
protocol. photographs diagnosis probably changes probably...
28
NEUROFIBROMATOSIS TYPE 1 IN CHILDREN BY George R. Beauchamp, MD ABSTRACT Objective: To document ophthalmic and general characteristics of Neurofibromatosis Type 1 (NFI) in children; and to characterize the iris changes of NF1 including their variability and reliability. Design and Patients: One hundred and ninety-six patients with NF1 were evaluated for general characteristics; 156 patients underwent prospective eye evaluations; and 151 NF1 patients and controls had iris photography in accordance with a protocol. Masked evaluation of photographs compared to a known diagnosis, and interobserver reliability tests were performed. Main Outcome Measures: Incidence of iris changes consistent with NF1 (Lisch nodules, plus) by age group; spectrum of iris changes found; correla- tion of iris findings to known diagnosis and Kappa coefficients for interobserver reliability. Results: Iris changes were common in children over age 5, and increased with age; masked evaluation of photographs compared to a known diagnosis yielded fair to poor correlation (Kappa = -.02 to .50); interobserver reliabil- ity was poor (Kappa = -.02 to .24, overall .174); iris changes found in NF1 are more diverse than classic descriptions of Lisch nodules, and including a broader spectrum probably increases the sensitivity and decreases the speci- ficity of their diagnostic value. Conclusion: Iris changes as a diagnostic marker for NFL may need rethink- ing; this study calls to question their reliability and validity. INTRODUCTION Of the neurocutaneous syndromes, neurofibromatosis type 1 (NF1) occurs once in about 3,000 births.' Mark Akenside was probably the first person to describe neurofibromatosis in 1768, but the disorder carries the name of von Recklinghausen because in 1882 he was first to recognize the neural origin of the tumors.2 In recent times, Riccardi3 classified the disorder into 7 types, and in 1987 the Neurofibromatosis Study Group4 established diag- nostic criteria for type 1, the most common, based on clinical evidence. The presence of two or more of the following defining features must be present to establish a diagnosis of neurofibromatosis type 1: 1. Six or more caf6-au-lait macules, the greatest diameter of which is
Transcript of protocol. photographs diagnosis probably changes probably...

NEUROFIBROMATOSIS TYPE 1 IN CHILDREN
BY George R. Beauchamp, MD
ABSTRACT
Objective: To document ophthalmic and general characteristics ofNeurofibromatosis Type 1 (NFI) in children; and to characterize the irischanges of NF1 including their variability and reliability.
Design and Patients: One hundred and ninety-six patients with NF1 wereevaluated for general characteristics; 156 patients underwent prospectiveeye evaluations; and 151 NF1 patients and controls had iris photography inaccordance with a protocol. Masked evaluation of photographs comparedto a known diagnosis, and interobserver reliability tests were performed.
Main Outcome Measures: Incidence of iris changes consistent with NF1(Lisch nodules, plus) by age group; spectrum of iris changes found; correla-tion of iris findings to known diagnosis and Kappa coefficients forinterobserver reliability.
Results: Iris changes were common in children over age 5, and increasedwith age; masked evaluation ofphotographs compared to a known diagnosisyielded fair to poor correlation (Kappa = -.02 to .50); interobserver reliabil-ity was poor (Kappa = -.02 to .24, overall .174); iris changes found in NF1are more diverse than classic descriptions of Lisch nodules, and including abroader spectrum probably increases the sensitivity and decreases the speci-ficity of their diagnostic value.
Conclusion: Iris changes as a diagnostic marker for NFL may need rethink-ing; this study calls to question their reliability and validity.
INTRODUCTION
Of the neurocutaneous syndromes, neurofibromatosis type 1 (NF1) occursonce in about 3,000 births.' Mark Akenside was probably the first person todescribe neurofibromatosis in 1768, but the disorder carries the name ofvon Recklinghausen because in 1882 he was first to recognize the neuralorigin of the tumors.2 In recent times, Riccardi3 classified the disorder into7 types, and in 1987 the Neurofibromatosis Study Group4 established diag-nostic criteria for type 1, the most common, based on clinical evidence.
The presence oftwo or more ofthe following defining features must bepresent to establish a diagnosis of neurofibromatosis type 1:
1. Six or more caf6-au-lait macules, the greatest diameter ofwhich is

Beauchamp
more than 5 mm in prepubertal and more than 15 mm inpostpubertal patients
2. Two or more neurofibromas of any type, or one plexiformneurofibroma
3. Freckling in the auxillary or inguinal region4. Optic glioma5. Two or more Lisch nodules6. A distinctive osseous lesion, such as sphenoid dysplasia or
pseudarthrosis7. A first-degree relative with NF1, according to the preceding criteria
The NSG claims that a clinician using these criteria can appropriately diag-nose 94% of children with NFL at age 6 years or younger at initial examina-tion. Bolande5 has pointed to the role ofthe neural crest in the pathogenicityof the disease, which has such remarkable diversity that clinicians often un-derestimate its severity, especially in children, because it is progressive.16 -10
Table Ill-"3 attests to the clinical diversity of NF1 in childhood. Thisvariability may be ascribed to genetic, developmental, and pathophysiologicaspects (see Appendix for a review of the literature54-83).
According to Riccardi and Eichner,24 halfofthe patients by age 15 havereached grade 3 disease, which compromises health without shortening lifespan, and one fourth of patients by age 30 have reached grade 4 disease,which may shorten life.
Iris changes, specifically Lisch nodules, are associated with the disease.They are found in virtually all adults with confirmed disease. They are alsoused as a marker in evaluation of unaffected family members. Their preva-lence in children is lower than in adults.
The author's inability as a clinician to consistently determine ophthalmicmanifestations ofNF1 in children prompted this study. Observations, espe-cially of Lisch nodules, were not corroborated by colleagues. Furthermore,the morphology of iris changes varied more than unequivocal "textbook"descriptions, suggesting that the specificity of these observations in chil-dren may be low.
This study, therefore, was designed (1) to quantify interobserver differ-ences in identification of iris changes in suspected and confirmed NF1, (2)to explain the difficulty in diagnosing the disease by ophthalmic examina-tion using current methods, and (3) to suggest categories for the atypicalvariations seen in iris morphology of children with suspected NFL. Theresults show that the clinician relying on Lisch nodules to detect NF1 inchildren should consider a broader spectrum of iris changes that carry alower specificity.
MATERIALS AND METHODS
Table II lists the general characteristics of the 196 patients studied. Ofthese, 95 children and 44 adults were confirmed as NF1, and 17 childrenand 40 adults were suspected as NFL but did not meet all diagnostic crite-
446

Neurofibromatosis Type 1
TABLE I: DIAGNOSTIC AND ASSOCIATED FEATURES OF NF1 IN CHILDHOOD
FEATURE % OF CHILDREN WITH NF1 POSSESSING
Diagnostic:Cafe-au-lait macules'9 9719Intertriginous freckling'9 81'sNeurofibromas2" 15 (tinder 6)2'Osseous lesions
Spheniod dysplasia22 (uncommon)9Pseudarthrosis (tibia)22 23 322,23Scoliosis/other spinal'9 60'Pectus excavatum 4324
Lisch nodtules"' 7 50-90+ (depending on age)"'Optic gliomas2' 1525
Associated:Malignancy26
Netirofibrosarcoma24Leukemia27Julvenile xanthogranuloma21Neuroblastoma (?)'Rhabdomyosarcoma2"Wilms' tumor3"
Cognitive impairment'Intellectual delay' 5-259Visual perceptual disability"t1-3 56-883'-33Cortical dvsgenesis34Learning disabilities9 30-5035
Precocious ptuberty (?)'
McCtine-Albright syndrome confusion (?)
Noonan svndrome'6-39Growth delay/short statture2441'Macrocephaly22.4'.42 2524Vascutlar manifestations9
Occlusion or stenosisHypertension: Renal artery stenosis, pheochromocytomaStroke
Congenital heart disease9Gastrointestinal disease4'
Abdominal pain, constipation 3-424Endocrine distuirbance/development44Multiple endocrine neoplasia syndrome (overlap)9
Other ocular45Plexiform neuroma of evelid,
glauicoma, and ipsilateral optic gliomaCongenital ptosis 94hProminent corneal nerves
Choroidal hamartomata47Buphthalmos4995'Glauicoma49 5'Retinal tumors52
Other neurologic9Behavior disturbances 18;3Seizures 3-139Clutmsiness 40 35Headache 256
NFI, Neurofibromatosis type 1.
447

Beauchamp
ria. In addition, 40 patients evaluated for NF1 because of a positive familyhistory or possible positive findings were declared negative and 33 individu-als (presumed normal; ie, not possessing NF1) were evaluated as controlsfor photographic iris analysis. The 112 NFl-positive children were exam-ined before their 18th birthday and diagnosed by a pediatric neurologist,examined by either a pediatric ophthalmologist or a neuro-ophthalmologist,and ophthalmologically examined between July 1, 1983, and December 31,1990. In the same period, the 44 adults were examined personally by theauthor, whose diagnosis of NF1 was confirmed by a neurologist. Althoughthe author examined all patients, the "Supreme Court" for diagnosis ofNF1were pediatric neurologists, functioning as neurofibromatosis consultantswho used established criteria. A registry of patients categorized as "con-firmed NFl" or "suspected NFl" was (and is) managed exclusively by apediatric neurologist. An appendix contains the data on which this report isbased. A number of analyses were performed:
1. A statistical comparison of the symptoms of children and adults, todiscern differences to qualify confirmed and suspected NF1 pa-tients as more or less typical of accumulated reported series
2. A detailed examination of the ophthalmic findings of 156 patientswith confirmed or suspected NF1, 40 NFl-negative patients, andthe 33 controls. The author also clinically examined 77 childrenand 44 adults with confirmed NF1, 16 children with suspected NF1,and the 40 NFl-negative patients.
3. Eye photography of 151 selected patients and controls using anestablished protocol. This group included 41 children with con-firmed NF1, 4 children with suspected NFL, 40 adults, 33 NFl-negative patients, and the 33 controls.
4. A masked review of all photographs for the presence or absence ofLisch nodules and "atypical" iris findings. Their number and loca-tion, iris color, and other anterior segment anomalies were observed,recorded, and subsequently correlated with clinical disease category,age, and race.
5. Design and assignment of the following morphologic categories:lumpy bumpy (including doughy, or indistinctly thickened); pea-nut butter splat (as if it had been flicked upon the surface of theiris); moonscape (with or without craters, finely granulated ordustlike surface, or coarsely spatulated); peppercorn; fishy (eitherpale fish flesh or translucent fish eggs); cotton-wool surface pat-terns; hypopigmentation defects; and confluent, when changes wereso extensive that a category could not be assigned.
6. Assessment by panel. From 1 to 3 representative photographsfrom 70 individuals were selected for masked assessment bya panel of 7 observers for the presence or absence of Lischnodules (ie, findings indicative of NF1). Of the 7 observers,
448

Neurofibromatosis Type 1
mIn -- mI
O> Cn 05 dce cs 1 cq
00 C-1 ~~ ~ ~ ~ ~ ~~~~~0
Z r--4C-1 cocl1 -
'~~j4 ~~ c~~1 -~00
z
UQO OC%11 (CO 000c4
-z
z
0~~~~~~~~C11
ucli o cq c0 VC- z _ --liI
kf~~~~ 0~0
z
r-T.~"
I
e-- iz v *,- s --
cQCi d
co 1-
coHz
0
-
E-i
U
0
k-
cn
*-
449
0
cU
UL)z
~14z
H
U-
.l
I
.
0
;
-

Beauchamp
3 were specialists (2 neuro-ophthalmologists and 1 pediatric oph-thalmologist), 2 residents, and 2 medical photographers. Possibleratings were positive (+), negative (-), or equivocal (?). No timelimit was given. Assessment was repeated for 5 of the 70 cases. Ofthe 70 individuals, 46 were NFl-positive, 21 NFl-negative, and 3NFL-suspected. The actual clinical diagnosis, established by a neu-rologist, was compared with each observer's masked rating. Sensi-tivity of the observer test was calculated as the percent of clinicallydiagnosed NFL-positive patients (N=46) who were positively (+)rated by the panel. Specificity was calculated as the percent ofNFL-negative (N=21) individuals who were negatively (-) rated.
7. Statistical analysis. Agreement between observers was calculated,as was agreement across time for the 5 repeated cases. Statisticalanalysis included calculation ofkappa coefficients of interobserveragreement using the McNemar and Wilcoxon rank sum tests.
RESULTS
Table III shows the prominent general features of the patients with con-firmed disease. Statistical comparisons showed significant differences be-tween children and adults for caf6-au-lait macules (Child {C} > Adult (A},P<.02), neurofibromas (C<A, P<.001), learning or school problems (C>A,P<.04), speech development problems (C>A, P<.02), and presence ofLischnodules (C<A, P<.01). Other comparisons were of marginal significance ornot significant.
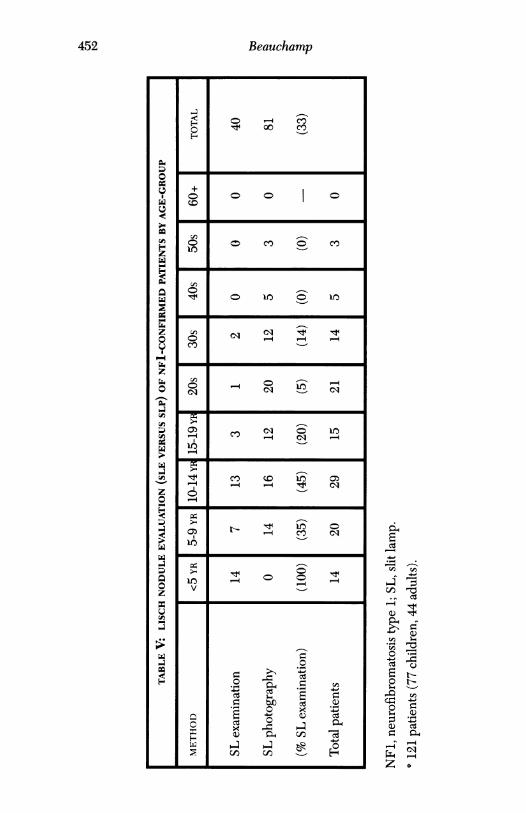
LISCH NODULE EVALUATION
The author determined the presence or absence of Lisch nodules or other"atypical" features ofNF1 and rated them as either present, absent, or equivo-cal. Table IV displays these ratings by 5-year groupings under age 20, andby decade thereafter. The method of diagnosis (clinical or photography) isgiven for each rating category in Table V. The apparent strength of photog-raphy to indicate the presence of Lisch nodules in comparison to clinicalobservation seems to reflect the obvious-photography is not possible forthose least likely to possess Lisch nodules (ie, the youngest children). Ingeneral, iris changes were least likely to be present under age 5 years, al-ways present over age 30 years, and unequivocally present in 61% of chil-dren and 89% of adults with confirmed NFL. Interestingly, 25% ofchildrenwith suspected NF1 and 18% of "negative" patients were determined tohave irish changes consistent with NFL.
Table VI gives a composite summary of positive findings of the photo-graphic iris analysis of 81 patients with confirmed NFL. Atypical findingsseem to augment the sensitivity of biomicroscopic determination of irischanges suggestive of NFL, particularly in the younger age-groups. While
450

Neurofibromatosis Type 1
TABLE III: PROMINENT NONOPHTHALMIC FEATURES OF NFl:95 CHILDREN AND 44 ADULTS WITH CONFIRMED DISEASE'
FEATURE j % CHILDREN I % ADULTS
Cutaneous manifestations 99 95Caf6-au-lait macules 96 82Freckling 47 45Neurofibroma 28 70
Orthopedic manifestation 24 30Spinal deformity (% scoliosis) 11(80) 23(100)Tibial disease 5 2Pectus excavatum 5 5
Neoplasia (noncutaneous) 21 27Neurofibroma 15 20Optic chiasmal glioma 4 2
Learning, school problems 33(38f) 7Learning disability 23 5Attention deficit disorder 15 2Hyperactivity 6 1
Hearing and speechdevelopment problems 23 4
Headache (migraine) 15(8/14t) 9(7/9t)
Neuromuscular control deficits 20 9Clumsy/awkward 9 2
Macrocephaly 12 0
Gastrointestinaldisease/complaint 7 5
Endocrine dysfunction 9 5
NF1, Neurofibromatosis type 1.
° For each feature, more than one of each subset types could be (and was in specific in-stances) present in a given patient. For example, freckling could accompany caf6-au-laitmacules, and freckling could be both axillary and inguinal.f If 14 patients less than 5 years of age are excluded.
I Migraine diagnosed/indicative, not diagnostic, of migraine.
451
Beauchamp
H0
o4 _0 co'l 00 co
O O0 0 Cl) 0 CIO
CO
CO _ 10
C-1 cli~~~~c1
- - - t4 Clco
CO Cl1
10
V -o
C~0
0
0~~~
0 0n-4 C ..I
E -6- Q)0 H1
452
000
z0
CA,0
0
Omi"
4
0
2
zCu
z
6o
(Au:
z
Om
;D;ow
z
uW)mmWN
S
_~
00VO4-
a4ad
04-
cq0 H-
Ci,;
0t tO .
a) c:E 0-OCl
0
0

Neurofibromatosis Type 1
6z
0H-
co 1- '-- '-
r-4r-
co-
r-q t, Cb C
00 01I
000 P-(40 -q C00~
m CIO . O COH O00
LOO O 00c) 000 )
CM, c>0 r-4CIO~ 000
cv)
o° V) CYo .* __c OC O O
(n ~~~~~~~--qOC1lt-1
00
~C-C1004-- 00kf-4- C1 qt 00C
(7)LHom cli' CIO -i 00 0 0
VC,-
>" o4)4 4)0 O~-- - * Q = *0 c c"O &O Q O 0 O
O,Q= aazXa_ =Z Z Z _ =~~~~C Z Z Z _ Z ZZ zZZZv b;; J N s; X JN4;4J ;;JZb ~Z-
453
00
0CL
0ma;
mz0
80z
u
U0-
U
U
.
D00C;I: -- SC! ;6:
0O.. a
Z v
11-11-1 I.-I (MCD C-1 I..cC.0 ".-I "-4 _,"-4 I.-I I.-I
co 01 "-4"..4
I.-II--, LN
C> 0 00 C.0cq "-q --,ll.r 1-1
CO t.- 1001

Beauchamp
NFl, Neurofibromatosis type 1.°81 Patients (41 children, 40 adults) with confirmed NFI.Two patients with atypical findings; one patient with insufficient typical findings.
t Two patients with atypical findings
66% of children had typical findings, inclusion of atypical findings permit-ted diagnosis in 83% of cases. The differential was less, though present(75% versus 88%) in adults. Overall, inclusion of atypical criteria raised theapparent sensitivity for the presence ofNF1 by iris evaluation to 85% (from70%), by masked iris-photographic evaluation of NFl-positive patients.
Table VII summarizes results of tests of association for iris changes withage-group, race, iris color, and location. Observations supported by the datainclude the following. The number ofatypical changes was nearly equal across
age-groups. While atypical changes appeared more commonly in blacks, closerscrutiny suggests these changes are more likely to occur in pigmented irides,and racial effect therefore may be only on the iris color. The number ofLischnodules per irs tended to increase with age. Lisch nodules were more fre-quently observed in the inferior one half of the iris, and iris changes of NFIwere more frequently observed in the inner one half of the irides, especiallyso for atypical changes. Examples of typical, atypical, and confluent iris find-ings are illustrated in Fig 1 through 16. Atypical changes in a normal subjectand ectropian uvea are illustrated in Fig 17 and 18.
FABLE VI: LISCH NODULE PHOTOGRAPHIC ANALYSIS: COMPOSITE SUMMARY OF
POSITIVE FINDINGS BY TYPE OF FINDINGS
41 CHILDREN 40 ADULTS 81 NFICIIARACTERISTI(C NFL CONFIRMED NF1 CONFIRMED CONFIRMED
NO.(%) NO.(%) NO.(%)
All patients with typical LN 27(66) 30(75) 57(70)Typical only 20(49) 16(40) 36(44)Typical + atypical 7(17) 10(25) 17(21)Typical + confluent 0 4(10) 4(5)
Atypical only 7(17) 5(13) 12(15)
Typical + atypical 34(83) 35(88) 69(85)
Equivocal 5(t)(12) 2(t)(5) 7(9)
Negative 2(5) 3(8) 5(6)
454

Neurofibromatosis Type 1
TABLE VII: SUMMARY RESULTS: TESTS OF ASSOCIATION IN PATIENTSWITH CONFIRMED NFI
CHARACTERISTIC
Lisch nodule (positive)
Typical Lisch nodule
Typical Lisch nodule (child)
Typical Lisch nodule (child)
Atypical Lisch nodule
Atypical Lisch nodule (child)
Atypical Lisch nodule (adult)
% WITH CHARACTERISTIC
Age-Group (yr)<10 10-19 20-29 >29 P
52.9 70.5 71.4 100.0 .01
Race
White Black P
74.3 50.0 .21
46.2 75.0 .34
No. of Changes per Iris
<5 5-10 11-20 >20 P
75.0 40.0 40.0 38.1 .35
Iris ColorBlue/Green Brown P
77.8 2.2 <.001
75.0 52.3 .28
Location > Inner 1/2
No Yes P
40.6 100.0 <.001
455

Beauchamp
FIGURE 1
Typical Lisch nodules. Notegreater number inferiorly (x 10).
FIGURE 2"Lumpy bumpy" atypical irischange. Note that iris structure andsurface is obscured by indistinct tis-sue (x 10).
FIGURE 3 FIGURE 4"Lumpy bumpy" atypical change. "Peanut butter splat" plus moreFew nevi. Note that iris structure typical lesions (x 16).and surface is obscured by indis-tinct tissue (x 10).
A
FIGURE 5"Peanut butter splat" plus typicalnodules (x 16).
FIGURE 6"Moonscape." Note that surface isflattened and undulating (x 10).
456

Neurofibromatosis Type I
FIGURE 7 FIGURE 8"Moonscape." Note that surface is "Peppercorn" (x 25).flattened and undulating (x 25).
FIGURE 9 FIGURE 10"Peppercorn" (pale) (x 25). "Fish egg" (x 25).
FIGURE 11"Cotton wool." Note also typicaland peppercorn change (x 16).
FIGURE 12"Cotton wool," next to more typi-cal nodule (x 16).
457
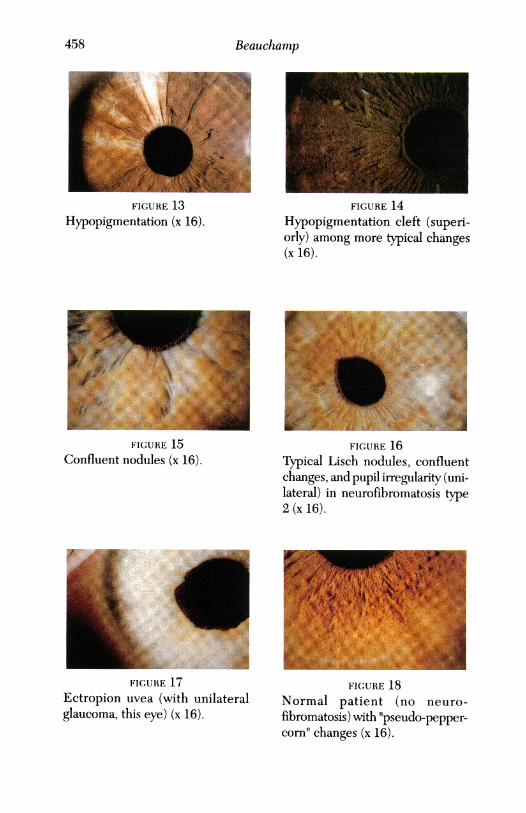
Beauchamp
FIGURE 13 FIGURE 14Hypopigmentation (x 16). Hypopigmentation cleft (superi-
orly) among more typical changes(x 16).
FIGURE 15
Confluent nodules (x 16)..~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~FIGURE 17
Ectropion uvea (with unilateralglaucoma, this eye) (x 16).
FIGURE 16Typical Lisch nodules, confluentchanges, and pupil irregularity (uni-lateral) in neurofibromatosis type2 (x 16).
FIGURE 18Normal patient (no neuro-fibromatosis) with "pseudo-pepper-corn' changes (x 16).
458

Neurofibromatosis Type 1
PANEL RESULTS
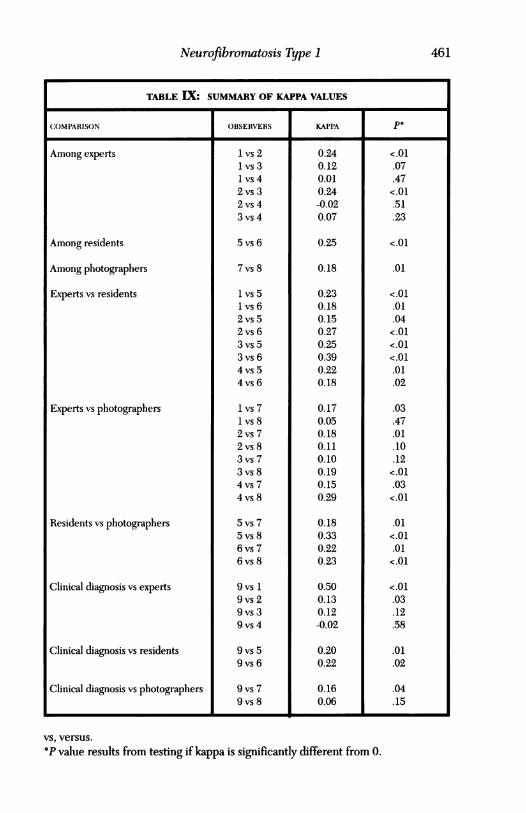
The study of iris changes and the ratings of photographs by a panel is sum-marized in Table VIII. The performance of each observer is displayed inrelation to the "true" clinical diagnosis. The kappa values ranged from -.02to .50, the sensitivity values ranged from 21.7% to 89.1%, and the specificityvalues ranged from 33.3% to 57.1%. More consistent agreement was seenbetween observer groups (experts versus residents, residents versus pho-tographers). Agreement with clinical diagnosis varied greatly as well. Fi-nally, even though many values were significantly higher than chance alone,only a few individual values approached the fair-to-good range of agree-ment. Table IX gives the rating ofvarious combinations of observers for the70 patients' photographs reviewed. Note that agreement patterns variedwidely among experts, with kappa values ranging from -.02 to .24. For eachpair of observers, the cross tabulation of ratings is given, along with thekappa statistic for that pair. The P value results from testing if kappa issignificantly different from zero. The best agreement comes from the ob-server pairs 1, 2, and 2, 3, each with kappa values of .24. While these valuesindicate agreement significantly greater than chance agreement, they arestill considered poor agreement. The results of McNemar's test indicatethe observer 1 (author) tends to rate more positively than the other observ-ers, and that observer 3 tends to rate more positively than observer 2.
Kappa values for various observer groups are given in Table X. TheWilcoxon rank sum test was used to test for differences in agreement be-tween groups. No strong differences were found. In addition to an overallmeasure ofagreement, kappa statistics for individual categories (+, -, ?) werecalculated and are given in Table XI for various combinations of observergroups. As would be expected, agreement on the - and + categories wasbetter than on the ? category. The overall kappa value for all 8 observerswas .174. Note that, consistent with previous results, the kappa values, whilegreater than chance, were still quite low.
Data from the 5 cases that were repeated by the 8 observers are listedin Table XII. Of the 5, 4 had a + clinical diagnosis and 1 had a - clinicaldiagnosis. The first case listed is the first reading of the case, and the sec-ond case listed is the second reading of the case. Of the 7 observers, 1observer (a photographer) had agreement between the 2 readings on all 5cases; 3 observers (2 experts, 1 resident) had agreement from time 1 to time2 on 4 of 5 cases, and three observers (1 expert, 1 resident, 1 photographer)had agreement from time 1 to time 2 on 3 of 5 cases. Of the pairs thatagreed, the number that in turn agreed with the clincal diagnosis variedwidely.
In general, there was no clear pattern of agreement within or betweentypes of observers. In addition, agreement with clinical diagnosis was notconsistent across observer. In most cases the level of agreement was quitelow.
459

Beauchamp
TABLE VIII: IRIS PHOTOGRAPHIC RATINGS BY A PANEL: AGREEMENT
WITH CLINICAL DIAGNOSIS
CLINICAL
OBSERVER RATING DIAGNOSIS STATISTICS
- I ?. I +
Author 1 - 9 0 1 kappa=0.50 (P<.Ol)t? 8 2 4 Sensitivity=89.1%+ 4 1 41 Specificity=42.9%
Experts 2 - 11 3 13 kappa=0.13 (P=.03)? 8 0 16 Sensitivity=37.0%+ 2 0 17 Specificity=52.4%
3 - 9 3 14 kappa=0.12 (P=.12)? 2 0 3 Sensitivity=63.0%+ 10 0 29 Specificity=42.9%
4 - 9 1 17 kappa=-0.02 (P=.01)? 3 0 11 Sensitivity=39.1%+ 9 2 18 Specificity=42.9%
Residents 5 - 12 2 13 kappa=0.20 (P=.01? 5 0 9 Sensitivity=52.2%+ 4 1 24 Specificity=57.1%
6 10 3 13 kappa=0.22 (P=.02)? 3 0 1 Sensitivity=69.69%+ 8 0 32 Specificity=47.6%
Photographers 7 - 7 2 5 kappa=0.16 (P=.04)? 3 0 8 Sensitivity=71.7%+ 11 1 33 Specificity=33.3%
8 - 12 2 13 kappa=0.06 (P=.15)? 3 1 23 Sensitivity=21.7%+ 6 0 10 Specificity=57.1%
NFl,neurofibromatosis type 1.The ? category for clinical diagnosis was not incorporated into sensitivity/specificity calculations.
fP value results from testing if kappa is significantly different from 0.
460
Neurofibromatosis Type 1
TABLE IX: SUMMARY OF KAPPA VALUES
COMPARISON OBSERVERS KAPPA PO
Among experts
Among residents
Among photographers
Experts vs residents
Experts vs photographers
Residents vs photographers
Clinical diagnosis vs experts
Clinical diagnosis vs residents
Clinical diagnosis vs photographers
1 vs 21 vs 31 vs 42 vs 32 vs 43 vs 4
5 vs 6
7vs 8
1 vs 51 vs 62 vs 52 vs 63 vs 53 vs 64 vs 54 vs 6
1 vs 71 vs 82 vs 72 vs 83 vs 73 vs 84 vs 74 vs 8
5 vs 75 vs 86 vs 76 vs 8
9vs 19 vs 29 vs 39 vs 4
9 vs 59 vs 6
9 vs 79 vs 8
0.240.120.010.24-0.020.07
0.25
0.18
0.230.180.150.270.250.390.220.18
0.170.050.180.110.100.190.150.29
0.180.330.220.23
0.500.130.12-0.02
0.200.22
0.160.06
vs, versus.
°P value results from testing if kappa is significantly different from 0.
<.01.07.47<.01.51.23
<.01
.01
<.01.01.04<.01<.01<.01.01.02
.03
.47
.01
.10
.12<.01.03<.01
.01<.01.01<.01
<.01.03.12.58
.01
.02
.04
.15
461

Beauchamp
KAPPA
COMPARISON N MEAN SD MEDIAN MINIMUM MAXIMUM
1. Among experts 6 0.11 0.11 0.06 -0.002 0.24
2. Among residents/photographers 2 0.21 0.05 0.21 0.18 0.25Wilcoxon test P value (1 vs 2)=.24
3. Experts vs residents 8 0.23 0.08 0.23 0.15 0.39
4. Experts vs photographers 8 0.15 0.08 0.16 0.005 0.29
5. Residents vs photographers 4 0.24 0.06 0.22 0.18 0.33Kruskal-Wallis test P value (3 vs 4)=.06
6. Clinical vs experts 4 0.18 0.22 0.13 -0.02 0.50
7. Clinical vs residents 2 0.21 0.02 0.21 0.20 0.22
8. Clinical vs photographers 2 0.11 0.07 0.11 0.06 0.16Wilcoxon test P value (6 vs 7,8)=.67
vs, versus.Other comparisons (Wilcoxon test P values):Among experts vs experts and other (1 vs 3,4)=0.17Among others vs between others (2 vs 5)=0.82
TABLE XI: KAPPA STATISTICS FOR INDIVIDUAL CATEGORIES'*
Disease Category Overall
Raters Kappvalue) Kapa(Pvlue) Kappa(Pvalue) Kappa(PvIue)
All 8 0.205(<.01) 0.030(.19) 0.241(<.01) 0.174(<.01)
4 Experts 0.104(.04) 0.0(-) 0.155(<.01) 0.096(.01)
2 Residents 0.302(.01) -0.144(.23) 0.343(<.01) 0.249(<.01)
2 Photographers 0.207(.09) -0.012(-) 0.100(.42) 0.181(<.01)
°P values result from testing in kappa is significantly different from 0. Where no P valueis given, the value of kappa was too small to warrant testing.
TABLE X: COMPARISON OF KAPPA VALUES BETWEEN OBSERVER GROUPS
462

Neurofibromwtosis Type 1
CL. + + c'r. c. c. + + + co -
+ + + + + + + + ++ir+
+l+ +c. ++++ + v
CL. + +CIL. + + + CO)
+ F + + aL XL + + ' t S
C F ' + + + + + +++ C_
C' f F + + a + CO CO __-~~~~~~~~~~~'4 + + + ++ + + + CI.CL
L)~~~~~~~~~~~~
H--
H'
0
r. Cda
Ein
+++ + I Z 0.~~~~~~~~b ~
"~cd
463
Beauchamp
OTHER OBSERVATIONS
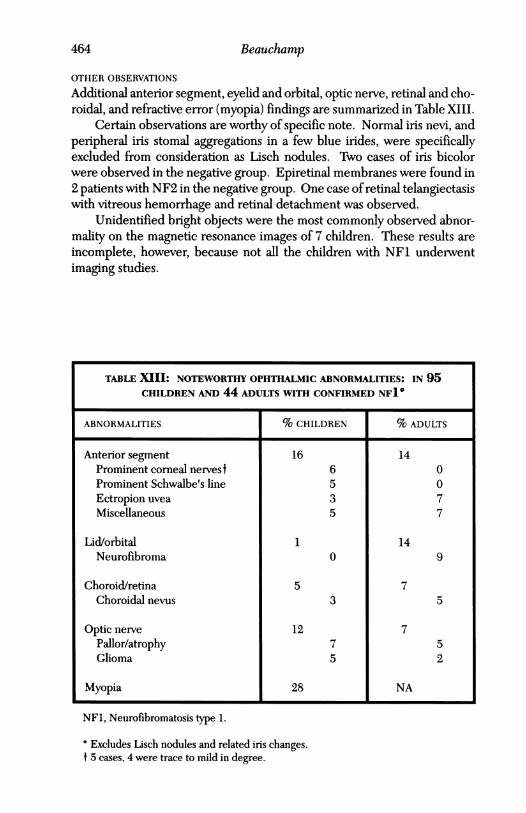
Additional anterior segment, eyelid and orbital, optic nerve, retinal and cho-roidal, and refractive error (myopia) findings are summarized in Table XIII.
Certain observations are worthy of specific note. Normal iris nevi, andperipheral iris stomal aggregations in a few blue irides, were specificallyexcluded from consideration as Lisch nodules. Two cases of iris bicolorwere observed in the negative group. Epiretinal membranes were found in2 patients with NF2 in the negative group. One case of retinal telangiectasiswith vitreous hemorrhage and retinal detachment was observed.
Unidentified bright objects were the most commonly observed abnor-mality on the magnetic resonance images of 7 children. These results areincomplete, however, because not all the children with NFL underwentimaging studies.
TABLE XIII: NOTEWORTHY OPHTHALMIC ABNORMALITIES: IN 95CHILDREN AND 44 ADULTS WITH CONFIRMED NFI'
ABNORMALITIES % CHILDREN % ADULTS
Anterior segment 16 14Prominent comeal nervest 6 0Prominent Schwalbe's line 5 0Ectropion uvea 3 7Miscellaneous 5 7
Lid/orbital 1 14Neurofibroma 0 9
Choroid/retina 5 7Choroidal nevus 3 5
Optic nerve 12 7Pallor/atrophy 7 5Glioma 5 2
Myopia 28 NA
NF1, Neurofibromatosis type 1.
o Excludes Lisch nodules and related iris changes.t 5 cases, 4 were trace to mild in degree.
464

Neurofibromatosis Type 1
DISCUSSION
The results reported here suggest that a diagnosis of NF1 based on theidentification of Lisch nodules in children may miss the mark, because de-tecting the presence of these nodules may be difficult or produce equivocalfindings. Specifically, children ofvarying ages permit and require examina-tions appropriate to their age and circumstances. To diagnose NFL reliablyin children, the clinician should look at a spectrum of ophthalmic manifes-tations that is wider than "classic" Lisch nodules. Identifying atypicalmorphologic clues, such as those presented in this thesis, can improve theclinician's performance in diagnosis.
This study is the first to include patients having incomplete or atypicalcharacteristics of NFL. It is also unique in documenting differences in per-formance within and between "expert" observers of iris photographs ofNFLpatients, suspected NF1 patients, and normal individuals.
GENERAL CHARACTERISTICS
Except for a high incidence ofmigraine, the general characteristics found inpatients with established NFL were typical of those reported elsewhere.The number of children studied here was comparable or larger than thatreported in extant reviews.
OPHTHALMIC MANIFESTATIONS
This study confirms the overall rapid rise in prevalence ofophthalmic mani-festations of NF1 that commence in early childhood. But the picture wasnot as clear-cut in the children studied here as in those reported elsewhere.Other studies reported neither the diverse expression ofthe trait nor equivo-cation by observers. This study characterized a variety of atypical changesin the iris and found interobserver equivocation in identifying Lisch noduleformation.
NONIRIS FINDINGS
Retinal and other findings not involving the iris were relatively uncommon inthis study, but three are noteworthy.
1. The incidence ofmyopia in the group ofchildren was 28%. Althoughno age-matched data exist to compare this percentage to a population,this incidence seems high and deserves further study.
2. One child experienced vitreous hemorrhage and subsequent retinaldetachment, events apparently associated with abnormal peripheralretinal vessels. The patient's course followed that reported by others.52
3. Two patients, 1 with unilateral Lisch nodules, were later confirmed ashaving neurofibromatosis type 2 (NF2) and epiretinal membrane for-mation. Such membranes have been observed in two thirds of pa-tients with NF2.`°
465

Beauchamp
IRIS CHANGES
Patient iris changes were found over a continuous spectrum in this study.Some changes appear as typical Lisch nodules, and others blend into theatypical; each variation may appear frequently in the same iris. When re-cording atypical changes, the author characterized them knowing that expe-rience builds "gestalt" reactions. Across the variations comes the conclu-sion that iris changes of NF1 literally "blur" the normal architecture ofstrands, crypts, collarette, etc, and they are difficult to specify. Others mightfind the "types" chosen arbitrary.
Taken with the interobserver data, discussed further in the next sec-tion, the data suggest that including a wider spectrum ofatypical iris changesduring examination of children suspected of having NF1 increases the sen-sitivity but decreases the specificity of diagnosing the disease. Neither thesensitivity nor specificity ofobservations is particularly trustworthy for a singleobserver, however, because of the high variability of interobserver correla-tions. Therefore, the spectrum of iris changes to be observed in childrenshould be much broader than that ofobserving "classic" Lisch nodules. Whilethese nodules are valuable markers for NF1, they cannot always be dis-cerned with certainty-and not necessarily because the irides are "normal."All extant studies report the presence or absence of Lisch nodules (and insome cases their numbers), without qualification or acknowledgment of in-decision. The author admits not being able to routinely make a binary deci-sion about iris changes in NFL, and experience has only increased perplex-ity. The interobserver data show that others make incorrect decisions. In-deed, the validity of Lisch nodule evaluation as a defining feature in thediagnosis of the disease may need rethinking; in general, obvious disease inolder children and adults tends to be accompanied by obvious iris changes,and less obvious disease by no change or less obvious (including "atypical")changes. The clinician's diagnosis mirrors (1) the heterogeneity of clinicalexpression of NF1, (2) the diversity of biologic phenotypic expression, and(3) the perceptive differences inherent in multiple human observers in theophthalmic evaluation of suspected NFL patients.
INTEROBSERVER VALIDATION OF IRIS CHANGES
Only one prior study has used photographic analysis to augment clinicalevaluation ofNF1, but that study did not test for interobserver differences.'5When both patient and "extraneous clues" (such as the clinical record orreferral information) were hidden, neither the author nor an assembled panelreviewing photographs fared as well as reported in that study. Interobserverstatistics showed that masked photographic analysis, review, and rating bymultiple raters introduced diversity and uncertainty into the diagnosis.
The use of photographic analysis to evaluate iris changes has bothstrengths and weaknesses. Although photography lacks the dynamic quali-ties of in vivo examination, including stereopsis, the author tended to find
466

Neurofibromatosis Type 1
more Lisch nodules/changes when reviewing photographs than when view-ing patients clinically. Using photographs also permitted masked review,which tests the ability of a clinician to evaluate NF1 without clues from casehistory of the parent or patient, and physical findings that might influenceobservations and conclusions. For example, a relatively small child with alarge head, dusky color, unusual speech tonality, and caf6-au-lait spots whowas referred by the pediatric neurologist for "Lisch nodule evaluation" mightbe more likely to evoke a concordant (positive) assessment by an ophthal-mologist than if those factors were hidden. Use of photographs permitsinterobserver comparison of such assessments. In this study, kappa coeffi-cients of interobserver reliability were poor.
The panel of "experts" (pediatric and neuro-ophthalmologists) per-formed unfavorably when compared with the known diagnosis and a panelofnovices (residents and photographers) who viewed selected photographs.The author reviewed all photographs for each patient. Poor ratings by otherobservers may have been intrinsic to the method: Insufficient views to drawappropriate conclusions, inappropriate case selection, confusion by the ob-server induced by an unfamiliar process, or inattention to the process byobservers. Nevertheless, the difference in ratings between observers in dis-cerning a known diagnosis begs for clarification. In addition, as the mostaccurate ofthe observers utilizing masked photographic analysis, the authorwas only "fair" in correctly discerning known diagnosis.
ORIGINS OF CLINICAL DIVERSITY
The basic science of neurofibromatosis, its genetics, and its cell biology arestill being studied, and the reasons for its remarkable clinical diversity arespeculative (perhaps a "megagene"). In 1981 Bolande5 compellingly ad-vanced the concept of NF1 as neurocristopathy-a generalized disorder ofneural crest origin. The neural crest arises early in embryogenesis, prolifer-ates, migrates, and differentiates in response to microenvironmental stimulialong predetermined routes. The multiple cell types so derived includesensory and sympathetic neurons, neurosecretory cells, melanocytes, partsof the skeleton and connective tissue, and most of the mesenchymal struc-tures of the face, neck, and eye.
The complexity of genetic (and possibly environmental) influences ondevelopmental biologic and pathophysiologic mechanisms may be inferredto be the bases for clinical diversity in NF1 (an appendix reviews this litera-ture). This diversity is apparently reflected in the spectrum of iris findingsseen in NF1 and the problematic specificity (for diagnostic purposes) theyrepresent.
APPENDIX
REVIEW OF SELECTED LITERATURE
The history, diagnostic and associated features ofNF1 in childhood are sum-marized in the introduction"0, and in Table I of the main thesis.'153 They
467

Beauchamp
attest to the clinical diversity of the NFL in childhood. This variability maybe ascribed to genetic, developmental, and pathophysiologic aspects.T4M80
In 1937, Lisch8l proposed tha association ofiris changes with NFL. Nod-ules, named for Lisch and characterized as hamartomata of the iris, are his-tologically described as condensations of irregularly composed spindle-shaped cells on the anterior iris surface. When pigmented, an underlyingnevus is present, confirming their melanocytic origin. They are found invirtually all adults with confirmed disease. They are also used as a marker inevaluating unaffected family members. Most authorities confirm a lowerprevalence of Lisch nodule formation in early childhood, which rapidly risesto nearly 100% by age 20."'-17
According to Riccardi and Eichner,24 half ofthe patients by age 15 havereached grade 3, disease that compromises health without shortening lifespan, and one fourth of patients by age 30 have reached grade 4, diseasethat may shorten life.
Only one study before this one has used photographic analysis to aug-ment clinical evaluation of NFL, but that study did not test for interobserverdifferences.'5 Color illustrations shown in a recent article are claimed toenable the cinican to readily differentiate hamartomas from other iris le-sions.82
The complexity of genetic (and possibly environmental) inferences ondevelopmental biologic and pathophysiologic mechanisms may be inferredto be the bases for clinical diversity in NFL. This diversity is apparentlyreflected in the spectrum of iris findings seen in NF1 and the problematicspecificity (for diagnostic purposes) they represent.
GENETICS OF ATYPICAL EXPRESSION
The conclusion that NF1 is a dominantly inherited autosomal disorder, nowconfirmed, dates to the work of Preiser and Davenport.54 The nature andcourse ofmaternally and paternally transmitted disease is apparently equiva-lent.5fi Yet half of NFL occurring in childhood is unaccompanied by evi-dence ofparental disease. Penetrance ofgene expression approaches 100%,but expressivity is highly variable even among affected siblings.
A suspected NF1 "megagene" presumably accounts for a very high mu-tation rate (100 x average for a single gene) and the incidence of sporadicdisease. Alternatively, the locus may be particularly fragile or subject todisruption.' Mutations are associated with increased paternal age; malesolder than 35 years of age have twice the risk of fathering a child with NF1than those under age 35.55
Utilizing linkage analysis, researchers have identified the gene locus onchomosome 17.57' 58 Pooling ofdata permitted mapping to 17ql 1.2 and iden-tified flanking DNA markers.5962 Despite the large size of the gene (200 to300 kb), no evidence for genetic heterogeneity exists.' Molecular cloning
468

Neurofibromwtosis Type 1
has permitted partial characterization of the gene. At least a part of thefunction ofthe gene is a guanosine diphosphatase-activating protein, whichevidently participates, through control ofras oncogenes, in control of cellu-lar proliferation.63
Three other genes-two species ofan ecotropic viral-insertion-site gene(EV12A and EV12B)-and an oligodendrocyte-myelin glycoprotein, arepresent on the gene.9 Finally, alterations at the 17pS3 locus are critical tothe progression of neurofibroma to neurofibrosarcoma.64
DEVELPOMETNAL BIOLOGY AND PATHOPHYSIOLOGY
In 1981 Bolande5 compellingly advanced the concept of NF1 asneurocristopathy-a generalized disorder of neural crest origin. The neu-ral crest arises early in embryogenesis, proliferates, migrates, and differen-tiates in response to microenvironmental stimuli along predeterminedroutes."" The multiple cell types so derived include sensory and sympa-thetic neurons, neurosecretory cells, melanocytes, parts of the skeleton andconnective tissue,' and most of the mesenchymal structures of the face,neck, and eye.67 The Lisch nodules are confirmed as being of melancyticorigin, consist of a condensation of spindle cells on the anterior iris surfaceand, when pigmented, and underlying stromal nevus is present.83
The mediation ofcellular response in NF1 by nerve growth factor (NGF)has been the subject ofsubstantial investigation.637' Other circulating mito-gens potentially implicated in NF1 abnormalities that have been identifiedinclude epidermal growth factor72 and heterogeneous substances, whichstimulate neuronal growth distinct from NGF,73 although they are not de-monstrably elevated in NFl.' On the other hand, glial growth factor is aSchwann cell mitogen, and elevations have been demonstrated with acous-tic neuromas; they require further study in NF1.74
The role of mast cells as important pathogenic mediators is the subjectof considerable interest and commends further investigation.76 Riccardi7'has postulated their key role on the basis of clinical pruritis and the highconcentration of histamine-containing mast cells in peripheralneurofibromas. Histamine and heparin (both elaborated from mast cells)are mitogens and also have been demonstrated to promote angiogenesis.
REFERENCES
1. Listernick R, Charrow J: Neurofibromatosis type 1 in childhood. J Pediatr, 1990;116(6):845-853.
2. Von Recklinghausen F: Uber die multiplen Fibrome der Haut und ihre Beziehungen zuden Neuromen (Concerning Multiple Fibromas of the Skin and Their Relationship toNeuromas). Berlin, August Hirschwald, 1882.
3. Riccardi VM: Neurofibromatosis: Clinical heterogeneity. CurrProbl Cancer 1982; 7(2):1-34.
4. Neurofibromatosis. National Institutes Consensus Development Conference Statement.Bethesda, Md: National Institutes of Health; 1987:6.
469

470 Beauchamp
5. Bol3n.lde RP: Neuirof'lbromatosis. The quintessential neurocristopathy: Pathogeneticconcepts an(l relationships. Adv Neturol 1981; 29:67-75.
6. Riccardi \TM: von Recklinglhauisen neiirofibromatosis. N EnglJ Med 1981; 305(27):1617-1627.
7. Mulvihill jj, Pairry DM, Shermlan JL et al: Neuirofibromatosis 1 (Recklinghauisen dis-ease) and Neurofibromatosis 2 (hilateral acoustic neurofibrom-atosis). Ann Intern AMed1990; 1 13(1):39-52.
8. Riccardi VM: Neurofibromnatosis: Past, present, and fuiture. N Engl J Med 1991;324(18): 1283-1285.
9. Diunnl D\V: Neurofibromiiatosis in childhood. Coirr Probl Pediatr 1987; 17(8):451-497.10. liusoni SM: Recent developments in the diagnosis and management ofneuirofibromatosis.
Arcli Dis Chiild 1989; 64:745-749.11. Lewis RA, Riccardi VM: von Recklinghauisen neurofibromatosis. Ophlthalmology 1981;
88:348-354.12. Fluieler U, Boltshauiser E, Kilchhofer A: Iris hamartomata as diagnostic criterion in
neurofibromatosis. Neu ropediatrics 1986; 17:183-185.13. Zehavi C, Roinano A, Goodman RM: Iris (Lisch) nodules in neurofibromatosis. Clin
Geuiet 1986; 29:51-55.14. Hulson S, Jones D, Beck L: Ophthalmic manifestations of neurofibromatosis. Br J
Ophlthlaltiool 1987; 71:235-238.15. Kilelhofer A, Boltshauser E, Flueler U: Aspekt and Inzidenz der Irishamartome bei
Neuirofibromatosis v. Recklinghausen (Appearance and Incidlence of Iris Haoarfooinfs inNeiurofibroniatosis). Klin Monatsbl Augeoheilkd 1986; 188:416-417.
16. Toonstra J, Dandrieui MR, Ippel PF, et al: Are Lisch nodules an ocular marker of theneturofibromatosis gene in otherwise unaffected family members? Denattologica 1987;174:232-235.
17. Lubs M, Bauer MS, Formas ME et al: Lisch nodtules in neurofibromatosis type 1. NEnglJ Med 1991; 324(18):1264-1266.
18. Crowe FW, Schull WJ: Diagnostic importance of cafe-au-lait spots in neurofibromatosis.Arcli Intern Med 1953; 91:758-766.
19. Crowe FW: Axillary freckling as a diagnostic aid in neurofibromatosis. Ann Intern Med1964; 61:1142-1143.
20. Harkin JC: Pathology of nerve sheath tumors. Ann NY Acad Sci 1986; 486: 147-154.21. Obringer AC, Meadows AT, Zackai EH: The diagnosis of neurofibromatosis-1 in the
child under the age of 6 years. Am J Dis Child 1989; 143:717-719.22. Ilolt JF: Neurofibromnatosis in children. AomJ Roentgenol 1978; 130:615-639.23. Crawford AH, Bagamery N: Osseouis manifestations of neurofibromatosis in childhood.
J Pediatr Orthop 1986; 6:72-88.24. Riccardi VM, Eichner JE: Neturofibrooiatosis: Phenotype, Natuiral Historl, and Patho-
genesis. Baltimnore, Johns Hopkins University Press, 1986.25. Listernick R, Charrow J, Greenwald MJ, et al: Optic gliomas in children vith
neurofibromnatosis type 1. J Pediatr 1989; 114:788-792.26. Sorensen SA, Mulvihill JJ, Nielsen A: Long-term follow-iip of von Recklinghallsen
neurofibrom-atosis: Survival and malignant neoplasmns. N Enlgl J Med 1986; 314:1010-1015.
27. Bader JL, Miller RW: Neurofibromatosis and childhlood leukemiiia. J Pediatr 1978; 92:925-929.
28. Cooper PEI, Frierson IIF, Kayne AL, et al: Association ofjmmvenile xanthogranluloma wAithjuvenile myeloid leukemia. Arc/i Denoaitol 1984; 120:371-375.
29. McKeen EA, Bodurtha J, Meadows AT, et al: Rhabdomyosarcoma complicating mulltipleneuirofibromnatosis. J Pediatr 1978; 93:992-993.
30. Stay EJ, Vawter C: The relationship between nephroblastomlia and neurofibromlatosis(von Reckhinghauisen's disease). Canicer 1977; 39:2550-2555.
31. Eldridge R, Denckla M, Bien E, et al: Neurofibromatosis type 1 (Recklinghauisen's dis-ease): Neuirologic and cognitive assessmiient with sibling controls. Anii J Dis Chlild 1989;143:833-837.
32. Eliason MJ: Neurofibromiiatosis: Imnplications for learning and behavior. Dev BeliavPediatr 1986; 7:175-179.

Neurofibromatosis Type 1 471
33. Varnblagen CK, Lewrin S, Das JP, et al: Neurofibromatosis and psylchological processes.Dev Belhav Pediatr 1988; 9:257-265.
34. Rosmnan NP, Pearce J: The brain in mulltiple neuirofibromnatosis (von Recklinghauisen'sdisease): A suiggested neuiropathological basis for the associated mental defect. Brain1967; 90:829-838.
35. Aron AM: Learning disabilities in children witb NF. Netvsletter of the NationalNet rofibromaultosi.s Fountdatiotn. XVinter/spring, 1984.
36. Allanson JE, liall JG, Vman Allen MI: Noonan phenotype associated1 withl nelirofi)ronlatosis.Aiii jAmed Geniet 1985; 21:4571-462.
37. Mendez II M: The neurofibromnatosis-Noonan syndrome. AmiJMed Geniet 1985; 21:471-476.
38. Opitz JM, Weaver DD: The neuirofibromatosis-Noonan syndrome. AmlJ M\ed Genlet1985; 21:477-490.
39. Meinecke P: Evidence that the "neuirofibromatosis-Noonan syndrome" is a variant ofvon Recklinghanisen neurofibromatosis. Aml J Mfed Genet 1987; 26:741-745.
40. RiccardiXM: The pathophysiology of neurofibromatosis: IN' Dermatologic insiglhts intoheterogeneity and pathogenesis. J Aml Acad Deoiiatol 1980; 3:157-166.
41. Norman ME: Neuirofibromiiatosis in a family. AmiJ Dis Chiild 1972; 123:159-160.42. Weichert KA, Dine MS, Benton C, et al: Macrocraniumin and neuirofibromlatosis. Radiol-
ogy 1973; 107:163-166.43. Petersen JM, Ferguison DR: Gastrointestinal nenrofibromatosis. J Clini Gastrocenterol
1984; 6:529-534.44. Saxena KM: Endocrine manifestations of neurofibromatosis in children. Am]iJ Dis Chiild
1970; 120:265-271.45. Duinn DW, P'uriin VA: Ophtlhalmlologic mlanifestations of nenirofibromatosis. In: Smith
JL, Katz B, edls: Neuro-oplhthalmology Enters tle 90s, Miamiii, Fla, Neuro-Ophlthalbnol-ogy Tapes, 1990, pp 153-170.
46. flall BD: Congenital lid ptosis associated with neurofibromatosis. Am] Mfed Cenet 1986;25:595-597.
47. SavinoPPJ, (laserJS, LixenbergMN: Pulsatingenophthlilmosandchoroidal hamartomilas:Two rare stigmata of neurofibromatosis. Br] Ophlthalmol 1977; 61:483-488.
48. Satran L, Letson RD, Seljeskog EL: Neuirofibromatosis with congenital glaucoma andbuiphthalminos in a newborn. Am]iJ Dis Chiild 1980; 134:182-183.
49. Bardelli AM, Iladjistilianoni T: Buiphthalimios an(d progressive elephantiasis inneuirofibromlatosis: A report of tlhree cases. Oph1thalmt1ic Pediatr Geniet, 1989: 10(4):279-286.
50. Castillo M, Quiencer RM, Glaser J, et al: Congenital glauicoma and buplithalmos in achildl withi neurofibromatosis. ] Clio Neiuro Ophlthalmiiol 1988; 8(1):69-71.
51. Grant WVM, Walton DS: Distinctive gonioscopic findings in glaullcomia diie toneuirofibromatosis. Archi Ophithalmol 1968; 79:127.
52. Destro M, DlAmilico DJ, Gragouidas ES, et al: Retinal manifestations of neurofibromatosis:Diagnosis and management. Archi Ophithlalmtiol 1991; 1()9:662.
53. Samiuiielsson B, Axelsson R: Neurofibromiatosis. Acta Denrn Venereol Stippl (Stokh) 1981;95:67.
54. Preiser SA, Davenport CB: Muiltiple neuirofibromlatosis (von Becklinghausen's (lisease)and its inheritance: With description of a case. Am]iJ Mled Sci 1918; 156:507-540.
55. Riccardi VM, Dobson CE, Chakraborty,R et al: The pathophysiolog of neurofibromatosis:IX. Paternal age as a factor in the origini of new miutations. Amii J Med Genet 1984;18:169-176.
56. Riccardi VM, Wald JS: Discouinting an adverse mi.aterinal effect of severity ofneurofibromatosis. Pediatrics 1987; 79:386-393.
57. Barker D, Wrighit E, Nguyen K, et al: (;ene for von Becklighauisen neurofibromatosis isin the pericentroinetric region of chromilosomile 17. Sci(ence 1987; 236:1100-1102.
58. Seizinger BB, Boileain GA, Ozeliuis LJ, et al: Genetic linkage of von Reckliughialusenneurofibroinatosis to the nerve gro\wth factor receptor gene. Cell 1987; 49:589-594.
59. Schmid(t MA, Michiels VVY Deward WV: Cases of neiirofibromatosis \ith reairraingemenitsof clromiiosomile 17 invol\ing hand 17 (11 1.2. Am] MJ1 ed CGenet 1987; 28:771-777.
60. Ledbetter 1)11, Bich DC, O(Connell P, et al: Precise localization of NFI to 17(111.2 by

Beauchamp
balanced translocation. Am J Humti Genet 1989; 44:38-40.61. Goidgar DE, Green P, Parry DM, et al: Multipoint linkage analysis in neurofibromatosis
type 1: An international collaboration. Am J Hum Cenet 1989; 44:6-12.62. Wallace MR, Marchuk DA, Andersen LB, et al: Type 1 neurofibromatosis gene: Identi-
fication of a large transcript disrupted in three NFI patients. Science 1990; 249:181-186.63. Xu G, O'Connell P, Viskochil D, et al: The neurofibromatosis type 1 gene encodes a
protein related to GAP. Cell 1990; 62:599-608.64. Menon AG, Anderson KM, Riccardi VM, et al: Chromosome 17p deletions and p 53
gene mutations associated with the formation of malignant neurofibromnatosis in vonRecklinghausen neurofibromnatosis. Proc Natl Acad Sci USA 1990; 87:5435-5439.
65. Weston JA: The regulation of normal and abnormal neural crest cell development. AdvNeurol 1981; 29:77-96.
66. LeDouarin NM: Investigations on the neural crest: Methodologic aspects and recentadvances. Ainn NY Acad Sci 1986; 486:66-68.
67. Beauchamp GR, Knepper PA: Role of the neural crest in anterior segment developmentand disease. J Pediatr Ophthalttml Strabismrlus 1985; 22(4):149-155.
68. Yanker BA, Shooter EM: The biology and mechanism of action of nerve growth factor.Antn Rev Biochem 1982; 51:845-868.
69. Sonnefeld KH, Bernd P, Sobue G, et al: Nerve growth factor receptors on dissociatedneurofibroma Schwann-like cells. Canicer Res 1986; 46:1446-1452.
70. Taniuchi M, Clark HB, Johnson EM: Induction of nerve growth factor receptors inSchwann cells after axotomy. Proc Natl Acad Sci USA 1986; 83:4094-4098.
71. Schenkein I, Beuker ED, Helson L, et al: Increased nerve-growth stimulating activity indisseminated neurofibromatosis. N EnglJ Med 1974; 290:613-614.
72. Zelkowitz M: Neurofibromatosis fibroblasts: Abnormal growth and binding to epider-mal growth factor. Adv Neurol 1981; 29:173-189.
73. Riopelle Rj, Riccardi VM, Faulkner S, et al: Serum neuronal growth factor levels in vonRecklinghausen's neurofibromatosis. Ann Neurol 1984; 16:54-59.
74. Brockes JP: Glial growth factor-like activitv in Schwann cell tumors. Anni Neurol 1986;20:317-322.
75. Riccardi VM: Pathophysiology of neurofibromatosis. J Am Acad Dentatol 1980; 3:15-7 -166.
76. Riccardi VM: Mast-cell stabilization to decrease neurofibroma growth. Arch Dermtatol1987; 123:1011-1016.
77. Lewis RA, oral commutnication, May, 1987.78. Thompson JA: Diagnosis and treatment of headache in the pediatric patient. Curr Probl
Pediatr 1980; 10:1-52.79. Liang JC, Juarez CP, Goldberg MF: Bilateral bicolored irides with Hirschsprung's dis-
ease. Arclh Ophthalmtol 1983; 101:69-73.80. Kaye LM, Rothner AD, Beauchamp GR, et al: Ocular findings associated with
neurofibromatosis type II. Ophtlhalmology 1992; 99:1424-1429.81. Lisch K: Ueber Beteiligung der Augen, insbesondere das Vorkommen von Irisknotchen
bei der Neuirofibromatose (Recklinghausen). Z Augenlheilkd 1937; 93:137-143.82. Ragge NK, Falk RE, (Cohen WE, et al: Images of Lisch nodules across the spectrumll.
Eye 1993; 7(pt 1):95-101.83. Williamson TH, Garner A, Moore AT: Structure of Lisch nodules in neurofibromatosis
type 1. Oplithamol Pediatr Gentet 1991; 12(1):11-17.
472


















