
Provided by - Cure4Kids · of them developed acute myeloid leukemia (AML). A new classification for...
28
Provided by Myelodysplastic Syndromes Lead contributors: Dr. Luiz Fernando Lopes Brazilian Cooperative Group for Pediatric Myelodysplastic Syndrome São Paulo, Brazil A. Introduction The number of myelodysplasia cases in children has increased during recent years. Because of this increase, the need to create interdisciplinary cooperative groups consisting of professionals with expertise in different aspects of the disease is critical for the development of effective therapies. The Brazilian Cooperative Group for Pediatric Myelodysplastic Syndrome (GCB-SMD- PED) was formed in January 1997. The group’s objective is to study cases of confirmed or suspected myelodysplasia in patients younger than 18 years of age.
-
Upload
dangkhuong -
Category
Documents
-
view
215 -
download
0
Transcript of Provided by - Cure4Kids · of them developed acute myeloid leukemia (AML). A new classification for...

Provided by
Myelodysplastic Syndromes
Lead contributors:
Dr. Luiz Fernando Lopes
Brazilian Cooperative Group for Pediatric Myelodysplastic Syndrome
São Paulo, Brazil
A. Introduction
The number of myelodysplasia cases in children has increased during
recent years. Because of this increase, the need to create interdisciplinary
cooperative groups consisting of professionals with expertise in different
aspects of the disease is critical for the development of effective therapies. The
Brazilian Cooperative Group for Pediatric Myelodysplastic Syndrome (GCB-SMD-
PED) was formed in January 1997. The group’s objective is to study cases of
confirmed or suspected myelodysplasia in patients younger than 18 years of
age.

Page 2 of 28
B. History
The myelodysplastic syndromes (MDS), initially thought to be restricted to
adult patients, comprise a heterogeneous group of hematological diseases. In
1949, Hamilton-Patterson 1 described three patients who presented with an
anemic phase prior to acute leukemia and named this condition preleukemic
anemia. In 1953, Block et al. 2 introduced the term preleukemia to identify a
group of 12 patients with hematological disorders who presented with a
peripheral blood cytopenia of poorly-defined and variable chronicity, that
evolved into acute leukemia. In 1958, Dameshek and Baldini 3 described
common morphologic alterations among the diverse pathologies of patients
displaying symptoms of leukemia, such as a low blast cell percentage in the
bone marrow.
In the early 1960s, acute leukemia characterized by a low blast percentage
(between 5% and 10%) and a slow evolution was termed smoldering or oligoblastic
leukemia. 4 During the 1970s, based on the observation of patients with
myelomonoblastic acute leukemia Linnan and Bagby 5 used the term preleukemia or
hematopoietic dysplasia when referring to patients who exhibited anemia
refractory to iron treatment and dysplasia of one of the hematopoietic cell
lineages and who carried less than 5% blast cells in their bone marrow. Dreyfus
et al. 6,7 also described refractory anemia (RA) with an excess of myeloblasts in a

Page 3 of 28
group of 26 patients. On average, these patients survived 20 months and 28%
of them developed acute myeloid leukemia (AML).
A new classification for acute leukemia was proposed and accepted by the
French-American-British (FAB) Cooperative Group during their 1976 meeting.
The proposal was accepted and was utilized until the present time. The FAB
classification system subdivided acute leukemias into two groups: a prevalent,
acute group, which included leukemic patients with more than 50% blast cells in
their bone marrow who required immediate treatment, and a second group that
included rarer, acute leukemias with different features diagnosed in patients
older than 50 years. These latter leukemias were designated MDS; patients with
these syndromes typically presented with symptoms of variable duration, had
less than 30% blasts in their bone marrow, and did not need urgent therapy. An
increase in the bone marrow blast count was most commonly evidenced by
increased numbers of myeloblastic and promyeloblastic cells. These patients
were initially classified and subdivided into two groups: RA patients with excess
blasts (RAEB) and patients with chronic myelomonocytic leukemia. 8
Many cases that presented with morphologic alterations both in bone
marrow and in peripheral blood cells were described between 1976 and 1980.
They were all classified as MDS according to their morphological findings,
despite variations in the risk of a blastic phase transformation.

Page 4 of 28
B. References
1 Hamilton-Patterson JL. Preleukemia anaemia. Acta Haematol 1949;2:309–16.
2 Block M, Jacobson LO, Bethard WF. Preleukemic acute human leukemia. JAMA
3 Dameshek W, Baldini M. The Di Guglielmo Syndrome. Blood 1958;13:192–4.
4 Rheingold JJ, Kaufman R, Adelson E, Lear A. Smoldering acute leukemia. N Engl J Med 1963;268:812–5.
5 Linnan JW, Bagby GC. The preleukemic syndrome (hemopoietic dysplasia). Cancer 1978;42:852–64.
6 Dreyfus B, Rochant H, Sultan C, Clauvil JP, Yvart J, Chesneau AM. Less anemies refractaires avec exces de myeloblasts dans Lamoelle: etude de onze observations. Nouv Presse Med 1970;78:359– 64.
7 Dreyfus B. Preleukemic states. I. Definition and classification. II. Refractory anemia with an excess of myeloblasts in the bone marrow (smoldering acute leukemia). Nouv Rev Fr Hematol Blood Cells 1976;17:33–55.
8 Bennett JM, Catovsky D, Daniel MT et al. (The French-American-British Cooperative Group - FAB). Proposals for the classification of the acute leukaemias. Br J Haematol 1976;33:455–8.
C. Definition and Diagnosis
C. 1 Morphology and Cytogenetics
MDS comprise a group of bone marrow clonal disorders originating in the
stem cells and characterized by heterogeneous clinical and laboratory findings.
These features can lead to a numerical and/or functional failure of bone
marrow, independent from the cellularity of the marrow.
Although this entity is less prevalent in children than in adults, clinical
presentation in childhood has distinctive and peculiar features that allows for a

Page 5 of 28
systematic and comprehensive approach to diagnosis and treatment of the
disease based on established inclusion and classification criteria.
MDS are included among the myeloproliferative diseases. Because of their
common features, differential diagnosis may be difficult. At the acute pole of
this class, we find more proliferative, less accumulative disorders such as AML.
At the chronic pole are the more accumulative, less proliferative disorders such
as the chronic myeloproliferative disorders. Differential diagnosis must also
include other primary hematological disorders and/or systemic diseases with
secondary bone marrow involvement.
Characteristically, these myelodysplastic disorders demonstrate dysplastic
morphologic features, both in bone marrow and peripheral blood cells of
erythrocytic, monocytic, megakaryocytic, or granulocytic lineage. These
features have not been uniformly defined by hematopathologists. Dysplastic
features include
• Erythrocytic lineage: megaloblastoid maturation, nuclear budding, and
multinucleated forms appear in the bone marrow; polychromasia may be
seen in peripheral blood.
• Monocytic lineage: an increased number of monocytes in bone marrow
and peripheral blood, abnormal granulation with persistence of
azurophilic granules, and hemophagocytosis and abnormal nuclei are
seen in the bone marrow.

Page 6 of 28
• Megakaryocytic lineage: giant platelets in peripheral blood and bone
marrow abnormal megakaryocytic nuclei are present in the bone marrow.
• Granulocytic lineage: increased number of myeloblasts are seen in the
bone marrow, along with dysgranulopoiesis, nuclear hyposegmentation,
and megaloblastoid maturation; dysgranulopoiesis, hypogranulation, and
Pelger–Huet-like abnormalities may be observed in peripheral blood.
In spite of technological advances, a diagnostic marker for these
syndromes does not yet exist. Diagnostic criteria are still based on an
exhaustive clinical examination and a rigorous peripheral blood and bone
marrow examination. Diagnosis of MDS is even more difficult because cell
lineage dysplasia may be found as an epiphenomenon in other clinical
conditions as well. The more frequent conditions are inflammation, infections
(especially viral infections), use of drugs or medicines (anticonvulsants),
exposure to chemical agents or heavy metals, mitochondrial diseases, and
anemia due to B12 vitamin/folate or iron deficiency. The more common
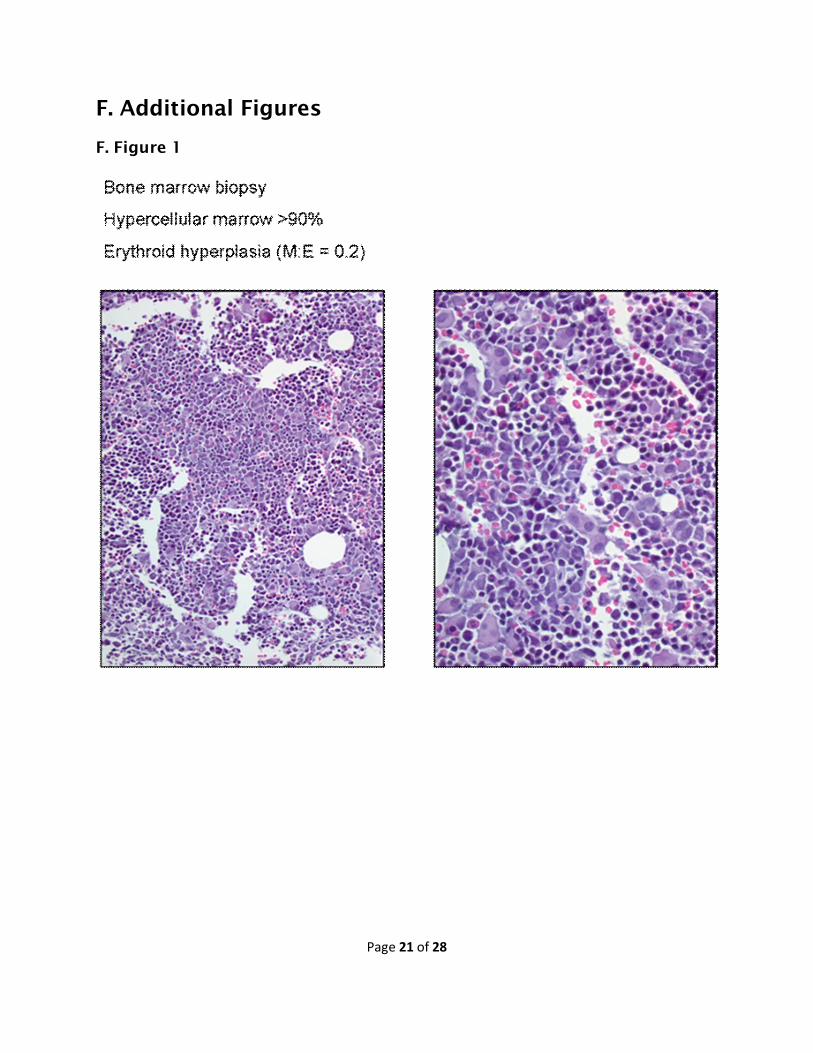
laboratory and morphological findings are (Figures 1 & 2):
1. Peripheral blood cytopenia and a generally hypercellular bone marrow
2. Peripheral blood cytopenia and a hypocellular bone marrow, although
with one or more lineage dysplasias
3. Reactive (nonclonal) peripheral blood monocytosis

Page 7 of 28
4. Borderline MDS, a bone marrow with a low blast count that is not enough
to be characterized as a clearly established leukemia
Cytopenia of one or more lineages and monocytosis are the two most
frequent findings in peripheral blood counts. Neutropenia (defined as an
absolute neutrophil count [ANC] < 1000 per mm 3 of blood) and
thrombocytopenia (ANC <100.000 per mm 3 of blood) usually lead to a bone
marrow assessment. Anemia as an isolated finding requires ruling out several
etiologies, and bone marrow assessment is usually performed when anemia
persists despite treatment.
Neutropenia in a previously healthy patient is usually secondary to
infection stress. Bone marrows in these patients show findings compatible with
vitamin B12 and/or folate deficiency. Adequate supplementation leads to white
blood cell count recovery within the first 24 to 48 hours. These findings can
also be seen in children carrying the genetic mutation for Down syndrome.
Monocytosis, which frequently occurs during infections or immune-
mediated diseases, must be distinguished from MDS subtypes such as juvenile
myelomonocytic leukemia and Down syndrome-associated transitory
myeloproliferative disorder.
In cooperative European studies, clonal cytogenetic abnormalities were found in
about 50% of cases with a predominance of chromosome 7 involvement

Page 8 of 28
(monosomy or deletion [-7/7q-]). In a US study that enrolled 101 children with
MDS or myeloproliferative syndrome , clonal abnormalities were found in 80%
and were most frequently associated with 7/7q (52.4%) and 21 trisomy (+21,
constitutional or acquired). Results of the GCB-SMD-PED for 77 patients (47 with
confirmed MDS) are shown in Figure 3.
C. 2 Pediatric classifications and prognostic scores
A study from the Hematology and Oncology Department of the Hospital
for Sick Children, University of Toronto, Toronto, Canada, 1 has proposed a
pediatric classification system called the “CCC Classification” (category,
cytology, and cytogenetic) based on three main characteristics related to their
de novo origin categories: secondary and/or associated to constitutional
alterations; cytological criteria, with or without evidence of dysplasia; and
cytogenetic criteria (Table 1). Patients’ results were related to prognostic
features; patients with an advanced form of the disease associated with a
cytogenetic abnormality had the worst prognosis. The study concluded that the
use of this classification system would allow standardization of the clinical
approach to the disease and criteria for scientific publications.
Hasle et al. 2 published a pediatric classification proposal for MDS and
myeloproliferative disorders based on the World Health Organization (WHO)
classification 3 and determined the minimum criteria for childhood MDS
diagnosis (Tables 2 & 3). Three published studies used the IPSS (International

Page 9 of 28
Prognostic Score System) score for pediatric MDS. 4-6 The Japanese group (Sasaki
et al. 2001 4 ), the United Kingdom group (Passmore et al. 5 ), and the European
group (Hasle et al. 6 ) concluded that IPSS scoring has many limitations and that
none of its criteria represented significant prognostic factors.
C. Figure 1
Bone Marrow aspirate: atypia findings.
Erythrocytic lineage
Granulocytic lineage

Page 10 of 28
Megakaryocytic lineage
C. Figure 2
Bone Marrow Biopsy: Histology
Refractory Anemia
Trilineage Dysplasia

Page 11 of 28
Cellularity Heterogeneity
Reticulin Fibers
C. Figure 3
77 children with cytogenetic studies enrolled in the GCB-SMD-PED from 08/1997 to 08/2006. 71 available, 6 without metaphases.
Diagnosis Number available/total
clonal abnormality
MDS 42/47 28.6% 7/7q, +8. other Down syndrome + AML
5/5 60% +8, complex
AML 4/5 50% T(8;21), 7q MPS (2 CML) 3/3 100% Ph, t(1;5) Aplasia 2/2 0% Reactive, Normal 6/6 0% Inconclusive 10 10% 17, +21: 3 cases

Page 12 of 28
C. Table 1
Category Cytology Cytogenetic
Idiopathic de novo Refractory cytopenia with 1 or more dysplastic series and with ringed sideroblast (RCRS)
Abnormal
Constitutional alterations Refractory cytopenia with 1 or more mildly dysplastic series (RC)
Normal
Secondary myelodysplastic syndromes (MDS)
Refractory cytopenia with 1 or more severely dysplastic series (DRC)
Undetermined
Any of the previous with 5– 30% blast count (EB)
(RCRSEB/RCEB/DRCEB)
Abnormality to be specified
C. Table 2
At least two of the criteria below:
Unexplained cytopenia
Morphology with two dysplastic lineages
Acquired clonal cytogenetic abnormality of the stem cells
Increased blast count ? 5%

Page 13 of 28
C. Table 3
Myelodysplastic/Myeloproliferative Disorder
I. Juvenile myelomonocytic leukemia (JMML)
II. Chronic myelomonocytic leukemia, only secondary
III. Chronic myeloid leukemia- BCR-ABL negative; chronic myelogenous leukemia (CML) Ph-
Disease in Down syndrome patients
I. Transitory abnormal myelopoiesis
II. Myeloid leukemia
Myelodysplastic Syndromes
I. Refractory cytopenia (RC) - < 2% peripheral blood blast count, < 5% bone marrow blast count
II. Refractory anemia with excess blast (RAEB) – 2% to 19% peripheral blood blast count, 5% to 19% bone marrow blast count
III. RAEB in transformation (RAEB-t) – peripheral blood and bone marrow with 20% to 29%
blast count
C. References
1 Mandel K, Dror Y, Poon A, Freedman MH. A practical comprehensive classification for pediatric myelodysplastic syndrome: the CCC System. J Pediatric Hematol Oncol 2002;24:343–52.
2 Hasle H, Niemeyer CM, Chessels JM et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia 2003;17:277–82.
3 Brunning RD, Bennett JM, Flandrin G et al. Myelodysplastic syndromes. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid tissues, Lyon: WHO, International Agency for Research on Cancer (IARC); 2001. p. 61– 73.
4 Sasaki H, Manabe A, Kojima S et al. Myelodysplastic syndrome in childhood: a retrospective study of 189 patients in Japan. Leukemia 2001;15:1713–20.
5 Passmore SJ, Chessells JM, Kempski HM et al. Paediatric MDS and LMMJ in the UK: a population based study of incidence and survival. Br J Haematol 2003;121:758–67.

Page 14 of 28
6 Hasle H, Baumann I, Bergstrasser E et al. International prognostic scoring system (IPSS) for childhood myelodysplastic syndrome (MDS) and juvenile myelomonocitic leukemia (JMML). [Abstract] Blood 2000b; 624a.
D. Incidence
There are very few published reports of pediatric myelodysplasia cases
before 1992. In 1997, the European Group for Pediatric Myelodysplasia (EWOG-
MDS) organized its first meeting. In that same year, the first International
Congress for Childhood Myelodysplasia and Leukemia was organized in São
Paulo by the Hospital do Cancer. Since then, reports about pediatric MDS
incidence have begun to appear in the literature. The Surveillance,
Epidemiology, and End Results (SEER) study from the USA showed an annual
incidence of seven cases per million population for de novo AML in children
under the age of 15 years. 1 According to Smith and Woods, 2 pediatric MDS
incidence in the USA is about 10% of AML incidence, which is about 0.8 cases
per a population of 1 million.Results from several international studies showing
case distribution according to FAB classification are shown in Table 4.

Page 15 of 28
D. Table 4
French-American-British (FAB) classification in several pediatric myelodysplastic syndromes (MDS) international studies.
FAB Classification (%) Study Group
RA RAEB RAEB-t JMML
Inconclusive FAB*(%)
Belarus 26 (Savva and Aleinkova, 2003)
28.2 17.3 15.2 39 ---
USA 44 (Woods et al., 2002)
2.7 44.5 35.1 17.5 ---
China/Hong- Kong 27 (Chan et al., 2003)
20.8 8.3 20.8 45.8 4.1 (RARS)
EWOG 28 (Stary, 2003)
36 17 13 34 ---
Greece 29
(Polychronopoulou, 2003)
59.3 6.2 18.7 15.6 8
Japan 30 (Manabe and Nakahata,
2003)
21 11.6 16.9 8.7 41.5
USA/Canada (Luna-Fineman, 1999)
6.8 16.7 7.4 37.2 31.6
Groupe Français (1997)
11.3 25 11.3 13.6 38.6
The Turkish National Pediatric MDS Study Group 32 (2003)
22.8 28.9 13.5 32.5 2.1
United Kingdom 22
(Passmore et al., 2003)
26.6 14 9.6 49.6 ---
Italy/Germany (Creutzig et al., 1987)
--- 19 76.1 4.7 ---
Poland 31 (Wöjcik and Ussowicz, 2003)
17.2 32.1 24.1 22.9 3.4

Page 16 of 28
D. References
1 Smith FO, Woods WG. Myeloproliferative and myelodysplastic disorders. In: Pizzo PA, Poplack DG, ed. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia: Lippincott Williams and Wilkins; 2002. p. 615–36.
2 Bennett JM, Catovsky D, Daniel MT et al. (The French-American-British Cooperative Group - FAB). Proposals for the classification of the acute leukaemias. Br J Haematol 1976;33:455–8.
E. Treatment
The therapeutic dilemma regarding MDS in children is reflected in the
absence of universally established approaches in the global literature. This
difficulty is mainly related to the fact that MDS in children comprise a
heterogeneous spectrum of clonal disorders grouped under the same
denomination. Therefore, therapy must have a common basis, though it should
be tailored to peculiarities related to these diverse disease subgroups.
The GCB-SMD-PED analyzed survival rates of the first 97 children who
enrolled in their study. At that time, every patient was treated according to the
policy of his or her referring physician, and most of the participants were not
treated with bone marrow transplant (BMT). Survival according bone marrow
blast percentage and the number of lineages that presented dysplastic features
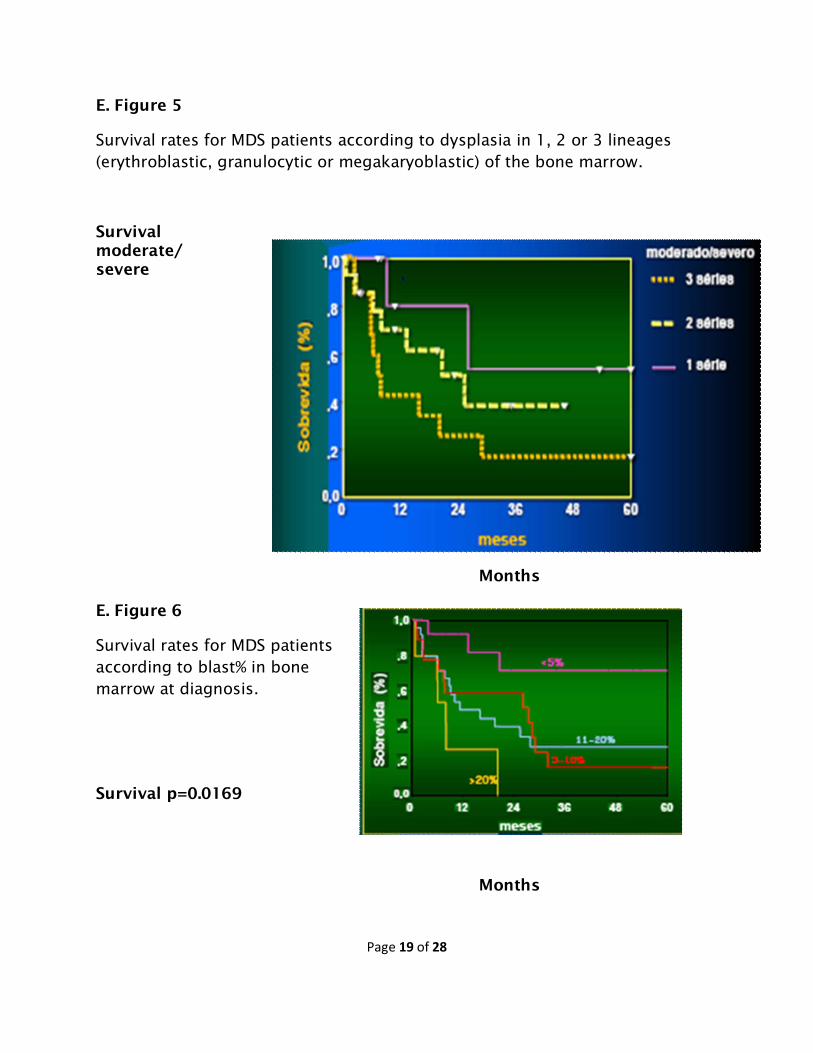
(moderate or severe) at diagnosis may be seen in Figures 5 and 6.

Page 17 of 28
E. 1 Bone Marrow Transplantation
Due to the failure of conventional treatment, allogeneic BMT was initially
indicated for both children and adults with MDS. Since the 1990s, many studies
have demonstrated the success of allogeneic BMT for MDS treatment. Most of
these studies included pediatric patients who formed a small part of the adult
series. This age mixture generated obstacles for data interpretation and
reliability since BMT is considered to be the only curative therapy for several
childhood MDS subtypes.
Patient and familial human leukocyte antigen (HLA) typing is the first step
after a diagnosis of MDS has been established. If an HLA-matched donor is
available, the patient must be referred to an allogeneic BMT center with
experience in pediatric cases. Otherwise, a search for an unrelated donor/cord
blood donor must be performed.
Early allogeneic BMT is the current indication for children with MDS with a
matched, related donor. Results from the EWOG-MDS point to clonal evolution
from fat cells even for the RA cases. Patients with RAEB and RAEB-t (RAEB in
transformation) have the worst survival rates among all MDS subtypes.
Nevertheless, allogeneic BMT is not the first choice of treatment for all
MDS patients. Children carrying constitutional abnormalities represent about
20% of pediatric MDS cases. Pathologies such as Down syndrome, Fanconi
anemia, Kostmann syndrome, and type 1 neurofibromatosis, among others,

Page 18 of 28
have an increased risk for MDS. The therapeutic approach taken for each type
of case should be decided by each cooperative group. A diagnosis of AML must
be considered for patients with MDS and cytogenetic alterations such as t(8;21),
t(15;17), and inv(16), and they should be treated with disease-specific
protocols.
Stem cell sources have diversified during the last few years. It is now
possible to obtain stem cells from allogeneic bone marrow aspirate, allogeneic
or autologous peripheral blood and stem cells from cord blood. BMT with
autologous or allogeneic peripheral blood stem cells is still considered an
experimental protocol for this disorder.
Conditioning regimens vary according to the different clinical trial protocols.
E. 2 Supportive Therapy
Historically, several therapeutic schemes have been developed to promote
an increase in survival, achieve partial remission, or improve quality of patient
life. Pediatric treatments derived from adult protocols have rarely been
validated by a rigorous methodology. Table 5 reviews the different treatment
modalities for pediatric MDS, the most pertinent studies, and their clinical
recommendations. Transfusional support and a wide spectrum of antibiotic
therapies are universal recommendations for patients with febrile neutropenia.
Page 19 of 28
E. Figure 5
Survival rates for MDS patients according to dysplasia in 1, 2 or 3 lineages (erythroblastic, granulocytic or megakaryoblastic) of the bone marrow.
Survival moderate/ severe
Months
E. Figure 6
Survival rates for MDS patients according to blast% in bone marrow at diagnosis.
Survival p=0.0169
Months

Page 20 of 28
E. Table 5
Treatment References Recommendations
Subcutaneous cytarabine
39,40 Not routinely recommended in pediatrics
Low-dose chemotherapy
5-azacytidine 41 Hypomethylating agent. Encouraging results in adults, experimental in pediatrics
AML induction and variations
41–44 Some responses for RAEB and RAEB-t. Controversial data, lack of pediatric controlled studies.
High-dose chemotherapy
Erythropoietin 42–45 May reduce transfusional requirements in adults. Recommendable for
specific situations
G-CSF and GM-CSF 41,42 Recommended only for severe neutropenia with serious infections. Temporary use, potentates erythropoietin effect
Stimulating factors
Interleukin 11 41 May increase platelet level in adult MDS. Experimental in pediatrics
Cytoprotectant Amifostine 42,46 May improve adult-MDS cytopenias. Described in children. Experimental in pediatrics
Differentiating agents
Retinoic acids 42,47 Some role for JMML treatment. Experimental in pediatrics
AML, acute myeloid leukemia; MDS, Myelodysplastic syndromes; RAEB, refractory
anemia with excess blasts; RAEB-t, refractory anemia with excess blasts in transformation); JMML, juvenile myelomonocytic leukemia
Page 21 of 28
F. Additional Figures
F. Figure 1

Page 22 of 28
F. Figure 2

Page 23 of 28
F. Figure 3

Page 24 of 28
F. Figure 4

Page 25 of 28
F. Figure 5

Page 26 of 28
F. Figure 6
G. Further Reading 1 The 2nd International Symposium on Myelodysplastic Syndromes in Childhood, Hindsgavl Castle, Funen, Denmark, May 11–14, 2000. Leukemia 2000;14:956– 971.
2 Lopes LF, Lorand-Metze I. Childhood myelodysplastic syndromes in a Brazilian population. Pediatr Hematol Oncol 1999;16:347–53.
3 Lorand-Metze I. Meira DG, Vassallo J, Lima CSP, Metze K. The differential diagnosis between aplastic anemia and hypocellular myelodysplasia in patients with pancytopenia. Haematologica 1999;84:564–565.
4 Magalhães SMM, Vassallo J, Lorand-Metze I. Heterogeneity of bone marrow lymphoid nodules in myelodysplastic syndromes. Virchows Archiv 1999;435(3):177.

Page 27 of 28
5 Niero-Melo L, Franco M, Gushiken T, Guilherme EL, de Abreu Machado PE. Myelodysplastic syndromes: clinical, hematological and histopathological evaluation of the bone marrow in 23 cases. Rev Assoc Med Bras 1987;33(3–4):53–6.
6 Souto EX, Chauffaile MD, Moncau JE, Niero-Melo L, Braga GW, Silva MR, Kerbauy J. Myelodysplastic syndromes (MDS): prognostic factors and scoring systems. Rev Paul Med 1997;115(5):1537–41.
7 Novaretti MC, Sopelete CR, Velloso ER, Rosa MF, Dorlhiac-Llacer PE, Chamone DA. Immunohematological findings in myelodysplastic syndrome. Acta Haematol 2001;105(1):1–6.
8 Bennett JM, Catovsky D, Daniel MT et al. (The French-American-British Cooperative
Group - FAB). Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 1982;51:189–99.
9 Souto EX, Kerbauy DMB, Lee MLM et al. Importancia de las características morfológicasen los síndromes mielodisplásicos (SMD): niños matriculados en el Grupo Cooperativo Brasileño de SMD: Enero de 1997–Febrero 2003 [Abstract]. Rev Chil Cancerol Hematol 2003;13:166.
10 Smith MA, Ries LA, Gurney JG et al. Leukemia. In: Ries LA, Smith MA, Gurney JG et al. editors. Cancer Incidence and Survival Among Children and Adolescents: United States SEER Program 1975–1995. Bethesda: National Cancer Institute; 1999. p.17–34. (SEER Program: NIH PUB. n° 99–4649).
11 Savva NN, Aleinikova OV. Experiences on MDS and JMML from Belarus. In: Lopes LF, Hasle H, eds. Myelodysplastic and Myeloproliferative Disorders in Children. São Paulo: Lemar;2003. p. 247–58.
12 Chan GCF. Experiences on MDS and JMML from China/Hong Kong. In: Lopes LF, Hasle H, eds. Myelodysplastic and Myeloproliferative Disorders in Children. São Paulo: Lemar;2003. p. 271–6.
13 Stary J. Experiences on MDS and JMML from the EWOG-MDS. In: Lopes LF, Hasle H, eds. Myelodysplastic and Myeloproliferative Disorders in Children. São Paulo: Lemar;2003. p. 277– 96.
14 Polychronopoulou S. Experiences on MDS and JMML from Greece. In: Lopes LF, Hasle H, eds. Myelodysplastic and Myeloproliferative Disorders in Children. São Paulo: Lemar;2003. p. 297– 316.
15 Manabe A, Nakahata T. Experiences on MDS e JMML from Japan. In: Lopes LF, Hasle H, eds. Myelodysplastic and Myeloproliferative Disorders in Children. São Paulo: Lemar;2003. p. 317– 24.
16 Wójcik D, Ussowicz M. Experiences on MDS and JMML from Poland. In: Lopes LF, Hasle H, eds. Myelodysplastic and Myeloproliferative Disorders in Children. São Paulo: Lemar;2003. p. 325– 30.
17 The Turkish National Pediatric MDS Study Group. Experiences on MDS and JMML from Turkey. In: Lopes LF, Hasle H, eds. Myelodysplastic and Myeloproliferative Disorders in Children. São Paulo: Lemar;2003. p. 331–8.
18 Webb DKH. Experiences on MDS and JMML from the United Kingdom. In: Lopes LF, Hasle H, eds. Myelodysplastic and Myeloproliferative Disorders in Children. São Paulo: Lemar;2003. p. 339–44.

Page 28 of 28
19 Mathew P, Woods W. Experiences on MDS from the United States. In: Lopes LF, Hasle H, eds. Myelodysplastic and Myeloproliferative Disorders in Children. São Paulo: Lemar;2003. p. 345– 60.
20 Paula MJA. Caracterização clínica e laboratorial dos pacientes pediátricos com síndromes mielodisplásicas e o estudo das diferentes classificações propostas na literatura. São Paulo; 2004. [Dissertação de Mestrado-Fundação Antônio Prudente].
21 Kardos G, Baumann I, Fenu S et al. Refractory anemia (RA) in childhood: a study of the European Working Group of MDS in childhood. Leukemia 2000;(14):967.
22 Witherspoon RP, Deeg HG. Allogenic bone marrow transplantation for secondary leukemia or myelodysplasia. Haematologica 1999;(84):1085–1087.
23 Niemeyer CM, Locatelli F, Nollke P et al. Primary MDS in childhood: AML-like chemotherapy prior to stem cell transplantation does not improve event-free survival. Leukemia 2000;(14):971.
24 Miller KB, Kyungmann K, Morrison FS et al. The evaluation of low-dose cytarabine in the treatment of myelodysplastic syndromes: a phase III intergroup study. Ann Hematol 1989;(65):162–168.
25 Chesen BD. Standard and low-dose chemotherapy for the treatment of myelodysplasic syndromes. Leuk Research 1998;(17):s17–s21.
26 Greenberg PL, Young NS, Gattermann N. Myelodysplastic syndromes. Hematology 2002 – American Society of Hematology. p. 136–161.
27 Valera ET, Lee MLM, Marques MGA et al. Instituição de protocolo de tratamento padronizado pelo Grupo Cooperativo Brasileiro de Síndrome Mielodisplásica em Pediatria. Rev Bras Hematol Hemoter 2002;24(3):236–241.
28 Woods W, Buckley J, Lange B et al. The treatment of children with myelodysplasic syndrome (MDS): The Children’s Cancer Group experience. J. Pediat Hematol Oncol 1997;(19):356a.
29 Woods WG, Barnard DR, Alonzo TA et al. Prospective study of 90 children requiring treatment for juvenile myelomonocitic leukemia or myelodysplastic syndrome: a report from the Children’s Cancer Group. J. Clin. Oncol 2002;20(2):434–440.
30 Miller K. Erythropoietin, with and without granulocyte-colony stimulating factor (G-CSF), in the treatment of myelodysplasic syndrome patients. Leuk Research 1998;(22):s13–s16.
31 List A F. Hematopoietic stimulation by amifostina and sodium phenylbutyrate: what is the potential in MDS? Leuk Research 1998;(22):ps7–s11.
32 Aul C, Runde V, Gattermann N. All-transretinoic acid in patients with myelodysplastic syndrome: results of a pilot study. Blood 1993;(82):1489.


















