
Promoting Direct CO Conversion to DME over Zeolite-Based ...
8
508 ISSN 0965-5441, Petroleum Chemistry, 2020, Vol. 60, No. 4, pp. 508–515. © The Author(s), 2020. This article is an open access publication, corrected publication 2021. Promoting Direct CO 2 Conversion to DME over Zeolite-Based Hybrid Catalysts L. Frusteri a , G. Bonura b , C. Cannilla b , S. Todaro c , G. Giordano c , M. Migliori c , and F. Frusteri b, * a University of Messina, Dip. Ingegneria Elettronica, Chimica ed Ingegneria Industriale INSTM/CASPE, Messina, 98166 Italy b CNR-ITAE, Istituto di Tecnologie Avanzate per l’Energia “Nicola Giordano”, Messina, 98126 Italy c Università della Calabria, Dip. Ing. Ambiente Territorio e Ing. Chimica, Rende, 87036 Italy *e-mail: [email protected] Received November 17, 2019; revised November 17, 2019; accepted December 10, 2019 Abstract—The direct hydrogenation of CO 2 into dimethyl-ether (DME) has been studied in presence of zeo- lite-based hybrid catalysts, prepared through gel-oxalate coprecipitation of copper, zinc and zirconium pre- cursors (in an atomic ratio of 60 : 30 : 10 respectively) in a solution containing different home-made zeolites (i.e., Sil-1, MFI, Y, FER, BEA, MOR), for a final CuZnZr/zeolite weight composition of 1:1. All the samples were properly characterized with different techniques for determining the textural, structural and morpho- logical nature of the catalytic surface. N 2 physisorption highlighted a variation both in the specific surface area and in the pore size distribution from the parent zeolites to the hybrid catalyst. TEM analyses disclosed how the pre-formed zeolite architecture affects the distribution of the oxides on the surface, significantly controlling not only the activity-selectivity pattern under the adopted experimental conditions (T R , 200– 260°C; P R , 30 bar, GHSV: 8.800 NL/kg cat /h), but also the catalyst stability during time on-stream. Keywords: hybrid catalysts, zeolites, CO 2 conversion, DME production DOI: 10.1134/S0965544120040076 1. INTRODUCTION In the last years, many novel technological approaches are being implemented with the aim of recycling anthropogenic CO 2 as a cheap and non-toxic building-block for carbon source synthetic fuels [1– 3]. Indeed, the utilization of CO 2 represents one of the hot topics of the current International environmental programmes [4, 5] and recent literature report on sev- eral studies dealing with CO 2 conversion into methane [6–8], methanol [9–16], higher alcohols [17], gaso- line [18], dimethyl ether [19–27] as well as liquid fuels [28]. In particular, dimethyl ether (DME) is a promis- ing alternative fuel due to its high efficiency of com- bustion, high cetane number, sulfur-free property and low emission of NO x [29–31]. The conventional DME synthesis includes a first step of methanol synthesis on copper-based catalyst (Eq. (1)) and a second step of methanol dehydration to DME over solid acid catalyst (Eq. (3)). The reverse water gas shift reaction (RWGS) is an inevitable side reaction under the experimental conditions (Eq. (2)). (1) (2) (3) Nevertheless, considering the capital and operating expenditures as well as thermodynamic limitations behind a traditional two step process, nowadays sev- eral studies report the economic sustainability of pro- ducing DME from one-step CO 2 hydrogenation pro- cesses rather than in traditional multi-step processes. From a catalytic point of view, the direct DME synthesis is boosting an intense research effort in the development and application of novel multi-func- tional hybrid systems, wherein a mix of different active sites are in close interaction among them to facilitate the rate of mass transfer of a reaction intermediate towards the final product. In particular, it is known that Cu 0 provides active sites for H 2 dissociation, while some carrier oxides (mainly ZnO, Al 2 O 3 , and ZrO 2 [32]) possess specific sites generated at the metal- oxide interface, suitable for CO 2 adsorption and acti- vation [33–35]. Recently CuO–ZnO–ZrO 2 was proposed as an efficient methanol synthesis catalyst from CO 2 /H 2 mixtures due to weak hydrophilicity of ZrO 2 in com- parison to Al 2 O 3 [36, 37]. Apart from this, the intro- duction of ZrO 2 was also recognized to increase the surface basicity for improving the CO 2 adsorption 2 2 3 2 CO 3H CH OH H O, ← + → + 2 2 2 CO H CO H O, ← + → + 3 3 3 2 2CH OH CH OCH H O. ← → +
Transcript of Promoting Direct CO Conversion to DME over Zeolite-Based ...

ISSN 0965-5441, Petroleum Chemistry, 2020, Vol. 60, No. 4, pp. 508–515. © The Author(s), 2020. This article is an open access publication, corrected publication 2021.
Promoting Direct CO2 Conversion to DME over Zeolite-Based Hybrid Catalysts
L. Frusteria, G. Bonurab, C. Cannillab, S. Todaroc, G. Giordanoc, M. Miglioric, and F. Frusterib, *aUniversity of Messina, Dip. Ingegneria Elettronica, Chimica ed Ingegneria Industriale INSTM/CASPE, Messina, 98166 Italy
bCNR-ITAE, Istituto di Tecnologie Avanzate per l’Energia “Nicola Giordano”, Messina, 98126 ItalycUniversità della Calabria, Dip. Ing. Ambiente Territorio e Ing. Chimica, Rende, 87036 Italy
*e-mail: [email protected] November 17, 2019; revised November 17, 2019; accepted December 10, 2019
Abstract—The direct hydrogenation of CO2 into dimethyl-ether (DME) has been studied in presence of zeo-lite-based hybrid catalysts, prepared through gel-oxalate coprecipitation of copper, zinc and zirconium pre-cursors (in an atomic ratio of 60 : 30 : 10 respectively) in a solution containing different home-made zeolites(i.e., Sil-1, MFI, Y, FER, BEA, MOR), for a final CuZnZr/zeolite weight composition of 1:1. All the sampleswere properly characterized with different techniques for determining the textural, structural and morpho-logical nature of the catalytic surface. N2 physisorption highlighted a variation both in the specific surfacearea and in the pore size distribution from the parent zeolites to the hybrid catalyst. TEM analyses disclosedhow the pre-formed zeolite architecture affects the distribution of the oxides on the surface, significantlycontrolling not only the activity-selectivity pattern under the adopted experimental conditions (TR, 200–260°C; PR, 30 bar, GHSV: 8.800 NL/kgcat/h), but also the catalyst stability during time on-stream.
Keywords: hybrid catalysts, zeolites, CO2 conversion, DME productionDOI: 10.1134/S0965544120040076
1. INTRODUCTIONIn the last years, many novel technological
approaches are being implemented with the aim ofrecycling anthropogenic CO2 as a cheap and non-toxicbuilding-block for carbon source synthetic fuels [1–3]. Indeed, the utilization of CO2 represents one of thehot topics of the current International environmentalprogrammes [4, 5] and recent literature report on sev-eral studies dealing with CO2 conversion into methane[6–8], methanol [9–16], higher alcohols [17], gaso-line [18], dimethyl ether [19–27] as well as liquid fuels[28]. In particular, dimethyl ether (DME) is a promis-ing alternative fuel due to its high efficiency of com-bustion, high cetane number, sulfur-free property andlow emission of NOx [29–31].
The conventional DME synthesis includes a firststep of methanol synthesis on copper-based catalyst(Eq. (1)) and a second step of methanol dehydrationto DME over solid acid catalyst (Eq. (3)). The reversewater gas shift reaction (RWGS) is an inevitable sidereaction under the experimental conditions (Eq. (2)).
(1)
(2)
(3)Nevertheless, considering the capital and operating
expenditures as well as thermodynamic limitationsbehind a traditional two step process, nowadays sev-eral studies report the economic sustainability of pro-ducing DME from one-step CO2 hydrogenation pro-cesses rather than in traditional multi-step processes.
From a catalytic point of view, the direct DMEsynthesis is boosting an intense research effort in thedevelopment and application of novel multi-func-tional hybrid systems, wherein a mix of different activesites are in close interaction among them to facilitatethe rate of mass transfer of a reaction intermediatetowards the final product. In particular, it is knownthat Cu0 provides active sites for H2 dissociation, whilesome carrier oxides (mainly ZnO, Al2O3, and ZrO2[32]) possess specific sites generated at the metal-oxide interface, suitable for CO2 adsorption and acti-vation [33–35].
Recently CuO–ZnO–ZrO2 was proposed as anefficient methanol synthesis catalyst from CO2/H2mixtures due to weak hydrophilicity of ZrO2 in com-parison to Al2O3 [36, 37]. Apart from this, the intro-duction of ZrO2 was also recognized to increase thesurface basicity for improving the CO2 adsorption
2 2 3 2CO 3H CH OH H O,←+ → +
2 2 2CO H CO H O,←+ → +
3 3 3 22CH OH CH OCH H O.←→ +
508

PROMOTING DIRECT CO2 CONVERSION 509
Table 1. Gel composition and textural properties of the zeolite samples
(a) Langmuir model, considering monolayer adsorption.(b) Micropore Volume estimated from t-plot.
Zeolitesample SDA Gel composition (mol/mol) SA (a), m2/g Vmicro
(b), cm3/g
Sil-1 TPABr 0.08NaOH : 1.0SiO2 : 0.08TPABr : 20H2O 407 ± 17 0.094MFI TPABr 0.10Na2O :1 .0SiO2 : 0.08TPABr : 0.03Al2O3 : 20H2O 368 ± 3 0.098Y TMA+ 0.01Na2O : 1.0SiO2 : 0.55(TMA)2O : 0.07Al2O3 : 60H2O 1027 ± 6 0.267FER C2DN 0.12Na2O : 1.0SiO2 : 1.3C2DN : 0.07Al2O3 : 40H2O 417 ± 5 0.124BEA TEAOH 0.10Na2O : 1.0SiO2 : 0.2TEAOH : 0.02Al2O3 :1 0H2O 455 ± 2 0.115MOR – 0.20Na2O : 1.0SiO2 : 0.02Al2O3 : 20H2O 348 ± 3 0.152
[38]. Accordingly, this system can be advantageouslyhybridized with an acidic matrix to shift equilibriumtowards the formation of DME, by methanol dehydra-tion. Whilst conventional γ-Al2O3 can optimally fulfillthis role at atmospheric pressure [39, 40], however itsstrong affinity to water adsorption during the reactionprevents its potential application in processes carriedout at high reaction pressure. For this reason, zeolitesappear as more promising crystalline acidic carriers tobe integrated with a methanol phase, owing to a tun-able system of voids (openings and spatial orientationof channels, size and location of cages, etc.) and con-centration, distribution, strength and location of acidsites (both of Brønsted and Lewis type) affecting theoverall activity, product selectivity and deactivation ofthe catalyst [41].
Herein, the catalytic behaviour of hybrid systemscharacterized by the interaction between metal-oxidesites from an optimized CuZnZr formulation andacidic sites of different home-made zeolites isassessed. Peculiar structure-activity relationships areaccordingly defined suitable to understand how a pre-formed zeolite architecture can drive the generation ofspecific metallic properties for an efficient catalyticsystem operating in the direct CO2-to-DME hydroge-nation reaction.
2. EXPERIMENTAL
2.1. Hybrid Systems for One-step CO2-to-DME Hydrogenation Reaction
The procedure followed for the preparation of thehybrid systems was to generate a methanol phasedirectly in a solution containing a zeolite finely dis-persed (particle size <100 μm). In Table 1, a list of thehome-made zeolites used as metal-oxide carriers isreported. All of the molecular sieves were preparedunder hydrothermal conditions in a PTFE lined stain-less steel autoclaves at 175°C for 10 days in tumblingconditions (20 rpm). Then, the samples were activatedin acidic form by calcination at 550°C for 8 h in order
PETROLEUM CHEMISTRY Vol. 60 No. 4 2020
to eliminate the structure-directing agent (SDA).Samples synthesized with sodium-containing synthe-sis gel, were exchanged with NH4Cl solution and cal-cined again at 550°C for 8 h in order to eliminateammonia and to obtain catalyst in acid-form.
For the preparation of the hybrid catalysts, thepowdered home-made zeolites were dissolved in anethanolic solution, containing a suitable amount ofoxalic acid for quantitative coprecipitation of theCuZnZr nitrate precursors (60/30/10 at %). The solu-tion of metal precursors was added, at room tempera-ture and in a slow and constant mode, to the vigor-ously stirred ethanolic solution in order to get a finaloxide composition of 1 : 1 by weight with the zeolite.The precipitate was stirred for 3 h, aged overnight,then filtered and dried at 110°C for 16 h, before of cal-cination at 350°C for 4 h.
2.2. Catalyst CharacterizationThe analytical composition of catalysts was deter-
mined by XRF, using a Bruker AXS-S4 Explorer spec-trometer, equipped with a rhodium X-ray source (Rhanode and 75 μm Be-window), a LiF 220 crystal ana-lyzer and a 0.12° divergence collimator. Samples wereanalyzed at the solid state, taking into account theemission value of Cu–Kα1, Zn–Kα1 and Al–Kα1transitions.
The textural properties of the catalysts were deter-mined from nitrogen adsorption/desorption isothermsat –196°C, using a Micromeritics ASAP 2020 gasadsorption device. The isotherms were elaboratedaccording to the Langmuir method for surface areacalculation (SA), while Barrett–Joyner–Halenda(BJH) and Horvath-Kawazoe (HK) methods wereused for meso- and micro-porosity evaluation.
Metallic properties were obtained by N2Ochemisorption measurements at 90°C, assuming aCu : N2O = 2 : 1 titration stoichiometry and a surfaceatomic density of 1.46 × 1019 Cuat/m2. Before “single-pulse” N2O-titration of the metal copper sites, thesamples were reduced in situ at 300°C in f lowing H2

510 FRUSTERI et al.
Table 2. Metallic, basic and acidic properties of the investigated hybrid samples
(a)From N2O titration @ 90°C.(b)Cumulative uptake in the range of 100—700°C.
SAMPLE N2O uptake(a), mmol/gcat CO2 uptake(b), mmol/gcat NH3 uptake(b), mmol/gcat
OX/Sil-1 0.440 0.205 0.032OX/MFI 0.247 0.123 0.717OX/Y 0.217 0.272 1.602OX/FER 0.172 0.132 0.733OX/BEA 0.317 0.187 1.142OX/MOR 0.301 0.124 0.893
(100 STP mL/min) for 1 h, then “flushed” at 310°C innitrogen carrier f low (15 min) and further cooleddown at 90°C.
Surface concentrations of base sites were deter-mined by CO2-TPD. The catalysts (≈100 mg) werereduced at 300°C, at atmospheric pressure, by f lowinghydrogen (100 STP mL/min) in a linear quartz micro-reactor (l, 200 mm; i.d., 4 mm) for 60 min and thensaturated for 60 min in f low of a gas mixture contain-ing 20 vol % of CO2/He (50 mL/min). After purging inhelium flow, the measurements were performed in thetemperature range 100–600°C, at a rate of 10°C/minusing helium (50 STP mL/min) as the carrier f low.The evolved gases were detected by an on-line ther-mal-conductivity detector, calibrated by the peak areaof known pulses of CO2.
TEM images of the hybrid catalysts were acquiredand elaborated by a Philips CM12 instrumentequipped with a high-resolution camera. Powderedsamples were dispersed in 2-propanol under ultra-sound irradiation and the resulting suspension putdrop-wise on a holey carbon-coated support grid.
2.3. Catalytic Testing
Catalytic testing was carried out in in CO2 hydroge-nation at 30 bar (CO2/H2/N2 = 23/69/8), in a tubularfixed catalyst bed reactor (i.d., 4 mm; l., 200 mm),jacketed within a stainless steel rod (o.d., 14 mm; l.,180 mm). The catalytic measurements were performedbetween 200 and 280°C, at a space velocity of 8.800NL/kgcat/h. Before the testing, the catalysts werereduced in situ at 300°C under “pure” hydrogen atatmospheric pressure for 1 h. The reaction stream wasanalyzed by a GC equipped with a double-columnsystem connected to a f lame ionized detector (CH4,CH3OH, CH3OCH3) and thermal conductivity detec-tor (H2, N2, CO, CO2) respectively.
3. RESULTS AND DISCUSSIONAccording to the graphs displayed in Fig. 1, the tex-
tural properties of the hybrid systems are strongly
depressed with respect to the parent zeolites, probablydue to a partial pore blocking to the presence ofCuZnZr particles. It is evident that an extended mes-oporosity on the hybrid systems is generated, suggest-ing that metallic components distribute over the outersurface of the zeolites, giving catalysts characterized bymore or less expanded pore volumes depending on thecorresponding features of the bare zeolites.
In particular, on the Sil-1, MFI, FER, BEA, andMOR zeolites, the coprecipitation of metal precursorsleads to a significant reduction of micropore volume(49–56%), being even more marked on the OX-Yhybrid sample in which the final values of 0.071 cm3/gaccounts for a decrease around 75% in respect of thebare zeolite.
As reported in Table 2, the hybrid catalystsobtained by oxalate co-precipitation method are char-acterized by different metallic, basic and acidic prop-erties. Indeed, the highest value of N2O uptake on sil-icalite (0.440 mmol/gcat) signals a higher copper sur-face area and dispersion in the correspondent hybridsample, as the result of particles with a smaller diame-ter. On the contrary, ferrierite brings to the lowestvalue of N2O uptake (0.172 mmol/gcat), accounting fora minor dispersion and larger copper particles gener-ated during preparation.
As for the basic capacity, it can be easily observedas all the hybrid CuZnZr/zeolite systems exhibit aquite variable concentration of surface basic sites, witha maximum CO2 uptake of 0.272 mmol/gcat for theOX/Y sample and the minimum amount of0.124 mmol/gcat recorded for OX/MOR. Evidently,irrespective of the same metal loading on the zeolitecarriers, the zeolite architecture significantly controlsthe exposure and accessibility of active sites in specificpositions, with a direct influence on the CO2 activa-tion process.
Regarding the NH3-TPD measurements, thecumulative acid capacity (with no regard if of Brønstedor Lewis type) was seen to depend on different topol-ogy of the zeolites in the hybrid catalysts. Indeed, withan approximately 1 : 1 ratio between the powderedzeolite and the methanol composition, the number of
PETROLEUM CHEMISTRY Vol. 60 No. 4 2020

PETROLEUM CHEMISTRY Vol. 60 No. 4 2020
PROMOTING DIRECT CO2 CONVERSION 511
Fig. 1. Cumulative volume and PSD of the prepared hybrid catalysts.
Cum
ulat
ive
volu
me,
cm
3 g–
1
Rel
ativ
e vo
lum
e, %
0
Dpore, Å
0.4
0.2
0.1
0.3
20
15
10
5
OX/Sil-1
103102101
Cum
ulat
ive
volu
me,
cm
3 g–
1
Rel
ativ
e vo
lum
e, %
0
Dpore, Å
0.4
0.2
0.1
0.3
20
15
10
5
OX/MFI
103102101
Cum
ulat
ive
volu
me,
cm
3 g–
1
Rel
ativ
e vo
lum
e, %
0
Dpore, Å
0.4
0.2
0.1
0.3
20
15
10
5
OX/Y
103102101
Cum
ulat
ive
volu
me,
cm
3 g–
1
Rel
ativ
e vo
lum
e, %
0
Dpore, Å
0.4
0.2
0.1
0.3
20
15
10
5
OX/BEA
103102101
Cum
ulat
ive
volu
me,
cm
3 g–
1
Rel
ativ
e vo
lum
e, %
0
Dpore, Å
0.4
0.2
0.1
0.3
20
15
10
5
OX/FER
103102101
Cum
ulat
ive
volu
me,
cm
3 g–
1
Rel
ativ
e vo
lum
e, %
0
Dpore, Å
0.4
0.2
0.1
0.3
20
15
10
5
OX/MOR
103102101
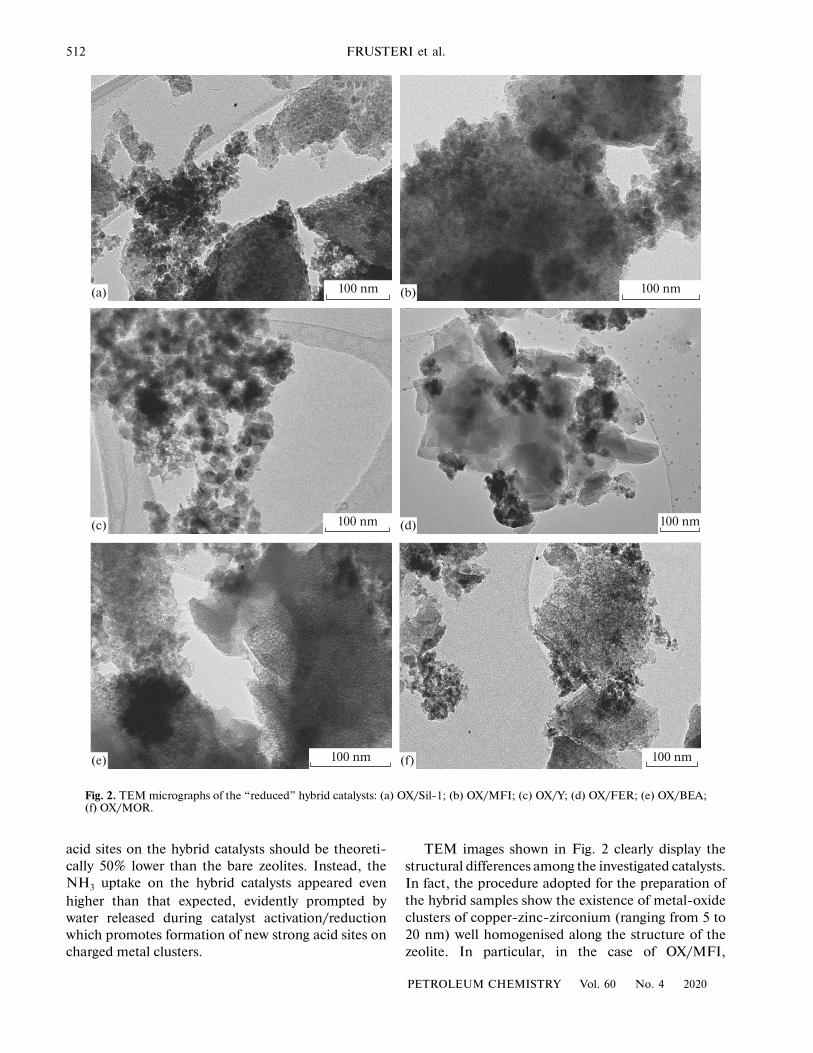
512 FRUSTERI et al.
Fig. 2. TEM micrographs of the “reduced” hybrid catalysts: (a) OX/Sil-1; (b) OX/MFI; (c) OX/Y; (d) OX/FER; (e) OX/BEA;(f) OX/MOR.
100 nm(а) 100 nm(b)
100 nm(c) 100 nm(d)
100 nm(e) 100 nm(f )
acid sites on the hybrid catalysts should be theoreti-cally 50% lower than the bare zeolites. Instead, theNH3 uptake on the hybrid catalysts appeared evenhigher than that expected, evidently prompted bywater released during catalyst activation/reductionwhich promotes formation of new strong acid sites oncharged metal clusters.
TEM images shown in Fig. 2 clearly display thestructural differences among the investigated catalysts.In fact, the procedure adopted for the preparation ofthe hybrid samples show the existence of metal-oxideclusters of copper-zinc-zirconium (ranging from 5 to20 nm) well homogenised along the structure of thezeolite. In particular, in the case of OX/MFI,
PETROLEUM CHEMISTRY Vol. 60 No. 4 2020

PROMOTING DIRECT CO2 CONVERSION 513
Fig. 3. Catalytic data under direct CO2-to-DME hydrogenation conditions (PR: 3.0 MPa; TR, 260°C; GHSV: 8.800 NL/kgcat/h;CO2/H2/N2: 23/69/8).
Time on stream, min
CO
2 con
vers
ion,
%
22
19
16
13
10
600012006000 5400480042003600300024001800
DM
F y
ield
, %
0
600012006000 5400480042003600300024001800
10
8
6
4
2
9
7
5
3
1
OX/YOX/MFIOX/Sil-1
OX/MOROX/BEAOX/FER
Time on stream, min
OX/FER catalysts, with a larger size, the metal oxidesare randomly deposited over the external surface of thezeolite, evidencing a minor interdispersion among theactive sites.
Moreover, when the pore size in the catalystsincreases (as for OX/Y), a core-shell type structure iswell visible, with a core of Cu–Zn–Zr metal-oxidesand an outer shell composed by the zeolite.
The activity pattern over the investigated hybridcatalysts is reported in Fig. 3 as a function of the timeon stream at 30 bar and 260°C. No hydrocarbons orcoke have been found on the used catalysts under theadopted experimental conditions, the only C-contain-ing products resulting DME, methanol and CO.
PETROLEUM CHEMISTRY Vol. 60 No. 4 2020
From a first analysis, the results show that, with theexception of the OX/Sil-1 catalyst, the initial conver-sion of CO2 is always quite high (>20%), although ittends to decrease over time mainly in the first phase ofreaction (TOS = 0–1200 min). Interestingly, theOX/MOR catalyst is the only one that maintains ahigh conversion of CO2 even after 100 h of reaction(from 23 to 20%). Instead, the conversion decay of theremaining hybrids is clearly more pronounced, withthe OX/Y system that shows the largest decrease inactivity over time, with a final conversion value below13%, recording a net decrease of over 9%.
In terms of yield, on OX/Sil-1 there is practicallyno DME formation, while on the other hybrid cata-

514 FRUSTERI et al.
Fig. 4. Space-time yield (STY) over the OX/MOR hybridcatalyst in the range of 200–280°C (PR, 3.0 MPa; GHSV:8.800 NL/kgcat/h; CO2/H2/N2: 23/69/8).
600
250
200
150
100
280260240220200TR, �C
550
500
450
400
350
300
R2 = 0.994
STY
DM
E[g
DM
E/K
g cat
/h]
lysts the trend of DME yield (comprised between 3.5and 8.5%) over time reflects somewhat the decay ofCO2 conversion. Only, on OX/Y the observed trend ofDME yield is quite f lat, suggesting another mecha-nism upon DME formation. Obviously, this aspectrequires further investigation.
Such catalytic behavior may be explained by con-sidering the cooperation among metal-oxide-acidsites. In fact, as the reactivity of the investigated sys-tems seems to be directly related neither to the metalCu exposure or surface basicity or surface acidity, itemerges that the direct CO2-to-DME hydrogenationon CuZnZr/zeolite hybrid systems is to be considereda multi-site reaction, where a balanced distribution ofdifferent sites on catalyst surface becomes determi-nant. In particular, mordenite (MOR) is a 12-MRzeolite with only one-dimensional channels that evi-dently favours not only a certain interdispersionamong the active sites, leading to a final relativelyhigher DME productivity, but also a suitable degree ofanchorage of metal-oxides over the zeolite structure,determining a better exposition and accessibility ofsuch sites.
The progressive loss of activity was mainly to becorrelated to the “negative” role of water formedduring catalytic run, leading to a more and more
extensive wetting of catalytic sites, that favours theincipient occupation of oxygen vacancies responsiblefor CO2 activation with a complete blockage of theactive sites. Thus, the occupancy of oxygen vacanciesby water leads to a weakened activity in methanoldehydration into DME. This does not exclude that thewater formed during the reaction can also be absorbedon the acid sites of the sample, but the absence of cokein the spent catalyst suggests it as a minor phenome-non. After that time, the metal-oxygen pairs and zeo-lite protons, resulting more available, start to drivecompetitive reaction pathways finally leading to adynamic equilibrium that, despite a continuousdecrease of the CO2 conversion, maintains unchangedthe selectivity values of CO, MeOH and DME, as aresult of a balance among the reactions ofMeOH/DME formation or decomposition from andto CO.
On the light of the best activity-stability behaviourof the MOR-based hybrid system, a definitive assess-ment of the catalyst performance is better visible look-ing at DME productivity (STY), reported in Fig. 4.
As displayed, the OX/MOR catalyst features asteady rise of the DME space-time yield with tem-perature, from 181 (200°C) to a maximum of559 gDME/Kgcat/h (280°C).
This result prompts new research efforts to designmore hydrophobic and stable catalytic systems for thedirect CO2-to-DME hydrogenation reaction wherethe formation of water certainly represents a criticalissue.
4. CONCLUSIONSZeolites of different topology were synthesized to
be used as carriers on which a CuZnZr methanolphase was coprecipitated to get hybrid catalysts usefulfor direct DME production via CO2 hydrogenation.BET analysis, N2O chemisorption, TPD results andTEM investigations suggested that the zeolite nature(namely Sil-1, MFI, Y, FER, BEA or MOR) and itsspecific architecture directly affect not only the metal-lic properties, but also the basic and acid capacity, byexerting a synergetic role during catalytic runs. Amongthe prepared zeolites, mordenite revealed to be a veryefficient carrier for metal-oxide sites, leading to anactive and selective hybrid catalyst under the adoptedexperimental conditions, attaining a very high DMEproductivity of ca. 600 gDME/Kgcat/h (TR, 280°C; PR,3.0 MPa). In terms of stability, the progressive block-age of the active sites due to the formation of waterduring the catalytic experiments was recognized as theprimary cause affecting the catalyst stability.
OPEN ACCESS
This article is licensed under a Creative CommonsAttribution 4.0 International License, which permits use,
PETROLEUM CHEMISTRY Vol. 60 No. 4 2020

PROMOTING DIRECT CO2 CONVERSION 515
sharing, adaptation, distribution and reproduction in anymedium or format, as long as you give appropriate credit tothe original author(s) and the source, provide a link to theCreative Commons licence, and indicate if changes weremade. The images or other third party material in this articleare included in the article’s Creative Commons licence,unless indicated otherwise in a credit line to the material. Ifmaterial is not included in the article’s Creative Commonslicence and your intended use is not permitted by statutoryregulation or exceeds the permitted use, you will need toobtain permission directly from the copyright holder. Toview a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
REFERENCES
1. W. Wang, S. P. Wang, X. B. Ma, and J. L. Gong, Chem.Soc. Rev. 40, 3703 (2011).
2. M. Aresta, A. Dibenedetto, and E. Quaranta, J. Catal.343, 2 (2016).
3. G. A. Olah, Angew. Chem. Int. Ed. Engl. 52, 104(2013).
4. S. Brynolf, M. Taljegard, M. Grahn, and J. Hansson,Renew. Sust. Energy Rev. 81, 1887 (2018).
5. S. Kar, J. Kothandaraman, A. Goeppert, andG.K.S. Prakash, J. CO2 Util. 23, 212 (2018).
6. H. H. Shin, L. Lu, C. J. Yang, et al., ACS Catal. 6, 2811(2016).
7. F. Frusteri, L. Frusteri, F. Costa, et al., Appl. Catal., A,545C, 24 (2017).
8. Y. Li, Z. Wang, Z. He, et al., Bioresourc. Technol. 295,122296 (2020).
9. K. Kobl, L. Angelo, Y. Zimmermann, et al., C. R.Chim. 18, 302 (2015),
10. J. Graciani, K. Mudiyanselage, F. Xu, et al., Science345, 546 (2014).
11. F. Arena, K. Barbera, G. Italiano, et al., J. Catal. 249,185 (2007).
12. F. Arena, G. Italiano, K. Barbera, et al., Appl. Catal., A350, 16 (2008).
13. F. Arena, G. Italiano, K. Barbera, et al., Catal. Today143, 80 (2009),
14. G. Bonura, M. Cordaro, C. Cannilla, et al., Appl.Catal., B 152–153, 152 (2014).
15. G. Bonura, A. A. Khassin, T. M. Yurieva, et al., Catal.Today (2018). https://doi.org/10.1016/j.cattpd.2018.10.038
16. G. Bonura, C. Cannilla, L. Frusteri, et al., Catal. To-day. (2019). https://doi.org/10.1016/j.cattpd.2019.08.014
17. J. H. He, Q. L. Qian, J. Ma, et al., Angew. Chem., Int.Ed. Engl. 55, 737 (2016).
18. J. Wei, Q. J. Ge, R. W. Yao, et al., Nat. Commun. 8,15174 (2017).
19. R. W. Liu, Z. Z. Qin, H. D. Ji, and T. M. Su, Ind. Eng.Chem. Res. 52, 16648 (2013).
20. G. Bonura, M. Cordaro, L. Spadaro, et al., Appl.Catal., B 140–141, 16 (2013).
21. G. Bonura, M. Cordaro, C. Cannilla, et al., Catal. To-day 228, 51 (2014).
22. F. Frusteri, M. Cordaro, C. Cannilla, and G. Bonura,Appl. Catal., B 162, 57 (2015).
23. F. Frusteri, G. Bonura, C. Cannilla, et al., Appl. Catal.,B 176–177, 522 (2015).
24. G. Bonura, F. Frusteri, C. Cannilla, et al., Catal. Today277, 48 (2016).
25. G. Bonura, C. Cannilla, L. Frusteri, et al., Catal. Today281, 337 (2017).
26. F. Frusteri, M. Migliori, C. Cannilla, et al., J. CO2 Util.18, 353 (2017).
27. E. Catizzone, G. Bonura, M. Migliori, et al., Mole-cules 23, 31 (2018).
28. T. Numpilai, T. Witoon, N. Chanlek, et al., Appl.Catal., A 547, 219 (2017),
29. G. Bonura, M. Migliori, L. Frusteri, et al., J. CO2 Util.24, 398 (2018).
30. I. Miletto, E. Catizzone, G. Bonura, et al., Materials11, 2275 (2018).
31. E. Catizzone, G. Bonura, M. Migliori, et al., Ann.Chim.–Sci. Mat. 43, 141 (2019).
32. G. Bonura, C. Cannilla, L. Frusteri, and F. Frusteri,Appl. Catal., A, 544C, 21 (2017).
33. M. Gentzen, W. Habicht, D. E. Doronkin, et al., Catal.Sci. Technol. 6, 1054 (2016).
34. C. Tisseraud, C. Comminges, T. Belin, et al., J. Catal.330, 533 (2015).
35. O. Martin, C. Mondelli, D. Curulla-Ferre, et al., ACSCatal. 5, 5607 (2015).
36. K. Kobl, S. Thomas, Y. Zimmermann, et al., Catal. To-day 270, 31 (2016).
37. L. Angelo, M. Girleanu, O. Ersen, et al., Catal. Today270, 59 (2016).
38. X. S. Dong, F. Li, N. Zhao, et al., Appl. Catal., B 191,8 (2016).
39. E. Catizzone, A. Aloise, M. Migliori, and G. Giorda-no, Microporous Mesoporous Mater. 243, 102 (2017).
40. E. Catizzone, A. Aloise, M. Migliori, and G. Giorda-no, Appl. Catal., A 502, 215 (2015).
41. A. Corma, J. Catal. 216, 298 (2003).
PETROLEUM CHEMISTRY Vol. 60 No. 4 2020


















