
Ps9004 Complete for Pharmaceutical
128
The Institute of Quality Assurance Pharmaceutical Quality Group A Guide to the GMP requirements of PS 9000:2001 Pharmaceutical packaging materials
-
Upload
valmik-soni -
Category
Documents
-
view
96 -
download
2
description
Pharmaceutical standards,industry
Transcript of Ps9004 Complete for Pharmaceutical

The Institute of Quality Assurance
Pharmaceutical Quality Group
A Guide to the GMP requirements of PS 9000:2001 Pharmaceutical packaging materials

The Institute of Quality Assurance
Pharmaceutical Quality Group
PS 9004
A Guide to the GMP requirements
of PS 9000:2001 Pharmaceutical packaging materials
September 2004
i

ii

PS 9
004
guid
e PS 9004 guide
PS 9004 guide
ISO 9000+
GMPfor Pharmaceutical
packaging materials=
PS 9000
Figure 1. Model of PS 9004: a Guide to the GMP requirements of PS 9000
iii
The symbol is used in Part 1 to identify risk areas.

iviv
ISBN 0-906810-79-5
PS 9004: 2004
For further information about the Pharmaceutical Quality Group (PQG) and
any additional information or updates on PS 9000 or PS 9004 visit the PQG
website on: www.pqg.org
PS 9004 is available from:IQA Information Service
12 Grosvenor Crescent
London SW1X 7EE
Tel: + 44 (0)20 7245 6722
Fax: + 44 (0)20 7245 6788
e-mail: [email protected]
website: www.iqa.org

v
COPYRIGHT
PS 9004 copyright © 2004 Institute of Quality Assurance (IQA)
This PS 9004 guide is copyright protected by IQA/Pharmaceutical Quality
Group(PQG). However in pursuit of their objectives to promote quality
throughout the pharmaceutical manufacturing and supply industries,
permission is given to use the contents for quality system improvement
in education, training, auditing or for certification purposes, provided
acknowledgement of the source of the material (PS 9004 IQA/PQG Copyright)
is given. Reproduction,storage in a retrieval system,transmission in any form
or by any means,electronic, photocopying, recording or otherwise FOR ANY
OTHER PURPOSE without prior permission being secured is prohibited.
Requests for permission to reproduce should be addressed to the IQA at the
address below:
IQA Information Service
12 Grosvenor Crescent
London SW1X 7EE
Tel: + 44 (0)20 7245 6722
Fax: + 44 (0)20 7245 6788
e-mail: [email protected]
Reproduction may be subject to royalty payments or a licensing agreement.
Violators may be prosecuted.
v

vi

vii
MHRA FOREWORD
The EU Guide to Good Manufacturing Practice (GMP) emphasizes the need for
pharmaceutical manufacturers and assemblers to ensure that the packaging
materials that they use are of the appropriate quality. Not only is this in the
interests of patient safety, but also in the pharmaceutical industry where
the increasing use of automated packaging processes relies heavily on the
consistent quality of packaging components.
Each year the Medicines and Healthcare products Regulatory Agency
(MHRA) receives and investigates a number of reports of quality defects
concerning medicinal products. A significant proportion of these concern
packaging errors, some of which are attributable to the use of packaging
materials not of the desired quality. The introduction of PS 9000 in 2001 by
the Pharmaceutical Quality Group of the Institute of Quality Assurance was
a key step in promoting the understanding of GMPs relevant to the suppliers
of packaging materials to the pharmaceutical industry. PS 9000 focused on
the development and implementation, by suppliers, of a quality management
system designed to provide assurance of the quality of their products and to
enhance customer satisfaction. Its contribution was welcomed by both industry
and the Regulator. PS 9004 is aimed at increasing the awareness of the quality
management system requirements embodied in PS 9000. This comprehensive
and easy to follow guide explains further the GMP requirements of PS 9000.
It adopts a risk assessment approach to the identification and implementation
of preventive action and uses case studies to illustrate areas of GMP risk.
The MHRA encourages the introduction of PS 9004 designed to improve
product and service quality in the supply of packaging materials for medicinal
products, for the benefit of patients and the pharmaceutical industry as a
whole.
John Taylor Quality and Systems Manager
Inspection and Enforcement Division, MHRA

viii

ix
GENERAL INTRODUCTION
What is PS 9004?This guide aims to assist suppliers of pharmaceutical packaging materials to
understand and implement the Quality Management System (QMS) of ISO
9001 and the application standard of PS 9000. It supplements the following
documents:
• ISO 9001 which specifies the requirements of the international QMS
standard
• ISO 9004 gives guidance on ISO 9001
• PS 9000 is an application standard written by the PQG for suppliers of
pharmaceutical packaging materials that integrates ISO 9001 and ISO
9004 together with additional Good Manufacturing Practices (GMP)
requirements particular to these suppliers
Companies complying with PS 9000 will comply with ISO 9001 and also the
additional GMP requirements endorsed by the highly regulated pharmaceutical
industry.
This guide is constructed around the clause structure of PS 9000 (and therefore
ISO 9001) to:
• clearly explain the GMP requirements of PS 9000
• list many of the risk areas associated with packaging processes and identify
relevant ISO 9001 and PS 9000 clauses
• provide examples of process inputs and process outputs typical of suppliers
of pharmaceutical packaging materials, and relate these to the QMS
requirements of ISO 9001 and the additional GMP requirements of PS
9000
• give examples of actual case histories of problems arising from the supply of
defective pharmaceutical packaging materials and where the consequences
would have been avoided by correct application of the GMP requirements
of PS 9000. These case histories help to explain the requirements and
provide a valuable training resource

x
How to use this GuidePS 9004 has been organized in the following way:
• Part 1 consists of a selection of process schematics, illustrating typical
operations of a packaging supplier, and giving examples of some of the
more significant risks caused by inadequate manufacturing practices
• Part 2 gives guidance on the clauses of PS 9000, by taking the user through
each major section and providing examples to illustrate suggested process
inputs and outputs
• Annex A gives expanded explanatory notes on several key concepts used in
PS 9000
• Annex B gives a listing of other external regulatory references, which are
the background to pharmaceutical GMP and which are directly relevant to
PS 9000 requirements
• the text is primarily black but blue is used for PS 9000 related GMP text • red text is used to highlight the ‘Risk Areas’
The user can cross refer between the schematics in Part 1 and the process information in Part 2. In addition, the user can move from Part 2 to the additional explanations in the annexes.
The annexes contained within this Guide are a stand-alone explanation of key concepts and external GMP references. The PS 9000 cross-references are for illustrative purposes and are not a definitive list of clauses or references.
The How to use section, which follows the contents list, shows the user how to
navigate between the different sections of the Guide.
As the range of organizations that may implement PS 9000 is great, both in
terms of size and type of business, the guidance given here is illustrative and
advisory.
Why was the PS 9004 Guide written?There are three main reasons:
• to guide professional quality specialists towards the implementation and
inspection of a Quality Management System (QMS) in accordance with PS
9000
• to support the training of all the various stakeholders in the PS 9000 process
(top management, quality professionals, staff at all levels of packaging

xi
material suppliers and their customers)
• to provide those people who are not quality specialists with a better
understanding of the aims, objectives and implementation of PS 9000
How was the PS 9004 Guide written? A group of experienced quality professionals selected from the packaging supply
industry and pharmaceutical customers was recruited by the Pharmaceutical
Quality Group Partners Team and given the task of preparing this Guide.
Many of the people who contributed to the writing of PS 9000 also participated
in the creation of this Guide.
Specific acknowledgements are given for the contributions of the following people: Roy Evans PS 9004 Team Leader
David Abraham Supplier
Ian Harwood Pharmaceutical
Jill Jenkins Pharmaceutical
Daniel Peek Supplier
Richard Rowlett Supplier
Mike Shorten Pharmaceutical
Steve Taylor Pharmaceutical
Additional contributions: Caroline Fowler Pharmaceutical
Peter Gough Pharmaceutical
Ian Holloway Regulator
Steve Merrit Pharmaceutical
Ashley McCraight Consultant
Jeff Monk Training
Dominic Parry Training
Steve Pike Accredited certification
Norman Randall Consultant
Trevor Stabb Consultant
Paul Stockbridge Consultant
QA review: Tony Harper Consultant and accredited certification
Afshin Hosseiny Consultant

xii
QA review (con’t): Jean Lanet Consultant
Iain Moore Supplier
Steve Moss Pharmaceutical
Ashok Chand Pharmaceutical
John Turner Consultant
The PQG Partners Team and Steering Committee: Roy Evans
Tony Harper
Femi Ibironke
Jill Jenkins
Ashley McCraight (PT Chairman)
Steve Moss
Norman Randall (SC Chairman)
Ian Richardson
In addition, the PQG is appreciative of the management of Patheon UK
Limited. for the use of their resources and facilities.

xiii
CONTENTS
Section Page
How to use this guide xvPart 1 – Process schematics and case studies 1 Overview of business processes 2 Schematic 1 Top level business processes 2 Schematic 2 From the customer 4 Schematic 3 Planning 6 Schematic 3 (a) Warehouse (purchased materials and in-process) 8 Schematic 4 Preparation 10 Schematic 4 (a) Print impression media controls 12 Schematic 5 Production 14 Schematic 6 Checking and final release 16 Schematic 7 Distribution 18 Case studies 21
Part 2 – Guidance on PS 9000 GMP clauses 35 Clause 4 Quality management system 37 Clause 5 Management responsibility 39 Clause 6 Resource management 45 Clause 7 Product realization 49 Clause 8 Measurement, analysis and improvement 59 Clause 9 Contamination control 66 Clause 10 Printed materials 68 Clause 11 Origination/artwork 70 Clause 12 Print impression media 72 Clause 13 Print and conversion processes 74

xiv
Section Page
Annex A – Additional Explanation of Key Concepts 77 Admixture 78 Change control 80 Counterfeit 82 Identification and traceability 84 Line clearance 86 Risk assessment 88 Segregation controls 92 Validation (and Qualification) 94
Annex B – Other external GMP References 99 B1 - US Code of Federal Regulations with corresponding PS 9000 references 100 B2 - Rules and Guidance for Pharmaceutical Manufacturers and Distributors 2002 with corresponding PS 9000 references 101 B3 - MHRA Defect Investigation reference table 103
Index 104

xv
HOW TO USE THIS GUIDE
STEP 1
Choose a schematic from the ‘Overview of business process’ diagram in Part 1 of this guide. (e.g. Schematic 3(a) is illustrative)
STEP 2
From your schematic, choose a particular stage of the process to examine, e.g. Store material (see below).
SCHEMATIC 3 (a) - WAREHOUSE
STEP 3
Consider the Risk Areas shown in red and the real life case study (if shown) - these are detailed at the end of Part 1, following the schematics.
Delivery checks
• Document errors• Missing documents
7.4.3
7.5.3
Quality inspection
• Labelling errors• Incorrect goods
7.4
7.4.3
Store material
• Pest Infestation
Case study G
7.5.5:Preservation of product
9:Contamina-tion control
Issue material
• Incorrect materials
7.5.3
See Part 2 for more details
Risk areas

xvi
STEP 4
Using the references shown in the schematic, move to the corresponding section in Part 2, entitled ‘Guidance on PS 9000 GMP clauses’. This gives reasons for the ‘extra’ GMPs and shows examples of processes, inputs and outputs.
General synopsis: Clause 9 Contamination controlThe capability of facilities and equipment, achieved through their design, location, etc.
Reasons for the ‘extra’ GMP requirementsThe highest priority of the pharmaceutical industry is... etc.
Process inputs Processes Process outputs and evidence
Cross refer to Schematic/Annex
Specification for design of new facilities, processes, or environment
Specialist support for pest control
Standards for environmental conditions
Processes which identify/control:• operating areas• pest control• cleaning proce-
dures
Records of:• processing • cleaning
schedules• material disposal• incidents/audit
reports
Schematic 3(a)
Schematic 4(a)
Schematic 5
Annex A - all parts
STEP 5
Use the cross references to move back to a relevant process schematic, alterna-tively refer to Annex A or B for further guidance.
ANNEX A
Additional Explanation of Key Concepts
ANNEX B
Other external GMP references.

1
The Institute of Quality Assurance
Pharmaceutical Quality Group
PS 9004
A Guide to the GMP requirements
of PS 9000:2001 Pharmaceutical packaging materials
PART 1 - PROCESS SCHEMATICS
PS 9000 GMP references are reproduced in blue Risk areas are reproduced in red

2
OVERVIEW OF BUSINESS PROCESSES
Schematic 1: Top level business processes
Schematic 2: From the customer
Schematic 3: Planning
Schematic 4: Preparation
Schematic 3 (a): Warehouse
Schematic 4 (a): Print impression media controls
SCHEMATIC 1: TOP LEVEL BUSINESS PROCESSES
Qualitymanagementsystem
Management commitment
• Inadequate incorporation of PS 9000 requirements
• QMS not communicated and/or supported by senior management
4 QMS5.1 Management
commitment5.5
Responsibility, authority and communication
Objectives
• Unclear business targets/goals
• No clear direction as objectives not published
5.3 Quality Policy
5.4 Planning
Resource allocation
• Too few people for quality compliant operation
• Wrong/ inadequate equipment
• Inadequate premises
• Inadequate training
6.1 Provision of resources
6.2 Human resources
Refer to the hypothetical examples which are located after the schematics.
See Part 2 for more details
Risk areas

3
OVERVIEW OF BUSINESS PROCESSES
Schematic 5: Production
Schematic 6: Checking and final release
Schematic 7: Distribution
This diagram shows the main business processes from customer requirements through to delivery of final product and provides an index to the schematics
Audit and measurement
• Inadequate performance measures
• Poorly controlled processes
• Unreliable information for management decisions
• Poor compliance to internally established processes/procedures
• Insufficient proof of continuous improvement
8.1 Measurement, analysis and improvement - general
Management review
• Quality System measures not reviewed
• Lack of senior management involvement/leadership
• No continual process improvement
5.1 Management commitment
5.6 Management review
8.5 Improvement
Quality management system
Feedback/communication
SCHEMATIC 1: TOP LEVEL BUSINESS PROCESSES
Refer to the hypothetical examples which are located after the schematics.

4
SCHEMATIC 2: FROM THE CUSTOMER
Organization Customer request/requirements
• Requirements unclear
• Requirements incorrectly specified
• Requirements misunderstood
• Requirements incorrectly accepted
See case study A
5.2 Customer focus
7.2 Customer-related processes
Supply requirements (what, how, when)
• Unclearly specified
• Failure to meet customer expectation
• Rejection by customer
Customer design
• Poor design• Inadequate detail• Failure to
function• Poor
performance at customers site
• Rejection by customer
• Potential risk to patient
• Inadequate testing/validation
See case studies A and B
Feedback
7.2 Customer-related processes7.3 Design and development
See Part 2 for more details
Risk areas
Refer to the case studies which are located after the schematics.

5
SCHEMATIC 2: FROM THE CUSTOMER
Assessment of capability
• Incorrect assessment of capability e.g. resource, technology, systems, environment
• Fail to meet delivery/quality
6.1 Provision of resources
6.3 Infrastructure6.4 Work
environment7.1 Planning
of product realization
Agreed contract/agreement and confirmed order
• Lack of formal contract/agreement
• Failure to agree contract requirements
• Poor communication
7.2.3 Customer-related processes
Planning
Refer to the case studies which are located after the schematics.

6
SCHEMATIC 3: PLANNING
Receive order from customer
Order review and customer feedback (where required)
• Incorrect specification chosen
• Inadequate communication with customer
• Order details incorrect• Supply incorrect product• Supply error (quantity/
schedule)• Proof approval error• Specification not
approved
See case studies C and D
5.2 Customer focus7.2 Customer-related
processes7.3 Design and
development
Availability checks on documentation, materials, tooling and equipment
• Incorrect material checked or issued (e.g. wrong foil gauge)
• Wrong print media (version or item)
• Non-availability of equipment (e.g. maintenance/validation status)
• Unapproved production process (e.g. not approved by customer)
• Unsuitable/unapproved material/changes
• Missing/incorrect documentation (e.g. missing mould inspection record)
7.4 Purchasing7.5 Production and service
provision11 Origination/artwork12 Print impression media
See Part 2 for more details
Risk areas
Refer to the case studies which are located after the schematics.
7
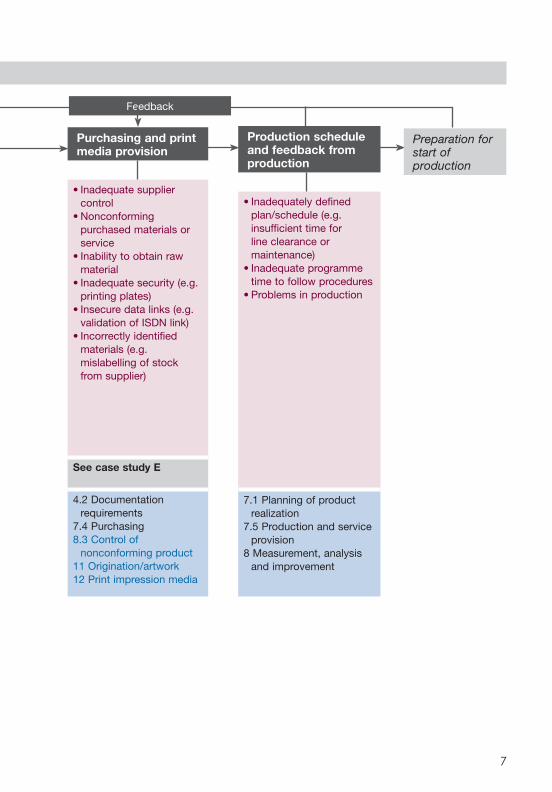
SCHEMATIC 3: PLANNING
Purchasing and print media provision
• Inadequate supplier control
• Nonconforming purchased materials or service
• Inability to obtain raw material
• Inadequate security (e.g. printing plates)
• Insecure data links (e.g. validation of ISDN link)
• Incorrectly identified materials (e.g. mislabelling of stock from supplier)
See case study E
4.2 Documentation requirements
7.4 Purchasing8.3 Control of
nonconforming product11 Origination/artwork12 Print impression media
Production schedule and feedback from production
• Inadequately defined plan/schedule (e.g. insufficient time for line clearance or maintenance)
• Inadequate programme time to follow procedures
• Problems in production
7.1 Planning of product realization
7.5 Production and service provision
8 Measurement, analysis and improvement
Preparation for start of production
Feedback
Availability checks on documentation, materials, tooling and equipment
• Incorrect material checked or issued (e.g. wrong foil gauge)
• Wrong print media (version or item)
• Non-availability of equipment (e.g. maintenance/validation status)
• Unapproved production process (e.g. not approved by customer)
• Unsuitable/unapproved material/changes
• Missing/incorrect documentation (e.g. missing mould inspection record)
7.4 Purchasing7.5 Production and service
provision11 Origination/artwork12 Print impression media
Refer to the case studies which are located after the schematics.

8
SCHEMATIC 3 (a): WAREHOUSE (purchased materials and in-process)
Delivery from supplier/sub-contractor
Delivery documentation and off-loading checks
• Errors not identified between delivery documentation & purchase order
• Missing delivery/QC documentation
• Transit damage or wet boxes not identified before off-loading
• Incorrect goods
7.4.3 Verification of purchased product
7.5.3 Identification and traceability
Goods checks and quality inspection
• Inadequate checks performed
• Labelling errors not identified
• Incorrect goods sent• Rogue box within delivery• Inadequate batch coding• Non representative sample
taken or provided• Transit/water damage not
detected• Inadequate packaging/
sealing not detected• Incorrect quality status
input to system
See case study F
7.4 Purchasing7.4.3 Verification of
purchased product
See Part 2 for more details
Risk areas
Refer to the case studies which are located after the schematics.

9
SCHEMATIC 3 (a): WAREHOUSE (purchased materials and in-process)
Store material
• Water damage• Pest infestation• Dirt contamination• Mix-up due to
inadequate segregation• Goods stored incorrectly
(e.g. on the floor, temperature, humidity)
• Incorrect quantity/ location recorded
See case study G
7.5.5 Preservation of product
9 Contamination control
Issue material
• Incorrect materials dispensed eg. wrong batch, version
• Incorrect quantity issued• Mixed batch issued but
not identified
7.5.3 Identification and traceability
To production
Goods checks and quality inspection
• Inadequate checks performed
• Labelling errors not identified
• Incorrect goods sent• Rogue box within delivery• Inadequate batch coding• Non representative sample
taken or provided• Transit/water damage not
detected• Inadequate packaging/
sealing not detected• Incorrect quality status
input to system
See case study F
7.4 Purchasing7.4.3 Verification of
purchased product
Refer to the case studies which are located after the schematics.
Return of unused material or semi-finished product

10
SCHEMATIC 4: PREPARATION
Production plan
Preparation/pre-production checks of purchased raw materials
• Error in checking raw material
• Quality status incorrect
• Availability status incorrect
Materials management
Errors in:• Co-ordination
of all required resources
• Combining/ assembly/ Bills of Material
• Timing/change management
• Machine allocation
7.1 Planning of product realization
7.2 Customer related processes
Preparation/pre-production checks of tooling and equipment
• Error in checking tooling/equipment
• Change control error
• Validation status error
• Maintenance omission/ error
• Availability status incorrect
See case study H
7.4 Purchasing7.5 Production and service provision10 Printed materials11 Origination/artwork12 Print impression media13 Print and conversion process
See Part 2 for more details
Risk areas
Refer to the case studies which are located after the schematics.
11
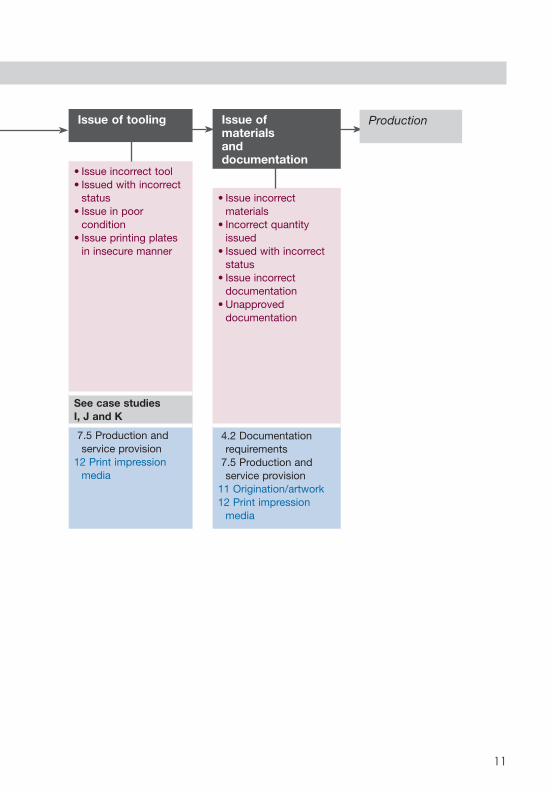
SCHEMATIC 4: PREPARATION
Issue of tooling
• Issue incorrect tool• Issued with incorrect
status• Issue in poor
condition• Issue printing plates
in insecure manner
See case studies I, J and K
7.5 Production and service provision
12 Print impression media
Issue of materials and documentation
• Issue incorrect materials
• Incorrect quantity issued
• Issued with incorrect status
• Issue incorrect documentation
• Unapproved documentation
4.2 Documentation requirements
7.5 Production and service provision
11 Origination/artwork12 Print impression
media
Production
Refer to the case studies which are located after the schematics.

12
SCHEMATIC 4 (a): PRINT IMPRESSION MEDIA CONTROLS
Customer supplied design text
New print media design
• Errors in media undetected by checks
• Individual plate incorrectly coded
• Set of plates incorrectly coded
• Failure to secure customer proof approval
• Failure to log customer approval
See case study L
11 Origination/ artwork
12.1 General12.2 Matched
plates
Secure storage and controlled issue of plates
• Mix-up with plates from another product
• Uncontrolled access of personnel
• Physical damage to plates
• Inadequate records of issue introducing admixture risk in production
Controls over partial re-designs and obsolete designs
• Incorrect plates retained
• Insecure disposal• Superseded plates
not rendered unusable
• Incorrect plate identification
• Error in replacement plate/ matching set
• Documentation errors
• Customer approval error
See case study M
12.2 Matched plates12.3 Copy / design change12.5 Quarantine and destruction13.2 Changeover systems
See Part 2 for more details
Risk areas
Refer to the case studies which are located after the schematics.

13
SCHEMATIC 4 (a): PRINT IMPRESSION MEDIA CONTROLS
Line use of plates
Failure to :• Check design• Clear line of
previous plates• Perform start-up
printing checks• Record checks• Keep samples
See case study N
9.1.7 Line clearance
12.4 Verification13.1 Print
machine set-up (make-ready)
13.2 Changeover systems
13.3 Retained samples
Controls over replacement plate (same design)
• A new first off check is not performed or recorded (e.g. plate made from existing approved source)
• New origination & new batch checks not performed or recorded (e.g. plate created from a new source)
13.4 Replacement print media
Line clearance and controlled return of plates to store
• Plate left on machine in error
• Inadequate records of return to store
9.1.7 Line clearance
12.1 General
Production
Refer to the case studies which are located after the schematics.

14
SCHEMATIC 5: PRODUCTION
Preparation Line clear check
• Admixture from previous batch
• Erroneous use of previous documents
• Use of a plate from previous job
7.5 Production and service provision
9.1.7 Line clearance
10 Printed materials
11 Origination/ artwork
Documents/materials/tooling to line
• Use of incorrect documents/ materials
• Failure to use documents correctly
• Incorrect quantities
• Defective moulds/equipment/plates
See case study O
4.2 Documentation requirements
7.5 Production and service provision
Set up checks and production start
• Errors in data input to machine, e.g. settings
• Setting error undetected
• Start up waste inadequately segregated from main batch
• Unauthorized start up
5.5 Respon- sibility, authority and communication
7.6 Control of monitoring and measuring devices
9 Contamination controls
10 Printed materials
12 Print impression media
See Part 2 for more details
Risk areas
Refer to the case studies which are located after the schematics.

15
SCHEMATIC 5: PRODUCTION
Production/in process control
• Quality variation due to environment e.g. temperature, humidity
• Contamination e.g. insects, dust
• Defective / out of spec or wrong materials
• Visual defects not detected
• Inadequate IPC of transit packaging
• Labelling errors• IPC errors• Material usage error• Failure to identify/
segregate non conforming product
• Recording error
See case studies P, Q
6.4 Work environment7.5 Production and
service provision8.2 Monitoring and
measurement8.3 Control of
nonconforming product11 Origination/artwork
Production completion and line clearance
Risk to next job due to:• lack of GMP training• failure to clear line• inadequate waste disposal• documentation not signed
off therefore status unclear• clearance of electronic
files/settings
See case study R
6.2.2 Competence, awareness and training
8.2 Monitoring and measurement
9 Contamination controls10 Printed materials13 Print and conversion
processes
Checking and final release
Refer to the case studies which are located after the schematics.
Feedback

16
SCHEMATIC 6: CHECKING AND FINAL RELEASE
Production End product QC testing
• Inadequate training• Test method not
validated• Out of Specification
results not investigated• Inadequate procedures• Release decision
contradicts data• Inconsistent sampling/
risk• Equipment/test
equipment inadequate• Calibration errors/
omissions
See case study S
6.2.2 Competence, awareness and training
7.6 Control of monitoring and measuring devices
8 Measurement, analysis and improvement
8.4 Analysis of data
Review of batch documents and QC testing
• Missing/incorrect document
• Missing/incorrect sample• Signature omissions,
unauthorized check signature
• Errors of information/data entry
• Checks out of sequence• Abnormal events not
reviewed
See case study T
5.5 Responsibility, authority and communication
6.2 Human resources4.2 Documentation
requirements13 Print and conversion
process
See Part 2 for more details
Risk areas
Refer to the case studies which are located after the schematics.

17
SCHEMATIC 6: CHECKING AND FINAL RELEASE
Create certificate of test or conformance
• Errors / omissions• Signature invalid• Inappropriate data
supplied• Incorrect specifications/
tolerances• Incorrect release decision• Certificate text differs
from batch details
See case study U
8.2 Monitoring and measurement
8.3 Control of nonconforming product
Check box labels against batch information
• Incorrect/missing label• Label data error/
omission• Label reconciliation• Incorrect QC status label
See case studies V and W
7.5 Production and service provision
DistributionReview of batch documents and QC testing
• Missing/incorrect document
• Missing/incorrect sample• Signature omissions,
unauthorized check signature
• Errors of information/data entry
• Checks out of sequence• Abnormal events not
reviewed
See case study T
5.5 Responsibility, authority and communication
6.2 Human resources4.2 Documentation
requirements13 Print and conversion
process
Refer to the case studies which are located after the schematics.

18
SCHEMATIC 7 - DISTRIBUTION
Checking and final release
Receipt of product into storage/holding (goods out warehouse)
• Error in system transaction (receipt)
• Inadequate segregation from other goods
• Storage damage
7.5 Production and service provision
10 Printed materials
Order preparation (picking and transit packaging)
• Picking wrong product• Damage to product due
to inappropriate transit packaging
• Pallets not suitable for customer handling
• Error in transaction (issue)
See case study X
7.5 Production and service provision
See Part 2 for more details
Risk areas
Refer to the case studies which are located after the schematics.
19
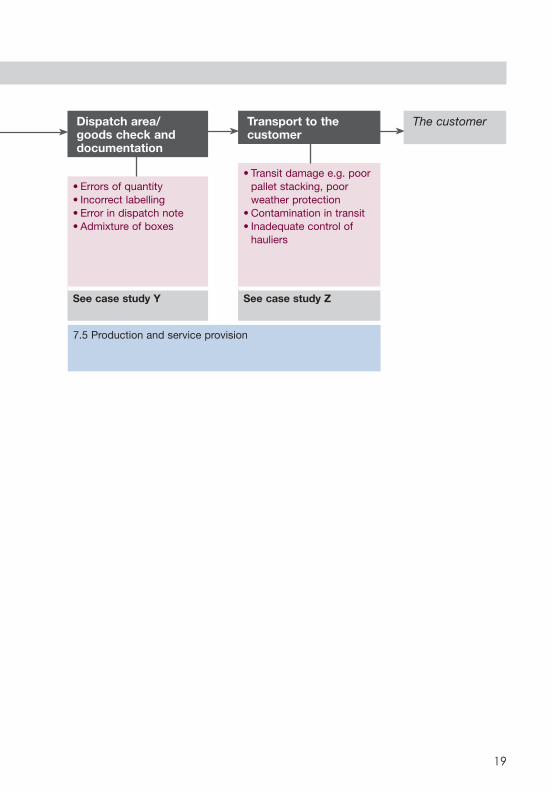
SCHEMATIC 7 - DISTRIBUTION
Dispatch area/goods check and documentation
• Errors of quantity• Incorrect labelling• Error in dispatch note• Admixture of boxes
See case study Y
Transport to the customer
• Transit damage e.g. poor pallet stacking, poor weather protection
• Contamination in transit• Inadequate control of
hauliers
See case study Z
The customer
7.5 Production and service provision
Order preparation (picking and transit packaging)
• Picking wrong product• Damage to product due
to inappropriate transit packaging
• Pallets not suitable for customer handling
• Error in transaction (issue)
See case study X
7.5 Production and service provision
Refer to the case studies which are located after the schematics.

20

21
The Institute of Quality Assurance
Pharmaceutical Quality Group
PS 9004
A Guide to the GMP requirements
of PS 9000:2001 Pharmaceutical packaging materials
PART 1 - CASE STUDIES

22

23
Part 1 – Examples and Case studies
These examples and ‘real life’ case studies should be used with the relevant Schematic, to illustrate specific GMP risk areas and the appropriate QMS processes involved.
HYPOTHETICAL EXAMPLE FOR SCHEMATIC 1 – TOP LEVEL BUSINESS PROCESSES
The senior management of a booklet printing company believes that PS 9000 certification will aid company profitability and smooth throughput because of consistent demand from the pharmaceutical industry, which is not adversely affected by recessions. The company has an enthusiastic Quality Manager who has produced a comprehensive manual with up to date and relevant procedures. However, the Quality Manager is beginning to feel like a ‘voice in the wilderness’ as there is little evidence that senior management is driving the whole organization to become aligned to the requirements of the standard. The lack of senior management commitment to the continual improvement philosophy and the Good Manufacturing Practices of the pharmaceutical industry means that only superficial actions are taken in areas considered highly visible to visiting auditors. Key individu-als are unclear about the standards required of them. The level of customer complaints remains high, some of these complaints being serious mix-ups. Productivity stays unchanged with a high level of wastage. Workforce moral is low and many staff are demotivated.
The company is then purchased by another company that already supplies cartons to the pharmaceutical industry - allowing them to offer a better product range to the customer. The corporate re-organization results in key changes in senior management at the booklet factory. The new managers know their prime objective is to encourage each and every employee to understand the particular needs of their pharmaceutical customers. They understand the value of PS 9000 in a competitive market, but know that the real benefits are much deeper than merely achieving the ‘badge’. The Qual-ity Manager begins to feel encouraged and genuinely part of the team. The frequency of management review meetings is increased with the emphasis changed to progressing preventive actions for nonconformities and review-

24
ing clear measures of performance for key parameters such as complaint rates. The senior management commitment begins to permeate throughout the staff and becomes evident in many areas such as resourcing, training, customer service, and improved product quality. The board of directors soon recognises that the PS 9000 implementation process is enabling them to profitably expand their business, improve relationships with their custom-ers, whilst still significantly reducing their cost of quality.
CASE STUDIES FOR SCHEMATIC 2 – FROM THE CUSTOMER
A Focus - Customer request/requirementsBar coded product labels could not be read by customer’s electronic read-ers due to shading of background colour, caused by inadequate testing and validation. The packing orders at the pharmaceutical company were delayed which threatened the availability of the medicinal products to the end customers. The labels had to be destroyed, incurring the direct expense of replacement. The product was redesigned with solid background, and all graphics designers were made aware of the issue and to consider the pos-sible need for trials on change of design or introduction of a new design.
B Focus – Customer designA market need for a needle protection mechanism was specified, but translation of this into a final design was lengthy and complicated. The component did not perform reliably due to limited experimental validation. The problems were addressed through increased communication with the supplier to make the design more robust, involving revalidation and testing. This extended work caused a delay in providing the improved safety prod-uct to the patient. An organizational review followed to ensure that in future, adequate resource is devoted at the design and testing stages of product development.
CASE STUDIES FOR SCHEMATIC 3 – PLANNING
C Focus – Order review and customer feedbackThe wrong material was ordered by the supplier’s UK sales office from their own factory. Normally, orders were placed for 40 micron PVDC coated PVC

25
(for tablet blister packing), but on this occasion a new order for 60 micron coated film was incorrectly notified by the sales office to the factory in Ger-many as 40 micron; the incorrect material was delivered and used causing a regulatory noncompliance and potentially inadequately protecting the tablets from moisture. Significant work was necessary by the pharmaceuti-cal company to assess stability data to determine the acceptability of the packed tablet batches. The root cause was established through customer audit of the ordering system at the sales office. An independent check of order details was implemented to prevent repetition.
D Focus – Order review and customer feedbackThe printer’s proof was incorrectly read by the supplier as ‘approved’ when it was actually ‘rejected’; also the wrong file was used to make replace-ment printing plates. Cartons for a heart condition medicinal product were printed and used showing on one end flap 14 tablets instead of 28. The consequence of this was that stock had to be recalled from the distribu-tion centres to enable a rework. This delayed the critical medicinal product reaching the market. Working together, customer and organization, the complete sequence of events was reconstructed. In addition to awareness training, the rules regarding issuing artworks and file management between organization and platemaker were made more robust.
E Focus – Purchasing and print media provisionLeaflets with a different paper weight were supplied, due to problems with availability of normal paper. This caused insertion difficulties on the phar-maceutical packing line, high wastage of components and loss of some pharmaceutical product. The supplier was advised of the existing change control clause in the quality agreement/contract and procedures were revised. The supplier managed to obtain further supplies of normal paper.
CASE STUDIES FOR SCHEMATIC 3 (a) – WAREHOUSE (PURCHASED MATERIALS & IN-PROCESS)
F Focus – Goods checks and quality inspectionSelf-adhesive labels were failing to stick to glass vials at the customer’s premises because a different adhesive had been used. This had been caused by the supplier switching their source of unprinted label material.

26
The raw material was thought to be of equivalent quality and no extra qual-ity checks were performed at goods-inwards. The change was introduced without informing the customer therefore no formal change control or a risk assessment could be performed. The problem caused chaos on the customer’s packing line and it was not certain how well the labels were affixed to ‘apparently’ satisfactory packed vials. The label printing company pursued the quality issue with its supplier and proper trials of alternatives were subsequently conducted with the pharmaceutical company. In addi-tion, revisions were made to the change control system.
G Focus – Store materialTwo dead mice were found inside the pallet wrapping of a consignment of plastic bottles to be used for an antiseptic liquid product. This caused major disruption to the output of the pharmaceutical filling plant. All the stock of bottles had to be manually inspected for contamination. It was necessary to audit the supplier and its remote warehouse. Immediate action involved the relocation of bottle storage back into the main factory. The root cause was inadequate warehouse pest controls at the supplier’s remote warehouse. It prompted an extensive programme of work to improve the standards at the site. The product was a low margin item for the pharmaceutical company and it has since been sold off to another company. Although many other factors were involved, this episode only added to the reasons to sell this part of the business.
CASE STUDIES FOR SCHEMATIC 4 – PREPARATION
H Focus – Preparation/pre-production checks of tooling and equipmentMould tool used to produce parts for a syringe was used in production when the validation documentation had not been fully approved and signed off. This contributed to high variability in functional performance of the assembled syringe with some batches of parts rejected, and it necessitated ongoing improvement work. It also delayed the launch of the pharmaceuti-cal product which in this case was a life saving cancer product. The opera-tional procedures were revised and the operators were retrained.

27
I Focus – Issue of toolingSyringe labels were printed correctly, but were cut with corners of the wrong profile/shape due to the issue of an incorrect cutter to the press. The labels were rejected by the pharmaceutical company at the expense of the print-ers. The time needed to replace the labels meant rescheduling of packing was necessary at the pharmaceutical company. The cutter identification process was reviewed & storage standards were improved.
J Focus – Issue of toolingCartons were produced with a varnish free area in the wrong position due to the issue of an incorrect varnish plate. The purpose of a varnish free area is to ensure the printing of variable data (e.g. lot & expiry) is clear and per-manent. This error was identified at the pharmaceutical company. The risk existed that variable data applied by the pharmaceutical company could be easily smudged or erased when in the market and this would clearly be a critical problem. The supplier reviewed the varnish plate issuing process and made improvements to the system which linked varnish plate usage to print plate usage.
K Focus – Issue of toolingMould tool for a syringe plunger rod was issued to production despite being overdue for refurbishment. This resulted in minor plastic ‘flash’ being present on the mouldings. If this problem became more serious then loose particles of plastic could break off and contaminate the pharmaceutical syringe pack. The supplier applied for a concession from the customer and this was granted for a limited period because of the extent of the problem. The case highlighted the need for proper planned preventive maintenance and sufficient resources allocated to facilitate it.
CASE STUDIES FOR SCHEMATIC 4 (a) – PRINT IMPRESSION MEDIA CONTROLS
L Focus – New print media designArtwork supplier made a “cut & paste” error which resulted in cards being part printed with Polish text in the main body of Portuguese text. The cards were for an anti-cancer injection. Fortunately the error was noticed dur-ing in-process checks of printing. The error had the potential to become

28
a product recall if it had reached the end user. An extensive review of the artwork supplier’s procedures was completed and a further proof-reading check was introduced.
M Focus – Controls over partial redesigns & obsolete designsThe printed foil for tablet blister strips (used to treat a heart condition) was incorrectly printed with two different identifying codes on the foil (different by one digit). The error occurred during the ‘step and repeat’ process to generate a single replacement cylinder. The error was only found on inspec-tion at the pharmaceutical company. In this case, no packed stock was lost, but the error could have been more serious. The supplier took the issue up with its cylinder manufacturer and reviewed its inspection procedure for first off sample inspection following cylinder replacement.
N Focus – Line use of platesA printing plate was left on the leaflet press by the operator after leaving the area to retrieve a spare part. Upon return the operator continued printing, but against the job documentation for the next job. The item was visually very similar and all subsequent checks by operators and QC failed to spot the admixture that had been created. The problem was found on goods inwards checks by the customer, and resulted in a detailed audit of proce-dures by the customer. Line clearance checks were reviewed, procedures around bar code scanning at the suppliers were enhanced and refresher training was delivered.
CASE STUDIES FOR SCHEMATIC 5 – PRODUCTION
O Focus – Documents/materials/tooling to lineDuring the folding stage of leaflet production two orders were mixed up. Job A was folded, scanned, labelled and released against Job B documenta-tion, and Job B documentation was folded, scanned, labelled and released against Job A documentation. The mixed-up orders were then delivered to the pharmaceutical company where the error was discovered. The potential for incorrect leaflets to be packed into pharmaceutical packs was increased. The supplier’s investigation identified the root cause as human failure to fol-low established procedures. In addition the codes for the bar code scanner are now input from an independent source (a PS 9000:2001 requirement).

29
P Focus – Production/in-process controlA delivery of poorly printed foil, used for tablet blister strips, was caused by inadequate ‘flagging’ of defective material during printing. The ‘flagged’ material is removed during later stages of slitting and rewinding. The defec-tive material was detected on the customer’s packing line and lead to the scrapping of some packed pharmaceutical product. The supplier’s printers and the operators responsible for removal of the ‘flagged’ material were given refresher training to ensure all defective material is removed, up to and around the ‘flagged’ point in the reel.
Q Focus – Production/in-process controlPlastic tablet bottles were manufactured with a wall thickness that was below specification. This was caused by poor appreciation of the cus-tomer’s requirements, inadequate in-process monitoring and an inability to segregate product that was out of specification. Packed pharmaceutical product had to be quarantined until all aspects of the problem had been assessed by the pharmaceutical company’s QA Department. The issue was resolved through improved specification regarding the criticality of certain dimensions, increased in-process measuring regime and enhanced segre-gation procedures.
R Focus – Production completion & line clearanceOn a reel of label/leaflet combinations, six ‘rogue’ components (of similar appearance) were found at intermittent points in the reel, by the pharmaceu-tical customer. This was despite a scanning operation being in place at the supplier. The root cause was human error involving inadequate line clear-ance and replacing incorrect labels back onto the web during rewinding/scanning. Although the rule existed which forbade this practice, the operator had recently been transferred from a department supplying non-pharmaceutical customers where this rule was not in place. The supplier commenced a fundamental review of procedures and operator retraining.
CASE STUDIES FOR SCHEMATIC 6 – CHECKING & FINAL RELEASE
S Focus – End product QC testing An out of specification result on a test machine record was transcribed incorrectly as satisfactory on the summary sheet, causing an incorrect

30
release decision of a batch of syringes. Although, in this case no harm was caused, it highlighted a risk area, should the error have occurred on another safety related function of the syringe. Refresher training was delivered to the operator concerned.
T Focus – Review of Batch documents & QC testingCarton barcode scanner was switched off during the night shift without QA authorization and not noticed upon review of documentation. This meant that the product had been made without the safeguards required by, and contractually agreed with the customer. The product was therefore inadequately checked for the absence of rogue components. The affected order was rejected and scrapped. A detailed audit was performed by the pharmaceutical company and numerous significant procedural enhance-ments were made.
U Focus – Create Certificate of Test or ConformanceIrradiated syringes were released for use despite radiation dose figures being omitted from the certificate of test. In this case the dosage was actually satisfactory, however the omission / error had the potential to threaten the sterility of the syringes thus endangering the patient, and to compromise the pharmaceutical sterile filling facility. The certificates are now double checked before release from the irradiation contractor.
V Focus – Check box labels against batch informationA box of shampoo closures was delivered amongst a consignment of tablet bottle closures, caused by box labelling error & poor segregation practices. This resulted in admixture on the filling line at the pharmaceutical company, line downtime, and some lost product. The supplier had been audited on numerous occasions but lack of commitment to eliminate such problems ultimately lead to a change of supplier for the component.
W Focus – Check box labels against batch informationA keying-in error of leaflet product identity code to outer box label printer caused rejection upon delivery to customer. Apart from the disruption caused by the rejection, there was a risk that the material could have been received into stock as a different item, i.e. admixture. The preventive action eventually implemented was to link the box label printer to the mainframe

31
computer thus removing the necessity to manually key the data into a standalone label printer.
CASE STUDIES FOR SCHEMATIC 7 – DISTRIBUTION
X Focus – Order preparation (picking & transit packaging)An admixture of boxes of labels was found on receipt by the customer caused by a picking error made due to poor warehouse lighting and lack of a double check. In another incident an admixture of batch of cartons occurred, caused by a picking error due to poor batch segregation in the warehouse. Both batch defectives were found during the customer’s receipt checking, but they caused disruption to the product scheduling programmes because of the need to return the material to the supplier for sorting/replacement. The corrective and preventive actions, in both cases, involved a fundamental review of the conditions (and checks made) in the warehouse and implementation of improved lighting and segregation mea-sures.
Y Focus – Despatch area/goods check & documentationDespatch note details accompanying the delivery differed from physical quantity delivered. This resulted in delays in product release and schedul-ing disruption while the supplier was contacted. The supplier’s despatch checking procedures were reviewed, no fundamental weaknesses were found. In this case, a one-off error had occurred and the issue was dis-cussed with the individual concerned.
Z Focus – Transport to the customerGoods were damaged in transit due to movement of the load and inade-quate control while the vehicle was being loaded. In another incident, goods were damaged by water penetration due to a poorly closed and maintained “curtain” of a flexible sided vehicle. Both examples resulted in the need for product inspection at the customer, and the return of damaged material to the supplier leaving too few components to meet requirements. In the first case, the checks made on loading were increased and in the second exam-ple the company decided it was feasible to switch to hard sided vehicles to eliminate the risk of water penetration.

32
Using the Case Studies - Guidance for Trainers
Introduction
The case studies that have been described here can be used as valuable aides to training programmes, for use either by a supplier or a pharmaceutical company. It is recommended that you select one or more of the case studies that are appro-priate to your business and design an activity that will encourage your trainees to discuss the particular subject you would like them to understand. An example of a training exercise protocol is provided below based on a case study selected from Schematic No. 3. Developing a short set of questions similar to those illustrated below should help improve understanding. It will probably be beneficial if such Q & A sets were developed jointly after discussion between pharmaceutical customers and their suppliers.
It will certainly be helpful to provide explanations and visual aides relevant to the case study. For example in the example illustrated below it would be helpful if: i) For a laminate that you use or manufacture explain which illness(es) the tablets,
that are packed within it, are used to treat.ii) A copy of an analogous page of a Product Authorisation (Licence) was shown
and the legal basis of such licences explained.iii) The function of PVDC in the laminate was explained.
Training Exercise
Case study No. C Title: Mix-up in the weight of PVDC on a PVC based blister film In this case study, the wrong material was ordered from the factory. Nomally, orders were placed for 40 micron PVDC coated PVC (for tablet blister packing) - and etc.
Training objectivesDiscuss the case study in groups and decide your answers to the following ques-tions:Q 1. Which requirement(s) of PS 9000 do you consider had not been effectively
implemented?
State the clause numbers and your reasons.

33
Q 2. Why do you think that the thickness of the PVDC could affect the stability of the tablets that were to be packed in the laminate?
Q 3. If this mistake had not been detected what do you think the implications
might have been to: i) The manufacturer of the laminate film? ii) The pharmaceutical customer?

34

35
The Institute of Quality Assurance
Pharmaceutical Quality Group
PS 9004
A Guide to the GMP requirements
of PS 9000:2001 Pharmaceutical packaging materials
PART 2 - GUIDANCE ON PS 9000 GMP CLAUSES
PS 9000 GMP related text is reproduced in blue

36

37
Gen
eral
syn
op
sis
- C
laus
e 4
Qua
lity
man
agem
ent
syst
em
A Q
MS
pro
vid
es a
bas
is fo
r co
ntin
uous
imp
rove
men
t of t
he o
rgan
izat
ion’
s ef
fect
iven
ess
and
effi
cien
cy, w
hils
t stil
l con
sid
erin
g th
e va
rious
nee
ds
of a
ll st
akeh
old
ers
(e.g
. cu
stom
ers,
sup
plie
rs,
inve
stor
s, e
mp
loye
es).
Cla
use
4.1
Gen
eral
req
uire
men
ts
Det
aile
d c
onsi
der
atio
n is
req
uire
d o
f th
e ne
eds
of s
take
hold
ers,
the
pro
cess
es t
o m
eet
thos
e ne
eds
and
to
mea
sure
per
form
ance
.
Ther
e ar
e no
‘ex
tra’
PS
900
0 G
MP
s w
ithin
thi
s cl
ause
. Th
ere
are
imp
orta
nt G
MP
prin
cip
les
in t
his
area
, b
ut t
hey
are
adeq
uate
ly
exp
ress
ed in
ISO
900
1 an
d IS
O 9
004.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• Id
entif
ied
pro
cess
es
that
mee
t or
gani
zatio
n st
rate
gic
obje
ctiv
es
• C
usto
mer
req
uire
men
ts•
Reg
ulat
ory
req
uire
men
ts
Pro
cess
es w
hich
:•
def
ine,
imp
lem
ent
and
mai
ntai
n Q
MS
p
roce
sses
(inc
lud
ing
man
agem
ent
and
op
erat
iona
l act
iviti
es)
• d
eter
min
e p
erso
nnel
req
uire
d t
o ca
rry
out
pro
cess
es a
nd t
rain
ing
need
s•
pro
vid
e p
rem
ises
and
eq
uip
men
t fo
r th
e p
roce
sses
• d
efin
e an
d im
ple
men
t co
ntro
ls t
o m
easu
re
per
form
ance
of
QM
S p
roce
sses
incl
udin
g ou
tsou
rcin
g
• W
ritte
n p
roce
dur
es
esse
ntia
l to
Q
ualit
y S
yste
m•
Org
aniz
atio
n st
ruct
ure
that
sup
por
ts Q
MS
• A
deq
uate
pre
mis
es
and
eq
uip
men
t•
Dat
a fr
om p
roce
sses
an
d m
easu
rem
ent
syst
ems
Sch
emat
ic 1
38
Cla
use
4.2
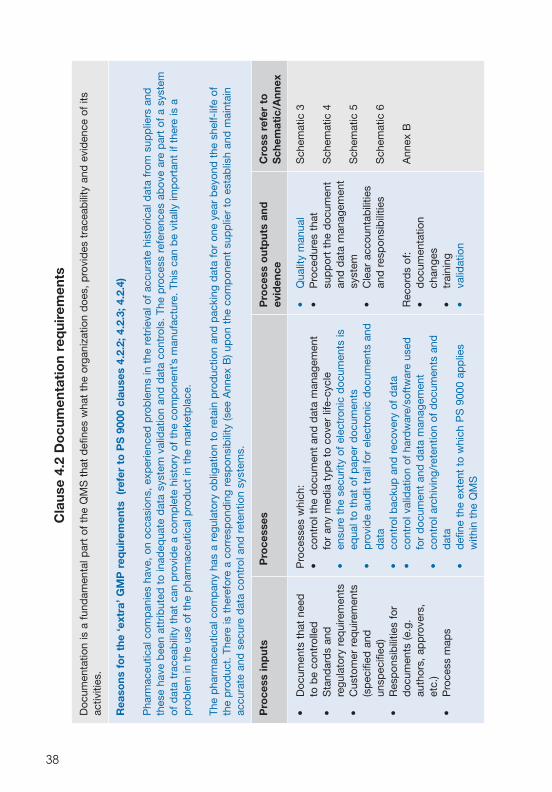
Do
cum
enta
tio
n re
qui
rem
ents
Doc
umen
tatio
n is
a f
und
amen
tal p
art
of t
he Q
MS
tha
t d
efin
es w
hat
the
orga
niza
tion
doe
s, p
rovi
des
tra
ceab
ility
and
evi
den
ce o
f its
ac
tiviti
es.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
(r
efer
to
PS
900
0 cl
ause
s 4.
2.2;
4.2
.3; 4
.2.4
)
Pha
rmac
eutic
al c
omp
anie
s ha
ve,
on o
ccas
ions
, ex
per
ienc
ed p
rob
lem
s in
the
ret
rieva
l of
accu
rate
his
toric
al d
ata
from
sup
plie
rs a
nd
thes
e ha
ve b
een
attr
ibut
ed t
o in
adeq
uate
dat
a sy
stem
val
idat
ion
and
dat
a co
ntro
ls.
The
pro
cess
ref
eren
ces
abov
e ar
e p
art
of a
sys
tem
of
dat
a tr
acea
bili
ty t
hat
can
pro
vid
e a
com
ple
te h
isto
ry o
f th
e co
mp
onen
t’s m
anuf
actu
re.
This
can
be
vita
lly im
por
tant
if t
here
is a
p
rob
lem
in t
he u
se o
f th
e p
harm
aceu
tical
pro
duc
t in
the
mar
ketp
lace
.
The
pha
rmac
eutic
al c
omp
any
has
a re
gula
tory
ob
ligat
ion
to r
etai
n p
rod
uctio
n an
d p
acki
ng d
ata
for
one
year
bey
ond
the
she
lf-lif
e of
th
e p
rod
uct.
The
re is
the
refo
re a
cor
resp
ond
ing
resp
onsi
bili
ty (s
ee A
nnex
B) u
pon
the
com
pon
ent
sup
plie
r to
est
ablis
h an
d m
aint
ain
accu
rate
and
sec
ure
dat
a co
ntro
l and
ret
entio
n sy
stem
s.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• D
ocum
ents
tha
t ne
ed
to b
e co
ntro
lled
• S
tand
ard
s an
d
regu
lato
ry r
equi
rem
ents
• C
usto
mer
req
uire
men
ts
(sp
ecifi
ed a
nd
unsp
ecifi
ed)
• R
esp
onsi
bili
ties
for
doc
umen
ts (e
.g.
auth
ors,
ap
pro
vers
, et
c.)
• P
roce
ss m
aps
Pro
cess
es w
hich
: •
cont
rol t
he d
ocum
ent
and
dat
a m
anag
emen
t fo
r an
y m
edia
typ
e to
cov
er li
fe-c
ycle
• en
sure
the
sec
urity
of
elec
tron
ic d
ocum
ents
is
equa
l to
that
of
pap
er d
ocum
ents
• p
rovi
de
aud
it tr
ail f
or e
lect
roni
c d
ocum
ents
and
d
ata
• co
ntro
l bac
kup
and
rec
over
y of
dat
a•
cont
rol v
alid
atio
n of
har
dw
are/
soft
war
e us
ed
for
doc
umen
t an
d d
ata
man
agem
ent
• co
ntro
l arc
hivi
ng/r
eten
tion
of d
ocum
ents
and
d
ata
• d
efin
e th
e ex
tent
to
whi
ch P
S 9
000
app
lies
with
in t
he Q
MS
• Q
ualit
y m
anua
l•
Pro
ced
ures
tha
t su
pp
ort
the
doc
umen
t an
d d
ata
man
agem
ent
syst
em•
Cle
ar a
ccou
ntab
ilitie
s an
d r
esp
onsi
bili
ties
Rec
ord
s of
:•
doc
umen
tatio
n
chan
ges
• tr
aini
ng•
valid
atio
n
Sch
emat
ic 3
Sch
emat
ic 4
Sch
emat
ic 5
Sch
emat
ic 6
Ann
ex B

39
Gen
eral
Syn
op
sis
- C
laus
e 5
Man
agem
ent
resp
ons
ibili
ty
Man
agem
ent’s
res
pon
sib
ility
to
ensu
re a
n ef
fect
ive
QM
S w
hich
rec
ogni
ses
the
need
s of
the
org
aniz
atio
n, c
usto
mer
s an
d
othe
r ke
y st
akeh
old
ers.
Cla
use
5.1
Man
agem
ent
com
mit
men
t
Top
man
agem
ent
def
ines
the
org
aniz
atio
n’s
stra
tegy
and
is r
esp
onsi
ble
for
dem
onst
ratin
g co
mm
itmen
t to
the
QM
S.
Con
tinua
l im
pro
vem
ent
of t
he s
yste
m a
nd it
s p
roce
sses
are
nec
essa
ry t
o en
sure
cus
tom
er a
nd r
egul
ator
y ne
eds
are
fully
sat
isfie
d.
Ther
e ar
e no
‘ex
tra’
PS
900
0 G
MP
’s w
ithin
thi
s cl
ause
. Th
ere
are
imp
orta
nt G
MP
prin
cip
les
in t
his
area
, b
ut t
hey
are
adeq
uate
ly
exp
ress
ed in
ISO
900
1 an
d IS
O 9
004.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
Sch
emat
ic/A
nnex
• C
omp
any
stra
tegy
/p
urp
ose
• A
deq
uate
res
ourc
es t
o m
eet
req
uire
men
ts
Pro
cess
es w
hich
:•
esta
blis
h q
ualit
y p
olic
y •
esta
blis
h q
ualit
y ob
ject
ives
• re
qui
re a
Man
agem
ent
Rev
iew
as
a m
eans
of
cont
inua
l im
pro
vem
ent
• en
sure
s th
e co
mm
unic
atio
n w
ith s
taff
to h
elp
th
em u
nder
stan
d a
nd c
ontr
ibut
e to
QM
S
imp
lem
enta
tion
(e.g
. co
ntin
uous
imp
rove
men
t ob
ject
ives
, cu
stom
er s
atis
fact
ion
targ
ets,
etc
.)
• E
vid
ence
of
staf
f un
der
stan
din
g an
d
com
mitm
ent
• C
usto
mer
sat
isfa
ctio
n m
easu
res
Cle
arly
doc
umen
ted
:•
qua
lity
pol
icy
• q
ualit
y ob
ject
ives
•
imp
rove
men
t p
lans
Sch
emat
ic 1

40
Cla
use
5.2
Cus
tom
er f
ocu
s
A k
ey r
equi
rem
ent
to p
rovi
de
suita
ble
pre
mis
es,
peo
ple
, an
d p
roce
sses
, in
ord
er t
o en
hanc
e cu
stom
er s
atis
fact
ion.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
Pha
rmac
eutic
al In
dus
try
exp
erie
nce
show
s th
at f
rom
the
pro
duc
t d
evel
opm
ent
stag
e on
war
ds,
com
plia
nt p
harm
aceu
tical
qua
lity
req
uire
s fa
cilit
ies
and
sta
ff to
be
‘fit
for
pur
pos
e’,
i.e.
‘sui
tab
le f
acili
ties
and
com
pet
ent
staf
f’,
and
tha
t th
is G
MP
req
uire
men
t is
eq
ually
ap
plic
able
to
the
sup
plie
r of
pha
rmac
eutic
al p
acka
ging
com
pon
ents
.
Pro
duc
t se
curit
y an
d t
he a
void
ance
of
cros
s co
ntam
inat
ion
und
erp
ins
all p
harm
aceu
tical
qua
lity,
sin
ce t
hese
are
bas
ic is
sues
tha
t ca
n le
ad t
o p
eop
le t
akin
g th
e w
rong
med
icin
e, d
amag
ing
the
pha
rmac
eutic
al c
omp
any’
s b
usin
ess,
and
pub
lic c
onfid
ence
in it
. (S
ee a
lso
Ann
ex A
, ‘C
ount
erfe
it’ t
hat
cove
rs in
det
ail t
he n
eed
s fo
r co
ntro
ls in
are
as in
volv
ing
dis
pos
al o
f ar
twor
k, p
roce
ss d
efec
tives
and
was
te
mat
eria
ls,
etc.
).
Beg
inni
ng w
ith it
s re
gula
tory
insp
ecto
rs,
the
who
le p
harm
aceu
tical
ind
ustr
y is
com
plia
nce
driv
en,
usin
g au
dits
as
a m
easu
re t
hat
GM
P p
ract
ices
, as
par
t of
a q
ualit
y sy
stem
, ar
e in
pla
ce.
Cen
tral
to
this
aud
it fo
cus,
is t
he ‘
Rul
es a
nd G
uid
ance
for
Pha
rmac
eutic
al
Man
ufac
ture
rs a
nd D
istr
ibut
ors’
. C
hap
ter
9, In
tern
al a
udits
, re
qui
res
self
insp
ectio
n b
y th
e p
harm
aceu
tical
man
ufac
ture
r an
d A
nnex
8.5
, re
qui
res
that
sam
plin
g p
lans
for
the
ass
essm
ent
of p
acka
ging
mat
eria
l del
iver
ies
shou
ld,
thro
ugh
aud
its,
take
into
acc
ount
kno
wle
dge
of
the
sup
plie
r’s q
ualit
y as
sura
nce
syst
em.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
evi
den
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• C
usto
mer
inp
ut/
req
uire
men
tsP
roce
sses
whi
ch:
• co
nsid
er c
usto
mer
req
uire
men
ts
in o
rder
to
del
iver
a s
ecur
ely
man
ufac
ture
d p
rod
uct
to s
pec
ifica
tion
• p
erm
it cu
stom
er a
udits
• en
sure
sec
ure
dis
pos
al o
f al
l prin
t re
late
d m
ater
ial
• S
uita
ble
fac
ilitie
s•
Evi
den
ce o
f G
MP
tra
inin
g an
d
task
com
pet
ency
• Im
pro
vem
ent
pla
ns f
rom
cu
stom
er a
udits
•
Phy
sica
l con
trol
and
rec
ord
s of
sec
ure
dis
pos
al o
f sp
ecifi
ed m
ater
ials
Sch
emat
ic 2
Sch
emat
ic 3
Ann
ex A
Ann
ex B

41
Cla
use
5.3
Qua
lity
po
licy
The
qua
lity
pol
icy
is a
top
-lev
el d
ocum
ent,
key
to
com
mun
icat
ing
and
lead
ing
cont
inuo
us im
pro
vem
ent
to s
atis
fy t
he n
eed
s of
the
or
gani
zatio
n, c
usto
mer
s an
d o
ther
sta
keho
lder
s.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
All
ISO
900
1 ce
rtifi
cate
d c
omp
anie
s ar
e re
qui
red
to
have
a ‘
qua
lity
pol
icy’
. Th
e av
oid
ance
of
cont
amin
atio
n an
d m
aint
aini
ng a
co
ntro
lled
env
ironm
ent
in o
rder
to
ensu
re p
rod
uct
secu
rity
and
inte
grity
is o
f su
ch im
por
tanc
e to
the
pha
rmac
eutic
al in
dus
try
that
the
se
two
req
uire
men
ts h
ave
to b
e in
clud
ed in
the
sup
plie
r’s q
ualit
y p
olic
y.
Whi
le P
S 9
000:
2001
is p
rimar
ily f
or s
upp
liers
of
pha
rmac
eutic
al p
acka
ging
mat
eria
ls,
som
e or
gani
zatio
ns s
upp
ly d
iffer
ent
ind
ustr
y se
ctor
s (e
.g.
food
, ag
roch
emic
al,
cosm
etic
s).
Ther
efor
e, f
or t
he p
urp
oses
of
clar
ity,
the
exte
nt t
o w
hich
thi
s ap
plic
atio
n st
and
ard
is
emp
loye
d w
ithin
the
org
aniz
atio
n ne
eds
to b
e d
ocum
ente
d.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• O
rgan
izat
ion
stra
tegy
/p
urp
ose
• C
usto
mer
nee
ds
incl
udin
g p
rod
uct
secu
rity
req
uire
men
ts
and
pre
vent
ion
of
cros
s-co
ntam
inat
ion
• E
xten
t to
whi
ch P
S
9000
is u
sed
with
in t
he
orga
niza
tion
Pro
cess
es w
hich
:•
def
ine
qua
lity
pol
icy
• re
view
pol
icy
to e
nsur
e it
mee
ts u
ser
need
s•
revi
ew q
ualit
y ob
ject
ives
• co
mm
unic
ate
qua
lity
pol
icy,
incl
udin
g co
ntam
inat
ion
cont
rol,
to in
tern
al s
taff
and
ex
tern
al p
eop
le,
as a
pp
rop
riate
• U
p-t
o-d
ate
qua
lity
pol
icy
Rec
ord
s of
:•
awar
enes
s/co
mm
unic
atio
n ac
tiviti
es•
pro
duc
t se
curit
y an
d
envi
ronm
ent
cont
rols
• im
ple
men
tatio
n
Sch
emat
ic 1

42
Cla
use
5.4
Pla
nnin
g
Str
ateg
ic b
usin
ess
pla
nnin
g is
ess
entia
l to
ensu
re t
hat
the
orga
niza
tion
mov
es f
orw
ard
s in
a w
ay t
hat
satis
fies
the
need
s of
all
stak
ehol
der
s. T
his
sect
ion
cove
rs t
he f
orm
al p
lann
ing
pro
cess
es n
eed
ed t
o re
alis
e or
gani
zatio
nal s
trat
egy
thro
ugh
gene
ratio
n of
ob
ject
ives
with
in t
he q
ualit
y m
anag
emen
t sy
stem
.
Ther
e ar
e no
‘ex
tra’
PS
900
0 G
MP
s w
ithin
thi
s cl
ause
. Th
ere
are
imp
orta
nt G
MP
prin
cip
les
in t
his
area
, b
ut t
hey
are
adeq
uate
ly
exp
ress
ed in
ISO
900
1 an
d IS
O 9
004.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• S
hort
and
long
ter
m
orga
niza
tion
stra
tegy
/go
als
• R
egul
ator
y re
qui
rem
ents
• N
eed
s of
the
or
gani
zatio
n an
d
mar
kets
• M
anag
emen
t re
view
fin
din
gs•
Cur
rent
pro
duc
t an
d
pro
cess
per
form
ance
• R
esou
rce
info
rmat
ion
• R
esul
ts f
rom
sel
f-as
sess
men
ts a
nd o
ther
au
dits
• In
dus
try
info
rmat
ion/
ben
chm
arki
ng
Pro
cess
es w
hich
:•
pla
n th
e im
ple
men
tatio
n, a
nd c
ontin
ual
imp
rove
men
t of
the
QM
S•
pro
vid
e a
fram
ewor
k fo
r th
e se
ttin
g of
m
easu
rab
le q
ualit
y ob
ject
ives
, w
hich
sup
por
t th
e q
ualit
y p
olic
y an
d s
trat
egic
goa
ls
• P
erfo
rman
ce
mea
sure
men
t d
ata
Def
ined
and
doc
umen
ted
:•
skill
s an
d k
now
led
ge
• re
spon
sib
ilitie
s an
d a
utho
rity
for
imp
lem
enta
tion
of
imp
rove
men
t p
lans
Qua
lity
obje
ctiv
es:
• at
org
aniz
atio
nal
leve
ls (e
.g.
man
ager
s,
sup
ervi
sors
, op
erat
ives
)•
for
func
tiona
l are
as
(e.g
. p
rod
uctio
n,
adm
inis
trat
ion)
Sch
emat
ic 1
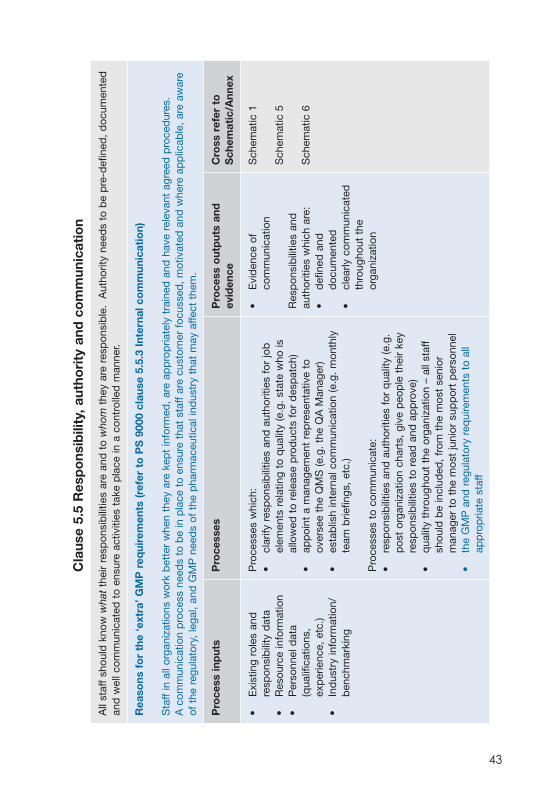
43
Cla
use
5.5
Res
po
nsib
ility
, aut
hori
ty a
nd c
om
mun
icat
ion
All
staf
f sh
ould
kno
w w
hat
thei
r re
spon
sib
ilitie
s ar
e an
d t
o w
hom
the
y ar
e re
spon
sib
le.
Aut
horit
y ne
eds
to b
e p
re-d
efin
ed,
doc
umen
ted
an
d w
ell c
omm
unic
ated
to
ensu
re a
ctiv
ities
tak
e p
lace
in a
con
trol
led
man
ner.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
(re
fer
to P
S 9
000
clau
se 5
.5.3
Int
erna
l co
mm
unic
atio
n)
Sta
ff in
all
orga
niza
tions
wor
k b
ette
r w
hen
they
are
kep
t in
form
ed,
are
app
rop
riate
ly t
rain
ed a
nd h
ave
rele
vant
agr
eed
pro
ced
ures
.A
com
mun
icat
ion
pro
cess
nee
ds
to b
e in
pla
ce t
o en
sure
tha
t st
aff
are
cust
omer
foc
usse
d,
mot
ivat
ed a
nd w
here
ap
plic
able
, ar
e aw
are
of t
he r
egul
ator
y, le
gal,
and
GM
P n
eed
s of
the
pha
rmac
eutic
al in
dus
try
that
may
affe
ct t
hem
.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• E
xist
ing
role
s an
d
resp
onsi
bili
ty d
ata
• R
esou
rce
info
rmat
ion
• P
erso
nnel
dat
a (q
ualif
icat
ions
, ex
per
ienc
e, e
tc.)
• In
dus
try
info
rmat
ion/
ben
chm
arki
ng
Pro
cess
es w
hich
:•
clar
ify r
esp
onsi
bili
ties
and
aut
horit
ies
for
job
el
emen
ts r
elat
ing
to q
ualit
y (e
.g.
stat
e w
ho is
al
low
ed t
o re
leas
e p
rod
ucts
for
des
pat
ch)
• ap
poi
nt a
man
agem
ent
rep
rese
ntat
ive
to
over
see
the
QM
S (e
.g.
the
QA
Man
ager
)•
esta
blis
h in
tern
al c
omm
unic
atio
n (e
.g.
mon
thly
te
am b
riefin
gs,
etc.
)
Pro
cess
es t
o co
mm
unic
ate:
• re
spon
sib
ilitie
s an
d a
utho
ritie
s fo
r q
ualit
y (e
.g.
pos
t or
gani
zatio
n ch
arts
, gi
ve p
eop
le t
heir
key
resp
onsi
bili
ties
to r
ead
and
ap
pro
ve)
• q
ualit
y th
roug
hout
the
org
aniz
atio
n –
all s
taff
shou
ld b
e in
clud
ed,
from
the
mos
t se
nior
m
anag
er t
o th
e m
ost
juni
or s
upp
ort
per
sonn
el•
the
GM
P a
nd r
egul
ator
y re
qui
rem
ents
to
all
app
rop
riate
sta
ff
• E
vid
ence
of
com
mun
icat
ion
Res
pon
sib
ilitie
s an
d
auth
oriti
es w
hich
are
:•
def
ined
and
d
ocum
ente
d•
clea
rly c
omm
unic
ated
th
roug
hout
the
or
gani
zatio
n
Sch
emat
ic 1
Sch
emat
ic 5
Sch
emat
ic 6

44
Cla
use
5.6
Man
agem
ent
revi
ew
Sen
ior
man
agem
ent
mus
t p
erio
dic
ally
rev
iew
the
QM
S t
o en
sure
it r
emai
ns e
ffect
ive
and
to
asse
ss im
pro
vem
ent
opp
ortu
nitie
s.
This
sho
uld
be
don
e in
the
con
text
of
imp
rovi
ng t
he p
erfo
rman
ce o
f th
e w
hole
org
aniz
atio
n.
Ther
e ar
e no
‘ex
tra’
PS
900
0 G
MP
s w
ithin
thi
s cl
ause
. Th
ere
are
imp
orta
nt G
MP
prin
cip
les
in t
his
area
, b
ut t
hey
are
adeq
uate
ly
exp
ress
ed in
ISO
900
1 an
d IS
O 9
004.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• Q
ualit
y p
olic
y an
d
obje
ctiv
es•
Qua
lity
aud
it re
sults
• P
rod
uct
qua
lity
info
rmat
ion
and
co
nfor
mity
dat
a•
Op
por
tuni
ties
to
imp
rove
• Fe
edb
ack
from
cu
stom
ers
• P
roce
ss p
erfo
rman
ce
info
rmat
ion
• C
orre
ctiv
e an
d
pre
vent
ive
actio
ns•
Cha
nges
tha
t m
ight
af
fect
the
QM
S•
Pre
viou
s Q
MS
rev
iew
s
Pro
cess
es w
hich
:•
revi
ew a
nd e
valu
ate
the
per
form
ance
of
the
QM
S•
asse
ss w
heth
er t
he Q
MS
req
uire
s im
pro
vem
ent
• ev
alua
te b
eyon
d t
he Q
MS
itse
lf, t
o th
at o
f th
e w
hole
org
aniz
atio
n –
its a
ctiv
ities
, p
erfo
rman
ce
and
effi
cien
cy
• R
evie
wed
qua
lity
pol
icy
and
ob
ject
ives
• In
form
atio
n fo
r st
rate
gic/
orga
niza
tiona
l d
ecis
ion-
mak
ing
Act
ions
to:
•
com
mun
icat
e fin
din
gs
of r
evie
w•
imp
rove
the
QM
S•
imp
rove
pro
cess
es•
imp
rove
pro
duc
ts•
add
ress
res
ourc
e ne
eds
Sch
emat
ic 1

45
Gen
eral
Syn
op
sis
- C
laus
e 6
Res
our
ce m
anag
emen
t
The
pro
visi
on o
f al
l res
ourc
es, i
nclu
din
g p
eop
le, i
nfra
stru
ctur
e, w
ork
envi
ronm
ent
and
info
rmat
ion,
nee
ded
for
op
erat
ion
and
im
pro
vem
ent
of t
he q
ualit
y sy
stem
to
assu
re c
omp
lianc
e w
ith t
he r
equi
rem
ents
of
cust
omer
s.
Cla
use
6.1
Pro
visi
on
of
reso
urce
s
It is
imp
orta
nt t
o ha
ve p
roce
sses
, w
hich
det
erm
ine
and
pro
vid
e th
e re
sour
ces
need
ed b
y th
e or
gani
zatio
n.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
Gai
ning
cer
tific
atio
n to
PS
900
0 re
qui
res
a fu
ll an
d t
horo
ugh
det
erm
inat
ion
of r
esou
rce
req
uire
men
ts.
This
will
ena
ble
the
im
ple
men
tatio
n of
a Q
MS
tha
t sa
tisfie
s th
e ex
pec
tatio
ns o
f th
e p
harm
aceu
tical
cus
tom
er,
with
the
ben
efits
of
cont
inuo
us im
pro
vem
ent
and
cus
tom
er a
udit
succ
esse
s.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
evi
den
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• Q
MS
pro
cess
re
qui
rem
ents
• P
roce
ss in
form
atio
n,
dat
a an
d r
equi
rem
ents
(p
eop
le,
pre
mis
es,
equi
pm
ent,
etc
.)•
Cur
rent
org
aniz
atio
n p
eop
le,
pre
mis
es,
equi
pm
ent
• P
S 9
000
spec
ific
add
ition
al r
equi
rem
ents
Pro
cess
es w
hich
:•
iden
tify
and
ap
poi
nt s
uffic
ient
per
sonn
el
• p
rovi
de
reso
urce
s ne
eded
to
imp
rove
cu
stom
er s
atis
fact
ion
• p
rovi
de
enou
gh r
esou
rces
to
fulfi
l the
re
qui
rem
ents
of
PS
900
0
• D
efin
ed o
rgan
izat
ion
stru
ctur
e an
d p
erso
nnel
and
re
spon
sib
ilitie
s•
Ad
equa
te p
erso
nnel
to
enab
le im
pro
vem
ent
pro
gram
mes
and
foc
us o
n cu
stom
er r
equi
rem
ents
• Q
uant
ified
ass
essm
ent
of
reso
urce
nee
ds
• A
utho
risat
ion
to p
rocu
re
reso
urce
Sch
emat
ic 1
Sch
emat
ic 2

46
Cla
use
6.2
Hum
an r
eso
urce
s
Det
erm
ine,
pro
vid
e an
d d
evel
op p
eop
le t
o su
pp
ort
the
orga
niza
tion’
s q
ualit
y p
olic
y an
d o
bje
ctiv
es
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
(re
fer
to P
S 9
000
clau
se 6
.2.2
Co
mp
eten
ce, a
war
enes
s an
d t
rain
ing
)
GM
P is
fun
dam
enta
l to
effe
ctiv
e co
ntro
l of
pro
duc
t q
ualit
y an
d r
equi
res
that
GM
P t
rain
ing
be
pro
vid
ed o
n an
ong
oing
bas
is.
Pha
rmac
eutic
al c
usto
mer
s lo
ok f
or e
vid
ence
of
this
at
thei
r su
pp
liers
.
Cro
ss c
onta
min
atio
n in
pac
kagi
ng c
an le
ad t
o th
e p
atie
nt b
eing
giv
en t
he w
rong
med
icat
ion
with
fat
al c
onse
que
nces
. W
hen
item
s ar
e no
t m
anuf
actu
red
usi
ng c
ontr
olle
d s
yste
ms
the
chan
ce o
f a
mix
-up
or
prin
t er
ror
is s
igni
fican
tly g
reat
er,
and
the
ris
k ca
nnot
be
acce
pte
d b
y th
e p
harm
aceu
tical
ind
ustr
y.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• Id
entif
ied
bus
ines
s p
roce
sses
• A
vaila
ble
peo
ple
to
carr
y ou
t p
roce
sses
• R
equi
red
co
mp
eten
cies
/leve
l of
tra
inin
g fo
r Q
MS
p
roce
sses
• R
ecru
itmen
t re
qui
rem
ents
: ed
ucat
ion,
tra
inin
g,
skill
s an
d e
xper
ienc
e
Pro
cess
es w
hich
:•
iden
tify
trai
ning
and
aw
aren
ess
need
s (g
ap
anal
ysis
)•
del
iver
tra
inin
g an
d a
war
enes
s p
rogr
ams
• ev
alua
te e
ffect
iven
ess
of t
rain
ing
and
aw
aren
ess
• en
sure
tra
inin
g co
vers
GM
P, t
he r
isk
and
d
ange
r of
cro
ss-c
onta
min
atio
n an
d t
he
imp
orta
nce
of f
ollo
win
g p
roce
dur
es•
ensu
re p
erio
dic
ref
resh
er t
rain
ing
is p
rovi
ded
• D
efin
ed a
nd
doc
umen
ted
GM
P
trai
ning
sch
edul
e
Rec
ord
s of
:•
trai
ning
•
per
sonn
el in
form
atio
n•
curr
icul
a vi
taru
m
(CV
s)•
ind
ivid
ual
dev
elop
men
t p
lans
Sch
emat
ic 1
Sch
emat
ic 6

47
Cla
use
6.3
Infr
astr
uctu
re
Ap
pro
pria
te e
qui
pm
ent,
pre
mis
es,
serv
ices
and
env
ironm
ent
are
fund
amen
tal r
equi
rem
ents
of
GM
P.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
Pac
kagi
ng d
efec
ts,
incl
udin
g m
isla
bel
ling/
mix
-up
s, a
re t
he m
ajor
cau
se o
f ap
pro
xim
atel
y 25
% o
f d
efec
tive
med
icin
es a
nd b
atch
rec
alls
(s
ee A
nnex
B 3
). It
mus
t b
e em
pha
size
d t
hat
the
caus
es o
f th
is p
rob
lem
are
not
sol
ely
attr
ibut
able
to
the
pac
kagi
ng s
upp
ly in
dus
try
sinc
e al
l par
ts o
f th
e su
pp
ly c
hain
and
man
ufac
turin
g p
roce
ss c
an b
e im
plic
ated
. Th
is a
lso
incl
udes
the
dis
trib
utio
n fr
om t
he s
upp
lier
to
the
cust
omer
, d
urin
g us
e in
the
cus
tom
er’s
pro
cess
es a
nd a
lso
the
final
mar
ket
dis
trib
utio
n.
Whi
lst
the
pro
duc
t sh
ould
be
des
igne
d t
o m
inim
ise/
avoi
d c
ontr
ibut
ing
to t
he r
isk,
the
infr
astr
uctu
re o
f m
anuf
actu
ring
and
sup
por
t p
roce
sses
at
the
sup
plie
r m
ust
also
be
des
igne
d,
mai
ntai
ned
and
op
erat
ed in
com
plia
nce
with
the
prin
cip
les
of G
MP
to
assu
re p
rod
uct
secu
rity.
It is
bec
ause
of
the
cont
inui
ng h
igh
pro
por
tion
of m
arke
t re
calls
tha
t ar
e ca
used
by
def
ectiv
e p
acka
ging
tha
t p
harm
aceu
tical
sup
plie
r au
dits
will
alw
ays
asse
ss h
ow t
he in
fras
truc
ture
may
influ
ence
pro
duc
t co
ntam
inat
ion.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
evi
den
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• B
usin
ess
pro
cess
re
qui
rem
ents
for
p
rem
ises
, eq
uip
men
t,
utili
ties
and
oth
er
serv
ices
incl
udin
g ha
rdw
are
and
sof
twar
e•
Mai
nten
ance
man
uals
an
d s
ched
ules
• R
egul
ator
y re
qui
rem
ents
Pro
cess
es w
hich
:•
iden
tify,
pro
vid
e an
d m
aint
ain
pre
mis
es,
equi
pm
ent,
util
ities
and
ot
her
serv
ices
incl
udin
g ha
rdw
are
and
so
ftw
are
and
tra
nsp
ort
• en
sure
tha
t G
MP
req
uire
men
ts
are
inco
rpor
ated
to
avoi
d p
rod
uct
cont
amin
atio
n
• A
deq
uate
pre
mis
es,
faci
litie
s,
equi
pm
ent
and
ser
vice
s•
Inte
rnal
rev
iew
s an
d c
usto
mer
fe
edb
ack
• R
ecor
ds
of o
ngoi
ng
mai
nten
ance
and
rev
iew
• A
ctio
n p
lans
(sho
rt a
nd lo
ng
term
)•
Cro
ss-c
onta
min
atio
n av
oid
ance
Sch
emat
ic 2
Ann
ex A
:S
egre
gatio
n co
ntro
ls

48
Cla
use
6.4
Wo
rk e
nvir
onm
ent
The
wor
k en
viro
nmen
t is
crit
ical
to
ensu
ring
not
only
pro
duc
t co
nfor
mity
, b
ut a
lso
the
abili
ty a
nd d
esire
of
peo
ple
to
per
form
effe
ctiv
ely.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
Pre
vent
ing
cont
amin
atio
n of
med
icin
al p
rod
ucts
may
req
uire
tha
t th
e co
mp
onen
ts b
e m
anuf
actu
red
und
er p
artic
ular
sta
ndar
ds
of
clea
nlin
ess,
whi
ch c
ould
ulti
mat
ely
invo
lve
clea
nroo
ms
(i.e.
con
trol
led
env
ironm
ents
). Th
ese
enab
le t
he p
harm
aceu
tical
pro
duc
t an
d
pat
ient
to
be
pro
tect
ed.
This
may
ap
ply
eve
n w
hen
the
com
pon
ents
und
ergo
a c
lean
ing
pro
cess
at
the
pha
rmac
eutic
al m
anuf
actu
rer’s
si
te.
For
som
e p
rod
uct
cont
act
com
pon
ents
, m
icro
bia
l con
tam
inat
ion
leve
ls (t
he b
iob
urd
en) n
eed
to
be
kep
t at
the
low
est
leve
ls p
ossi
ble
. Th
is s
till a
pp
lies
even
whe
n th
e p
harm
aceu
tical
pro
cess
ing
invo
lves
a s
teril
izat
ion
oper
atio
n.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• C
ateg
orie
s/st
and
ard
s fo
r en
viro
nmen
tal
cond
ition
s•
Pro
duc
t/m
anuf
actu
ring
pro
cess
es•
Cus
tom
er r
equi
rem
ents
• In
form
atio
n fr
om
ongo
ing
envi
ronm
enta
l m
onito
ring
• P
eop
le n
eed
s/co
nsid
erat
ion
• R
egul
ator
y re
qui
rem
ents
Pro
cess
es w
hich
:•
iden
tify
nece
ssar
y w
ork
envi
ronm
ent
• id
entif
y fa
ctor
s ne
eded
to
ensu
re p
rod
ucts
m
eet
req
uire
men
ts•
man
age
and
mai
ntai
n w
ork
envi
ronm
ent
incl
udin
g cl
eani
ng,
valid
atio
n an
d p
roce
sses
• in
corp
orat
e ap
pro
pria
te ‘
clea
nroo
m’
cond
ition
s fo
r th
e ty
pe
of p
rod
uct
bei
ng m
ade
• A
pp
rop
riate
en
viro
nmen
t fo
r p
rod
uct
bei
ng m
ade
• R
ecor
ds
of
envi
ronm
enta
l m
onito
ring,
val
idat
ion
and
cle
anin
g
Sch
emat
ic 2
Sch
emat
ic 5
Ann
ex A
:R
isk
asse
ssm
ent

49
Gen
eral
Syn
op
sis
- C
laus
e 7
Pro
duc
t re
aliz
atio
n
Mak
ing
pro
duc
t in
volv
es m
any
inte
rrel
ated
pro
cess
es,
the
inp
uts
and
out
put
s of
whi
ch s
houl
d b
e an
alys
ed a
nd m
anag
ed.
Cla
use
7.1
Pla
nnin
g o
f p
rod
uct
real
izat
ion
Man
agem
ent
shou
ld p
lan
the
pro
cess
es n
eed
ed f
or p
rod
uct
real
izat
ion.
Ther
e ar
e no
‘ex
tra’
PS
900
0 G
MP
s w
ithin
thi
s cl
ause
. Th
ere
are
imp
orta
nt G
MP
prin
cip
les
in t
his
area
but
the
y ar
e ad
equa
tely
ex
pre
ssed
in IS
O 9
001
and
ISO
900
4.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• Q
ualit
y re
qui
rem
ents
fo
r th
e p
rod
uct
• P
rod
uct
spec
ifica
tions
• C
usto
mer
req
uire
men
ts•
Pro
duc
t re
qui
rem
ents
–
valid
atio
n, m
onito
ring,
te
st c
riter
ia.
• R
esou
rce
req
uire
men
ts
spec
ific
to t
he p
rod
uct
Pro
cess
es w
hich
:•
pla
n th
e p
rod
uctio
n p
roce
sses
• p
lan
the
sup
por
t p
roce
sses
• m
anag
e ch
ange
s•
cont
rol p
roce
ss/p
rod
uct
valid
atio
n
• Q
ualit
y p
lan
• C
lear
ly id
entif
ied
p
roce
sses
• A
n op
erat
ing
pla
n to
m
anag
e p
roce
sses
• Id
entif
ied
res
ourc
e re
qui
rem
ents
to
sup
por
t th
e op
erat
ion
and
mon
itorin
g of
p
roce
sses
• C
hang
e p
roce
dur
e an
d r
ecor
ds
• Va
lidat
ion
reco
rds
Sch
emat
ic 2
Sch
emat
ic 3
Sch
emat
ic 4
Ann
ex A
:C
hang
e co
ntro
l
Valid
atio
n (a
nd
Qua
lific
atio
n)

50
Cla
use
7.2
Cus
tom
er r
elat
ed p
roce
sses
The
orga
niza
tion
shou
ld h
ave
effe
ctiv
e p
roce
sses
for
com
mun
icat
ing
with
its
cust
omer
s an
d o
ther
inte
rest
ed p
artie
s.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
(re
fer
to P
S 9
000
clau
se 7
.2.3
Cus
tom
er c
om
mun
icat
ion)
The
pha
rmac
eutic
al in
dus
try
is h
eavi
ly r
egul
ated
by
regu
lato
ry a
utho
ritie
s, t
hrou
ghou
t th
e w
orld
. C
omp
anie
s ar
e re
qui
red
to
obta
in a
m
arke
ting
auth
oriz
atio
n (M
A)/
Pro
duc
t Li
cenc
e (P
L) t
o p
lace
a m
edic
inal
pro
duc
t on
the
mar
ket.
The
ap
plic
atio
n fo
r an
MA
/PL
mus
t b
e su
pp
orte
d b
y ex
tens
ive
dat
a in
clud
ing
info
rmat
ion
on t
he p
acka
ging
mat
eria
ls t
o b
e us
ed.
Com
pan
ies
are
not
at li
ber
ty t
o m
ake
any
chan
ges
to t
he p
rod
uct,
its
com
pon
ents
or
its m
etho
d o
f p
rod
uctio
n w
ithou
t ap
pro
val f
rom
the
reg
ulat
ory
auth
ority
. Th
e lic
ensi
ng o
f p
harm
aceu
tical
pro
duc
ts r
equi
res
that
the
pha
rmac
eutic
al c
omp
anie
s in
form
gov
ernm
ent
regu
lato
rs o
f vi
rtua
lly a
ll ch
ange
s. T
hey
are
not
at li
ber
ty t
o ch
ange
any
reg
iste
red
pro
duc
t d
etai
ls w
ithou
t go
ing
thro
ugh
an e
xten
sive
and
leng
thy
sub
mis
sion
pro
cess
.
One
par
ticul
ar r
easo
n fo
r th
is c
omm
unic
atio
n p
roce
ss is
tha
t ch
ange
s ca
n re
qui
re t
rial w
ork
(e.g
. st
abili
ty t
rial)
at t
he p
harm
aceu
tical
co
mp
any
or o
ther
act
ions
, su
ch a
s m
achi
ne a
ltera
tions
, w
ith im
plic
atio
ns f
or d
esig
n an
d p
lann
ing.
In o
rder
to
give
due
con
sid
erat
ion
to e
ach
pro
pos
ed c
hang
e, t
here
sho
uld
be
a cl
ear
agre
emen
t b
etw
een
the
pha
rmac
eutic
al c
usto
mer
and
org
aniz
atio
n of
wha
t co
mm
unic
atio
n is
req
uire
d f
or v
ario
us t
ypes
of
chan
ge.

51
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
Sch
emat
ic/A
nnex
• C
usto
mer
st
ated
re
qui
rem
ents
• E
xter
nal
mar
ket
dat
a•
Sta
tuto
ry a
nd
regu
lato
ry
req
uire
men
ts
Pro
cess
es w
hich
:•
enab
le c
omm
unic
atio
n w
ith c
usto
mer
s (a
bou
t p
rod
uct
info
rmat
ion,
sal
es e
nqui
ries,
com
pla
ints
, et
c.)
• re
view
the
cus
tom
er’s
ord
er,
pro
duc
t re
qui
rem
ents
b
efor
e p
rod
ucts
are
sup
plie
d (e
.g.
cont
ract
rev
iew
)•
req
uire
cha
nge
cont
rol m
easu
res
to b
e in
pla
ce t
o re
cord
and
mon
itor
chan
ges
to p
roce
sses
• re
view
rel
ated
info
rmat
ion
– m
arke
t re
sear
ch,
com
pet
itor
anal
ysis
, st
atut
ory
req
uire
men
ts•
spec
ifica
lly id
entif
y th
ose
typ
es o
f ch
ange
s w
hich
re
qui
re p
rior
app
rova
l by,
or
notif
icat
ion
to t
he
cust
omer
• D
efin
ed p
rod
uct
req
uire
men
ts•
Doc
umen
tatio
n d
enot
ing
cust
omer
ap
pro
val
• E
ffect
ive
dat
a ex
chan
ge
cove
ring
pro
duc
ts,
amen
dm
ents
, fe
edb
ack
and
com
pla
ints
• P
ossi
ble
inp
uts
to
man
agem
ent
pla
nnin
g p
roce
ss
(e.g
. re
sour
ce/
mac
hine
ry im
plic
atio
ns)
Rec
ord
s of
:•
cust
omer
co
mm
unic
atio
n (e
.g.
pro
duc
t sp
ecifi
catio
ns,
ord
er c
onfir
mat
ions
, co
mp
lain
t in
vest
igat
ions
, et
c.)
•
ord
er/p
rod
uct
revi
ew
Sch
emat
ic 2
Sch
emat
ic 3
Sch
emat
ic 4
Ann
ex A
:C
hang
e co
ntro
l

52
Cla
use
7.3
Des
ign
and
dev
elo
pm
ent
Pla
nnin
g an
d c
ontr
ollin
g d
esig
n an
d d
evel
opm
ent
activ
ities
are
fun
dam
enta
l to
mak
ing
pro
duc
t th
at f
ully
mee
ts t
he r
equi
rem
ents
of
cust
omer
s.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
(re
fer
to P
S 9
000
clau
ses
7.3.
1, 7
.3.2
, 7.3
.5, 7
.3.6
and
7.3
.7)
The
orga
niza
tion
ofte
n ha
s sp
ecia
list
know
led
ge r
egar
din
g p
rod
uct
des
ign
that
can
ass
ist
in e
valu
atin
g ris
ks t
o th
e en
d u
ser
or p
atie
nt.
This
wou
ld n
orm
ally
for
m p
art
of c
usto
mer
/org
aniz
atio
n co
mm
unic
atio
n at
the
pro
duc
t d
evel
opm
ent
stag
e. T
echn
ical
dat
a fr
om t
he
des
ign
and
dev
elop
men
t st
ages
of
a co
mp
onen
t m
ay b
e re
qui
red
as
par
t of
the
pha
rmac
eutic
al c
omp
any’
s su
bm
issi
on t
o re
gula
tory
au
thor
ities
whe
n ap
ply
ing
for
per
mis
sion
to
mar
ket
a m
edic
inal
pro
duc
t. T
his
is m
and
ator
y fo
r a
com
pon
ent
that
will
be
in p
hysi
cal
cont
act
with
the
med
icin
al p
rod
uct,
as
the
com
pos
ition
of
the
com
pon
ent
mat
eria
l can
influ
ence
the
med
icin
al p
rod
uct’s
com
pos
ition
an
d s
tab
ility
and
the
saf
ety
of t
he p
atie
nt.
The
avai
lab
ility
of
valid
atio
n d
ocum
ents
at
this
sta
ge g
ives
rec
ord
ed a
ssur
ance
tha
t th
e d
esig
n p
rod
uced
has
bee
n th
orou
ghly
tes
ted
an
d is
sui
tab
le f
or lo
ng-t
erm
pro
duc
tion.
Thi
s, in
con
junc
tion
with
the
tes
ting
and
ap
pro
val o
f sa
mp
les
by
the
cust
omer
, en
able
s th
e d
esig
n to
pro
gres
s in
to p
rod
uctio
n in
a f
orm
al c
ontr
olle
d m
anne
r.
Sim
ilar
cont
rols
are
exp
ecte
d w
hen
des
ign
and
dev
elop
men
t ac
tiviti
es a
re in
volv
ed in
mak
ing
mod
ifica
tions
to
the
pro
duc
t or
p
rod
uctio
n m
etho
ds.
It is
man
dat
ory
that
the
det
ails
of
the
med
icin
al p
rod
uct
lod
ged
with
reg
ulat
ory
auth
oriti
es a
re a
ccur
ate
and
m
aint
aine
d (‘
The
Rul
es a
nd G
uid
ance
for
Pha
rmac
eutic
al M
anuf
actu
rers
and
Dis
trib
utor
s 20
02,’
Art
icle
5 E
urop
ean
Com
mis
sion
D
irect
ives
200
3/94
/EC
and
91/
412/
EE
C).

53
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
Inte
rnal
inp
uts,
e.g
. •
com
pet
ence
re
qui
rem
ents
for
peo
ple
p
erfo
rmin
g d
esig
n an
d
dev
elop
men
t•
reco
rds
and
dat
a on
ex
istin
g p
roce
sses
and
p
rod
ucts
• ou
tput
s fr
om o
ther
p
roce
sses
Ext
erna
l inp
uts,
e.g
. •
cust
omer
/mar
ketp
lace
ne
eds
• us
er in
put
to
achi
eve
rob
ust
des
ign
and
d
evel
opm
ent
• st
atut
ory
req
uire
men
ts•
inte
rnat
iona
l sta
ndar
ds
• in
dus
try
cod
es o
f p
ract
ice
• ot
her
req
uire
men
ts
for
safe
op
erat
ion,
ha
ndlin
g, s
tora
ge•
envi
ronm
enta
l/dis
pos
al
cons
trai
nts
for
the
pro
duc
ts o
r p
roce
sses
Pro
cess
es w
hich
:•
cont
rol p
lann
ing
the
des
ign
and
d
evel
opm
ent
of p
rod
ucts
• co
ntro
l pro
duc
t d
esig
n an
d
dev
elop
men
t•
cont
rol p
rod
uctio
n p
roce
ss d
esig
n an
d
dev
elop
men
t•
eval
uate
pot
entia
l ris
ks o
f th
e p
rod
uct
des
ign
to c
usto
mer
and
ulti
mat
ely
the
pat
ient
• p
rovi
de
tech
nica
l/mat
eria
l dat
a to
th
e p
harm
aceu
tical
cus
tom
er a
nd/o
r m
edic
inal
reg
ulat
ory
auth
oriti
es a
nd t
o no
tify
of a
ny c
hang
es a
risin
g fr
om t
he
sup
ply
cha
in•
cove
r te
stin
g, r
etai
ning
and
gai
ning
cu
stom
er a
pp
rova
l of
sam
ple
s re
sulti
ng f
rom
new
pro
duc
t d
esig
ns•
pro
duc
e an
d p
rovi
de
valid
atio
n d
ocum
enta
tion
to t
he c
usto
mer
• co
ntro
l mod
ified
or
refu
rbis
hed
eq
uip
men
t an
d t
oolin
g an
d p
rod
uct
• R
epor
ts o
f va
lidat
ion/
test
s•
Info
rmat
ion
for
othe
r fu
nctio
ns
(e.g
. p
urch
asin
g)•
Dat
a to
ena
ble
ver
ifica
tion
of d
esig
n an
d d
evel
opm
ent
pro
cess
out
put
s to
the
p
roce
ss in
put
s•
Evi
den
ce o
f cu
stom
er
app
rova
l of
chan
ges
Iden
tific
atio
n of
:•
revi
ew,
verif
icat
ion
and
va
lidat
ion
app
rop
riate
to
each
d
esig
n an
d d
evel
opm
ent
stag
e•
resp
onsi
bili
ties
for
des
ign
and
d
evel
opm
ent
Doc
umen
ted
:•
chan
ge c
ontr
ol•
risk
asse
ssm
ents
• sp
ecifi
catio
ns f
or p
rod
uct,
p
roce
ss,
mat
eria
l, te
stin
g,
safe
ty c
hara
cter
istic
s•
trai
ning
req
uire
men
ts a
nd
reco
rds
Sch
emat
ic 2
Sch
emat
ic 3
Ann
ex A
:C
hang
e co
ntro
l
Ris
k as
sess
men
t
Valid
atio
n (a
nd
Qua
lific
atio
n)

54
Cla
use
7.4
Pur
chas
ing
Man
agem
ent
has
a re
spon
sib
ility
to
def
ine
and
imp
lem
ent
effe
ctiv
e p
urch
asin
g p
roce
sses
to
ensu
re p
urch
ased
mat
eria
ls m
eet
stat
ed
req
uire
men
ts.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P p
roce
sses
(re
fer
to P
S 9
000
clau
ses
7.4.
1 an
d 7
.4.3
)
The
pha
rmac
eutic
al c
omp
any
need
s to
kno
w w
here
all
par
ts o
f a
man
ufac
turin
g p
roce
ss a
re p
erfo
rmed
. Th
ese
det
ails
, in
clud
ing
sub
-co
ntra
cted
ele
men
ts,
may
be
‘reg
iste
red
’ as
par
t of
the
mar
ketin
g au
thor
isat
ion,
and
thu
s ne
ed c
usto
mer
ap
pro
val.
The
pha
rmac
eutic
al
com
pan
y m
ust
be
satis
fied
tha
t th
ere
are
no o
ther
ris
ks t
o p
rod
uct
secu
rity
or q
ualit
y in
volv
ed w
ith s
ubco
ntra
cted
par
ts o
f th
e m
anuf
actu
ring
pro
cess
. P
rod
uct
qua
lity
can
also
be
affe
cted
by
man
y co
ntra
cted
out
ser
vice
s (e
.g.
clea
ning
, m
aint
enan
ce,
calib
ratio
n),
and
evi
den
ce o
f go
od m
anag
emen
t an
d c
ontr
ol o
f th
ese
is e
xpec
ted
.
Qua
rant
ine
of p
urch
ased
pro
duc
t un
til a
pp
rove
d f
or u
se e
limin
ates
the
ris
k of
usi
ng u
nap
pro
ved
or
unsu
itab
le m
ater
ial.

55
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
evi
den
ceC
ross
ref
er t
o
Sch
emat
ic/A
nnex
• P
rod
uct
spec
ifica
tions
• C
ontr
acts
, w
arra
ntie
s,
pric
es•
Logi
stic
req
uire
men
ts•
Pro
duc
t id
entif
icat
ion
and
tra
ceab
ility
• S
upp
lier
per
form
ance
d
ata
– q
ualit
y, p
rice,
d
eliv
ery,
res
pon
se•
Aud
its o
f su
pp
liers
• C
omm
erci
al
dep
end
abili
ty o
f su
pp
liers
• S
upp
lier’s
cap
abili
ties
• C
ertif
icat
ion
• P
urch
asin
g p
roce
ss t
hat
satis
fies
the
orga
niza
tion’
s ne
eds
whi
lst
cons
ider
ing
cost
s an
d o
ther
ap
pro
pria
te p
erfo
rman
ce
mea
sure
s
Pro
cess
es w
hich
:•
cont
rol s
upp
liers
• en
able
ver
ifica
tion
of p
urch
ased
pro
duc
ts
Pro
cess
es w
hich
:•
gain
cus
tom
er a
pp
rova
l of
sub
cont
ract
ed
par
ts o
f th
e m
anuf
actu
ring
pro
cess
• co
ntro
l con
trac
ted
out
ser
vice
s th
at c
an
affe
ct p
rod
uct
qua
lity
• q
uara
ntin
e p
urch
ased
pro
duc
t un
til
app
rove
d f
or u
se
• C
o-or
din
ated
pur
chas
ing
info
rmat
ion,
e.g
. req
uire
men
ts
for
app
rova
l of
pro
duc
t,
sup
plie
r’s p
roce
dur
es,
pro
cess
es a
nd e
qui
pm
ent,
ot
her
per
sonn
el o
r q
ualit
y sy
stem
s re
qui
rem
ents
Rec
ord
s of
:•
insp
ectio
n ch
ecks
mad
e on
in
com
ing
item
s•
the
rele
ase
(for
acce
pta
ble
ite
ms)
or
reje
ctio
n (fo
r un
acce
pta
ble
item
s) o
f p
urch
ased
goo
ds
Evi
den
ce o
f:•
a su
pp
lier
app
rova
l sch
eme
in p
lace
(e.g
. an
ap
pro
ved
su
pp
lier
list
and
aud
it re
por
ts)
• th
e m
onito
ring
of
sub
cont
ract
ors
Sch
emat
ic 3
Sch
emat
ic 3
(a)
Sch
emat
ic 4
Ann
ex A
:C
hang
e co
ntro
l

56
Cla
use
7.5
Pro
duc
tio
n an
d s
ervi
ce p
rovi
sio
n
Pro
duc
tion
(or
serv
ice
pro
visi
on) w
here
key
ele
men
ts a
re t
he c
ontr
ol a
nd v
alid
atio
n of
pro
duc
tion
cond
ition
s, a
nd t
race
abili
ty o
f m
ater
ials
and
eq
uip
men
t us
ed.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
(r
efer
to
PS
900
0 cl
ause
s 7.
5.2,
7.5
.3, 7
.5.4
and
7.5
.5)
The
spec
ial n
eed
s of
pha
rmac
eutic
al p
acka
ging
mea
n th
at c
erta
in p
roce
sses
are
crit
ical
to
pro
duc
t q
ualit
y, s
afet
y, a
nd f
unct
ion.
The
se
‘crit
ical
’ p
roce
sses
req
uire
val
idat
ion
to b
e ce
rtai
n th
at t
hey
func
tion
relia
bly
and
con
sist
ently
. It
is im
por
tant
tha
t va
lidat
ion
is p
erfo
rmed
as
nec
essa
ry d
urin
g d
evel
opm
ent
and
on
star
t-up
in p
rod
uctio
n an
d a
lso
per
form
ed w
hen
ther
e ar
e ch
ange
s to
pro
duc
tion
pro
cess
es.
Iden
tific
atio
n an
d t
race
abili
ty is
ess
entia
l to
enab
le a
rec
onst
ruct
ion
of e
vent
s w
hen
inve
stig
atin
g a
qua
lity
pro
ble
m.
To b
e ab
le t
o in
vest
igat
e an
y p
rob
lem
with
a m
edic
ine
in t
he m
arke
tpla
ce,
the
pha
rmac
eutic
al c
omp
any
is o
blig
ed t
o re
tain
rec
ord
s fo
r (a
t le
ast)
a ye
ar b
eyon
d t
he li
fe o
f th
e m
edic
ines
it is
mak
ing.
The
re is
a c
orre
spon
din
g re
spon
sib
ility
on
the
par
t of
the
com
pon
ent
sup
plie
rs t
o b
e ab
le t
o su
pp
ort
any
such
inve
stig
atio
n th
roug
h its
ret
aine
d r
ecor
ds
of m
anuf
actu
re e
.g.
trac
eab
ility
of
mat
eria
ls,
equi
pm
ent
and
p
erso
nnel
invo
lved
in t
he c
omp
onen
ts m
anuf
actu
re (s
ee A
nnex
A).
The
pha
rmac
eutic
al c
omp
any
pla
ces
trus
t in
its
sup
plie
rs t
o p
rote
ct it
s p
rop
erty
. Th
e m
ain
cons
ider
atio
n is
pro
tect
ion
from
the
ft
and
sub
seq
uent
fra
udul
ent
use,
una
utho
rized
sup
ply
and
use
in c
ount
erfe
it m
edic
ines
. Th
is p
artic
ular
ly a
pp
lies
to a
rtw
ork
or d
ata
in
elec
tron
ic f
orm
due
to
its e
ase
of t
rans
mis
sion
.
Follo
win
g m
anuf
actu
re u
nder
con
trol
led
con
diti
ons,
the
con
tinui
ng p
rote
ctio
n of
the
pro
duc
t is
imp
orta
nt t
o p
reve
nt d
amag
e an
d
cont
amin
atio
n.
Acc
urat
e id
entif
icat
ion
of t
he f
inal
pro
duc
t is
vita
l in
ord
er t
o m
inim
ise
risk
of a
dm
ixtu
re b
oth
with
in t
he o
rgan
izat
ion
and
dur
ing
rece
ipt
at t
he p
harm
aceu
tical
com
pan
y. T
his
is a
lway
s im
por
tant
, b
ut p
artic
ular
ly s
o w
here
the
re a
re d
irect
‘su
pp
ly t
o lin
e’ a
rran
gem
ents
, w
ith
min
imal
goo
ds-
inw
ard
s ch
ecki
ng in
pla
ce a
t th
e p
harm
aceu
tical
site
.

57
Pro
cess
Inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• R
aw m
ater
ial b
atch
re
cord
s•
Sp
ecifi
catio
ns
– p
rod
uct,
mat
eria
ls•
Man
ufac
turin
g m
etho
d/p
roce
ss
inst
ruct
ions
• P
rovi
sion
of
suita
ble
re
sour
ces
– m
achi
nery
, p
eop
le•
Trai
ning
of
pro
cess
op
erat
ors
• E
nviro
nmen
tal
inst
ruct
ions
• P
acki
ng in
stru
ctio
ns•
Valid
atio
n p
lans
Pro
cess
es w
hich
:•
ensu
re p
rod
uctio
n/se
rvic
e p
rovi
sion
act
ivity
is
und
er c
ontr
olle
d c
ond
ition
s, i.
e. s
uita
ble
eq
uip
men
t w
ith a
pp
rop
riate
mon
itorin
g an
d
cont
rols
• en
sure
val
idat
ion
of p
roce
sses
whe
re t
he
resu
lting
out
put
can
not
be
chec
ked
by
sub
seq
uent
mea
sure
men
t•
ensu
re c
ontin
ual i
mp
rove
men
t of
rea
lizat
ion
pro
cess
es,
e.g.
red
ucin
g w
aste
, im
pro
ving
m
etho
ds,
pre
vent
ing
pro
ble
ms,
imp
rovi
ng y
ield
• va
lidat
e cr
itica
l pro
cess
es t
hat
coul
d a
ffect
p
rod
uct
qua
lity
incl
udin
g sp
ecifi
c ex
amp
les
and
re
valid
atio
n re
qui
rem
ents
• en
sure
iden
tific
atio
n an
d t
race
abili
ty o
f m
ater
ials
and
eq
uip
men
t us
ed t
hrou
ghou
t p
rod
uctio
n•
pro
tect
and
car
e fo
r cu
stom
er p
rop
erty
(e.g
. m
ould
s, a
rtw
ork,
ele
ctro
nic
dat
a)•
pro
tect
and
cle
arly
iden
tify
the
final
pro
duc
t
• C
ontin
ual
imp
rove
men
t ac
tiviti
es
Rec
ord
s of
:•
pro
duc
tion
(e.g
. b
atch
sh
eets
) •
pro
cess
val
idat
ion/
re
valid
atio
n•
mat
eria
l usa
ge
• w
areh
ouse
and
d
isp
atch
tra
nsac
tions
• p
rod
uct
iden
tific
atio
n
Sch
emat
ic 3
Sch
emat
ic 4
Sch
emat
ic 5
Sch
emat
ic 6
Sch
emat
ic 7
Ann
ex A
:C
ount
erfe
it
Ad
mix
ture
Seg
rega
tion
cont
rols
Valid
atio
n (a
nd
Qua
lific
atio
n)

58
Cla
use
7.6
Co
ntro
l of
mo
nito
ring
and
mea
sure
men
t d
evic
es
Con
trol
of
such
eq
uip
men
t is
crit
ical
to
good
con
trol
of
man
ufac
turin
g p
roce
sses
.
Ther
e ar
e no
‘ex
tra’
PS
900
0 G
MP
s w
ithin
thi
s cl
ause
. Th
ere
are
imp
orta
nt G
MP
prin
cip
les
in t
his
area
but
the
y ar
e ad
equa
tely
ex
pre
ssed
in IS
O 9
001
and
ISO
900
4.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
Sch
emat
ic/A
nnex
• O
utp
uts
from
des
ign
and
dev
elop
men
t ac
tiviti
es -
e.g
. d
esig
n to
lera
nces
• P
rod
uct
spec
ifica
tions
• C
usto
mer
re
qui
rem
ents
• C
alib
ratio
n st
and
ard
s
Pro
cess
es w
hich
:•
det
erm
ine
the
mon
itorin
g/m
easu
ring
and
eq
uip
men
t ne
eded
• en
sure
iden
tific
atio
n an
d p
rote
ctio
n of
eq
uip
men
t•
cont
rol c
alib
ratio
n •
cont
rol t
he m
onito
ring/
mea
surin
g ac
tiviti
es t
o ve
rify
pro
duc
ts (a
nd p
rod
uctio
n p
roce
sses
)•
cont
rol n
onco
nfor
min
g eq
uip
men
t/d
evic
e•
cont
rol t
he c
heck
s on
com
put
er s
oftw
are
whe
n us
ed in
mon
itorin
g an
d m
easu
rem
ent
• Li
st o
f m
onito
ring/
mea
surin
g eq
uip
men
t•
Sta
tus
lab
elle
d
equi
pm
ent
• C
alib
ratio
n re
cord
s
Pro
ced
ures
for
:•
mon
itorin
g•
mea
surin
g •
calib
ratio
n
Sch
emat
ic 5
Sch
emat
ic 6

59
Gen
eral
Syn
op
sis
- C
laus
e 8
Mea
sure
men
t, a
naly
sis
and
imp
rove
men
t
Mea
sure
men
t in
form
atio
n is
vita
l to
soun
d d
ecis
ion
mak
ing.
The
org
aniz
atio
n sh
ould
mon
itor
its p
roce
sses
, pro
duc
ts a
nd a
ll as
pec
ts o
f th
e Q
MS
in o
rder
to
pro
vid
e d
ata
for
cont
inua
l im
pro
vem
ent
in p
erfo
rman
ce.
Cla
use
8.1
Mea
sure
men
t, a
naly
sis
and
imp
rove
men
t: G
ener
al
Thes
e ac
tiviti
es a
re o
f su
ch im
por
tanc
e th
at c
aref
ul p
lann
ing
is n
eed
ed o
n ho
w it
sha
ll b
e p
erfo
rmed
.
Ther
e ar
e no
‘ex
tra’
PS
900
0 G
MP
s w
ithin
thi
s cl
ause
. Th
ere
are
imp
orta
nt G
MP
prin
cip
les
in t
his
area
, b
ut t
hey
are
adeq
uate
ly
exp
ress
ed in
ISO
900
1 an
d IS
O 9
004.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
Sch
emat
ic/A
nnex
• P
roce
ss a
naly
sis
wor
k•
Com
pan
y ob
ject
ives
• C
usto
mer
nee
ds
Pro
cess
es w
hich
:•
pla
n ho
w t
o m
onito
r/m
easu
re p
roce
sses
and
p
rod
ucts
• d
evel
op s
uita
ble
imp
rove
men
t m
echa
nism
s•
cont
rol t
he im
ple
men
tatio
n of
the
ab
ove
• Fo
rmal
mea
sure
men
t sy
stem
s•
Sta
tistic
al t
echn
ique
s•
Pro
ced
ures
• R
epor
ts
Sch
emat
ic 3
Sch
emat
ic 6

60
Cla
use
8.2
Mo
nito
ring
and
mea
sure
men
t
Mon
itorin
g th
e sa
tisfa
ctio
n of
cus
tom
ers,
oth
er in
tere
sted
par
ties,
mon
itorin
g p
rod
ucts
, p
roce
sses
and
sys
tem
s.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
(re
fer
to P
S 9
000
clau
se 8
.2.4
Mo
nito
ring
and
mea
sure
men
t o
f p
rod
uct)
ISO
900
1:20
00 r
equi
res
qua
lity
des
igne
d a
nd s
yste
m c
omp
liant
pro
cess
es,
and
com
plim
enta
ry t
o th
is in
PS
900
0 is
the
nee
d f
or
pro
cess
es t
o b
e op
erat
ed in
a w
ay w
hich
mai
ntai
ns p
rod
uct
secu
rity
and
avo
ids
cont
amin
atio
n (s
ee a
lso
5.2
and
5.3
).
Sin
ce p
roce
ss a
sses
smen
t is
oft
en b
ased
up
on p
rod
uct
sam
ple
s, t
hese
mus
t b
e re
pre
sent
ativ
e of
the
pro
cess
with
the
sam
plin
g sc
hem
e d
efin
ed.
Whi
lst
sam
ple
s ex
amin
ed o
n th
e lin
e ar
e co
nsid
ered
sec
ure,
a c
onta
min
atio
n or
ad
mix
ture
ris
k ex
ists
if t
here
are
in
adeq
uate
con
trol
s on
the
sec
urity
han
dlin
g of
sam
ple
s re
mov
ed f
rom
the
pro
duc
tion
area
.
To a
void
thi
s ris
k, P
S 9
000
spec
ifica
lly r
equi
res
the
secu
re d
isp
osal
aft
er e
xam
inat
ion,
of
all s
amp
les
that
hav
e b
een
rem
oved
fro
m t
he
imm
edia
te p
rod
uctio
n ar
ea a
nd a
dd
ition
ally
stip
ulat
es t
hey
cann
ot b
e re
inco
rpor
ated
into
the
pro
duc
t (s
ee a
lso
‘Rul
es a
nd G
uid
ance
fo
r P
harm
aceu
tical
Man
ufac
ture
rs a
nd D
istr
ibut
ors
2002
’, C
hap
ter
5.54
)
It is
gen
eral
ly k
now
n th
at p
roce
sses
wor
k b
ette
r w
hen
they
hav
e st
abili
zed
fol
low
ing
star
t-up
and
are
run
ning
con
tinuo
usly
. H
ence
w
hen
equi
pm
ent
bre
aks
dow
n or
the
re is
an
unsc
hed
uled
inte
rrup
tion
that
sto
ps
the
pro
cess
, q
ualit
y va
riab
ility
can
occ
ur a
nd
add
ition
al q
ualit
y m
onito
ring
is r
equi
red
on
reco
mm
ence
men
t.

61
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• C
usto
mer
sur
veys
• Fo
cus
grou
p d
ata
• Fe
edb
ack
on p
rod
ucts
• C
usto
mer
re
qui
rem
ents
/con
trac
ts•
Mar
ket
need
s•
Del
iver
y d
ata
• Fi
nanc
ial d
ata/
cost
s of
no
ncon
form
ities
• M
arke
t b
ench
mar
king
an
d b
est
pra
ctic
e
Pro
cess
es t
hat
iden
tify
(and
imp
lem
ent)
suita
ble
m
etho
ds
to:
• m
onito
r an
d m
easu
re c
usto
mer
sat
isfa
ctio
n•
mon
itor
and
mea
sure
the
sat
isfa
ctio
n of
in
tere
sted
par
ties
(oth
er t
han
cust
omer
s) e
.g.
emp
loye
es,
inve
stor
s, s
upp
liers
and
soc
iety
• m
onito
r an
d m
easu
re p
rod
uctio
n p
roce
sses
•
verif
y th
at p
rod
uct
is m
eetin
g re
qui
rem
ents
at
app
rop
riate
sta
ges
of p
rod
uctio
n
Pro
cess
es w
hich
:•
cont
rol i
nter
nal a
uditi
ng –
pro
gram
me,
p
roce
dur
es,
aud
itors
• d
efin
e a
stat
istic
ally
val
id s
amp
ling
sche
me
• p
reve
nt s
amp
les
from
bei
ng r
etur
ned
to
a b
atch
-
keep
sep
arat
e th
en d
estr
oy/d
isp
ose
secu
rely
• ap
ply
ad
diti
onal
mon
itorin
g, a
fter
a m
achi
ne
bre
akd
own
or s
top
pag
e
Cor
rect
ive
actio
ns:
• w
hen
pro
cess
es f
ail
to a
chie
ve p
lann
ed
resu
lts•
in r
esp
onse
to
cust
omer
info
rmat
ion
(com
pla
ints
, su
rvey
s et
c.)
Rec
ord
s of
:•
pro
duc
t m
onito
ring
and
mea
surin
g ac
tiviti
es (e
.g.
test
re
por
ts)
• au
dit
rep
orts
/act
ions
/fo
llow
-up
act
ions
Feed
bac
k to
:•
cust
omer
s•
othe
r in
tere
sted
p
artie
s
Sch
emat
ic 5
Sch
emat
ic 6

62
Cla
use
8.3
Co
ntro
l of
nonc
onf
orm
ing
pro
duc
t
It is
ess
entia
l tha
t p
rod
uct
not
mee
ting
spec
ifica
tion
be
segr
egat
ed f
rom
sui
tab
le p
rod
uct
until
cor
rect
ed,
des
troy
ed,
or r
ecla
ssifi
ed.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
Ther
e is
an
assu
mp
tion
that
all
pro
cess
es w
ork
effic
ient
ly w
ithou
t p
rob
lem
s an
d t
hat
mat
eria
l will
be
unifo
rm,
cons
iste
nt a
nd f
ully
co
mp
liant
with
the
sp
ecifi
catio
n. In
rea
lity
pro
ble
ms
can
occu
r re
sulti
ng in
sho
rt p
erio
ds
whe
n th
e p
roce
ss m
ay n
ot b
e in
con
trol
and
p
rod
uces
non
conf
orm
ing
pro
duc
t. T
his
can
resu
lt in
pro
ble
ms
in t
he m
arke
t, c
usto
mer
com
pla
ints
and
ret
urne
d p
rod
uct.
For
GM
P r
easo
ns a
nd g
ood
cus
tom
er r
elat
ions
, th
e p
harm
aceu
tical
ind
ustr
y ne
eds
to b
e aw
are
of a
ny q
ualit
y is
sues
dur
ing
the
man
ufac
ture
of
its p
acka
ging
mat
eria
ls.
The
cust
omer
sho
uld
alw
ays
be
notif
ied
of
nonc
onfo
rmin
g or
ass
ocia
ted
pro
duc
t, w
hich
may
ha
ve a
lread
y le
ft t
he o
rgan
izat
ion,
so
that
the
pha
rmac
eutic
al c
omp
any
can
cons
ider
any
imp
licat
ions
to
pro
cess
es,
stoc
k or
pac
ked
p
rod
uct.
Sus
pec
t p
rod
uct
reta
ined
by
the
sup
plie
r or
ret
urne
d f
rom
the
cus
tom
er,
req
uire
s fo
rmal
GM
P c
ontr
ols
on it
s se
cure
sto
rage
, ha
ndlin
g an
d r
emed
ial r
ewor
k ac
tions
, in
clud
ing
a ris
k as
sess
men
t to
ens
ure
that
the
rew
orki
ng p
roce
ss d
oes
not
intr
oduc
e fu
rthe
r q
ualit
y p
rob
lem
s.
All
of t
hese
issu
es r
equi
re g
ood
doc
umen
tatio
n to
pro
vid
e tr
acea
bili
ty o
ver
time
and
to
reco
nstr
uct
even
ts if
req
uire
d.

63
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• P
rod
ucts
not
mee
ting
spec
ifica
tion
• C
usto
mer
rej
ects
or
com
pla
ints
•
Res
ults
of
insp
ectio
ns
or in
-pro
cess
tes
ting
activ
ities
• O
bse
rvat
ions
by
mem
ber
s of
sta
ff
Pro
cess
es w
hich
:•
revi
ew id
entif
ied
non
conf
orm
ing
pro
duc
t an
d
actio
ns t
o b
e d
eter
min
ed•
cont
rol t
he s
ecur
e p
hysi
cal (
or e
lect
roni
c)
qua
rant
inin
g of
item
s/d
ata
• co
ntro
l rew
ork
• re
-ver
ify o
r in
spec
t co
rrec
ted
pro
duc
t (e
.g.
follo
win
g re
wor
k)•
notif
y cu
stom
ers
if p
rob
lem
s ar
e fo
und
aft
er
del
iver
y•
iden
tify
and
con
trol
any
bat
ches
of
pro
duc
t th
at
are
rela
ted
to
any
nonc
onfo
rmin
g p
rod
uct
that
ha
s b
een
retu
rned
fro
m t
he c
usto
mer
• D
ocum
ente
d r
isk
asse
ssm
ent
for
any
bat
ches
tha
t ar
e re
wor
ked
to
reco
ver
stoc
k fr
om a
rej
ecte
d
or q
uara
ntin
ed b
atch
• R
ecor
ds
of c
usto
mer
co
mm
unic
atio
ns/
notif
icat
ions
• D
efin
ed
resp
onsi
bili
ties/
auth
oriti
es f
or d
ealin
g w
ith n
onco
nfor
min
g p
rod
uct
Pro
ced
ures
: •
det
ailin
g w
hat
to
do
in t
he e
vent
of
nonc
onfo
rmin
g p
rod
uct
bei
ng
det
ecte
d•
for
hand
ling
reje
ctio
ns
and
com
pla
ints
• fo
r ha
ndlin
g re
cove
ry/
rew
orks
Sch
emat
ic 3
Sch
emat
ic 5
Sch
emat
ic 6
Ann
ex A
:R
isk
asse
ssm
ent

64
Cla
use
8.4
Ana
lysi
s o
f d
ata
In a
dd
ition
to
mon
itorin
g ac
tiviti
es,
the
sens
ible
ana
lysi
s an
d in
terp
reta
tion
of d
ata
is c
ritic
al t
o en
able
goo
d d
ecis
ions
to
be
take
n an
d
bus
ines
s im
pro
vem
ents
to
be
mad
e.
Ther
e ar
e no
‘ex
tra’
PS
900
0 G
MP
s w
ithin
thi
s cl
ause
. Th
ere
are
imp
orta
nt G
MP
prin
cip
les
in t
his
area
, b
ut t
hey
are
adeq
uate
ly
exp
ress
ed in
ISO
900
1 an
d IS
O 9
004.
Pro
cess
inp
uts
Req
uire
d p
roce
sses
Pro
cess
out
put
s an
d
evid
ence
Cro
ss r
efer
to
Sch
emat
ic/A
nnex
• O
utp
uts
from
M
onito
ring
and
M
easu
rem
ent
(8.2
) and
ot
her
pro
cess
es•
Dat
a fr
om r
elev
ant
sour
ces
Pro
cess
es w
hich
det
erm
ine
the
dat
a re
qui
red
, co
llect
and
ana
lyse
it,
to:
• ev
alua
te t
he Q
MS
• m
onito
r an
d m
easu
re it
s su
itab
ility
and
ef
fect
iven
ess
• im
pro
ve it
Pro
cess
es f
or:
• ro
ot c
ause
ana
lysi
s of
pro
ble
ms
• an
alys
is o
f d
ata
whi
ch u
ses
valid
met
hod
s an
d
app
rop
riate
sta
tistic
al t
echn
ique
s•
dec
isio
n-m
akin
g b
ased
on
resu
lts o
f lo
gica
l an
alys
is,
bal
ance
d w
ith e
xper
ienc
e an
d
know
led
ge
Tren
ds,
info
rmat
ion
and
d
ata
abou
t:•
cust
omer
s •
sup
plie
rs•
pro
duc
ts•
pro
cess
es•
QM
S
Sch
emat
ic 6

65
Cla
use
8.5
Imp
rove
men
t
Con
tinua
l im
pro
vem
ent
of t
he Q
MS
is a
t th
e he
art
of t
he IS
O 9
001
and
PS
900
0 p
hilo
sop
hy.
Ther
e ar
e no
‘ex
tra’
PS
900
0 G
MP
s w
ithin
thi
s cl
ause
. Th
ere
are
imp
orta
nt G
MP
prin
cip
les
in t
his
area
, b
ut t
hey
are
adeq
uate
ly
exp
ress
ed in
ISO
900
1 an
d IS
O 9
004.
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
Sch
emat
ic/A
nnex
• A
udit
resu
lts•
Qua
lity
dat
a•
Qua
lity
pol
icy
and
ob
ject
ives
• M
anag
emen
t re
view
s •
Pre
viou
s co
rrec
tive
and
p
reve
ntiv
e ac
tions
• Va
lidat
ion
dat
a•
Pro
cess
and
pro
duc
t m
easu
rem
ents
• D
ata
from
sel
f as
sess
men
ts•
Req
uire
men
ts f
rom
cu
stom
ers/
inte
rest
ed
par
ties
• Fi
nanc
ial a
nd s
ervi
ce
dat
a
Pro
cess
es t
o as
sess
pot
enti
al n
onco
nfor
miti
es,
stud
y th
eir
effe
cts
and
:•
eval
uate
s th
e ne
ed t
o ta
ke p
reve
ntiv
e ac
tion
• d
evel
ops
suita
ble
act
ions
to
elim
inat
e ca
uses
• ex
amin
es t
he e
ffect
iven
ess
of t
hem
Pro
cess
es t
o d
etec
t ac
tual
non
conf
orm
ities
whi
ch:
• ev
alua
tes
the
need
to
take
cor
rect
ive
actio
n•
dev
elop
s su
itab
le a
ctio
ns t
o el
imin
ate
caus
es•
exam
ines
the
effe
ctiv
enes
s of
the
m
Pro
cess
tha
t p
lans
for
loss
es s
usta
ined
by
the
orga
niza
tion
in o
rder
to
miti
gate
the
ir p
oten
tial
effe
cts
(e.g
. fir
e or
wea
ther
dam
age)
• R
isk
asse
ssm
ent
rep
orts
• Im
pro
vem
ent
pla
ns•
Pro
ced
ure
for
nonc
onfo
rman
ce
man
agem
ent
• P
reve
ntiv
e an
d
corr
ectiv
e ac
tions
ta
ken
Rec
ord
s:•
of t
hese
act
ions
• of
the
effe
ctiv
enes
s of
th
ese
actio
ns
Sch
emat
ic 1
Ann
ex A
:R
isk
asse
ssm
ent

66
Gen
eral
Syn
op
sis
- C
laus
e 9
Co
ntam
inat
ion
cont
rol
The
cap
abili
ty o
f fa
cilit
ies
and
eq
uip
men
t, a
chie
ved
thr
ough
the
ir d
esig
n, l
ocat
ion,
con
stru
ctio
n, a
nd m
aint
enan
ce,
to
min
imis
e th
e ris
k of
pro
cess
err
ors
and
avo
id c
ross
-con
tam
inat
ion
affe
ctin
g p
roce
ss a
nd p
rod
uct.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
The
high
est
qua
lity
prio
rity
of t
he p
harm
aceu
tical
ind
ustr
y is
for
the
com
pon
ent
mat
eria
l to
mee
t sp
ecifi
catio
ns a
nd m
eet
or e
xcee
d
def
ined
GM
P c
ontr
ols.
Pha
rmac
eutic
al p
rod
uct
safe
ty,
effic
acy,
and
sta
bili
ty c
an a
ll b
e co
mp
rom
ised
by
chem
ical
con
tam
inat
ion
of t
he p
rimar
y p
acka
ging
m
ater
ial.
Par
ticul
ate
cont
amin
atio
n m
ay r
educ
e th
e vi
sual
qua
lity
of t
he p
rod
uct
as w
ell a
s in
terf
ere
with
its
func
tion;
it c
an a
lso
be
a ‘c
arrie
r m
ediu
m’
for
mic
rob
ial c
onta
min
atio
n w
ith a
pot
entia
l inf
ectio
n ris
k to
the
pat
ient
.
GM
P c
ontr
ols
and
pro
ced
ures
pro
vid
e th
e sa
fegu
ard
s to
pre
vent
cro
ss-c
onta
min
atio
n (a
dm
ixtu
re).
A s
ingl
e ‘r
ogue
’ co
mp
onen
t w
ithin
a
bat
ch c
ould
cre
ate
a lif
e-th
reat
enin
g ha
zard
to
the
pat
ient
(see
Ann
ex A
- A
dm
ixtu
re).
Sin
ce c
onta
min
atio
n ca
n ta
ke m
any
form
s an
d b
e in
trod
uced
any
whe
re,
pre
vent
ive
mea
sure
s (e
.g.
good
fac
ility
des
ign,
Sta
ndar
d
Op
erat
ing
Pro
ced
ures
, tr
aini
ng,
area
cle
aran
ce,
segr
egat
ion
cont
rols
, et
c.),
mus
t b
e ap
plie
d t
hrou
ghou
t th
e fa
cilit
y. In
ad
diti
on,
sim
ilar
mea
sure
s sh
ould
be
incl
uded
in t
he d
esig
n of
the
pro
cess
.
PS
900
0 sp
ecifi
es G
MP
req
uire
men
ts a
cros
s th
e m
anuf
actu
ring
oper
atio
n as
wel
l as
the
envi
ronm
enta
l con
diti
ons
need
ed f
or
pro
cess
ing
pac
kagi
ng m
ater
ials
for
use
with
med
icin
al p
rod
ucts
. Th
ree
diff
eren
t cl
eanl
ines
s ca
tego
ries
are
def
ined
dep
end
ent
upon
the
m
ater
ial a
nd it
s ap
plic
atio
n.

67
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• S
pec
ifica
tion
for
des
ign
of n
ew f
acili
ties,
p
roce
sses
, eq
uip
men
t an
d e
nviro
nmen
t•
Sp
ecia
list
pes
t an
d
cont
amin
atio
n co
ntro
l•
Cus
tom
er r
equi
rem
ents
• S
tand
ard
s fo
r en
viro
nmen
tal
cond
ition
s
Pro
cess
es w
hich
iden
tify/
cont
rol:
• op
erat
ing
area
s/ac
tiviti
es t
hat
cont
ribut
e to
co
ntam
inat
ion
• en
viro
nmen
tal c
ateg
ory
app
rop
riate
to
the
pro
duc
t (a
gree
with
cus
tom
er)
• ha
ndlin
g of
mat
eria
l and
seg
rega
tion
cont
rols
• go
od p
ract
ices
to
avoi
d c
onta
min
atio
n d
urin
g op
erat
ion,
sup
por
t, a
nd m
aint
enan
ce•
pes
t co
ntro
l•
clea
ning
pro
ced
ures
and
che
cks
• tr
aini
ng o
f st
aff
and
vis
itors
in c
onta
min
atio
n av
oid
ance
and
sta
ndar
ds
of d
ress
• m
anag
emen
t an
d c
ontr
ol o
f w
aste
mat
eria
l•
line
clea
ranc
e•
chan
ge c
ontr
ol•
envi
ronm
enta
l mon
itorin
g
• In
cid
ents
/aud
it re
por
ts•
Dra
win
gs (e
.g.
floor
p
lans
)•
Ap
pro
pria
te
envi
ronm
enta
l ca
tego
ry e
stab
lishe
d
for
pro
duc
t (a
gree
d
with
cus
tom
er)
Pro
ced
ures
to:
•
cont
rol p
roce
sses
• m
onito
r an
d c
ontr
ol
envi
ronm
ents
Rec
ord
s of
:•
pro
cess
ing
• cl
eani
ng s
ched
ules
• cl
eani
ng•
trai
ning
• m
ater
ial d
isp
osal
Sch
emat
ic 3
(a)
Sch
emat
ic 4
(a)
Sch
emat
ic 5
Ann
ex A
: al
l par
ts

68
Gen
eral
Syn
op
sis
- C
laus
e 10
Pri
nted
mat
eria
ls
The
app
licat
ion
of c
ontr
ols
spec
ific
to d
iffer
ent
typ
es o
f p
rinte
d m
ater
ials
in
ord
er t
o en
sure
id
entit
y, m
axim
ise
pro
duc
t se
curit
y an
d p
reve
nt c
ross
-con
tam
inat
ion.
It in
clud
es t
he u
se a
nd v
erifi
catio
n of
var
ious
typ
es o
f se
curit
y co
de
syst
ems
and
as
soci
ated
sof
twar
e.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
This
sec
tion
inte
ntio
nally
fol
low
s th
e se
ctio
n on
Con
tam
inat
ion
Con
trol
bec
ause
it c
over
s in
dep
th t
he s
pec
ific
GM
P r
equi
rem
ents
for
se
cure
ly p
rintin
g co
mp
onen
ts t
o p
reve
nt a
dm
ixtu
re a
nd c
ross
-con
tam
inat
ion.
The
risk
asso
ciat
ed w
ith a
dm
ixtu
re is
tha
t an
inco
rrec
t p
rinte
d c
omp
onen
t, o
ften
sim
ilar
in d
esig
n to
the
cor
rect
com
pon
ent,
will
be
used
to
lab
el o
r co
ntai
n th
e p
harm
aceu
tical
pro
duc
t. T
his
can
lead
to
the
pat
ient
usi
ng t
he w
rong
pro
duc
t or
usi
ng it
in a
n in
app
rop
riate
m
anne
r.
(For
mor
e d
etai
l ref
er t
o A
nnex
A -
Ad
diti
onal
Exp
lana
tions
of
Key
Con
cep
ts).
Ther
e ar
e m
any
stag
es in
the
man
ufac
ture
of
prin
ted
mat
eria
ls,
all o
f w
hich
can
ben
efit
from
goo
d c
ontr
ol p
roce
dur
es.
The
adva
ntag
e of
sec
urity
bar
cod
es s
yste
ms
is t
hat
auto
mat
ed d
etec
tion
chec
ks c
an b
e in
corp
orat
ed in
to k
ey p
roce
ss s
tage
s at
the
sup
plie
r or
gani
zatio
n an
d c
usto
mer
.
The
pha
rmac
eutic
al in
dus
try
bel
ieve
s th
at,
alth
ough
sec
urity
bar
cod
e sy
stem
s ca
n ap
pro
ach
100%
sec
urity
con
fiden
ce,
they
are
not
a
sub
stitu
te f
or o
ther
goo
d m
anuf
actu
ring
pra
ctic
es.

69
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• P
rod
uct
req
uire
men
ts
incl
udin
g le
afle
ts,
cart
ons,
re
el-f
ed m
ater
ials
, la
bel
s an
d p
rod
uct
prin
ting
usin
g d
igita
l tec
hnol
ogy
• A
ll p
rod
ucts
mad
e b
y as
sem
bly
of
sub
-co
mp
onen
ts (e
.g.
lab
el/
leaf
let
com
bin
atio
ns)
• G
rap
hic
des
ign
(e.g
. in
clus
ion
of s
ecur
ity
bar
cod
e in
orig
inat
ion/
artw
ork)
• M
achi
ne d
esig
n/op
erat
iona
l set
tings
• C
usto
mer
req
uire
men
ts
and
agr
eem
ent
on
qua
ntity
tol
eran
ces
• S
pec
ifica
tions
for
ree
l-fe
d m
ater
ials
incl
udin
g sp
lice
join
s an
d h
ow
reel
mat
eria
ls s
hall
be
iden
tifie
d•
Ele
ctro
nic
and
/or
mec
hani
cal m
achi
ne
enha
ncem
ents
(e.g
. op
tical
, d
oub
le s
heet
d
etec
tors
)•
Pac
king
and
sto
rage
sp
ecifi
catio
ns
Pro
cess
es w
hich
con
trol
:•
the
issu
e an
d a
lloca
tion
of s
ecur
ity
bar
cod
es
• th
e se
t-up
and
use
of
scan
ners
to
reje
ct
unsu
itab
le m
ater
ial
• eq
uip
men
t th
at c
an,
und
er c
erta
in c
ond
- iti
ons
prin
t w
ith m
issi
ng c
olou
rs o
r te
xt
• an
y ov
erp
rintin
g p
roce
ss a
nd it
s ve
rific
atio
n •
the
scan
ning
and
off-
line
verif
icat
ion
of
bar
cod
es w
hen
nece
ssar
y an
d c
halle
nge
of t
he e
ffect
iven
ess
of s
cann
ing
• th
e ch
ecki
ng o
f sp
lices
in r
eels
• co
untin
g/se
que
ntia
l num
ber
ing
Pro
cess
es w
hich
:•
pro
hib
it th
e re
pla
cem
ent
of m
issi
ng la
bel
s on
a r
eel
• p
reve
nt p
artia
lly p
rinte
d o
r b
lank
leaf
lets
• en
sure
tha
t th
e lin
e cl
eara
nce
pro
cess
es
incl
ude
com
put
er a
rtw
ork
file
rem
oval
(w
here
ap
plic
able
) •
ensu
re t
hat
dig
ital f
ile a
cces
s is
sec
ure,
va
lidat
ed a
nd p
reve
nts
acci
den
tal u
se o
f in
corr
ect
orig
inat
ion/
artw
ork
files
•
ensu
re s
ecur
e as
sem
bly
(e
.g.
lab
el/
leaf
lets
) with
sec
ure
pro
duc
tion
of a
ll su
b-
com
pon
ents
•
cont
rol v
alid
atio
n ac
tiviti
es•
cont
rol p
acki
ng a
nd s
tora
ge a
ctiv
ities
• R
egis
ter
of s
ecur
ity
bar
cod
es•
Acc
urat
e co
unts
det
aile
d
on t
he o
uter
car
ton
lab
els
• Tr
acea
bili
ty o
f p
rod
uct
(aud
it tr
ail)
• G
ood
pac
king
line
p
erfo
rman
ce o
f co
mp
onen
ts w
hen
in u
se
by
cust
omer
(com
pla
ints
/fe
edb
ack)
• E
stab
lishe
d s
ettin
gs t
o en
sure
rep
rod
ucib
ility
Verif
ied
and
sec
urel
y p
rinte
d
pro
duc
t th
at is
:•
free
fro
m a
dm
ixtu
re•
free
fro
m m
issi
ng p
rint/
erro
rs•
accu
rate
ly c
ount
ed•
corr
ectly
ass
emb
led
(e.g
. la
bel
/leaf
lets
)
Sys
tem
s p
rove
n to
be
effe
ctiv
e, r
ecor
ds
of:
• va
lidat
ion
and
qua
lific
atio
n•
verif
icat
ion
chec
ks•
scan
ning
rej
ects
and
re
view
of
them
• eq
uip
men
t te
stin
g
Sch
emat
ic 4
Sch
emat
ic 5
Sch
emat
ic 7
Ann
ex A
:A
dm
ixtu
re
Iden
tific
atio
n an
d
trac
eab
ility
Line
cle
aran
ce
Valid
atio
n (a
nd
Qua
lific
atio
n)

70
Gen
eral
Syn
op
sis
– C
laus
e 11
Ori
gin
atio
n/ar
two
rk
The
gene
ratio
n of
new
or
amen
ded
orig
inat
ion
is th
e st
art o
f the
man
ufac
turin
g p
roce
ss fo
r p
rinte
d it
ems
and
req
uire
s co
ntro
l fr
om c
once
pts
to
orig
inat
ion
and
thr
ough
to
final
prin
ting.
A
ll or
igin
atio
n an
d p
rinte
d m
edia
mus
t b
e co
ntro
lled
to
ensu
re
secu
rity
and
inte
grity
of
the
final
prin
ted
pro
duc
t.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
Bot
h p
acka
ging
sup
plie
rs a
nd p
harm
aceu
tical
com
pan
ies
have
exp
erie
nce
of c
omm
on p
rob
lem
s th
at o
ccur
in t
he p
rep
arat
ion
of
orig
inat
ion/
artw
ork,
whi
ch c
ould
be
avoi
ded
by
good
man
ufac
turin
g p
ract
ices
.
Orig
inat
ion
pro
ble
ms
occu
r in
the
sam
e w
ay a
s th
ose
for
prin
ting
of t
he p
acka
ging
mat
eria
l and
GM
P p
reve
ntiv
e co
ntro
ls t
o ad
dre
ss
thes
e, f
ollo
w t
he s
ame
pat
tern
, (e
.g.
def
ined
wor
k ar
eas,
seg
rega
tion,
line
cle
aran
ce,
reco
rded
che
cks,
etc
.).
Ele
ctro
nic
syst
ems
are
wid
ely
used
to
pro
duc
e p
harm
aceu
tical
orig
inat
ion;
it is
imp
orta
nt t
hat
the
syst
ems
are
secu
re a
nd h
ave
cont
rolle
d a
cces
s (s
ee P
S 9
000
4.2.
3).
Sin
ce o
rigin
atio
n/ar
twor
k is
com
par
able
to
a d
esig
n p
roce
ss,
erro
rs b
uilt
into
the
des
ign
cann
ot b
e re
mov
ed b
y la
ter
insp
ectio
n an
d t
here
fore
qua
lity,
i.e.
GM
P, m
ust
be
bui
lt in
to t
he c
reat
ion
pro
cess
. T
his
star
ts w
ith u
niq
ue
iden
tific
atio
n of
the
mat
eria
l and
iden
tity
chec
ks a
t al
l pro
cess
sta
ges
with
in t
he o
rgan
izat
ion
and
dur
ing
tran
sfer
to
the
cust
omer
.

71
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• A
ll or
igin
atio
n m
ater
ial
• C
usto
mer
re
qui
rem
ents
• C
usto
mer
des
igns
/sp
ecifi
catio
ns•
Cus
tom
er c
omp
onen
t co
des
Pro
cess
es w
hich
: •
seek
agr
eem
ent
with
cus
tom
er (w
here
req
uire
d)
to s
ign
off
a co
py
of t
he f
inal
pro
of b
efor
e p
rod
uctio
n is
aut
horiz
ed•
req
uire
the
iden
tific
atio
n of
all
orig
inat
ion
mat
eria
l (e.
g. e
nsur
ing
that
pro
ofs
are
mar
ked
w
ith c
omp
onen
t nu
mb
ers
and
issu
e d
ates
)•
cont
rol o
rigin
atio
n m
ater
ials
and
cov
ers
stor
age,
is
sue,
ret
urn
and
line
cle
aran
ces
• en
sure
und
erst
and
ing
of h
ow c
odes
are
use
d
by
cust
omer
s an
d a
pp
licat
ion
of a
pp
rop
riate
co
ntro
l mea
sure
s
• re
qui
re t
he c
heck
ing
of c
opy
(ele
ctro
nic
or
hard
out
put
) aga
inst
orig
inal
sp
ecifi
catio
ns a
nd
cust
omer
ap
pro
ved
ver
sion
s
• A
sys
tem
of
cont
rol f
or a
rtw
ork
that
pre
vent
s th
e in
adve
rten
t us
e of
in
corr
ect
item
s
Rec
ord
s of
:•
app
rova
l by
cust
omer
(e
.g.
app
rova
l of
artw
ork)
•
in-p
roce
ss c
heck
s p
erfo
rmed
on
artw
orks
• lin
e cl
eara
nces
•
chec
ks m
ade
on
in-h
ouse
pro
duc
ed
item
s ag
ains
t a
cust
omer
ap
pro
ved
m
aste
r •
cust
omer
ap
pro
ved
m
aste
rs
Sch
emat
ic 3
Sch
emat
ic 4
Sch
emat
ic 4
(a)
Sch
emat
ic 5
Ann
ex A
:C
hang
e co
ntro
l
Iden
tific
atio
n an
d
trac
eab
ility
Line
cle
aran
ce

72
Gen
eral
Syn
op
sis
– C
laus
e 12
Pri
nt im
pre
ssio
n m
edia
Life
-cyc
le m
anag
emen
t of p
rint i
mp
ress
ion
med
ia is
ess
entia
l and
sho
uld
inco
rpor
ate
good
com
mun
icat
ion
with
the
cust
omer
to
und
erst
and
the
des
ign
and
ap
plic
atio
n of
the
pro
duc
t.
Key
is t
he t
race
abili
ty f
rom
cus
tom
er d
esig
n th
roug
h to
del
iver
y of
al
l prin
ted
item
s.
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
Prin
ting
of p
acka
ging
mat
eria
ls is
nor
mal
ly a
thr
ee-s
tage
pro
cess
, in
volv
ing
crea
tion
of t
he o
rigin
atio
n/ar
twor
k, p
rod
uctio
n of
the
prin
t im
pre
ssio
n m
edia
, i.e
. ‘t
he p
late
s’ a
nd la
stly
use
of
the
pla
tes
to c
reat
e th
e te
xt a
nd im
ages
on
the
pac
kagi
ng m
ater
ial.
The
GM
P r
equi
rem
ents
for
all
thes
e st
ages
mus
t th
eref
ore
be
very
sim
ilar
sinc
e th
ey h
ave
the
sam
e ob
ject
ive,
i.e.
com
plia
nce
with
the
cu
stom
er’s
sp
ecifi
catio
n an
d a
void
ance
of
cros
s co
ntam
inat
ion/
mix
-up
s.
Prin
t d
esig
ns c
an b
e ve
ry s
imila
r an
d d
iffic
ult
to d
iffer
entia
te,
par
ticul
arly
with
mod
ern
com
pan
y ‘h
ouse
’ d
esig
ns o
r fo
r a
fam
ily r
ange
of
pro
duc
t p
rese
ntat
ions
. P
rint
imp
ress
ion
med
ia m
ust
ther
efor
e b
e st
rictly
con
trol
led
with
uni
que
iden
tific
atio
n co
des
, an
d c
heck
s in
corp
orat
ed in
to a
ll us
age,
tog
ethe
r w
ith s
ecur
e an
d c
ontr
olle
d d
isp
osal
of
imp
ress
ion
med
ia w
hen
even
tual
ly s
uper
sed
ed.
For
mor
e in
form
atio
n se
e A
nnex
A -
Ad
diti
onal
Exp
lana
tions
of
Key
Con
cep
ts.
Als
o ‘R
ules
and
Gui
dan
ce f
or P
harm
aceu
tical
Man
ufac
ture
rs a
nd
Dis
trib
utor
s 20
02’,
Cha
pte
r 5.
43.
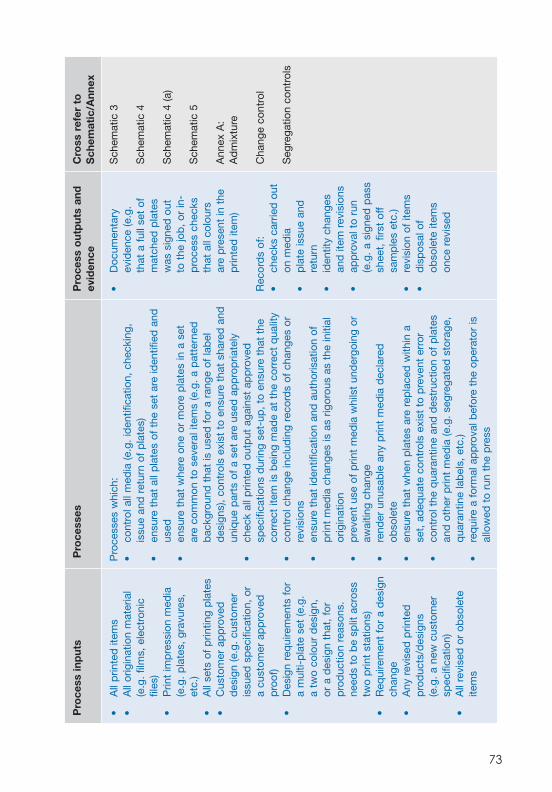
73
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
S
chem
atic
/Ann
ex
• A
ll p
rinte
d it
ems
• A
ll or
igin
atio
n m
ater
ial
(e.g
. fil
ms,
ele
ctro
nic
files
)•
Prin
t im
pre
ssio
n m
edia
(e
.g.
pla
tes,
gra
vure
s,
etc.
)•
All
sets
of
prin
ting
pla
tes
• C
usto
mer
ap
pro
ved
d
esig
n (e
.g.
cust
omer
is
sued
sp
ecifi
catio
n, o
r a
cust
omer
ap
pro
ved
p
roof
)•
Des
ign
req
uire
men
ts f
or
a m
ulti-
pla
te s
et (e
.g.
a tw
o co
lour
des
ign,
or
a d
esig
n th
at,
for
pro
duc
tion
reas
ons.
ne
eds
to b
e sp
lit a
cros
s tw
o p
rint
stat
ions
) •
Req
uire
men
t fo
r a
des
ign
chan
ge
• A
ny r
evis
ed p
rinte
d
pro
duc
ts/d
esig
ns
(e.g
. a
new
cus
tom
er
spec
ifica
tion)
•
All
revi
sed
or
obso
lete
ite
ms
Pro
cess
es w
hich
:•
cont
rol a
ll m
edia
(e.g
. id
entif
icat
ion,
che
ckin
g,
issu
e an
d r
etur
n of
pla
tes)
•
ensu
re t
hat
all p
late
s of
the
set
are
iden
tifie
d a
nd
used
• en
sure
tha
t w
here
one
or
mor
e p
late
s in
a s
et
are
com
mon
to
seve
ral i
tem
s (e
.g.
a p
atte
rned
b
ackg
roun
d t
hat
is u
sed
for
a r
ange
of
lab
el
des
igns
), co
ntro
ls e
xist
to
ensu
re t
hat
shar
ed a
nd
uniq
ue p
arts
of
a se
t ar
e us
ed a
pp
rop
riate
ly•
chec
k al
l prin
ted
out
put
aga
inst
ap
pro
ved
sp
ecifi
catio
ns d
urin
g se
t-up
, to
ens
ure
that
the
co
rrec
t ite
m is
bei
ng m
ade
at t
he c
orre
ct q
ualit
y•
cont
rol c
hang
e in
clud
ing
reco
rds
of c
hang
es o
r re
visi
ons
• en
sure
tha
t id
entif
icat
ion
and
aut
horis
atio
n of
p
rint
med
ia c
hang
es is
as
rigor
ous
as t
he in
itial
or
igin
atio
n •
pre
vent
use
of
prin
t m
edia
whi
lst
und
ergo
ing
or
awai
ting
chan
ge•
rend
er u
nusa
ble
any
prin
t m
edia
dec
lare
d
obso
lete
•
ensu
re t
hat
whe
n p
late
s ar
e re
pla
ced
with
in a
se
t, a
deq
uate
con
trol
s ex
ist
to p
reve
nt e
rror
• co
ntro
l the
qua
rant
ine
and
des
truc
tion
of p
late
s an
d o
ther
prin
t m
edia
(e.g
. se
greg
ated
sto
rage
, q
uara
ntin
e la
bel
s, e
tc.)
• re
qui
re a
for
mal
ap
pro
val b
efor
e th
e op
erat
or is
al
low
ed t
o ru
n th
e p
ress
• D
ocum
enta
ry
evid
ence
(e.g
. th
at a
ful
l set
of
mat
ched
pla
tes
was
sig
ned
out
to
the
job
, or
in-
pro
cess
che
cks
that
all
colo
urs
are
pre
sent
in t
he
prin
ted
item
)
Rec
ord
s of
:•
chec
ks c
arrie
d o
ut
on m
edia
• p
late
issu
e an
d
retu
rn
• id
entit
y ch
ange
s an
d it
em r
evis
ions
•
app
rova
l to
run
(e.g
. a
sign
ed p
ass
shee
t, f
irst
off
sam
ple
s et
c.)
• re
visi
on o
f ite
ms
• d
isp
osal
of
obso
lete
item
s on
ce r
evis
ed
Sch
emat
ic 3
Sch
emat
ic 4
Sch
emat
ic 4
(a)
Sch
emat
ic 5
Ann
ex A
:A
dm
ixtu
re
Cha
nge
cont
rol
Seg
rega
tion
cont
rols

74
Gen
eral
Syn
op
sis
– C
laus
e 13
Pri
nt a
nd c
onv
ersi
on
pro
cess
es
This
cla
use
cove
rs t
he G
MP
s re
qui
red
to
ensu
re a
deq
uate
con
trol
thr
ough
out
the
mak
e-re
ady
and
pro
duc
tion
pro
cess
es o
f p
rinte
d m
ater
ials
Rea
sons
fo
r th
e ‘e
xtra
’ GM
P r
equi
rem
ents
This
cla
use
add
ress
es a
num
ber
of
spec
ific
asp
ects
of
the
prin
t p
roce
ss t
hat
are
asso
ciat
ed w
ith k
now
n ris
k ar
eas,
i.e.
ris
k of
ser
ious
p
rint
erro
rs a
nd/o
r cr
oss-
cont
amin
atio
n.
The
det
ails
in t
his
sect
ion
of P
S 9
000
rep
rese
nt c
urre
nt b
est
pra
ctic
e, r
outin
ely
seen
by
pha
rmac
eutic
al a
udito
rs d
urin
g au
dits
of
pro
gres
sive
sup
plie
rs w
ho a
re a
war
e of
the
GM
P n
eed
s of
the
pha
rmac
eutic
al in
dus
try.
Thi
s m
eans
tha
t th
e m
ost
dan
gero
us o
f p
ract
ices
, fo
r ex
amp
le ‘
Gan
g p
rintin
g’,
is e
xclu
ded
bec
ause
of
the
hist
oric
al a
ssoc
iatio
n w
ith m
ix-u
ps.
Whi
le it
is a
ccep
ted
tha
t th
e ris
k m
ight
now
be
stat
istic
ally
low
, it
is n
ot z
ero
and
the
con
seq
uenc
es c
anno
t b
e ac
cep
ted
by
the
pha
rmac
eutic
al in
dus
try.
(For
mor
e in
form
atio
n se
e A
nnex
A -
Ad
diti
onal
Exp
lana
tions
of
Key
Con
cep
ts).
How
ever
, in
cer
tain
situ
atio
ns f
or e
xam
ple
, us
ing
prin
ted
mat
eria
l to
set-
up t
he m
achi
ne,
or w
here
tot
al li
ne c
lear
ance
may
be
imp
ract
ical
due
to
the
mac
hine
tec
hnol
ogy,
an
app
roac
h in
volv
ing
a d
ocum
ente
d a
sses
smen
t of
ris
k w
ith s
uita
ble
alte
rnat
ive
secu
rity
chec
ks in
trod
uced
, is
allo
wed
.

75
Pro
cess
inp
uts
Pro
cess
esP
roce
ss o
utp
uts
and
ev
iden
ceC
ross
ref
er t
o
Sch
emat
ic/A
nnex
• C
usto
mer
sp
ecifi
catio
n/d
esig
n re
qui
rem
ents
• A
ny s
pec
ial c
usto
mer
ag
reed
req
uire
men
ts•
Sui
tab
le e
qui
pm
ent,
m
ater
ials
, tr
aine
d s
taff
• B
raill
e sp
ecifi
catio
ns
whe
re r
equi
red
Pro
cess
es w
hich
:•
limit
the
use
of p
rinte
d m
ater
ial f
or m
achi
ne
set-
up p
urp
oses
• in
volv
e a
risk
asse
ssm
ent
of c
hang
eove
r sy
stem
s (e
.g.
whe
re t
he n
atur
e of
the
m
achi
nery
doe
s no
t p
erm
it a
tota
l lin
e-
clea
ranc
e)
• co
ntro
l any
sam
ple
s ta
ken
• ve
rify
any
chan
ges
of p
rint
med
ia b
efor
e co
ntin
uing
pro
duc
tion,
to
ensu
re c
onsi
sten
t q
ualit
y•
ensu
re w
hen
new
pla
tes
are
mad
e fr
om a
new
so
urce
file
, th
e su
bse
que
nt w
ork
is t
reat
ed a
s a
new
bat
ch•
ensu
re w
hen
new
pla
tes
are
mad
e fr
om a
fix
ed/a
pp
rove
d f
ile,
the
sub
seq
uent
wor
k m
ust
und
ergo
a f
irst
off
chec
k•
ensu
re a
ny B
raill
e sy
mb
ols
mee
t sp
ecifi
catio
n•
cont
rol s
tora
ge t
o en
sure
sec
urity
, in
tegr
ity a
nd
trac
eab
ility
•
cont
rol t
he h
old
ing
of s
tock
(whe
n co
ntra
ctua
lly a
gree
d)
• ex
clud
e ga
ng p
rintin
g d
ue t
o p
oten
tial f
or
cros
s-co
ntam
inat
ion
• G
ood
logi
cal f
low
of
mat
eria
ls a
nd c
lear
se
greg
atio
n
Rec
ord
s of
:•
pro
duc
tion
cont
rols
/ch
ecks
• al
l rep
lace
men
t of
p
late
s•
sam
ple
s ta
ken
and
d
isp
ositi
on
Sch
emat
ic 4
Sch
emat
ic 4
(a)
Sch
emat
ic 6
Ann
ex A
:A
dm
ixtu
re
Ris
k as
sess
men
t
Seg
rega
tion
cont
rols

76

77
The Institute of Quality Assurance
Pharmaceutical Quality Group
PS 9004
A Guide to the GMP requirements
of PS 9000:2001 Pharmaceutical packaging materials
ANNEX A - ADDITIONAL EXPLANATION OF KEY CONCEPTS
PS 9000 GMP related text is reproduced in blue

78
Ref: Definition/References Explanation
3.1
3.14
6.2.2
6.3
8.3
9.1.7
9.1.8
9.1.11
10.1.5
10.2
10.3
13.5
PS 9000 Definition The presence within the batch of one or more items not of the nature of that specified by the product description, i.e. cross contamination. Other terms used include ‘mix-up’, ‘rogue’ or ‘stranger’
PS 9000 ReferencesCross-contamination
Competence, awareness and training
Infrastructure
Control of non- conforming product
Line clearance (cross-contamination control)
Segregation controls (cross-contamination control)
Personal hygiene and security
Converter’s flap code
Reel fed materials (general)
Reel fed self-adhesive labels
Gang Printing
Data supplied by the MHRA on their investigations into defective medicine shows that 12 % involve mislabelling in its widest sense (i.e. the product container is printed, or labelled, cartoned or has a descriptive leaflet which is either incorrect or has text which is not compatible with the use of the product), see Table Annex B3.
The source of admixture can originate at the supplier, the pharmaceutical manufacturer and the distribution chain or at the point of dispensing. Some causes might be outside the control of the pharmaceutical manufacturer, e.g. the pharmacist dispensing the product. However the pharmaceutical manufacturer needs to take all possible precautions to prevent mix-up, including:• bar coded items (and detectors)• visual codes• colour/text differentiation• procedural controls at supplier (i.e. the
focus of PS 9000) and at the pharmaceutical manufacturer (i.e. GMP)
• physical segregation
Mislabelling of the medicinal product introduces the risk that the patient will:• use the wrong product• use the product in an inappropriate manner Furthermore, there can be serious damage to market confidence in the company’s products or pharmaceutical products in general. Product recalls are very serious, and may cause withdrawal of a critical medicinal product, otherwise urgently needed in the market. However, the risk of causing patient harm cannot be over-emphasised and should never be forgotten.
One historically recognised cause of admixtures is via ‘gang printed’ sources where two or more different designs are printed on the same matrix at the same time.
This conflicts with a strict GMP principle, which can
Admixture

79
Ref: Definition/References Explanation
be paraphrased, as “it is bad practice for a process to deliberately mix product in the expectation that technology or procedures can and will later differentiate and separate them”. In the USA gang printing can be permitted according to 21 CFR 211.122(f) where “the labelling from gang printed sheets is adequately differentiated by size, shape or color”.
This contrasts with the view in Europe where it is known that colour differences have proved inadequate to prevent mix-ups and best practice therefore prohibits gang printing.
These two approaches might seem contradictory but the requirement detailed in PS 9000 is strictly in keeping with Europe’s view of ‘Risk Assessment’ where the possibility of a critical admixture, even though small, cannot be accepted.

80
Ref: Definition/References Explanation
Change control
The development of a new component or material is a period of extensive design change, with controls and liaison between customer and supplier. This results in specifications approved by customer and supplier, covering raw materials, sources, processes, and tests.
Later in the product life-cycle, variability can occur through uncontrolled changes.
The following are examples of changes which could affect component dimensions, constituents or other characteristics (e.g. surface finish), creating operational problems for the customer, or incompatibility during the shelf-life of the medicinal product:• refurbishment of injection moulds• use of a different production line• raw materials sourced from a new supplier• reprogramming of equipment controlling
computers• replacement equipment
To avoid the risk of uncontrolled changes PS 9000 requires the use of a Change Control process which:• documents all planned changes• assesses potential problems (e.g. Risk
Assessment)• revalidates where required• includes an independent change review and
authorisation• notifies the customer and schedules the
introduction of the change • provides traceability on the change• retains a record
The bottom line of Change Control is to plan and document change to avoid potential issues that could affect supplier and/or customer.
For further information see ‘Rules and Guidance for Pharmaceutical Manufacturers and Distributors 2002’, Section A15.43, A15.44.
PS 9000 Definition A process that ensures changes to materials, methods, equipment and software are prop-erly documented, vali-dated, approved and traceable.
PS 9000 ReferencesRisk Assessment
Control of documents (electronic)
Customer communication
Design and development inputs
Control of design and development changes
Validation of processes for production and service provision
Materials (starting, ancillary and intermediate)
Copy/design change
Replacement print media
PS 9004 ReferencesRisk assessment
Validation
3.7
3.31
4.2.3
7.2.3
7.3.2
7.3.7
7.5.2
9.1.6(b)
12.3
13.4
Annex A
Annex A

81
Ref: Definition/References Explanation

82
Ref: Definition/References Explanation
3.13
5.2
5.3
7.4.1
9.1.10
11.1
12.1
12.5
PS 9000 DefinitionA copy produced without authority with the intention of deceiving a user as to its true origin
PS 9000 ReferencesCustomer focus
Quality Policy
Purchasing process
Waste material (cross-contamination control)
Origination procedures and work areas
Print impression media – General
Quarantine and destruction
Most people will have heard of counterfeit consumer products, such as watches and perfumes, but may not be aware that in some parts of the world there is a serious problem involving counterfeit medicinal products.
The scale of the problem can amount to many billions of dollars, and governments are even sensitive that this practice might eventually pose a serious risk to the normal commercial supply of medicinal products. This is evidenced by the May 2003 FDA directive to pharmaceutical companies supplying the US market, requiring notification within five days of the detection of a counterfeit product.
In the UK the practice is rare but not unknown, as “the last example of a counterfeit product entering the legal supply chain was approximately 20 years ago”. However data from the World Health Organisation, (WHO), shows that “between 1984 and 1999, there were 771 reports of counterfeit drugs, the majority (78%) of these reports came from developing countries”. Similarly “a recent report from Russia showed that 12% of medicines in circulation were counterfeit products and it was as high as 40% in the Ukraine”. (Reference 1).
To illustrate the risk posed to the patient by counterfeit drugs, it is necessary not only to consider the level of incidents but also the lack of drug efficacy or potential toxicity of these products, e.g. for the years 2000-01, of the cases reported to the WHO, “43% had no active ingredients, while a further 21% had low content, 7% had the wrong ingredient and 24% were of generally poor quality” (Reference 2).
It will be easy to understand that while the possible consequences of buying a counterfeit watch are simply disappointment or embarrassment, using a counterfeit drug may carry serious, or even fatal, health risks. Some counterfeit drugs are very crude copies of the original product while others are less so. However the counterfeiter is always keen to
Counterfeit

83
Ref: Definition/References Explanation
use ‘quality image’ packaging that is as close as possible to the original pharmaceutical packaging because people usually believe what is printed on the carton or label.
For obvious reasons there is pressure from enforcement agencies that “the pharmaceutical industry, importers, distributors and consumer organization should adopt a shared responsibility in the fight against counterfeit drugs”. (Reference 2).
In some markets “a variety of anti-counterfeiting measures have been put into place by pharmaceutical manufacturers, for example, the use of holograms and other devices on the packaging”. (Reference 1).
It is therefore clear why the pharmaceutical industry attaches such importance to basic security of print media at the suppliers, to ensure that:• all print media is securely stored.• there is controlled access to the print media
stores• obsolete and no longer required media is made
unusable and disposed of securely• all waste and scrap material is stored securely
and rendered unusable prior to disposal. (see 21CFR 211.122)
By preventing access to legitimate print media, the packaging supplier removes the opportunity for the counterfeiter to use authentic and genuine materials, for the good of the pharmaceutical supplier, customer and product user.
Reference 1 British Pharmaceutical Conference, Harrogate, UK, September 2003.Reference 2 International Pharmaceutical Federation Congress, Sydney, Australia, September 2003.
Acknowledgement for the above quotations is made to The Pharmaceutical Journal (Volume 271, pages 453-454, 465-466).

84
Ref: Definition/References Explanation
Identification and traceability
A fundamental requirement when an operational problem occurs at the pharmaceutical manufacturer, (e.g. material usage problem, market complaint, etc.), is an effective QMS, which assists problem resolution. Operational problems can occur for many reasons, but should the problem be related to a supplier’s material, then it is essential that the total inter-company record system enables traceability of the material on site from delivery and during manufacture at the supplier organization.
Through GMP guidance via the regulatory organizations, e.g. Medicines and Healthcare products Regulatory Agency (MHRA), all pharmaceutical companies are required to use unique and unambiguous lot numbers and material identification codes, since it is the cornerstone of traceability, avoids potential confusion or misunderstanding and allows referral to:• the delivery date and supplier• the quantity received• receipt check results• remaining stock• batch and market usage
A requirement for medicinal products is that records are retained for at least one year after the expiry of the batch in which they were used. For this reason, and that packaging materials may not be used immediately, PS 9000 requires suppliers to keep quality records for at least “5 years from the last date of supply of the batch of packaging material”.Similarly, to provide complete traceability, supplier’s documented quality systems require:• unique material identifications, for raw materials
used in the product, as well as for the product itself
• records showing batch history (e.g. process dates, quantity made, in-process test results)
• change control records• record retention system
This system can then allow tracking back to the same quality data as that for the pharmaceutical
PS 9000 Definition There is no PS 9000 definition for identification
Traceability - The ability to follow data/records logically forwards or backwards within and between organizations, to allow reconstruction of events.
PS 9000 ReferencesQuality Records
Identification and traceability
Preservation of product
Control of non- conforming product
Waste material (cross-contamination control)
Security barcode systems - General
Reel fed materials (General)
Origination procedures and work areas
Print impression media - General
Matched plates
Copy/design change
3.35
3.29
7.5.3
7.5.5
8.3
9.1.10
10.1.1
10.2
11.1
12.1
12.2
12.3

85
Ref: Definition/References Explanation
manufacturer detailed above, e.g.• raw material data, quantity received, receipt test
data • supply source details• where else the same batch of raw material may
have been used• how it was stored• personnel employed on processing and testing• possible environmental monitoring data, (if
relevant)• customer shipment data
Whilst the main purposes of an effective identification and traceability system are to control use and to assist in problem analysis, the supplier also possesses the means to operate a continual improvement process, which aids company financial and quality efficiency.
Retained samples
Batched production and stock holding
13.3
13.7

86
Ref: Definition/References Explanation
Line clearance
3.19
9.1.7
10.7
11.1
13.2
Annex A
PS 9000 Definition The assurance that a production facility (line), and its associated working area is completely cleared of all materials, waste, products, samples, documentation, etc. associated with the current production, prior to the introduction of new documentation and materials required for commencement of the next production run.
PS 9000 ReferencesLine clearance (cross-contamination control)
Digital printing
Origination procedures and work areas
Changeover systems
PS 9004 ReferenceAdmixture
A fundamental requirement in the practice of avoiding mix-ups is ensuring that only the materials, documents, etc. as required for that product, are on a line or machine, or in a working area at any one time. This material or document has to be that allocated and issued for that particular batch or job. Virtually all investigations following a mix-up, focus on the batch that preceded the incident product, or was being processed or handled in the vicinity, e.g. an adjacent line.
The obvious risk of inadequate line clearance is that once a rogue material is introduced into a batch it is virtually impossible to reliably detect and remove in subsequent operations. The only practical quality measure is therefore not detection, but prevention, using procedures, checks, counterchecks and training.
The least serious effect of a mix-up is processing difficulties on the pharmaceutical manufacturer’s packaging line, e.g. the rogue components are too large or too small to process properly and they block hoppers and feed chutes. However, even this can disrupt a smooth running process and cause product variability.
The most serious consequences involve a rogue component or material, which is incorporated into the pharmaceutical manufacturer’s product, with potential implications ranging from product instability to patient confusion on usage instructions and market recall.
Line clearance is an essential element in product mix-up prevention and needs to focus on:• input materials on the line from the previous
batch• samples and waste from the previous batch• documents on the line from the previous batch
(e.g. process instructions or test procedures)
Line clearance activities need to be documented and cover all areas, not just the machine or line, i.e.

87
Ref: Definition/References Explanation
• the whole machine, including hoppers, conveyors, reject stations, etc.
• the floor around the line• benches, cupboards, shelves, etc. around the
line• pallets, etc.
PS 9000 calls for a dual approach to line clearance, i.e.• a recorded line clearance at the end of each
production run• an independent recorded check immediately
prior to the next run.
Similar security precautions are needed when there are temporary line stoppages to ensure that other operations, e.g. maintenance, do not accidentally introduce rogue materials, which, because the process is in the middle of a run, is thought to be secure.

88
Ref: Definition/References Explanation
Risk assessment
3.31
4.2.3 (d)
7.3.1
8.3
9.1.1
9.1.2
9.1.4
9.1.5
9.1.10
9.1.11
9.2.1 d)
13.2
PS 9000 DefinitionA documented assessment of potential failures or faults arising from new or changed processes, materials, equipment or facilities, which could affect product quality, performance or use
PS 9000 References(References to risk and/or risk assessment)
Control of documents (electronic)
Design and development planning
Control of non- conforming product
General
Facilities design
Cleaning
Pest Control
Waste material (cross-contamination control)
Personal hygiene and security
Minimum environmental conditions
Changeover systems
Most companies will have experienced the situation where a response to a process problem, or a change, results in a second, sometimes more serious problem. Often, the secondary problem could have been avoided if the change or the original process problem had been given proper consideration and planning.
This consideration is known as Risk Assessment, and its application is best explained by two typical examples: 1. Batch Reworking/Sorting
When tests revealed a ‘cosmetic’ defect in a printed component, reworking the batch introduced a critical ‘rogue’ component, due to inadequate rework area cleandown from the previous job.
The initial defect was minor, but the ultimate consequences of an undetected rogue component reaching the patient could be fatal.
A risk assessment of the rework process must cover the areas specified in PS 9000 Clause 8.3. However, the rework/sorting process may introduce other GMP risks related to, e.g. infrastructure, competency, work environment, and so an assessment should consider risks of:• a mix-up with other materials• contamination or damage from further handling• incomplete rework due to poor training• difficulty in detecting the difference between
good and bad product• inadequate defect description• poor lighting, poor job separation, poor labelling
in inspection area• discontinuous sorting process (over several days
or by different staff)• poor separation between sorted/unsorted
material• poor labelling of sorted/unsorted product
The risk assessment should include complete batch

89
Ref: Definition/References Explanation
ISO 9004 ReferenceProduct and process validation and changes
PS 9004 ReferencesChange control
Validation
7.1.3.3
Annex A
Annex A
documentation, i.e. details of the product and problems, lot number and quantity, defects involved, deadline, etc. From this a suitable plan of rework can be generated which includes:• identification and segregation of the batch,
(before, during and after sorting)• inspection area cleandown checks• special lighting needs, tools and equipment to
assist rework• documentation of the sorting process, the
objective and data collection form• training of staff (using a controlled example of
the defective to illustrate the problem)• secure area for removed defectives• repackaging (and relabelling) of sorted product• area clean down, on completion and recorded
defectives disposal• final process release check (matching that for a
new batch)• records (of rework details included in batch file)• process revision (where practical)• notification to customers (where required)• completion reviews
It is important that management ensures that the urgency to act quickly to rectify defective product does not compromise documented procedures designed to avoid admixtures or introduce other risks to product quality.
2. Process Change
A refurbished or replacement tool/mould is returned to production use without a “full” examination of its potential effect on the product, resulting in surface finish differences compared with pre-change product. This change affected the customer’s process, e.g. the fit with other components, processing at normal speed.
This type of ongoing change or upgrade is not uncommon, and it is known that a similar surface finish problem can be triggered by supplier changes to running speeds, cycling conditions, the

90
Ref: Definition/References Explanation
software of process controllers, i.e. computers or programmable logic controllers (PLC’s).
As for the first example, assessment of the change should involve:• documentation details of the change including
hardware, software, reasons for change etc.• risk assessment technique to be used, e.g.
failure mode and effect analysis (FMEA)• plan of controlled change to include risk
assessment of both the supplier’s process and customer’s process, e.g.i. Implications to the supplier’s process:
- installation/changeover- process speeds - process conditions - yield wastage - staff retraining - tool identification - specification and procedural revisions
ii. Implications to the customer’s process:- phase in (old and new requirement) - compatibility (with product, other components)- differences (visual, performance, dimensions)- change schedule (old stock, etc.)- notification (to regulators, market, etc.)- specification (revisions to documents)- machine set-up (changes)
Details of the eventual change introduction can include:• management (responsibility for change
implementation and customer notification)• documentation (details of the change, reason or
purpose e.g. what, how, when, why)• review (past specification, performance
characteristics, issues, requirements, change schedule)
• reference or involvement of customer• risks (documented, classified, with responses,
actions and cures)

91
Ref: Definition/References Explanation
• compliance of the ‘agreed’ specification, to meet:- organizational needs- customer’s needs
• change implementation plan
Outputs from the change implementation can include:• testing (at customer or supplier)• trials and/or revalidation (at customer or supplier,
in market)• revisions (and additions to requirements)• records (of performance, new specification,
controls, etc.)• modifications (details)• phase in (changeover schedules)• phase in (controls and schedule to monitor and
measure performance compliance)• change of identification numbers (reference
numbers to control new item)• revised specifications (supplier and customer
agreed)• completion review
For further information see 21CFR 211.115 Reprocessing

92
Ref: Definition/References Explanation
3.37
8.3
9.1.2
9.1.8
11.1
13.1
PS 9000 Definition Note: There is no definition in PS 9000
PS 9000 ReferencesWorking (work) areas
Control of nonconforming product
Facilities Design
Segregation controls (cross-contamination control)
Origination procedures and work areas
Print machine set up (make-ready).
To avoid the risk of product cross-contamination, it is essential that, in addition to normal good material handling disciplines, there is adequate process segregation during manufacture.
PS 9000 states, “working areas should be defined and segregated”.
Often in practice, a separation gap between processes of 1 – 2 metres is maintained, with the actual process areas identified by lines marked on the floor. This can be wasteful of space and is often abused when pallet materials straddle the delineating segregation lines.
A more absolute method, routinely used in the pharmaceutical industry, is to use a physical barrier between processes - PS 9000 states:• “Where physical barriers are used to segregate
working areas they shall be a minimum of 1.5 metres high and continuous from floor level.”
• “Inspection and sorting areas shall be defined and segregated by physical barriers of a minimum of 1.5 metres high and continuous from floor level.”
The purpose behind these requirements is that most working conveyor belts are about 1m high and similarly pallets are stacked to about 1m high, hence, a 1.5 metre barrier effectively prevents items falling over the barrier. Making it continuous from floor level, i.e. sealed to the floor prevents the transfer of items passing under the barrier. Making the barrier solid, with no openings or holes, prevents passage of items through the barrier (if mesh barriers are used, the hole size needs to be sufficiently small to prevent product passing through).
These measures have been proven effective in preventing cross transfer i.e. preventing admixture, mix-ups, rogues or strangers.
For further information see 21 CFR 211.130(a).
Segregation controls

93
Ref: Definition/References Explanation

94
Ref: Definition/References Explanation
Validation (and Qualification)
QualificationBefore implementing a validation program it is necessary to link into the more comprehensive ‘qualification system’ which is shown in the diagram below:
ValidationThrough experience of operational and in-market problems, the pharmaceutical industry has found that validation is the only dependable method to fully understand the process capability and to ensure process control. It should be an essential tool at the supply organization and is a requirement of PS 9000, for reasons detailed below.
Inconsistent product quality can often be traced to an inadequately controlled process which has not been validated and where process parameters have not been fully defined.
Validation can be viewed as a key part of a defect prevention system and as such requires no justification when management objectives are for an efficient process. However, the sound operational
3.36
4.2.3
7.3.6
7.5.2
9.2.3
10.2
10.4
10.5
10.6
10.7
PS 9000 Definition Documented confirmation that a procedure, process, equipment, material, activity or system, performs as intended and achieves the expected results in accordance with predefined acceptance criteria.
PS 9000 ReferencesControl of documents (electronic)
Design and development validation
Validation of processes for production and service provision
Enhanced cleanroom conditions
Reel fed materials (general)
Leaflets
Board products (cartons, cards, wallets, catch cards etc.)
Combination products
Digital printing
Specification Qualification (SQ)
Installation Qualification (IQ)
Operation Qualification (OQ)
PerformanceQualification (PQ)
Determine what is requiredSpecify requirement
Check on receiptInstall as appropriate
Trial processes
Evaluate first three batches for:• process performance• process compliance• product compliance

95
Ref: Definition/References Explanation
reasons for validation includes ensuring:• the process is controllable and operates
consistently with defined control settings• compliance with the product requirements,
i.e. variability only occurs within the agreed specification
• compliance with known regulatory requirements, (where applicable)
• change is controlled• prevention of problems for the organization and/
or customer
Validation is not simply for production processes, but is relevant to test equipment used on line or in the laboratory, as well as computer hardware and software.
From the above detail it is clear that there is a need for a validation protocol before introducing new equipment. To establish a comprehensive and focused programme, it is beneficial but not essential for the organization to liaise with the pharmaceutical customer. Here there is a wealth of validation experience and the motivation exists to assist suppliers in defect and problem prevention.
A general validation procedure is practical and often used, (e.g. an across the process Validation Master Plan or VMP), which can define the broad scope of the validation approach. This can then be customised for any specific item needing assessment. The VMP can cover:1. The scope• New process or test equipment that contributes
to product quality or process efficiency, e.g.i. a new machine (e.g. an injection blow
moulding machine)ii. a new machine tool, (e.g. a 24 impression
mould replacing a 16 impression mould) • New process software (e.g. new process
software from the existing supplier)• New starting materials, (e.g. replacement
equivalent grade from a new supplier)• Others where appropriate
PS 9004 ReferencesChange control
Risk assessment
Annex A
Annex A

96
Ref: Definition/References Explanation
PS 9000, reference 7.5.2 identifies specific examples where validation and revalidation are required.
Similarly, control of change (see Annex A-Change Control), needs to be incorporated within the validation system, when the risk assessment, (see Annex A-Risk Assessment), identifies that change may introduce a significant risk of problems (to organization or customer).
The VMP should include changes to, e.g.:• equipment hardware, e.g.
i. refurbished moulds or toolsii. full maintenance strip down and rebuildiii. relocation of equipment to a new position
• process materials, e.g.i. revised specification material from the
existing supplierii. the same specification material but made
through a different process.iii. process materials delivered in a different
format, (e.g. quantity, presentation etc.)
2. Responsibilities for, e.g.:• the validation protocol• the trials• the testing• final acceptance authorisation
3. Equipment parameters, e.g.:• operating conditions - according to theory or
practice• control settings - running speeds/cycle times,
e.g. temperature/pressure
4. Product Specification, e.g.:• confirmation - against existing specification• compliance - against a draft specification• variability - inside or outside specification
5. Trial conditions, e.g.:• duration• output quantity/units to be produced• environmental controls

97
Ref: Definition/References Explanation
6. Sample evaluation, e.g.:• Tests, (e.g. visual/measurement/function) • units examined, e.g. from beginning, middle and
end of trial)• test comparison with previous data• test location, e.g. on site and/or at customer• numbers retained, e.g. for future reference
7. Documentation responsibilities, e.g.:• validation protocol preparation • report preparation• report circulation (to customer where
appropriate)• report and protocol archiving• revised operating/test procedures (where
applicable)
8. Training, e.g.:• revised training where applicable, (and revised
training documentation)
9. Revision authorisation, e.g.:• to process conditions and settings (organization)• to product specification (organization and
customer)• to procedures
10. Validation review, e.g.:• completion review including any lessons from
the validation process
The success of an operational validation system can be measured through the lack of problems encountered following the introduction of new hardware /software or after a change.
However even where a minor problem might be encountered, the details can be reviewed and the lessons built into the validation practice, as an example of continual improvement.

98

99
The Institute of Quality Assurance
Pharmaceutical Quality Group
PS 9004
A Guide to the GMP requirements
of PS 9000:2001 Pharmaceutical packaging materials
ANNEX B - OTHER EXTERNAL GMP REFERENCES

100
B1. Code of Federal Regulations 21 Part 211 Current Good Manufacturing Practice for Finished Pharmaceuticals
(www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm (select CFR Part No. 211 and search))
Ref.21 CFR
Summary PS 9000 Cross Reference
211.42(a) Building design to facilitate cleaning and maintenance
9.1 Facilities, equipment and operating conditions
9.1.2 Facilities design
211.42(b) Adequate building space for orderly placement of equipment and material to prevent mix-ups
9.1.2 Facilities design
211.63 Equipment design to facilitate cleaning and maintenance
9.1.3 Equipment and maintenance
211.67 Equipment cleaning and maintenance
9.1.4 Cleaning
211.68(b) Electronic equipment change control
4.2.3 Control of documents (Electronic)
4.2.4 Control of records
211.80(b) Off floor material storage 9.1.2 Facilities design
211.115 Procedures for reprocessing 9.1.10 Waste material (cross- contamination control)
8.3 Control of non-conforming product
211.122(e) Disposal of obsolete material 9.1.10 Waste material (cross-contamination control)
12.5 Quarantine and destruction
211.122(f) Use of gang printed materials 13.5 Gang printing
211.130(a) Prevention of mix-ups and cross-contamination by physical or spatial separation
9.1.2 Facilities design 9.1.8 Segregation controls (cross-
contamination control)
211.180 General Requirements (records and reports)
4.2.4 Control of records
211.182 Equipment cleaning and use log 9.1.7 Line clearance (cross-contamination control)
11.1 Origination procedures and work areas
211.204 Returned products 8.3 Control of non-conforming product

101
EU Guide to Pharmaceutical GMP
Summary PS 9000 Cross ReferenceGeneral information
Chapter 3.4 Protection against infestation
9.1.5 Pest control
Chapter 3.8 Avoidance of cross-contamination
9.1 Facilities, equipment and operating conditions
9.1.1 General9.1.2 Facilities design
Chapter 3.36 Equipment cleaning 9.1.7 Line clearance (cross-contamination control)
9.1.4 Cleaning
Chapter 4.8 Retention of records 4.2.4 Control of records
Chapter 4.22 Sampling procedures and precautions
8.2.4 Monitoring and measurement of product
Chapter 5.22/5.23 Process changes 7.5.2 Validation of processes for production and service provision
Chapter 5.35/5.45 Line clearance checks 9.1.7 Line clearance control (cross-contamination control)
Chapter 5.54 Sample disposal 8.2.4 Monitoring and measurement of product
Chapter 5.43 Obsolete material disposal
9.1.10 Waste control (cross-contamination control)
Chapter 5.44 Physical segregation to minimise risk of cross contamination
9.1.2 Facilities design9.1.8 Segregation controls (cross-
contamination control)
Chapter 5.62 Reprocessing of rejected product
8.3 Control of nonconforming product
Chapter 6.14 Sample retention 8.2.4 Monitoring and measurement of product
Chapter 8 Complaints 8.3 Control of nonconforming product
Annex 8 (3) and (5) Supplier GMP 5.2 Customer focus
Annex 11 (2) Computer system validation
4.2.3 Control of documents (electronic)7.3.6 Design and development
validation 7.5.2 Validation of processes for
production and service provision
B2. Rules and Guidance for Pharmaceutical Manufacturers and Distributors 2002
(http://pharmacos.eudra.org/F2/eudralex/vol-4/home.htm)

102
EU Guide to Pharmaceutical GMP
Summary PS 9000 Cross ReferenceGeneral information
Annex 15 (43) and (44) Change control 4.2.3 Control of documents (electronic)7.2.3 Customer communication7.3.2 Design and development inputs7.5.2 Validation of processes for
production and service provision
Annex 16 (8) Finished product batch certification that manufacture and testing are in compliance with the marketing authorisation
7.2 Customer related processes7.4 Purchasing
Annex 18 (9) Labelling of active pharmaceutical ingredients
7.4.3 Verification of purchased product7.5.3 Identification and traceability7.5.5 Preservation of product

103
Defect Type Report Received
This table includes all formal defect investigations carried out by the MHRA, whether they were confirmed as quality defects or not. Some of these investigations resulted in batch recalls.
It does not include all defects reported to the MHRA or all investigations carried out by licence holders and not reported to the MHRA.
Some headings are very similar, and in most cases could be combined, e.g. product mix-up is usually where the wrong tablets are found in a pack.
Acknowledgement to:The Defective Medicines Report CentreMHRAMay 2003
Year 02/03 Year 01/02 Year 00/01
Noncompliance to product specification (stability)
6 19 16
ADR (adverse drug reaction)
9 16 23
Product mix-up 3 14 18
Noncompliance to product specification (analytical)
11 12 9
Other 77 10 29
Other particulates 15 9 14
Wrong data 0 9 18
Label mix-up 6 8 13
Container 5 7 6
Leaflet Missing/mix-up 12 5 9
Closure Fault 1 4 3
Precipitation 2 4 1
Label details missing 6 4 0
Lack of efficacy 12 4 0
Lack of sterility assurance 5 3 2
Re-labelling error 21 3 5
Glass particulates 6 2 2
Adulteration 1 2 0
Under fill 1 2 5
Wrong data/poor print 3 1 0
Fibre particles 1 1 0
Solubility 0 1 0
Wrong fill 3 0 3
Bacteria contamination 3 0 2
Mould Contamination 1 0 1
Label Missing 2 0 1
Poor Print 0 0 3
Total 212 140 183
B3. MHRA Defect Investigations

104
INDEX PAGE
AAdmixture 12, 14, 28, 30, 31, 56, 60, 66, 68, 69, 78, 79, 86, 89, 92Agreements 5, 25, 50, 69, 71Archive 38Artwork 12, 25, 27, 56, 57, 69, 70, 71, 72, 92Audit 23, 25, 26, 28, 30, 38, 40, 42, 44, 45, 47, 55, 61, 65, 67, 69, 74
BBar code (see also codes) 24, 29, 69, 68Bar code (readers, scanners) 28, 30, 69, 78, 83Batch samples (see also samples)Bench mark 42, 43, 61Bioburden 48Blister (pack) 25, 28, 29, 32Business processes xv, 2Bottles 26, 29, 30
CCalibration 16, 54, 58Case studies ix, 21-32Certificate/certification 17, 30, 41, 45, 55Change (control) 6,10, 24, 25, 26, 49, 50, 51, 53, 56, 67, 73, 80, 84, 88, 89, 90,91, 94, 95, 96, 97, 100, 101Clearance (see also line clearance) 13, 15 Clean areas (rooms) 48, 94Cleaning xvi, 48, 54, 67, 88, 89, 100, 101Closures 30Codes (see also bar codes) 28, 30, 68, 71 , 72Code of Federal Regulations (CFR) xiv, 79, 83, 91, 92, 100Complaints 24, 51, 61, 62, 63, 69, 84, 101Contaminate (contamination) 15, 26, 27, 47,48, 56, 60, 66, 67, 68, 69, 70, 88

105
INDEX PAGE
Continual improvement 3, 23, 39, 41, 42, 45, 57, 59, 65, 85, 97Contract 5, 25, 30, 51, 61, 75Corrective (action) 31, 44, 61, 65Counterfeit 40, 56, 82, 83Cross contamination 40, 41, 46, 47, 65, 66, 68, 72, 74, 78, 82, 83, 86, 88, 92, 100, 101
DDefective Medicines Report Centre 103Defect (prevention) 94Design 4, 13, 24, 50, 52, 53, 58, 66, 67, 68, 70, 71, 72, 73, 80, 84, 88, 94, 100, 101, 102Development 40, 52, 53, 56, 58, 80, 94Disposal (dispose) 40, 72, 73, 83, 100, 101Distribution 18, 19
EEfficiency 44, 68, 85, 94,95Environment xvi, 5, 48, 53, 57, 67, 87Environmental (conditions/control) 41, 57, 66, 67, 85, 87, 88, 96Errors 8, 10, 12, 14, 16, 17, 18, 27, 28, 29, 30, 31, 46, 66, 69, 70, 73, 74, 103European Commission 52Expiry (see shelf life) 84
FFilm 24, 32, 33FMEA (Failure mode and effect analysis) 90Feedback 61, 69Foil 6, 28, 29Food and Drug Administration (FDA) 82
GGang printing 74, 75, 78, 79, 100

106
INDEX PAGE
Gap analysis 46Good Manufacturing Practice (GMP) iii, vii, ix, xvi, 15, 23, 40, 43, 46, 47, 62, 66, 68, 70, 72, 74, 78, 84, 88, 100, 101
H
IIdentification (see also traceability) 7, 8, 27, 51, 55, 56, 57, 58, 67, 69, 70, 72, 73, 84, 85, 89, 91, 102Improve/improvement 32, 44, 45, 59, 64, 65Incidents 31, 67, Infestation xv, 9, 101In process controls 15, 27, 29, 63, 71, 73Insects 15Institute of Quality Assurance (IQA) iv, v, viiISO (International Standards Organization) ii, ix
J
K
LLabels/labelling xv, 7, 8, 15, 17, 19, 24, 25, 26, 27, 28, 29, 30, 31, 58, 69, 78, 83, 89, 102 Laminate 32, 33Leaflets 25, 28, 29, 30, 69Line clearance (see also cleaning) 7, 13, 15, 28, 29, 66, 67, 69, 70, 71, 74, 75, 78, 86, 87, 100, 101
MMaintenance 6, 7, 10, 27, 47, 54, 66, 67, 87, 96, 100Management review 23, 39, 42, 44, 65Marketing Authorisation (MA) 50, 54, 102Material disposal 67,73, 83

107
INDEX PAGE
Media (print) 6, 12, 38, 70, 72, 73, 75, 82, 83, 84MHRA (Medicines and Healthcare products Regulatory Agency) vii, xiv, 78, 84, 103Microbial contamination (moulds) 48, 66, 103Mislabelling 7, 78Mix-ups 9, 12, 23, 28, 46, 72, 74, 78, 79, 86, 88, 92, 100, 103 Moulds (see also tooling) 6, 14, 26, 27, 57, 89, 95, 96
N
OOrange Guide (see Rules and Guidance for Pharmaceutical Manufacturers and Distributors 2002)
PPartners Team xi, xiiPests xv, xvi, 9, 26, 67, 88, 101Pharmaceutical Quality Group i, iv, v, vii, xiiPlan/planning 7, 42, 80Preparation 10Preventive (actions) vii, 23, 27, 30, 31, 44, 65, 66, 70, 86, 94Print (Printing) 12, 13, 14, 15, 18, 25, 26, 27, 28, 29, 68, 69, 70Print impression/ media 12, 13, 70, 72, 73Product Authorisation/ Licence (PL) 32, 50Project team (PS 9004) xi, xiiProcedures (see also SOPs) 3, 16, 26, 28, 29, 30, 31, 37, 38, 43, 46, 55, 61, 63, 66, 67, 86, 90, 97, 100
QQualification (and validation) 69, 94

108
INDEX PAGE
Quality Assurance (QA) 29, 30, 40, 43Quality Control (QC) 8, 17, 28Quality Management System (QMS) ix, x, 2, 37, 38, 39, 42, 43, 44, 45, 46, 59, 64, 65, 84Quality manual 38Quality plan 49Quality policy 39, 41, 42, 44, 65, 82Quarantine 29, 54, 55, 63, 73, 82, 100
RRecall 25, 28, 47, 78, 86, 103Reconciliation 17Records xvi, 12, 13, 29, 40, 49, 51, 53, 55, 56, 57, 58, 61, 63, 65, 67, 69, 71, 73, 75, 80, 84, 87, 89, 91, 100, 101Recovery (reprocessing/rework) 25, 62, 63, 88, 89, 101Regulations/regulatory vii, x, 25, 38, 40, 42, 43, 47, 48, 50, 51, 52, 53, 84, 90, 95Registered product 50Returned product 62, 63, 100Risk vii, 4, 27, 30, 47, 52, 53, 54, 56, 60, 66, 68, 74, 78, 82 ,89, 90Risk areas ix, x, xv, 46, 52, 53, 60, 74, 92Risk assessment 26, 53, 62, 63, 65, 75, 79, 80, 88, 89, 90, 96Rogues (see also admixture, mix-ups, strangers) 8, 29, 30, 66, 78, 86, 87, 88, 92Rules and Guidance for Pharmaceutical Manufacturers and Distributors 2002 xiv, 52, 60, 72, 80, 101
SSafety 24, 52, 53, 56, 66Samples/sampling 8, 13, 16, 28, 52, 53, 60, 61, 73, 75, 86, 97, 101

109
INDEX PAGE
Security 7, 40, 41, 47, 54, 60, 61, 62, 63, 68, 69, 70, 72, 74, 75, 87Segregate/segregation 14, 15, 18, 29, 30, 31, 47, 62, 66, 67, 70, 73, 75, 78, 89, 92, 100, 101Separation 88, 992, 100Shelf life 38, 56, 80Specification xvi, 6, 17, 29, 40, 49, 51, 53, 55, 57, 58, 63, 66, 69, 71, 72, 73, 75, 80, 90, 91, 96, 97, 103Stability 25, 33, 50, 52, 66, 86Standard Operating Procedures (SOPs) (see also procedures) 3, 66Storage 18, 26, 27, 53, 62, 69, 71, 75, 83, 100Sterile/sterilization 30, 48Syringe 26, 27, 30Sub-contract 54, 55
TThe Pharmaceutical Journal 83Tool/tooling 10, 11, 26, 27, 53, 89, 90, 95, 96Traceability (see also identification) 38, 56, 57, 62, 69, 71, 72, 75, 80, 84, 85Training ix, x, 2, 15 ,16, 24, 25, 28, 29, 30, 32, 37, 38, 40, 43, 46, 53, 57, 67, 75, 78, 86, 89, 90, 97Transit (damage) 8, 9, 18, 19, 31
U
VValidation (see also qualification) 4, 10, 16, 24, 26, 38, 48, 49, 52, 53, 56, 57, 65, 69, 80, 89, 91, 94, 95, 96, 97, 101, 102Vials 25, 26

110
INDEX PAGE
WWarehouse 8, 26, 31, 57Waste (materials) 14, 15, 23, 25, 40, 57, 67, 82, 83, 84, 86, 88, 90, 101World Health Organisation (WHO) 82

The Institute of Quality AssurancePharmaceutical Quality Group
A Guide to the GMP requirements of PS 9000:2001 Pharmaceutical
packaging materials


















