-subject variability occurs [372]. Other advantages listed include
aspects such as the accessibility of the skin; a relatively large surface area for
absorption and the fact that it is non-invasive make it more patient compliant [373].
Thus, an external formulation suitable for cosmetic use (skin hyperpigmentation, acne
and wrinkle protection) and for medicinal use as an anti-inflammatory gel was
derived. The formulation was prepared with aloe vera gel as a base due to the well
known skin and medicinal properties of aloe [374]. Orange oil was used as studies
have shown the limonene (moneterpene) content has a high ability to enhance in vitro
percutaneous transport greater than conventional lipophilic penetration enhancing
FRPSRXQGV HJ 6SDQ Į-bisabolol, oleic acid) and hydrophilic penetration
enhancing compounds (e.g., ethanol, polyethylene glycol 600, 1,8-cineole) [375].
Further studies on the efficacy of the gel and penetration test are required.

310
11.5 Conclusion
For the first time, eight compounds (scopoletin, scoparone, (+)-catechin, lioxin,
cynaroside, hyperin, vomifoliol and syringic acid) were isolated from Canarium
patentinervium Miq. Three of these compounds (vomifoliol, lioxin and syringic acid)
were isolated for the first time from the genus Canarium. These compounds were then
subjected to a range of biological assays, all reported for the first time from this plant.
Canarium patentinervium Miq. is a rare plant from the family of Burseraceae and
genus Canarium found in Asia Pacific region previously recorded for its usage in
wound healing by the indigenous people of Malaysia. One of the objectives of this
study was to ascertain the pharmacological and phytochemical aspect for the
ethnopharmacological usage. Wound healing involves manifold inflammatory
processes of which notably the massive release of leukotrienes from arachidonic acid
via the 5-lipoxygenase pathway (5-LOX) and the generation of nitric oxide (NO) from
inducible nitric oxide synthase (iNOS). Of note, nitric oxide (NO) is a free radical,
and the generation of cytokines involves a reactive oxygen species (ROS) outburst.
Therefore, agents able to block the enzymatic activity of 5-LOX and to scavenge free
radical are of immense interest against inflammatory conditions which englobe not
only epidermal insults but also neurodegeneration and obesity.
Cells produce superoxide anion (O2í), peroxide anion (HO2
í), and hydroxyl ion (HOí)
as part of the physiological aerobic metabolism which are quickly scavenged by
cytoplasmic antioxidant defense system. However, in the event of ageing or
pathologies, the antioxidant defense system is overwhelmed, and cells suffer massive

311
oxidative stress leading eventually to carcinogenesis or apoptosis. In fact, oxidative
stress is the main causative factor for cholinergic and dopaminergic neurons apoptosis
hence $' DQG 3DUNLQVRQ¶V GLVHDVH 3'. In addition, there is a growing body of
evidence that point to the fact that 5-LOX inhibitors are of immense therapeutic
values. The various isolated secondary metabolites clearly demonstrate various potent
biological activities that substantiate the ethnopharmacological claim.
Given the aforementioned evidence it is tempting to speculate that Canarium
patentinervium Miq. represents an exciting scaffold from which to develop leads for
treatment of neurodegenerative diseases.

312
CHAPTER 12
GENERAL CONCLUSIONS
The objectives of the study were to:
i. Extraction of leaves and barks of plant with solvents of different
polarity to obtain crude extract with solvents of different polarity.
ii. Investigation of the possible secondary metabolite types present in
the crude extracts of leaves and barks with qualitative
phytochemical analysis.
iii. Isolation and identification of the chemical compounds responsible
for the antibacterial and antioxidant activity in the most active
extract through bioassay-guided fractionation using liquid-liquid
extraction or solvent partioning, column chromatography, thin
layer chromatography (TLC) and nuclear magnetic resonance
(NMR).
iv. Investigation of the antioxidant activity of the crude extracts of
both leaves and barks using the 2,2-diphenyl-1-picrylhydrazyl
(DPPH) radical scavenging assay, ferric reducing ability of plasma
)5$3DVVD\¶-Azino-bis(3-ethyl-14 benzthiazoline-6-sulfonic
DFLG $%76DVVD\ ȕ-carotene bleaching assay and superoxide
dismutase assay.

313
v. Investigation of the anti-inflammatory potential of the crude
extracts against 5-lipoxygenase and cycloxygenase systems in
vitro.
vi. Investigation of the anti AChE potential of the crude extracts in
vitro.
vii. Investigation of the in vitro antibacterial activity of the crude
extracts of leaves and barks using the disc diffusion assay and
determination of minimum inhibitory concentration, minimum
bactericidal concentration and death kill rate against four ATCC
bacterial strains namely Gram-positive bacteria Staphylococcus
aureus, Bacillus cereus, methicillin-resistant Staphylococcus
aureus and Gram-negative Pseudomonas aeruginosa and ten
clinical strain bacteria.
viii. Investigation of the in vitro antifungal activity of the crude extracts
of leaves and barks using the disc diffusion assay against three
clinical strain fungi namely, Candida parapsilosis, Candida
glabrata and Candida albicans.
ix. Investigation of the anticancer activity of the crude extracts of the
leaves and barks against human derived cell lines [HCT 116, colon
cancer cell line and MCF-7 (ER+), MDA 468 (ER-) breast
carcinoma] using MTT assay.
x. Investigation of the in vitro anti-parasitic activity of the crude
extracts of leaves and barks against Leishmania. donovani
promastigotes, Entamoeba histolytica and Giardia intestinalis.

314
xi. Bioactivity screening of all isolated compounds using the same
bioassay as mentioned in iii-ix.
The following appropriate conclusions were drawn based on the experimental data
obtained.
i. Extraction of samples
Extraction was done with maceration with solvents of increasing
polarity.The yield for the hexane, chloroform and ethanol extract of leaves
were 1.25 %, 1.11 % and 6.45 % respectively. The yield for the hexane,
chloroform and ethanol extract of barks was 1.04 %, 0.4 % and 2.61 %.
Crude extracts were kept at -20oC until further use.
ii. Phytochemical analysis
Phytochemical analysis of Canarium patentinervium Miq. revealed
presence of tannins and flavonoids in the ethanol extract of leaves and
barks. Steroids were detected in the ethanol extract of leaves and all tested
bark extracts. Results from our phytochemical analysis revealed that the
ethanol extract of leaves and barks of Canarium patentinervium Miq.
accumulate substantial amounts of flavonoids and tannins which could be
possibly result in antioxidant or antibacterial activity.
iii. Isolation of biologically active compounds
Eight compounds were isolated for the first time from Canarium
patentinervium Miq. using various isolation techniques such as TLC, CC,
HPLC and identified with NMR method. Compounds isolated scopoletin,

315
scoparone, (+)-catechin, hyperin, cynaroside, lioxin and syringic acid were
phenolics while vomifoliol was a norsesquiterpene with a cyclohexenone
ring. Lioxin, syringic acid and vomifoliol were isolated from this genus
Canarium for the first time.
iv. Antioxidant activity
The ethanol extract of leaves and barks displayed superior antioxidant
capacities. The EC50 values of the samples were consistently low in SET
methods (ABTS, DPPH and FRAP assay) superior to standard as opposed
WR +$7 PHWKRG ȕ-carotene bleaching assay). The present study
demonstrates that Canarium patentinervium Miq. is a potent source of
antioxidants that exhibits its antioxidant activity predominantly via the
SET method.
Compounds (+)-catechin and hyperin exhibited the most consistent free
radical scavenging capability across the five antioxidant assay. Flavanols,
namely here (+)-catechin and flavonol (hyperin) are more effective free
radical scavengers than flavones (cynaroside). Hyperin and (+)-catechin
have significantly lower IC50 (0.75±0.03 µg/ml and 0.94±0.27 µg/ml
respectively) compared to SOD enzyme. Between both coumarins,
scopoletin exhibited potent antioxidant activity compared to scoparone. It
had significantly lower IC50 values in ABTS (IC50 1.08±0.03 µg/ml)
compared to ascorbic acid (IC50 1.54±0.03 µg/ml) and lower values in
FRAP assay (IC50 49.00±0.64 µg/ml) than quercetin (IC50 86.00±0.24
µg/ml) and ascorbic acid (IC50 347.00±0.23 µg/ml). Vomifoliol displayed

316
low DPPH activity (IC50 >100 µg/ml), moderate activity is noted in ABTS
(IC50 JPO DQG SRWHQW ȕ-carotene bleaching activity was
noted (IC50 6.85±0.37 µg/ml).
v. Anti-inflammatory activity
In view of dual inhibition of 5-LOX and COX activity, the ethanol extract
of leaves seemed to have moderate inhibition against 5-LOX and good
inhibition against COX enzymes (selective COX-2). Chloroform extract of
the bark though had good 5-LOX inhibition but did not have activity
DJDLQVW&2;HQ]\PHVEHORZȝJPO
In the 5-LOX inhibition assay, all isolated compounds displayed
significantly lower IC50 compared to positive control NDGA. Both
scoparone and scopoletin (IC50 0.20±0.01 µg/ml and 0.34±0.01 µg/ml
respectively) had the most potent 5-LOX enzyme inhibition (Figure 11.2)
which was more than eightyfold compared to NDGA (IC50 29.19±0.02
µg/ml). This was followed by syringic acid (IC50 1.38±0.03 µg/ml),
hyperin (IC50 3.02±0.02 µg/ml), vomifoliol (IC50 8.72±0.02 µg/ml), (+)-
catechin (IC50 16.10±0.03 µg/ml), lioxin (IC50 17.79±0.03 µg/ml) and
cynaroside (IC50 18.12±0.04 µg/ml).
vi. Anti AChE activity
Chloroform extract of the barks displayed the best activity (IC50 =
ȝJPODVRSSRVHGWRJDODQWKDPLQH,&50 ȝJPO

317
The ethanol extract of barks and leaves follow through with IC50 =
ȝJPO DQG ,&50 ȝJPO UHVSHFWLYHO\+H[DQH
extracts of bark and leaves and the chloroform extract of leaves had the
lowest enzyme inhibition activity (IC50 ȝJPO ,&
ȝJPO DQG ,&50 ȝJPO UHVSHFWLYHO\ The
potency of activity against AChE was BC > BE > LE > BH >LH > LC.
Only 4 compounds showed good to moderate enzyme inhibition namely
syringic acid (IC50 29.53±0.19 µg/ml), scopoletin (IC50 51.00±0.02 µg/ml),
scoparone (IC50 86.58±0.05 µg/ml) and vomifoliol (IC50 96.64±0.09
µg/ml). Rest of the compounds showed poor enzyme inhibition with IC50
values >100 µg/ml.
vii. Antimicrobial activity
All crude extracts of Canarium patentinervium Miq. under investigation
exhibited exceptional concentration-dependent antimicrobial activity
against both Gram-positive and Gram-negative bacteria. The most
promising activity was displayed against Gram-positive bacteria
Staphylococcus aureus, MRSA, Bacillus cereus and Gram-negative
bacteria and Pseudomonas aeruginosa, Klebsiella sp, Klebsiella
pneumoniae ESBL and yeast Candida parapsilopsis. The antimicrobial
activity exhibited by the bark extracts and ethanol extract of leaves against
MSSA and MRSA indicates exceptional antimicrobial activity. All the
tested bacterias were susceptible to either bark or leaf extracts.

318
All isolated compounds tested against Staphylococcus aureus ATCC
11632 showed bacterial growth inhibition. Scopoletin, scoparone, hyperin,
cynaroside and syringic acid had bactericidal effect <100 µg/ml. Only
scopoletin had bactericidal effect and complete kill at MBC 50.00 µg/ml.
viii. Antifungal activity
The most promising activity was displayed against yeast Candida
parasilopsis. The ethanol extracts and hexane extract of barks also showed
inhibition against Candida parasilopsis with a cidal and static action
respectively.
ix. Anticancer activity
The chloroform and ethanol extract of barks showed good antitumor
activities with GI50 values of 23.44±0.05 ȝJPODQG±0.21 ȝJPO7KH
most susceptible cell lines were found to be the breast cancer cell line,
MDA 468.
Scopoletin displayed potent anticancer effect against breast cancer cell line
MDA 468 (GI50 0.09±0.25 µg/ml) and colorectal cancer cell line HT-29
(GI50 0.17±0.05 µg/ml), the latter being more significant that positive
control doxorubicin (GI50 0.66±0.60 µg/ml).
x. Anti parasitic activity
The hexane extract of leaves showed moderate antileishmanial activity
with IC50 YDOXHVRIȝJPO7KLVFRXOGEHGXHWRHVVHQWLDORLOV

319
present in the hexane extracts as shown previously in the family of
Burseraceae [216]. Only ethanol extracts showed activity against Giardia
intestinalis and Entamoeba histolytica at concentration of 500 ȝJPO
Other extracts were inactive against these two intestinal parasites.
Scopoletin was tested against all three parasite amd it was more potent
against Leishmania donovani (IC50 163.30±0.32 µg/ml) and MIC of >200
µg/ml for both Giardia intestinalis and Entamoeba histolytica. Sensitivity
of Leishmania donovani is then followed by with the presence of lioxin
(IC50 211.48±0.32 µg/ml), vomifoliol (IC50 302.80±0.33 µg/ml), scoparone
(IC50 329.90±0.32 µg/ml) and (+)-catechin (IC50 478.93±0.28 µg/ml).
In Chinese folk medicine, Canarium album (Lour.) Raeusch has been used for
treatment of various diseases ranging from cardiovascular, gastrointestinal and
toxicities while Canarium schweinfurthii Engl. has been used by traditional healers as
a remedy for diabetes mellitus in southern Senegal while in Congo and Central
African Republic the plant is used in fever, as stimulant, emollient, in post-partum
pain, constipation, malaria, diarrhoea, sexual infections and rheumatism (Chapter 1)
to name a few. The fact only 12 % of 75 species of Canarium have been studied for
their pharmacological activities promises an unopened crypt of various secondary
metabolites as lead compounds and biological effects that needs to be uncovered and
investigated. Thus, by using an ethnopharmacological approach, the investigation of
Canarium patentinervium Miq. a plant from Malaysia that has been used by the orang
asli (indigenous people) for healing wounds was done. Crude extracts and isolated

320
secondary metabolites were investigated for their various activities.. Three out of
eight compounds were isolated for the first time from this genus of 75 species
(Canarium). Six kinds of major biological effects were evident in the crude and
compounds namely, antioxidant, antibacterial, anti-inflammatory, anti AChE, anti
parasitic, and anticancer all of which were reported for the first time from this plant.
This study is therefore a small leap in the field of an important endeavor for the
discovery of potent biologically active molecules for the treatment of various
diseases. This discovery also opens forth a pathway where biologically active
molecules isolated from biosynthesis of plants can now pose as lead molecules in the
field of synthetic chemistry for drug discovery.
As modern cultures and scientific advances spread around the world, the depth of the
knowledge store of traditional use still remains crucial. The full significance of the
indigenous knowledge forfeited may not be realised. It is thus important that the
knowledge be documented and the traditional use be given some credence through
modern scientific studies. Canarium patentinervium Miq. is such an example.

321
CHAPTER 13
RECOMMENDATIONS FOR FUTURE WORK
This study intends to contribute towards the knowledge base of plant species with
therapeutic potential. Material for this study was only collected from a single site. To
account for possible geographical and chemotypic variation material should be
studied from several populations.
i. Antioxidant activity
The isolated compounds should be tested for synergistic or antagonistic effect by the
construction of an isobologram.
ii. Antimicrobial activity
,VRODWLRQ DQG VWUXFWXUDO HOXFLGDWLRQ RI WKH FRPSRXQGV from the tannin fraction
responsible for the antimicrobial activity of Canarium patentinervium Miq. should be
investigated.
'HWHUPLQDWLRQRIV\QHUJLVWLFDQWDJRQLVWLFor additive effects should be investigated.
iii. Anti-inflammatory Activity
The actual efficacy of an anti-inflammatory activity needs to be validated by testing
the isolated bioactive compounds in a cell-based environment (in vitro) and in a
biological system (in vivo).

322
The effects of these active compounds on the suppression of the COX-2 gene
expression, requires investigation and clarification. The effectiveness of interaction of
these extracts and isolated compounds, including the flavonoid derivatives, with other
pro-inflammatory biochemical pathways may be assessed, and the possible structure-
activity relationships determined. This will provide a better understanding of the
possible mode of action.
iv. Anti AChE activity
More secondary metabolites should be isolated and tested in vitro as it may lead to
the development of novel treatments in the global struggle against neurodegenerative
diseases.
All the compounds should also be subjected to cell based study to investigate the
mechanism of actions.
v. Anticancer
7KLV VWXG\ KDV ZLWKRXW D Goubt, proven the existence of other compound or
compounds (besides scopoletin) with potential in vitro anticancer activity in different
species and extracts of Canarium patentinervium Miq. Activity-guided fractionation,
isolation and identification of these compounds is imperative and may lead to the
development of novel treatments in the global struggle against cancer and cancer-
related ailments.
vi. Antiparasitic activity
More secondary metabolites should be isolated and tested for the efficacy against
parasitic kill and it should be tested against various clinical strains of parasites.

323
vii. Toxicity
:KLOHLQGLJHQRXVCanarium patentinervium Miq. has edible fruit, it is important to
note that in order to establish a toxicity profile, other cell lines should be investigated
such as the liver HepG2 cells, as well as in vivo studies.
viii. Phytochemical investigation
5HVHDUFK RQ WKH QRQ-volatile compounds warrant further study. Further analysis
must be conducted using different detectors and chromatographic techniques such as
liquid chromatography ± mass spectrometry, to provide a greater insight of the
phytochemical composition of this species.
3ODQWV SURGXFH D ZLGH UDQge of secondary metabolites, which may prove to be
invaluable in development of drugs, flavours, fragrances, dyes, anti-oxidants and
insecticides. It is thus important to locate and determine the role of these secondary
metabolites in plants and unravel their biosynthesis.
Finding new leads for drug development, and determining the biosynthesis of such
products, thus requires a different approach which researchers are developing.
Metabolomics investigates the end products of cellular functions. The levels of these
metabolites are viewed as a response of the biological systems to environmental or
genetic manipulation. The use of metabolomics in plant studies will enable the
characterisation and differentiation of genotypes and phenotypes based on the levels
of metabolites, and also aid in the rapid screening of multiple extracts giving an
exceptionally broad overview of the chemistry. This may also provide a means of
improving the production of certain metabolites in plants through genetic engineering.

324
REFERENCES
[1] -'3KLOOLSVRQ³3K\WRFKHPLVWU\DQGSKDUPDFRJQRV\´Phytochem. 68 2960±2972, vol. 68, pp. 2960±2972, 2007.
[2] . 6 /DP ³1HZ DVSHFWV RI QDWXUDO SURGXFWV LQ GUXJ GLVFRYHU\´ TRENDS Microbiol., vol. 15, no. 6, 2007.
[3] 6 6 +DQGD ³6WDWXV RI 0HGLFLQDO DQG $URPDWLF 3ODQWV 0$3V 8WLOL]DWLRQGlobally Including Issues of Quality Control and Technologies for Large Scale 3URGXFWLRQRI3ODQW%DVHG3URGXFWV´United Nations Ind. Dev. Organ., 2007.
[4] ''6RHMDUWR ³(WKQRJUDSKLF FRPSRQHQW DQGRUJDQLVPGRFXPHQWDWLRQ LQ DQHWKQRSKDUPDFRORJ\ SDSHU $µPLQLPXP¶ VWDQGDUG´ J. Ethnopharmacol., vol. 100, pp. 27±29, 2005.
[5] $ :HHNV ' & 'DO\ DQG % % 6LPSVRQ ³7KH SK\ORJHQHWLF KLVWRU\ DQGbiogeography of the frankincense and myrrh family (Burseraceae) based on QXFOHDU DQG FKORURSODVW VHTXHQFH GDWD´Mol. Phylogenet. Evol., vol. 35, pp. 85±101, 2005.
[6] - + /DQJHQKHLP ³3ODQW UHVLQV&KHPLVWU\ (YROXWLRQ (FRORJ\ DQG(WKQRERWDQ\´Timber Press. Portl. , Oregon, USA, 2003.
[7] ' ' )DEULFDQW DQG 1 5 )DUQVZRUWK ³7KH 9DOXH RI 3ODQWV 8VHG LQ7UDGLWLRQDO0HGLFLQHIRU'UXJ'LVFRYHU\´Environ. Heal. Perspect, vol. 109, pp. 69±75, 2001.
[8] 3:/HHQKRXWV³%XUVHUDFHDH´Flora Malesiana, vol. 5, no. 1, pp. 209±296, 1956.
[9] 3: /HHQKRXWV ³ 5HYLVLRQ RI WKH %XUVHUDFHDH RI WKHMalaysian area in a wider sense Canarium Stickm´Blumea, vol. 9, no. 2, pp. 275±647, 1959.
[10] %(YDQV ³Canarium nuts-D QHZ FDVK FURS IRU WKH6RORPRQ ,VODQGV´Trop. For. Mgt. Updat., vol. 3, no. 2, pp. 7±9, 1993.
[11] - ,:KHDWOH\ ³$*XLGH WR WKH&RPPRQ7UHHVRI9DQXDWX´Dep. For. Port Vila, Vanuatu, 1992.
[12] &:LDUW ³0HGLFLQDO SODQWV RI$VLD DQG WKH 3DFLILF´CRC Press Taylor Fr. Gr., 2006.
[13] R. E. CRURQHO ³3LOL QXW Canarium ovatum Engl. Promoting the conservation DQGXVH RI XQGHUXWLOL]HG DQGQHJOHFWHG FURSV´ Inst. Plant Genet. Crop Plant Res. Gatersleben/International Plant Genet. Resour. Institute, Rome, Italy. , 1996.

325
[14] (6WLFNPDQ³7UHH)ORUDRI0DOD\D´YROSS±127, 1972.
[15] 53HUQHW³3K\WRFKLPLHGHV%XUVHUDFHHV´Lloydia, vol. 35, p. 280, 1972.
[16] - -XOHV DQG5 ( 3DXOO ³7KH HQF\FORSHGLD RI IUXLW DQG QXWV´ SS ±207, 2006.
[17] +=KL\RQJ;:HQVKXLDQG-&KHQ³,VRODtion and structure elucidation of phenolic compounds in Chinese olive (Canarium album / IUXLW´Eur Food Res Technol, vol. 226, pp. 1191±1196, 2008.
[18] B. X. Zhao, A. S. Chen, S. J. Yong, L. W. Guang, L. X. Xiang, and W. Chen, ³3KHQROLF&RQVWLWXHQWVRI Canarium album´Chem. Nat. Compd., vol. 46, no. 1, 2010.
[19] P. Kamtchouing, S. M. Kahpui, P. D. Dzeufiet, T. Djomeni, L. Asongalem, and 7 'LPR ³$QWL-diabetic activity of methanol/methylene chloride stem bark extracts of Terminalia superba and Canarium schweinfurthii on streptozotocin-LQGXFHGGLDEHWLFUDWV´J. Ethnopharmacol., vol. 104, no. 3, pp. 306±309, 2006.
[20] - .RXGRX $ $ $EHQD 3 1JDLVVRQD DQG - 0 %HVVLHUH ³&KHPLFDOcomposition and pharmacological activity of essential oil of Canarium schweinfurthii´Fitoterapia, vol. 76, pp. 700±703, 2005.
[21] &:LDUW³0HGLFLQDO3ODQWV2I7KH$VLD-Pacific 'UXJVIRUWKH)XWXUH´World Sci. Publ. Co. Pte. Ltd., vol. British Li, pp. 380±382, 2006.
[22] D. J. Guo, H. L. Cheng, S. W. Chan, and 3+)<X³$QWLR[LGDWLYHDFWLYLWLHVDQG WKH WRWDO SKHQROLF FRQWHQWV RI WRQLF &KLQHVH PHGLFLQDO KHUEV´Inflammopharmacology, vol. 16, no. 5, pp. 201±207, 2008.
[23] / / =KDQJ DQG < 0 /LQ ³7DQQLQV IURP Canarium album with potent antioxidant activity´J. Zhejiang Univ. B, vol. 9, no. 5, pp. 407±415, 2008.
[24] M. Ito, H. Shimura, N. Watanabe, M. Tamai, K. Hanada, and A. Takahashi, ³+HSDWRSURWHFWLYH &RPSRXQGV IURP Canarium album and Euphorbia nematocypha´Chem. Pharm. Bull, vol. 38, no. 8, pp. 2201±2203, 1990.
[25] L. C. Obame, J. Koudou, B. S. Kumulungui, H. N. Ismael, E. Prosper, S. O. $ERXEDNDU DQG 6 7 $OIUHG ³$QWLR[LGDQW DQG DQWLPLFURELDO DFWLYLWLHV RICanarium schweinfurthii (QJO (VVHQWLDO RLO IURP &HQWUDIULFDQ 5HSXEOLF´African J. Biotechnol., vol. 6, no. 20, pp. 2319±2323, 2007.
[26] K. N. Prasad, L. Y. Chew, H. E. Khoo, B. Yang, A. Azrina, and I. Amin, ³&DURWHQRLGV DQG DQWLR[LGDQW FDSDFLWLHV IURPCanarium odontophyllum Miq. IUXLW´Food Chem., vol. 124, no. 4, pp. 1549±1555, 2010.

326
[27] $ , 3UDVDG .1 &KHZ /< .KRR +( <DQJ % $]ULQD $ ³$QWLR[LGDQWCapacities of Peel, Pulp and Seed Fractions of Canarium Odontophyllum Miq. )UXLW´J. Biomed. Biotechnol., p. 8 pg, 2010.
[28] M. J. Moshi, E. Innocent, P. J. Masimba, D. F. Otieno, A. Weisheit, P. 0EDED]L$/\QHV.0HDFKHP$+DPLOWRQDQG,8UDVVD³$QWLPLFURELDOand brine shrimp toxicity of some plants used in traditional medicine in Bukoba district, north- :HVWHUQ7DQ]DQLD´Tanzan. J. Health Res., vol. 11, no. 1, pp. 23±28, 2009.
[29] .$QDQG91*XSWD95DQJDUL%6LQJKDQG%.&KDQGDQ³6WUXFWXUHand Hepatoprotective Activity of a Biflavonoid from Canarium manii´Planta Med., vol. 58, no. 6, pp. 493±495, 1992.
[30] M. Tamai, N. Watanabe, M. Someya, H. Kondoh, S. Omura, P. L. Zhang, C. 5DRDQG&0LQJ³1HZ+HSDWRSURWHFWLYH7ULWHUSHQHVIURPCanarium album´Planta Med., vol. 55, pp. 44±47, 1987.
[31] P. M. . Dongmo, T. François, N. Bernadin, A. Wilson, S. Bertrand, H. A. Z. 3DXODQG0&KDQWDO³&KHPLFDOFKDUDFterization, antiradical, antioxidant and anti-inflammatory potential of the essential oils of Canarium schweinfurthii and Aucoumea klaineana %XUVHUDFHDHJURZLQJLQ&DPHURRQ´Agric. Biol. J. N. Am., vol. 1, no. 4, pp. 606±611, 2010.
[32] R. Bhuvanendran, : 0DQVRQ DQG ) 6 6SULQJ ³7ULWHUSHQH 5HVLQROV DQGRelated Acids: Isolation of a Triterpene Diol from Canarium schweinfurthii UHVLQ´J. Chem. Soc., pp. 3472±3482, 1950.
[33] + =KL\RQJ;:HQVKXL /4LQJKXD DQG - &KHQ ³,GHQWLILFDWLRQ RI D QHZphenolic compound from Chinese olive (Canarium album /IUXLW´Eur Food Res Technol, vol. 228, pp. 339±343, 2009.
[34] + =KL\RQJ DQG ; :HQVKXL ³$QDO\VLV RI SKHQROLF FRPSRXQGV LQ &KLQHVHolive (Canarium album L.) fruit by RPHPLC±DAD±ESI±0´ Food Chem., vol. 105, pp. 1307±1311, 2007.
[35] 30*LDQJ$.:LOIULHG DQG763KDQ ³&KHPLFDO&RPSRVLWLRQRI WKHResin Essential Oil Of Canarium album )URP9LHWQDP´Chem. Nat. Compd., vol. 42, no. 5, pp. 523±524, 2006.
[36] : 0 %DQGDUDQD\DNH ³7HUSHQRLGV of Canarium Zeylanicum´Phytochemistry, vol. 19, pp. 225±257, 1980.
[37] 50&DUPDQDQG'(&RZOH\ ³&DQDULF DFLG- A 3,4-Secotriterpenes Acid From Canarium Muelleri´Tetrahedron Lett., vol. 12, pp. 627±629, 1964.

327
[38] + =KL\RQJ DQG ; :HQVKXL ³3UHSarative separation and purification of phenolic compounds from Canarium album /E\PDFURSRURXV UHVLQV´J Sci Food Agric, vol. 88, pp. 493±498, 2008.
[39] K. N. Prasad, L. Y. Chew, H. E. Khoo, B. Yang, A. Azrina, and I. Amin, ³&DURWHQRLGV DQG DQWLR[LGDQW capacities from Canarium odontophyllum Miq. IUXLW´Food Chem., vol. 124, no. 4, pp. 1549±1555, 2010.
[40] ' 0 9LQFHQ]L ' $ 9LQFHQ]L DQG 0 6LODQR ³&RQVWLWXHQWV RI DURPDWLFSODQWVHOHPLFLQ´Fitoterapia, vol. 75, no. 6, pp. 615±618, 2004.
[41] H. WHL : 3HQJ < 0DR % /LX DQG 6 /L ³6WXGLHV RQ WKH &KHPLFDOConstituents in the fruit of Canarium album /RXU 5DHXVFK´ Zhongguo Zhong Yao Za Zhi, vol. 24, no. 7, pp. 421±423, 1999.
[42] 2-$ED\HK$.$EGXOUD]DTDQG52ODRJXQ³4XDOLW\&KDUDcteristics of Canarium schweinfurthii (QJORLO´Plant Foods Hum. Nutr., vol. 54, pp. 43±48, 1999.
[43] &'%UXFHDQG11%HQHGLFWD³7KHOLSLGDQGIDWW\DFLGSURILOHRIWKHIUXLWof Canarium schweinfurthii (QJO´ S. Afr. J. Sci., vol. 99, no. July/August, 2003.
[44] '%LOOHW6+HLW]'5DXODLVDQG$0DWVFKHQNR³&RQVWLWXDQWV WHUSHQLTXHVde Canarium boivinii HQJO´Phytochemistry, vol. 10, no. 7, pp. 1681±1683, 1971.
[45] 5 %KXYDQHQGUDQ 5 & 9DVLVWD DQG 6 'XWW ³&KHPLFDO ([DPLQDWLRQ RICanarium Bengalense 5R[E´Curr. Sci., vol. 32, pp. 162±163, 1963.
[46] 0 / 6WHYHQV 5 0 %RXUNH DQG % 5 (YDQV ³6RXWK 3DFLILF ,QGLJHQRXV1XWV´ Proc. a Work. Le Lagon Resort, Port Vila, Vanuatu, 31 Oct. ± 4 Novembe, 1994.
[47] B. Evans and L. J. ThomsoQ³Canarium indicum var. indicum and C. harveyi FDQDULXPQXW%XUVHUDFHDH WRUFKZRRG IDPLO\´Species Profiles Pacific Isl. Agrofor., 2006.
[48] S. D. Edited by 6DUNHU /DWLI = *UD\ $, ³([WUDFWLRQ RI 3ODQW 6HFRQGDU\0HWDEROLWHV´Nat. Prod. Isol., vol. 2nd ed, pp. 339±342, 2006.
[49] )0RMDE0.DPDOLQHMDG1*KDGHULDQG+59DKLGLSRXU³3K\WRFKHPLFDO6FUHHQLQJRI6RPH6SHFLHVRI,UDQLDQ3ODQWV´Iran. J. Pharm. Res., pp. 77±82, 2003.
[50] -%+DUERUQH³3K\WRFKHPLFDOPHWKRGV´The Terpenoids, p. 129, 1998.

328
[51] -%+DUERUQH DQG&$:LOOLDPV ³5HYLHZ$GYDQFHV LQ)DYRQRLG UHVHDUFKVLQFH´Phytochemistry, vol. 55, pp. 481±504, 2000.
[52] 5$UWLFOH³)ODYRQRLGVௗDUHYLHZRISUREDEOHPHFKDQLVPVRIDFWLRQDQG´SS418±425, 2001.
[53] $51GKODOD00R\RDQG-9DQ6WDGHQ³1DWXUDODQWLR[LGDQWVIDVFLQDWLQJRU P\WKLFDO ELRPROHFXOHV"´ Molecules, vol. 15, no. 10, pp. 6905±30, Jan. 2010.
[54] 1-/DUNLQV³)UHHUDGLFDOELRORJ\DQGSDWKRORJ\´J. Equine Vet. Sci., vol. 19, pp. 84±89, 1999.
[55] A. V Badarinath, K. M. Rao, C. M. S. Chetty, S. Ramkanth, T. V. S. Rajan, and .*QDQDSUDNDVK³$5HYLHZRQ,Q-YLWUR$QWLR[LGDQW0HWKRGVௗ&RPSDULVLRQV&RUUHODWLRQVDQG&RQVLGHUDWLRQV´YROQRSS±1285, 2010.
[56] K. Joon and S. 7DND\XNL ³$QWLR[LGDQW $VVD\V IRU 3ODQW DQG )RRG&RPSRQHQWV´J. Agric. Food Chem., vol. 57, no. 1655±1666, 2009.
[57] $ .DUDGDJ % 2]FHOLN DQG 6 6DQHU ³5HYLHZ RI 0HWKRGV WR 'HWHUPLQH$QWLR[LGDQW&DSDFLWLHV´Food Anal. Methods, vol. 2, pp. 41±60, 2009.
[58] R. R. Hafidh, A. S. Abdulamir, F. Abu Bakar, F. Abas, F. Jahanshiri, and Z. 6HNDZL³$QWLR[LGDQW5HVHDUFKLQ$VLDLQWKH3HULRGIURP-´Am. J. Pharmacol. Toxicol., vol. 4, no. 3, pp. 48±66, 2009.
[59] E. Middleton, C. Kandaswami, and T. C. 7KHRKDULGHV ³7KH HIIHFWV RI SODQWflavonoids on mammalian cells: implications for inflammation, heart disease DQGFDQFHU´Pharmacol. Rev., vol. 52, no. 4, pp. 673±751, 2000.
[60] 0%$UQDR$&DQRDQG0$FRVWD³0HWKRGVWRPHDVXUHWKHDQWL-oxidant DFWLYLW\LQSODQWPDWHULDO$FRPSDUDWLYHGLVFXVVLRQ´Free Radic. Res., vol. 31, pp. S89±S96, 1999.
[61] + 0DHGD DQG 7 $NDLNH ³5HYLHZ 1LWULF R[LGH DQG R[\JHQ UDGLFDOV LQLQIHFWLRQ LQIODPPDWLRQ DQGFDQFHU´Biochem., vol. 63, no. 7, pp. 854±1007, 1998.
[62] 5-3HUU\-5+RGJHVDQG3:DWVRQ³7KHQDWXUHDQGVWDJLQJRIDWWHQWLRQG\VIXQFWLRQ LQ HDUO\ PLQLPDO DQG PLO $O]KHLPHU¶V GLVHDVH UHODWLRQVKLS WRHSLVRGLF DQG VHPDQWLFPHPRU\ LPSDLUPHQW´Neuropsychologia, vol. 38, pp. 252±271, 2000.
[63] M. Ito, S. Fukushima, A. Hassegawa, M. Shibata, and T. Ogiso, ³&DUFLQRJHQLFLW\ RI EXW\ODWHG K\GUR[\DQLVROH LQ ) UDWV´ J. Natl. Cancer Inst, vol. 70, pp. 343±347, 1983.

329
[64] C. P. Jayalakshmi 6KDUPD -' ³(IIHFW RI %XW\ODWHG +\GUR[\DQLVROH %+$and But\ODWHG+\GUR[\WROXHQH%+7RQ5DW(U\WKURF\WHV´Environ. Res., vol. 41, pp. 235±238, 1986.
[65] 3 0RO\QHX[ ³7KH XVH RI WKH VWDEOH IUHH UDGLFDO GLSKHQ\OSLFU\OK\GUD]\O'33+ IRU HVWLPDWLQJ DQWLR[LGDQW DFWLYLW\´Songklanakarin J. Sci. Technol., vol. 26, no. 2, pp. 211±219, 2004.
[66] 3 6FDUWH]]LQL DQG ( 6SHURQL ³5HYLHZ RQ VRPH ,QGLDQ WUDGLWLRQDOPHGLFLQHZLWK DQWLR[LGDQW DFWLYLW\´ J. Ethnopharmacol., vol. 71, no. 1±2, pp. 23±43, 2000.
[67] L. L. Zhang and L. Yi-PLQJ ³7DQQLQV IURP Canarium album with potent DQWLR[LGDQWDFWLYLW\ ´J Zhejiang Univ Sci B, vol. 9, no. 5, pp. 407±415, 2008.
[68] .3UDVDG/&KHZ+.KRR%<DQJ$$]ULQDDQG,$PLQ³$QWLR[LGDQWCapacities of Peel, Pulp and Seed Fractions of Canarium Odontophyllum Miq. )UXLW´J. Biomed. Biotechnol., p. 8 pg, 2010.
[69] '+XDQJ%2XDQG5/3ULRU³7KHFKHPLVWU\EHKLQGDQWLR[LGDQWFDSDFLW\DVVD\V´J. Agric. Food Chem., vol. 53, no. 6, pp. 1841±56, Mar. 2005.
[70] 5 / 3ULRU ; :X DQG . 6FKDLFK ³6WDQGDUGL]HG PHWKRGV IRU WKHdetermination of antioxidant capacity and phenolics in foods and dietary VXSSOHPHQWV´J. Agric. Food Chem., vol. 53, no. 10, pp. 4290±302, May 2005.
[71] M. Juan-Badaturuge, S. Habtemariam DQG 0 - . 7KRPDV ³$QWLR[LGDQWcompounds from a South Asian beverage and medicinal plant, Cassia auriculata´Food Chem., vol. 125, pp. 221±225, 2011.
[72] 06%ORLV ³$QWLR[LGDQWVGHWHUPLQDWLRQE\ WKHXVHRID VWDEOH IUHH UDGLFDO´Nature, vol. 181, pp. 1199±1200, 1958.
[73] $5HúDW*.XELOD\'%LUVHQg0XVWDID (&6DOLKD%%XUFX. ,úÕO%HUNHU DQGg'LOHN³&RPSDUDWLYH(YDOXDWLRQRI9DULRXV7RWDO$QWLR[LGDQW&DSDFLW\$VVD\V$SSOLHGWR3KHQROLF&RPSRXQGVZLWKWKH&835$&$VVD\´Molecules, vol. 12, pp. 1496±1547, 2007.
[74] : %UDQG :LOOLDPV 0 ( &XYHOLHU DQG & %HUVHW ³8VH RI D IUHH UDGLFDOPHWKRG WRHYDOXDWHDQWLR[LGDQWDFWLYLW\´Food Sci. Technol., vol. 28, pp. 25±30, 1995.
[75] N. J. Miller, C. A. Rice-Evans, M. J. Davies, V. GRSLQDWKDQDQG$0LOQHU³$novel method for measuring antioxidant capacity and its application to PRQLWRULQJWKHDQWLR[LGDQWVWDWXVLQSUHPDWXUHQHRQDWHV´Clin. Sci., vol. 84, pp. 407±412, 1993.

330
[76] C. A. Rice-(YDQV ³)RUPDWLRQ RI IUHH UDGLFDOV DQGPHFhanisms of action in QRUPDOELRFKHPLFDOSURFHVVHVDQGSDWKRORJLFDOVWDWHV´New Compr. Biochem., vol. 28, pp. 131±153, 1994.
[77] R. Roberta, P. Ananth, M. Yang, C. Rice- Evans, and N. Pellegrini, ³$QWLR[LGDQW $FWLYLW\ $SSO\LQJ $Q ,PSURYHG $%76 5DGLFDO &DWion 'HFRORUL]DWLRQ$VVD\´Free Radic. Biol. Med., vol. 26, pp. 1231±1237, 1999.
[78] 5*XSWD06KDUPDDQG5/DNVKP\³,PSURYHGPHWKRGRIWRWDODQWLR[LGDQWDVVD\´YROQR)HEUXDU\SS±129, 2009.
[79] ,--%HQ]LHDQG--6WUDLQ³7KH)HUULF5HGXFLQJ$ELOLW\RI3ODVPD)5$3DV D0HDVXUH RI µ³$QWLR[LGDQW 3RZHU´¶ 7KH )5$3$VVD\´Anal. Biochem., vol. 239, pp. 70±76, 1996.
[80] 6 +DEWHPDULDP DQG & -DFNVRQ ³$QWLR[LGDQW DQG F\WRSURWHFWLYH DFWLYLW\ of OHDYHV RI3HOWLSK\OOXPSHOWDWXP 7RUU(QJO´Food Chem., vol. 105, no. 2, pp. 498±503, 2007.
[81] 6ZLW]HUODQG³62'GHWHUPLQDWLRQNLW´Sigma-Aldrich Chemie GmbH · Ind. 25 · Postfach ·, vol. CH-9471 Bu.
[82] 9 / 6LQJOHWRQ DQG - $ 5RVVL ³&RORULPHtry of total phenolics with phosphomolybdic-SKRVSKRWXQJVWLFDFLGUHDJHQWV´Am. J. Enol. Vitic., vol. 16, pp. 144±158, 1965.
[83] T. Froehlicher, T. Hennebelle, F. Martin-Nizard, P. Cleenewerck, J. Hilbert, H. 7URWLQDQG6*UHF³3KHQROLFSURILOHVDQGDQtioxidative effect of hawthorn cell VXVSHQVLRQVIUHVKIUXLWVDQGPHGLFLQDOGULHGSDUWV´Food Chem., vol. 115, pp. 897±903, 2009.
[84] & &KDQJ0 <DQJ +:HQ DQG - &KHUQ ³(VWLPDWLRQ RI 7RWDO )ODYRQRLGContent in Propolis by Two Complementary ColorimHWULF0HWKRGV´YRO no. 3, pp. 178±182, 2002.
[85] '+XDQJ%2XDQG5/3ULRU³7KHFKHPLVWU\EHKLQGDQWLR[LGDQWFDSDFLW\DVVD\V´J. Agric. Food Chem., vol. 53, no. 6, pp. 1841±56, Mar. 2005.
[86] A. Dapkevicius, R. Venskutonis, T. A. Van Beek, and J. P. H. Linssen, ³$QWLR[LGDWLYH DFWLYLW\ RI H[WUDFWV REWDLQHG E\ GLIIHUHQW LVRODWLRQ SURFHGXUHVIURPVRPHDURPDWLFKHUEVJURZQLQ/LWKXDQLD´J. Sci. Food Agric., vol. 77, pp. 140±146, 1998.
[87] P. Dhar, a B. Tayade, P. K. Bajpai, V. K. Sharma, S. K. Das, O. P. Chaurasia, 5 % 6ULYDVWDYD DQG 6 % 6LQJK ³$QWLR[LGDQW &DSDFLWLHV DQG 7RWDOPolyphenol Contents of Hydro-ethanolic Extract of Phytococktail from Trans-+LPDOD\D´J. Food Sci., vol. 77, no. 2, pp. C156±61, Feb. 2012.

331
[88] N. Abdullah, S. M. Ismail, N. Aminudin, A. S. Shuib, and B. F. Lau, ³(YDOXDWLRQ RI 6HOHFWHG &XOLQDU\-Medicinal Mushrooms for Antioxidant and $&( ,QKLELWRU\ $FWLYLWLHV´ Evid. Based. Complement. Alternat. Med., vol. 2012, p. 464238, Jan. 2012.
[89] M. M. Kulisic T, RadoniF$.DWDOLQLF9³8VHRIGLIIHUHQWPHWKRGVIRUWHVWLQJDQWLR[LGDWLYHDFWLYLW\RIRUHJDQRHVVHQWLDORLO´Food Chem., vol. 85, no. 4, pp. 633±640, 2004.
[90] R. Mogana, K. Teng--LQ DQG&:LDUW ³,Q9LWUR$QWLPLFURELDO$QWLR[LGDQWActivities and Phytochemical Analysis of Canarium patentinervium Miq. from 0DOD\VLD´Biotechnol. Res. Int., vol. 2011, p. 768673, 2011.
[91] G. Rusak, H. O. Gutzeit, and J. L. MuelOHU ³6WUXFWXUDOO\ UHODWHG IODYRQRLGVwith antioxidative properties differentially affect cell-cycle progression and DSRSWRVLVRIKXPDQDFXWHOHXNHPLDFHOOV´Nutr. Res., vol. 25, no. 2, pp. 141±153, 2005.
[92] / ):DQJ DQG+ < =KDQJ ³$ WKHRUHWLFDO LQvestigation on DPPH radical VFDYHQJLQJPHFKDQLVPRI HGDUDYRQH´Bioorganic Med. Chem. Lett., vol. 13, no. 21, pp. 3789±3792, 2003.
[93] 0-7-$UWV-6'DOOLQJD9+3*500+DHQHQDQG$%DVW³$critical appraisal of the use of the antioxidant capacity (TEAC) assay in GHILQLQJ RSWLPDO DQWLR[LGDQW VWUXFWXUHV´Food Chem., vol. 80, pp. 409±414, 2003.
[94] 03DUDVNHYDD5YDQ=\O+'DYLGVD$9LOMRHQEDQG6YDQ9XXUHQD³7KHin vitro biological activity of selected South African Commiphora VSHFLHV´J. Ethnopharmacol., vol. 119, pp. 673±679, 2008.
[95] .5REDUGV3'3OHQ]HU*7XFNHU36ZDWVLWDQJDQG:*ORYHU³3KHQROLFFRPSRXQGV DQG WKHLU UROH LQR[LGDWLYHSURFHVVHV LQ IUXLWV´Food Chem., vol. 66, pp. 401±436, 1999.
[96] A. Rojas, L. Hernandez, Pereda-0LUDQGD DQG 5 0DWD ³6FUHHQLQJ IRUantimicrobial activity of crude drug extracts and pure natural products from 0H[LFDQPHGLFLQDOSODQWV´J. Ethnopharmacol., vol. 35, pp. 275±283, 1992.
[97] J. D. Levine and D. B. Reichling ³3HULSKHUDO PHFKDQLVPV RI LQIODPPDWRU\SDLQ´LQTextbook of pain, Fourth Edi., 1999, pp. 59±84.
[98] -%&DOL[WR0)2WXNLDQG$566DQWRV³$QWL-inflammatory compounds of plant origin. Part I. Action on arachidonic acid pathway, nitric oxide and nuclear factor kappa B (NF-NDSSD%´Planta Med., vol. 69, no. 11, pp. 973±83, Nov. 2003.

332
[99] %6DPXHOVVRQ³$QHOXFLGDWLRQRIWKHDUDFKLGRQLFDFLGFDVFDGH'LVFRYHU\RISURVWDJODQGLQV WKURPER[DQHDQG OHXNRWULHQHV´Drugs, no. Suppl 1, pp. 2±9, 1987.
[100] 0-DQJDQG-03H]]XWR³$VVHVVPHQWRIF\FORR[\JHQDVHLQKLELWRUVXVLQJLQYLWURDVVD\V\VWHPV´Methods Cell Sci., vol. 31, no. March, pp. 25±31, 1997.
[101] V. Kumar, Pathological Basis of Disease, 8th ed. 2010.
[102] H. P. Kim, K. H. Son, H. :&KDQJDQG66.DQJ³&ULWLFDO5HYLHZ$QWL-LQIODPPDWRU\ 3ODQW )ODYRQRLGV DQG &HOOXODU $FWLRQ 0HFKDQLVPV´ J. Pharmacol. Sci., vol. 245, no. 3, pp. 229 ± 245, 2004.
[103] B. Samuelsson, S. E. Dahlen, J. A. Lindgren, C. A. Rouzer, and C. N. Serhan, ³/HXNRWULHQHV DQG OLSR[LQV VWUXFWXUHV ELRV\QWKHVLV DQG ELRORJLFDO HIIHFWV´Science (80-. )., vol. 237, no. 4819, pp. 1171±1176, 1987.
[104] $$6SHFWRU-$*RUGRQDQG6$0RRUH³+\GUR[\HLFRVDWHWUDHQRLFDFLGV+(7(V´Prog. Lipid Res., vol. 27, no. 4, pp. 271±323, 1988.
[105] 2 :HU] DQG ' 6WHLQKLOEHU ³7KHUDSHXWLF RSWLRQV IRU -Lipoxygenase LQKLELWRUV´Pharmacol. Ther., vol. 112, pp. 701±718, 2006.
[106] M. G. A. W. Ford-Hutchinson DQG 5 1 <RXQJ ³-/,32;<*(1$6(´Annu. Rev. Biochem., vol. 63, pp. 383±417, 1994.
[107] +3.LP.+6RQ+:&KDQJDQG66.DQJ³$QWL-inflammatory plant IODYRQRLGVDQGFHOOXODUDFWLRQPHFKDQLVPV´J. Pharmacol. Sci., vol. 96, no. 3, pp. 229±245, 2004.
[108] +(&ODHVVRQDQG6('DKOHQ³$VWKPDDQG leukotrienes: Antileukotrienes as novel anti-DVWKPDWLFGUXJV´J. Intern. Med., vol. 245, no. 3, pp. 205±227, 1999.
[109] 5 $ /HZLV . ) $XVWHQ DQG 5 - 6REHUPDQ ³/HXNRWULHQHV DQG RWKHUproducts of the 5-lipoxygenase pathway. Biochemistry and relation to SDWKRELRORJ\LQKXPDQGLVHDVHV´N. Engl. J. Med., vol. 323, no. 10, pp. 645±655, 1990.
[110] J. M. Schwab, H. J. Schluesener, R. Meyermann, and C. N. Serhan, ³3URVWDJODQGLQV/HXNRW(VVHQW´Fat. Acids, vol. 69, p. 339, 2003.
[111] A. K. Gadad, M. B. Palkar, K. Anand, M. N. Noolvi, T. S. Boreddy, and J. :DJZDGH³%LRRUJ´Med. Chem., vol. 16, p. 276, 2008.
[112] -*LHUVHDQG&.REROGW³&\FORR[\JHQDVHDVVD\V´Curr. Protoc. Pharmacol., no. 3.1, 1998.

333
[113] M. H. Yang, K. D. Yoon, Y.-: &KLQ - + 3DUN DQG - .LP ³3KHQROLFcompounds with radical scavenging and cyclooxygenase-2 (COX-2) inhibitory activities from Dioscorea opposita´Bioorg. Med. Chem., vol. 17, no. 7, pp. 2689±94, Apr. 2009.
[114] S. Fiorucci, i R. Mel, M. Bucci, DQG * &LULQR ³'XDO LQKLELWRUV RIcyclooxygenase and 5-lipoxygenase. A new avenue in anti-inflammatory WKHUDS\"´Biochem Pharmacol, vol. 62, pp. 1433±1438, 2001.
[115] T. Kirchner, B. Aparicio, i D. C. Argentier, C. Y. Lau, and D. M. Ritchie, ³(IIHFWV Rf tepoxalin, a dual inhibitor of cyclooxygenase/5-lipoxygenase and cyclooxygenase-1 and 2 on events associated with NSAID-induced JDVWURLQWHVWLQDO LQIODPPDWLRQ´Prostaglandins Leukot Essent Fat. Acids, vol. 56, pp. 417±423, 1997.
[116] I. F. Cellot and T 'XUDQG ³7KH PHWDEROLF HIIHFWV RI LQKLELWRUV RI - lipoxygenase and of cyclooxygenase 1 and 2 are an advancement in the efficacy and safety of anti-LQIODPPDWRU\WKHUDS\´Prostaglandins Other Lipid Mediat, vol. 71, no. 147±162, 2003.
[117] M. M. Shelly DQG&-+DZNH\³&R[-Lox inhibition: current evidence for an HPHUJLQJQHZWKHUDS\´Int J Clin Pr., vol. 57, pp. 301±304, 2003.
[118] J. M. Alvaro-*DUFLD ³/LFRIHORQH-clinical update on a novel LOX/COX LQKLELWRUIRUWKHWUHDWPHQWRIRVWHRDUWKULWLV´Rheumatol., vol. 43 (Supp 1, pp. 21±25, 2004.
[119] . %UXQH ³6DIHW\ RI DQWL-inflammatory treatments-QHZ ZD\V RI WKLQNLQJ´Rheumatol., vol. 43 (Supp 1, pp. 116±120, 2004.
[120] - & 6LUFDU & ) 6KZHQGHU DQG ( $ -RKQVRQ ³6R\EHDQ OLSR[\JHQDVHinhibition by nonsteroidal anti-LQIODPPDWRU\GUXJV´Prostaglandins, vol. 25, pp. 393±396, 1983.
[121] $ 7 (YDQV ³$FWLRQV RI FDQQDELV FRQVWLWXHQWV RQ HQ]\PHV RI DUDFKLGRQDWHmetabolism anti-LQIODPPDWRU\ SRWHQWLDO´Biochem. Pharmacol., vol. 36, pp. 2035±2037, 1987.
[122] * 3 3 .DPDWRX $ 0 9LOMRHQ DQG 3 6WHHQNDPS ³$QWLR[LGDQWantiinflammatory activities and HPLC analysis of South African Salvia VSHFLHV´Food Chem., vol. 119, pp. 684±688, 2010.
[123] 6%D\ODF DQG 3 5DFLQH ³,QKLELWLRQ RI -lipoxygenase by essential oils and RWKHU QDWXUDO IUDJUDQW H[WUDFWV´ Int. J. Aromatheraphy, vol. 13, pp. 138±142, 2003.
[124] A. A. Vieira, M. G. D. A. Oliveira, I. C. José, N. D. Piovesan, S. T. D. 5H]HQGH0$0RUHLUDDQG(*DQG%DUURV³%LRFKHPLFDOevaluation

334
RIOLSR[\JHQDVHSDWKZD\RIVR\EHDQSODQWVVXEPLWWHGWRZRXQGLQJ´Rumos da Fisiol. Veg. no Bras., vol. 13, no. 1, pp. 5±12, 2001.
[125] 6=VFKRFNH6('UHZHV.3DXOXV5%DXHUDQG-YDQ6WDGHQ³$QDO\WLFDOand pharmacological investigation of Ocotea bullata (black stinkwood) bark DQGOHDYHV´J. Ethnopharmacol., vol. 71, no. 219±230, 1999.
[126] ( 2VKHU * :HLVLQJHU 5 /LPRU . 7RUGMPDQ DQG 1 6WHUQ ³7KH -lipoxygenase system in the vasculature: emerging role in health and diseasH´Mol. Cell. Endocrinol., vol. 252, no. 1±2, pp. 201±2±6, 2006.
[127] J. H. Dwyer, H. Altayee, K. M. Dwyer, H. Fan, J., Wu, R. Mar, A. J. Lusis, and 00³$UDFKLGRQDWH-lipoxygenase promoter genotype, dietary arachidonic DFLGDQGDWKHURVFOHURVLV´N. Engl. J. Med., vol. 350, no. 1, pp. 29±37, 2004.
[128] A. Helgadottir, A. Manolescu, G. Thorleifsson, S. Gretarsdottir, H. Jonsdottir, G. Thorsteinsdottir, U. Samani, N.J. Gudmundsson, S. F. A. Grant, G. Thorgeirsson, H. Sveinbjornsdottir, S. Valdimarsson, E.M. Matthiasson, S.E. Johannsson, M. Gudmundsdottir, O. Gurney, M.E., Sainz, J. Thorhallsdottir, M. Andresdottir, H. Frigge, M.L., Topol, E.J. Kong, A., Gudnason, V. +DNRQDUVRQ - 5 *XOFKHU DQG . 6WHIDQVVRQ ³7KH JHQH HQFRGLQJ -lipoxygenase activating protein confers risk of myocardial infarction and VWURNH´Nat. Genet., vol. 36, no. 3, pp. 233±239, 2004.
[129] S. Helgadottir, A. Gretarsdottir, D. St. Clair, A. Manolescu, J. Cheung, G. Thorleifsson, A. Pasdar, S. F. Grant, L. J. Whalley, U. Hakonarson, H. Thorsteindottir, A. Kong, J. Gulcher, K. Stefansson, and M. J. MacLeod, ³$VVRFLDWLRQEHWZHHQWKHJHQHHQFRGLQJ-lipoxygenase activating protein and VWURNHUHSOLFDWHGLQD6FRWWLVKSRSXODWLRQ´Am. J. Hum. Genet., vol. 76, no. 3, pp. 505±509, 2005.
[130] A. Helgadottir, A. Manolescu, A. Helgason, U. Thorleifsson, G. Thorsteinsdottir, G. Gudbjartsson, D.F. Gretarsdottir, S. Magnusson, K.P. *XGPXQGVVRQ$+LFNV7-RQVVRQ6)$*UDQW-6DLQ]62¶%ULHQ6-Sveinbjornsdottir, S. E. Valdimarsson, E.M. Matthiasson, A. I. Levey, J. L. Abramson, M. P. Reilly, V. Vaccarino, M. L. Wolfe, V. Gudnason, A. A. Quyyumi, E. J. Topol, D. J. Rader, G. Thorgeirsson, J. R. Gulcher, H. +DNRQDUVRQ $ .RQJ DQG . 6WHIDQVVRQ ³$ YDULDQW RI WKH JHQH HQFRGLng leukotriene A4 Hydrolase confers ethnicity-specific risk of myocardial LQIDUFWLRQ´Nat. Genet., vol. 38, no. 1, pp. 68±74, 2006.
[131] % 3 %XUQHWW 4 -LD < =KDR DQG 5 0 /HY\ ³$ PHGLFLQDO H[WUDFW RIScutellaria baicalensis and Acacia catechu acts as a dual inhibitor of cyclooxygenase and 5-OLSR[\JHQDVH WR UHGXFH LQIODPPDWLRQ´ J. Med. Food, vol. 10, no. 3, pp. 442±51, Sep. 2007.

335
[132] & : &KRL ³$QWL-oxidant activity and free radical scavenging capacity between Korean medicinal plants and flDYRQRLGVE\DVVD\JXLGHGFRPSDULVRQ´Plant Sci., vol. 163, pp. 1161±1168, 2002.
[133] G. A. Alitonou, F. Avlessi, D. K. Sohounhloue, H. Agnaniet, J. M. Bessiere, DQG&00HQXW³,QYHVWLJDWLRQVRQWKHHVVHQWLDORLORICymbopogon giganteus from Benin for its potential use as an anti-LQIODPPDWRU\ DJHQW´ Int. J. Aromatheraphy, vol. 16, pp. 37±41, 2006.
[134] 3-+RXJKWRQDQG0-+RZHV³1DWXUDO3URGXFWVDQG'HULYDWLYHVDIIHFWLQJQHXURWUDQVPLVVLRQ UHOHYDQW WR $O]KHLPHU¶V DQG 3DUNLQVRQ¶V GLVHDVH´Neurosignals, vol. 14, pp. 6±22, 2005.
[135] .+RVWHWWPDQQ$8%RUOR]DQG$0DUVWRQ³1DWXUDO3URGXFW,QKLELWRUVRI$FHW\OFKROLQHVWHUDVH´Curr. Org. Chem., vol. 10, pp. 825±847, 2006.
[136] %)UDQNDQG6*XSWD³$UHYLHZRIDQWLR[LGDQWV DQG$O]KHLPHU¶VGLVHDVH´Ann. Clin. Psychol., vol. 17, pp. 269±286, 2005.
[137] +0+RXJKWRQ3-5HQ< ³$FHW\OFKROLQHVWHUDVH LQKLELWRUV IURPSODQWV DQGIXQJL´Nat. Prod. Rep., vol. 23, pp. 181±199, 2006.
[138] C. M. Beard, E. Kökmen, and L. T. KurlaQG ³3UHYDOHQFH RI GHPHQWLD LVFKDQJLQJRYHUWLPHLQ5RFKHVWHU0LQQHVRWD´Neurology, vol. 45, pp. 75±79, 1995.
[139] ,2UKDQ%6HQHU0,&KRXGKDU\DQGD.KDOLG³$FHW\OFKROLQHVWHUDVHDQGbutyrylcholinesterase inhibitory activity of some Turkish mHGLFLQDOSODQWV´J. Ethnopharmacol., vol. 91, no. 1, pp. 57±60, Mar. 2004.
[140] - 5 +RGJHV ³$O]KHLPHU¶V FHQWHQQLDO OHJDF\ RULJLQV ODQGPDUNV DQG WKHFXUUHQWVWDWXVRINQRZOHGJHFRQFHUQLQJFRJQLWLYHDVSHFWV´Brain, vol. 129, pp. 2811±2822, 2006.
[141] T. M. Hung, T. M. Ngoc, U. J. Youn, B. S. Min, M. Na, P. T. Thuong, and K. %DH ³$QWL-amnestic activity of pseudocoptisine from Corydalis WXEHU´Biol. Pharm. Bull., vol. 31, pp. 159±162, 2008.
[142] 66DQFKHWL%+8PDQG6<6HR³-penta-Ogalloyl- ȕ-D-glucose: A cholinesterase inhibitor from Terminalia chebula´S. Afr. J. Bot., vol. 76, no. 2, pp. 285±288, 2010.
[143] 0-+RZHVDQG3-+RXJKWRQ³3ODQWVXVHGLQ&KLQHVHDQG,QGLDQWUDGLWLRQDOmedicine for improvement of memory and FRJQLWLYH IXQFWLRQ´ Pharmacol. Biochem. Behav., vol. 75, pp. 513±527, 2003.
[144] 3 . 0XNKHUMHH 9 .XPDU DQG 3 - +RXJKWRQ ³$FHW\OFKROLQHVWHUDVHLQKLELWRUVIURPSODQWV´Phytomedicine, vol. 14, no. 4, pp. 289±300, Apr. 2007.

336
[145] H. Rang, M. DaleDQG-5LWWHU³&KROLQHUJLFWUDQVPLVVLRQ´LQPharmacology, 3rd ed., Churchil Livingstone, 1995, pp. 117±146.
[146] . 6 6:RQ +\XN 6XK DQG <RR-+XQ 6XK ³7KHUDSHXWLF $JHQWV IRU$O]KHLPHU¶V'LVHDVH´Curr. Med. Chem. ± Cent. Nerv. Syst. Agents,, vol. 5, pp. 259±269, 2005.
[147] $ 7ULSDWKL DQG 8 6ULYDVWDYD ³$FHW\OFKROQHVWHUDVH $ YHUVDWLOH HQ]\PH RIQHUYRXVV\VWHP´Ann. Neurosci., vol. 15, no. 4, pp. 1±14, 2008.
[148] < 6XK DQG ) &KHFOHU ³$P\ORLG 3UHFXUVRU 3URWHLQ 3UHVHQLOLQV DQGհ -SynuFOHLQௗ 0ROHFXODU 3DWKRJHQHVLV DQG 3KDUPDFRORJLFDO $SSOLFDWLRQV LQ$O]KHLPHU¶V'LVHDVH´Pharmacol. Rev., vol. 54, no. 3, pp. 469±525, 2002.
[149] ' - 6HONRH ³$O]KHLPHU ¶ V 'LVHDVHௗ *HQHV 3URWHLQV DQG 7KHUDS\´Pharmacol. Rev., vol. 81, no. 2, pp. 741±766, 2001.
[150] */(OOPDQ'&RXUWQH\9-$QGUHVDQG50)HDWKHUVWRQH³$1HZDQG5DSLG &RORULPHWULF 'HWHUPLQDWLRQ RI $FHWK\OFKROLQHVWHUDVH $FWLYLW\´Biochem. harmacology, vol. 7, pp. 88±95, 1961.
[151] P. J. Houghton, M.-J. Howes, C. C. /HHDQG*6WHYHQWRQ³8VHVDQGDEXVHVRILQ YLWUR WHVWV LQ HWKQRSKDUPDFRORJ\ YLVXDOL]LQJ DQ HOHSKDQW´ J. Ethnopharmacol., vol. 110, pp. 391±400, 2007.
[152] *(PLOLHQ.%H\UHXWKHU&/0DVWHUDQG-00DORWHDX[³3URVSHFWVIRUpharmacological inWHUYHQWLRQLQ$O]KHLPHUGLVHDVH´Arch. Neurol., vol. 57, pp. 454±459, 2000.
[153] 1 7DEHW ³$FHW\OFKROLQHVWHUDVH LQKLELWRUV IRU $O]KHLPHU¶V GLVHDVHDQWLLQIODPPDWRULHV LQ DFHW\OFKROLQH FORWKLQJ´Age Ageing, vol. 35, pp. 336±338, 2006.
[154] K. J. BarnhaP&/0DVWHUVDQG$,%XVK³1HXURGHJHQHUDWLYHGLVHDVHVDQGR[LGDWLYHVWUHVV´Nat. Rev. Drug Disc., vol. 3, pp. 205±214, 2004.
[155] / 9 %RURYLNRYD DQG 0 ,YDQRYD 6 =KDQJ ³9DJXV QHUYH VWLPXODWLRQattenuates the systemic inflammatory response to endo-WR[LQ´ Nature, vol. 405, pp. 458±462, 2000.
[156] ,2UKDQ0.DUWDO ) 7RVXQ DQG%6HQHU ³6FUHHQLQJ RI YDULRXV SKHQROLFDFLGV DQG IODYRQRLG GHULYDWLYHV IRU WKHLU DQWLFKROLQHVWHUDVH SRWHQWLDO´ Z. Naturforsch. C., vol. 62, no. 11±12, pp. 829±32, 2007.
[157] :+2³&RQWDLQLQJ$QWLPLFURELDO5HVLVWDQFH´
[158] -(%DQGRZ+%URW]DQG,/HLFKHUW³3URWHRPLFDSSURDFKWRXQGHUVWDQGLQJDQWLELRWLFDFWLRQ´Antimicrob Agents Chemother, vol. 47, pp. 948±955, 2003.

337
[159] M. A. Pfaller, R. N. JoQHV*9*RHUQDQG..XJOHU³%DFWHULDO3DWKRJHQVIsolated from Patients with Bloodstream Infection: Frequencies of Occurrence and Antimicrobial Susceptibility Patterns from the SENTRY Antimicrobial Surveillance Program (United States and Canada, 1997)´Antimicrob. agent Chemother., pp. 1762±1770, 1998.
[160] L. M. Prescott +DUOH\-3DQG.OHLQ'$³1R7LWOH´Microbiology, vol. 3rd Ed. Th, no. Inc. United States of America, 1996.
[161] 7(&ORHWH³5HVLVWDQFHPHFKDQLVPVRIEDFWHULDWRDQWLPLFURELDOFRPSRXQGV´Int. Biodeterior. Biodegrad., vol. 51, pp. 277±282, 2003.
[162] :,ZX$5'XQFDQDQG&22NXQML³1HZDQWLPLFURELDOVRISODQWRULJLQ´Janick J ed. Perpectives New Crop. New Uses. Alexandria, VA ASHS Press, pp. 457±462, 1999.
[163] 00&RZDQ³3ODQWSURGXFWVDVDQWLPLFURELDODJHQWV´Clin. Microbiol. Rev., vol. 12, no. 4, pp. 564±582, 1999.
[164] I. H. Burkhill, A Dictionary of the Economic Products of the Malay Peninsula. Kuala Lumpur. Ministry of Agriculture and Co-Operatives, 1966, p. 424.
[165] 0 . /DOLWKD ³0DQXDO RQ $QWLPLFURELDO 6XVFHSWLELOLW\ 7HVWLQJ´ (Under auspices Indian Assoc. Med. Microbiol.
[166] 1&&/6³3HUIRUPDQFH6WDQGDUGVIRU$QWLPLFURELDO'LVN6XVFHSWibility Tests; Approved Standard----Ninth Edition-1&&/6´ Perform. Stand. Antimicrob. susceptibility testing. 8th Informational Suppl. M100S12. Natl. Comm. Clin. Lab. Stand. Villanova, Pa, 2002.
[167] K. Samidurai 6DUDYDQDNXPDU$ ³$QWLEDFWHULDO$FWLYLW\ RIPemphis Acidula )RUVW´Glob. J. Pharmacol., vol. 3, no. 2, pp. 113±115, 2009.
[168] $6KDUPD9.3DWHO65DZDW35DPWHNHDQG59HUPD³,GHQWLILFDWLRQRIThe Antibacterial Component of Some Indian Medicinal Plants Against Klebsiella Pneumoniae´Int. J. Pharm. Pharm. Sci., vol. 2, no. 3, pp. 123±127, 2010.
[169] -(ORII ³$VHQVLWLYH DQGTXLFNPLFURSODWHPHWKRG WRGHWHUPLQH WKHPLQLPDOinhibitory concentration of plant extracts for bacteriD´Planta Med., vol. 64, pp. 711±713, 1998.
[170] , :LHJDQG . +LOSHUW DQG 5 ( : +DQFRFN ³$JDU DQG EURWK GLOXWLRQmethods to determine the minimal inhibitory concentration (MIC) of DQWLPLFURELDOVXEVWDQFHV´Nat. Protoc., vol. 3, no. 2, pp. 163±75, Jan. 2008.

338
[171] 6 2]WXUN DQG 6 (UFLVOL ³&KHPLFDO FRPSRVLWLRQ DQG LQ YLWUR DQWLEDFWHULDOactivity of Seseli libanotis´World J. Microbiol. Biotechnol., vol. 22, no. 3, pp. 261±265, Jan. 2006.
[172] 5 /L ³1HZ SKDUPDFRG\QDPLF SDUDPHWHUV IRU DQWLPLFURELDO DJHQWV´ J. Antimicrob. Agents, vol. 13, pp. 229±235, 2000.
[173] 97DP$6FKLOOLQJDQG01LNRODRX³0RGHOOLQJWLPH-kill studies to discern WKHSKDUPDFRG\QDPLFVRIPHURSHQHP´J. Antimicrob. Chemother., vol. 55, pp. 699±706, 2005.
[174] A. Sartoratto, A. L. M. Machado, C. Delarmelina, G. M. Figueira, M. C. T. 'XDUWH DQG 9 / * 5HKGHU ³&RPSRVLWLRQ DQG DQWLPLFURELDO DFWLYLW\ RIHVVHQWLDORLOVIURPDURPDWLFSODQWVXVHGLQ%UD]LO´Brazilian J. Microbiol., vol. 35, no. 4, pp. 275±280, Dec. 2004.
[175] 3 3HWHUVHQ 7 :DQJ 5 'XVKLQ DQG 3 %UDGIRUG ³&RPSDUDWLYH LQ YLWURactivities of AC98-6446, a novel semisynthetic glycopeptide derivative of the natural product mannopeptimycin alpha, and other antimicrobial agents against gram-positive clinical LVRODWHV´Antimicrob Agents Chemother, vol. 48, no. 3, pp. 739±746, 2004.
[176] 0(/HYLVRQ³3KDUPDFRG\QDPLFVRIDQWLPLFURELDOGUXJV´Infect. Dis. Clin. North Am., vol. 18, no. 3, pp. 451±65, vii, Sep. 2004.
[177] T. T. Benjamin, J. A. Adebare, R. RHPL5DPRWDDQG.5DFKDHO³(IILFLHQF\RI6RPH'LVLQIHFWDQWVRQ%DFWHULDO:RXQG3DWKRJHQV´Life Sci. J., vol. 9, no. 2, p. 2012, 2012.
[178] *)*LVOHQH-/RFDWHOOL3&3UHLWLVDQG*/6LOYD³$QWLEDFWHULDODFWLYLW\of plant extracts and phytocKHPLFDOVRQDQWLELRWLFUHVLVWDQWEDFWHULD´Brazilian J. Microbiol. 31(4) 20-33., vol. 31, no. 4, pp. 20±33, 2000.
[179] $.XGL/2(GXYLH-*HIXDQG-88PRK³6FUHHQLQJRIVRPH1LJHULDQPHGLFLQDOSODQWVIRUDQWLEDFWHULDODFWLYLW\´J. Ethnopharmacol., vol. 67, 1999.
[180] :)DEU\322NHPRDQG5$QVRUJ³$QWLEDFWHULDODFWLYLW\RI(DVW$IULFDQPHGLFLQDOSODQWV´J. Ethnopharmacol., vol. 60, pp. 79±84, 1998.
[181] 1$OLJLDQQLV(.DOSRW]DNLV60LWDNXDQG,%&KLQRX³&RPSRVLWLRQDQd antimicrobial activity of the essential oils of two Origanum VSHFLHV´J. Agric. Food Chem., vol. 40, pp. 4168±4170, 2001.
[182] M. C. T. Duarte, G. M. Figueira, A. Sartoratto, V. L. G. Rehder, and C. 'HODUPHOLQD ³$QWL-Candida activity of Brazilian medLFLQDO SODQWV´ J. Ethnopharmacol., vol. 97, pp. 305±311, 2005.

339
[183] - %RZHUVR[ ³([SHULPHQWDO 6WDSK 9DFFLQH %URDGO\ 3URWHFWLYH LQ $QLPDO6WXGLHV´
[184] . 5RJHUV 3 )H\ DQG 0 5XSS ³&RDJXODVH-negative staphylococcal LQIHFWLRQ´Infect Dis Clin North Am, vol. 23, no. 1, pp. 73±98, 2009.
[185] .RWLUDQWD./RXQDWPDDDQG0+DDSDVDOR³(SLGHPLRORJ\DQGSDWKRJHQHVLVof Bacillus cereus LQIHFWLRQV´Microbes Infect., vol. 2, no. 2, pp. 189±198, 2000.
[186] 5 3RGVFKXQ DQG 8 8OOPDQQ ³.OHEVLHOOD VSS DV QRVRFRPLDO SDWKRJHQVHSLGHPLRORJ\ WD[RQRP\ W\SLQJ PHWKRGV DQG SDWKRJHQLFLW\ IDFWRUV´ Clin. Microbiol. Rev., vol. 11, no. 4, pp. 589±603, Oct. 1998.
[187] 0$3IDOOHU51-RQHV*9*RHUQDQG..XJOHU³%DFWHULDO3DWKRJHQVIsolated from Patients with Bloodstream Infection: Frequencies of Occurrence and Antimicrobial Susceptibility Patterns from the SENTRY Antimicrobial Surveillance Program (United SWDWHV DQG &DQDGD ´Antimicrob. agent Chemother., vol. 42, no. 7, pp. 1762±1770, 1998.
[188] 6'HQ\HUDQG-0DLOODUG³&HOOXODULPSHUPHDELOLW\DQGXSWDNHRIELRFLGHVDQVantibiotics in Gram-QHJDWLYHEDFWHULD´J. Appl. Microbiol. Symp. Suppl., vol. 92, p. 35S±45S, 2002.
[189] R. G. Linfield, W.M., Micich, T.J., Montville, T.J., Simon, J.R., Murray, E.B. DQG %LVWOLQH ³$QWLEDFWHULDOO\ DFWLYH VXEVWLWXWHG DQLOLGHV RI FDUER[\OLF DQGVXOIRQLFDFLGV´J. Med. Chem., vol. 26, no. 12, pp. 1741±1746, 1982.
[190] '7URID$*iFVHUDQG-'1RVDQFKXN³Candida parapsilosis, an emerging IXQJDOSDWKRJHQ´Clin. Microbiol. Rev., vol. 21, no. 4, pp. 606±25, Oct. 2008.
[191] T. D. Machado, I. C. R. Leal, A. C. F. Amaral, K. R. N. Santos, M. G. da Siva, and R. 0 .XVWHU ³$QWLPLFURELDO HOODJLWDQQLQ RI 3XQLFD JUDQXWXPIUXLWV´ J Braz Chem Soc, vol. 13, pp. 606±610, 2002.
[192] $6KHU³$QWLPLFURELDO$FWLYLW\RI1DWXUDO3URGXFWV)URP0HGLFLQDO3ODQWV´Gomal J. Med. Sci. January-June 2009, Vol. 7, No. 1, vol. 7, no. 1, pp. 72±78, 2009.
[193] 0 / &KDQFH ³1HZ GHYHORSPHQWV LQ WKH FKHPRWKHUDS\ RI OHLVKPDQLDVLV´Ann.Trop.Med. Parasitol., vol. 1, pp. 37±43, 1995.
[194] 0:LQN³0HGLFLQDOSODQWVDVRXUFHRIDQWL-SDUDVLWLFVHFRQGDU\PHWDEROLWHV´Molecules, vol. 17, no. 11, pp. 12771±91, Jan. 2012.
[195] W. Peters and G. Pasvol, Atlas of Tropical Medicine and Parasitology, 6th ed. Mosby-Elsevier: Philadelphia, PA, USA, 2007.

340
[196] :+2³&RQWURORI/HLVKPDQLDVLV´Rep. WHO Expert Comm., vol. Technical , 1990.
[197] $*7HPSRQH&0GH2OLYHLUDDQG5*6%HUOLQFN³&XUUHQWDSSURDFKHVWRGLVFRYHUPDULQHDQWLOHLVKPDQLDOQDWXUDOSURGXFWV´Planta Med., vol. 77, pp. 572±585, 2011.
[198] 7 3RORQLR DQG 7 (IIHUWK ³/HLVKPDQLDVLV 'UXJ UHVLVWDQFH DQG QDWXUDOprodXFWVUHYLHZ´Int. J. Mol. Med., vol. 22, pp. 277±286, 2008.
[199] .1XVVEDXP-+RQHN&&DGPXVDQG7(IIHUWK³7U\SDQRVRPDWLGSDUDVLWHVFDXVLQJQHJOHFWHGGLVHDVHV´Curr. Med. Chem., vol. 17, pp. 1594±1617, 2007.
[200] N. Sawangjaroen, S. Phongpaichit, S. Subhadhirasakul, M. Visuthi, N. 6ULVXZDQ DQG 1 7KDPPDSDOHUG ³7KH DQWL-amoebic activity of some PHGLFLQDOSODQWVXVHGE\$,'6SDWLHQWVLQVRXWKHUQ7KDLODQG´Parasitol Res, vol. 98, pp. 588±592, 2006.
[201] S. Laishram, G. Kang, and S. S. AjjDPSXU ³*LDUGLDVLV $ UHYLHZ RQDVVHPEODJH GLVWULEXWLRQ DQG HSLGHPLRORJ\ LQ ,QGLD´ Indian J Gastroenterol, vol. 31, pp. 3±12, 2012.
[202] 1,WRK10XUDRND+6DHNL0$RNLDQG7,WDJDNL³3UHYDOHQFHRI Giardia intestinalis infection in dogs of breHGLQJNHQQHOVLQ-DSDQ´J Vet Med Sci, vol. 67, pp. 717±718, 2005.
[203] 1 ,WRK10XUDRND -.DZDPDWD0$RNL DQG7 ,WDJDNL ³3UHYDOHQFH RIGiardia intestinalis LQIHFWLRQLQKRXVHKROGFDWVRI7RKRNXGLVWULFWLQ-DSDQ´J Vet Med Sci, vol. 68, pp. 161±163, 2006.
[204] J. A. Castro-Hermida, C. Carro-Corral, M. Gonzalez-Warleta, and M. Mezo, ³3UHYDOHQFH DQG LQWHQVLW\ RI LQIHFWLRQ RI Cryptosporidium spp. and Giardia duodenalis LQGDLU\FDWWOHLQ*DOLFLD1:6SDLQ´J Vet Med B Infect Dis Vet Public Heal., vol. 53, pp. 244±246, 2006.
[205] T. K. Graczyk, R. C. Thompson, R. Fayer, P. Adams, U. M. Morgan, and E. J. /HZLV³Giardia duodenalis cysts of genotype A recovered from clams in the &KHVDSHDNH%D\VXEHVWXDU\5KRGH5LYHU´Am J Trop Med Hyg, vol. 61, pp. 526±529, 1999.
[206] M. Lalle, E. Pozio, G. Capelli, F. Bruschi, D. Crotti, and S. M. Caccio, ³*HQHWLF KHWHURJHQHLW\ DW WKH EHWD-giardin locus among human and animal isolates of Giardia duodenalis and identification of potentially zoonotic subgenoW\SHV´Int J Parasitol, vol. 35, pp. 207±213, 2005.
[207] 0 6 /HJDWRU 7 + &RQQRU DQG 0 6WRHFNHO ³'HWHFWLRQ RI PXWDJHQLFDFWLYLW\RIPHWURQLGD]ROHDQGQLWULGD]ROHLQERG\IOXLGVRIKXPDQVDQGPLFH´Science (80-. )., vol. 188, pp. 1118±1119, 1975.

341
[208] 05XVWLDDQG36KXELN³,QGXFWLRQRIOXQJWXPRUVDQGPDOLJQDQWO\PSKRPDVLQPLFHE\PHWURQLGD]ROH´J Natl Cancer Inst, vol. 48, pp. 721±729, 1972.
[209] 36KXELN³&XUUHQWVWDWXVRIFKHPLFDOFDUFLQRJHQV´Proc Natl Acad Sci U S A, vol. 69, pp. 1052±1055, 1972.
[210] -0/OLEUH-7RU-00DQWHUROD&&DUERQHOODQG0)R]³Blastocystis hominis FKURQLF GLDUUKRHD LQ$,'6 SDWLHQWV´Lancet, vol. 333, no. 8631, p. 221, 1989.
[211] 3 - -RKQVRQ ³0HWURQLGD]ROH DQGGUXJ UHVLVWDQFH´Parasitol Today, vol. 9, pp. 183±186, 1993.
[212] M. Van Wyk and B. E. Wink, Medicinal Plants of the World: An Illustrated Scientific Guide to Important Medicinal Plants and Their Uses. Timber Press: Portland, OR, USA, 2004.
[213] D. J. Newman and G. M. Cragg, ³1DWXUDO SURGXFWV DV VRXUFHV RI QHZ GUXJVRYHUWKHODVW\HDUV´J. Nat. Prod., vol. 70, pp. 461±477, 2007.
[214] N. Sawangjaroen, S. Subhadhirasakul, S. Phongpaichit, C. Siripanth, K. -DPMDURHQDQG.6DZDQJMDURHQ³7KHLQYLWURDQWL-giardial activity of extracts from plants that are used for self-medication by AIDS patients in Southern 7KDLODQG´Parasitol Res, vol. 95, pp. 17±21, 2005.
[215] 7 0RVPDQQ ³5DSLG FRORULPHWULF DVVD\ IRU FHOOXODU JURZWK DQG VXUYLYDODSSOLFDWLRQWRSUROLIHUDWLRQDQGF\WRWR[LFLW\DVVD\V´J Immunol Methods, vol. 65, pp. 55±63, 1983.
[216] C. Siripanth, T. Chintana, Y. Tharaphan, and A. /HNNUD ³&ORQLQJ RI 7KDLstrain Giardia intestinalis´Asian Pac J Allergy Immunol, vol. 13, no. 1, pp. 71±73, 1995.
[217] /6'LDPRQG&*&ODUNDQG&&&XQQLFN³<,-S, a casein-free medium for axenic cultivation of Entamoeba histolytica, related Entamoeba, Giardia intestinalis and Trichomonas vaginalis´ J Eukaryot Microbiol, vol. 42, pp. 277±278, 1995.
[218] - $ 8SFURIW DQG 3 8SFURIW ³'UXJ VXVFHSWLELOLW\ WHVWLQJ RI DQDHURELFSURWR]RD´Antimicrob Agents Chemother, vol. 45, pp. 1810±1814, 2001.
[219] 7(IIHUWK)+HUUPDQQ$7DKUDQLDQG0:LQN³&\WRWR[LFDFWLYLW\WRZDUGVcancer cells of secondary constituents derived from Artemisia annua L. in FRPSDULVRQ WR LWV GHVLJQDWHG DFWLYH FRQVWLWXHQW DUWHPLVLQLQ´Phytomedicine, vol. 18, pp. 959±969, 2011.

342
[220] 00 6DOHP DQG . $:HUERYHW] ³1DWXUDO SURGXFWV IURP SODQWV DV GUXJFDQGLGDWHV DQG OHDG FRPSRXQGV DJDLQVW OHLVKPDQLDVLV DQG WU\SDQRVRPLDVLV´Curr. Med. Chem., vol. 13, pp. 2571±2598, 2006.
[221] 0:LQN ³(YROXWLRQDU\ DGYDQWDJH DQd molecular modes of action of multi-FRPSRQHQWPL[WXUHVXVHGLQSK\WRPHGLFLQH´Curr. Drug. Metab., vol. 9, pp. 996±1009, 2008.
[222] M. Wink and B. E. van Wyk, Mind-Altering and Poisonous Plants of the World. Timber Press: Portland, OR, USA, 2008.
[223] / & &KDQJ DQG$ ' .LQJKRUQ ³)ODYRQRLGV DV FDQFHU FKHPRSUHYHQWDWLYHDJHQWV´Bioact. Compd. from Nat. Sources, Isol. Characterisation Biol. Prop., vol. Corrado-Tr, 2001.
[224] / 5HGG\ % 2GKDY DQG . ' %KRROD ³1DWXUDO SURGXFWV IRU FDQFHUpreventioQ D JOREDOSHUVSHFWLYH´ Pharmacol. Ther. 99 1-13. (referenced Denmark- Wahnefried al., 2001)., 2003.
[225] W. Fan, K. Ogusu, K. K. Kouda, H. Nakamura, Satoh, H. Ochi, and H. 7DNHXFKL ³5HGXFHG R[LGDWLYH '1$ GDPDJH E\ YHJHWDEOH MXLFH LQWDNH $controlled tULDO´J. Physiol. Anthropol. Appl. Human Sci., vol. 19, no. 6, pp. 287±289., 2000.
[226] *0&UDJJDQG'-1HZPDQ³3ODQWVDVDVRXUFHRIDQWLFDQFHUDJHQWV´J. Ethnopharmacol., vol. 100, pp. 72±79, 2005.
[227] 15)DUQVZRUWK DQG& -.DDV ³$Q$Sproach Utilizing Information From Traditional Medicine To Identify Tumor-,QKLELWLQJ 3ODQWV´ J. Ethnopharmacol., vol. 3, 1981.
[228] 6 6KLVKRGLD DQG % % $JJDUZDO ³*XJJXOVWHURQH LQKLELWV 1)-ț% DQG ,ț%Įkinase activation, suppresses expression of anti-apoptotic gene products, and HQKDQFHVDSRSWRVLV´J. Biol. Chem., vol. 279, no. 45, pp. 47148±47158., 2004.
[229] ' 5 $ 0DQV $ 'D 5RFKD DQG * 6FKZDUWVPDQQ ³$QWL-cancer drug discovery and development in Brazil: targeted plant collection as a rational strategy to acquire candidate anti-FDQFHUFRPSRXQGV´Oncologist, vol. 5, pp. 185±198., 2000.
[230] V. Srivastava, A. S. Negi, J. K. Kumar, M. M. Gupta, and S. P. S. Khanuja, ³3ODQW-based anticancer molecules: A chemical and biological profile of some imSRUWDQWOHDGV´Bioorganic Med. Chem., vol. 13, pp. 5892±5908., 2005.
[231] .+/HH³$QWLFDQFHUGUXJGHVLJQEDVHGRQSODQW-GHULYHGQDWXUDOSURGXFWV¶´J. Biomed. Sci., vol. 6, pp. 236±250, 1999.

343
[232] J. M. Chapdelaine and I. Pharmakon Research InternaWLRQDO³077UHGXFWLRQ- a tetrazolium-EDVHG FRORULPHWULF DVVD\ IRU FHOO VXUYLYDO DQG SUROLIHUDWLRQ´Appl. Note 5 Maxcline Microplate Read., vol. Molecular.
[233] C. G. Mortimer, G. Wells, J. P. Crochard, E. L. Stone, T. D. Bradshaw, M. F. G. Stevens, and $ ' :HVWZHOO ³$QWLWXPRU %HQ]RWKLD]ROHV -(3,4-Dimethoxyphenyl)-5-fluorobenzothiazole (GW 610, NSC 721648), a Simple Fluorinated 2-Arylbenzothiazole, Shows Potent and Selective Inhibitory Activity against Lung, Colon, and Breast Cancer Cell Lines,´J. Med. Chem., vol. 49, no. 1, pp. 179±185, 2006.
[234] -+DUWZHOO³7\SHVRI$QWLFDQFHU$JHQWV,VRODWHG)URP3ODQWV´Cancer Treat. Rep., vol. 60, no. 8, 1976.
[235] R. R. Mendonça-)LOKR ³%LR-active phytocompounds: new approaches in the phytosciences LQ0RGHUQ SK\WRPHGLFLQHV´ LQTurning medicinal plants into drugs., In: Turnin., M. Aqil, A.F. and Owais, Ed. Wiley-VCH, Weinheim, 2006.
[236] - ( %HDUW + / 7HUHQFH DQG ( +DVODP ³3ODQW 3RO\SKHQROV-Secondary Metabolism And Chemical Defence: Some ObVHUYDWLRQV´ Phytochemisrry, vol. 24, no. 1, pp. 33±38, 1985.
[237] 6=ZHQJHUDQG&%DVX³3ODQWWHUSHQRLGVௗDSSOLFDWLRQVDQGIXWXUHSRWHQWLDOV´vol. 3, no. February, pp. 1±7, 2008.
[238] T. S. Agostini-costa, R. F. Vieira, H. R. Bizzo, D. Silveira, and M. A. Gimenes, ³6HFRQGDU\ 0HWDEROLWHV´ LQ Chromatography and Its Applications, Dr. Sasikumar Dhanarasu, Ed. 2012, pp. 131±164.
[239] /95R]H$&KDQGDDQG-(/LQ]³&RPSDUWPHQWDOL]DWLRQDQGPROHFXODUtraffic in secondary metabolism: a new undestanding of established cellular SURFHVVHV´Fungal Genet. Biol., vol. 48, pp. 35±48, 2011.
[240] 5 &URWHDX 70 .XWFKDQ DQG 1 * /HZLV ³1DWXUDO SURGXFWV VHFRQGDU\PHWDEROLWHV´LQBiochemistry and molecular biology of plants, J. R. Buchanan B, Gruissem W, Ed. Rockville, MD: American Society of Plant Physiologists, 2000, pp. 1250±1318.
[241] A. Gurib-)DNLP ³0HGLFLQDO SODQWV 7UDGLWLRQV RI \HVWHUGD\ DQG GUXJV RIWRPRUURZ´Mol. Asp. Med. 27 1±93, vol. 27, pp. 1±93, 2006.
[242] J. Lin, A. R. Opoku, M. Geheeb-Keller, A. D. Hutchings, S. E. Terblanche, J. $. DQG - YDQ 6WDGHQ ³3UHOLPLQDU\ VFUHHQLQJ RI VRPH WUDGLWLRQDO =XOXmedicinal plants for anti-LQIODPPDWRU\ DQG DQWLPLFURELDO DFWLYLWLHV´ J. Ethnopharmacol., vol. 68, pp. 267±274, 1999.

344
[243] 5 3 : 6FRWW ³7HFKQLTXHV DQG SUDFWLFHV RI FKURPDWRJUDSK\´ [Online]. Available: http://www.uv.mx/mca/02-intro.pdf#search. . [Accessed: 15-Jun-2012].
[244] K. Hostettmann, A. Marston, and M. Hostettmann, Preparative Chromatography Techniques: Applications in Natural Product Isolation, 2nd ed. Springer-Verlag Berlin Heidelberg, 1998.
[245] L. Wade Jr., Organic Chemistry. Upper Saddle River, NJ. Prentice Hall., 2006.
[246] 0.DUDPDü$.RVLĔVND$5\EDUF]\NDQG5$PDURZLF]³(xtraction and chromatographic separation of tannin fractions from tannin-rich plant PDWHULDO´YROQRSS±474, 2007.
[247] A. E. Hagerman, Tannin Handbook. 2002.
[248] 5 ' +DUWOH\ ³+3/& WKH VHSDUDWLRQ DQG GHWHUPLQDWLRQ RI SKHQROLFcompoundVLQSODQWV´LQHigh Performance Liquid Chromatography in Plant Sciences., Linskens H., .Springer-Verlag, New York., 1986.
[249] & :LDUW ³(WKQRSKDUPDFRORJ\ RI PHGLFLQDO SODQWVௗ $VLD DQG WKH 3DFLILF´Humana Press Inc., 2006.
[250] C. Shaw, C. Chen, C+VX&&KHQDQG<7VDL ³$QWLR[LGDQW3URSHUWLHVRIScopoletin Isolated from Sinomonium acutum´ YRO QR0D\ SS823±825, 2003.
[251] T.-N. Chang, J.-S. Deng, Y.-C. Chang, C.-Y. Lee, L. Jung-Chun, M.-M. Lee, W. H. Peng, S.-S. Huang, and G.-- +XDQJ ³$PHOLRUDWLYH (IIHFWV RIScopoletin from Crossostephium chinensis against Inflammation Pain and Its 0HFKDQLVPVLQ0LFH´Evid. Based. Complement. Alternat. Med., vol. 2012, p. 595603, Jan. 2012.
[252] S. Prachayasittikul, S. Suphapong, A. Worachartcheewan, R. Lawung, S. 5XFKLUDZDW DQG 9 3UDFKD\DVLWWLNXO ³%LRDFWLYH PHWDEROLWHV IURP Spilanthes acmella 0XUU´Molecules, vol. 14, no. 2, pp. 850±67, Jan. 2009.
[253] 51RYHPEHU DQG$0DUFK ³&RXPDULQV)URPCynanchum Acutum´YRO no. November 2008, pp. 65±69, 2009.
[254] N. N. Rumzhum, H. Sohrab, and M. A. Al-PDQVXU ³6HFRQGDU\0HWDEROLWHVfrom Jatropha Podagrica +RRN´YROQRSS±37, 2012.
[255] Y.-P. Lin, T.-Y. Chen, H.-W. Tseng, M.-H. Lee, and S.-7&KHQ³1HXUDOFHOOprotective compounds isolated from Phoenix hanceana var. formosana´Phytochemistry, vol. 70, no. 9, pp. 1173±81, Jun. 2009.

345
[256] G. H. Meulenbeld, H. Zuilhof, a van Veldhuizen, R. H. van den Heuvel, and S. +DUWPDQV³(QKDQFHG-catechin transglucosylating activity of Streptococcus mutans GS-5 glucosyltransferase-'GXH WR IUXFWRVH UHPRYDO´Appl. Environ. Microbiol., vol. 65, no. 9, pp. 4141±7, Sep. 1999.
[257] P. K. Sharma, M. He, J. Jurayj, D.-M. Gou, R. Lombardy, L. J. Romanczyk, DQG+6FKURHWHU ³(QDQWLRVHOFtive syntheses of sulfur analogues of flavan-3-2OV´Molecules, vol. 15, no. 8, pp. 5595±619, Aug. 2010.
[258] D. Lee, H. Lyu, H. Kwak, L. Jung, Y. Lee, D. Kim, I. Chung, S. Kim, and N. %DHN³,VRODWLRQRI)ODYRQRLGVIURPWKH)UXLWVRICornus kousa %XUJ´J. Appl. Biol. Chem., vol. 50, no. 3, pp. 144±147, 2007.
[259] -/LX³'LYHUVLW\RI&KHPLFDO&RQVWLWXHQWVIURPSaxifraga montana +´SS863±870, 2008.
[260] K. K. Chiruvella, A. Mohammed, G. Dampuri, R. G. Ghanta, and S. C. 5DJKDYDQ ³3K\WRFKHmical and Antimicrobial Studies of Methyl Angolensate and Luteolin-7-O-glucoside Isolated from Callus Cultures of Soymida febrifuga´Int. J. Biomed. Sci., vol. 3, no. 4, pp. 269±78, Dec. 2007.
[261] 0 5]\ZDF] * 2QLND 5 $ZURxVND DQG 7 . $GHXV] ³)lavonoids and Coumarins From Hieracium Pilosella /$VWHUDFHDH´YROQRSS±195, 2009.
[262] G. C. and Z. M. S. Hammami , H. Ben Jannet , A. Bergaoui , L. Ciavatta, ³,VRODWLRQ DQG 6WUXFWXUH (OXFLGDWLRQ RI D )ODYDQRQH D )ODYDQRQH *O\FRVLGHand Vomifoliol from Echiochilon Fruticosum *URZLQJLQ7XQLVLD´Molecules, vol. 9, pp. 602±608, 2004.
[263] </XDQG/<)RR³,GHQWLILFDWLRQDQGTXDQWLILFDWLRQRIPDMRUSRO\SKHQROVLQDSSOHSRPDFH´Food Chem., vol. 59, no. 2, pp. 187±194, Jun. 1997.
[264] 0 %RJGDQ & * )ORDUH DQG $ 3vUQDX ³ + 105 LQYHVWLJDWLRQ RI VHOI-DVVRFLDWLRQ RI YDQLOOLQ LQ DTXHRXV VROXWLRQ´ J. Phys. Conf. Ser., vol. 182, p. 012002, Aug. 2009.
[265] E. H. Hansen, B. L. Møller, G. R. Kock, C. M. Bünner, C. Kristensen, O. R. JHQVHQ)72NNHOV&(2OVHQ060RWDZLD DQG -+DQVHQ ³'HQRYRbiosynthesis of vanillin in fission yeast (Schizosaccharomyces pombe) and EDNHU¶V\HDVWSaccharomyces cerevisiae´Appl. Environ. Microbiol., vol. 75, no. 9, pp. 2765±74, May 2009.
[266] F. A. Abbas, S. M. Al-Massarany, S. Khan, T. a Al-Howiriny, J. S. Mossa, and (D$ERXUDVKHG³3K\WRFKHPLFDODQGELRORJLFDOVWXGLHVRQ6DXGLCommiphora opobalsamum /´Nat. Prod. Res., vol. 21, no. 5, pp. 383±391, May 2007.

346
[267] S. Mcdonald, P. D. 3UHQ]OHU 0 $QWRORYLFK DQG . 5REDUGV ³3KHQROLFFRQWHQWDQGDQWLR[LGDQWDFWLYLW\RIROLYHH[WUDFWV´YROSS±84, 2001.
[268] 06FKXVWHU1&KULVWLDQVHQ& -DNXSRYLF -0XQJDO ³$QXQXVXDO >@cycloadduct of terpenoid coumarin from Ethulia vernonioide´Phytochemistry, vol. 34, pp. 1179±1181, 1993.
[269] ,1DJDU³%LRGHJUDGDWLRQRI&DWHFKLQ´YROQRSS±370, 2003.
[270] 78VLD+,ZDWD$+LUDWVXNDDQG7:DWDEH³6HVTXLWHUSHQHVDQGIODYRQROglycosides from Zingiber aromaticum and their CYP3A4 and CYP2D6 LQKLELWRU\DFWLYLWLHV´J. Nat. Prod., vol. 67, pp. 1079±1083, 2004.
[271] &+LUREH=64LDR.7DNH\DDQG+,WRNDZD³&\WRWR[LFIODYRQRLGVIURPVitex agnus-castus´Phytochemistry., vol. 46, pp. 521±524, 1997.
[272] +=KDQJDQG<&KHQ³3ODQWWHUSHQHV´Encylopedia Life Support Syst.
[273] 70DLPRQHDQG,$,QWURGXFWLRQ³&ODVVLF7HUSHQH6\QWKHVHV´
[274] =*QDWD/:LUWK-pUpPLH:*XRDQG5/%DXPHV³&-Norisoprenoid Aglycon Composition of Leaves and Grape Berries from Muscat of Alexandria DQG 6KLUD] &XOWLYDUV´ LQ Carotenoid-Derived Aroma Compounds. ACS Symposium Series 802, 2001, p. 255.
[275] '03HUHLUD39DOHQWmR-D3HUHLUDDQG3%$QGUDGH³3KHQROLFV)URPChemistry to %LRORJ\´Molecules, vol. 14, no. 6, pp. 2202±2211, Jun. 2009.
[276] L. P. S. Quideau, D. Deffieux, C. Douat-&DVDVVXV ³3ODQW SRO\SKHQROVFKHPLFDO SURSHUWLHV ELRORJLFDO DFWLYLWLHV DQG V\QWKHVLV´Angew. Chem. Int., vol. 50, pp. 586±621, 2011.
[277] 9&KH\QLHU* &RPWH.0'DYLHV9 /DWWDQ]LR DQG 60DUWHQV ³3ODQWphenolics: Recent advances on their biosynthesis, genetics, and HFRSK\VLRORJ\´Plant Physiol. Biochem., May 2013.
[278] -%+DUERUQH ³*HQHUDO SURFHGXUHV DQGPHDVXUHPHQWRI WRWDO SKHQROLFV´ LQMethods in Plant Biochemistry, Plant Phenolics, , vol. 1, Academic Press, London, UK, 1989.
[279] P. Valentão, E. Fernandes, F. Carvalho, P. B. Andrade, R. M. Seabra, and M. / %DVWRV ³+\GUR[\O UDGLFDO DQG K\SRFKORURXV DFLG VFDYHQJLQJ DFWLvity of small centaury (Centaurium erythraea) infusion. A comparative study with green tea (Camellia sinensis´Phytomedicine, vol. 10, pp. 517±522, 2003.
[280] $-3DUUDQG-3%ROZHOO³3KHQROVLQWKHSODQWDQGLQPDQ7KHSRWHQWLDOIRUpossible nutritional enhancement of the diet by modifying the phenols content RUSURILOH´J. Sci. Food Agric. \, vol. 80, pp. 985±1012, 2002.

347
[281] C. S. Yang, J. M. Landau, M.-7+XDQJ DQG+/1HZPDUN ³,QKLELWLRQRIcarcinogenesis by dietary polyphenolic compoXQGV´Annu. Rev. Nutr., vol. 21, pp. 381±406, 2001.
[282] .'&URIW³7KHFKHPLVWU\DQGELRORJLFDOHIIHFWVRIIODYRQRLGVDQGSKHQROLFDFLGV´Ann. N. Y. Acad. Sci., vol. 854, pp. 435±442, 1988.
[283] P. Cos, L. Ying, M. Calomme, J. P. Hu, K. Cimanga, B. V. Poel, L. Pieters, A. - 9OLHWLQFN DQG ' 9 %HUJKH ³6WUXFWXUH-activity relationship and classification of flavonoids as inhibitors of xanthine oxidase and superoxide VFDYHQJHUV´J. Nat. Prod. 1988, 61, 71-76., vol. 61, pp. 71±76, 1988.
[284] C. A:LOOLDPVDQG5-*UD\HU³$QWKRF\DQLQVDQGRWKHUIODYRQRLGV´J. Nat. Prod. Reports, vol. 21, pp. 539±573, 2004.
[285] 737&XVKQLHDQG$-/DPE³$QWLPLFURELDODFWLYLW\RIIODYRQRLGV´Int. J. Antimicrob. Agents, vol. 26, pp. 343±356, 2005.
[286] 3%$QGUDGH'03HUHLUD))HUUHUHVDQG39DOHQWmR³5HFHQW WUHQGVLQKLJK WKURXJKSXW DQDO\VLV DQG DQWLR[LGDQW SRWHQWLDO VFUHHQLQJ IRU SKHQROLFV´Curr. Pharm. Anal., vol. 4, pp. 137±150, 2008.
[287] B. G. . Raymond, K. S. Marie, F. Paulo, and & 6WHIDQ ³7KH 9LVLEOH)ODYRQRLGV RU $QWKRF\DQLQV )URP 5HVHDUFK WR $SSOLFDWLRQV´ Recent Adv. Polyphen. Res.
[288] 1 & &RRN DQG 6 6DPPDQ ³)ODYRQRLGV- Chemistry, metabolism, FDUGLRSURWHFWLYHHIIHFWVDQGGLHWDUWVRXUFHV´Nutr. Biochem., vol. 7, no. 2, pp. 66±76, 1996.
[289] 3 0 $URQ DQG - $ .HQQHG\ ³)ODYDQ-3-ols: Nature, occurrence and ELRORJLFDODFWLYLW\´Mol. Nutr. Food Res. 2008, 52, 79-10, vol. 52, pp. 79±100, 2008.
[290] D. B. Bagchi, C. K. Sen, S. D. Ray, D. K. Das, M. Bagchi, H. G. Preuss, and J. $9LQVRQ³0ROHFXODUPHFKDQLVPVRIFDUGLRSURWHFWLRQE\DQRYHOJUDSHVHHGSURDQWKRF\DQLGLQH[WUDFW´Mutat. Res., vol. 523, pp. 87±97, 2003.
[291] 5$'L[RQDQG')HUUHLUD³*HQLVWHLQ´Phytochemistry, vol. 60, pp. 205±211, 2002.
[292] S. Lafay and A. Gil-,]TXLHUGR³%LRDYDLODELOLW\RI3KHQROLFDFLGV´Phytochem. Rev., vol. 7, pp. 301±311, 2008.
[293] 5 - 5REELQV ³3KHQROLF DFLGV LQ IRRGV $Q RYHUYLHZ RI DQDO\WLFDOPHWKRGRORJ\´J. Agr. Food Chem., vol. 51, pp. 2866±2887, 2003.

348
[294] S. .KDGHP DQG 5 - 0DUOHV ³0RQRF\FOLF SKHQROLF DFLGV K\GUR[\- and SRO\K\GUR[\EHQ]RLF DFLGV RFFXUUHQFH DQG UHFHQW ELRDFWLYLW\ VWXGLHV´Molecules, vol. 15, no. 11, pp. 7985±8005, Nov. 2010.
[295] J. B. Harborne, H. Baxter, and G. P. Moss, Phytochemical Dictionary²A Handbook of Bioactive Compounds from Plants, 2nd ed. Taylor & Francis: London, UK, 1999, p. 976.
[296] J. B. Harborne, Plant Phenolics. Academic Press: San Diego, CA, USA, 1989.
[297] J. Bruneton, Pharmacognosy, Phytochemistry, Medicinal Plants., 2nd ed. Intercept Ltd., Hampshire, UK., 1999.
[298] T. Ojala, Biological Screening of Plant Coumarins, no. Aleksanterinkatu 5. 2001, p. 62.
[299] R. G. Kalkhambkar, G. M. Kulkarni, C. M. Kamanavalli, N. Premkumar, S. M. % $VGDT DQG 0 6XQ ³6\QWhesis and biological activities of some new fluorinated coumarins and 1-D]DFRXPDULQV´Eur. J. Med. Chem., vol. 43, pp. 2178±2188, 2008.
[300] 7 2 6RLQH ³1DWXUDOO\ RFFXUULQJ FRXPDULQV DQG UHODWHG SK\VLRORJLFDODFWLYLWLHV´J. Pharm. Sci., vol. 53, p. 231, 1964.
[301] &6/LQR0/7DYHLUD*6%9LDQDDQG)-$0DWRV³$QDOJHVLFDQGantiinflammatory activities of Justicia pectoralis Jacq. and its main FRQVWLWXHQWV FRXPDULQV DQG XPEHOOLIHURQH´ Phyther. Res., vol. 11, p. 211, 1997.
[302] J 5 6 +RXOW 0 $ 0RURQH\ DQG 0 3D\i ³$FWLRQV RI IODYRQRLGV DQGcoumarins on lipoxygenase and cyclo-R[\JHQDVH´ Methods Enzymol., vol. 234, p. 443, 1994.
[303] *%LVLJQDQR56DQRJR$0DULQR5$TXLQR9'¶$QJHOR03*HUPDQzC. De, and R. 3DVTXDOH³$QWLPLFURELDODFWLYLW\RI0LWUDFDUSXVVFDEHUH[WUDFWDQGLVRODWHGFRQVWLWXHQWV´Lett. Appl. Microbiol., vol. 30, p. 105, 2000.
[304] S. Sardari, Y. Mori, K. Horita, R. G. Micetich, S. Nishibe, and M. Daneshtalab, ³6\QWKHVLVDQGDQWLIXQJDODFWLYLW\RIFRXPDULQVDQGDQJXODUIXUDQRFRXPDULQV´Bioorganic Med. Chem., vol. 7, no. 9, p. 1933, 1999.
[305] + +|QLJVPDQQ $ 7DQHZ - %UFNH DQG % 2UWHO ³3KRWRVHQVLWL]LQJFRPSRXQGV LQ WKH WUHDWPHQW RI SVRULDVLV´ LQ Ciba Foundation Symposium, 1989, pp. 146±159.
[306] A. Goodman and A. Gilman, The Pharmacological Basis of Therapeutics, 9th ed. McGraw-Hill Companies, USA., 1996.

349
[307] 07&RQFRQL) 0RQWHVL DQG333DUQLJRWWR ³$QWLSUROLIHUDWLYH DFWLYLW\and phototoxicity of some methyl derivatives of 5-methoxypsoralen and 5- PHWKR[\DQJHOLFLQ´Pharmacol. Toxicol., vol. 82, p. 193, 1998.
[308] '(JDQ52¶.HQQHG\(0RUDQ'&R[(3URVVHUDQG'7KRUQHV³7KHpharmacology, metabolism, analysis and applications of coumarins and coumarinrelDWHGFRPSRXQGV´Drug Metab. Rev., vol. 22, no. 5, p. 503.
[309] T. Iizuka, S. Nagumo, H. Yotsumoto, H. Moriyama, and M. Nagai, ³9DVRUHOD[DQWHIIHFWVRIAcer nikoense extract and isolated coumarinolignans RQUDWDRUWLFULQJV´Biol. Pharm. Bull., vol. 30, pp. 1164±1166, 2007.
[310] T. L. G. Lemos, L. L. Machado, J. S. N. Souza, A. M. Fonseca, J. L. Maia, and 2'/3HVVRD³$QWLR[LGDQWLFWK\RWR[LFLW\DQGEULQHVKULPSOHWKDOLW\WHVWVRIMagonia glabrata´Fitoterapia, vol. 77, pp. 443±445, 2006.
[311] 3 ( %RQLOOD 5LYHUD 2 /RFN GH 8JD] DQG + -XUXSH &KLFR ³&KHPLFDO-biological study of Werneria dactilophylla´Bol. Soc. Quim. Peru, vol. 57, pp. 181±188, 1991.
[312] P. D. Moon, B. H. Lee, H. J. Jeong, H. J. An, S. J. Park, H. R. Kim, S. G. Ko, J. Y. 8P 6 + +RQJ DQG + 0 .LP ³8VH RI VFRSROHWLQ WR LQKLELW WKHproduction of inflammatory cytokines through inhibition of the IkB/NF-kB signal cascade in the human mast cell line HMC-´Eur. J. Pharmacol., vol. 555, pp. 218±225, 2007.
[313] C. Delporte, N. Backhouse, R. Negrete, P. Salinas, P. Rivas, B. K. Cassels, and $ 6DQ )HOLFLDQR ³$QWLS\UHWLF K\SRWKHUPLF DQG DQWLLQIODPPDWRU\ DFWLYLWLHVand metabolites from Solanum ligustrinum /RRG´Phytother. Res. 1998, 12, 118-122, 1998.
[314] Y. Okada, N. Miyauchi, K. Suzuki, T. Kobayashi, C. Tsutsui, K. Mayuzumi, S. 1LVKLEH DQG 7 2NX\DPD ³6HDUFK IRU QDWXUDOO\ RFFXUULQJ VXEVWDQFHV WRprevent the complications of diabetes. II. Inhibitory effect of coumarin and flavonoid derivatives on bovine lens aldose reductase and rabbit platelet DJJUHJDWLRQ´Chem. Pharm. Bull. (Tokyo), vol. 43, pp. 1385±1387, 1995.
[315] D. Son, P. Lee, J. Lee, S. Lee, S. Y. Choi, J. W. Lee, and S. Y. Kim, ³1HXURSURWHFWLYH HIIHFW RI VFRSROHWLQ IURPAngelica dahurica on oxygen and glucose deprivation-H[SRVHGUDWRUJDQRW\SLFKLSSRFDPSDOVOLFHFXOWXUH´Food Sci. Biotechnol., vol. 16, pp. 632±635, 2007.
[316] A. N. Guantai and I. Addae-0HQVDK³&DUGLRYDVFXODUHIIHFWRIArtemisia Afra DQGLWVFRQVWLWXHQWV´YROQo. 37, pp. 351±356.
[317] M. G. Manuele, G. Ferraro, M. L. Barreiro Arcos, P. Lopez, G. Cremaschi, and &$QHVLQL³&RPSDUDWLYH LPPXQRPRGXODWRU\HIIHFWRIVFRSROHWLQRQ WXPRUDODQGQRUPDOO\PSKRF\WHV´Life Sci., vol. 79, pp. 2043±2048, 2006.

350
[318] S. PanGD DQG $ .DU ³(YDOXDWLRQ RI WKH DQWLWK\URLG DQWLR[LGDWLYH DQGantihyperglycemic activity of scopoletin from Aegle marmelos leaves in K\SHUWK\URLGUDWV´Phytother. Res., pp. 1103±1105, 2006.
[319] H. C. Huang, C. R. Lee, Y. I. Weng, M. C. Lee, and Y. T/HH³9DVRGLODWRUeffect of scoparone (6,7-GLPHWKR[\FRXPDULQ IURP D &KLQHVH KHUE´Eur. J. Pharmacol., vol. 218, no. 1, pp. 123±128, 1992.
[320] P. D. Huei-&KHQ6 6+XDQJ DQG3'&KX ³9DVRUHOD[DQWV IURP&KLQHVHherbs, emodin and scoparone, posseVVLPPXQRVXSSUHVVLYHSURSHUWLHV´Eur. J. Pharmacol., vol. 198, no. 2±3, pp. 211±213, 1991.
[321] N. Saul, K. Pietsch, R. Menzel, S. R. Stürzenbaum, and C. E. Steinberg, ³&DWHFKLQ LQGXFHG ORQJHYLW\ LQ C. elegans: from key regulator genes to disposable somD´Mech. Ageing Dev., vol. 130, no. 9, pp. 477±486, 2009.
[322] S. Auclair, D. Milenkovic, C. Besson, S. Chauvet, E. Gueux, C. Morand, A. 0D]XUDQG$6FDOEHUW³&DWHFKLQUHGXFHVDWKHURVFOHURWLFOHVLRQGHYHORSPHQWin apo E-GHILFLHQWPLFH$WUDQVFULSWRPLFVWXG\´Atherosclerosis, vol. 204, no. 2, pp. 21±27, 2009.
[323] )1\IHOHU8.0RVHUDQG3:DOWHU³6WHUHRVSHFLILFHIIHFWVRI- DQGí-FDWHFKLQ RQ JO\FRJHQ PHWDEROLVP LQ LVRODWHG UDW KHSDWRF\WHV´ Biochim. Biophys. Acta - Mol. Cell Res. 763 50, vol. 763, p. 50, 1983.
[324] M. J. Weyant, A. M. Carothers, A. J'DQQHQEHUJDQG00%HUWDJQROOL³-Catechin inhibits intestinal tumor formation and suppresses focal adhesion NLQDVH DFWLYDWLRQ LQ WKHPLQPRXVH´Cancer Res., vol. 61, no. 1, pp. 118±125, 2001.
[325] H. Mangiapane, J. Thomson, A. Salter, S. Brown, G. D. Bell, and D. A. White, ³7KH LQKLELWLRQRI WKHR[LGDWLRQRI ORZGHQVLW\ OLSRSURWHLQE\ -catechin, a QDWXUDOO\RFFXUULQJIODYRQRLG´Biochem. Pharmacol., vol. 43, no. 3, pp. 445±450, 1992.
[326] 6%/RWLWRDQG&*)UDJD³-Catechin prevents KXPDQSODVPDR[LGDWLRQ´Free Radic. Biol. Med., vol. 24, no. 3, pp. 435±441, 1998.
[327] M. López-/i]DUR ³'LVWULEXWLRQ DQG ELRORJLFDO DFWLYLWLHV RI WKH IODYRQRLGOXWHROLQ´Mini Rev. Med. Chem., vol. 9, no. 1, pp. 31±59, Jan. 2009.
[328] A. Basile, S. Giordano, J. A. Lopez-6DH]DQG5&&RELDQFKL³$QWLEDFWHULDODFWLYLW\RISXUHIODYRQRLGVLVRODWHGIURPPRVVHV´Phytochemistry, vol. 52, pp. 1479±1482, 1999.
[329] $/ /LX% /LX+/4LQ 60/HH<7:DQJ DQG*+'X ³$QWL-Influenza Virus Activities of Flavonoids from the Medicinal Plant Elsholtzia UXJXORVD´Planta Med, vol. 74, pp. 847±851, 2008.

351
[330] M. P. De Campos, F. V. Cechinel, R. Z. Da Silva, R. A. Yunes, S. Zacchino, S. -XDUH]5&%HOOD&UX]DQG&$%HOOD³(YDOXDWLRQRIantifungal activity of Piper solmsianum C. DC. var. solmsianum 3LSHUDFHDH´Biol. Pharm. Bull., vol. 28, pp. 1527±1530, 2005.
[331] / %LWLV 6 .XOWDU * 0HOLNRJOX 1 2]VR\ DQG $ &DQ ³)ODYRQRLGV DQGantioxidant activity of Rosa agrestis OHDYHV´Nat. Prod. Radiance, vol. 24, pp. 580±589, 2003.
[332] *5 5REDN - ³)ODYRQRLGV DUH VFDYHQJHUV RI VXSHUR[LGH DQLRQV´Biochem Pharmacol., vol. 37, no. 5, pp. 837±841, 1988.
[333] =/LX;7DR&=KDQJ</XDQG':HL³3URWHFWLYHHIIHFWVRIK\SHURVLGH(quercetin-3-O-galactoside) to PC12 cells against cytotoxicity induced by hydrogen peroxide and tert-EXW\OK\GURSHUR[LGH´Biomed Pharmacother., vol. 59, pp. 481±490, 2005.
[334] L. Gombau, F. Garcia, A. Lahoz, M. Fabre, P. Roda-Navarro, P. Majano, J. L. Alonso-Lebrero, J. P. Pivel, J. V. Castell, M. J. Gomez-Lechon, and S. *RQ]DOH] ³Polypodium leucotomos extract: antioxidant activity and GLVSRVLWLRQ´Toxicol. Vitr., vol. 20, pp. 464±471, 2006.
[335] 773KDQ/:DQJ36HH5-*UD\HU6<&KDQDQG67/HH³3KHQROLFcompounds of Chromolaena odorata protect cultured skin cells from oxidative GDPDJH LPSOLFDWLRQ IRU FXWDQHRXVZRXQG KHDOLQJ´Biol. Pharm. Bull., vol. 24, pp. 1373±1377, 2001.
[336] 4;=KDQJ:+/XR+/LDQG=;/LQ³7KHHIIHFWVRIILYHFRPSRXQGVRQ GHR[\ULERQXFOHLF DFLG R[LGDWLRQ GDPDJH´Aibian Jibian Tubian, vol. 18, pp. 12±15, 2006.
[337] / %LURVRYD 0 0LNXODVRYD DQG 6 9DYHUNRYD ³Antimutagenic effect of SKHQROLFDFLGV´Biomed. Pap., vol. 149, pp. 489±491, 2005.
[338] *&<HQ&<+XQJDQG<-&KHQ³$QWLR[LGDQWSURSHUWLHVRI+VLDQ-tsao (Mesona procumbens +HPVO´LQACS Symp. Series, 2003, pp. 202±214.
[339] W. K. Kong, Y. L. Zhao, L. M. Shan, X. H. Xiao, and W. Y. Guo, ³7KHUPRFKHPLFDOVWXGLHVRQWKHTXDQWLW\ - Antibacterial effect relationship of four organic acids from Radix Isatidis on Escherichia coli JURZWK´ Biol. Pharm. Bull., vol. 31, pp. 1301±1305, 2008.
[340] A. Itoh, K. Isoda, M. Kondoh, M. Kawase, A. Watari, M. Kobayashi, M. 7DPHVDGD DQG . <DJL ³+HSDWRSURWHFWLYH (IIHFW RI 6\ULQJLF $FLG DQGVanillic Acid on CCl4-,QGXFHG/LYHU,QMXU\´Biol. Pharm. Bull., vol. 33, pp. 983±987, 2010.

352
[341] L. Zhang, Y.-X. Cheng, A.-L. Liu, H.-D. Wang, Y.-L. Wang, and G.-H. Du, ³$QWLR[LGDQW DQWL-inflammatory and anti-influenza properties of components from Chaenomeles VSHFLRVD´Molecules, vol. 15, no. 11, pp. 8507±17, Nov. 2010.
[342] L. H. Yao, Y. M. Jiang, J. Shi, F. A. Tomás-Barberán, N. Datta, R. 6LQJDQXVRQJDQG66&KHQ³)ODYRQRLGVLQIRRGDQGWKHLUKHDOWKEHQHILWV´Plant Foods Hum. Nutr., vol. 59, pp. 113±122, 2004.
[343] ( + .HOO\ - % 'HQQLV DQG 5 7 $QWKRQ\ ³)ODYRQRLG DQWLR[LGDQWVchemistry, metabolism and structure-DFWLYLW\UHODWLRQVKLSV´J. Nutr. Biochem., vol. 13, pp. 572±584, 2002.
[344] '$PL''DYLGRYL'%H95DVWLMD% /X DQG1 7ULQDMVWL ³6$5 DQG46$5RIWKH$QWLR[LGDQW$FWLYLW\RI)ODYRQRLGV´SS±845, 2007.
[345] O. Firuzi, A. LDFDQQD53HWUXFFL*0DUURVXDQG/6DVR³(YDOXDWLRQRIWKHDQWLR[LGDQWDFWLYLW\RIIODYRQRLGVE\µIHUULFUHGXFLQJDQWLR[LGDQWSRZHU¶DVVD\DQGF\FOLFYROWDPPHWU\´Biochim. Biophys. Acta, vol. 1721, no. 1±3, pp. 174±84, Jan. 2005.
[346] Y. Sawai and . 6DNDWD ³105 $QDO\WLFDO $SSURDFK WR FODULI\ WKHDQWLR[LGDWLYHPROHFXODUPHFKDQLVPRI FDWHFKLQV XVLQJ'33+´ J Agric Food Chem, vol. 46, no. 1, pp. 111±114, 1998.
[347] *:*:3OXPE.53ULFH³$QWLR[LGDQWSURSHUWLHVRIIODYRQROJO\FRVLGHVfrom teD´Redox Rep, vol. 4, pp. 13±16, 1999.
[348] M. N. J. L. S.A.B.E. Van Acker, M.J. De Groot, D.J. van den Berg and A. B. 7URPS *'2 GHQ .HOGHU :-) YDQ GHU 9LMJK ³$ TXDQWXP FKHPLFDOH[SODQDWLRQRIWKHDQWLR[LGDQWDFWLYLW\RIIODYRQRLG´Chem Res Toxicol, vol. 6, pp. 1305±1312, 1996.
[349] 0 - $ $ 0RUD 0 3D\D -/ 5LRV ³6WUXFWXUH-activity relationships of polymethoxyflavones and other flavonoids as inhibitors of non-enzymic lipid SHUR[LGDWLRQ´Biochem Pharmacol, vol. 40, pp. 793±797, 1990.
[350] 0 6 : %RUV : +HOOHU & 0LFKHO ³)ODYRQRLGV DV DQWLR[LGDQWVdetermination of radical-VFDYHQJLQJ HIILFLHQFLHV´Methods Enzym., vol. 186, pp. 343±355, 1990.
[351] G. P. C.A. Rice-(YDQV1-0LOOHU³6WUXFWXUH-antioxidant activity relationships RIIODYRQRLGVDQGSKHQROLFDFLGV´Free Radic Biol Med ., vol. 20, pp. 933±956, 1996.
[352] H.-C. Lin, S.-H. Tsai, C.-S. Chen, Y.-C. Chang, C.-M. Lee, Z.-Y. Lai, and C.-M. LiQ ³6WUXFWXUH-activity relationship of coumarin derivatives on xanthine

353
oxidase-inhibiting and free radical-VFDYHQJLQJ DFWLYLWLHV´ Biochem. Pharmacol., vol. 75, no. 6, pp. 1416±25, Mar. 2008.
[353] A. F. Welton, L. D. Tobias, C. Fiedler-Nagy, W. Anderson, W. Hope, and K. 0H\HU ³(IIHFW RI IODYRQRLGV RQ DUDFKLGRQLF DFLG PHWDEROLVP´ LQ Plant flavonoids in biology and medicine., H. J. Cody V, Middleton E, Ed. New York: Alan R. Liss, 1986, pp. 231±224.
[354] M. G. L. Hertog, E. J. M. Feskens, P. C. H. Hollman, M. B. Katan, and D. .URPKRXW³'LHWDU\DQWLR[LGDQWIODYRQRLGVDQGULVNRIFRURQDU\KHDUWGLVHDVHWKH=XWSKHQHOGHUO\VWXG\´Lancet., pp. 1007±1011, 1993.
[355] -</HH<:-DQJ+6.DQJ+0RRQ666LPDQG&-.LP³$QWL-inflammatory action of phenolic compounds from Gastrodia elata URRW´Arch Pharm Res., vol. 29, no. 10, pp. 849±858, 2006.
[356] 56WDQLNXQDLWH6 ,.KDQ -07UDSSHDQG6D5RVV ³&\FORR[\JHQDVH-2 inhibitory and antioxidant compounds from the truffle Elaphomyces granulatus´Phytother. Res., vol. 23, no. 4, pp. 575±8, Apr. 2009.
[357] S. D. Bhagavathula, Synthetic approaches and biological applications of coumarins: A review. 2013, p. 33.
[358] $10D\HNDU³6\QWKHVLV&KDUDFWHUL]DWLRQDQG$QWLPLFURELDO6WXdy of Some 1HZ&\FORKH[HQRQH'HULYDWLYHV´YROQRSS±123, 2010.
[359] M. Alipour, M. Khoobi, A. Foroumadi, H. Nadri, A. Moradi, A. Sakhteman, 0 *KDQGL DQG $ 6KDILHH ³1RYHO FRXPDULQ GHULYDWLYHV EHDULQJ 1-benzyl pyridinium moiety: potent and dual binding site acetylcholinesterase LQKLELWRUV´Bioorg. Med. Chem., vol. 20, no. 24, pp. 7214±22, Dec. 2012.
[360] 3 $QDQG % 6LQJK DQG 1 6LQJK ³$ UHYLHZ RQ FRXPDULQV DVDFHW\OFKROLQHVWHUDVHLQKLELWRUVIRU$O]KHLPHU¶VGLVHDVH´Bioorg. Med. Chem., vol. 20, no. 3, pp. 1175±80, Feb. 2012.
[361] &5DPDVVDP\³(PHUJLQJUROHRISRO\SKHQROLFFRPSRXQGVLQWKHWUHDWPHQWRIQHXURGHJHQHUDWLYH GLVHDVHV $ UHYLHZ RI WKHLU LQWUDFHOOXODU WDUJHWV´ Eur. J. Pharmacol., vol. 545, 2006.
[362] M. K. Wassef, T. B )LRUHWWL DQG'0'Z\HU ³/LSLG DQDO\VHV RI LVRODWHGVXUIDFH PHPEUDQHV RI /HLVKPDQLD GRQRYDQL SURPDVWLJRWHV´ Lipids 20:108-115., vol. 20, pp. 108±115, 1985.
[363] N. S. Vila-nova, S. M. Morais, M. J. C. Falcão, C. M. L. Bevilaqua, F. C. M. Rondon, M. E. Wilson, I. G. P. Vieira, H. F. Andrade, A. V. N. S, S. M. Morais, M. J. C. Falcão, C. M. L. Bevilaqua, and F. C. M. Rondon, ³/HLVKPDQLFLGDODQGFKROLQHVWHUDVHLQKLELWLQJDFWLYLWLHVRISKHQROLFFRPSRXQGV

354
of Dimorphandra gardneriana and Platymiscium floribundum , native plants from Caatinga biome ´YROQRSS±1168, 2012.
[364] N. S. Vila-Nova, S. M. Morais, M. J. C. Falcão, L. K. A. Machado, C. M. L. Bevilaqua, I. R. S. Costa, N. V. G. P. Brasil, and H. F. Andrade, ³/HLVKPDQLFLGDO DFWLYLWy and citotoxicity of compounds from two Annonacea VSHFLHV FXOWLYDWHG LQ1RUWKHDVWHUQ%UD]LO´Revta Soc. Bras. Med. Trop., vol. 44, pp. 567±571, 2011.
[365] *6HQ60XNKRSDGK\D\05D\DQG7%LVZDV³4XHUFHWLQLQWHUIHUHVZLWKiron metabolism in Leishmania donovani and targets ribonucleotide reductase WRH[HUWOHLVKPDQLFLGDODFWLYLW\´J. antimicrob. Chemother., vol. 61, pp. 1066±1075, 2008.
[366] ;//LX/=KDQJ;/)X.&KHQDQG%&4LDQ³(IIHFWRIVFRSROHWLQon PC3 cell proliferation DQG DSRSWRVLV´Acta Pharmacol. Sin., vol. 22, pp. 929±933, 2001.
[367] E.-K. Kim, K.-B. Kwon, B.-C. Shin, E.-A. Seo, Y.-R. Lee, J.-S. Kim, J.-W. Park, B.-H. Park, and D.-* 5\X ³6FRSROHWLQ LQGXFHV DSRSWRVLV LQ KXPDQpromyeloleukemic cells, accompanied by activations of nuclear factor kappaB and caspase-´Life Sci., vol. 77, no. 7, pp. 824±36, Jul. 2005.
[368] S. D. Bhagavathula, Synthetic Approaches and Biological Applications of Coumarins: A Review. pp. 1±43.
[369] J. L. Hwang, S. K. Hyun, D. L. Kyung, H. N. Sang, H. P. Ki, and S. Y. Min, ³&RPSDULVRQRI7\URVLQDVH,QKLELWRU\(IIHFWRIWKH1DWXUDO$QWLR[LGDQWVIURPCedrela sinensis´Agric. Chem. Biotechnol., vol. 48, no. 3, pp. 144±147, 2005.
[370] R. J. Yin, X. F. Xiao, Y. Y. Xu, C. Y. Li, C. X. Liu, and D. Y. Wang, ³5HVHDUFKLQIRUPDWLRQDQGUHYLHZRQWKHOHDYHVRIDiospyros kaki L II ´Asian J. Pharmacodyn. Pharmacokinet., vol. 10, no. 4, pp. 271±285, 2010.
[371] 6 ' 6KUXWKL DQG < / 5DPDFKDQGUD ³5%3-J as a therapeutic target to rheumotoid arthritis- DQLQVLOLFRVWXG\´Int. J. Preclin. Pharm. Res., vol. 2, no. 1, pp. 38±44, 2011.
[372] 6'5R\³3UHIRUPXODWLRQDVSHFWVRIWUDQVGHUPDOGUXJGHOLYHU\V\VWHPV´LQTransdermal and Topical Drug Delivery Systems, Ghosh, T.K., Interpharm Press: Buffalo Grove, IL, US, 1997, pp. 139±166.
[373] $1DLN<1.DOLDDQG5+*X\³7UDQVGHUPDOGUXJGHOLYHU\2YHUFRPLQJWKH VNLQ¶V EDUULHU IXQFWLRQ´ Pharm. Sci. Tech. Today, vol. 3, pp. 318±325, 2000.
[374] N. M. Feily A, ³$ORHYHUD LQGHUPDWRORJ\DEULHI UHYLHZ´G Ital Dermatol Venereol., vol. 2009, no. 1, pp. 85±91, 2009.

355
[375] /7)R[0*HUEHU-'X3OHVVLVDQG-++DPPDQ³7UDQVGHUPDO'UXJ'HOLYHU\(QKDQFHPHQWE\&RPSRXQGVRI1DWXUDO2ULJLQ´Molecules, vol. 16, no. 12, pp. 10507±10540, Dec. 2011.

356
APPENDIX A: NMR DATA
Figure A1: 1H NMR spectrum of compound 1

357
Figure A2: 13C NMR spectrum of compound 1

358
Figure A3: 1H NMR spectrum of compound 2

359
Figure A4: 13C NMR spectrum of compound 2

360
Figure A5: 1H NMR spectrum of compound 3

361
Figure A6: 13C NMR spectrum of compound 3

362
Figure A7: 1H NMR spectrum of compound 4

363
Figure A8: 13C NMR spectrum of compound 4

364
Figure A9: 1H NMR spectrum of compound 5

365
Figure A10: 13C NMR spectrum of compound 5

366
Figure A11: 1H NMR spectrum of compound 6

367
Figure A12: 13C NMR spectrum of compound 6

368
Figure A13: HMBC spectrum of compound 7

369
Figure A14: COSY spectrum of compound 7

370
Figure A15: 1H NMR spectrum of compound 7

371
Figure A16: 13C NMR spectrum of compound 7

372
Figure A17: 1H NMR spectrum of compound 8

373
Fiigure A18: 13C NMR spectrum of compound 8

374
APPENDIX B: HPLC CHROMATOGRAMS
Figure B1: HPLC chromatogram for compound 6

375
Figure B2: HPLC chromatogram for compound 7

376
Figure B3: HPLC chromatogram for compound 8

377
APPENDIX C: PUBLICATIONS
SAGE-Hindawi Access to Research Biotechnology Research International Volume 2011, Article ID 768673, 5 pages doi:10.4061/2011/768673
Research Article In Vitro Antimicrobial, Antioxidant Activities and Phytochemical Analysis of Canarium patentinervium Miq. fromMalaysia R.Mogana,1 Khoo Teng-Jin,1 and C.Wiart2 1 School of Pharmacy, Faculty of Science, University of Nottingham, Malaysia Campus, JlnBroga, Semenyih, 43500 Selangor, Malaysia 2 School of Biomedical Science, Faculty of Science, University of Nottingham, Malaysia Campus, JlnBroga, Semenyih, 43500 Selangor, Malaysia Correspondence should be addressed to R. Mogana, [email protected] Received 3 March 2011; Revised 7 April 2011; Accepted 2 May 2011 Academic Editor: Goetz Laible Copyright © 2011 R. Mogana et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Six different extracts of Canarium patentinervium Miq. (Burseraceae) leaves and barks were screened for their phytochemical composition, and antimicrobial and free radical scavenging activities. Among the different extracts tested, the ethanol extract of leaves showed significant antimicrobial and radical scavenging activities. The most susceptible micro-organisms were found to be Gram-positive bacteria (Staphylococcus aureus, methicillin-resistant Staphylococcus aureus or MRSA) and Gram-negative bacteria (Pseudomonas aeruginosa). Phytochemical analysis of the extracts revealed that the antimicrobial and the radical scavenging activities are mainly due to the presence of tannins and flavonoids. The results obtained suggest that Canarium patentinervium Miq. could be exploited in the management of various infectious diseases.

378

379
R.Mogana et al. / Journal of Pharmacy Research 2011,4(8),2482-2489
2482-2489
Review Article ISSN: 0974-6943
Available online through
www.jpronline.info
Canarium L. : A Phytochemical and Pharmacological Review
R.Mogana1* and C.Wiart2
1School of Pharmacy, Faculty of Science, University of Nottingham(Malaysia Campus), Jln Broga, Semenyih, 43500, Selangor Darul Ehsan, Malaysia
2School of Biomedical Science, Faculty of Science, University of Nottingham(Malaysia Campus), Jln Broga, Semenyih, 43500, Selangor Darul Ehsan, Malaysia
Received on: 17-05-2011; Revised on: 12-06-2011; Accepted on:16-07-2011
ABSTRACT The genus Canarium L. consists of 75 species of aromatic trees which are found in the rainforests of tropical Asia, Africa and the Pacific. The medicinal uses, botany, chemical constituents and pharmacological activities are now reviewed. Various compounds are tabulated according to their classes their structures are given. Traditionally Canarium L. species have been used to treat a broad array of illnesses. Pharmacological actions for Canarium L. as discussed in this review include antimicrobial, antioxidant, anti-inflammatory, hepatoprotective and antitumor activity.
Keywords: Canarium L., Burseraceae, antibacterial, antioxidant, pharmacology, secondary
metabolites

380
Planta Med 2011; 77 - PL9
DOI: 10.1055/s-0031-1282658
The first antibacterial activity report of three selected Malaysian
rainforest medicinal plants
A Nematollahi 1, N Aminimoghadamfarouj 1, M Rajagopal 1, TJ Khoo 1, C Wiart 2
1School of Pharmacy, Faculty of Science, Nottingham University, 43500, Malaysia
2School of Biomedical Science, Faculty of Science, Nottingham University, 43500, Malaysia
Despite increasing resistance among clinically important gram-negative and gram-positive pathogens
to many common antibacterial agents, many large pharmaceutical companies are showing decreased
interest in this product area that is so critical to public health. This downturn in antibacterial discovery
and development, in turn, is leaving us vulnerable to emerging resistance, particularly to recently
arrived vancomycin-resistant Staphylococcus aureus and multiply resistant, gram-negative bacilli for
which we do not have adequate antimicrobial therapy in the future. Rainforest plants possess
therapeutic potential, including antimicrobial activity [1,2]. Therefore, this study is a screening
program of several extracts from three endemic medicinal plants in Malaysian rainforest which have
not been fully discovered and investigated for their antimicrobial properties against several Gram-
positive and Gram-negative bacteria strains. These plants included families namely Annonaceae,
Ebenaceae and Burseraceae. The antibacterial activity of hexane, chloroform and ethanol fractions of
some parts of Uvaria grandiflora Roxb. (Annonaceae), Diospyros wallichii King & Gamble
(Ebenaceae) and Canarium patentinervium Miq. (Burseraceae) was determined against Gram-positive
bacteria Bacillus cereus ATCC10876, Staphylococcus aureus ATCC11632, Methicilin resistant
Staphylococcus aureus ATCC43300 and Gram- negative bacteria Pseudomonas aeruginosa
ATCC10145 and Escherichia coli ATCC10536 using the disk diffusion method. Results showed that
the bark ethanol fraction of U. grandiflora, the fruit hexane fraction of D. wallichii and the leaf ethanol
fraction of C.patentinervium are active (Table 1). The results indicate that these medicinal herbs can be
used as active and potent ingredients in the formulation of natural antibacterial products.
References:
1- Wiart C (2006) Medicinal Plants of the Asia-Pacific: Drugs from the Future, World Scientific,
Singapore.
2. Harvey AL (1999) Trends Pharmacol Sci 20: 196-198.

381
Hindawi Publishing Corporation Evidence-Based Complementary and Alternative Medicine Volume 2013, Article ID 734824, 7 pages http://dx.doi.org/10.1155/2013/734824
Research Article Anti-Inflammatory, Anticholinesterase, and Antioxidant Potential of Scopoletin Isolated from Canarium patentinervium Miq. (Burseraceae Kunth) R. Mogana, K. Teng-Jin, and C.Wiart Center for Natural and Medicinal Products Research, School of Pharmacy, Faculty of Science, University of Nottingham (Malaysia Campus), Jalan Broga, 43500 Semenyih, Selangor Darul Ehsan, Malaysia. Correspondence should be addressed to R. Mogana; [email protected] Received 13 March 2013; Accepted 14 June 2013 Academic Editor: Mohd Roslan Sulaiman Copyright © 2013 R. Mogana et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Bioassay guided fractionation of an ethanol extract of leaves of Canarium patentinervium Miq. (Burseraceae Kunth.) led to the isolation of scopoletin. The structure of this coumarin was elucidated based on spectroscopic methods including nuclear magnetic resonance (NMR-1D and 2D) and mass spectrometry. Scopoletin inhibited the enzymatic activity of 5-lipoxygenase and acetyl cholinesterase with an IC50 equal to 1.76 ± 0.01 ߤM and 0.27 ± 0.02mM, respectively, and confronted oxidation in the ABTS, DPPH, FRAP, and ߚ-carotene bleaching assay with EC50 values equal to 5.62 ± 0.03 ߤM, 0.19 ± 0.01 mM, 0.25 ± 0.03mM and 0.65 ± 0.07 mM, respectively. Given the aforementioned evidence, it is tempting to speculate that scopoletin represents an exciting scaffold from which to develop leads for treatment of neurodegenerative diseases.

382
Hindawi Publishing Corporation ISRN Biotechnology Volume 2013, Article ID 986361, 8 pages http://dx.doi.org/10.5402/2013/986361
Research Article The Medicinal Timber Canarium patentinervium Miq. (Burseraceae Kunth.) Is an Anti-Inflammatory Bioresource of Dual Inhibitors of Cyclooxygenase (COX) and 5-Lipoxygenase(5-LOX) R. Mogana, K. Teng-Jin, and C.Wiart Center for Natural and Medicinal Products Research, School of Pharmacy, Faculty of Science, University of Nottingham (Malaysia Campus), Jln Broga, 43500 Semenyih, Selangor Darul Ehsan, Malaysia Correspondence should be addressed to R. Mogana; [email protected] Received 10 July 2013; Accepted 19 August 2013 Academic Editors: A. O. Ballesteros, A. Maggio, and D. Pant Copyright © 2013 R. Mogana et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The barks and leaves extracts of Canarium patentinerviumMiq. (Burseraceae Kunth.) were investigated for cyclooxygenase (COX) and 5-lipoxygenase (LOX) inhibition via in vitro models.The corresponding antioxidative power of the plant extract was also tested via nonenzyme and enzyme in vitro assays.The ethanolic extract of leaves inhibited the enzymatic activity of 5-LOX, COX-1, and COX-2 with IC50 equal to 49.66±0.02 ߤg/mL, 0.60±0.01 ߤg/mL, and 1.07±0.01 ߤg/mL, respectively, with selective COX-2 activity noted in ethanolic extract of barks with COX-1/COX-2 ratio of 1.22.The ethanol extract of barks confronted oxidation in the ABTS, DPPH, and FRAP assay with EC50 values equal to 0.93 ± 0.01 ߤg/mL, 2.33 ± 0.02 ߤg/mL, and 67.00 ± 0.32 ߤg/mL, respectively, while the ethanol extract of leaves confronted oxidation in ߚ-carotene bleaching assay and superoxide dismutase (SOD) assay with EC50 value of 6.04 ± 0.02 ߤg/mL and IC50 value of 3.05 ± 0.01 ߤg/mL. The ethanol extract acts as a dual inhibitor of LOX and COXenzymes with potent antioxidant capacity.The clinical significance of these data is quite clear that they support a role for Canarium patentinervium Miq. (Burseraceae Kunth.) as a source of lead compounds in the management of inflammatory diseases.

383
APPENDIX D: CONTINUOUS EDUCATION
x 2SWLPL]LQJ <RXU /DERUDWRU\¶V 3URGXFWLYLW\ YLD LPSURYHG 6DPSOH 3UHSDUDWLRQ
Technique and Advanced Column Technology, Monash University, 12th Oct
2010.
x Project Management for Research, Graduate School Training Course, Nottingham
University Malaysia Campus, 4th Oct 2010
x Getting Going on Your Thesis, Graduate School Training Course, Nottingham
University Malaysia Campus, 5th Oct 2010
x Lab Demonstrations (Science) , Graduate School Training Course, Nottingham
University Malaysia Campus, 17th Jan 2011.
x Back To Nature symposium, Prof Iqbal Choudhary, UITM Puncak Alam, Jan
2011.
x NMR Theory & Applications: Natural products, UITM Puncak Alam, 17-18th Feb
2011.
x HPLC column determination, UITM Puncak Alam, April 2011.
x Workshop 2011: NMR for Intermediates and Metabolomics, UITM Puncak Alam,
17-19th Nov 2011.
x Technical Writing and Getting Published, Graduate School Training Course,
Nottingham University Malaysia Campus, 14th March 2012.
x Finishing Your Thesis, Graduate School Training Course, Nottingham University
Malaysia Campus, 30th Nov 2012.
x NMR Spectroscopy Workshop, UiTM Puncak Alam, 1-3rd March 2013.

384

385








![Original Article Canarium album extract restrains lipid ... · album [9]. Tannins, isolated from leaves, twigs and stem bark of Canarium album, were pro- ved to be with antioxidant](https://static.fdocuments.in/doc/165x107/5ee0e01fad6a402d666bf4cb/original-article-canarium-album-extract-restrains-lipid-album-9-tannins.jpg)









