
PROTOCOL AS0007 AMENDMENT 3 MULTICENTER, OPEN-LABEL …
105
UCB 22 Jan 2020 Clinical Study Protocol Certolizumab pegol AS0007 Confidential Page 1 of 113 PROTOCOL AS0007 AMENDMENT 3 MULTICENTER, OPEN-LABEL STUDY TO ASSESS THE EFFECTS OF CERTOLIZUMAB PEGOL ON THE REDUCTION OF ANTERIOR UVEITIS FLARES IN AXIAL SPONDYLOARTHRITIS SUBJECTS WITH A HISTORY OF ANTERIOR UVEITIS (C-VIEW) PHASE 4 EudraCT Number: 2016-000343-14 Sponsor: UCB Biopharma SPRL Allée de la Recherche 60 1070 Brussels BELGIUM Protocol/Amendment number Date Type of amendment Final protocol 14 Apr 2016 Not applicable Protocol amendment 1 03 Jun 2016 Non-substantial Protocol amendment 1.1 (Spain) 28 Oct 2016 Non-substantial Protocol amendment 2 17 Jan 2017 Substantial Protocol amendment 2.1 (Spain) 06 Apr 2017 Substantial Protocol amendment 3 22 Jan 2020 Non-substantial Confidential Material Confidential This document is the property of UCB and may not – in full or in part – be passed on, reproduced, published, or otherwise used without the express permission of UCB. B B BELG BELG umbe umb ED COPY herch herch Bruss rus This document cannot be c be use ol am ol am ed ame me e to end end o supp dmen dmen pport ent 1.1 nt 1. t pp any a marketing er r rke m authorization sels els GIUM IUM au application e 60 e 60 and any extensions or variations F F thereof.
Transcript of PROTOCOL AS0007 AMENDMENT 3 MULTICENTER, OPEN-LABEL …
UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 1 of 113
PROTOCOL AS0007 AMENDMENT 3
MULTICENTER, OPEN-LABEL STUDY TO ASSESS THE EFFECTS OF CERTOLIZUMAB PEGOL ON THE REDUCTION
OF ANTERIOR UVEITIS FLARES IN AXIAL SPONDYLOARTHRITIS SUBJECTS WITH A HISTORY OF
ANTERIOR UVEITIS (C-VIEW)
PHASE 4
EudraCT Number: 2016-000343-14
Sponsor:
UCB Biopharma SPRL
Allée de la Recherche 60
1070 BrusselsBELGIUM
Protocol/Amendment number Date Type of amendment
Final protocol 14 Apr 2016 Not applicable
Protocol amendment 1 03 Jun 2016 Non-substantial
Protocol amendment 1.1 (Spain) 28 Oct 2016 Non-substantial
Protocol amendment 2 17 Jan 2017 Substantial
Protocol amendment 2.1 (Spain) 06 Apr 2017 Substantial
Protocol amendment 3 22 Jan 2020 Non-substantial
Confidential Material
Confidential
This document is the property of UCB and may not – in full or in part – be passed on, reproduced, published, or otherwise used without the express permission of UCB.
BBBELGBELG
umbeumbED
COPYherchherch
Brussrus
This do
cumen
t can
not b
e cbe
use
ol amol amed
amemee
to endendosu
ppdmendmenppo
rt ent 1.1nt 1.tpp
any
amark
eting
err
rkem
autho
rizati
on
selselsGIUMIUM
au
appli
catio
n
e 60e 60
and a
ny ex
tensio
ns or
varia
tions
F F
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 2 of 113
STUDY CONTACT INFORMATION Sponsor UCB Biopharma SPRL
Allée de la Recherche 60
1070 Brussels
BELGIUM
Principal/Coordinating Investigator
Name: Dr.
Affiliation: VUMC afd. Reumatologie
Address: VUMC afd. ReumatologieKamer 3A- 64Postbus 7057 1007MB AmsterdamNETHERLANDS
Phone:
Fax:
Sponsor Study Physician
Name: , MD
Address: UCB Biosciences GmbHAlfred-Nobel-Str 1040789 Monheim/RheinGERMANY
Phone:
Fax:
REienieR
EDACTED
MDMD AC
RE
COPY PY
C
This do
cumen
t can
not b
e use
d to to
supp
ouuuupsu
port RRrt
ppo
u
any MM
RMANRMANmark
eting
nces Gces G
obelbel--StSMonhMonh
gg autho
rizati
on
tio
th
appli
catio
n
ca
ap
and a
ny ex
tensio
ns
ns
ns
ex
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 3 of 113
Clinical Project Manager
Name:
Address: UCB Biopharma SPRLAllée de la Recherche 601070 BrusselsBELGIUM
Phone:
Fax:
Clinical Trial Biostatistician
Name: Dr.
Address: UCB Biosciences GmbHAlfred-Nobel-Strasse 1040789 MonheimGermany
Phone:
Fax:
Clinical Monitoring Contract Research Organization
Name: PRA Health Sciences
Address: 500 South Oak WayGreen ParkReadingBerkshire, RG2 6AD UNITED KINGDOM
Phone: +44 (118) 918 1000
Fax: +44 (118) 918 1001
REDACTED DCT
ReseReseR
COPY OP
C
This do
cumen
t can
not b
e :b
used
us
to tosu
pport
ReadieadBerkBerUsu
anyn Par Pa
dingin
markth Oakh Oakk
ketin
g ea
Scienccienng
rke
autho
rizati
on
tio
orarcharch
appli
catio
n
ap
and d an
y a
exten
sions
io
e
or rova
riatio
ns
v
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 4 of 113
SERIOUS ADVERSE EVENT REPORTING
Serious adverse event reporting (24h)
Fax: +32 2 386 24 21
Email: [email protected]
REDACTED COPY
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on ap
plica
tion a
nd an
y exte
nsion
s or v
ariati
ons t
hereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 5 of 113
TABLE OF CONTENTS LIST OF ABBREVIATIONS.................................................................................................. 10 1 SUMMARY..................................................................................................................... 13 2 INTRODUCTION ........................................................................................................... 14 2.1 Natural history of axial spondyloarthritis .................................................................... 14 2.2 Uveitis in axial spondyloarthritis ................................................................................. 15 2.3 Diagnosing axial spondyloarthritis in clinical practice................................................ 16 2.4 Current management of axial spondyloarthritis and uveitis ........................................ 17 2.5 Rationale ...................................................................................................................... 18 3 STUDY OBJECTIVE(S) ................................................................................................. 19 3.1 Primary objective ......................................................................................................... 19 3.2 Secondary objectives ................................................................................................... 19 3.3 Other objectives ........................................................................................................... 19 3.4 Safety objective............................................................................................................ 19 4 STUDY VARIABLES..................................................................................................... 19 4.1 Efficacy variables......................................................................................................... 19
4.1.1 Primary efficacy variable..................................................................................... 19 4.1.2 Secondary efficacy variables ............................................................................... 19 4.1.3 Other efficacy variables ....................................................................................... 20
4.2 Safety variables............................................................................................................ 21 4.2.1 Secondary safety variable .................................................................................... 21 4.2.2 Other safety variables .......................................................................................... 21
5 STUDY DESIGN............................................................................................................. 21 5.1 Study description ......................................................................................................... 21
5.1.1 Study duration per subject ................................................................................... 22 5.1.2 Planned number of subjects and sites .................................................................. 22 5.1.3 Anticipated regions and countries ....................................................................... 22
5.2 Schedule of study assessments..................................................................................... 22 5.3 Schematic diagram....................................................................................................... 27 5.4 Rationale for study design and selection of dose......................................................... 28 6 SELECTION AND WITHDRAWAL OF SUBJECTS................................................... 28 6.1 Inclusion criteria .......................................................................................................... 28 6.2 Exclusion criteria ......................................................................................................... 29 6.3 Withdrawal criteria ...................................................................................................... 34 7 STUDY TREATMENT(S) .............................................................................................. 35 7.1 Description of investigational medicinal product(s).................................................... 35 7.2 Treatment(s) to be administered .................................................................................. 36 7.3 Packaging..................................................................................................................... 36
REDACTED .................
..........bleble ....
COPY ...............
This do
cumen
t 6.26.266
cann
ot ELECELECII
betionationaus
edof sof
matic datic to
atf stustu
supp
ortn perper
umber omberted reed re
any .........
er sur s
marketi
ng ..........
......................
autho
rizati
on .....
....................
..........
appli
catio
n ...................
.................
and .......... an
y ...........ex
tensio
ns
....................
.........
or ........ varia
tions
. 16161
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 6 of 113
7.4 Labeling ....................................................................................................................... 36 7.5 Handling and storage requirements ............................................................................. 36
7.5.1 Storage at the site................................................................................................. 36 7.5.2 Storage at the subject’s home .............................................................................. 37
7.6 Drug accountability...................................................................................................... 37 7.7 Procedures for monitoring subject compliance............................................................ 37 7.8 Concomitant medication(s)/treatment(s) ..................................................................... 38
7.8.1 Permitted concomitant treatments (medications and therapies) .......................... 38 7.8.2 Prohibited concomitant treatments (medications and therapies) ......................... 39 7.8.3 Rescue medication ............................................................................................... 39
7.9 Blinding........................................................................................................................ 39 7.10 Enrollment into treatment and numbering of subjects ................................................. 39 8 STUDY PROCEDURES BY VISIT ............................................................................... 40 8.1 Visit 1 (Week -5 to -1) Screening ................................................................................ 40 8.2 Visit 2 (Week 0) Baseline Visit ................................................................................... 42 8.3 Visit 3 and 4 (Week 2 and 4) ....................................................................................... 43 8.4 Visits 5 and 6, and Visits 8 to 12 (Weeks 12, 24, 36, 48, 60, 72, and 84) ................... 44 8.5 Visit 7 (Week 32)......................................................................................................... 45 8.6 Visit 13 (Week 96/Withdrawal Visit) .......................................................................... 45 8.7 Visit 14 (Follow-Up Visit)........................................................................................... 46 8.8 Unscheduled Visit ........................................................................................................ 46 9 ASSESSMENT OF EFFICACY...................................................................................... 47 9.1 Assessment of primary and secondary efficacy variables ........................................... 47
9.1.1 Assessment of AU flares ..................................................................................... 47 9.1.2 Ankylosing spondylitis disease activity score (ASDAS) .................................... 47 9.1.3 Bath ankylosing spondylitis disease activity index (BASDAI)........................... 48 9.1.4 ASAS20, ASAS40, ASAS5/6, and ASAS PR..................................................... 49 9.1.5 Physician’s global assessment of disease activity (PhGADA) ............................ 49 9.1.6 Swollen and tender joint counts (44 joints evaluation)........................................ 49 9.1.7 Patient’s global assessment of disease activity (PtGADA) (NRS)...................... 50 9.1.8 Total and nocturnal spinal pain NRS................................................................... 50 9.1.9 Bath ankylosing spondylitis functional index (BASFI)....................................... 50 9.1.10 Extra-articular assessments.................................................................................. 51
9.2 Assessment of other efficacy variables........................................................................ 51 9.2.1 Ankylosing spondylitis quality of life (ASQoL) ................................................. 51 9.2.2 ASAS HI Questionnaire ...................................................................................... 51 9.2.3 Short-Form 36-Item Health Survey (SF-36)........................................................ 52
10 ASSESSMENT OF SAFETY.......................................................................................... 52
REDACTED )) .........
.......................
YY
COPY 2, 24, 2, 24,
........
This do
cumen
t9.9.9.29.2
cann
ot 1.9.911
bePaPaT
used
icicwollenwollenatiti
toian’iansu
pport
nsing sping s
, ASAASA
any lala
dylitidylitimark
eting
..............
secondeconaresares
autho
rizati
on ............
.....................
....
appli
catio
n ..
........................
36, 486, 48
and .......
.....an
y ...........
exten
sions
...................
..........
or ...... va
riatio
ns
388383
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 7 of 113
10.1 Adverse events ............................................................................................................. 52 10.1.1 Definition of adverse event.................................................................................. 52 10.1.2 Procedures for reporting and recording adverse events....................................... 53 10.1.3 Description of adverse events .............................................................................. 53 10.1.4 Follow up of adverse events ................................................................................ 53 10.1.5 Rule for repetition of an adverse event ................................................................ 53 10.1.6 Pregnancy ............................................................................................................ 54 10.1.7 Overdose of investigational medicinal product ................................................... 54 10.1.8 Safety signal detection ......................................................................................... 55
10.2 Serious adverse events ................................................................................................. 55 10.2.1 Definition of serious adverse event ..................................................................... 55 10.2.2 Procedures for reporting serious adverse events ................................................. 56 10.2.3 Follow up of serious adverse events .................................................................... 56
10.3 Adverse events of interest ............................................................................................ 57 10.4 Immediate reporting of adverse events ........................................................................ 57 10.5 Anticipated serious adverse events .............................................................................. 57 10.6 Laboratory measurements ............................................................................................ 58
10.6.1 Pregnancy testing................................................................................................. 60 10.6.2 Physical assessments ........................................................................................... 60 10.6.3 MRI assessments ................................................................................................. 60 10.6.4 Tuberculosis assessments .................................................................................... 60
10.6.4.1 Tuberculosis test........................................................................................ 60 10.6.4.2 Chest x-ray ................................................................................................ 62 10.6.4.3 Tuberculosis questionnaire........................................................................ 62
11 STUDY MANAGEMENT AND ADMINISTRATION ................................................. 63 11.1 Adherence to protocol.................................................................................................. 63 11.2 Monitoring ................................................................................................................... 63
11.2.1 Definition of source data ..................................................................................... 63 11.2.2 Source data verification ....................................................................................... 64
11.3 Data handling ............................................................................................................... 64 11.3.1 Case Report Form completion ............................................................................. 64 11.3.2 Electronic reporting outcome .............................................................................. 65 11.3.3 Database entry and reconciliation........................................................................ 65 11.3.4 Subject Screening and Enrollment Log/Subject Identification Code list ............ 65
11.4 Termination of the study.............................................................................................. 65 11.5 Archiving and data retention........................................................................................ 66 11.6 Audit and inspection .................................................................................................... 66 11.7 Good Clinical Practice ................................................................................................. 67
REDACTED ............
...........ss ........
COPY ..........
........
This do
cumen
t1111cann
ot 11.3.2.3.2
beta hata ha
C
used
iiource urce an
totiontionsu
pport
Ntocolocol
............
any ququ
NT ANNT ANmark
eting
..............
............uestiues
autho
rizati
on ............
.....................
....
appli
catio
n ..
........................
...........
and .......
.....an
y ...........
exten
sions
...................
..........
or ...... va
riatio
ns
5454545
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 8 of 113
12 STATISTICS ................................................................................................................... 67 12.1 Definition of analysis sets............................................................................................ 67 12.2 General statistical considerations................................................................................. 67 12.3 Planned efficacy analyses ............................................................................................ 68
12.3.1 Analysis of the primary efficacy variable............................................................ 68 12.3.2 Other efficacy analyses........................................................................................ 68
12.4 Planned safety analyses................................................................................................ 68 12.4.1 Safety analyses..................................................................................................... 68
12.5 Handling of protocol deviations................................................................................... 68 12.6 Handling of dropouts or missing data.......................................................................... 69 12.7 Planned interim analysis and data monitoring ............................................................. 69 12.8 Determination of sample size....................................................................................... 69 13 ETHICS AND REGULATORY REQUIREMENTS...................................................... 69 13.1 Informed consent ......................................................................................................... 69 13.2 Subject identification cards.......................................................................................... 70 13.3 Institutional Review Boards and Independent Ethics Committees.............................. 70 13.4 Subject privacy............................................................................................................. 71 13.5 Protocol amendments................................................................................................... 71 14 FINANCE, INSURANCE, AND PUBLICATION ......................................................... 71 15 REFERENCES ................................................................................................................ 71 16 APPENDICES ................................................................................................................. 77 16.1 Modified NY criteria for ankylosing spondylitis......................................................... 77 16.2 ASAS classification criteria for axSpA ....................................................................... 78 16.3 Corticosteroid equivalent doses ................................................................................... 79 16.4 Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ............................... 80 16.5 PtGADA (NRS) ........................................................................................................... 81 16.6 Total spinal pain NRS and nocturnal spinal pain NRS................................................ 82 16.7 Bath Ankylosing Spondylitis Functional Index (BASFI) ............................................ 83 16.8 Ankylosing Spondylitis Quality of Life (ASQoL) questionnaire ................................ 85 16.9 ASAS Health Index Questionnaire .............................................................................. 87 16.10 Short-Form (SF-36) Questionnaire .............................................................................. 92 16.11 Tuberculosis Sample Worksheet.................................................................................. 98 17 APPENDICES ................................................................................................................. 99 17.1 Protocol Amendment 1 ................................................................................................ 99 17.2 Protocol Amendment 2 .............................................................................................. 101 17.3 Protocol Amendment 3 .............................................................................................. 110 18 DECLARATION AND SIGNATURE OF INVESTIGATOR ..................................... 112 19 SPONSOR DECLARATION ........................................................................................ 113
REDACTED LICATLICA
.......................
yloyl
COPY ..........
........
This do
cumen
t 1771717
cann
otShorhor1 TuTAA
beAS HAS us
ed yloyl
osing sing HH
toosinosinsu
pport
on............
ain NRain NR
any t dt d
ndylitdylitmark
eting
..osing ssing s
a for axfor axdosedos
autho
rizati
on ............
TIONTION...........
....
appli
catio
n ..
............CommComm
...........
and .......
.....an
y ...........
exten
sions
...................
..........
or ...... va
riatio
ns
6868686
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 9 of 113
LIST OF TABLES Table 5-1: Schedule of assessments .................................................................................. 23
Table 6‒1: Concomitant Medications (Prior to Baseline and Study Visits) ...................... 31
Table 9‒1: Swelling and tenderness grading ..................................................................... 50
Table 10‒1: Anticipated serious adverse events in population independent of drug exposure ........................................................................................................... 58
Table 10‒2: Laboratory measurements ................................................................................ 59
Table 16‒1: Corticosteroid equivalent doses (with reference to prednisone 10mg dose) (Meikle and Tyler, 1977) ................................................................................. 79
LIST OF FIGURES
Figure 2‒1: ASAS classification criteria for axSpA ........................................................... 17
Figure 5-2: Schematic diagram........................................................................................... 27
REDACTED COPY ...
..........
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on ..... ap
plica
tion
............
.....
and a
ny ex
tensio
ns ..........
mg domg do..........
or .... va
riatio
ns
5858
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 10 of 113
LIST OF ABBREVIATIONS ABA abatacept
ADA adalimumab
AE adverse event
AS ankylosing spondylitis
ASAS Assessment of SpondyloArthritis international Society
ASDAS Ankylosing Spondylitis Disease Activity Score
ASQoL Ankylosing Spondylitis Quality of Life
AU anterior uveitis
axSpA axial spondyloarthritis
AZA azathioprine
BASDAI Bath Ankylosing Spondylitis Disease Activity Index
BASFI Bath Ankylosing Spondylitis Functional Index
CD20 cluster of differentiation 20
CDMS clinical data management system
CI clinical improvement
CII clinically important improvement
CPMP Committee for Proprietary Medicinal Products
CRF Case Report Form
CRO contract research organization
CRP C-reactive protein
CZP certolizumab pegol
DMARD disease-modifying antirheumatic drug
DS Drug Safety
ECG electrocardiogram
eCRF electronic Case Report Form
ePRO electronic patient reported outcome
ES Enrolled Set
ETN etanercept
EudraCT European Union Drug Regulating Authorities Clinical Trials
FAS Full Analysis Set
FU Follow-Up
mentment
portanportan
ee foree for
COPY 2020
ment sment
This do
cumen
t ESESca
nnot
ROO
be us
ed to
supp
ort
certoceran
y reactireactmark
eting
r ProPro
ort Forort Fo
ct resect res
autho
rizati
onsystesyste
nt impt imp
opop
appli
catio
nActivcti
tional iona
and
tiviivi
any e
xtens
ions o
r vari
ation
s the
reof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 11 of 113
GCP Good Clinical Practice
GOL golimumab
HCQ hydroxychloroquine
HI Health Index
HIV human immunodeficiency virus
HLA-B27 human leukocyte antigen B27
HRQoL health-related quality of life
ia intra-articular
IBD inflammatory bowel disease
ICF informed consent form
ICH International Council for Harmonisation
ID inactive disease
IEC Independent Ethics Committee
IFX infliximab
IGRA Interferon-Gamma Release Assay
IMP investigational medicinal product
IRB Institutional Review Board
iv intravenous(ly)
IWRS interactive web response system
JIA juvenile idiopathic arthritis
LFN leflunomide
LTBI latent tuberculosis infection
MCID minimal clinically important difference
MI major improvement
mNY modified New York (classification criteria)
MRI magnetic resonance imaging
MTX methotrexate
nr-axSpA nonradiographic axSpA
NRS Numerical Rating Scale
NSAID nonsteroidal anti-inflammatory drug
PsA psoriatic arthritis
PEG polyethylene glycol
edicinedicin
ReviewRevie
ous(ly)ous(ly)
COPY eleaselease
This do
cumen
tnrnr--aa
NN
cann
ot XX
be us
ed to
supp
ort
latenlatean
y unomunommark
eting
)
e web rweb
le idiope idio
autho
rizati
one Asse Ass
nal pronal pro
w Boaw Bo
appli
catio
n
say
and a
ny ex
tensio
ns or
varia
tions
there
of.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 12 of 113
PhGADA Physician’s Global Assessment of Disease Activity
PR partial remission
PtGADA Patient’s Global Assessment of Disease Activity
Q2W every 2 weeks (every other week)
RA rheumatoid arthritis
RCT randomized controlled trial
SAE serious adverse event
SAP Statistical Analysis Plan
sc subcutaneous(ly)
SD standard deviation
SF-36 Short-Form 36-Item Health Survey
SIJ sacroiliac joint
SOP Standard Operating Procedure
SpA spondyloarthritis
SPC Summary of Product Characteristics
SS Safety Set
SSZ sulfasalazine
TB tuberculosis
TNF tumor necrosis factor
TNFα tumor necrosis factor alpha
ULN upper limit of normal
WBC white blood cell
WD withdrawal
VEGF vascular endothelial growth factor
VAS visual analog scale
sissis
COPY CharaChara
This do
cumen
t can
not b
e use
d to s
uppo
rt whitwhian
y per limper limark
eting
rosis faosis f
necrosnecro
autho
rizati
onacteracter appli
catio
n
isti
and a
ny ex
tensio
ns or
varia
tions
there
of.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 13 of 113
1 SUMMARY AS0007 is a multicenter, open-label, Phase 4 study to evaluate the effects of certolizumab pegol (CZP) on the incidence of anterior uveitis (AU) flares per year in subjects with both active axial spondyloarthritis (axSpA) and a history of AU by comparing the historical AU flare rate that occurred prior to CZP treatment with the AU flare rate occurring while under CZP treatment.
Approximately 130 subjects will be screened in Europe in order to enroll 86 subjects into the study.
The study duration from the start of treatment will be 96 weeks from Baseline onwards, and a follow-up visit will be performed at Week 104; 10 weeks after the last dose at Week 94.
The study includes 3 periods as follows:
Period 1 (Screening Period): 1 to 5 weeks before Baseline
Subjects will be informed about the study and sign the informed consent form (ICF). Eligibility will be evaluated and assessments will be performed. The Screening Period should be kept as short as possible but can be extended to 5 weeks if certain medications need to be washed out or to allow to obtain information from the subject’s ophthalmologist. For subjects who start a prophylactic treatment for latent tuberculosis infection (LTBI) the Screening Period can be extended up to 12 weeks; see Section 10.6.4.1.
Period 2 (Treatment Period): Week 0 to Week 96
Eligible subjects will receive a dose of CZP 400mg subcutaneously (sc) at Baseline, Week 2, and Week 4 followed by CZP 200mg sc every 2 weeks (Q2W) starting at Week 6 until Week 94.
All subjects will be trained at the beginning of the study on self-administration before the subjects start self-administration with the study drug (relative or caregiver may also perform the injections). The injection schedule will provide the sequence of self-administration and site visits including injection at the site.
Period 3 (Follow-Up [FU] Period): 10 weeks from the final dose of study medication received.
All subjects will have a FU Visit at Week 104 or earlier in case of an early withdrawal (WD), 10 weeks after the final administration of CZP administration received within the study.
A Week 48 analysis will be performed after all subjects have either completed the Week 48 Visit or have prematurely withdrawn prior to the Week 48 Visit. The final analysis will be conducted at the end of the study after all study data is locked.
The primary objective of the study will be to demonstrate the effect of CZP treatment on the reduction of AU flares in subjects with both active axSpA and a documented history of AU. The secondary objectives of the study will be to assess the effect of CZP treatment on 1) the reduction of AU flares in subjects with axSpA having at least 1 documented history of AU within 12 months prior to Baseline and 2) axSpA disease activity. Other objectives and safety objective are listed in Section 3.3 and Section 3.4, respectively.
REDACTED 400m400m
y 2 wy 2 w
ginninginninh thh th
COPY ek 96k 96
This do
cumen
t edudusecosecorere
cann
ot d oo
primaprimauctiouctio
be matmat
of thof thus
ed
alysis lysisatureture
tofinafina supp
ort
a FU Va FU al aal
any ] Peri] Permark
eting
ng og ohe stude stud
will proill pr
autho
rizati
on
mg submg subweeks (eeks
of tof t
appli
catio
n tiononist. Forst. Fo
BI) the I) the
andPerioPerio
ons nns n
any form form
odd
exten
sions
94.94. orandandvaria
tions
d ad
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 14 of 113
2 INTRODUCTION 2.1 Natural history of axial spondyloarthritis Spondyloarthritis (SpA) is an umbrella term applied to a family of rheumatic diseases that have features in common with and distinct from other inflammatory arthritides, particularly rheumatoid arthritis (RA).
The Assessment of SpondyloArthritis international Society (ASAS) working group established classification criteria distinguishing 2 broad categories of SpA: peripheral and axial SpA (Rudwaleit et al, 2011; Rudwaleit, 2010; Rudwaleit et al, 2009b). This division is based on the body part predominantly involved in the inflammatory process and those areas of the body that may respond similarly well to medication. Therefore, peripheral SpA includes diseases affecting mainly peripheral joints, such as reactive arthritis and psoriatic arthritis, whereas axSpA comprises those diseases with mainly axial involvement (sacroiliac joints [SIJs] and spine), including ankylosing spondylitis (AS) and nonradiographic axSpA (nr-axSpA).
Patients with AS have definitive evidence of structural changes in the SIJ (sacroiliitis) on x-ray, fulfilling the modified NY (mNY) classification criteria (mNY-positive) (van der Linden, 1984);whereas, those with nr-axSpA structural changes on conventional radiographs do not meet the mNY classification criteria (mNY-negative) (Appendix 16.1) (Rudwaleit et al, 2005; Dougados et al, 1991).
In patients with axSpA, the disease typically originates in the SIJs, then progresses to the spine. In the SIJs and the spine, active inflammation results in erosions, sclerosis, and fatty lesions. However, the most characteristic feature is new bone formation leading to ankylosis of the SIJs and syndesmophytes attached to the vertebral bodies. As a result of extended syndesmophyte formation, the spine may become fused over time. The majority of patients with axSpA have inflammatory back pain. Other objective signs of inflammation, such as enthesitis, dactylitis, peripheral arthritis, or uveitis; genetic features, such as the presence of human leukocyte antigen B27 (HLA-B27); and laboratory parameters, such as elevated C-reactive protein (CRP), may also be present (Braun, 2012; Rudwaleit et al, 2009a; Braun, 2007). Disability in axSpA is related to both the degree of inflammatory activity, causing pain, stiffness, fatigue, and poor quality of sleep, and to the degree of bony ankylosis, causing loss of spinal mobility.
The natural history of axSpA is characterized by a variable disease course. Over time, patients develop structural damage or radiographic abnormalities involving their SIJs and may fulfill the mNY classification criteria for AS. However, the rate of development of structural damage varies among patients (Rudwaleit, 2012). Some patients develop bilateral sacroiliitis, some unilateral sacroiliitis, and others may never develop definitive sacroiliitis on x-ray despite significant disease burden and other signs and symptoms of the disease, such as spinal lesions, uveitis, enthesitis, and peripheral arthritis. Approximately 10% of patients with nr-axSpA (25%, if CRP levels are elevated) develop definitive evidence of sacroiliitis on x-ray within 2 years (Sieper and van der Heijde, 2013).
Spondyloarthritides and inflammatory bowel disease (IBD) are chronic, idiopathic inflammatory disorders of, respectively, the axial and peripheral joints/entheses, and the intestinal tract, affecting up to 1% of our population. Typically, SpA manifests between adolescence and the age of 40. There is clinical and genetic evidence supporting some degree of overlap between the pathogenesis of these 2 entities. In SpA, microscopic gut inflammation can be present as an acute
REDACTEDigg
n resun resus new new
ertebraertebrased ovsed ovtivti
COPY
ginateinate
This do
cumen
t veieif CRif CR(S(S
anno
tal sal saficantficant
itis, tis,
beng png pacac
used
al dadacationationpa
to of aof
damdamsu
pport
matoratorof bonof bo
f axSaxS
anypp
waleitwaleiryry
marketi
ngover tver
ve signve signtic featic fea
paramaram
autho
rizati
ontes in es in ults in ults in
w bonew boneal bodl bod
timtim
appli
catio
nositivsitinal radal rad
)) (Rud(Ru
ande SIJ SIJ
iveive
anySpA)SpA
(
exten
sionse bodbod
eases asesas axSas axS
IJs] aJs] a
oon ondd
varia
tions
ed ed
thth
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 15 of 113
or a chronic inflammation. Normal histology of the gut or acute lesions, mimicking a bacterial colitis, were predominantly found in patients presenting with transient arthritis, whereas in patients with chronic intestinal lesions similar to Crohn’s disease, a more persistent joint inflammation was perceived (Mielants et al, 1995). Microscopic gut inflammation was observed in about half of SpA patients originally in the 1980s, and this prevalence has been confirmed in the Gent Inflammatory Arthritis and Spondylitis cohort T, which focused on new-onset forms of SpA using the recently described ASAS classification criteria (Van Praet et al, 2013). Although about 6.5% of SpA patients evolved into IBD over 5 years, in patients exhibiting the chronic type of gut inflammation resembling Crohn’s disease, this number rose to 20%.
2.2 Uveitis in axial spondyloarthritis Uveitis is the third leading cause of blindness in developed countries. The annual incidence rate is estimated between 17 and 52 per 100,000 persons, and the prevalence is 38 to 714 per 100,000 persons (Wakefield and Chang, 2005). Males and females are generally equally affected overall, but sex preponderance may be observed in some uveitis groups, such as a male predominance HLA-B27 associated uveitis and a female preponderance in juvenile idiopathic arthritis (JIA)-related uveitis. Uveitis may occur at any age, but most commonly affects the population aged between 20 and 59 years.
Uveitis is the most common extra-articular manifestation experienced by patients with axSpA (Boonen et al, 2002) and affects patients with both AS and nr-axSpA (Boonen et al, 2003). Although limited information is available regarding the overall axSpA population, 1 study reported the prevalence of ever experiencing a uveitis flare to be 19.3% for AS patients with disease duration of ≤5 years and 12.4% for nr-axSpA patients (Braun et al, 2006). Reports from patients with established AS suggest that approximately 40% of patients will experience 1 or more uveitis flares during the course of their disease (Braun and Sieper, 2007), and observational data sources (such as cohort studies and patient registries) have reported that 10% to 30% of axSpA patients classified using the ASAS criteria have a history of uveitis (Boonen et al 2003, Braun et al, 2011). Moreover, an attack of acute AU can be the first presenting symptom that leads to a diagnosis of axSpA (Braun, 2012).
The burden of uveitis on affected patients is high, as it is commonly associated with photophobia, pain, and in some cases blurred vision (Braun, 2012). In axSpA patients, AU can range from a single flare to an episodic and recurrent course. Flares of uveitis are typically of sudden onset in a unilateral fashion (Callhoff et al, 2015). Regardless of disease course, in patients developing uveitis, impaired visual performance and reduced health-related quality of life is reported (Cantini et al, 2013). Acute attacks usually resolve completely with topical corticosteroid treatment. For axSpA patients who develop episodic and recurrent uveitis flares, treatment is limited. Nonsteriodal anti-inflammatory drugs (NSAIDs) may relieve symptoms for a short period of time (Cuirea et al, 2013), and the disease-modifying antirheumatic drugs (DMARDs) sulfasalazine (SSZ) and methotrexate (MTX) have been used, although available evidence relating to a possible decrease in flares of uveitis is limited to observational reports (Dougados et al, 1991; Dougados et al, 2013; Feldtkeller et al, 2000: Haibel et al, 2005). Given the lack of treatment options and the fact that a substantial proportion of axSpA patients are resistant to conventional therapies, new treatment avenues have been explored to gain control of uveitis flares in these patients (Haibel et al, 2013).
REDACTEDdi
a uvea uvor nrr nr--a
hat apphat appe of the of thanan
COPY tiontioh AS aAS a
ing thng t
This do
cumen
tshh(DM(DMevev
anno
teroeroment ient i
hort port p
berted rtedoidoid
used
a ununopingoping
(C(C
to flafla
nilanilasu
pportaffecteaffect
d in somin soare tare
any(Brau(Bramark
eting
heir deir nd patied patie
ASASASASattackattac
autho
rizati
onan
the ovhe oveitis fleitis fl
axSpAaxSpAproximproximdidi
appli
catio
nmost ost
experiexperiend nrd nr
and
,nce ince int coco
any eraera
such asuch j
exten
sions
al incidincid
to 71to 71allyally
or va
riatio
ns
h h c typetype
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 16 of 113
Analyses of uveitis flares from multiple randomized controlled trials (RCTs), in which patients received anti-tumor necrosis factor (TNF) therapy for the treatment of AS, have demonstrated a significant reduction in the incidence of acute AU flares in AS patients compared to that demonstrated in those patients who received placebo (Huscher et al, 2006). Furthermore, data from a large open-label non-controlled study demonstrated a significant reduction in the rate of AU flares following adalimumab (ADA) treatment of AS patients (Kobelt et al, 2004). A UCB study by Van Denderen et al, 2014, a pooled data analysis (Huscher et al, 2006), and a recent review (Rosenbaum et al, 2014) have suggested that the monoclonal antibodies ADA and infliximab (IFX) are equally effective in reducing AU flares, and are superior to etanercept (ETN) in this regard. However, at present, there are no RCT data available demonstrating the efficacy of anti-TNF in the reduction of uveitis flares in an axSpA population.
2.3 Diagnosing axial spondyloarthritis in clinical practice The diagnosis of AS and/or axSpA should be based on clinical assessments, considering typical signs and symptoms but also excluding other diseases that may have similar presentations. The mNY classification criteria, often used to support the diagnosis of AS, excludes patients whose SIJ x-rays do not have definitive evidence of sacroiliitis (Rostom, 2010). For a definitive classification of AS, the mNY classification criteria require radiographic evidence of sacroiliitis grade ≥2 bilaterally or sacroiliitis grade 3 to 4 unilaterally PLUS at least 1 of the following clinical criteria: low back pain and stiffness for ≥3 months, limitation of lumbar spine motion, or limitation of chest expansion. These criteria were designed for classification of patients in clinical studies rather than for diagnostic purposes. However, they have in fact been used for diagnosis resulting in diagnosis being delayed until irreversible structural damage documented on SIJ x-rays. Several publications have documented that the time from symptom onset to diagnosis of AS ranges from 5 to 10 years (Feldtkeller et al, 2003; Feldtkeller et al, 2000; van der Linden et al, 1984), thus demonstrating that radiographic or x-ray changes lag far behind other signs and symptoms.
Due to the problem of delayed disease recognition, ASAS recently developed new classification criteria for axSpA that do not require the presence of definitive sacroiliitis on x-ray, thus identifying a nr-axSpA subpopulation (Rudwaleit et al, 2009b; Rudwaleit et al, 2009c). These criteria establish standards that apply to patients with or without radiographic sacroiliitis, enabling the conduct of clinical studies in patients with both nr-axSpA and AS. In patients with a history of chronic back pain for ≥3 months and age of onset <45 years, a classification of axSpA can be made based on either 1) current evidence of sacroiliitis on imaging (radiographs or magnetic resonance imaging [MRI]) plus ≥1 typical SpA feature or 2) the presence of HLA-B27 plus ≥2 typical SpA-features (Figure 2‒1). In these criteria, sacroiliitis is defined as MRI evidence of SIJ inflammation or radiographic evidence of sacroiliitis meeting the mNY classification criteria (Rostom, 2010).
REDACTEDosesoseyed unyed un
documdocuyears years
ing ing
COPYmonthmonte desig desis
This do
cumen
t can
not ypyp
nce of ce of ssificasifica
be son
picalpicalus
edbb
ased osed onannan
tobackacksupp
ort bpopupop
rds thads thof clinof cli
anyrequirrequ
pulapu
marketi
ng(F(
that rathat r
sease ease
autho
rizati
ongnedgned
HoweHowentil irrntil irr
umentementFeldFeld
appli
catio
n20120diograpiogra
LUS aLUS ahs, limis, lim
d fod
andS, exc, exc
01010
any , c, c
ilar prilar pl
exten
sions
tin
cticeticeconcon
orpp
ng thg thva
riatio
ns
tt
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 17 of 113
Figure 2‒1: ASAS classification criteria for axSpA
ASAS=Assessment of SpondyloArthritis international Society; axSpA=axial spondyloarthritis; CRP=C-reactive protein; HLA-B27=human leukocyte antigen B27; mNY=modified New York (classification criteria); MRI=magnetic resonance imaging; NSAID=nonsteroidal anti-inflammatory drug; SpA=spondyloarthritis. Source: Rudwaleit et al, 2009c
2.4 Current management of axial spondyloarthritis and uveitis Based on the current treatment recommendations developed by ASAS and the European League Against Rheumatism for axSpA, first-line therapy consists of NSAIDs and nonpharmacologic treatment, such as patient education and regular exercise/physiotherapy (Braun et al, 2011). Nonsteroidal anti-inflammatory drugs usually have a rapid initial effect for the symptoms pain and stiffness of axSpA (Poddubnyy et al, 2012; Poddubnyy et al, 2013), but many patients lose symptomatic response and structural damage often progresses despite their use. Conventional DMARDs (eg, MTX and SSZ) have limited efficacy in axial disease but may benefit patients with peripheral joint disease (Braun et al, 2006; Haibel et al, 2005; Haibel et al 2007). Therefore, DMARDs are recommended only in patients with predominantly peripheral manifestations (Braun et al, 2011).
Patients who are intolerant of or have inadequately responded to NSAIDs, or those in whom NSAIDs are contraindicated, have limited treatment options. Tumor necrosis factor alpha (TNFα) inhibitors (CZP, ADA, ETN, IFX, and golimumab [GOL]) are currently the only effective and approved treatment options.
Uveitis can be caused by infectious and noninfectious etiologies. The precise diagnosis is crucially important to establish the most accurate therapy.
For most noninfectious uveitis, the control of inflammation is the key to treatment success. The normal option is a stepladder approach and the treatment includes local corticosteroids, systemic corticosteroids, and systemic immune modulators, often sequentially starting with topical therapy. Noninfectious uveitis is often associated with other systemic conditions, such as HLA-B27-related spondyloarthropathies, inflammatory bowel disease, JIA, Behcet’s Disease, and sarcoidosis. The treatment of systemic symptoms may also improve the ocular inflammation.
OR
SpA features• Inflammatory back pain• Arthritis• Enthesitis (heel)• Uveitis• Dactylitis• Psoriasis
• Crohn’s/colitis• Good response to NSAIDs• Family history for SpA• HLA-B27• Elevated CRP
*Sacroiliitis on imaging N = 649 patients with back painOverallSensitivity: 82.9%; Specificity: 84.4%Imaging arm aloneSensitivity: 66.2%; Specificity: 97.3%
HLA-B27 PLUS≥ 2 Other SpA features
Sacroiliitis on imaging*PLUS ≥ 1 SpA feature
Patients with back pain ≥ 3 months & age of onset < 45 years
• Active (acute) inflammation on MRI highly suggestive of sacroiliitis associated with SpA
OR• Definitive radiographic sacroiliitis
according to mNY criteria
REDACTEDns
erapyerapyegulargular
usuallusualet al, et al, dd
COPYl spo sps devdev
This do
cumen
tor
normnormcoco
cann
otan
lly imy im
mosmos
ben be n be us
edrs (Cs (C
approapproto
indiindCZCZ
supp
ort
erant orant odicatica
anyonly inonly mark
eting
201201damageamage
e limitlimiun et un et
autho
rizati
onvelopeelopey consicons
r exerr exerly havy hav1212
appli
catio
n teriaria
ondylndy
anditis; CRs; CR
a); Ma); M
any e
xtens
ions
97.3%.3%
xtens
ion
xtens
ionor
varia
tions
there
of.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 18 of 113
Systemic immunomodulatory medications used for ocular inflammatory conditions include conventional immunosuppressive agents and biologic response modifiers. The agents include antimetabolites, inhibitors of T-cell signaling, and alkylating agents.
Biologic therapy can be an alternative in patients with inadequate response to or intolerance of conventional immunotherapy. Biologicals were developed and approved to treat systemic inflammatory diseases or to prevent organ transplantation rejection. Several biologicals are approved for the treatment of axSpA, however none have been approved in the USA or the EU specifically for the treatment of uveitis but have been used off-label to treat uveitis or ocular inflammation.
2.5 Rationale Noninfectious AU is characterized by a genetic disposition expressed by positive HLA-B27. Spondyloarthritis (SpA) represents the most frequent extra-ocular manifestation observed in patients with HLA-B27 associated AU. The prevalence of SpA in subjects with HLA-B27 positive AU reportedly ranges from 13% to 58.3% (Monnet et al, 2004; Rosenbaum, 1989).
The prevalence of AU is variable between the different types of SpA. Considering the 3 most frequent SpA (AS, psoriatic arthritis [PsA], and IBD-associated SpA), it is generally understood that in patients with AS, there is a greater frequency of AU in patients with AS compared to the other 2 disorders. Anterior uveitis at onset or complicating the disease course has been reported in 33% of patients with AS, while the reported percentages are lower in PsA and IBD-associated SpA (Zeboulon et al, 2008). Therefore, AU represents the most frequent extra-articular manifestation and is often also the starting point to recognizing the spinal involvement and AS.
The extra-articular manifestations in AS are seen in more than one-third of the AU patients and, within this set of manifestations, uveitis represents the largest group with 51% of the extra-articular manifestations. This means that 20% of the axSpA patients are confronted with uveitis at some point during their lifetime.
While uveitis flares are usually infrequent and are managed with topical therapies, an important subset has frequent treatment refractory flares.
A significant amount of data exists for ADA and IFX for the treatment of uveitis and the prevention of flares, but neither agent is approved (ADA in Phase 3) at the moment for this indication. Although CZP belongs to the same group of agents, less data are available.
Certolizumab pegol, a PEGylated Fragment crystallizable-free anti-TNF, has been shown to improve spinal and extra-articular manifestations, such as enthesitis and peripheral arthritis, in patients with axSpA (including both AS and nr-axSpA patients) (Landewé et al, 2014).
Emerging data from a retrospective case series indicate a treatment effect of CZP in active refractory uveitis (Myasoedova et al, 2010) and justifies further research. Further data are required to determine the effectiveness of CZP in reducing the incidence of uveitis flares in axSpA patients, an important facet of disease symptomatology (Olivieri et al, 2013).
AS0007 will investigate the effect of CZP treatment on the frequency of AU flares in subjects with axSpA.
REDACTEDpresprespoint tpoint t
SS are sare seitis reeitis remeame
COPYicatinicatiercentaercentsensen
This do
cumen
t efrafrarequrequaxax
cann
ot withwit
rging gingactoacto
be inalnal
th ath aus
ed
pegol,egol,al anl an
togh Ch Csupp
ort
data exata ebut neitut neiCZPCZ
any nfrnf
refracreframark
eting
eprepreans thaans tha
fetime.etime
frequfreq
autho
rizati
onagesge
nts thets theto reco rec
seen ieen ireseese
appli
catio
n pA.Ad SpA)SpA
n patien patig the dg the es ares a
and 4; R R
A. C. C
anys wit wi
RoseRos
exten
sions
ve HLve HLion oon othh
or va
riatio
ns
U U ar r
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 19 of 113
3 STUDY OBJECTIVE(S) The following key definition is provided as a reference for understanding the study objectives and is used throughout the protocol.
A flare is defined as being a new episode of AU that, based on the judgment of an ophthalmologist, requires specific treatment. A flare is considered a new episode if a gap of at least 3 months occurs between 2 flares.
3.1 Primary objective The primary objective of the study will be to demonstrate the effect of CZP treatment on the reduction of AU flares in subjects with active axSpA and a documented history of AU.
3.2 Secondary objectives The secondary objectives of the study will be to assess the effect of CZP treatment on:
The reduction of AU flares in subjects with active axSpA having at least 1 documented history of AU within 12 months prior to Baseline
AxSpA disease activity
3.3 Other objectives The other objectives of the study will be to assess the effect of CZP treatment on:
Physical function
Signs and symptoms of axSpA
Morning stiffness
Fatigue
Extra-articular manifestations of axSpA
Duration and severity of AU flares
3.4 Safety objective The safety objectives of the study will be to evaluate the safety and tolerability of CZP therapy.
4 STUDY VARIABLES 4.1 Efficacy variables 4.1.1 Primary efficacy variable The primary efficacy variable will be the count of distinct episodes of AU flares during the Treatment Period.
4.1.2 Secondary efficacy variables The following secondary efficacy variables will be assessed at Week 48 and Week 96:
Number of AU flares per 100 patient-years in subjects with active axSpA and a history of AU
REDACTED COPY
s the es the e
This do
cumen
tTrearea
44
cann
ot e primprim
t
be us
edSTUTUEfEf
to s
UDUDsu
pport
objectbjec
of theof th
any AU flaAU flmark
eting
ons ofons o
autho
rizati
oneffecffecap
plica
tion
ect oct o
and t lele an
y eatea
east east
exten
sions
onAU.AU.
atmetme
orn then thva
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 20 of 113
Number of AU flares per 100 patient-years in subjects with active axSpA and at least 1 AU episode within 12 months prior Baseline
Change from Baseline in Ankylosing Spondylitis Disease Activity Score (ASDAS)
Change from Baseline in Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)
Assessment of ASAS 20, 40, 5/6, and partial remission (PR) response rates
Change from Baseline in tender and swollen joint count (44 joint count); Physician’s Global Assessment of Disease Activity (PhGADA).
Change from Baseline in the respective components of the ASAS response criteria, assessed by:
Patient’s Global Assessment of Disease Activity (PtGADA)
Pain assessment (total spinal pain Numerical Rating Scale [NRS])
Function (represented by Bath Ankylosing Spondylitis Functional Index [BASFI])
Inflammation (the mean of the BASDAI questions 5 and 6 concerning )
4.1.3 Other efficacy variables The following other efficacy variables will be assessed at the timepoints indicated in Table 5-1:
Duration of AU flares
Severity of AU flares
Change from Baseline in ASDAS
Change from Baseline in BASDAI
ASAS 20, 40, 5/6, and PR response rates
Change from Baseline in tender and swollen joint count (44 joint count), PhGADA.
Change from Baseline in the respective components of the ASAS criteria, assessed by:
PtGADA
Pain assessment (total spinal pain NRS)
Function (represented by BASFI)
Inflammation (the mean of the BASDAI questions 5 and 6 concerning )
Change from Baseline in ASDAS disease activity (clinical improvement [CI], major improvement [MI], inactive disease [ID], clinically important improvement [CII]), and BASFI) (including Weeks 48 and 96)
Change from Baseline in (NRS) (from BASDAI) (including Weeks 48 and 96)
Change from Baseline in Ankylosing Spondylitis Quality of Life (ASQoL) (including Weeks 48 and 96)
REDACTED
SS
COPY sessesesse
This do
cumen
t CCca
nnot InflaInfla
anbenctionctious
ed
sessmessmto
supp
ort n ten t
ine in tne in
any spsp
endeendmark
eting
AIAI
ponspon
autho
rizati
oned at td at tapp
licati
on6 con6 con
h
andional onal an
y In
exten
sionsia, asa, a or ss
varia
tions
obal bal
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 21 of 113
Change from Baseline in ASAS Health Index (HI) questionnaire (including Weeks 48 and 96)
Change from Baseline in Short-Form 36-Item Health Survey (SF-36) (including Weeks 48 and 96)
Number of IBD exacerbations
Number of psoriasis exacerbations (in subjects with concurrent psoriasis)
4.2 Safety variables 4.2.1 Secondary safety variable The secondary safety variable to be assessed is treatment-emergent adverse events (TEAEs).
4.2.2 Other safety variables The following other safety variables to be assessed are:
Blood pressure
Physical examination
Clinical laboratory values (hematology, biochemistry, and urinalysis)
5 STUDY DESIGN 5.1 Study description AS0007 is a multicenter, open-label, Phase 4 study to evaluate the effect of CZP on the incidence of AU flares per year in subjects with both active axSpA and a history of AU by comparing the historical AU flare rate that occurred prior to CZP treatment with the AU flare rate occurring while under CZP treatment.
The study duration from the start of treatment will be 96 weeks from Baseline onwards, and a FU Visit will be performed at Week 104, 10 weeks after the final dose. A schedule of study assessments is provided in Table 5-1.
The study includes 3 periods as follows:
Period 1 (Screening Period): 1 to 5 weeks before Baseline
Subjects will be informed about the study and sign the ICF. Eligibility will be evaluated and assessments will be performed as listed in Table 5-1. The Screening Period should be kept as short as possible but can be extended to 5 weeks if certain medications need to be washed out or to allow to obtain information from the subject’s ophthalmologist. For subjects who start a prophylactic treatment for LTBI, the Screening Period can be extended up to 12 weeks (see Section 10.6.4.1).
Period 2 (Treatment Period): Week 0 to Week 96
Eligible subjects will receive a dose of CZP 400mg sc administered at Baseline, Week 2, and Week 4 followed by CZP 200mg sc Q2W (starting at Week 6 until Week 94).
All subjects will be trained at the beginning of the study on self-administration before the subjects start with self-administration (relative or caregiver may also perform the injections). The
REDACTED se 4 stse 4 s
both aboth ad priod prio
COPY strtr
This do
cumen
tSectiectican
not poso
w to ow to ophylaphylaioi
be w
ssibssibus
ede infoe infowill bwill b
toenineninsu
pport
Taba
eriods ariods
ng Png
anyk 10k 10
bleble 5mark
etingor to or to
of treaof tre0404
autho
rizati
on
study ttudy activective
CC
appli
catio
n
nd nd urinuri
and a
ny ex
tensio
ns
ts (TEs (TE
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 22 of 113
injection schedule will provide the sequence of self-administration and site visits including injection at the site.
Period 3 (FU Period): 10 weeks from the final dose of study medication received.
All subjects will have a FU Visit at Week 104 or earlier in case of an early WD, 10 weeks after the final CZP administration received within the study.
A Week 48 analysis will be performed after all subjects have either completed the Week 48 Visit or have prematurely withdrawn prior to the Week 48 Visit. The final analysis will be conducted at the end of the study after all study data is locked.
5.1.1 Study duration per subject For each subject, the study will last a maximum of 109 weeks, as follows:
Up to 5 weeks of Screening Period (see Section 10.6.4.1 for details on an extended Screening Period of 12 weeks)
A Treatment Period of 96 weeks
A FU Visit at Week 104
The end of the study is defined as the date of the last study FU Visit.
5.1.2 Planned number of subjects and sites Approximately 130 subjects will be screened in order to enroll 86 subjects into the study.
The estimated recruitment period will be approximately 12 months. This takes into account an estimate of a 35% screen failure rate between Screening and Baseline. Prematurely discontinued randomized subjects will not be replaced.
Subjects are planned for enrollment at approximately 20 to 30 sites.
5.1.3 Anticipated regions and countries The study will be conducted in Europe.
5.2 Schedule of study assessments The Schedule of Assessments is shown in Table 5-1. A study schematic is presented in Figure 5-2.
REDACTEDin orin o
approxpproxetweenetween
aced. aced.
COPY tutu
and snd srdrd
This do
cumen
t can
not b
e use
d to
e supp
ort
le of e ofessmessme
any n Eurn Eurmark
eting
at approt appr
ons aons
autho
rizati
on site
der to eer to
ximatximatn Scren Scre
appli
catio
n
FU VisFU Vi
eses
and a
ny ex
tensio
ns
n exteext
or va
riatio
ns
Visit sit ucted cted
thereo
f.

UC
B22
Jan
2020
Clin
ical
Stu
dy P
roto
col
Cer
toliz
umab
peg
olA
S000
7
Con
fiden
tial
Page
23
of 1
13
Tabl
e 5-
1:
Sche
dule
of a
sses
smen
ts
Stud
y Pe
riod
Peri
od 1
(S
cree
ning
)
Peri
od 2
(Ope
n-L
abel
)Pe
riod
3 FU
Wee
k (W
)Pr
otoc
ol A
ctiv
ity-5
to -1
a0 BL
24
1224
3236
4860
7284
96/
WD
104
Vis
it1
23
45
67
89
1011
1213
14
Writ
ten
info
rmed
co
nsen
tbX
Ass
essm
ents
for
incl
usio
n/ex
clus
ion
crite
riaX
X
Dem
ogra
phic
dat
aX
Med
ical
his
tory
(in
clud
ing
axSp
A
hist
ory,
and
toba
cco
use)
and
co
ncom
itant
dis
ease
s
X
Prio
rand
conc
omita
nt
med
icat
ionc
XX
XX
XX
XX
XX
XX
XX
Uve
itis h
isto
ryX
Uve
itis f
lare
ev
alua
tion
X
XX
XX
XX
XX
XX
X
Vita
l sig
nsd
XX
XX
XX
XX
XX
XX
XX
Phys
ical
ex
amin
atio
neX
X
XX
XX
XX
XX
XX
X
Extra
-arti
cula
r as
sess
men
tsf
XX
XX
XX
XX
XX
REDACTED ED
R
CTCOPY
OPCOOOO
This do
cumen
t meenee
cann
ot nn
be eeebeus
ed
XXed
u
to tosu
pport
rtrtrtrt ort
Xupp
any XX
marketi
ng g
rkma
autho
rizati
on n
azazaza
a
za
appli
catio
n ntio
pp
and dan
y yyyyya
exten
sions
60
sio sio sio sio
e
or va
riatio
ns
on
ari
thereo
f.
22

UC
B22
Jan
2020
Clin
ical
Stu
dy P
roto
col
Cer
toliz
umab
peg
olA
S000
7
Con
fiden
tial
Page
24
of 1
13
Stud
y Pe
riod
Peri
od 1
(S
cree
ning
)
Peri
od 2
(Ope
n-L
abel
)Pe
riod
3 FU
Wee
k (W
)Pr
otoc
ol A
ctiv
ity-5
to -1
a0 BL
24
1224
3236
4860
7284
96/
WD
104
Vis
it1
23
45
67
89
1011
1213
14
Hem
atol
ogy/
ur
inal
ysis
/bi
oche
mis
try/
Xg
XX
XX
XX
XX
XX
X
CR
P X
XX
XX
XX
XX
XX
XX
Preg
nanc
y te
stin
ghX
X
XX
Che
st x
-rayi
X
TB te
stj
X
XX
TB q
uest
ionn
aire
X
XX
XX
XX
XX
X
MR
I (nr
-axS
pA
subj
ects
onl
y)k
X
X-ra
y (A
S su
bjec
ts
only
)lX
BA
SDA
IX
X
XX
XX
XX
XX
XX
X
BA
SFI
X
XX
XX
XX
XX
XX
ASQ
oLX
XX
XX
XX
XX
XX
ASA
S H
I qu
estio
nnai
reX
XX
XX
XX
XX
XX
SF-3
6X
XX
XX
XX
XX
XX
PtG
AD
AX
XX
XX
XX
XX
XX
X
REDACTED TTTTECTAC
R
COPY PPPPYOPC
This do
cumen
t can
not ot
anbe
be
used
XX u
to ototototosu
pport
t
X
ttt ort
XXppan
y XXy amark
eting
g eti
ma m
autho
rizati
on
onza
XX
izzzzth
a
appli
catio
n n
XX
nnn nca
pppppppppp
and XX
any yyyyyex
tensio
ns
7272
sio
1010ten
e
or ova
riatio
ns
ononthe
reof.
22

UC
B22
Jan
2020
Clin
ical
Stu
dy P
roto
col
Cer
toliz
umab
peg
olA
S000
7
Con
fiden
tial
Page
25
of 1
13
Stud
y Pe
riod
Peri
od 1
(S
cree
ning
)
Peri
od 2
(Ope
n-L
abel
)Pe
riod
3 FU
Wee
k (W
)Pr
otoc
ol A
ctiv
ity-5
to -1
a0 BL
24
1224
3236
4860
7284
96/
WD
104
Vis
it1
23
45
67
89
1011
1213
14
Tota
l and
noc
turn
alsp
inal
pai
nX
XX
XX
XX
XX
XX
Swol
len
and
tend
er
join
t cou
nts
XX
XX
XX
XX
XX
X
PhG
AD
AX
XX
XX
XX
XX
XX
AEs
XX
XX
XX
XX
XX
XX
X
IWR
SX
XX
XX
XX
XX
XX
XX
X
Stud
y dr
ug sc
in
ject
ions
mX
XX
XX
XX
XX
XX
Sche
dule
ap
poin
tmen
t for
nex
t vi
sit
XX
XX
XX
XX
XX
XX
X
AE=
adve
rse
even
t; A
S=an
kylo
sing
spon
dylit
is; A
SAS=
Ass
essm
ent o
f Spo
ndyl
oArth
ritis
inte
rnat
iona
l Soc
iety
; ASQ
oL=A
nkyl
osin
g Sp
ondy
litis
Qua
lity
of L
ife;
axSp
A=
axia
l spo
ndyl
oarth
ritis
; BA
SDA
I=B
ath
Ank
ylos
ing
Spon
dylit
is D
isea
se A
ctiv
ity In
dex;
BA
SFI=
Bat
h A
nkyl
osin
g Sp
ondy
litis
Fun
ctio
nal I
ndex
; B
L=B
asel
ine;
CR
P=C
reac
tive
prot
ein;
FU
=Fol
low
-up;
HI=
Hea
lth In
dex;
HLA
-B27
= hu
man
leuk
ocyt
e an
tigen
B27
; IB
D=i
nfla
mm
ator
y bo
wel
dis
ease
; IW
RS=
Inte
ract
ive
Web
Res
pons
e Sy
stem
; LTB
I=la
tent
tube
rcul
osis
infe
ctio
n; m
NY
=mod
ified
New
Yor
k (c
lass
ifica
tion
crite
ria);
MR
I=m
agne
tic re
sona
nce
imag
ing;
nr-a
xSpA
=non
radi
ogra
phic
axS
pA; P
hGA
DA
=Phy
sici
an’s
Glo
bal A
sses
smen
t of D
isea
se A
ctiv
ity;P
tGA
DA
=Pat
ient
’s G
loba
l Ass
essm
ent o
f Dis
ease
A
ctiv
ity; Q
2W=e
very
2 w
eeks
(eve
ry o
ther
wee
k); s
c=su
bcut
aneo
us(ly
); SF
-36=
Shor
t-For
m 3
6-Ite
m H
ealth
Sur
vey;
SIJ
=sac
roili
ac jo
int;
TB=t
uber
culo
sis;
W
D=W
ithdr
awal
REDACTED
XX
T
XX
TTTECT
RER
COPY P
XX
PPPYOPC
This do
cumen
t can
not b
e use
d we
w
to
PhG
wee
k)ee
k
supp
ort
osin
gsi
ngup
; HI
up; H
=lat
ent t
late
ntG
AD
GA
D
any
sess
mse
ssm
ng S
png
S
marketi
ng
XX
g
ma
g
mmm
autho
rizati
on
XX tioza
XX
riz
aa
appli
catio
n
XX
n
Xicapp apppp
and XX an
any y
XX
yyyyexten
sions
7272
sio
1010ten
e
or ova
riatio
ns
ononthe
reof.
22

UC
B22
Jan
2020
Clin
ical
Stu
dy P
roto
col
Cer
toliz
umab
peg
olA
S000
7
Con
fiden
tial
Page
26
of 1
13
Stud
y Pe
riod
Peri
od 1
(S
cree
ning
)
Peri
od 2
(Ope
n-L
abel
)Pe
riod
3 FU
Wee
k (W
)Pr
otoc
ol A
ctiv
ity-5
to -1
a0 BL
24
1224
3236
4860
7284
96/
WD
104
Vis
it1
23
45
67
89
1011
1213
14a
For s
ubje
cts w
ith L
TBI w
ho in
itiat
e a
prop
hyla
ctic
trea
tmen
t and
nee
d to
hav
e 8
wee
ks o
f pr
ophy
lact
ic tr
eatm
ent p
rior t
o ra
ndom
izat
ion,
the
Scre
enin
g Pe
riod
can
be e
xten
ded
to 1
2 w
eeks
. For
oth
er su
bjec
ts, t
he S
cree
ning
Per
iod
shou
ld b
e ke
pt a
s sho
rt as
pos
sibl
e, b
ut c
an b
e up
to 5
wee
ks if
cer
tain
med
icat
ions
nee
d to
be
was
hed
out o
r to
allo
w to
obt
ain
info
rmat
ion
from
the
subj
ects
’ oph
thal
mol
ogis
t.b
Info
rmed
con
sent
: prio
r to
any
stud
y ac
tiviti
es, s
ubje
cts w
ill b
e as
ked
to re
ad a
nd si
gn th
e in
form
ed c
onse
nt fo
rm.
cPa
st m
edic
atio
ns w
ill re
cord
ed a
t Scr
eeni
ng. C
onco
mita
nt m
edic
atio
ns w
ill b
e re
cord
ed a
t all
othe
r vis
its.
dPu
lse
rate
, sys
tolic
and
dia
stol
ic b
lood
pre
ssur
es, a
nd te
mpe
ratu
re a
re to
be
mea
sure
d at
Scr
eeni
ng a
nd B
asel
ine,
ther
eafte
r sys
tolic
and
dia
stol
ic b
lood
pr
essu
res a
re to
be
mea
sure
d.
eW
eigh
t is t
o be
mea
sure
d at
Scr
eeni
ng, B
asel
ine,
Wee
k 48
, and
at c
ompl
etio
n at
Wee
k 96
/WD
Vis
it. H
eigh
t will
be
mea
sure
d at
the
Bas
elin
e V
isit
only
.f
Extra
-arti
cula
r ass
essm
ents
incl
udes
the
num
ber o
f IB
D e
xace
rbat
ions
and
num
ber o
f pso
riasi
s exa
cerb
atio
ns (i
n su
bjec
ts w
ith c
oncu
rren
t pso
riasi
s)g
Test
ing
to ru
le o
ut h
epat
itis B
cor
e an
tibod
y, h
epat
itis B
surf
ace
antig
en, h
epat
itis B
surf
ace
antib
ody,
ant
ibod
ies t
o he
patit
is C
, and
ant
ibod
ies t
o hu
man
im
mun
odef
icie
ncy
viru
s at S
cree
ning
onl
y. T
estin
g fo
r HLA
-B27
at S
cree
ning
, onl
y if
not p
erfo
rmed
bef
ore.
hPr
egna
ncy
test
ing
for w
omen
of c
hild
bear
ing
pote
ntia
l will
be
seru
m te
stin
g at
the
Scre
enin
g V
isit
and
urin
e di
pstic
k te
stin
gat
Bas
elin
e an
d W
eek
96/W
D V
isit
and
the
FU V
isit
(10
wee
ks a
fter t
he fi
nal d
ose
of st
udy
drug
). i
Che
st x
-ray
use
d fo
r scr
eeni
ng m
ust h
ave
been
don
e w
ithin
3 m
onth
s prio
r to
the
Scre
enin
g V
isit
and
shou
ld b
e re
peat
ed o
nly
if th
e TB
test
was
con
firm
ed
posi
tive
or a
ny fu
rther
evi
denc
e is
sugg
estiv
e of
pot
entia
l TB
infe
ctio
n (e
g, e
xpos
ure)
. If n
o ch
est x
- ray
is a
vaila
ble
with
in th
e 3
mon
ths p
rior t
o th
e Sc
reen
ing
Vis
it, th
e x-
ray
can
be d
one
durin
g th
e Sc
reen
ing
Perio
d.j
Qua
ntiF
ERO
N T
B G
OLD
test
. The
TB
test
s will
be
repe
ated
at W
eek
48 a
nd 9
6 (o
r at W
D V
isit
if m
edic
ally
indi
cate
d) fo
r sub
ject
s with
pre
viou
sly
nega
tive
TB te
st re
sult.
kFo
r sub
ject
s with
nr-
axSp
A, a
n M
RI o
f the
SIJ
s is t
o be
per
form
ed d
urin
g th
e Sc
reen
ing
Perio
d. A
n M
RI p
erfo
rmed
≤3
mon
ths p
rior t
o th
e B
asel
ine
Vis
it m
ay
be u
sed
as th
e B
asel
ine.
An
MR
I ass
essm
ent i
s not
nee
ded
for p
atie
nts w
ith A
S.l
AS
subj
ects
mus
t hav
e ev
iden
ce o
f sac
roili
itis o
n x-
ray
take
n pr
ior t
o B
asel
ine
mee
ting
the
mN
Y c
lass
ifica
tion
crite
ria a
ccor
ding
to th
e In
vest
igat
or. I
f not
av
aila
ble,
this
x-ra
y is
to b
e ta
ken
durin
g th
e Sc
reen
ing
Perio
d. If
an
x-ra
y w
as p
erfo
rmed
prio
r to
the
Scre
enin
g V
isit
and
the
mN
Y c
lass
ifica
tion
crite
ria w
ere
met
, the
x-r
ay is
not
to b
e re
peat
ed.
mO
nsite
stud
y dr
ug a
dmin
istra
tion
will
occ
ur a
fter a
ll ot
her v
isit
asse
ssm
ents
are
com
plet
ed a
nd la
bora
tory
sam
ples
are
dra
wn.
Stu
dy d
rug
inje
ctio
ns w
ill o
ccur
atho
me
Q2W
on
Wee
ks 6
, 8, a
nd 1
0, 1
4 to
22,
26
to 3
0, 3
4, 3
8 to
46,
50
to 5
8, 6
2 to
70,
74
to 8
2, a
nd 8
6 to
94.
At a
ll vi
sits
, an
even
num
ber o
f syr
inge
s (2
per
box)
will
be
assi
gned
(eg,
at W
eek
4 sy
ringe
s will
be
assi
gned
for W
eeks
6, 8
, and
10;
1 sy
ringe
will
be
retu
rned
by
the
subj
ect a
t Wee
k12
).
REDACTED
patit
ipa
titen
ing,
en
ing,
es
ting
estin
g
onth
s pr
nths
pfe
cti
fect
i
COPY
Wee
k 9
Wee
k 9
mbe
r of
ber o
isB
iB
This do
cumen
t can
not b
e use
d
ur
to 2
2,
22,
syrin
gsy
ring
toafte
afte
supp
ort
edede
ray
tak
ray
tare
enin
g en
in
any
erfo
rmer
for
ed fodfo
marketi
ng
rior
ior
on (e
g,
on (e
g,
at W
eek
t Wee
autho
rizati
on
fpso
rifp
sor
B su
rfa
surf
aon
ly if
only
iat
the
Sat
the
rto
rto
appli
catio
n
conson
er v
isits
r vis
ini
ng a
ndni
ng a
n
6/W
D V
/WD
as
and
bubu
nsensen
anyy
men
t pm
ent
ut c
anut
ca
yyyexten
sions
7272
sio
1010ten
e
or ova
riatio
ns
ononthe
reof.
22
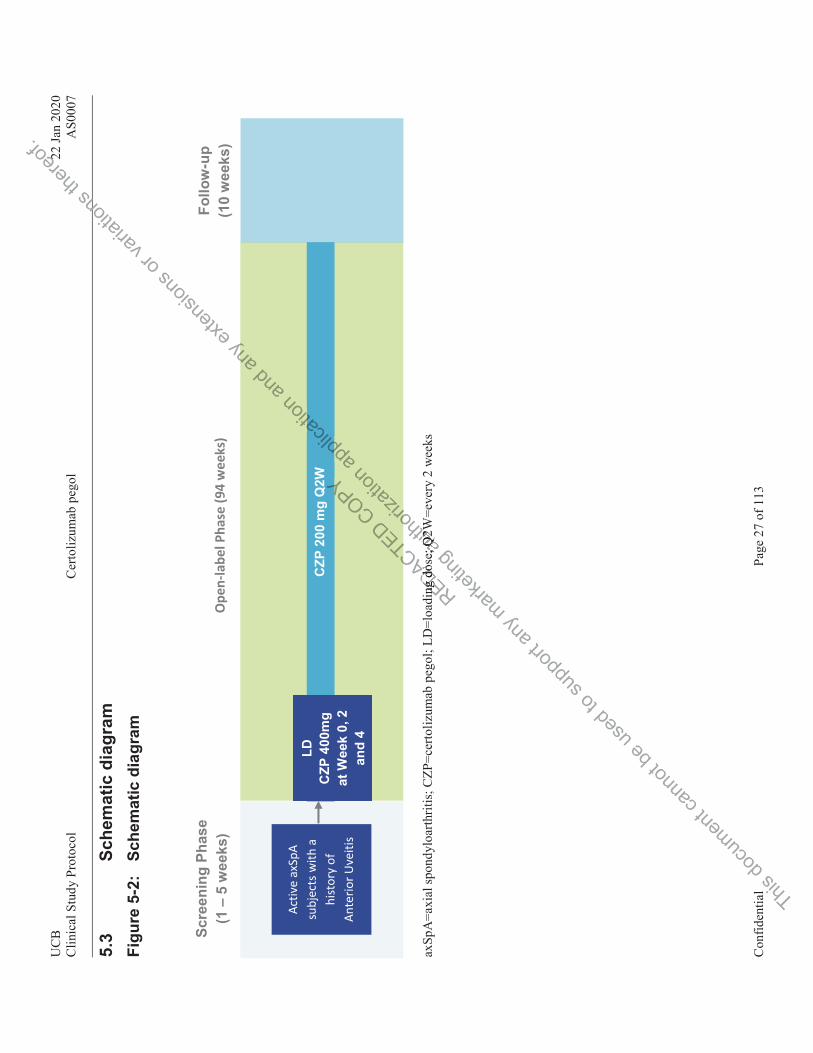
UC
B22
Jan
2020
Clin
ical
Stu
dy P
roto
col
Cer
toliz
umab
peg
olA
S000
7
Con
fiden
tial
Page
27
of 1
13
5.3
Sche
mat
ic d
iagr
am
Figu
re 5
-2:
Sche
mat
ic d
iagr
am
axSp
A=a
xial
spon
dylo
arth
ritis
; CZP
=cer
toliz
umab
peg
ol; L
D=l
oadi
ng d
ose;
Q2W
=eve
ry 2
wee
ks
Open
-labe
l Pha
se (9
4 w
eeks
)
Activ
e ax
SpA
subj
ects
with
a
hist
ory
of
Ante
rior U
veiti
s
Follo
w-u
p (1
0 w
eeks
) Sc
reen
ing
Phas
e (1
– 5
wee
ks)
CZP
200
mg
Q2W
LD
C
ZP 4
00m
g at
Wee
k 0,
2
and
4
REDACT
ing
dos
ing
do
CTED COPY YY
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
r
e; Q
2W; Q
2W
oriza
tion a
ppplic
at ation
pplic
atian
d ny
an
yexten
sions
or va
riatio
ns
onthe
reof.
22

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 28 of 113
5.4 Rationale for study design and selection of dose The aim of the study is to evaluate the flare rate of AU before the start of CZP treatment compared to that of a period with CZP treatment. Since the occurrence of flare is both subject and disease specific, each subject should act as his/her own control. Because an untreated uveitis flare can cause severe eye complication, it was deemed necessary to treat all subjects with an active drug.
Therefore, 1 treatment arm with 1 set of subjects is deemed the right approach to allow for the comparison of the history of flare occurrence with the flare occurrence on a biological treatment in the same subjects. The dose of CZP used in this study is the same as per label totreat axSpA subjects, which has been shown to be and effective for reducing the main axSpA symptoms.
6 SELECTION AND WITHDRAWAL OF SUBJECTS 6.1 Inclusion criteria To be eligible to participate in this study, all of the following criteria must be met:
1. An Institutional Review Board (IRB)/Independent Ethics Committee (IEC) approved written Informed Consent form is signed and dated by the subject.
2. Subject is considered reliable and capable of adhering to the protocol (eg, able to understand and complete diaries), visit schedule, or medication intake according to the judgment of the Investigator.
3. Subject is at least 18 years old at the Screening Visit.
4. Female subjects must be either postmenopausal for at least 1 year, surgically incapable of childbearing, or effectively practicing an acceptable method of contraception (including oral/parenteral/implantable hormonal contraceptives, intrauterine device or barrier and spermicide or contraception methods that are considered as at least as safe for contraception). Abstinence only is not an acceptable method. Subjects must agree to use adequate contraception during the study and for at least 5 months (in accordance with the Summary of Product Characteristics [SPC]) after the last dose of study treatment. Male subjects must agree to ensure that they or their female partner(s) use adequate contraception during the study and for at least 5 months (in accordance with the SPC), after the subject receives their last dose of study treatment.
5. Subjects must have a documented diagnosis of adult-onset axSpA with at least 3 months’ symptom duration and meet the ASAS criteria (according to Appendix 16.2).
6a. Subjects must have active disease at Screening as defined by
BASDAI score ≥ 4
Spinal pain ≥ 4 on a 0 to 10 NRS (from BASDAI item 2)
Nr-axSpA subjects must either have CRP > upper limit of normal (ULN) and/or current evidence of sacroiliitis on MRI (no confirmation by central reading) as defined by ASAS criteria
REDACTED
the Scthe Sc
er poser posracrac
COPY of adhof adh
heduhedu
This do
cumen
t can
notympymp
6a.a. SuSu
bejectect
mptomptus
ede subjsubj
ts mts
toon don dsupp
orttion on
duct Chduct Cagree tagree durd
anynce once
n dun du
marketi
ng stmtm
cticing ticinghormonormo
on metn meon
autho
rizati
onhe
ule, orule, o
creenicreeni
menmen
appli
catio
n cs Coms Comby the sy the
ering tring
and teria eria
any e
xtens
ions
perng the ng the
JECTECT
orr lablabva
riatio
ns
for or icalcal
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 29 of 113
AS subjects must have evidence of sacroiliitis on x-ray meeting the mNY classification criteria according to the Investigator
7. Subjects must have a documented history of AU diagnosed by an ophthalmologist and have at least 2 AU flares in the past, of which at least 1 AU flare was in the last 12 months prior to Baseline.
8. Subjects must be HLA-B27 positive (if known prior to Screening, no additional testing is to be performed; if unknown, testing is to be performed at Screening and checked at Baseline).
9. Subjects must have been intolerant to or had an inadequate response to at least 2 NSAIDs. Inadequate response to an NSAID is defined as lack of response to at least 14 days of continuous NSAID therapy at the highest tolerated dose of the administered NSAID.
6.2 Exclusion criteria Subjects are not permitted to enroll in the study if any of the following criteria is met:
1. The subject has previously participated in this study or has previously received CZP treatment in or outside of another clinical study. However rescreening is possible if prophylactic treatment for LTBI was to be given but the Screening Period exceeded the 12 weeks.
2. The subject has participated in another study of an investigational medicinal product (IMP) (or a medical device) within the previous 3 months or is currently participating in another study of an IMP (or a medical device).
3. The subject has a history of chronic alcohol or drug abuse.
4. The subject has any medical or psychiatric condition that, in the opinion of the Investigator, could jeopardize or would compromise the subject’s ability to participate in this study.
5. The subject has a known hypersensitivity to any components of CZP or a history of an adverse reaction to polyethylene glycol (PEG).
Axial SpA-disease-related exclusions
6. Subjects must not have any other inflammatory arthritis (eg, RA, systemic lupus erythematosus, sarcoidosis, or fibromyalgia).
7. Subjects must not have a secondary, noninflammatory condition that, in the Investigator’s opinion, is symptomatic enough to interfere with evaluation of the effect of study drug on the subject’s primary diagnosis of axSpA.
Ophthalmic exclusion criteria
8. Any history of uveitis (eg, posterior, panuveitis) except for AU associated with axSpA.
9a. Any condition or complicating factor that may interfere with the AU assessment, for example:
a. History of cataract surgery within 6 months prior to Baseline
REDACTEDr studr stuthe prthe p
medicalmedica
chronchron
COPYven bven
d
This do
cumen
t can
notSubjubj
opiopit
be hem
jecje
seds musmus
matomat
to aseasesu
pport
knownnown to polto po
see--rer
any
wn
marketi
ngnic alnic al
or psycor psydize orize o
autho
rizati
on
dy of ay of aprevioueviou
al devidevi
ll
appli
catio
n has preas prewever rewever r
but the ut the
and owingowing
any e
xtens
ions st
se to ae to ae adme adm
or va
riatio
ns
is is the
reof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 30 of 113
b. Corneal or lens opacity
c. Proliferative or severe nonproliferative diabetic retinopathy or clinically significant macular edema due to diabetic retinopathy
d. Neovascular/wet age-related macular degeneration
e. History of scleritis
f. History of intraocular surgery, with the exception of phacoemulsification
10. Subject has Retisert® or Iluvien® (glucocorticosteroid implant) within 3 years prior to the Baseline Visit or has had complications related to the device. Subject has had Retisert or Iluvien (glucocorticosteroid implant) removed within 90 days prior to the Baseline Visit or has had complications related to removal of the device.
11. Subject has received intraocular or periocular corticosteroids within 90 days prior to the Baseline visit.
12. Subject has received Ozurdex® (dexamethasone implant) within 6 months prior to the Baseline Visit.
13. Subject on cyclophosphamide within 30 days prior to the Baseline Visit.
14. Subject has received intravitreal MTX within 90 days prior to the Baseline Visit.
15. Subject has received intravitreal anti-vascular endothelial growth factor (VEGF) therapy:a. Within 45 days of the Baseline visit for Lucentis® (ranibizumab) or Avastin®
(bevacizumab)
or
b. Within 60 days of the Baseline visit for anti-VEGF Trap Zaltrap® (aflibercept)
Prior medications exclusions 16a. Subjects must not have used the following medications in the manner as detailed by the
exclusion criteria in Table 6‒1.
REDACTED isit foisit fo
COPYn 90 n 90
cular ular
This do
cumen
t can
not b
e use
d to s
uppo
rt TabTa anye usee use
ableabl
marketi
ng
eline viline v
d
autho
rizati
onendoendor Lucr Luc
appli
catio
n the Bahe B
days prays p
th
andhin 6 in 6
any e
xtens
ions RetRet
Baselinaselin
90 da0 da
or r r
tisetiseari
ation
s
to thto th
thereo
f.
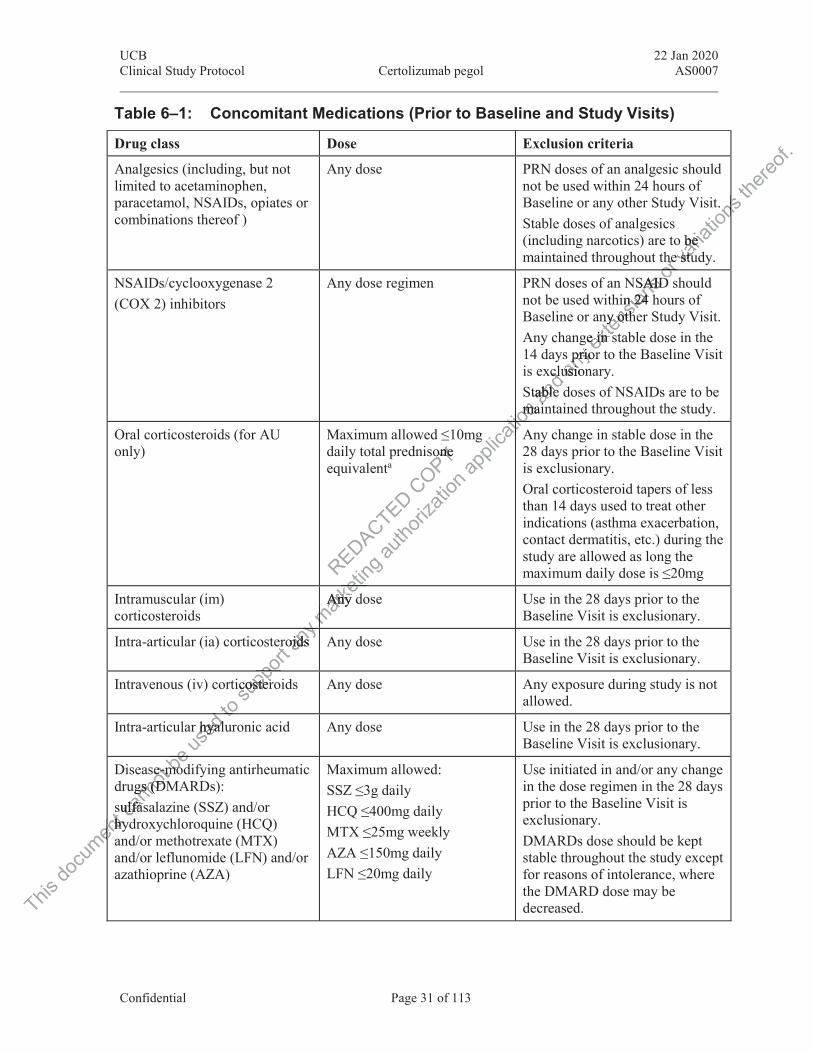
UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 31 of 113
Table 6‒1: Concomitant Medications (Prior to Baseline and Study Visits)
Drug class Dose Exclusion criteria
Analgesics (including, but not limited to acetaminophen, paracetamol, NSAIDs, opiates or combinations thereof )
Any dose PRN doses of an analgesic should not be used within 24 hours of Baseline or any other Study Visit. Stable doses of analgesics (including narcotics) are to be maintained throughout the study.
NSAIDs/cyclooxygenase 2 (COX 2) inhibitors
Any dose regimen PRN doses of an NSAID should not be used within 24 hours of Baseline or any other Study Visit.Any change in stable dose in the 14 days prior to the Baseline Visit is exclusionary.Stable doses of NSAIDs are to be maintained throughout the study.
Oral corticosteroids (for AU only)
Maximum allowed ≤10mg daily total prednisone equivalenta
Any change in stable dose in the 28 days prior to the Baseline Visit is exclusionary.Oral corticosteroid tapers of less than 14 days used to treat other indications (asthma exacerbation, contact dermatitis, etc.) during the study are allowed as long the maximum daily dose is ≤20mg
Intramuscular (im) corticosteroids
Any dose Use in the 28 days prior to the Baseline Visit is exclusionary.
Intra-articular (ia) corticosteroids Any dose Use in the 28 days prior to the Baseline Visit is exclusionary.
Intravenous (iv) corticosteroids Any dose Any exposure during study is not allowed.
Intra-articular hyaluronic acid Any dose Use in the 28 days prior to the Baseline Visit is exclusionary.
Disease-modifying antirheumatic drugs (DMARDs):sulfasalazine (SSZ) and/or hydroxychloroquine (HCQ) and/or methotrexate (MTX) and/or leflunomide (LFN) and/or azathioprine (AZA)
Maximum allowed:SSZ ≤3g dailyHCQ ≤400mg dailyMTX ≤25mg weeklyAZA ≤150mg dailyLFN ≤20mg daily
Use initiated in and/or any change in the dose regimen in the 28 days prior to the Baseline Visit is exclusionary.DMARDs dose should be kept stable throughout the study except for reasons of intolerance, where the DMARD dose may be decreased.
REDACTED COPY one one
This do
cumen
tshyhyen
cann
otsee--mmugs (Dgs (D
sulfasulfas
bemomobebe us
edr hyaluhyaldto to
supp
ort er
icostercosterpo
any
oidsoidsyy mark
eting
Any dAny ket
m
autho
rizati
on ap
plica
tionmama
tiotitiotiotioan
d xcluclu
table able i
anyprioprio
usionusio
exten
sionsSAIDAID
in 24 n 24 ny othny oth
ge in se in s
or hehe
Dorva
riatio
ns t.
o be be e stue stu
nsthe
reof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 32 of 113
Table 6‒1: Concomitant Medications (Prior to Baseline and Study Visits)
Drug class Dose Exclusion criteria
DMARDs:cyclosporine, cyclophosphamide, mycophenolic acid, apremilast
Any dose Use within 28 days prior to the Baseline Visit is exclusionary.Must not be started during the study.
Biologicals:infliximab (IFX)adalimumab (ADA)etanercept (ETN)golimumab (GOL)
Any dose For IFX, ADA, and GOL, any use within the 3 months prior to the Baseline Visit.
For ETN, use within the 28 days prior to the Baseline Visit.Only 1 previous biological is allowed.
Other biologicals eg,abatacept (ABA)anti CD20tocilizumabustekinumabsecukinumabcertolizumab pegol (CZP)
Any dose Any exposure history.Must not be started during the study.
AU= anterior uveitis; DMARD=disease modifying antirheumatic drug; NSAID=nonsteroidal anti-inflammatory drug; PRN=as needed; STJ= swollen and tender joint
a A table of corticosteroid equivalent doses is provided in Appendix16.3, Table 16‒1.
Previous clinical studies and previous biological therapy exclusions
17. Subjects must not have received any nonbiological therapy for axSpA not listed in Table 6‒1 within or outside of a clinical study in the 3 months or within 5 half lives prior to the Baseline Visit (whichever is longer).
18. Subjects must not have received any experimental biological agents (defined as those agents unlicensed for use in axSpA in the EU or the USA).
19. Subjects must not have received previous treatment with a PEGylated compound that resulted in a severe hypersensitivity reaction or an anaphylactic reaction.
20. Subjects must not have been exposed to more than one TNF antagonist prior to the baseline visit and may not be a primary failure to any TNF antagonist therapy (defined as no response within the first 12 weeks of the TNF antagonist treatment).
Medical history exclusions21. Female subjects who are breastfeeding, pregnant, or plan to become pregnant during the
study or within 5 months (or longer, if required by local regulation) following the final dose of the investigational product.
REDACTED
modifymodifyand teand te
DA
COPY
This do
cumen
t can
not
su
0. SubSubbb
beec
ultedultedus
ednliceice
cts mts m
to ust nst nensens
supp
orr outout
Visit (wVisit (
not not
anye recee rece
utsidts
marketi
ngin
ender jender jes is pros is pr
d previprev
i
autho
rizati
on
ng ang anut
appli
catio
n AnAnMMtio
andowedwed
nynyanan
y previprevdd
exten
sions
se withe wite BasBas
oror tor t vari
ation
s ns
, any any ria
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 33 of 113
22. Subjects with a history of chronic or recurrent infections, excluding uveitis (more than 3 episodes requiring antibiotics or antivirals during the preceding year), recent serious or life–threatening infection within the 6 months prior to the Baseline Visit (including hospitalization for any infection in the last 6 months or any current sign or symptom that may indicate an infection).
23. Subjects with a history of herpes zoster infection within 6 months prior to the Baseline Visit.
24. Subjects with known tuberculosis (TB) infection, at high risk of acquiring TB infection, or LTBI.
a. Known TB infection whether present or past is defined as:
Active TB infection or clinical signs and symptoms suspicious for TB (pulmonary or extrapulmonary)
History of active TB infection involving any organ system or findings in other organ systems consistent with TB infection
Any evidence by radiography or other imaging modalities consistent with previously active TB infection that is not reported in the subject’s medical history.
b. High risk of acquiring TB infection is defined as:
Known exposure to another person with active TB infection within the 3 months prior to Screening
Time spent in a healthcare delivery setting or institution where individuals infected with TB are housed and where the risk of transmission of infection is high.
c. Latent TB infection (unless appropriate prophylaxis is initiated prior to study treatment and continued to completion of prophylaxis) is defined as:
The absence of signs, symptoms (ie, evidence of organ-specific involvement), or physical findings suggestive of TB infection with a positive interferon-gamma release assay (IGRA; or 2 indeterminate IGRA test results) and a chest x-ray (or other imaging) without evidence of TB infection. If the result of the IGRA is indeterminate, the particular IGRA test previously performed may be repeated once; if positive or indeterminate on retest, the subject may not be randomized to study medication without further evaluation, treatment, and discussion with Study Physician, if LTBI is identified. (If active TB is identified, subject must undergo appropriate study-specified withdrawal procedures.) The retest must be done during the protocol-defined Screening window.
Note: If available, respiratory or other specimens must also be smear and culture negative for TB (Centers for disease control diagnosis of LTBI, http://www.cdc.gov/TB/topic/testing/default.htm)
Detailed information on TB definition, clinical signs, diagnosis, documentation, and treatment will be available in this protocol.
25. Subjects with current acute or chronic viral hepatitis B or C or with human immunodeficiency virus (HIV) infection.
REDACTED witwi
elivereliverwhere where
COPYfined fined
ith aith
This do
cumen
t can
notssb
eidenidenuse
dtermermthoutthout
t
toiculculmini
supp
ort s sus
or 2 inor 2 iwithoutthoular Ilar
anys, sys, s
uggeuggmark
eting
theth
s approappd to comto co
ymym
autho
rizati
on
activective
ry setty setthe rie ri
appli
catio
n alities litiesject’s mect’s
as:as:
and
or fr any
findifinex
tensio
ns
TB (pTB (p
or va
riatio
ns
ctiontion
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 34 of 113
26. Subjects with current or a history of active infection with Histoplasma, Coccidiodes,Paracoccidioides, Pneumocystis, nontuberculous mycobacteria, Blastomyces, or Aspergillus.
27. Subjects with a history of an infected joint prosthesis at any time.
28. Subjects receiving any live (includes attenuated) vaccination within the 8 weeks prior to Baseline (eg, inactivated influenza and pneumococcal vaccines are allowed, but nasal influenza vaccination is not allowed).
29. Subjects who in the Investigator’s opinion have a high risk of infection (eg, subjects with leg ulcers, indwelling urinary catheter, persistent or recurrent chest infections, and subjects who are permanently bedridden or wheelchair bound).
30. Subjects with a history of a lymphoproliferative disorder, including lymphoma or current signs and symptoms suggestive of lymphoproliferative disease.
31. Current malignancy or a history of malignancy (although subjects with less than 3 completely excised basal cell carcinomas or with cervical carcinoma in situ successfully surgically treated more than 5 years prior to Screening may be included).
32. Subjects with Class III or IV congestive heart failure as per the New York Heart Association 1964 criteria.
33. Subjects with a history of, or suspected, demyelinating disease of the central nervous system (eg, multiple sclerosis or optic neuritis).
34. Subjects who have had major surgery (including joint surgery) within 8 weeks prior to Screening, or have planned surgery within 6 months of the Screening Visit.
35. Subjects with a history of or a current, as determined by the Investigator, severe, progressive, and/or uncontrolled renal, hepatic, hematological, endocrine, pulmonary, cardiac, or neurological disease.
36. Subjects with significant laboratory abnormalities at Screening including but not limited to:
Liver function tests >2.0 x ULN
Estimated Glomerular Filtration Rate<60 mL/min/1.732 as measured by Chronic Kidney Disease Epidemiology Collaboration (Levey et al, 2009)
White blood cell (WBC) count <3.0x109/L.
37. Subjects with any other condition which, in the Investigator’s judgment, would make the subject unsuitable for inclusion in the study
6.3 Withdrawal criteria Subjects will be free to withdraw from the study at any time, without prejudice to their continued care.
At the Investigator’s discretion, the subject may be withdrawn from the study at any time (eg, to receive alternative treatment for the disease).
REDACTEDneurneu
gery (iery (igery wgery w
a cua cu
COPY demyeemye
ritit
This do
cumen
t66cann
otSubjSubjsusu
beWhWh us
edney Dney D
hitehite
toed Ged Gsupp
ort
on teson te
GloG
anynt labnt lamark
etingurrent,urrent
led rened resease.ease.
b
autho
rizati
onelinaelinaitis).is).
includncludwithinwithin
appli
catio
nay bey be
as per as per
tinti
and
ctstsrcinomcinome ii
any
s withwithex
tensio
ns, andan
mphommphom
orctsct
nddva
riatio
ns
ts ws w
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 35 of 113
Subjects should be withdrawn from the study if any of the following events occur:
1. Subject develops an illness that would interfere with his/her continued participation.
2. Subject is noncompliant with the study procedures or medications in the opinion of the Investigator.
3. Subject takes prohibited concomitant medications as defined in this protocol.
4. Subject withdraws his/her consent.
5. There is confirmation of a pregnancy during the study, as evidenced by a positive pregnancy test.
6. The regulatory agency requests withdrawal of the subject.
7. The Sponsor requests withdrawal of the subject for safety reasons.
8. Subject’s subsequent TB test was confirmed positive or any further evidence is suggestive of potential TB infection (eg, exposure), and further examinations result in a diagnosis of active TB or in case of LTBI, no prophylactic treatment was initiated (see Section 10.6.4).
9. Subject who prematurely discontinues treatment for latent TB or, in the opinion of the Investigator or Sponsor, is noncompliant with anti TB therapy, must be withdrawn.
Once withdrawn from study treatment, subjects must return for a Withdrawal Visit and perform all assessments as defined for the W96/WD Visit in Table 5-1, and complete a final FU Visit, 10 weeks after the last dose of study medication is administered.
Investigators should attempt to obtain information on subjects in the case of withdrawal or discontinuation. For subjects considered lost to follow up, the Investigator should make an effort (at least 1 phone call and 1 written message to the subject), and document his/her effort (date and summary of the phone call and copy of the written message in the source documents), to complete the final evaluation. All results of these evaluations and observations, together with a narrative description of the reason(s) for removing the subject, must be recorded in the source documents. The Case Report Form (CRF) must document the primary reason for withdrawal or discontinuation.
Investigators should contact the Medical Monitor, whenever possible, to discuss the withdrawal of a subject in advance.
Subjects withdrawn from the study will not be replaced.
7 STUDY TREATMENT(S) 7.1 Description of investigational medicinal product(s) For the purpose of the protocol IMP refers to CZP. Investigational medicinal products will be supplied under the responsibility of the UCB Clinical Supply Unit. The frequency at which the IMP will be supplied to each individual site will be adapted to the recruitment capacity of that site and to the expiry date of the IMP and will be managed by the interactive web response system (IWRS).
REDACTEDec
he W9e W9of stuof stu
btain intain insidenside
COPYth anth an
cts mcts m
This do
cumen
t77cann
ot ct
7 17
bets wits wus
edof a sof a tohouloul s
uppo
rtitht
he soue souwithdwithd
dd
anye fine fin
h a nh a nmark
etingin
ered lered lwrittenwritte
ne call ne calnalna
autho
rizati
onmust rmust r6/WD6/WD
udy mudy m
nformform
appli
catio
n
latent Tatent i TB tTB
andr exex
eatmeatman
yr evir evxamxam
exten
sions
id
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 36 of 113
Certolizumab pegol is an engineered humanized monoclonal antibody Fab´ fragment with specificity for human TNFα, manufactured in E. coli. The antibody fragment is subsequently purified and conjugated with high molecular weight PEG (40kDa).
Certolizumab pegol will be supplied as a sterile, clear, colorless to-slightly yellow liquid solution with a pH of approximately 4.7 in 1mL single-use glass prefilled syringe with a thin-walled needle for sc injection. Each syringe will contain an extractable volume of 1mL at a concentration of 200mg/mL of CZP in 10mM sodium acetate buffer and 125mM sodium chloride as a tonicity agent.
7.2 Treatment(s) to be administered The IMPs will be administered as sc injections into either the lateral abdominal wall or the upper outer thigh. Each injection should be administered at a separate injection site, and rotation between the injection sites should be observed. Before injection, the PFS should be brought to room temperature. Certolizumab pegol will be administered as 400mg at Baseline and Weeks 2 and 4 followed by 200mg Q2W (starting at Week 6 until Week 94). Subjects will be trained by the qualified, designated, site personnel and be provided with written instructions on the correct injection technique. Once the subject is trained at the start of the study the subject will have the opportunity to self-inject IMP. Site personnel must investigate whether the subjects have adequate storage conditions at home and are willing and able to perform the injection of IMP. Subjects who opt for self-administration may also request that a designated caregiver/family member administers the IMP. This person must attend all protocol-required training and be tested and judged as capable of administering the study medication to the subject.
7.3 Packaging Certolizumab pegol is packaged and labeled according to Good Manufacturing Practice guidelines and applicable laws or regulations. It is suitably packaged in such a way as to protect the IMP from deterioration during transport and storage.
7.4 Labeling Certolizumab pegol packaging will be labeled in accordance with the current International Council for Harmonisation (ICH) guidelines on Good Clinical Practice (GCP) and Good Manufacturing Practice and will include any locally required statements. If necessary, labels will be translated into the local language.
7.5 Handling and storage requirements 7.5.1 Storage at the site The Investigator (or designee) is responsible for the safe and proper storage of IMP at the site. The IMP stored by the Investigator should be kept in a secured area with limited access.
The IMP containers should be under conditions as detailed in the IMP manual. In case an out-of-range temperature is noted, it must be immediately communicated within the IWRS and UCB as described in the IMP manual before further use of the IMP.
Appropriate storage conditions (storage of CZP is to be done at 2 to-8ºC) must be ensured and completion of a temperature log in accordance with local requirements must be done on
REDACTEDin
and juand ju
anan
COPY tiontiot for st for s
nisterister
This do
cumen
tss cann
ot .11The The i
be us
edgg
slatedlatedtoPracPra
d
supp
ort gg
packagpackaonisationisatctit
any m
arketi
ng
nd labed labeor regur regu
ation dation
autho
rizati
onse
rs the s the udged dged
appli
catio
nect isect isMP. SiMP. S
at homat homelflf--fff admad
and ntinti
e provprovan
yd as 4d as il Wl W
exten
sions
al walll wall
tion sition sithe Pthe P44
or va
riatio
ns
um um
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 37 of 113
a regular basis (eg, once a week), but at least once per working day showing minimum and maximum temperatures reached over the time interval. In case of an out of range temperature, based on discussion with a UCB Quality Assurance representative, the Drug Supply Coordinator will then provide the site with instructions regarding use of the IMP and adjust the status of the IMP as needed in the IWRS.
The Investigator (or designee) will instruct subjects to store the IMP following the instructions on the label.
7.5.2 Storage at the subject’s home The Investigator (or designee) will instruct the subject how to handle the IMP during transport and how to store the IMP as per the label. Specific emphasis on the transport from site to home is required and instructions on the use of cold bags for CZP syringes must be followed. The subject is instructed to put the syringes as quickly as possible into his/her refrigerator.
In the case of a malfunctioning refrigerator or damaged or lost syringes, the subject must inform the site immediately and new syringes will be provided.
7.6 Drug accountability A Drug Accountability form will be used to record IMP dispensing and return information on a by-subject basis and will serve as source documentation during the course of the study. Details of any IMP lost (due to breakage or wastage), not used, disposed of at the study site, or returned to the Sponsor or designee must also be recorded on the appropriate forms. All supplies and pharmacy documentation must be made available throughout the study for UCB (or designee) to review.
The Investigator (or designee) is responsible for retaining all used, unused, and partially used containers of IMP until return or destruction.
The Investigator may assign some of the Investigator’s duties for drug accountability at the study site to an appropriate pharmacist/designee.
The Investigator must ensure that the IMP is used only in accordance with the protocol.
7.7 Procedures for monitoring subject compliance At each on-site visit after IMP is dispensed, subjects must return all used and unused IMP to the site.
Drug accountability must be recorded on the drug accountability form.
Doses of IMP that were missed due to a reasonable interfering AE that does not allow administration of an anti-TNF due to safety reasons will not be considered for the evaluation of subject compliance. Evaluation of the reasonability of the AE must be discussed immediately with the Medical Monitor.
REDACTED r wr wmust almust al
n musn mus
resres
COPYrd IMrd Icumencume
wastwas
This do
cumen
tooca
nnot ac
Doses ooses oadmadm
beccouccouus
edite vte vto visvis
supp
ort
t ensurensu
ocedoce
any
sosoe phare pha
marketi
ng
sponsibponsidestrudestr
ome ome
autho
rizati
on ntattat
tage), age), lso beso be
st be mst be m
appli
catio
n
MP dispP dispationtion
andringeingea
ny
es
exten
sions
ring ingranspoanspo
ringesngesble inble in
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 38 of 113
7.8 Concomitant medication(s)/treatment(s) 7.8.1 Permitted concomitant treatments (medications and therapies) The following concomitant medications are permitted during the study for uveitis:
Corticosteroids (see Section 7.8.2 for prohibited corticosteroids): Oral chronic stable doses are allowed as long as the maximum daily total prednisone equivalent dose is ≤10mg (see Table 6‒1).
Oral corticosteroid tapers of less than 14 days used to treat other indications (asthma exacerbation, contact dermatitis, etc) are allowed as long the maximum daily dose is ≤20mg. The taper must end 1 week before a study visit.
The following concomitant medications are permitted during the study for axSpA:
NSAIDs/cyclooxygenase 2 (COX-2) inhibitors
Stable doses of analgesics established at Baseline (including, but not limited to acetaminophen, paracetamol, NSAIDs, opiates or combinations) will be permitted except that ad hoc (prn) usage is prohibited within the 14 days prior to Baseline or 24 hours prior to any post-Screening assessments.
Corticosteroids (see Section 7.8.2 for prohibited corticosteroids): Oral chronic stable doses are allowed as long as the maximum daily total prednisone equivalent dose is ≤10mg (see Table 6‒1).
Oral corticosteroid tapers of less than 14 days used to treat other indications (asthma exacerbation, contact dermatitis, etc) are allowed as long the maximum daily dose is ≤20mg. The taper must end 1 week before a study visit.
Intra-articular (ia) administration of corticosteroids is permissible in peripheral joints; however, after an ia injection the joint will no longer be evaluated for data regarding swollen and tender joints.
Intravenous (iv) administration of corticosteroid will be permitted for the purposes of stress dosing for a surgical procedure under general or spinal anesthesia. Furthermore, iv administration of corticosteroids may be used during the study for acute illnesses as long as the dose is not given within 1 week prior to Week 12, Week 24, Week 48, or Week 96, and the underlying disease does not present a contraindication to the subject remaining in the study. Indications might include dermatitis, gastroenteritis, asthma exacerbation, and pneumonia.
Disease-modifying antirheumatic drugs (DMARDs) (only SSZ and/or hydroxychloroquine [HCQ] and/or MTX and/or azathioprine [AZA] and/or leflunomide [LFN]: maximum SSZ ≤3g daily; HCQ ≤400mg daily; MTX ≤25mg weekly; AZA ≤150mg daily; LFN ≤20mg daily) are allowed. No change in dose or dose regimen is allowed during the study except for reasons of intolerance, where the DMARDs dose may be reduced or discontinued. No change is permitted in the route of administration for MTX (sc or oral) during the study.
REDACTED ss thans than
atitis, eatitis, end 1 wnd 1 w
COPYited cited m dailym dail
This do
cumen
t can
not
DisDishh
beub
asthasthus
edWeek eek bjectbject
toas thas t9
supp
ortadmadm
for a sfor a trationtrationheh
any ointoin
dminmi
marketi
ngweewee
strationstratioection ection
nts.nts.
autho
rizati
ony toto
n 14 d14 detc) artc) aekek
appli
catio
ne 14 de 14 d
orticosrticootal otal
and
g, b, bnationation
dd
any
but nbutex
tensio
ns lyy
axSpAaxSpA
ordodova
riatio
ns
sthmathm
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 39 of 113
Osteoporosis medications (eg, risedronate, alendronate, ibandronate, denosumab, cathepsin K inhibitor, cinacalcet, calcitonin) with the exception of iv bisphosphonates are allowed without restriction.
7.8.2 Prohibited concomitant treatments (medications and therapies) Prior medication exclusions and washout periods are listed in Section 6.2. In addition, use of the following concomitant medications is prohibited during the study, except where indicated:
Corticosteroids (administered oral, ia, or iv) are permitted only as described in Section 7.8.1. Intramuscular corticosteroids and SIJ ia corticosteroid injections are not permitted.
Hyaluronic acid may be used as ia injection in the knee only.
Specific DMARDs (cyclosporine, cyclophosphamide, mycophenolic acid, apremilast). Biologicals (TNF antagonists: IFX, ADA, ETN, GOL; abatacept [ABA]; CZP; Rituximab or other anticluster of differentiation 20 [anti-CD20] antibodies; tocilizumab; ustekimumab; tofacitinib; or any other biological response modifiers that are not licensed for the treatment of AS or axSpA).
All iv bisphosphonates (zoledronic acid, ibandronate, pamidronate) are excluded.
Subjects must not participate in any other clinical study for any indication or receive any unauthorized medication during the Study Period.
If the subject requires any of the medications specified in this section, the subject must be withdrawn from the study prior to the initiation of these medications.
The administration of live vaccines is not recommended for subjects treated with TNF antagonists. Live vaccines should not be administered 8 weeks prior to Baseline. If immunization with a live organism-based vaccine is considered during the study, the clinician is urged to carefully weigh the risks vs benefits of immunization. If the subject is going to proceed with live organism-based immunization, the subject must be withdrawn from the study prior to administration of the vaccine. Such vaccines must be recorded in the electronic Case Report Form (eCRF).
7.8.3 Rescue medication A rescue medication is any medication other than prohibited medication that is used to treat axSpA or uveitis. The use of rescue medication is allowed during Screening through Week 96/WD Visit, except under the conditions noted in Section 7.8.2.
7.9 Blinding This will be an open-label study; therefore, blinding is not applicable.
7.10 Enrollment into treatment and numbering of subjects An IWRS will be used for assigning CZP treatment to eligible subjects. The IWRS will generate individual assignments for subject kits of IMP, as appropriate, according to the visit schedule.
REDACTEDStudy Study
dicatioicatiothe inthe i
nesnes
COPYndronndro
r clinclin
This do
cumen
t TTca
nnotekk 9696
7.97.9
beor uor u6/6/
used
RRmedicmedic
toResRessu
pport
ive ve to admo adm
eport Feport F
anylly wly w
orgor
marketi
ng
is not is notld not bd not
anismnismwe
autho
rizati
onnical snical sPeriodPeriod
ons spns spitiatiotiati
appli
catio
ne momo
ate, paate, p
and pt [t [
20] an0] andd
anylic aclic a
[AB[ABex
tensio
nsns ars ar or rere
varia
tions
there
of.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 40 of 113
To enroll a subject into the study (Visit 1), the Investigator or designee will contact the IWRS and provide brief details about the subject to be enrolled. Each subject will receive a 5-digit number assigned at Screening that serves as the subject identifier throughout the study. The subject number will be required in all communications between the Investigator or designee and the IWRS regarding a particular subject. Subject numbers and kit numbers will be tracked via the IWRS.
To enroll a subject into the Treatment Phase, the Investigator or designee will contact the IWRS and provide brief details about the subject to be enrolled. The IWRS will automatically allocate kit numbers to the subject based on the subject number during the course of the study.
8 STUDY PROCEDURES BY VISIT Section 5.2 (Schedule of assessments) provides a general overview of study assessments. A detailed listing of procedures to be undertaken at each visit is described below. During the study, the Investigator will assess each subject over the entire study period of up to 109 weeks including a FU Period of 10 weeks after the last dose of the study drug. For Week 2 to Week 96 the visit window is ±3 days from the actual date in Weeks from the initial administration of IMP (Week 0). For the FU Visit a window of 1-2 weeks is acceptable.
8.1 Visit 1 (Week -5 to -1) Screening Prior to any study activities, subjects will be asked to read and sign an informed consent form (ICF) for participation in the study. All ICFs will have been approved by an IEC/IRB and will comply with regulatory requirements. Subjects will be given adequate time to consider any information concerning the study given to them by the Investigator or designee. As part of the informed consent procedure, subjects will be given the opportunity to ask the Investigator any questions regarding potential risks and benefits of participation in the study.
Assessments at the Screening Visit include:
Confirm informed consent
Review inclusion/exclusion criteria
Demographic data (includes date of birth, gender, race/ethnicity)
Washout of excluded medications (3 months for TNFs)
Significant past medical and procedure history (including tobacco use) and concomitant disease (including axSpA history)
Prior and concomitant medication
Review of the uveitis history containing all documented UA
Vital signs (pulse rate, systolic and diastolic blood pressures, and temperature)
Physical examination (including weight) and extra-articular assessment (including the number of IBD exacerbations and number of psoriasis exacerbations [in subjects with concurrent psoriasis])
REDACTEDl be al be all ICl ICFF
ementementstudy study
ee
COPY creencree
This do
cumen
t can
not
ggdiseadisea
PP
benificnificus
ed
out of ut of tohic dic dsu
pport
onsons
on/excn/ex
datd
anyng Vg V
sensen
marketi
ngy givegiv
, subjesubjerding pding p
VisiVi
autho
rizati
onninnin
sked tsked tFs willFs will
ts. Subs. Subvenven
appli
catio
nual daual da windoa wind
ngng
andy pepe
of thof than
yd bed beperioeri
exten
sions
udy asdy al
ortheth varia
tions
hehe
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 41 of 113
Hematology, biochemistry, and urinalysis for clinical laboratory values
Hepatitis-associated disease markers assessment:
Hepatitis B core antibody (HBc-Ab)
Hepatitis B surface antigen (HBs-Ag)
Hepatitis B surface antibody (HBs-Ab)
Hepatitis C virus antibody (HCV-Ab)
Testing of antibodies to HIV
Testing for HLA-B27 if unknown at Screening Visit
CRP testing
Serum pregnancy testing for women of childbearing potential
TB evaluation
Screening chest x-ray must have occurred within 3 months prior to Screening and should be repeated only if the TB test was confirmed positive or any further evidence is suggestive of potential TB infection (eg, exposure).
TB: IGRA test (QuantiFERON TB GOLD test).
TB evaluation questionnaire (includes evaluation of signs and symptoms of active TB and risk factors for exposure to TB)
Chest x-ray used for screening must have been done within 3 months prior to the Screening Visit and should be repeated only if the TB test was confirmed positive or any further evidence is suggestive of potential TB infection (eg, exposure). If no chest x-ray is available within the 3 months prior to the Screening Visit, the x-ray can be done during the Screening Period.
For subjects with nr-axSpA, an MRI of the SIJs is to be performed during the Screening Period. An MRI performed ≤3 months prior to the Baseline Visit may be used as the Baseline. An MRI assessment is not needed for patients with AS.
AS subjects must have evidence of sacroiliitis on x-ray taken prior to Baseline meeting the mNY classification criteria according to the Investigator. If not available, this x-ray is to be taken during the Screening Period. If an x-ray was performed prior to the Screening Visit and the mNY classification criteria were met, the x-ray is not to be repeated.
BASDAI
Contact the IWRS to indicate the subject has been screened
Schedule appointment for next visit
The Screening chest x-ray should be read by an experienced TB imaging specialist, radiologist, pulmonologist or infectious disease specialist, who is specifically requested to look for signs of active TB or signs of past/inactive TB infection and must exclude evidence of TB. For subjects with LTBI who initiate a prophylactic treatment and need to have
REDACTEDudes udeso TB)TB)
g mustg musbe repbe rep
COPY , ex, e
OLD tOLD
This do
cumen
t can
notVisitVisit
BAB
bebe tae tait ait a
used
cts ts NY claY claakeake
to s mumusu
pport
axaxperformperfo
MRI asMRI a
any
xSpAxSp
marketi
ngpeatepeate of potof po
onths pnths
autho
rizati
ontest)est)
evaluaevalua
havehaved
appli
catio
nonthsonthed posed po
osure).sure)
t).)
and a
ny ex
tensio
ns or
varia
tions
there
of.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 42 of 113
8 weeks of prophylactic treatment prior to randomization, the Screening Period can be extended to 12 weeks. For other subjects, the Screening Period should be kept as short as possible, but can be up to 5 weeks if certain medications need to be washed out or to allow to obtain information from the subjects’ ophthalmologist.
8.2 Visit 2 (Week 0) Baseline Visit Subjects qualifying for the study will have the following procedures performed/recorded prior to study medication administration:
Review of inclusion/exclusion criteria
Evaluation of uveitis flares that occurred since last visit
Prior and concomitant medications
Vital signs (pulse rate, systolic and diastolic blood pressure, and temperature)
Physical examination and extra-articular assessment
Height
Weight
Number of IBD exacerbations
Number of psoriasis exacerbations (in subjects with concurrent psoriasis)
Blood samples will be collected for
Hematology analyses
Biochemistry analyses
CRP testing
Urine will be collected for
Urinalysis
Urine pregnancy test for women of childbearing potential
TB questionnaire
Patient reported outcomes:
BASDAI
BASFI
ASQoL
ASAS HI questionnaire
SF-36
Total and nocturnal spinal pain
PtGADA
REDACTED COPY
n subjsub
This do
cumen
t can
not BbeBABA us
ed
reporrepor
S
tonnainnai supp
ort
ancy teancy t
ireire
any r r mark
eting
autho
rizati
onjects jects ap
plica
tion a
nd an
y mpp exten
sions
peraera
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 43 of 113
PhGADA
Swollen and tender joint counts
AEs
Contact IWRS to enroll subject and to obtain kit numbers
Study drug administration on-site (after all other visit assessments are completed and laboratory samples are drawn)
Schedule appointment for next visit
8.3 Visit 3 and 4 (Week 2 and 4) Assessments at these visits will include:
Evaluation of uveitis flares that occurred since last visit
Concomitant medications
Vital signs (systolic and diastolic blood pressures)
Physical examination
Blood samples will be collected for
Hematology analyses
Biochemistry analyses
CRP testing
Urine will be collected for urinalysis
Patient reported outcomes:
BASDAI
BASFI
ASQoL
ASAS HI questionnaire
SF-36
Total and nocturnal spinal pain
PtGADA
PhGADA
Swollen and tender joint counts
AEs
Contact IWRS to obtain kit numbers
REDACTED
rinalyrinaly
COPY
This do
cumen
t can
not PbeTotaTot us
ed
366toHI qHI q su
pport
quequ
any m
arketi
ng ysis ysis au
thoriz
ation
appli
catio
n and
any e
xtens
ions o
r vari
ation
s the
reof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 44 of 113
Study drug administration on-site. At Week 4, syringes of CZP are provided for home administration at Weeks 6, 8, and 10. The subject has to return 1 syringe at the Week 12 Visit.
Schedule appointment for next visit
8.4 Visits 5 and 6, and Visits 8 to 12 (Weeks 12, 24, 36, 48, 60, 72, and 84)
Assessments at these visits will include:
Evaluation of uveitis flares that occurred since last visit
Concomitant medications
Vital signs (systolic and diastolic blood pressures)
Physical examination (weight at Week 48 only) and extra-articular assessment (including the number of IBD exacerbations and number of psoriasis exacerbations [in subjects with concurrent psoriasis])
Blood samples will be collected for
Hematology analyses
Biochemistry analyses
CRP testing
Urine will be collected for urinalysis
TB test for subjects with previously negative TB test result (Week 48 only)
TB questionnaire
Patient reported outcomes:
BASDAI
BASFI
ASQoL
ASAS HI questionnaire
SF-36
Total and nocturnal spinal pain
PtGADA
PhGADA
Swollen and tender joint counts
AEs
Contact IWRS to obtain CZP kit numbers
Study drug administration onsite
REDACTED
ysis ysis
viouslviousl
COPY
This do
cumen
t can
notTTb
e SFF usedAS HIS H
3636
to Isu
pport
any m
arketi
ngly ney n
:
autho
rizati
on
negneg
appli
catio
n and
cerberbanyr asseass
batiobat
exten
sions
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 45 of 113
Provide subject with CZP for home administration Q2W until next scheduled site visit
Schedule appointment for next visit
8.5 Visit 7 (Week 32) Assessments at this visit will include:
Evaluation of uveitis flares that occurred since last visit
Concomitant medications
Vital signs (systolic and diastolic blood pressure)
CRP testing
BASDAI
PtGADA
AEs
Contact IWRS to obtain CZP kit numbers
Study drug administration onsite
Provide subjects with CZP for home administration Q2W until next scheduled site visit.
Schedule appointment for next visit
8.6 Visit 13 (Week 96/Withdrawal Visit) Assessments at this visit will include:
Evaluation of uveitis flares that occurred since last visit
Concomitant medications
Vital signs (systolic and diastolic blood pressures)
Physical examination (including weight) and extra-articular assessment (including the number of IBD exacerbations and number of psoriasis exacerbations [in subjects with concurrent psoriasis])
Blood samples will be collected for
Hematology analyses
Biochemistry analyses
CRP testing
Urine will be collected for
Urinalysis
Urine pregnancy test for women of childbearing potential
TB test for subjects with previously negative TB test result
TB questionnaire
REDACTED WithdWithd
de:de:
hathat
COPY istratiistrati
This do
cumen
t can
notBBb
e HemHem
BB
usedamplmpl
emama
to so
lessu
pportion (ion (i
exacerxaceoriasiriasi
anyd diad dia
inci
marketi
ng
occurroccur
ast
autho
rizati
on
drawraw
appli
catio
n
on Q2n Q2
and a
ny ex
tensio
ns or
varia
tions
there
of.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 46 of 113
Patient reported outcomes
BASDAI
BASFI
ASQoL
ASAS HI questionnaire
SF-36
Total and nocturnal spinal pain
PtGADA
PhGADA
Swollen and tender joint counts
AEs
Contact IWRS to confirm Week 96/WD Visit
Schedule appointment for next visit
8.7 Visit 14 (Follow-Up Visit) The FU visit will occur 10 weeks after the final dose. Assessments at this visit will include:
Concomitant medications
Vital signs (systolic and diastolic blood pressures)
Physical examination
Urine dipstick pregnancy testing for women of childbearing potential
AEs
Contact IWRS to indicate that subject has completed the FU
8.8 Unscheduled Visit It is at the Investigator’s discretion to initiate an Unscheduled Visit, if deemed necessary by the Investigator for the subject’s safety and well-being. At this visit, any of the following assessments may be performed depending on the presenting reason:
Concomitant medications
Vital signs
Blood samples for hematology, biochemistry analyses, other testing such as for TB or CRP
Urine for urinalysis and/or pregnancy testing (for women of childbearing potential)
Physical examination
Concomitant medication
REDACTED finfin
olic bloolic b
COPY
nal dnal
This do
cumen
t can
notConcon
VV
be mentent us
ed veses
gator fator nts mts m
ostigastigsu
pportndicandica
schesche
any
ate
marketi
ng
esting fsting
autho
rizati
on
dose. ose.
ood pod p
appli
catio
n and
any e
xtens
ions o
r vari
ation
s the
reof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 47 of 113
AEs
TB questionnaire
9 ASSESSMENT OF EFFICACY 9.1 Assessment of primary and secondary efficacy variables 9.1.1 Assessment of AU flares Subjects will be requested to contact an ophthalmologist when they experience an AU flare at any time during the study. The ophthalmologist will confirm the AU flare and determine the duration, severity, and treatment. The ophthalmologist will be provided with documentation to ensure all protocol-requested information is collected. The documentation will inform the ophthalmologist about the subject’s study participation and the requirements.
At each site visit, the subject will be questioned on the occurrence of uveitis flares since last visit. In case uveitis flares are confirmed by the subject the site will obtain the needed information from the ophthalmologist containing the diagnosis of AU, the affected eye, location of the uveitis in the affected eye, duration, severity, and treatment provided.
The following information will be collected:
Start and end date of each AU flare (episode)
Start date: when subject experience first signs of the AU episode
End date: when treatment of the AU is stopped
Grading
For uveitis flare, the highest grade noted during an episode of uveitis will be graded as follows:
Grade 0=None; Grade 1+=Faint; Grade 2+=Moderate, iris and lens details clear; Grade3+=Marked, iris and lens details hazy; Grade4+=Intense, fibrin or plastic aqueous; ND=Not done or not assessed; UNK=Unknown
For cell count, the highest cell count noted during an episode of uveitis will be graded as follows:
Grade 0=<1 cell; Grade 0.5+=1-5 cells; Grade 1+=6-15 cells; Grade 2+=16-25 cells; Grade 3+=26-50 cells; Grade 4+=>50 cells; ND=Not done or not assessed; UNK=Unknown
Treatment
9.1.2 Ankylosing spondylitis disease activity score (ASDAS) The ASDAS is comprised of a number of assessments that are scored by the subject and physician and multiplied by a proven formula (van der Heijde et al, 2009) as listed:
0.121 × (BASDAI Question 2 result; see Section 9.1.3)
0.058 × (BASDAI Question 6 result)
0.110 × PtGADA (see Section 9.1.7)
AU isAU is
hest ghest g
COPY ))
rst sirst si
This do
cumen
t can
not TreaTre
9.19.1
beUNUN usedde 0=e 0=
adeade 33to s
<<su
pport
Not dot d
unt, thent, the:
anyris aris a
dondon
marketi
nggradegrade
1+=F1+=Fan
autho
rizati
onigns oigns o
s stopstop
appli
catio
nnd td t
f
andf AUAU
treatre
any veivei
btain btaint
exten
sionsumeume
will inill ints.s.
itis tis
ormineminentt
varia
tions
flare alare ane tne
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 48 of 113
0.073 × (BASDAI Question 3 result)
0.579 × (natural logarithm of the CRP [mg/L] + 1, see Week 32 in Table 5-1)
PtGADA, are all assessed on a numerical scale (0 to 10 units) (Lukas et al, 2009).
The variables related to ASDAS disease activity are defined as follows:
ASDAS-Inactive Disease (ASDAS-ID): ASDAS <1.3
ASDAS-Moderate Disease (ASDAS-MD): ASDAS ≥1.3, <2.1
ASDAS-High Disease activity: ASDAS ≥2.1, ≤3.5
ASDAS-very High Disease activity: ASDAS >3.5
The variables related to ASDAS improvement are defined as follows:
ASDAS-CII: ASDAS reduction (improvement) of ≥1.1 relative to Baseline
ASDAS-Major Improvement: ASDAS reduction (improvement) of ≥2.0 relative to Baseline
The ASDAS will be calculated according to the assessment schedule in Table 5-1.
9.1.3 Bath ankylosing spondylitis disease activity index (BASDAI) The most common instrument used to measure the disease activity of AS from the subject’s perspective and in the broad axSpA population is the BASDAI (van Tubergen et al, 2015; Garrett et al, 1994). The BASDAI is a validated self-reported instrument which consists of six 10-unit horizontal NRSs to measure
over the last week. The final BASDAI score ranges from 0 to 10, with lower scores indicating lower disease activity. The minimal clinically important difference (MCID) used to interpret scores is 10mm on a visual analog scale (VAS) or 22.5% of the Baseline score (Pavy et al, 2005). An MCID of 1 unit will be selected for the NRS version (Appendix 16.4).
The BASDAI 50 is defined as an improvement of at least 50% in the BASDAI response.
The BASDAI is calculated as follows:
item of the BASDAI
as a major symptom of AS can effectively be measured with single item questions such as the BASDAI item (van Tubergen et al, 2002). This item has shown moderate to good reliability and responsiveness (van Tubergen et al, 2002). The same MCID will be used for the item of the BASDAI and for the total BASDAI score (ie, a change of 1 unit on the NRS).
REDACTEDasureasurpulapulatiotio
a valia valeasureasureRE
COPY ses
is diss disre tre t
This do
cumen
tnt ca
nanno
ttt
c
bbbeeeeeeeeeeeee se
d I isis
uuuuuuuuuuuussssssssssssse
to s cas casu
pport
al,al
is defis defi
anyoresore
, 200200mark
etin
st weekt weediseasediseases ises
ggtin
g auth
oriza
tion
sethe dishe di
on is ton is tidateddatedau
appli
catio
n me
ment schent sc
ease ase
and
tot
ent) ent) an
y o Baso Bas
exten
sions
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 49 of 113
The BASDAI assessments per visit are described in the schedule of study assessmentsTable 5-1.
9.1.4 ASAS20, ASAS40, ASAS5/6, and ASAS PR The ASAS20 is defined as an improvement of at least 20% and absolute improvement of at least 1 unit on a 0 to 10 NRS in at least 3 of the 4 following domains and absence of deterioration in the potential remaining domain [deterioration is defined as a relative worsening of at least 20% and an absolute worsening of at least 1 unit]:
PtGADA (see Section 9.1.7)
Pain assessment (the average of total and nocturnal spinal pain NRS scores)
Function (represented by BASFI, Section 9.1.9)
Inflammation (the mean of the BASDAI questions 5 and 6, [see Section 9.1.3])
The ASAS criteria for 40% improvement are defined as relative improvements of at least 40%, and absolute improvement of at least 2 units on a 0 to 10 NRS in at least 3 of the 4 domains and no worsening at all in the remaining domain.
The ASAS 5/6 response is defined as at least 20% improvement in 5 of 6 domains, including spinal mobility (lateral spinal flexion) and CRP as more objective measures (Brandt et al, 2004).
The ASAS PR response is defined as a score of ≤2 units on a 0 to 10 unit scale in all 4 domains listed above for ASAS20.
The assessments per visit are described in the schedule of study assessments Table 5-1.
9.1.5 Physician’s global assessment of disease activity (PhGADA) The Investigator will assess the overall status of the subject with respect to the axSpA signs and symptoms and the functional capacity of the subject using a VAS where 0 is “very good, asymptomatic and no limitation of normal activities” and 100 is “very poor, very severe symptoms that are intolerable, and the inability to carry out all normal activities.”
This assessment by the Investigator should be made without any knowledge of the PtGADA.
The PhGADA will be completed as described in the schedule of study assessments Table 5-1.
9.1.6 Swollen and tender joint counts (44 joints evaluation) The following 44 joints are to be examined for swelling and tenderness by the Investigator, another delegated physician, or an appropriately qualified medical professional (based on local requirements) who has had documented training on how to perform these assessments correctly. Preferably the same assessor should evaluate the subject at each arthritis assessment.
Upper body (4) – bilateral sternoclavicular and acromioclavicular joints.
Upper extremity (26) – bilateral shoulders, elbows, wrists (includes radiocarpal, carpal, and carpometacarpal bones considered as a single unit), metacarpophalangeals (MCPs) I,
REDACTED scorescore
scribescribe
COPY% im% imRP as mRP as
This do
cumen
t alca
nnot
6The fohe foanoano
be us
edDA wA w to t byby
isu
pport
imitamitntoleratoler
y they th
anyctionction
tatiotat
marketi
ng ed ind in
obal asbal ahe ove ove
autho
rizati
on mormo
e of ≤2of ≤2
n tt
appli
catio
n0 NR0 NR
provemrovemore ore o
and imprompro
RSRS
any i ex
tensio
ns
ion on 99
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 50 of 113
II, III, IV, and V, and thumb interphalangeals (IPs), and proximal IPs (PIPs) II, III, IV, and V.
Lower extremity (14) – bilateral knees, ankles, and metatarsophalangeals (I, II, III, IV, and V).
The assessment for swelling and tenderness is made on 44 joints from the above list. Artificial and ankylosed joints are excluded from swelling and tenderness assessments.
The assessments per visit are described in the schedule of study assessments Table 5-1.
9.1.7 Patient’s global assessment of disease activity (PtGADA) (NRS) For the PtGADA questionnaire, subjects will score their global assessment of their disease activity in response to the question “How active was your spondylitis on average during the last week?” using a NRS where 0 is “not active” and 10 is “very active” (van Tubergen et al, 2015) (Appendix 16.5).
The PtGADA assessments per visit are described in the schedule of study assessments Table 5-1.
9.1.8 Total and nocturnal spinal pain NRS The questionnaire for pain in the spine due to AS consists of 2 questions (ie, “How much pain of your spine due to spondylitis do you have?”; and “How much pain of your spine due to spondylitis do you have at night?”) (Sieper et al, 2009; van der Heijde et al, 2005; CPMP/EWP/556/95). Usually, a 10% difference (ie, a 1 point difference on a NRS ranging from 0 to 10) is considered the MCID used to interpret scores (Dworkin et al, 2008). Pain experienced by axSpA subjects has also been measured with this assessment (Haibel et al, 2008) (Appendix 16.6).
The pain NRS assessments per visit are described in the schedule of study assessments Table 5-1.
9.1.9 Bath ankylosing spondylitis functional index (BASFI) The BASFI is a validated disease-specific instrument for assessing physical function (van Tubergen et al, 2015; Calin et al, 1994; van der Heijde et al, 2005). The BASFI comprises 10 items relating to the past week. The NRS version will be used for the answering options of each item on a scale of 0 (“Easy”) to 10 (“Impossible”) (van Tubergen et al, 2002). The BASFI is the mean of the 10 scores such that the total score ranges from 0 to10, with lower scores indicating better physical function. The MCID used to interpret scores is 7mm on a 0 to 100mm VAS or 17.5% of the Baseline score (Pavy et al, 2005); an MCID of 1 unit will be used for the NRS version (Appendix 16.7).
The BASFI assessments per visit are described in the schedule of study assessments Table 5-1.
REDACTED cribcri
nal spnal spe spine spin
ii
COPY andand
ibedbe
This do
cumen
t eca
nnotBABA
Tubergubergitemitem
be
SFSFus
ed to
asseasse supp
ortA subjsubj6.66).).
essmess
any
y,yd the Md the jecj
marketi
ngne dune dutis do ytis do
ight?”)ght?”y, a 10, a 1
autho
rizati
on
d in thd in th
pinal inal
appli
catio
nal assal asspondyspond
0 is “veis “v
andtivityvitya
ny (a
ny ex
tensio
ns or
varia
tions
va
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 51 of 113
9.1.10 Extra-articular assessments Extra-articular assessments (eg, number of IBD exacerbations and number of psoriasis exacerbations [in subjects with concurrent psoriasis]) are to be performed at Screening, Baseline, Weeks 12, 24, 36, 48, 60, 72, 84, and at Completion at Week 96/WD Visit.
9.2 Assessment of other efficacy variables 9.2.1 Ankylosing spondylitis quality of life (ASQoL) The ASQoL, a validated disease specific 18 item questionnaire, has been developed specifically for measuring health-related quality of life (HRQoL) in subjects with AS (Doward et al, 2003). The ASQoL has been used and has shown to be responsive in axSpA (Barkham et al, 2009; Haibel et al, 2008). The ASQoL score ranges from 0 to 18 with higher score indicating worse HRQoL. A change of 1.8 points, which represents 10% of the possible score range, has been used as the MCID criteria to guide the interpretation of ASQoL score changes in previous studies with a TNF antagonist (van der Heijde et al, 2009; Davis et al, 2007). A change in ASQoL score of 2 points (ie, 10% of the total score range) will be used as the MCID to guide the interpretation of ASQoL score changes (Appendix 16.8).
9.2.2 ASAS HI Questionnaire The ASAS HI has been developed under the auspices of ASAS to assess health in subjects with all forms of SpA. The questionnaire contains 17 items measuring “functioning, disability and health” – a concept which is conceptualized in the International Classification of Functioning, Disability and Health. The International Classification of Functioning, Disability and Health, a model to systematically classify and describe functioning, disability, and health in human beings, has been used by ASAS as a basis to define a core set of items that are typical and relevant for subjects with AS. Based on this ICF core set for AS, an item pool has been developed containing various items which are linked to specific ICF categories including items related to functioning as well as environmental factors. The performance of the item pool has been tested and analyzed with Rasch Analysis. The best performing items have been included in the final measure. It can be used in clinical studies as a new composite index that captures relevant information on the health status of subjects with SpA (Klitz et al, 2015) (Appendix 16.9).
Each statement on the ASAS HI is given a score of 1=I agree OR 0=I do not agree. All item scores are summed up to give a total score that ranges from 0 (good functioning) to 17 (poorfunctioning).
It is to be noted that items 7 and 8 are not applicable for all subjects. For those subjects who ticked the response “not applicable,” the sum score is analyzed based on n=16 or n=15, respectively.
REDACTED concohe Inthe Int
ematiemateen useen us
subjesubje
COPYpicespiceainains 1s
oncenc
This do
cumen
t can
not o bo b
cked thked tresperespe
be
be nbe nus
edummmmng).g).
toon on supp
ortx thatthat015) (15) (AA
theth
any l hl h
en incen inct
marketi
ngects wects wing varng va
ted to fed to has bhas
autho
rizati
on 17 i7 i
eptualptualternaternat
icallyicallyed byed by
appli
catio
n ((AA
of ASAof ASitemitem
andscorscor
AppApp
any
onoal, 20al, 2re
exten
sionse in ae in a
o 18 w18 w10% o0% o
on ofn of
or S S varia
tions
there
of.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 52 of 113
Missing data:
A total score can be analyzed if no more than 20% of the data are missing. The total score is calculated as follows for respondents with 1 to a maximum of 3 missing responses:
Sumscore = x X17 x=Item summation score
17-m m=Number of missing items
Cases with more than 3 missing responses cannot be allocated a total score.
The ASAS HI questionnaire assessments per visit are described in Table 5-1.
9.2.3 Short-Form 36-Item Health Survey (SF-36) The SF-36 (Version 2, standard recall) is a 36-item generic HRQoL instrument that uses a recall period of 4 weeks. Items are grouped into 8 domains as follows: Physical Functioning (10 items), Role Physical (4 items), Bodily Pain (2 items), General Health (5 items), Vitality (4 items), Social Functioning (2 items), Role Emotional (3 items), Mental Health (5 items), and 1 item for perceived stability or change in health (Health Transition) during the last year. The concepts represented by these domains contribute to physical, mental, and social aspects of HRQoL.
In addition to domain scores, the Physical Component Summary and Mental Component Summary scores are calculated from the 8 domains (excluding the Health Transition item). Component scores appreciate the impact of each domain on physical and mental health status (Ware et al, 1994). Each of the 8 domain scores and the component summary scores range from 0 to 100, with a higher score indicating a better health status. The 2-component summary scores are standardized with a mean of 50 and a standard deviation (SD) of 10 in the general USA population. The SF-36 Version 2 manual (Ware et al,1994) states the following values: physical component summary, 2; mental component summary, 3; physical function, 3; role physical, 3; bodily pain, 3; general health, 2; vitality, 2; social functioning, 3; role emotional, 4; and mental health, 3 (Appendix 16.10).
The SF-36 has been used and has shown to be responsive in axSpA (van Tubergen et al, 2015; Haibel et al, 2008). The SF-36 will be administered per visit as described in the schedule of study assessments Table 5-1.
10 ASSESSMENT OF SAFETY 10.1 Adverse events 10.1.1 Definition of adverse event An AE is any untoward medical occurrence in a subject or clinical investigation subject administered a pharmaceutical product that does not necessarily have a causal relationship with this treatment. An AE can therefore be any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease temporally associated with the use of an IMP, whether or not related to the IMP.
To ensure complete safety data collection, all AEs occurring during the study (ie, after the signing of the ICF), including any pretreatment and posttreatment periods required by the protocol, must be reported in the eCRF even if no IMP was taken but specific study
ean scoren scor
dicatincatinwith a with a
e SFe SF 3
COPYonennemains (ains (ach dach d
This do
cumen
twwca
nnot 11
An AEn AEadmadm
be us
ed A
AtoASAS su
pportd ad
08). T08). Tassessmssess
SS
any
and hand hmark
eting
36 V36 Vnent suent su
dily padily ptal heal he
autho
rizati
on(e
domadomaes andes and
ng a beg a bemeanmeanVeVe
appli
catio
nsical,cal
SummSummexcludxclud
andansitionsitio
l ml m
anylth lth
ental entalex
tensio
ns
ent thant thahysicalysicalh (5h (5
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 53 of 113
procedures were conducted. This includes all AEs not present prior to the initial visit and all AEs that recurred or worsened after the initial visit.
Signs or symptoms of the condition/disease for which the IMP is being studied should be recorded as AEs only if their nature changes considerably or their frequency or intensity increases in a clinically significant manner as compared to the clinical profile known to the Investigator from the subject’s history or the Baseline Period.
10.1.2 Procedures for reporting and recording adverse events The subject will be given the opportunity to report AEs spontaneously. A general prompt will also be given at each study visit to detect AEs. For example:
“Did you notice anything unusual about your health (since your last visit)?”
In addition, the Investigator should review any self-assessment procedures (eg, diary cards) employed in the study.
10.1.3 Description of adverse events When recording an AE, the Investigator should use the overall diagnosis or syndrome using standard medical terminology, rather than recording individual symptoms or signs. The CRF and source documents should be consistent. Any discrepancies between the subject’s own words on his/her own records (eg, diary card) and the corresponding medical terminology should be clarified in the source documentation.
Details for completion of the Adverse Event CRF (including judgment of relationship to IMP) are described in the CRF Completion Guidelines.
10.1.4 Follow up of adverse events An AE should be followed until it has resolved, has a stable sequelae, the Investigator determines that it is no longer clinically significant, or the subject is lost to follow-up.
If an AE is ongoing at the end of the study for a subject, follow-up should be provided until resolution/stable level of sequelae is achieved, or until the Investigator no longer deems that it is clinically significant, or until the subject is lost to follow-up. If no follow up is provided, the Investigator must provide a justification. The follow-up will usually be continued for 70 days after the subject has discontinued his/her IMP.
10.1.5 Rule for repetition of an adverse event An increase in the intensity of an AE should lead to the repetition of the AE being reported with:
The outcome date of the first AE that is not related to the natural course of the disease being the same as the start date of the repeated AE, and the outcome of “worsening”
The AE verbatim term being the same for the first and repeated AE, so that the repeated AE can be easily identified as the worsening of the first one
REDACTEDvent Cvent Cletion etion
verseverseit hit
COPYnd thed thon.on.
This do
cumen
t can
not
ThTh
beeaseas us
edRRse ine in
to e su
Rsu
pportant, ont, o
ust provst prsubjeubje
any nd ond
equelequemark
eting
e has reshas res
linicallinical
of thof
autho
rizati
on
CRF (iRF (i GuidGuid
eveeve
appli
catio
nl diadiadual syual sy
panciesanciecorrescorre
and
iagnag
any e
xtens
ions
visit)?visit)
es (eg,s (eg
ormpm va
riatio
ns
mpt wmpt w
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 54 of 113
10.1.6 Pregnancy If an Investigator is notified that a subject has become pregnant after the first intake of any IMP, the Investigator must immediately notify UCB’s Drug Safety (DS) department by providing the completed Pregnancy Report and Outcome form (for contact details see Serious Adverse Event reporting information at the beginning of this protocol). The subject should be withdrawn from the study as soon as pregnancy is known (by positive pregnancy test), and the following should be completed:
The subject should return for an early discontinuation visit.
The subject should immediately stop the intake of the IMP.
A Safety Follow-Up Visit should be scheduled 10 weeks after the subject has discontinued her IMP.
The Investigator must inform the subject of information currently known about potential risks and about available treatment alternatives.
The progression of the pregnancy and the eventual birth (if applicable) must be followed up using the Pregnancy Report and Outcome form in which the Investigator has to report on the health of the mother and of the child. Every reasonable attempt should be made to follow the health of the child for 30 days after birth for any significant medical issues. In certain circumstances, UCB may request that follow-up is continued for a period longer than 30 days. If the subject is lost to follow-up and/or refuses to give information, written documentation of attempts to contact the subject needs to be provided by the Investigator and filed at the site. UCB’s DS department is the primary contact for any questions related to the data collection for the pregnancy, eventual birth, and follow-up.
In cases where the partner of a male subject enrolled in a clinical study becomes pregnant, the Investigator or designee is asked to contact the subject to request consent of the partner via the Partner Pregnancy Consent form that has been approved by the responsible IRB/IEC and should be available in the Investigator site file. In case of questions about the consent process, the Investigator may contact the UCB/contract research organization (CRO) contract monitor for the study. The Investigator will complete the Pregnancy Report and Outcome form and send it to UCB’s DS department (for contact details see Serious Adverse Event reporting information at the beginning of this protocol) only after the partner has agreed that additional information can be captured and has provided the signed Partner Pregnancy Consent form. UCB’s DS department is also the primary contact for any questions related to the data collection for the partner pregnancy, eventual birth, and follow-up.
A pregnancy becomes a serious adverse event (SAE) in the following circumstances: miscarriage, abortion (elective or spontaneous), unintended pregnancy after hormonal contraceptive failure (if the hormonal contraceptive was correctly used), ectopic pregnancy, fetal demise, or any congenital anomaly/birth defect of the baby. Those SAEs must be additionally reported using the Investigator SAE Report form.
10.1.7 Overdose of investigational medicinal product Excessive dosing (beyond that prescribed in the protocol and including overdose) should be recorded in the Study Drug Dosing module of the CRF. Any SAE or nonserious AE
REDACTED and/ande subje subj
nt is thnt is theventueven
malemale
COPYsignsignup is cup is
d/ord/o
This do
cumen
tcccann
ot pregnpregnmiscmisc
beo
a cola cous
ednformformorm. orm.
tomatimati sup
portr mm
y. The . Theo UCBo UCBion
any he Inhe I
may may
marketi
ng
e subjesubjesked toked t
nsent fsent n
autho
rizati
onconon
r refusrefusject neect ne
he prihe priual bial bi
appli
catio
n plic
e InvesInvettempt ttemp
ficicant mantntinntin
and
cablcaban
ywn awn exten
sions
t has has
abb
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 55 of 113
associated with excessive dosing must be followed as any other SAE or nonserious AE. These events are only considered AEs or SAEs if there are associated clinical signs and symptoms or if the act of taking the excess medicine itself is an AE or SAE (eg, suicide attempt).
10.1.8 Safety signal detection Selected data from this study will be reviewed periodically to detect as early as possible any safety concern(s) related to the IMP so that Investigators, clinical study subjects, regulatory authorities, and IRBs/IECs will be informed appropriately and as early as possible.
The Study Physician or medically qualified designee/equivalent will conduct an ongoing review of SAEs and perform ongoing SAE reconciliations in collaboration with the DS representative.
As appropriate for the stage of development and accumulated experience with the IMP, medically qualified personnel at UCB may identify additional safety measures (eg, AEs, vital signs, laboratory or electrocardiogram [ECG] results) for which data will be periodically reviewed during the course of the study.
10.2 Serious adverse events 10.2.1 Definition of serious adverse event Once it is determined that a subject experienced an AE, the seriousness of the AE must be determined. An SAE must meet 1 or more of the following criteria:
Death
Life-threatening
(Life-threatening does not include a reaction that might have caused death had it occurred in a more severe form.)
Significant or persistent disability/incapacity
Congenital anomaly/birth defect (including that occurring in a fetus)
Important medical event that, based upon appropriate medical judgment, may jeopardize the subject or subject and may require medical or surgical intervention to prevent 1 of the other outcomes listed in the definition of serious
(Important medical events may include allergic bronchospasm requiring intensive treatment in an emergency room or at home, blood dyscrasias that do not result in inpatient hospitalization, or the development of drug dependency or drug abuse.)
Initial inpatient hospitalization or prolongation of hospitalization
(A subject admitted to a hospital, even if he/she is released on the same day, meets the criteria for the initial inpatient hospitalization. An emergency room visit that results in admission to the hospital would also qualify for the initial inpatient hospitalization criteria. However, emergency room visits that do not result in admission to the hospital would not qualify for this criteria and, instead, should be evaluated for 1 of the other criteria in the definition of serious [eg, life-threatening adverse experience, important medical event].
REDACTEDof tof
ll
COPYeveevced aned an
hh
This do
cumen
t can
noteaea
inpainpabe
oratmeatme
sedcomom
rtantrtan
toor suor smeme
supp
ort y/biry/bir
ical evcal esubjsub
anydisadis
rthrth
marketi
ng
ude a rde a
sabsab
autho
rizati
onn AEn AEhe folle foll
appli
catio
n
nttE tE
andatata wa wa
nymeasumeaswili
exten
sions
ongongoh the h the
withwith
or oinoinva
riatio
ns
y yory ry
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 56 of 113
Hospitalizations for reasons not associated with the occurrence of an AE [eg, preplanned surgery or elective surgery for a pre-existing condition that has not worsened or manifested in an unusual or uncharacteristic manner] do not qualify for reporting. For example, if a subject has a condition recorded on his/her medical history and later has a preplanned surgery for this condition, it is not appropriate to record the surgery or hospitalization as an SAE, since there is no AE upon which to assess the serious criteria. Please note that, if the pre-existing condition has worsened or manifested in an unusual or uncharacteristic manner, this would then qualify as an AE and, if necessary, the seriousness of the event would need to be determined.)
10.2.2 Procedures for reporting serious adverse events If an SAE is reported, UCB must be informed within 24 hours of receipt of this information by the site (see contact information for SAE reporting listed in the Serious Adverse Event Reporting section at the front of the protocol). The Investigator must forward to UCB (or its representative) a duly completed “Investigator SAE Report Form for Development Drug” (SAE report form) provided by UCB, even if the data are incomplete, or if it is obvious that more data will be needed in order to draw any conclusions. Information recorded on this form will be entered into the global safety database.
An Investigator SAE report form will be provided to the Investigator. The Investigator SAE Report form must be completed in English.
It is important for the Investigator, when completing the SAE report form, to include the assessment as to a causal relationship between the SAE and the IMP administration. This insight from the Investigator is very important for UCB to consider in assessing the safety of the IMP and in determining whether the SAE requires reporting to the regulatory authorities in an expedited manner.
Additional information (eg, autopsy or laboratory reports) received by the Investigator must be provided within 24 hours. All documents in the local language must be accompanied by a translation in English, or the relevant information included in the same document must be summarized in the Investigator SAE report form.
The Investigator is specifically requested to collect and report to UCB (or its representative) any SAEs (even if the Investigator is certain that they are in no way associated with the IMP), up to 30 days from the end of the study (FU Visit) for each subject, and to also inform participating subjects of the need to inform the Investigator of any SAE within this period. Serious AEs that the Investigator thinks may be associated with the IMP must be reported to UCB regardless of the time between the event and the end of the study.
Upon receipt of the SAE report form, UCB will perform an assessment of expectedness of the reported SAE. The assessment of the expectedness of the SAE is based on the IB and product information/SPC/Product Monographs, as applicable.
10.2.3 Follow up of serious adverse events An SAE should be followed until it has resolved, has a stable sequelae, the Investigator determines that it is no longer clinically significant, or the subject is lost to follow-up.
Information on SAEs obtained after clinical database lock will be captured through the DS database without limitation of time.
ompometweenetween
importmporher theher th
COPY d tod t
mpleple
This do
cumen
t t cann
otB regreg
Upon Upon thethe
beAEAE
egaregarus
ed 0 d0 d
ng subg subEs tEs
toif
daydaysu
pport
stigtig
specifipeciff the Ithe
anyhe rele re
gatogat
marketi
ng
opsy psy oroAll doAll do
levl
autho
rizati
on
eting tting tn the n the
rtant fotant fSAESAE
appli
catio
nformorm
he InvesInve
andlete, ete,
rmatma
any rwrw
DeveDeveor
exten
sions
his infis inf
s AdveAdvwardwar
or va
riatio
ns
or or the
reof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 57 of 113
10.3 Adverse events of interest An AE of interest is any AE that a regulatory authority has mandated be reported on an expedited basis, regardless of the seriousness, expectedness, or relatedness of the AE to the administration of a UCB product/compound.
Adverse events of interest include:
Serious infections including opportunistic infections
Malignancies including lymphoma
Demyelinating-like disorders
Congestive heart failure
Aplastic anemia, pancytopenia, thrombocytopenia, neutropenia, and leucopenia
Serious bleeding events
Lupus and lupus-like illness
Serious skin reactions (eg, Stevens Johnson Syndrome, toxic epidermal necrosis, and erythema multiforme)
A confirmed LTBI must be reported as an AE of interest
Confirmed active TB is an AE of interest. In the event that an active TB occurrence meets the protocol’s SAE criteria, it is also an SAE.
Note : Potential Hy’s Law, defined as ≥3xULN alanine aminotransferase or aspartate aminotransferase with coexisting ≥2xULN total bilirubin in the absence of ≥2xULN alkaline phosphatase, with no alternative explanation for the biochemical abnormality, must ALWAYS be reported to UCB as an AE of interest (ie, without waiting for any additional etiologic investigations to have been concluded). Follow-up information should then be reported if an alternative etiology is identified during investigation and monitoring of the subject.
10.4 Immediate reporting of adverse events The following AEs must be reported immediately:
SAE: AE that the Investigator classifies as serious by the above definitions regardless of causality
Suspected transmission of an infectious agent via a medicinal product
AE of interest (see Section 10.3)
10.5 Anticipated serious adverse events The following list of Anticipated SAEs has been identified, as these events are anticipated to occur in the population studied in this protocol at some frequency that is independent of drug exposure. This original list will remain in effect for the duration of the protocol.
This list does not change the Investigator’s obligation to report all SAEs (includingAnticipated SAEs) as detailed in Section 10.2.2.
REDACTED st.st.is alsis als
as ≥3xas ≥3xg ≥2xUg ≥2xUexpexp
COPY AE of AE of
. InIn
This do
cumen
t 11ca
nnotSuspSusp
AA
besalitalitus
ed
AE thaE thaityty
toAEsAEssupp
ort
mediamedias ms m
anyiologiolo mark
eting
ULNULplanatiplanat
as an A an Ae beene beegy
autho
rizati
on
the ehe eso an So an S
xULNxULNLN tN t
appli
catio
n toxic eoxic
interentere
and a
ny ex
tensio
ns
leucopeucop
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 58 of 113
Table 10‒1: Anticipated serious adverse events in population independent of drug exposure
Population Anticipated SAE(s) Rheumatoid arthritisa Rheumatoid arthritis
Crohn’s diseasea Crohn’s disease Perianal abscessAbdominal pain
Ankylosing spondylitis Ankylosing spondylitis
Psoriatic arthritis Psoriatic arthritis
JIA Juvenile arthritis JIA=juvenile idiopathic arthritis; SAE=serious adverse event; RA=rheumatoid arthritisa The lists of anticipated SAEs were based on the treatment-emergent SAEs with an incidence ≥0.5% in
subjects who participated in UCB Sponsored placebo-controlled studies who received placebo (Crohn’s disease and rheumatoid arthritis safety pooling).
10.6 Laboratory measurements Hematology, biochemistry, and urinalysis samples will be taken at Screening, Baseline, Weeks 2, 4, 12, 24, 36, 48, 60, 72, 84, and 96/WD Visit. Testing for HBs-Ag, HBc-Ab,HBs-Ab, HCV-Ab, HIV, and HLA-B27 (if not yet known) will be performed at Screening.
Hepatitis –associated disease markers
For all subjects, blood samples will be collected by qualified site personnel for the determination of the following:
HBc-Ab
HBs-Ag
HBs-Ab
HCV-Ab
These samples will be collected at the Screening Visit.
Hepatitis B virus deoxyribonucleic acid and hepatitis C virus ribonucleic acid are available as needed to assess the clinical status of subjects if required during the study.
REDACTED 96/96/(if no(if no
arkersarkers
will bwill b
COPY mples wmples6/WD/W
This do
cumen
t can
not ed d be
s Bto ato a
sedes wes w
B virB vi
to wilwilsu
pport
any m
arketi
ngbe colbe co autho
rizati
on wilwil
WD VisD Viot yet t yet
llll
appli
catio
n
ill bell b
and
th hho reco rec
any ritisritis
an inan ini
exten
sions
iss on
thritishritisen
e
or orva
riatio
ns
iothe
reof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 59 of 113
The urinalysis will be performed with a dipstick, and in case of a positive outcome, on a clean catch urine sample sent to the central laboratory for analysis. The central laboratory will analyze and assess blood and urine samples for the following:
Table 10‒2 lists the laboratory parameters that will be measured:
Table 10‒2: Laboratory measurements
Hematology Chemistry Urinalysis OthersRed blood cells Sodium pH Hepatitis B surface
antigen
Hemoglobin Potassium Protein Antibodies to hepatitis C
Hematocrit Chloride Glucose Antibodies to HIV
Platelets Bicarbonate Blood HLA-B27
White blood cells Total calcium Esterase
Neutrophils Inorganic phosphorus Microscopy (WBC, RBC, casts, crystals, bacteria) (Microscopy will be performed only when there are abnormalities on dipstick)
Lymphocytes CRP
Monocytes Glucose Eosinophils Creatinine
Basophils Uric acid
Urea
Total protein
Albumin
Alkaline phosphatase Aspartate aminotransferase
Alanine aminotransferase
Bilirubin
Total cholesterol CRP= C-reactive protein; HIV= human immunodeficiency virus; HLA-B27= human leukocyte antigen B27; RBC=red blood cell; WBC=white blood cell
REDACTED
DARE
COPYBC
bactebactewilwi
CO
This do
cumen
t ntca
nnot no
c
be us
ed d
usto
supp
ort Urearea
ToTpoupsuuuu
any acidacid
anmark
eting
g
e kem
autho
rizati
onll bel be
only wonly wabnabn
oriuth
appli
catio
n y (WBC(WB
asts, crysts, crria) (Mia) (Me pee pe
onan
d ndan
y AnA
HHny
aex
tensio
ns
dies tdies tatitisatitis C
ns
Antibntibxt
orrfacrfa va
riatio
ns
tioioio
aceacear
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 60 of 113
10.6.1 Pregnancy testing Pregnancy testing must be carried out for women of childbearing potential and will consist of serum testing at Screening, and urine dipstick testing at Baseline, Week 96/WD Visit, and FU Visit.
10.6.2 Physical assessments A physical examination will be performed at Screening, Baseline, Weeks 2, 4, 12, 24, 36, 48, 60, 72, 84, 96/WD Visit, and at the FU Visit. Physical examination findings will be recorded in the eCRF only at Screening. Clinically important abnormal changes in subsequent physical examinations will be recorded as AEs. Physical examinations must be documented in source documentation.
The following body systems will be examined:
General Appearance
Ear, Nose, and Throat
Eyes
Hair and Skin
Respiratory
Cardiovascular
Gastrointestinal
Musculoskeletal
Hepatic
Neurological (including limb reflexes)
Mental status
In addition, the TB signs and symptoms questionnaire will be performed at Screening, Baseline, Weeks 12, 24, 36, 48, 60, 72, 84, 96/WD Visit.
Weight will be measured at Screening, Baseline, Week 48, and at Completion at Week 96/WD Visit. Height will be measured at the Baseline Visit only.
10.6.3 MRI assessments Magnetic resonance imaging of the SIJs will be performed at the Screening Visit only. AnMRI performed ≤3 months prior to the Baseline Visit may be used as the Baseline.
10.6.4 Tuberculosis assessments 10.6.4.1 Tuberculosis test The IGRA test that will be used by the central laboratory is the QuantiFERON®-TB Gold In-Tube (QFT-GIT) test or updated version (QuantiFERON® Plus). The test results will be reported as positive, negative, or indeterminate and must be reviewed by an experienced TB specialist, radiologist, or a pulmonologist.
REDACTED COPY
This do
cumen
t 1cann
otnetineti
MRI peRI pe
10.10
be
tic rticus
ed. HeHeto
meameHeigeig
supp
orns anns an
, 24, 3624, 3
easurasu
any
ndnd
marketi
ng
mb refleb refl
autho
rizati
on ap
plica
tion a
nd an
y exte
nsion
sd ind inor phph
n sn sva
riatio
ns
48, 48, orded rded hyshys
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 61 of 113
The TB test will be performed at the following timepoints:
At Screening, all subjects will have a QuantiFERON-TB GOLD In-Tube test.
At Week 48 and 96/WD Visit all subjects with a negative Quantiferon-TB GOLD In-Tube test at Screening will have the test repeated.
For subjects with a positive Quantiferon-TB GOLD In-Tube test at Screening, the test is not to be repeated at Weeks 48 and 96/WD Visit.
Per the judgment of the Investigator, a QuantiFERON-TB GOLD In-Tube test can be performed at unscheduled visits at any time during the study.
In case of an indeterminate result, a retest must be done shortly after, during the protocol-defined Screening window. If the test is positive or indeterminate on retest, the subject may not be randomized to IMP without further evaluation by a TB specialist. If LTBI is identified, appropriate treatment for TB prophylaxis and discussion with the study physician is required. If active TB is identified the subject cannot be randomized and is excluded from the study.
At any other timepoint during the study if the IGRA test result is indeterminate, the IGRA must be reported once shortly after.
If during the study the assessment by IGRA is positive or indeterminate on retest for subjects who were previously negative at Screening and not treated for LTBI, the subject may not continue study treatment without further evaluation by a TB specialist and discussion with the Medical Monitor. If LTBI is identified, study treatment can only be re-initiated after prophylactic TB treatment is initiated. If active TB is identified, the subject must undergo appropriate study-specified withdrawal procedures.
Conditions for including LTBI subjects in the study:
The Investigator must provide full documentation of the duration, and start and stop dates of LTBI treatments, and discuss with the study physician prior to allowing subject to screen (if the LTBI was discovered prior to subject screening) or prior to receiving study drug (if LTBI was identified at screening).
Subjects who initiate treatment for LTBI during the Screening Period must repeat initial screening laboratory parameters, examination, and questionnaires (after receiving at least 4 weeks duration of treatment for LTBI) prior to randomization in the study, and must plan to continue the full course of LTBI prophylaxis therapy. The Investigator must assess that the subject’s likelihood of completing the therapy is high and duly record his/her opinion in the subject’s record prior to randomizing the subject. It is suggested to obtain the opinion of a TB specialist, especially for subjects having been in contact with multi-resistant TB.
An extension of the Screening Period up to 12 weeks is allowed in order to allow for 8 weeks of LTBI treatment prior to study drug initiation. If the initial 8 weeks of prophylactic LTBI therapy cannot be accomplished with the extended Screening Period, the subject should be considered screen failed.
evaluevalufied, sfied, s
ed. If aed. If drawadrawa
COPY positivpositi
nd nond no
This do
cumen
t can
not ssese
his/his/be
n to to ssss
used
lablabks duras durao coco
too i
aborborsu
pport
waswasas identiden
initianitia
anyd disd dis
as dis d
marketi
ngal prp
bjects ibjects
vide fuvide fcu
autho
rizati
onot treot treation bation
study udy activeactiv
procroc
appli
catio
n t resultresu
ve or ine or it
and ct ct c an
yiscuiscucannoann
exten
sions
ng the g the
ate onte ona TB a TB
s
or va
riatio
ns
be be
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 62 of 113
Rescreening may occur only after discussion with and approval by the study physician (or Medical Monitor)
Procedure to be followed for subjects with conversion of IGRA or diagnosis of a new TB infection during the Study:
Conversion is defined as a positive IGRA or 2 indeterminate results at the same visit (scheduled or unscheduled) of the same IGRA method.
The subject should immediately discontinue study drug intake upon positive testconversion. Should evaluation of the subject by the appropriate TB specialist result in a diagnosis of LTBI, the study drug must be withheld until the appropriate prophylactic therapy has been received for at least 4 weeks and the subject is deemed likely to continue therapy to completion. The treatment must be discussed with the study physician before study drug is restarted.
If the subject’s evaluation reveals active TB infection during the course of the study, the subject must be immediately discontinued from study medication and a WD Visit must be scheduled as soon as possible, but no later than the next scheduled visit. The subject should be encouraged to keep the FU Visit as specified by the protocol.
Refer to Section 10.4, for instructions on reporting a confirmed LTBI or confirmed active TB occurrence (note that in the event that an active TB occurrence meets the protocol-defined SAE criteria, it is also an SAE).
10.6.4.2 Chest x-ray Screening chest x-ray must have occurred within 3 months prior to Screening Visit and should be repeated only if the TB test was confirmed positive or any further evidence is suggestive of potential TB infection (eg, exposure).
Imaging must be read by an experienced TB imaging specialist, radiologist, pulmonologist or infectious disease specialist, who is specifically requested to look for signs of active TB or signs of past/inactive TB infection.
The official report of the imaging must specifically indicate the absence of findings suggestive of current active TB or prior inactive TB.
Additional chest x-ray or other imaging test should be performed when positive signs and symptoms indicate pulmonary infection, including potential TB infection, or when close exposure to persons with TB is documented.
10.6.4.3 Tuberculosis questionnaire The questionnaire “Evaluation of Signs and Symptoms of Tuberculosis” should be used as a source document. The questionnaire will be completed at Screening, Baseline, and every 12 weeks thereafter until completion at Week 96/WD Visit.
The questionnaire will assist with the identification of subjects who may require therapy for TB. A subject who answers “Yes” to the question
at Screening is excluded. A “Yes” response to any of the other questions within the questionnaire at Screening should trigger further careful assessment to determine if subject
REDACTED an
curredcurredB test B test ionio
COPYrtinrtian actin acti
n SAn SA
This do
cumen
tss cann
ot6.4.3.4.3The qThe qo
bere toto
33us
ed eses
indicandicato po p
to st xt xsu
pport
f the the imi
ent actent ac
any whw
nfectinfectmark
etingwaswa
n (eg, en (eg, e
periencperienwho iwho
autho
rizati
on iv
AE).AE).
d withwithas cas c
appli
catio
nt schschby theby the
a confa conve TBe TB
andation ion
chehe
any coursecoursex
tensio
nsphylahylkekely tly t
the stuhe stu
orult ult ac
varia
tions
inin
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 63 of 113
has latent or active TB (see Exclusion Criterion 24, Section 6.2). A “Yes” response to any of the questions during the study should trigger further assessments to determine whether the subject has either LTBI or active TB infection (Appendix 16.11).
Subjects with a LTBI must receive prophylactic therapy prior to continuing study medication.
Subjects with active TB infection must be withdrawn from the study and should be referred to a TB specialist for further assessments.
11 STUDY MANAGEMENT AND ADMINISTRATION 11.1 Adherence to protocol The Investigator should not deviate from the protocol. However, the Investigator should take any measure necessary in deviation from or not defined by the protocol to protect clinical study subjects from any immediate hazard to their health and safety. In this case, this action should be taken immediately, without prior notification of the regulatory authority, IRB/IEC, or Sponsor.
After implementation of such a measure, the Investigator must notify the Clinical Project Manager of the Sponsor within 24 hours and follow any local regulatory requirements.
11.2 Monitoring UCB (or designee) will monitor the study to meet the Sponsor’s monitoring Standard Operating Procedures (SOPs), ICH-GCP guideline, and applicable regulatory requirements, and to ensure that study initiation, conduct, and closure are adequate. Monitoring of the study may be delegated by UCB to a CRO or a contract monitor.
The Investigator and his/her staff will be expected to cooperate with UCB (or designee) and to be available during the monitoring visits to answer questions sufficiently and to provide any missing information. The Investigator(s)/institution(s) will permit direct access to source data/documents for study-related monitoring, audits, IRB/IEC review, and regulatory inspection(s).
The Investigator will allow UCB (or designee) to periodically review all CRFs and corresponding source documents (eg, hospital and laboratory records for each study participant). Monitoring visits will provide UCB (or designee) with the opportunity to evaluate the progress of the study, verify the accuracy and completeness of CRFs, ensure that all protocol requirements, applicable authorities regulations, and Investigator’s obligations are being fulfilled, and resolve any inconsistencies in the study records.
11.2.1 Definition of source data All source documents must be accurate, clear, unambiguous, permanent, and capable of being audited. They should be made using some permanent form of recording (ink, typing, printing, optical disc). They should not be obscured by correction fluid or have temporary attachments (such as removable self-stick notes). Photocopies of CRFs are not considered acceptable source documents.
Source documents are original records in which raw data are first recorded. These may include hospital/clinic/general practitioner records, charts, diaries, x-rays, laboratory results,
REDACTEDto mo m
P guidP guidonductnduct
RO or RO or
f wf w
COPY
meeme
This do
cumen
tAAcann
otbeinein
1.2.11.2.1AlAl
beocolcong fng
used
MoMohe proge prool reol r
toou
onitonitsu
pport
ill allowl allo
urce drce d
anyelateelatem
arketi
ng
will be eill be oring vring
InvestInvesedd
autho
rizati
on
et the et the delineeline
t, and , and a concon
appli
catio
n must notust nolocal rlocal
and ulatoatoan
yn thisn thitory ory
exten
sions
igator igatoro proto prot
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 64 of 113
printouts, pharmacy records, care records, ECG or other printouts, completed scales, or quality of life questionnaires, for example. Source documents should be kept in a secure, limited access area.
Source documents that are computer generated and stored electronically must be printed for review by the monitor. Once printed, these copies should be signed and dated by the Investigator and become a permanent part of the subject’s source documents. The Investigator will facilitate the process for enabling the monitor to compare the content of the printout and the data stored in the computer to ensure all data are consistent.
Electronic data records must be saved and stored as instructed by UCB (or its designee).
The following data will be recorded directly in the electronic patient reported outcome (ePRO) device (Section 11.3.2) on site and will not appear in a source document as defined above:
Patient PROs: BASDAI, BASFI, ASQoL, ASAS HI questionnaire, SF-36, total and nocturnal spinal pain, and PtGADA,
PhGADA and swollen and tender joint counts
11.2.2 Source data verification Source data verification ensures accuracy and credibility of the data obtained. During monitoring visits, reported data should be reviewed with regard to being accurate, complete, and verifiable from source documents (eg, subject files, recordings from automated instruments, tracings [ECG], x-ray films, laboratory notes). All data reported on the CRF should be supported by source documents, unless otherwise specified in Section 11.2.1.
11.3 Data handling 11.3.1 Case Report Form completion This study will use remote data capture (RDC); and the Investigator is responsible for prompt reporting of accurate, complete, and legible data in the eCRF and in all required reports.
This study will also use an electronic device (Site Tablet) to capture the ePRO questionnaires (see Section 11.3.2).
Serious adverse event reporting will be done using the SAE form (Section 10.2) while also entering the event in the appropriate eCRF section. The safety database and the clinical database will be reconciled during the study and discrepancies will be corrected as needed.
The Investigator should maintain a list of personnel authorized to enter data into the eCRF. Access to the RDC will be given after training has been received. A training certificate will be provided and filed.
Detailed instructions on the use of the RDC will be provided in the eCRF Completion Guidelines.
Corrections made after the Investigator’s review and approval (by means of a password/electronic signature) will be re-approved by the Investigator. Any change or correction to the eCRF after saving must be accompanied by a reason for the change.
REDACTED,, subsubms, labms, lab
mentsments
gg
COPYredibiredibvieweiewebjj
This do
cumen
t can
notInvenve
Accessccessbe pbe p
be e w
vesves
sede eveneven
will bwill
toe eveev supp
ortse an se an ).)
en
anyplete,pletemark
eting
orm corm c
ta capa ca
autho
rizati
oned wied wiect filect fil
boratooratos, unle, unle
appli
catio
n
lity ofity othh
and a
ny
e, SFe, SFex
tensio
ns gn
outcomutcomment ment
ornee)ee)va
riatio
ns
the the
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 65 of 113
11.3.2 Electronic reporting outcome Compared to the paper patient questionnaires, the new electronic options have several advantages combining handheld devices in conjunction with online technologies in order tosend subject self assessments directly to a central server. The collected data could then be reviewed in real time for monitoring of subject symptoms and compliance. The ePRO possibilities will be used in this study.
This study will use an electronic Site Tablet having a large screen and intuitive fingertip data entry to ensure all questionnaire data are captured appropriately, completely, and on time.
Access to the system by site personnel will be given after training has been received. A training certificate will be provided and filed. The Investigator should maintain a list of personnel authorized to enter data into the electronic ePRO device.
11.3.3 Database entry and reconciliation Case Report Forms/external electronic data will be entered/loaded into a validated electronic database using a clinical data management system (CDMS). Computerized data cleaning checks will be used in addition to manual review to check for discrepancies and to ensure consistency of the data. Case Report Form data are entered into the clinical database using independent, double-data entry, with the exception of comment fields, which are verified by a second person. In the event that the study is performed using RDC, the data are entered into the eCRFs once and are subsequently verified.
An electronic audit trail system will be maintained within the CDMS to track all data changes in the database once the data have been saved initially into the system or electronically loaded. Regular backups of the electronic data will be performed.
11.3.4 Subject Screening and Enrollment Log/Subject Identification Code list
The subject’s screening and enrollment will be recorded in the Subject Screening and Enrollment Log.
The Investigator will keep a Subject Identification Code list. This list remains with the Investigator and is used for unambiguous identification of each subject.
The subject’s consent and enrollment in the study must be recorded in the subject’s medical record. These data should identify the study and document the dates of the subject’s participation.
11.4 Termination of the study UCB reserves the right to temporarily suspend or prematurely discontinue this study either at a single site, multiple sites, or at all sites at any time for reasons including, but not limited to, safety or ethical issues, inaccurate or incomplete data recording, noncompliance, or unsatisfactory enrollment with respect to quality or quantity.
If the study is prematurely terminated or suspended, UCB (or its representative) will inform the Investigators/institutions and the regulatory authority(ies) of the termination or suspension and the reason(s) for the termination or suspension, in accordance with applicable regulatory requirement(s). The IRB/IEC should also be informed and provided with reason(s)
REDACTED maintamainta
have havekups okups o
ningning
COPYn o
erformerformd.d.
This do
cumen
taacann
ot 4UCB UCB a sa s
be atious
edse dae daon.on.
toonseons supp
ortkeep akeep used fused
en
any m
arketi
ng
g andg and
nrollmnroll
autho
rizati
on
ained ined been been
of the f the
appli
catio
n discdiscd intinto tho
ommenmmemed used u
and
intnmputempute
crcr
any
to a vo a vex
tensio
nsved. ed.
n a lista lisor AA
varia
tions
data data me. me.
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 66 of 113
for the termination or suspension by the Sponsor or by the Investigator/institution, as specified by the applicable regulatory requirement(s). In addition, arrangements will be made for the return of all unused IMP and other material in accordance with UCB procedures for the study.
Upon completion of the study, the monitor will conduct the following activities in conjunction with the Investigator, as appropriate:
Return of all study data to the Sponsor or its representative
Data clarification and/or resolution
Accountability, reconciliation, and arrangements for used and unused study drugs
Review of site study records for completeness
Discussion/reminder on archiving responsibilities
11.5 Archiving and data retention The Investigator will maintain adequate records for the study, including CRFs, medical records, laboratory results, Informed Consent documents, drug dispensing and disposition records, safety reports, information regarding participants who discontinued, and other pertinent data.
All essential documents should be retained by the Investigator until at least 2 years after the last approval of a marketing application in an ICH region and until there are no pending or contemplated marketing applications in an ICH region, or at least 2 years have elapsed since the formal discontinuation of clinical development of the IMP. These documents should be retained for a longer period, however, if required by the applicable regulatory requirement(s) or by an agreement with UCB (CPMP/ICH/135/95, 2002 [Section 4.9.5]). The Investigator will contact UCB for authorization prior to the destruction of any study records or in the event of accidental loss or destruction of any study records. The Investigator will also notify UCB should he/she relocate or move the study-related files to a location other than that specified in the Sponsor’s study master file.
11.6 Audit and inspection The Investigator will permit study-related audits mandated by UCB, after reasonable notice, and inspections by domestic or foreign regulatory authorities.
The main purposes of an audit or inspection are to confirm that the rights and well-being of the subjects enrolled have been protected, that enrolled subjects (ie, signing consent and undergoing study procedures) are appropriate for the study, and that all data relevant for the evaluation of the IMP have been processed and reported in compliance with the planned arrangements, the protocol, investigational site, and IRB/IEC SOPs, ICH GCP, and applicable regulatory requirements.
The Investigator will provide direct access to all study documents, source records, and source data. If an inspection by a regulatory authority is announced, the Investigator will immediately inform UCB (or designee).
REDACTEDn an n an n an ICn an IC
al deval devwever, wever, CPMCPM
COPY y the y theIC
This do
cumen
taacann
ot ubjubj
ndergodergoevalueval
bein pn pjectject
usedons bns b
purpur
tor wr wbb
supp
ortr’s ss s
dit anit anwillil
anye or me or
studstu
marketi
ng if rf r
MP/ICHMP/ICion prioon pr
tructiotruct
autho
rizati
onInveInveCH reCH r
ICH reCH revelopmelopmreqreq
appli
catio
n incrug disug di
s who s who
t
and
cludcludan
y exte
nsion
sdrugsdrugsor
varia
tions
there
of.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 67 of 113
11.7 Good Clinical Practice Noncompliance with the protocol, ICH-GCP, or local regulatory requirements by the Investigator, institution, institution staff, or designees of the Sponsor will lead to prompt action by UCB to secure compliance. Continued noncompliance may result in the termination of the site’s involvement in the study.
12 STATISTICS A summary of statistical methods follows; more details will be provided in the Statistical Analysis Plan (SAP).
12.1 Definition of analysis sets Two analysis sets are defined for this study:
The Enrolled Set (ES) will consist of all subjects who have given informed consent.
The Safety Set (SS) will consist of all subjects in the ES who have received at least 1 dose of study medication.
Efficacy variables will be analyzed using the SS. In the unexpected case that subjects in the SS have an unknown incidence of AU flares for the prestudy period (ie, missing Baseline value for the primary efficacy variable), then a Full Analysis Set (FAS) would additionally be defined for efficacy analysis as all subjects in the SS with nonmissing Baseline value for the primary efficacy variable (ie, AU flare incidence data from prestudy period). In this situation, efficacy analysis would be conducted using the FAS rather than the SS.
Safety variables will be summarized using the SS.
12.2 General statistical considerations In general, summary statistics (n [number of available measurements], arithmetic mean, SD, median, minimum, and maximum) for quantitative variable and frequency tables for qualitative data will be presented. As there is a single treatment arm in this study, all subjectswill be presented together in a single treatment group (eg, as “All CZP” or similar).
Unless otherwise specified, the last valid measurement before study medication administration will be utilized as Baseline value.
All statistical analyses besides the primary efficacy analysis will be exploratory in nature only. Any p-values or confidence intervals produced in association with secondary or other analyses will be considered non-confirmatory.
The full details of the statistical analyses will be provided in the SAP. Any deviations from the final SAP as well as changes from the protocol will be detailed in the SAP and discussed in the Clinical Study Report.
All the analyses will be performed by using SAS Version 9.4 or above. The details regarding the presentation of statistical outputs will be provided in the SAP.
REDACTEDinciincnductenducte
d usind usin
sticastica
COPYFull AFull As in ths in thcidecid
This do
cumen
tinincan
not he fulle ful
the fithe fi
bey
es wies w usedal anal an
pp--vavato il
allsu
pport
her ier i
pecifieecifiell be ul be
anysenteent
in ain
marketi
ngal coal con [numb[num
mum) fmum)d
autho
rizati
onhe SSe S
dence dence ted usied usi
ng the g the
appli
catio
n xpectexpecttudy peudy p
AnalysisnalysSS wS w
andhaveavean
y foofff
e rece re
exten
sions
ormrm
or va
riatio
ns
cal al
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 68 of 113
12.3 Planned efficacy analyses 12.3.1 Analysis of the primary efficacy variable The primary efficacy analysis will consist of a comparison of the frequency of AU flares during the prestudy/historical period with that observed while in the study during CZP treatment. It is assumed that the frequency of AU flares follows a Poisson distribution. As such, the analysis will be performed as a generalized estimating equations analysis for Poisson outcome that will take into account the possible within-subject correlation (between the retrospective and prospective AU flare counts). The model will contain 2 records per subject corresponding to the frequency of AU flares before study and during the study, offset by log-time of each of the reporting periods. Disease duration will be included as a factor in the model, initially defined as <2 years and ≥2 years; the specific cutoffs for analysis may deviate from this based on the distribution seen in the data. The p-value for effect of CZP treatment on frequency of AU flares, along with rate ratio (CZP/historical) and 95% confidence interval, will be obtained from this model.
The above analysis will be repeated in the subgroup of subjects with at least 1 documented AU flare within 12 months prior to Baseline.
The primary efficacy analysis hypothesis will be tested based on significance level of alpha 0.05 and will be 2 sided. The entire alpha will be used for the primary efficacy analysis and analysis of the other secondary efficacy parameters will be supportive. Therefore, no alpha adjustment will be performed.
12.3.2 Other efficacy analyses The AU flare rate (event rate) per 100 patient-years of exposure, both before and during the study, along with 95% confidence intervals (Garwood, 1936) will be presented as a key supportive analysis to the primary efficacy analysis results.
Other efficacy variables will be summarized by time point including change from Baseline or shift from Baseline, with descriptive statistics.
12.4 Planned safety analyses 12.4.1 Safety analyses The frequency of all AEs will be presented by System Organ Class, high level term, and preferred term. The data will be displayed as number of subjects experiencing the AEs, percentage of subjects, and number of AEs. Data will also be corrected for exposure by 100 patient years.
Laboratory evaluations and vital signs will be analyzed over time in the SS for observed cases and at the end of treatment. The details will be elaborated in the SAP.
12.5 Handling of protocol deviations Important protocol deviations are deviations from the protocol which could potentially have a meaningful impact on study conduct, on the safety for an individual subject, or on the primary efficacy outcome for an individual subject. The criteria for identifying important protocol deviations will be defined within the appropriate document. All the important
REDACTED ysesses
100 p100 pce intce int
COPYwill bwill paramearam
This do
cumen
t can
not patiat
Laboraaboracasecase
be ageg
tienttientus
edy oy o
erm. Trm. Te of e of
toof aof asu
pported sed s
fety aety all
anyescriescr
sa
marketi
ngtervatervay efficay effic
be sume sumipti
autho
rizati
oneterter
atienttientll
appli
catio
n based oased
e usede usedrs wrs w
and with awith a
any cal)cal ex
tensio
nsud
as a faas a faanalyanaly
r effeeffeal) a) a
orpp
dy, ody, va
riatio
ns
ween een er er
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 69 of 113
protocol violations will be tracked, although no analysis will be performed based on per protocol population.
12.6 Handling of dropouts or missing data Analysis based on observed case and last observation carried forward for continuous variables or nonresponder imputation for categorical variables will be presented.
12.7 Planned interim analysis and data monitoring Regular monitoring of safety data collected during clinical studies will be performed as described in the Safety Signal Detection in the Ongoing Clinical Trials Charter for CZP.
A specific data monitoring, steering, or evaluation committee is not planned for this study.
An interim analysis is scheduled after the last subject has completed the Week 48 assessment. Details of the interim analysis will be included within the final SAP (or an interim SAP, if a separate SAP is needed specific to details of the interim analysis).
Although there is no intention to stop the study early due to efficacy or futility on the basis of these interim results, α=0.0001 will be spent in conjunction with this formal interim review of the data (Haybittle, 1971), and the final analysis of the primary efficacy variable will be conducted at the reduced 2-sided α-level of 0.049.
12.8 Determination of sample size Based on results from a previous study with CZP in axSpA subjects (AS001), the AU flare rate while treated with CZP is estimated to be 14.6 events per 100 subject-years of exposure. Assuming the rate while treated with CZP represents a reduction in the flare rate of approximately 50%, the AU flare rate for the prestudy period prior to initiation of CZP is estimated to be 29.2 events per 100 subject-years of exposure. A conservative estimate of the sample size for this study is obtained via derivation of the per treatment-group sample size for Poisson regression based on the approach for a 2-group setting initially described by Signorini (1991) and implemented within SAS (Hu, 2008). Assuming an average follow-upperiod of 1.5 years with 2-sided α-level=0.049 for the final analysis of the primary efficacy variable, a sample size of N=86 subjects per treatment group will have 80% statistical power for a 2 group comparison, corresponding to a total sample size of N=86 subjects for this study with 1 treatment group. Due to the expected increase in statistical efficiency from the fact that each subject serves as his/her own control in the primary efficacy analysis, the actual power for this study is assumed to be >80 percent.
13 ETHICS AND REGULATORY REQUIREMENTS 13.1 Informed consent Subject’s informed consent must be obtained and documented in accordance with local regulations, ICH-GCP requirements, and the ethical principles that have their origin in the principles of the Declaration of Helsinki.
Prior to obtaining informed consent, information should be given in a language and at a level of complexity understandable to the subject in both oral and written form by the Investigator (or designee). Each subject will have the opportunity to discuss the study and its alternatives with the Investigator prior to participation in the study.
ith Cith Cd to bd to b
h CZPh CZPe rate fe rate 0000
COPY e sizee sizZZ
This do
cumen
tSScann
ot 13.13.1
be us
edh subh subthis this
toeatmatmbjbj
supp
ort 22
ze of Ne of parisonparisomenme
anymenmen
sidesidmark
eting
forforsubjecsubje
ained vned on the n thentet
autho
rizati
on e
ZP in aP in abe 14.6e 14.6
P reprrepror thr th
appli
catio
n with thith tprimaryrimar
andacy oacy o
thth
anyerim aerim ex
tensio
ns C
or this r this
Week 4eek 4nal Sal S
orCZPZPva
riatio
ns
s s
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 70 of 113
Prior to participation in the study, the written ICF should be signed and personally dated by the subject, or his/her legal representative, and by the person who conducted the informed consent discussion (Investigator or designee). The subject or his/her legal representative must receive a copy of the signed and dated ICF. As part of the consent process, each subject must consent to give direct access to his/her medical records for study-related monitoring, auditing, IRB/IEC review, and regulatory inspection.
If the ICF is amended during the study, the Investigator (or the Sponsor, if applicable) must follow all applicable regulatory requirements pertaining to the approval of the amended ICF by the IRB/IEC and use of the amended form.
The subject may withdraw his/her consent to participate in the study at any time. A subject is considered as enrolled in the study when he/she has signed the ICF. A CRF must not be started, nor may any study specific procedure be performed for a given subject, without having obtained his/her written consent to participate in the study.
13.2 Subject identification cards Upon signing the ICF, the subject or his/her legal representative will be provided with a subject identification card in the language of the subject. The Investigator will fill in the subject identifying information and medical emergency contact information. The Investigator will instruct the subject to keep the card with him/her at all times.
13.3 Institutional Review Boards and Independent Ethics Committees
The study will be conducted under the auspices of an IRB/IEC, as defined in local regulations, ICH-GCP, and in accordance with the ethical principles that have their origin in the Declaration of Helsinki.
The Investigator/UCB will ensure that an appropriately constituted IRB/IEC that complies with the requirements of the current ICH-GCP version or applicable country-specific regulations will be responsible for the initial and continuing review and approval of the clinical study. Prior to initiation of the study, the Investigator/UCB will forward copies of the protocol, Informed Consent form, Investigator’s Brochure, Investigator’s curriculum vitae (if applicable), advertisement (if applicable), and all other subject-related documents to be used for the study to the IRB/IEC for its review and approval.
Before initiating a study, the Investigator will have written and dated full approval from the responsible IRB/IEC for the protocol. Approval from the authorities should also be obtained before study initiation.
The Investigator will also promptly report to the IRB/IEC all changes in the study, all unanticipated problems involving risks to human subjects or others, and any protocol deviations, to eliminate immediate hazards to subjects.
The Investigator will not make any changes in the study or study conduct without IRB/IEC approval, except where necessary to eliminate apparent immediate hazards to the subjects. For minor changes to a previously approved protocol during the period covered by the original approval, it may be possible for the Investigator to obtain an expedited review by the IRB/IEC as allowed.
REDACTED
he auspe ausordanordan
COPYm/herm/he
ardsards
This do
cumen
tucann
otre ste s
The Inhe Inunauna
besibleblestudstu
used
tiatingiatinge IRe I
tothe the supp
ortinitianiti
Consenonsentisemeiseme
IRI
anyible fible
iatioiat
marketi
ng
re that e thaurrenturren
f
autho
rizati
ons ans an
spices pices ce wie wi
appli
catio
n ve we whe Invee Inv
contactontacat all tat all
d
and
willwil
any e
xtens
ionse. A suA su
must nmust nbject, bject,
or va
riatio
ns
ust ust d ICF ICF
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 71 of 113
As part of the IRB/IEC requirements for continuing review of approved studies, the Investigator will be responsible for submitting periodic progress reports to the IRB/IEC(based on IRB/IEC requirements), at intervals appropriate to the degree of subject risk involved, but no less than once per year. The Investigator should provide a final report to the IRB/IEC following study completion.
UCB (or its representative) will communicate safety information to the appropriate regulatory authorities and all active Investigators in accordance with applicable regulatory requirements. The appropriate IRB/IEC will also be informed by the Investigator or the Sponsor, as specified by the applicable regulatory requirements in each concerned country. Where applicable, Investigators are to provide the Sponsor (or its representative) with evidence of such IRB/IEC notification.
13.4 Subject privacy UCB staff (or designee) will affirm and uphold the subject’s confidentiality. Throughout this study, all data forwarded to UCB (or designee) will be identified only by the subject number assigned at Screening.
The Investigator agrees that representatives of UCB, its designee, representatives of the relevant IRB/IEC, or representatives of regulatory authorities will be allowed to review that portion of the subject’s primary medical records that directly concerns this study (including, but not limited to, laboratory test result reports, ECG reports, admission/discharge summaries for hospital admissions occurring during a subject’s study participation, and autopsy reports for deaths occurring during the study).
13.5 Protocol amendments Protocol changes may affect the legal and ethical status of the study and may also affect the statistical evaluations of sample size and the likelihood of the study fulfilling its primary objective.
Significant changes to the protocol will only be made as an amendment to the protocol and must be approved by UCB, the IRB/IEC, and the regulatory authorities (if required), prior to being implemented.
14 FINANCE, INSURANCE, AND PUBLICATION Insurance coverage will be handled according to local requirements.
Finance, insurance, and publication rights will be addressed in the Investigator and/or CRO agreements, as applicable.
15 REFERENCES Barkham N, Keen HI, Coates LC, O'Connor P, Hensor E, Fraser AD, et al. Clinical and imaging efficacy of infliximab in HLA-B27-positive patients with magnetic resonance imaging-determined early sacroiliitis. Arthritis Rheum. 2009;60(4):946-54.
Boonen A, van der Heijde D, Landewé R, Guillemin F, Rutten-van Molken M, Dougados M, et al. Direct costs of ankylosing spondylitis and its determinants: an analysis among three European countries. Ann Rheum Dis. 2003;62:732-40.
REDACTEDsubsub
menmenlegalega
COPYthat thatts, ECts, ECbjebje
This do
cumen
tBBcann
otememe
155be
e, ininmenten
usedoveravera
nsunsu
to FINsu
pport
CB,CB
NANNAN
anyprotoproto
B, thth
marketi
ngal andl andize andize an
oc
autho
rizati
onCG rG r
ect’s sct’s s
tss
appli
catio
n ignee, gneerities wities
directlyirectlreporepo
and nly ly an
ytialityiality by y by
exten
sionswithwith
t
orountounhh
varia
tions
yy
entryntr
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 72 of 113
Boonen A, van der Heijde D, Landewé R, Spoorenerg A, Schouten H, Rutten-van Molken M, et al. Work status and productivity costs due to ankylosing spondylitis: comparison of three European countries. Ann Rheum Dis. 2002;61:429-37.
Brandt J, Listing J, Sieper J, Rudwaleit M, van der Heijde D, Braun J. Development and preselection of criteria for short term improvement after anti-TNFα treatment in ankylosing spondylitis. Ann Rheum Dis. 2004;63:1438-44.
Braun J. Axial spondyloarthritis: thoughts about nomenclature and treatment targets. Clin Exp Rheumatol. 2012;30(4 Suppl 73):S132-5.
Braun J, van den Berg R, Baraliakos X, Boehm H, Burgos-Vargas R, Collantes-Estevez E, et al. 2010 update of the ASAS/EULAR recommendations for the management of ankylosing spondylitis. Ann Rheum Dis. 2011;70: 896-904.
Braun J, Sieper J. Ankylosing spondylitis. Lancet. 2007;369:1379-90.
Braun J, Zochling J, Baraliakos X, Alten R, Burmester G, Grasedyck K, et al. Efficacy of sulfasalazine in patients with inflammatory back pain due to undifferentiated spondyloarthritis and early ankylosing spondylitis: a multicentre randomised controlled trial. Ann Rheum Dis. 2006;65: 1147-1153.
Calin A, Garrett S, Whitelock H, Kennedy LG, O'Hea J, Mallorie P, et al. A new approach to defining functional ability in ankylosing spondylitis: the development of the Bath Ankylosing Spondylitis Functional Index. J Rheumatol. 1994;21(12):2281-5.
Callhoff J, Sieper J, Weiß A, Zink A, Listing J. Efficacy of TNFα blockers in patients with ankylosing spondylitis and non-radiographic axial spondyloarthritis: a meta-analysis. Ann Rheum Dis. 2015;74(6):1241-8.
Cantini F, Niccoli L, Cassara E, Kaloudi O, Nannini C: Duration of remission after halving of the etanercept dose in patients with ankylosing spondylitis: a randomized, prospective, long-term, follow-up study, Biologics. 2013:7 1-6.
CPMP/EWP/556/95 Rev. 2. Clinical investigation of medicinal products other than non-steroidal anti-inflammatory drugs (NSAIDs) for treatment of rheumatoid arthritis. Released for second consultation Jun 2015.
CPMP/ICH/135/95 Note for guidance on Good Clinical Practice (EMEA) Jul 2002.
Cuirea A, Scherer A, Exer P, Bernhard J, Dudler J, Beyeler B, et al. Tumor necrosis factor α inhibition in radiographic and nonradiographic axial spondyloarthritis: results from a large observational cohort. Arthritis Rheum. 2013;65:3096-106.
Davis Jr JC, Revicki D, van der Heijde DMF, Rentz AM, Wong RL, Kupper H, et al. Health-related quality of life outcomes in patients with active ankylosing spondylitis treated with adalimumab: results from a randomized controlled study. Arthritis Rheum. 2007;57(6):1050-7.
Dougados M, van der Heijde D, Sieper J, Braun J, Maksymowych W, Citera G, et al. Clinical and imaging efficacy of etanercept in early non-radiographic axial spondyloarthritis: a 12-week, randomised, double-blind, placebo-conrolled trial. OP0108, Anals Rheum Dis. 2013;72, Suppl 3:87.
REDACTED J RJ R
ListingListingographograp
KK
COPYO'HeO'Hedylitisdyliti
RheuRhe
This do
cumen
trr cann
otervatrvat
DavisDavisrelrel
beon ion iatioatio
used
35
ScherScherinin
to5/955/95supp
ortv. .
matoryatoryation Jion
N
any
2. Cl2. Clmark
eting
Kaloudi aloud
ts withs withiologolog
autho
rizati
on s: th: t
umatomato
g J. EfJ. Efphic axhic ax
appli
catio
nntre rtre r
J, MalJ, Mathe dhe d
and
ck kifferefferera
any
K, eK, eex
tensio
nsstevstev
of ankyf ankor vezve
varia
tions
n
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 73 of 113
Dougados M, van der Linden S, Juhlin R, Huitfeldt B, Amor B, Calin A, et al. The European Spondylarthropathy Study Group preliminary criteria for the classification of spondylarthropathy. Arthritis Rheum. 1991;34:1218-27.
Doward LC, Spoorenberg A, Cook SA, Whalley D, Helliwell PS, Kay LJ, et al. Development of the ASQoL: a quality of life instrument specific to ankylosing spondylitis. Ann Rheum Dis. 2003;62(1):20-6.
Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain. 2008;9:105-21.
Feldtkeller E, Khan MA, van der Heijde D, van der Linden S, Braun J. Age at disease onset and diagnosis delay in HLA-B27 negative vs. positive patients with ankylosing spondylitis. Rheumatol Int. 2003;23:61-6.
Feldtkeller E, Bruckel J, Khan MA. Scientific contributions of ankylosing spondylitis patient advocacy groups. Curr Opin Rheumatol. 2000;12:239-247.
Garrett S, Jenkinson T, Kennedy LG, Whitelock H, Gaisford P, Calin A. A new approach to defining disease status in ankylosing spondylitis: the Bath Ankylosing Spondylitis Disease Activity Index. J Rheumatol. 1994;21(12):2286-91.
Garwood F. Fiducial limits for the Poisson distribution. Biometrika. 1936;28:437-42.
Haibel H, Brandt HC, Song IH, Brandt A, Listing J, Rudwaleit M, et al. No efficacy of subcutaneous methotrexate in active ankylosing spondylitis: a 16-week open-label trial. Ann Rheum Dis. 2007;66:419-21
Haibel H, Rudwaleit M, Braun J, Sieper J. Six months open label trial of leflunomide in active ankylosing spondylitis. Ann Rheum Dis. 2005;64:124-6.
Haibel H, Rudwaleit M, Listing J, Heldmann F, Wong RL, Kupper H, et al. Efficacy of adalimumab in the treatment of axial spondylarthritis without radiographically defined sacroiliitis: results of a twelve-week randomized, double-blind, placebo-controlled trial followed by an open-label extension up to week fifty-two. Arthritis Rheum. 2008;58(7):1981-91.
Haibel H, Heldmann F, Braun J, Listing J, Kupper H, Sieper J. Long-term efficacy of adalimumab after drug withdrawal and retreatment in patients with active non-radiographically evident axial pondyloarthritis who experience a flare. Arthritis Rheum. 2013;65(8):2211-3.
Haybittle JL. Repeated assessments of results in clinical trials of cancer treatment. Brit. J. Radiol. 1971. 44 (526):793-7.
Hu K. Sample Size Estimation for Trials of Recurrent Events. Amgen Inc. 2008. 1-8.
Huscher D, Merkesdal S, Thiele K, Zeidler H, Schneider M, Zink A. Cost of illness in rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis and systemic lupus erythematosus in Germany. Ann Rheum Dis. 2006;65:1175-1183.
REDACTED List Lisnkylosnkylos
, Siep, Siep
COPY ..
tributribu
tinti
This do
cumen
t can
not 656
HaybittaybittRadRad
be php
5(8)5(8)se
db afteb aftehicalhica
tomanmansup
port
wwlabel eabel
1..
nn
anyt of t o
elveelvemark
eting
per Jer Jn RheumRheu
ng J, Hg J, Hf axf a
autho
rizati
on tionon
ng J, Rng J, Rsing sping sp
JJ
appli
catio
nP, CaP, CAnkyAnkyl
on. Bn. B
and
Cal
anyosing osingex
tensio
ns
diseasdiseassing sping s
or va
riatio
ns
ting ing T T
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 74 of 113
Klitz U, van der Heijde D, Boonen A, Cieza A, Stucki G, Khan MA, et al. Development of a health index in patients with ankylosing spondylitis (ASAS HI): final result of a global initiative based on the ICF guided by ASAS. Ann Rheum Dis. 2015;830-5
Kobelt G, Andlin-Sobocki P, Brophy S, Jonsson L, Calin A, Braun J. The burden of ankylosing spondylitis and the cost-effectiveness of treatment with infliximab (Remicade). Rheumatology. 2004;43:1158-66.
Landewé R, Braun J, Deodhar A, Dougados M, Maksymowch P, Mease P, et al. Efficacy of certolizumab pegol on signs and symptoms of axial spondyloarthritis including ankylosing spondylitis: 24-week results of a double-blind randomised placebo-controlled Phase 3 study. Ann Rheum Dis. 2014; 73: 39-47.
Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604-12.
Lukas C, Landewé R, Sieper J, Dougados M, Davis J, Braun J, et al. Development of an ASAS-endorsed disease activity score (ASDAS) in patients with ankylosing spondylitis. Ann Rheum Dis. 2009;68(1):18-24.
Meikle AW and Tyler FH. Potency and duration of action of glucocorticoids. Effects of hydrocortisone prednisone and dexamethasone on human pituitary-adrenal function. Am J Med 1977;63;200-7
Mielants H, Veys EM, Cuvelier C, De Vos M, Goemaere S, De Clercq L, et al. The evolution of spondyloarthropathies in relation to gut histology. II. Histological aspects. J Rheumatol. 1995;22(12):2273-8.
Monnet D, Breban M, Hudry C, Dougados M, Brézin AP. Ophthalmic findings and frequency of extraocular manifestations in patients with HLA-B27 uveitis: a study of 175 cases. Ophthalmology. 2004;111:802-9.
Myasoedova E, Crowson CS, Kremers HM, Therneau TM, Gabriel SE. Is the incidence of rheumatoid arthritis rising?: results from Olmsted County, Minnesota, 1955-2007. Arthritis Rheum. 2010;62:1576-82.
Olivieri I, D’Angelo S, Padula A, Leccese P, Nigro P, Palazzi C. Can we reduce the dosage of biologics in spondyloarthritis?, Autoimmun Rev. 2013; 12: 691-3.
Pavy S, Brophy S, Calin A. Establishment of the minimum clinically important difference for the bath ankylosing spondylitis indices: a prospective study. J Rheumatol. 2005;32(1):80-5.
Poddubnyy D. Axial spondyloarthritis: is there a treatment of choice? Ther Adv Musculoskelet Dis. 2013;5:45-54
Poddubnyy D, Rudwaleit M, Haibel H, Listing J, Märker-Hermann E, Zeidler H, et al. Effect of non-steroidal anti-inflammatory drugs on radiographic spinal progression in patients with axial spondyloarthritis: results from the German Spondyloarthritis Inception Cohort. Ann Rheum Dis. 2012;71:1616-22.
Rosenbaum JT, Suhler EB, Lin P. The future of uveitis treatment. Opthamolology. 2014;121(1):365-76
REDACTED MMut histut his
DougaDougastatistati
COPY huh
M, GM, G
This do
cumen
tPPcann
otdubnubnMusculuscu
PoPo
be an
nyny
sedophy phy
nkylnkyl
tosponspo su
pport
82.2.
lo S, Po S, Pondynd
any
,?: res?: res
marketi
ng adoad
tions inons in:802802-9
KremKrem
autho
rizati
on
Goemaoemastologyolog
dos Mos M
appli
catio
n of glucf glu
an pitun pitu
andankynkyan
yDevelDeveylol
exten
sions
se
I, et a, et a0:604:604
or3 st3 sva
riatio
ns
y of of sing sing
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 75 of 113
Rosenbaum JT. Characterization of uveitis associated with spondyloarthritis. J Rheumatol. 1989;16:792-6.
Rostom S, Dougados M, Gossec L. New tools for diagnosing spondyloarthropathy. Joint Bone Spine. 2010;77:108-14.
Rudwaleit M, Sieper J. Referral strategies for early diagnosis of axial spondyloarthritis. Nat Rev Rheumatol. 2012;8:262-8.
Rudwaleit M, van der Heijde D, Landewé R, Akkoc N, Brandt J, Chou CT, et al. The Assessment of SpondyloArthritis International Society classification criteria for peripheral spondyloarthritis and for spondyloarthritis in general. Ann Rheum Dis. 2011;70:25-31.
Rudwaleit M. New approaches to diagnosis and classification of axial and peripheral spondyloarthritis. Curr Opin Rheumatol. 2010;22:375-80.
Rudwaleit M, Haibel H, Baraliakos X, Listing J, Märker-Hermann E, Zeidler H, et al. The early disease stage in axial spondylarthritis: results from the German Spondyloarthritis Inception Cohort. Arthritis Rheum. 2009a;60:717-27.
Rudwaleit M, Landewe R, van der Heijde D, Listing J, Brandt J, Braun J, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part I): classification of paper patients by expert opinion including uncertainty appraisal. Ann Rheum Dis. 2009b;68:770-6.
Rudwaleit M, Landewé R, van der Heijde D, Listing K, Akkoc N, Brandt J, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part II): validation and final selection. Ann Rheum Dis. 2009c;68:777-83.
Rudwaleit M, Khan MA, Sieper J. The challenge of diagnosis and classification in early ankylosing spondylitis: do we need new criteria? Arthritis Rheum. 2005;52:1000-8.
Sieper J, van der Heijde D. Review: Nonradiographic axial spondyloarthritis: new definition of an old disease? Arthritis Rheum. 2013;65:543-51.
Sieper J, Rudwaleit M, Baraliakos X, Brandt J, Braun J, Burgos-Vargas R, et al. The Assessment of SpondyloArthritis International Society (ASAS) handbook: a guide to assess spondyloarthritis. Ann Rheum Dis. 2009;68(Suppl 2):ii1-ii44.
Signorini DF. Sample size for Poisson regression. Biometrika. 1991. 78:446-50.
van der Heijde D, Dougados M, Davis J, Weisman MH, Maksymowych W, Braun J, et al. Assessment in Ankylosing Spondylitis International Working Group/Spondylitis Association of America recommendations for conducting clinical trials in ankylosing spondylitis. Arthritis Rheum. 2005;52(2):386-94.
van der Heijde D, Schiff MH, Sieper J, Kivitz A, Wong RL, Kupper H, et al. Adalimumab effectiveness for the treatment of ankylosing spondylitis is maintained for up to 2 years: long-term results from the ATLAS trial. Ann Rheum Dis. 2009;68:922-9.
van der Linden S, Valkenburg HA, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria. Arthritis Rheum. 1984;27:361-8.
REDACTEDD, LD, LyloArthloArth
validavalida
COPYof paof p;68:77;68:7
LiLi
This do
cumen
tAAcann
otsessmessmof Amof AmArtAr
ber Heir Heus
ed t
DF. SDF. Stop
is. Ais. Asupp
ort titi
M, BarM, BaondyloondyAn
anyRevRev
s Rhes Rhemark
eting
The chThe c
need neeed n
viewvie
autho
rizati
on 700
isting sting hritis hritis
ation aion a
appli
catio
nndt J, Bdt J, Btional Stional
per per pat6.6.
and
an n anyZeidZeid
SponSponex
tensio
ns 25--33
ripheripher
dledle
orphephe3131
varia
tions
heraera
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 76 of 113
van Denderen JC, Visman IM, Nurmohamed MT, Suttorp-Schulten MS, van der Horst-Bruinsma IE. Adalimumab significantly reduces the recurrence rate of anterior uveitis in patients with Ankylosing Spondylitis. J Rheumatol. 2014 Sep;41(9):1843-8.
van Praet L, van den Bosch FE, Jacques P, Carron P, Jans L, Colman R, et al. Microscopicgut inflammation in axial spondyloarthritis: a multiparametric predictive model. Ann Rheum Dis. 2013;72(3):414-7.
van Tubergen A, Black PM, Coteur G. Are patient-reported outcome instruments for ankylosing spondylitis fit for purpose for the axial spondyloarthritis patient? A Qualitative and Psychometric Analysis. Rheumatology. 2015; 54(10):1842-51.
van Tubergen A, Debats I, Ryser L, Londoño J, Burgos-Vargas R, Cardiel MH, et al. Use of a numerical rating scale as an answer modality in ankylosing spondylitis-specific questionnaires. Arthritis Rheum. 2002;47(3):242-8.
Wakefield D, Chang JH. Epidemiology of uveitis. Int Ophthalmol Clin. 2005;45(2):1-13.
Ware JE, Kosinski M, Keller SD. SF-36 Physical and Mental Health Summary scales: a user’s manual. 1994. Maruish, M. E. (Ed.) 2011.
Zeboulon N, Dougados M, Gossec L. Prevalence and characteristics of uveitis in the spondyloarthropathies: a systematic literature review. Ann Rheum Dis. 2008;67:955-9.
REDACTED COPYviewview
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on ap
plica
tion
aracteriaracterAnn RAnn R
andealth Salth S
anyin. 20in. 20ex
tensio
ns
H, et al, et aecificcific
or va
riatio
ns
ative tive
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 77 of 113
16 APPENDICES 16.1 Modified NY criteria for ankylosing spondylitis Subjects meeting the NY criteria in the context of this protocol are defined as subjects meeting the definite AS diagnosis according to the modified NY criteria below.
Modified NY criteria for ankylosing spondylitis
Diagnosis
1) Clinical criteria
a) Low back pain and stiffness for more than 3 months which improves with exercise, but is not relieved by rest.
b) Limitation of motion of the lumbar spine in both the sagittal and frontal planes.
c) Limitation of chest expansion relative to normal values corrected for age and sex.
2) Radiologic criterion
Sacroiliitis grade ≥2 bilaterally or sacroiliitis grade 3 to 4 unilaterally.
Grading
1) Definite ankylosing spondylitis if the radiologic criterion is associated with at least 1 clinical criterionNote: A second grading of “probably ankylosing spondylitis” is part of the modified NY criteria, but it is not applicable for this study. It is included here for completeness. The grading will be probable ankylosing spondylitis if three clinical criteria are present and the radiologic criterion is present without any signs or symptoms satisfying the clinical criteria (other causes of sacroiliitis should be considered).
REDACTEDing sping sphere fohere fo
resent esentria (othria (ot
DDCOPY gic crigic crYC
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
oondylitondylit
or compcompand thend th
er cauer cau
tion
tioap
plica
tionaterallyaterall
tio
terion erionpl
and
anan
yage anage aa
exten
sionsse, bue, bu
planes.lanes.ene
or ova
riatio
ns
tiotiotiotio
va
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 78 of 113
16.2 ASAS classification criteria for axSpA
ASAS classification criteria for axSpA(for subjects with chronic back pain ≥3 months and age at onset <45 years)
Imaging criteria ASAS clinical criteria for axSpA
Sacroiliitis (MRI or radiographsa) plus ≥1 SpA feature
HLA-B27 plus ≥2 other SpA features
SpA featuresb
Inflammatory back painc
Arthritis Enthesitis (heel) Uveitis Dactylitis
PsoriasisCrohn’s disease/ulcerative colitisGood response to NSAIDsFamily history for SpAHLA-B27Elevated CRP
ASAS=Assessment of SpondyloArthritis international Society; axSpA=axial spondyloarthritis; CRP=C-reactive protein; MRI=magnetic resonance imaging; HLA-B27=human leukocyte antigen B27; NSAID=nonsteroidal anti-inflammatory drug; SpA=spondyloarthritis
a Active inflammatory lesions of sacroiliac joints with definite bone marrow oedema/osteitis suggestive of sacroiliitis associated with spondyloarthritis in MRI or radiographic sacroiliitis Grade 2 to 4 bilaterally or grade 3 to 4 unilaterally according to modified NY criteria.
b Family history for SpA and Good response to NSAIDs are excluded as SpA feature criteria. c Inflammatory back pain according to ASAS criteria for axSpA defined as the presence of 4 out
of 5 of the following parameters:1) Age at onset <40 years2) Insidious onset3) Improvement with exercise4) No improvement with rest5) Pain at night (with improvement upon getting up)
REDACTEDilateilateponse tponse t
to ASto ASs:s:
COPY pps with s with
ndyloandyloaera
This do
cumen
t can
not b
e use
d to s
uppo
rtth imph impo
any h resth restmark
eting
sese
autho
rizati
onarthriarthrally acly ac
to NSAo NSASAS crAS c
appli
catio
nxSpASpAHLALA--B2B
pondyloondyldefiniteefinit
itisi
anAAand
anan
y exte
nsion
s or r va
riatio
ns
ioria
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 79 of 113
16.3 Corticosteroid equivalent doses Table 16‒1: Corticosteroid equivalent doses (with reference to prednisone
10mg dose) (Meikle and Tyler, 1977)
Prednisone (reference) 10mg
Cortisone 50mg
Hydrocortisone 40mg
Prednisolone 10mg
Triamcinolone 8mg
Methylprednisolone 8mg
Betamethasone 1.5mg
Dexamethasone 1.5mg
REDACTED COPY
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on ap
plica
tion a
nd d an
y ya
exten
sions
ion
tene
or ova
riatio
ns
iotioioioar
thereo
f.

The following information (eg, ) is copyrighted and UCB does not have permission todisclose the contents.
REDACTED COPY
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on ap
plica
tion a
nd an
y exte
nsion
s or v
ariati
ons t
hereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 81 of 113
16.5 PtGADA (NRS) NRS patient global disease activity
How active was your spondyloarthritis on average during the last week?
Please tick the box that represents your answer ( ie, 10 )
Not active very active
0 1 2 3 4 5 6 7 8 9 10
REDACTED COPY
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on ap
plica
tion a
nd
ndan
y exte
nsion
s
ensio
ns
ensio
ns9
or va
riatio
ns th
ereof.
th

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 82 of 113
16.6 Total spinal pain NRS and nocturnal spinal pain NRS NRS painnpPlease tick the box that represents your answer ( i.e. )
1. Total SpineePainnpHow much pain of your spine due to spondyloarthritis do you have?o
no painn most severe painn
2. Nocturnal SpineeePainnpHow much pain of your spine due to spondyloarthritis do you have at night?
no painn most severe painn
100
1 2 3 4 6 7 8 9 10050
1 2 3 4 6 7 8 9 10050
REDACTED COPY
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on ap
plica
tion a
nd an
y an
y exten
sions
exte
exte
eext
ex99xtext
nenxte
or va
riatio
ns
atatva
re painpainar
thereo
f.

The following information (eg, ) is copyrighted and UCB does not have permission todisclose the contents.
REDACTED COPY
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on ap
plica
tion a
nd an
y exte
nsion
s or v
ariati
ons t
hereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 87 of 113
16.9 ASAS Health Index Questionnaire
ASAS Health Index Name: Date:
Please answer all statements by placing one check mark per statement to indicate
which response best applies to you at this moment in time taking into account your
rheumatic disease (the term “rheumatic disease” contains all forms of spondyloarthritis
including ankylosing spondylitis) .
1. Pain sometimes disrupts my normal activities.
□ I agree
□ I do not agree
2. I find it hard to stand for long.
□ I agree
□ I do not agree 3. I have problems running.
□ I agree
□ I do not agree 4. I have problems using toilet facilities.
□ I agree
□ I do not agree 5. I am often exhausted.
□ I agree
□ I do not agree 6. I am less motivated to do anything that requires physical effort.
□ I agree
□ I do not agree 7. I have lost interest in sex.
□ I agree
REDACTED COPY
This do
cumen
t can
not
6.6.
be us
ed ftenen
□□to d
nn eesu
pport
e
doo nnoo
any nngg ttoomark
eting
e
autho
rizati
on ap
plica
tion a
nd an
y exte
nsion
s nt t yyoo
ondndyylloo
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 88 of 113
□ I do not agree
□ Not applicable, I do not want to answer 8. I have difficulty operating the pedals in my car.
□ I agree
□ I do not agree
□ Not applicable, I cannot / do not drive 9. I am finding it hard to make contact with people.
□ I agree
□ I do not agree 10. I am not able to walk outdoors on flat ground.
□ I agree
□ I do not agree 11. I find it hard to
concentrate.
□ I agree
□ I do not agree 12. I am restricted in traveling because of my mobility.
□ I agree
□ I do not agree 13. I often get frustrated.
□ I agree
□ I do not agree 14. I find it difficult to wash my hair.
□ I agree
□ I do not agree 15. I have experienced financial changes because of my rheumatic disease.
□ I agree
□ I do not agree 16. I sleep badly at night.
□ I agree
□ I do not agree
REDACTED
becaubecau
COPY
This do
cumen
t can
not
15.15.
be us
ed ddiif
□□to iffffiicc
supp
ort e
doo nnoot
any .mark
eting
u
e
autho
rizati
on
see oof
appli
catio
n and
any e
xtens
ions o
r vari
ation
s the
reof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 89 of 113
ASAS Health Index
17. I cannot overcome my difficulties.
□ I agree
□ I do not agree.
Thank you for answering this questionnaire
REDACTED COPY
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on ap
plica
tion a
nd an
y exte
nsion
s or v
ariati
ons t
hereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 90 of 113
Environmental factors relatedto ASAS Health Index
Date: Name:
Please answer all statements by placing one check mark per statement to indicate which response
best applies to you at this moment in time taking into account your rheumatic disease (the term
“rheumatic disease” contains all forms of spondyloarthritis including ankylosing spondylitis).
As a result of my rheumatic disease, my family / relatives take more responsibility for household tasks.
□ I agree
□ I do not agree I don’t like the way my friends act around me.
□ I agree
□ I do not agree I cannot count on my relatives to help me with my problems.
□ I agree
□ I do not agree I modify my home and work environments.
□ I agree
□ I do not agree I have difficulties getting worsening of my disease acknowledged by a health care professional.
□ I agree
□ I do not agree Treatment of my rheumatic disease is taking up time.
□ I agree
□ I do not agree
REDACTED
eenntts.s.
COPY pprrobob
This do
cumen
t can
not □□ be
I aa used
heueumm
aaggrr
to aagg
mmsu
pport
ggrreee
any niingng oomark
eting
autho
rizati
onemmssapp
licati
on
ss
and a
ny ex
tensio
ns yyl
ittyy ffoo
or ttiiss))va
riatio
ns
ponseonse
hee tteerr
thereo
f.
eth

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 91 of 113
My friends expect too much of me.
□ I agree
□ I do not agree No one pays much attention to me at home.
□ I agree
□ I do not agree My friends understand me.
□ I agree
□ I do not agree Thank you for answering this questionnaire Developed by Assessment of SpondyloArthritis International Society (ASAS)
REDACTED COPY
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on ap
plica
tion a
nd an
y exte
nsion
s or v
ariati
ons t
hereo
f.

The following information (eg, ) is copyrighted and UCB does not have permission todisclose the contents.
REDACTED COPY
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on ap
plica
tion a
nd an
y exte
nsion
s or v
ariati
ons t
hereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 99 of 113
17 APPENDICES 17.1 Protocol Amendment 1 Rationale for the amendmentThe purpose of this non-substantial amendment is to clarify the Screening requirement for MRI and history of tobacco use, and to incorporate the administration of IMP at Week 32.
Modifications and changes Global changesMagnetic resonance imaging of the spine at Screening is not required for this study. An MRI of the SIJs will be performed during the Screening Period, or an MRI of the SIJs performed ≤3 months prior to the Baseline Visit may be used as the Baseline.
The IMP will be administered onsite at Visit 7 (Week 32).
Specific changes
Change #1Table 5-1: Schedule of assessments
Row 4
Medical history (including axSpA history) and concomitant diseases
Has been changed to:Medical history (including axSpA history, and tobacco use) and concomitant diseases
Change #2 Table 5-1: Schedule of assessments
Row 29
“Study drug sc injections” has been checked for Visit 7 (Week 32).
Change #3 Table 5-1: Schedule of assessments
Footnote k
Magnetic resonance imaging of the spine and SIJs to be performed during the Screening Period. An MRI performed ≤3 months prior to the Baseline Visit may be used as the Baseline.
Has been changed to: Magnetic resonance imaging of the SIJs to be performed during the Screening Period. An MRI performed ≤3 months prior to the Baseline Visit may be used as the Baseline.
REDACTED
istory)istory
COPY
This do
cumen
t oooo
MaMaca
nnot 55--1:1:
otnotno
be #3#3us
edinjecnjecto
supp
ort
assesassean
y mark
eting
y) anan
A histohisto
autho
rizati
on
ndnd
appli
catio
n and
any IJsIJ ex
tensio
ns
studystudyJs pes p
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 100 of 113
Change #4 Section 8.1 Visit 1 (Week -5 to -1) Screening
Bullet point 5
Significant past medical and procedure history and concomitant disease (including axSpA history)
Has been changed to: Significant past medical and procedure history (including tobacco use) and concomitant
disease (including axSpA history)
Change #5 Section 8.1 Visit 1 (Week -5 to -1) Screening
Bullet point 17
MRI evaluation of the spine and SIJ. An MRI performed ≤3 months prior to the Baseline Visit may be used for assessing the eligibility at Baseline.
Has been changed to: MRI evaluation of the SIJ. An MRI performed ≤3 months prior to the Baseline Visit may be
used for assessing the eligibility at Baseline.
Change #6 Section 8.5 Visit 7 (Week 32)
Bullet point 9 has been added:
Study drug administration onsite
Change #7 Section 10.6.3 MRI assessmentsMagnetic resonance imaging of the spine and SIJs will be performed at the Screening Visit only. An MRI performed ≤3 months prior to the Baseline Visit may be used as the Baseline.
Has been changed to: Magnetic resonance imaging of the SIJs will be performed at the Screening Visit only. An MRI performed ≤3 months prior to the Baseline Visit may be used as the Baseline.
REDACTED ne.ne. COPY ed ≤3 d ≤3
This do
cumen
t can
notic rec re
rmed med be
chesoeso
sedmed ≤ed
hanhan
toe ime im≤3≤3
supp
ort
sessmeessmmagmag
any m
arketi
ng
ite ite
autho
rizati
onmonmonappli
catio
n montmont
ntht
and
tthh
any e
xtens
ions
mit rtantanva
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 101 of 113
17.2 Protocol Amendment 2 Rationale for the amendmentThe main purpose of this substantial amendment is to provide clarification on magnetic resonance imaging (MRI) and x-ray assessments. In addition, text was added to the protocol about information collected for the assessment of AU flares and about Hy’s Law cases, and other minor updates/clarifications were also done to the protocol (see global changes below). Details of each specific change are provided below.
Modifications and changes Global changes Clarification has been added that if no chest x-ray is available within the 3 months prior to
Screening, the x-ray can be done during the Screening Period. If the modified New York (mNY) classification criteria were met in the x-ray performed prior to Screening, the x-ray is not to be repeated.
Clarification has been added that an MRI assessment is not needed for subjects with AS and that AS subjects must have evidence of sacroiliitis on x-ray taken prior to Baseline meeting the mNY classification criteria according to the Investigator.
Minor updates have been made to ophthalmic exclusion criteria and prior concomitant medications exclusion table.
Minor edits have been made to text about storage conditions at the site.
Details about the information collected for the assessment of AU flares have been added to the protocol.
Text about Hy’s Law cases has been added to the Adverse Events of Interest section.
Minor corrections have been done to the table in Appendix 16.2.
Minor editorial and format corrections have been done to the protocol.
Specific changes
Change #1Table 5-1: Schedule of assessmentsRow 17
MRIk
Has been changed to:MRI (nr-axSpA subjects only)k
REDACTEDalmic almic
xt aboxt abo
lecle
COPYtis ontis ohe Invhe Inv
This do
cumen
tRow ow
MM
cann
ot ##
e 55--11w 11
be
#1#1us
ed
angesngesto
supp
ortbeebee
formaform
anyhashas
en den
marketi
ng
ctedted fof
s bes b
autho
rizati
onvestiest
excluexclu
ut stot sto
appli
catio
n
not neeot nexx--ray raytigatiga
and he me m
ior toor toan
yhe 3 he 3modmod
exten
sions
or va
riatio
ns
er er ils ls
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 102 of 113
Change #2 Table 5-1: Schedule of assessments
Row 18 has been added and checked for Period 1 Screening Visit:
X-ray (AS subjects only)l
Change #3 Table 5-1: Schedule of assessments, footnote ii Screening chest x-ray must have occurred within 3 months prior to Screening Visit and should
be repeated only if the TB test was confirmed positive or any further evidence is suggestive of potential TB infection (eg, exposure).
Has been changed to: i Chest x-ray used for screening must have been done within 3 months prior to the Screening
Visit and should be repeated only if the TB test was confirmed positive or any further evidence is suggestive of potential TB infection (eg, exposure). If no chest x-ray is available within the 3 months prior to the Screening Visit, the x-ray can be done during the Screening Period.
Change #4 Table 5-1: Schedule of assessments, footnote kk Magnetic resonance imaging of the SIJs to be performed during the Screening Period. An MRI
performed ≤3 months prior to the Baseline Visit may be used as the Baseline.
Has been changed to: k For subjects with nr-axSpA, an MRI of the SIJs is to be performed during the Screening
Period. An MRI performed ≤3 months prior to the Baseline Visit may be used as the Baseline. An MRI assessment is not needed for patients with AS.
Change #5 Table 5-1: Schedule of assessments, new footnote lFootnote l has been added (for new Row 18 and former footnote l has been renumbered to footnote m):l AS subjects must have evidence of sacroiliitis on x-ray taken prior to Baseline meeting the
mNY classification criteria according to the Investigator. If not available, this x-ray is to be taken during the Screening Period. If an x-ray was performed prior to the Screening Visit and the mNY classification criteria were met, the x-ray is not to be repeated.
REDACTED e ke kto be to be
seline seline
COPY
This do
cumen
tthethecann
otje
Y clasclasken duken de me m
bects mcts mus
edeen en to
n adad
supp
ort
of assof as
anyededede mark
eting
RI of thRI of
monthsmonthd fd
autho
rizati
on
perfoperfoVisit Visit
appli
catio
n os
est xest x-re durine durin
andhs prs pr
sitiveitivean
y riorrio
exten
sionst andand
is suggs sug
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 103 of 113
Change #6 Section 6.1 Inclusion Criteria
Inclusion criterion 6, bullets 3 and 4
Nr-axSpA subjects must either have CRP > upper limit of normal (ULN) and/or current evidence of sacroiliitis on MRI taken within 3 months prior to Baseline (no confirmation by central reading) as defined by ASAS criteria
AS subjects must have evidence of sacroiliitis on x-ray taken within 12 months prior to Baseline meeting mNY criteria according to the Investigator
Have been changed to: Nr-axSpA subjects must either have CRP > upper limit of normal (ULN) and/or current
evidence of sacroiliitis on MRI (no confirmation by central reading) as defined by ASAS criteria
AS subjects must have evidence of sacroiliitis on x-ray meeting the mNY classification criteria according to the Investigator
Change #7 Section 6.2 Exclusion Criteria
Ophthalmic exclusion criteria
Exclusion criterion 9
10.9.Any condition or complicating factor that may interfere with the AU assessment, for example:
a. Intraocular pressure of ≥25 mmHg
b. Corneal or lens opacity
c. Proliferative or severe nonproliferative diabetic retinopathy or clinically significant macular edema due to diabetic retinopathy
d. Neovascular/wet age-related macular degeneration
e. History of scleritis
Has been changed to: 11. 9.Any condition or complicating factor that may interfere with the AU assessment, for
example:
a. History of cataract surgery within 6 months prior to Baseline
b. Corneal or lens opacity
c. Proliferative or severe nonproliferative diabetic retinopathy or clinically significant macular edema due to diabetic retinopathy
d. Neovascular/wet age-related macular degeneration
REDACTED
g factg fact
COPY
This do
cumen
t aca
nnot Any cAny c
examexam
ben chn ch used
y of scof sc
tor/wer/w supp
ortvere nere ndue todue
wet aet
any yymark
eting
tor thor th
5 mmHmmH
autho
rizati
on
haha
appli
catio
neetinetinand
ingng
anyg) asg) a ex
tensio
ns
LN) aLN)
opriorrio varia
tions
on n
or t
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 104 of 113
e. History of scleritis
f. History of intraocular surgery, with the exception of phacoemulsification
Change #8 Exclusion criterion 16
Table 6-1: Concomitant Medications (Prior to Baseline and Study Visits)
Rows 8, 9, and 10:
Table 6-1: Concomitant Medications (Prior to Baseline and Study Visits)
Drug class Dose Exclusion criteria
DMARDs:hydroxychloroquine (HCQ) and/or methotrexate (MTX)
Maximum allowed:HCQ ≤400mg daily MTX ≤25mg weekly
Use initiated in and/or any change in the dose regimen in the 28 days prior to the Baseline Visit is exclusionary.DMARDs dose should be kept stable throughout the study except for reasons of intolerance, where the DMARD dose may be decreased but not discontinued.
DMARDs:sulfasalazine (SSZ), azathioprine, cyclosporine, cyclophosphamide, mycophenolic acid, apremilast
Any dose Use within 28 days prior to the Baseline Visit is exclusionary.Must not be started during the study.
DMARDs:leflunomide
Any dose Use in the 6 months prior to the Baseline Visit is exclusionary unless a cholestyramine washout has been performed. In case of a cholestryramine washout, use 28 days prior to the Baseline Visit is acceptable. Must not be started during the study.
REDACTED D
R
COPY
C
This do
cumen
t can
not b
e use
d se
to su
pport
any nymark
eting
y dosey doseing
ting au
thoriz
ation
tio
appli
catio
nDMADMAstabstafca
andto tho th
lusionusionan
yse regse regthe Bhe
exten
sions
riaiaio
in anin ate
or ova
riatio
ns th
ereof.
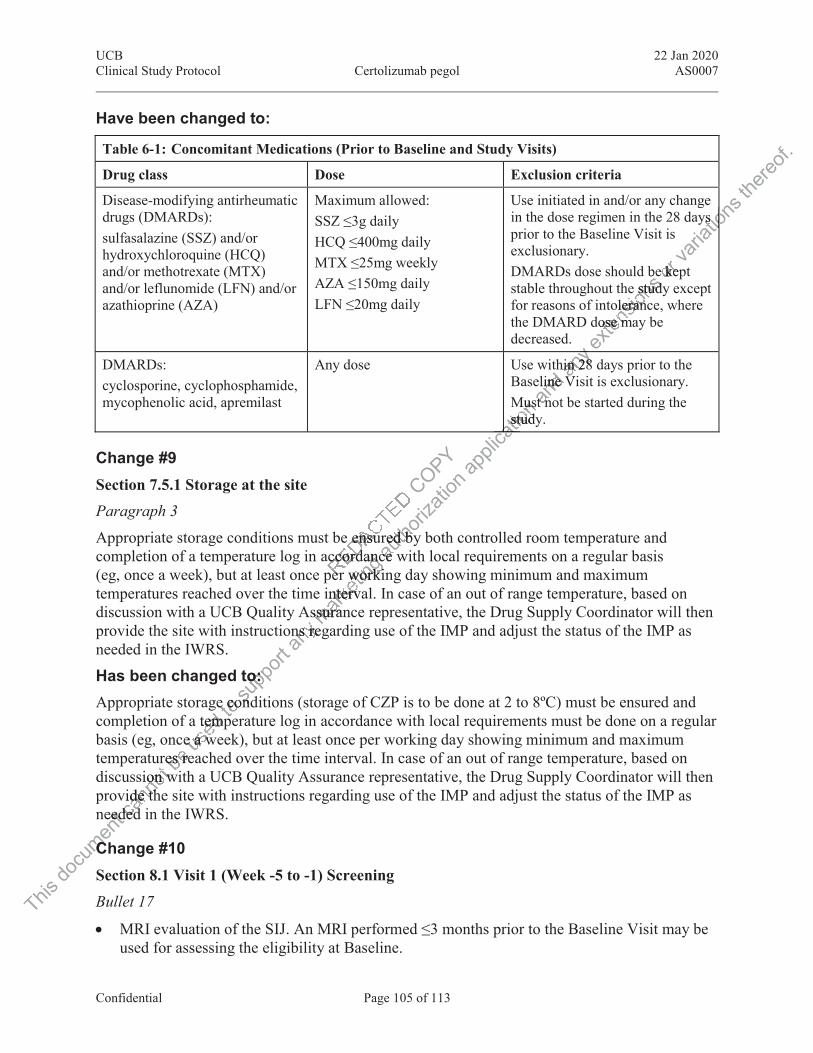
UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 105 of 113
Have been changed to:
Table 6-1: Concomitant Medications (Prior to Baseline and Study Visits)
Drug class Dose Exclusion criteria
Disease-modifying antirheumatic drugs (DMARDs):sulfasalazine (SSZ) and/or hydroxychloroquine (HCQ) and/or methotrexate (MTX) and/or leflunomide (LFN) and/or azathioprine (AZA)
Maximum allowed:SSZ ≤3g dailyHCQ ≤400mg dailyMTX ≤25mg weeklyAZA ≤150mg dailyLFN ≤20mg daily
Use initiated in and/or any change in the dose regimen in the 28 days prior to the Baseline Visit is exclusionary.DMARDs dose should be kept stable throughout the study except for reasons of intolerance, where the DMARD dose may be decreased.
DMARDs:cyclosporine, cyclophosphamide, mycophenolic acid, apremilast
Any dose Use within 28 days prior to the Baseline Visit is exclusionary.Must not be started during the study.
Change #9 Section 7.5.1 Storage at the siteParagraph 3
Appropriate storage conditions must be ensured by both controlled room temperature and completion of a temperature log in accordance with local requirements on a regular basis (eg, once a week), but at least once per working day showing minimum and maximum temperatures reached over the time interval. In case of an out of range temperature, based on discussion with a UCB Quality Assurance representative, the Drug Supply Coordinator will then provide the site with instructions regarding use of the IMP and adjust the status of the IMP as needed in the IWRS.
Has been changed to: Appropriate storage conditions (storage of CZP is to be done at 2 to 8ºC) must be ensured and completion of a temperature log in accordance with local requirements must be done on a regular basis (eg, once a week), but at least once per working day showing minimum and maximum temperatures reached over the time interval. In case of an out of range temperature, based on discussion with a UCB Quality Assurance representative, the Drug Supply Coordinator will then provide the site with instructions regarding use of the IMP and adjust the status of the IMP as needed in the IWRS.
Change #10 Section 8.1 Visit 1 (Week -5 to -1) Screening
Bullet 17
MRI evaluation of the SIJ. An MRI performed ≤3 months prior to the Baseline Visit may be used for assessing the eligibility at Baseline.
e ensure ensuraccordaccord
erer
COPY
This do
cumen
t eeedd
CC
cann
otsion won wvide thde thdedded
bees rees reus
edtempteme a we a w
toe coe co supp
ort
to:o:ondon
anyns rens remark
eting
dancancr workiworki
intervantervssuranssura
autho
rizati
on
ured byed bncece
appli
catio
nMustusstudystud
caacaca
andeline Vine
st nt n
any hin 28 in 28
Vi
y exten
sionsbe
e studystudoleranleran
dose mose m
ore kepe keva
riatio
ns
ys ys iothe
reof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 106 of 113
Has been changed to: For subjects with nr-axSpA, an MRI of the SIJs is to be performed during the Screening
Period. An MRI performed ≤3 months prior to the Baseline Visit may be used as the Baseline. An MRI assessment is not needed for patients with AS.
And the following bullets have been added above and below Bullet 17: Chest x-ray used for screening must have been done within 3 months prior to the Screening
Visit and should be repeated only if the TB test was confirmed positive or any further evidence is suggestive of potential TB infection (eg, exposure). If no chest x-ray is available within the 3 months prior to the Screening Visit, the x-ray can be done during the Screening Period.
AS subjects must have evidence of sacroiliitis on x-ray taken prior to Baseline meeting the mNY classification criteria according to the Investigator. If not available, this x-ray is to be taken during the Screening Period. If an x-ray was performed prior to the Screening Visit and the mNY classification criteria were met, the x-ray is not to be repeated.
Change #11 Section 9.1.1 Assessment of AU flaresSubjects will be requested to contact their ophthalmologist when they experience an AU flare at any time during the study. The ophthalmologist will confirm the AU flare and determine the duration, severity, and treatment. The subject’s ophthalmologist will be provided with documentation to ensure all protocol-requested information is collected. The documentation will inform the subject’s ophthalmologist about the subject’s study participation and the requirements.
At each site visit, the subject will be questioned on the occurrence of uveitis flares since last visit. In case uveitis flares are confirmed by the subject the site will obtain the needed information from the subject's ophthalmologist containing the diagnosis of AU, the affected eye, location of the uveitis in the affected eye, duration, severity, and treatment provided.
Has been changed to: Subjects will be requested to contact an ophthalmologist when they experience an AU flare at any time during the study. The ophthalmologist will confirm the AU flare and determine the duration, severity, and treatment. The ophthalmologist will be provided with documentation to ensure all protocol-requested information is collected. The documentation will inform the ophthalmologist about the subject’s study participation and the requirements.
At each site visit, the subject will be questioned on the occurrence of uveitis flares since last visit. In case uveitis flares are confirmed by the subject the site will obtain the needed information from the ophthalmologist containing the diagnosis of AU, the affected eye, location of the uveitis in the affected eye, duration, severity, and treatment provided.
REDACTEDgist wgist wject’s ect’s
equestequestt about abou
COPY almollmo
i
This do
cumen
t At eaevisitvisitinin
cann
ot ll prl pr
almolalmol
eachach
beeververprotprot
used
reqeqng the g the rityrity
toquesquessu
pporthe affe af
d to:to:
any onfonf
ophtophf
marketi
ngut tht th
be quese qufirmfirm
autho
rizati
onlogistlogisill conill con
ophthophthted infd inthehe
appli
catio
n re an
dr to to epepeata
anyble, tble,
theth
exten
sions
s avavhe Scrhe Scr
selineeline
or vailvaiva
riatio
ns
ning ing
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 107 of 113
The following information will be collected:
Start and end date of each AU flare (episode)
Start date: when subject experience first signs of the AU episode
End date: when treatment of the AU is stopped
Grading
For uveitis flare, the highest grade noted during an episode of uveitis will be graded as follows:
Grade 0=None; Grade 1+=Faint; Grade 2+=Moderate, iris and lens details clear; Grade3+=Marked, iris and lens details hazy; Grade4+=Intense, fibrin or plastic aqueous; ND=Not done or not assessed; UNK=Unknown
For cell count, the highest cell count noted during an episode of uveitis will be graded as follows:
Grade 0=<1 cell; Grade 0.5+=1-5 cells; Grade 1+=6-15 cells; Grade 2+=16-25 cells; Grade 3+=26-50 cells; Grade 4+=>50 cells; ND=Not done or not assessed; UNK=Unknown
Treatment
Change #12 Section 10.3 Adverse events of interest
The following text has been added as last paragraph:
Note : Potential Hy’s Law, defined as ≥3xULN alanine aminotransferase or aspartate aminotransferase with coexisting ≥2xULN total bilirubin in the absence of ≥2xULN alkaline phosphatase, with no alternative explanation for the biochemical abnormality, must ALWAYS be reported to UCB as an AE of interest (ie, without waiting for any additional etiologic investigations to have been concluded). Follow-up information should then be reported if an alternative etiology is identified during investigation and monitoring of the subject.
REDACTED
stst
s lass las
COPY
This do
cumen
t can
not b
e use
d to s
uppo
rt E oE
en conen conidentifdenti
anye exe ex
of inof mark
etingst part par
as ≥3xUs ≥3x≥2xUL≥2xUxplp
autho
rizati
on ap
plica
tioncells; Gells;
done odone
and a
ny
uveitisveitiex
tensio
ns
clearclearr plasr plas
or eded va
riatio
ns
d as d as
thereo
f.
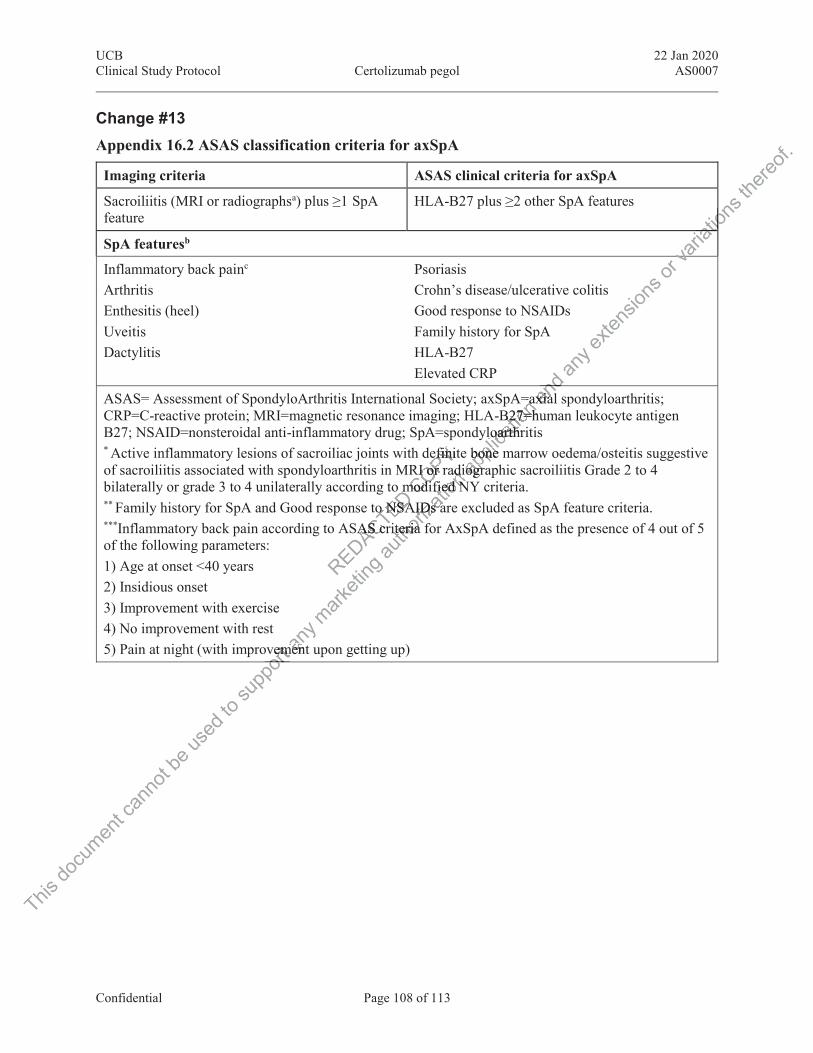
UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 108 of 113
Change #13 Appendix 16.2 ASAS classification criteria for axSpA
Imaging criteria ASAS clinical criteria for axSpA
Sacroiliitis (MRI or radiographsa) plus ≥1 SpA feature
HLA-B27 plus ≥2 other SpA features
SpA featuresb
Inflammatory back painc
Arthritis Enthesitis (heel) Uveitis Dactylitis
PsoriasisCrohn’s disease/ulcerative colitisGood response to NSAIDsFamily history for SpAHLA-B27Elevated CRP
ASAS= Assessment of SpondyloArthritis International Society; axSpA=axial spondyloarthritis; CRP=C-reactive protein; MRI=magnetic resonance imaging; HLA-B27=human leukocyte antigen B27; NSAID=nonsteroidal anti-inflammatory drug; SpA=spondyloarthritis* Active inflammatory lesions of sacroiliac joints with definite bone marrow oedema/osteitis suggestive of sacroiliitis associated with spondyloarthritis in MRI or radiographic sacroiliitis Grade 2 to 4bilaterally or grade 3 to 4 unilaterally according to modified NY criteria. ** Family history for SpA and Good response to NSAIDs are excluded as SpA feature criteria. ***Inflammatory back pain according to ASAS criteria for AxSpA defined as the presence of 4 out of 5 of the following parameters:1) Age at onset <40 years2) Insidious onset3) Improvement with exercise4) No improvement with rest5) Pain at night (with improvement upon getting up)
REDACTED o mo NSAo NSA
SAS crAS cr
COPYefinitefiniRI or raRI or
modimodi
This do
cumen
t can
not b
e use
d to s
uppo
rtvemeemor
any
mentmen
marketi
ng au
thoriz
ation
adiadiiffied Nied Nfff
AIDs aIDs ariteria iteria
appli
catio
n A=axaB27=huB27=h
loarthrloarthe bone mbone
diograogr
and
axiaxiand nd
any e
xtens
ions o
r vari
ation
s ioat
va
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 109 of 113
Has been changed to:
ASAS classification criteria for axSpA(for subjects with chronic back pain ≥3 months and age at onset <45 years)
Imaging criteria ASAS clinical criteria for axSpA
Sacroiliitis (MRI or radiographsa) plus ≥1 SpA feature
HLA-B27 plus ≥2 other SpA features
SpA featuresb
Inflammatory back painc
Arthritis Enthesitis (heel) Uveitis Dactylitis
PsoriasisCrohn’s disease/ulcerative colitisGood response to NSAIDsFamily history for SpAHLA-B27Elevated CRP
ASAS=Assessment of SpondyloArthritis international Society; axSpA=axial spondyloarthritis; CRP=C-reactive protein; MRI=magnetic resonance imaging; HLA-B27=human leukocyte antigen B27; NSAID=nonsteroidal anti-inflammatory drug; SpA=spondyloarthritis
a Active inflammatory lesions of sacroiliac joints with definite bone marrow oedema/osteitis suggestive of sacroiliitis associated with spondyloarthritis in MRI or radiographic sacroiliitis Grade 2 to 4 bilaterally or grade 3 to 4 unilaterally according to modified NY criteria.
b Family history for SpA and Good response to NSAIDs are excluded as SpA feature criteria. c Inflammatory back pain according to ASAS criteria for axSpA defined as the presence of 4 out of 5
of the following parameters:1) Age at onset <40 years2) Insidious onset3) Improvement with exercise4) No improvement with rest5) Pain at night (with improvement upon getting up)
REDACTED dylodyloaterallyaterally
nse to nse to ASAASA
COPYA=spoA=spwith dewith doartoar
This do
cumen
t can
not b
e use
d to s
uppo
rtrestrestimprovimproup
pan
y eestt
marketi
ng au
thoriz
ation
efinifinithritis hritis
y accoaccoNSAINSAI
S critecrit
appli
catio
n pA=axpA=axLAA--B27B2
ndyloardyloaite bite b
nn and a
ny ex
tensio
ns or
ova
riatio
ns
io
va
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 110 of 113
17.3 Protocol Amendment 3 Rationale for the amendmentThe purpose of this non-substantial amendment is to update the safety variables as a secondary safety variable and other safety variables.
Modifications and changes Global changes Updates have been made to the safety variables to include TEAEs as a secondary safety
variable and blood pressure, physician examination, and clinical laboratory values as other safety variables.
Study contact information was updated.
Minor editorial and format corrections have been done to the protocol.
Specific changes
Change #1 Study Contact Information
Clinical Trial Biostatistician
Name: , MS
Address: UCB Biosciences, Inc.8010 Arco Corporate DriveRaleigh, NC 27617USA
Phone:
Fax:
RRREDACTED
EDRE
COPY
This do
cumen
t can
not b
e use
d edto
supp
ort USASA
posusssu
ppan
yigh, Nigh, Nmark
eting, MSMS
ciencesienceco Coco C
tiniau
thoriz
ation
SSau
appli
catio
n andtocolocol a
ny ex
tensio
nsry safery safvalues alues
or va
riatio
ns th
ereof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 111 of 113
Has been changed to:
Clinical Trial Biostatistician
Name: Dr.
Address: UCB Biosciences GmbHAlfred-Nobel-Strasse 1040789 MonheimGermany
Phone:
Fax:
Change #2Section 4.2: Safety variables
Safety variables to be assessed are:
Adverse events (AEs)
Blood pressure
Physical examination
Clinical laboratory values (hematology, biochemistry, and urinalysis)
Has been changed to:Section 4.2: Safety variables
Section 4.2.1: Secondary safety variableThe secondary safety variable to be assessed is treatment-emergent adverse events (TEAEs).
Section 4.2.2: Other safety variables
The following other safety variables to be assessed are:
Blood pressure
Physical examination
Clinical laboratory values (hematology, biochemistry, and urinalysis)
REDACTED COPY
This do
cumen
tPPcann
ot owiw
Blood lood
PhPh
be
wing wing us
ed afetfet
: OthOthtoty vty v
supp
ort ablesbles
dary sdary s
any m
arketi
ng
matolomatolo
autho
rizati
on ap
plica
tion a
nd
ndan
y ny ex
tensio
ns
te
or rva
riatio
ns
v
thereo
f.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 112 of 113
18 DECLARATION AND SIGNATURE OF INVESTIGATOR I confirm that I have carefully read and understood this protocol and agree to conduct this clinical study as outlined in this protocol, according to current Good Clinical Practice and local laws and requirements.
I will ensure that all subInvestigators and other staff members read and understand all aspects of this protocol.
I have received and read all study-related information provided to me.
The objectives and content of this protocol as well as the results deriving from it will be treated confidentially, and will not be made available to third parties without prior authorization by UCB.
All rights of publication of the results reside with UCB, unless other agreements were made in a separate contract.
Investigator:
Printed name Date/Signature
REDACTED COP
DatDaOPY
OP
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on
te/Sige/Sigap
plica
tion
a
and a
nymenme ex
tensio
nsl be tbe rizatiozatio
entsnts
or tva
riatio
ns
f the
reof.

UCB 22 Jan 2020Clinical Study Protocol Certolizumab pegol AS0007
Confidential Page 113 of 113
19 SPONSOR DECLARATION I confirm that I have carefully read and understand this protocol and agree to conduct this clinical study as outlined in this protocol and according to current Good Clinical Practice.
REDACTED COPY
This do
cumen
t can
not b
e use
d to s
uppo
rt any
mark
eting
autho
rizati
on ap
plica
tion a
nd an
y exte
nsion
s or v
ariati
ons t
hereo
f.

Approval SignaturesName: as0007-protocol-amend-3
Version: 1 . 0
Document Number: CLIN-000148098
Title: AS0007 Protocol Amendment 3
Approved Date: 27 Jan 2020
Document Approvals
ApprovalVerdict: Approved
Name: Capacity: ClinicalDate of Signature: 26-Jan-2020 15:35:31 GMT+0000
ApprovalVerdict: Approved
Name: Capacity: ClinicalDate of Signature: 27-Jan-2020 09:27:27 GMT+0000
ApprovalVerdict: Approved
Name: Capacity: ClinicalDate of Signature: 27-Jan-2020 09:34:22 GMT+0000
DACTED
REDADA
OPYYAppAppCOOOCO
This do
cumen
t can
not b
e use
d to s
upppport
pp
any m
arketi
ng
mama
np
uthori
zatio
n n
oriNamCa
autut
appli
catio
n
provalovaa
and a
ny ex
tensio
ns or
varia
tions
there
of.


















