
Programa de trabajo 2003 - European Medicines Agency · Está disponible previa petición en todas...
59
Agencia Europea para la Evaluación de Medicamentos Programa de trabajo 2003 Adoptado por el Consejo de Administración el 19 de diciembre de 2002
Transcript of Programa de trabajo 2003 - European Medicines Agency · Está disponible previa petición en todas...

Agencia Europea para la Evaluación de Medicamentos
Programa de trabajo 2003
Adoptado por el Consejo de Administración el 19 de diciembre de 2002

The European Agency for the Evaluation of Medicinal Products
Público EMEA, 7 Westferry Circus, Canary Wharf, UK-London E14 4HB.
Tel. (44-20) 74 18 84 00 Fax (44-20) 74 18 84 16 E-mail: http://www.emea.eu.int
EMEA/MB/057/02/es/Final
Programa de trabajo de la
Agencia Europea para la Evaluación de Medicamentos
2003
Adoptado por el Consejo de Administración el 19 de diciembre de 2002

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 2/58
Este Programa de Trabajo correspondiente al período 2003es presentado al Consejo de Administración por el Director Ejecutivo de conformidad con el apartado 3 del artículo 57 del Reglamento (CEE) nº 2309/93 del Consejo. Se remite asimismo al Parlamento Europeo, al Consejo, a la Comisión y a los Estados miembros. Está disponible previa petición en todas las lenguas oficiales de la UE. El sistema europeo ofrece dos vías para la autorización demedicamentos. La EMEA interviene en ambas: El procedimiento centralizado es obligatorio para medicamentos obtenidos por biotecnología, y
los laboratorios pueden optar por él para otros productos nuevos e innovadores. Las solicitudes se envían directamente a la EMEA. Finalizada la evaluación científica, que la Agencia lleva a cabo en un plazo de 210 días, se transmite el dictamen del comité científico a la Comisión Europea, que lo convierte en una autorización de comercialización válida en toda la Unión Europea.
El procedimiento descentralizado (o procedimiento de reconocimiento mutuo) es válido para la
mayoría de los medicamentos convencionales y se basa en el principio del reconocimiento mutuo de las autorizaciones nacionales. Amplía las autorizaciones de comercialización concedidas por un Estado Miembro a otro o a varios de ellos, identificados por el solicitante. Cuando no es posible reconocer la autorización nacional original, las cuestiones en disputa se someten al arbitraje de la EMEA. La opinión del comité científico se transmite a la Comisión Europea.
La Comisión Europea adopta sus decisiones con la colaboración de un comité permanente formado por representantes de los Estados miembros.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 3/58
Contenido Introducción del Director Ejecutivo 4 1. La EMEA en el sistema europeo 5
1.1 Consejo de Administración 5 1.2 Autoridades nacionales competentes 5 1.3 Transparencia 6 1.4 Preparativos para la ampliación de la UE 6 1.5 Preparativos para la revisión del sistema europeo 7 1.6 Revisión de las tasas de la EMEA 7 1.7 Socios internacionales 8 1.8 Departamento Europeo para la Calidad de los Medicamentos 8 1.9 Gestión integral de la calidad y control financiero 8 1.10 Organización interna de la EMEA 9
2. Medicamentos de uso humano 10 2.1 Evaluación inicial 10 2.2 Actividades posteriores a la autorización 12 2.3 Actividades de farmacovigilancia y mantenimiento 13 2.4 Asesoramiento científico y asistencia de protocolo 14 2.5 Arbitraje y remisiones comunitarias 16 Distribución paralela 17 2.7 Actividades internacionales 17 2.8 Medicamentos huérfanos 18 2.9 Grupos de trabajo y grupos ad hoc 20 2.10 Grupo de facilitación del reconocimiento mutuo 21
3. Medicamentos veterinarios 22 3.1 Evaluación inicial 23 3.2 Establecimiento de límites máximos de residuos para sustancias antiguas 24 3.3 Actividades posteriores a la autorización 25 3.4 Actividades de farmacovigilancia y mantenimiento 26 3.5 Asesoramiento científico 26 3.6 Arbitraje y remisiones comunitarias 27 3.7 Partes interesadas 27 3.8 Actividades internacionales 27 3.9 Grupos de trabajo y grupos ad hoc 28 3.10 Grupo de facilitación del reconocimiento mutuo 28
4. Inspecciones 29 4.1 Inspecciones 29 4.2 Acuerdos de reconocimiento mutuo 30 4.3 Muestreos y ensayos 30 4.4 Certificados 31 4.5 Transposición de la Directiva relativa a los ensayos clínicos 31
5. Comunicaciones y redes 32 5.1 Aplicación de la estrategia comunitaria de telemática 32 5.2 TI y dirección de proyectos en la EMEA 34 5.3 Organización de reuniones y conferencias 34 5.4 Gestión y publicación de documentos 35
6. Administración 36 6.1 Personal y presupuesto 36 6.2 Servicios de infraestructura 36 6.3 Contabilidad 37
Anexos 38 Anexo 1 Plan de recursos humanos de la EMEA 2001 -2003 39 Anexo 2 Resumen de los presupuestos de la EMEA para el período 2001 -2003 40 Anexo 3 Documentos orientadores de la EMEA en 2003 41 Anexo 4 Puntos de contacto con la EMEA 45 Anexo 5 Perfil de las personalidades de la EMEA 47

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 4/58
Introducción del Director Ejecutivo Thomas Lönngren Un período de preparación para el cambio y la ampliación… El programa de trabajo para 2003 abarca un período en el que la EMEA y el conjunto de la Unión Europea se prepararán para un importante cambio. La Agencia y sus colaboradores en las autoridades nacionales se enfrentarán a la doble prueba de los cambios previstos en el sistema europeo de autorización y supervisión de medicamentos y los retos que plantea la incorporación de los nuevos Estados miembros a la Unión Europea. …al tiempo que se afrontan cambios en la carga de trabajo… Al mismo tiempo, la EMEA tendrá que seguir prestando atención a la gestión de su actividad básica de evaluación de nuevas solicitudes y supervisión de un número cada vez mayor de medicamentos autorizados. Al final de 2002 había unos 269 medicamentos de uso humano y veterinario autorizados por el procedimiento centralizado, a los que han de añadirse las 38 solicitudes para medicamentos de uso humano y 10 para medicamentos de uso veterinario que se esperan en 2003. …y se asumen nuevos cometidos en el terreno de le salud pública La sociedad civil y la clase política están pidiendo de forma creciente a la Comunidad Europea que asuma un papel de mayor relevancia en la protección y la promoción de la salud pública y veterinaria. Como organismo comunitario responsable de la evaluación y supervisión de los medicamentos, la EMEA tiene una clara función que desempeñar en este ámbito y ha visto cómo las instituciones de la UE y los Estados miembros añadían cometidos nuevos a las responsabilidades que ya tenía. Una de las tareas encomendadas a la Agencia es la puesta en práctica de la estrategia comunitaria de telemática para la regulación de los productos farmacéuticos. La estrategia abarca distintas actividades y operaciones que ayudan directamente a las autoridades competentes y a la industria farmacéutica, pero también iniciativas que permitan el acceso de los pacientes y los profesionales sanitarios a información sobre los medicamentos en toda la Unión Europea. Las terapias nuevas y los avances de las ciencias médicas impondrán nuevas demandas a la Agencia y a sus comités. Tendremos que mejorar la capacidad de la EMEA para ofrecer asesoramiento científico a las empresas durante sus investigaciones y sus actividades de desarrollo de nuevos medicamentos. La Agencia deberá tener también capacidad para vigilar y controlar el riesgo de estos medicamentos una vez que empiecen a utilizarse. La EMEA hizo una contribución esencial a la formulación de una política europea eficaz en materia de medicamentos huérfanos. La Agencia trabajará en estrecha colaboración con las instituciones europeas y con todas las partes interesadas para apoyar la formulación de una política sobre medicamentos de uso pediátrico, además de concluir los preparativos para la entrada en vigor de la Directiva sobre ensayos clínicos.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 5/58
1. La EMEA en el sistema europeo
1.1 Consejo de Administración El Consejo de Administración se reunirá 4 veces en 2003, durando cada reunión 1 día. El Consejo iniciará un nuevo mandato a principios de 2003 y elegirá a sus nuevos presidente y vicepresidente en la reunión de febrero. Se invitará a los observadores de los países candidatos a asistir a la reunión de junio del Consejo de Administración, conjuntamente con los responsables de las autoridades nacionales que no sean miembros del Consejo.
Reuniones del Consejo de Administración en 2003
20 de febrero 2 de octubre
5 de junio 18 de diciembre
El Consejo es responsable de garantizar la aplicación de unas normas adecuadas de gobierno corporativo, adoptar y vigilar la ejecución del programa de trabajo de la Agencia y vigilar sus indicadores del rendimiento. Sus prioridades específicas para 2003 seguirán consistiendo en asesorar al Director Ejecutivo sobre:
la revisión del sistema europeo de autorizaciones de comercialización la adhesión de nuevos Estados miembros a la Unión Europea la creación de un modelo de financiación a largo plazo para la Agencia
1.2 Autoridades nacionales competentes Algunos sitios web útiles: Responsables de las agencias de medicamentos de uso humano http://heads.medagencies.org Responsables de la agencias de medicamentos veterinarios http://www.hevra.org Se estrechará la colaboración entre la EMEA y las autoridades nacionales competentes para la planificación de los recursos necesarios para el funcionamiento de todo el sistema europeo de autorización y supervisión de medicamentos. La EMEA seguirá participando como miembro en las reuniones de los responsables de las agencias de medicamentos de uso humano y veterinario. La Declaración de los principios que rigen la colaboración entre las autoridades nacionales competentes y la EMEA, adoptada en 1997, se revisará en 2003. También se revisará el contrato tipo para la prestación de servicios científicos y de inspección en nombre de la EMEA.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 6/58
1.3 Transparencia A principios de 2003 está prevista la modificación de la legislación por la cual se creó la Agencia, para incluir a la EMEA en el ámbito de aplicación del Reglamento (CE) nº 1049/2001 sobre el acceso público a los documentos (DO L 145, 31.5.2001, pág. 43). En todos los organismos de la UE se realizarán cambios similares. Estas disposiciones reemplazarán a la Decisión del Director Ejecutivo de 3 de diciembre de 1997 sobre las normas de acceso a los documentos de la EMEA. No se espera que el Reglamento repercuta de manera importante en los recursos de la Agencia y la aplicación del mismo se vigilará durante todo el año. Además de este cambio legislativo, el Parlamento Europeo y el Consejo seguirán considerando una serie de propuestas para mejorar la transparencia de la EMEA como parte de la revisión de la legislación farmacéutica de la UE. Pendiente del resultado de estos debates políticos, la EMEA propondrá algunas iniciativas en 2003, destinadas sobre todo a mejorar la información sobre las actividades de la Agencia relacionadas con los medicamentos de uso humano y veterinario que ya se comercializan. En concreto, se publicarán resúmenes de los dictámenes cuando se aprueben nuevas indicaciones o información sobre cuestiones importantes relacionadas con la seguridad. Otras iniciativas consisten en la actualización periódica de los informes públicos europeos de evaluación (EPAR) para incluir información sobre las actividades posteriores a la autorización. La EMEA consultará a las partes interesadas antes de adoptar estas iniciativas. La principal herramienta de comunicación sigue siendo el sitio web de la EMEA – http://www.emea.eu.int – y se hará un esfuerzo para asegurar un desarrollo y un mantenimiento adecuados del sitio.
1.4 Preparativos para la ampliación de la UE Algunos sitios web útiles: Foro Paneuropeo de Regulación http://perf.eudra.org Dirección General de Ampliación de la Comisión Europea http://europa.eu.int/comm Autoridades nacionales competentes de los países candidatos para los medicamentos de uso humano http://www.cadreac.org Autoridades nacionales competentes de los países candidatos para los medicamentos veterinarios http://www.cavdri.info El Consejo de la Unión Europea ha fijado como fecha para la ampliación propuesta de la Unión Europea el 1 de mayo de 2004. Con esta ampliación, el número de Estados miembros que participen en el trabajo de la EMEA pasará de 15 a 25 (Chipre, República Checa, Estonia, Hungría, Letonia, Lituania, Malta, Polonia, República Eslovaca y Eslovenia), además de los estados del EEE-AELC Islandia, Liechtenstein y Noruega. La ampliación exigirá unos preparativos considerables. Habrá que adoptar las medidas necesarias para la incorporación gradual de los países candidatos a las actividades de la Agencia, empezando por su participación como observadores en las reuniones de los comités científicos y los grupos de trabajo de la EMEA en 2003. Por limitaciones presupuestarias, en 2003 no se han podido asignar recursos adicionales para el trabajo de la EMEA relacionado con la ampliación. La Agencia sigue comprometida a trabajar con sus colaboradores en los países candidatos y en 2003 se acometerá la tercera fase del Foro Paneuropeo para la Regulación de los Productos Farmacéuticos

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 7/58
(PERF III). El PERF se financia con cargo al programa PHARE de la Comisión Europea y tiene un presupuesto total de 1.430.000 euros. Las actividades incluidas en el PERF III se dividen en 6 áreas de acción prioritaria: Farmacovigilancia Buenas prácticas de fabricación Evaluación de expedientes
Cuestiones veterinarias Aplicación del acervo comunitario Telemática
En cada una de estas áreas de acción prioritaria se organizará un programa de formación práctica en el que participarán expertos de las autoridades competentes de la UE y de los países candidatos. Como parte de las cuatro áreas técnicas de acción prioritaria, se realizará también un estudio comparativo con la finalidad de establecer buenas prácticas reguladoras. El PERF III incluirá asimismo tres conferencias públicas sobre la industria farmacéutica (febrero de 2003), los medicamentos veterinarios (julio de 2003) y los pacientes y los profesionales de la salud (octubre de 2003).
1.5 Preparativos para la revisión del sistema europeo Algunos sitios web útiles: Dirección General de Empresas de la Comisión Europea/ Unidad de Productos Farmacéuticos http://pharmacos.eudra.org/f2 El programa de trabajo y el presupuesto para 2003 se han preparado partiendo del supuesto de que las propuestas de la Comisión Europea para la revisión del sistema europeo de autorización y supervisión de medicamentos no entrarán en vigor hasta 2005 (DO C 75 E, 26.3.2002, p. 189 et seq.). La EMEA contribuirá según sea necesario al trabajo de las instituciones de la UE en su examen de las propuestas. La Agencia vigilará también de cerca la posible repercusión de dichas propuestas en sus recursos y actividades.
1.6 Revisión de las tasas de la EMEA A principios de 2003 está previsto un aumento de las tasas abonadas a la EMEA por los solicitantes y titulares de autorizaciones de comercialización. Esta subida de las tasas se realizará mediante un Reglamento adoptado por la Comisión Europea. El nivel total de incremento solicitado por la EMEA es del 16,6%, para tener en cuenta la inflación desde la última revisión de 1998 y la necesidad de generar un nivel suficiente de ingresos para que la EMEA pueda funcionar. En 2003 se emprenderá una iniciativa paralela para revisar la estructura del sistema actual de tasas, teniendo en cuenta la repercusión de la revisión de la legislación farmacéutica europea y la ampliación.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 8/58
1.7 Socios internacionales Algunos sitios web útiles: Conferencia Internacional de Armonización http://www.ich.org Conferencia Veterinaria Internacional de Armonización http://vich.eudra.org Organización Mundial de la Salud http://www.who.int Dirección General de Investigación de la Comisión Europea http://europa.eu.int/comm Además de las actividades de la Agencia relacionadas con la futura ampliación de la UE, se intensificará la colaboración entre la EMEA y otras autoridades reguladoras internacionales. La Agencia mantendrá su compromiso con los procesos de la Conferencia Internacional para la Armonización de los Requisitos Técnicos para el registro de Medicamentos de Uso Humano y Veterinario (ICH y VICH) y, en particular, contribuirá a la sexta Conferencia ICH que se celebrará en Osaka, Japón, en 2003. Se estrechará la cooperación con autoridades sanitarias como la Food and Drug Administration (FDA) de Estados Unidos. Los programas de estancias de expertos proseguirán en 2003, acogiendo la EMEA a expertos de las autoridades canadienses y japonesas durante el año. Se reforzará también la cooperación con la Organización Mundial de la Salud, sobre todo en el campo de la farmacovigilancia. Asimismo, la Agencia apoyará las actividades englobadas en el sexto programa marco de la Unión Europea para la investigación y el desarrollo tecnológico en cuestiones relacionadas con el desarrollo de medicamentos dirigidos al Tercer Mundo.
1.8 Departamento Europeo para la Calidad de los Medicamentos Algunos sitios web útiles: Departamento Europeo para la Calidad de los Medicamentos/ Farmacopea Europea http://www.pheur.org El programa de muestreos y ensayos de medicamentos autorizados por el procedimiento centralizado en 2003 se llevará a cabo a través de la red de Laboratorios Oficiales de Control de Medicamentos del EDQM en la UE y los estados del EEE-AELC. El programa de 2003 será el más extenso hasta la fecha, con un 20% más de productos sometidos a muestreos y ensayos que en 2002. La Agencia seguirá colaborando con el trabajo de la Comisión de la Farmacopea Europea. Representantes del Departamento Europeo para la Calidad de los Medicamentos (EDQM) participarán también en algunos grupos de trabajo de la EMEA.
1.9 Gestión integral de la calidad y control financiero La atención se centrará en la implantación de los sistemas integrados de gestión con los objetivos de mejorar continuamente los procesos de la EMEA y crear un registro de riesgos operativos, apoyados por auditorías internas y formación para el desarrollo de capacidades. Continuará el estudio comparativo de las buenas prácticas reguladoras con colaboradores de las autoridades de la UE, el EEE-AELC y los países candidatos, además de las instituciones europeas.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 9/58
La función de control financiero y auditoría de la EMEA se adaptará al nuevo marco de la Comisión Europea para la administración de recursos y las auditorías internas. Se tendrán en cuenta las actividades ya existentes de auditoría interna y la opinión del Consejo de Administración sobre distintas alternativas para asegurar la continuidad del control financiero y las auditorías dentro de la Agencia en el marco del gobierno corporativo de la EMEA.
1.10 Organización interna de la EMEA La Agencia disfruta ya de una estructura interna estable, con 5 unidades que realizan las actividades científicas y las funciones de apoyo de la EMEA. En 2003 se espera introducir una función reforzada de apoyo a la gestión para atender la creciente diversidad de actividades de la EMEA y la necesidad de asegurar la coordinación interna dentro de la Agencia. Esta función se centrará especialmente en apoyar al equipo directivo interno del Director Ejecutivo y de los Jefes de Unidad y contribuirá también a coordinar las actividades internacionales, científicas y de comunicación de la Agencia.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 10/58
2. Medicamentos de uso humano Prioridades para medicamentos de uso humano en 2003: Hacer frente a la carga de trabajo y cumplir los plazos establecidos para las actividades previas y
posteriores a la autorización.
Facilitar y mejorar la transmisión electrónica de informes de casos individuales de seguridad (ICSRs) a través de la base de datos EudraVigilance implantada.
Finalizar y aplicar la estrategia de gestión de riesgos de la EMEA y colaborar con los responsables de las autoridades nacionales para la formulación de una estrategia europea de gestión de riesgos.
Hacer frente a la carga de trabajo, cumplir los plazos establecidos y mejorar el procedimiento de asesoramiento científico y asistencia de protocolo.
Prestar atención al concepto de gestión del ciclo de vida de los medicamentos, mejorando las actividades y los procedimientos (asesoramiento científico, dictámenes del CPMP y gestión de riesgos) y la memoria reglamentaria.
Mejorar la política de transparencia de la Agencia, perfeccionando sus actuales herramientas de comunicación.
Hacer frente a la carga de trabajo y cumplir los plazos establecidos para las actividades relacionadas con la designación y el seguimiento de los medicamentos huérfanos.
2.1 Evaluación inicial La evaluación inicial abarca las actividades de la EMEA desde el diálogo con los futuros solicitantes antes de presentar la solicitud hasta la evaluación del CPMP, la autorización del producto y la publicación del informe público europeo de evaluación (EPAR).
Solicitudes de evaluación inicial (Entrada)
46
19
22
12
12
16
0 20 40 60 80
2001
2002
2003
Solicitudes recibidas (medicamentos no huérfanos)Solicitudes recibidas (medicamentos huérfanos)

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 11/58
Tendencias: Mismo nivel de solicitudes iniciales de autorizaciones de comercialización, continuando la
tendencia observada en 2002, con una previsión de 38 solicitudes y muy pocas solicitudes múltiples.
Ligero aumento de solicitudes iniciales para medicamentos huérfanos. Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Creación de grupos de asesoramiento terapéutico en el CPMP para prestar asesoramiento
especializado a los ponentes, ponentes adjuntos y el CPMP sobre aplicaciones específicas y elaboración de directrices. Mayor participación del personal de la EMEA en apoyo de los grupos terapéuticos como coordinadores. En la primera etapa piloto se crearán grupos de asesoramiento terapéutico en tres áreas: antiinfecciosos, oncología y agentes diagnósticos.
El aumento del número de solicitudes para la designación de medicamentos huérfanos – cerca del 40 % de las solicitudes totales en 2003 – repercutirá en los procedimientos de trabajo. Estas solicitudes suelen entrañar una complejidad considerable y requieren más recursos que los medicamentos no huérfanos.
Formulación de una estrategia de la EMEA para la gestión de riesgos y formulación de una estrategia europea en colaboración con las autoridades nacionales.
Se prevén los primeros problemas relacionados con la exclusividad comercial durante 10 años de los medicamentos huérfanos.
Desde mediados de 2003 será obligatorio presentar las solicitudes utilizando el formato del documento técnico común (CTD) y se espera recibir un mayor número de solicitudes con el formato electrónico del CTD en el segundo semestre del año. Este formato se aprobó en 2002 en la Conferencia internacional para la armonización (ICH) trilateral de la UE, Japón y Estados Unidos. El personal y los expertos de la EMEA deberán recibir formación y familiarizarse con el nuevo formato.
El CPMP iniciará sus actividades de colaboración con las organizaciones de pacientes, estando previstas 4 reuniones en 2003.
Objetivos: Cumplimiento de los plazos de revisión por parte del CPMP Publicación de resúmenes de los dictámenes en el momento de su adopción por el CPMP Rápida publicación de los EPAR tras la decisión de la Comisión Europea de conceder una
autorización de comercialización. Seguimiento de la creación por el CPMP de los grupos de asesoramiento terapéutico y apoyo
activo prestado por la Secretaría de la Agencia. Esfuerzos continuados en apoyo de las crecientes actividades del CPMP en términos de carga de
trabajo, nuevos desafíos científicos y de salud pública, medicamentos huérfanos y nuevas terapias. Gestión y organización del CPMP Está previsto que el Comité de Especialidades Farmacéuticas (CPMP) se reúna 11 veces en 2003. Además, podrá celebrarse una reunión extraordinaria del CPMP en caso necesario. El CPMP seguirá revisando sus procedimientos de trabajo con objeto de realizar los cambios necesarios para mejorar el funcionamiento del Comité y del procedimiento centralizado, con la creación de nuevas herramientas de aseguramiento de la calidad y el seguimiento de las herramientas ya introducidas, las actividades de sus grupos satélite (Grupo de revisión de nombres arbitrarios, Grupo para asuntos de organización, reunión del presidente del CPMP y los grupos de trabajo). El CPMP proseguirá el debate sobre la puesta en práctica de la estrategia de gestión de riesgos de la EMEA, centrándose especialmente en la naturaleza y la extensión de los cambios que se introducirán en la organización para crear un sistema más adecuado y capaz de resolver los problemas de seguridad con eficiencia y rapidez, tanto antes como después de la autorización de los medicamentos, pudiendo tomar así decisiones más fundadas desde el punto de vista científico.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 12/58
Reuniones del CPMP en 2003
21-23 de enero 22-24 de julio
18-20 de febrero No se celebra reunión en agosto
18-20 de marzo 23-25 de septiembre
23-25 de abril 21-23 de octubre
20-22 de mayo 18-20 de noviembre
24-26 de junio 16-18 de diciembre
Se designarán ponentes y ponentes adjuntos en cada reunión
2.2 Actividades posteriores a la autorización Aquí se incluyen las actividades relacionadas con las variaciones, ampliaciones y transferencias de autorizaciones de comercialización. Los cambios en las autorizaciones de comercialización se denominan variaciones, y pueden ser de menor (de tipo I) o mayor (de tipo II) importancia. Esta clasificación de los cambios se establece en la legislación de la UE.
Solicitudes recibidas (Entrada)
421
468
500245
256
23637
13
22
0 100 200 300 400 500
2001
2002
2003
Variaciones tipo I Variaciones tipo II Ampliaciones
Dictámenes adoptados (Salida)
438
463
480
264
269
24514
14
20
0 100 200 300 400 500
2001
2002
2003
Variaciones tipo I Variaciones tipo II Ampliaciones

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 13/58
Tendencias: Estabilidad o ligero aumento de las solicitudes de variaciones tipo II y I, respectivamente, con la
legislación vigente. La misma tendencia se espera en las solicitudes de ampliaciones de línea.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: La entrada en vigor del nuevo Reglamento comunitario sobre las variaciones podría influir en el
número y el tipo de solicitudes tipo I y II en 2003, que se vigilarán de cerca. El nuevo Reglamento propone la introducción de un nuevo tipo de variación cuya responsabilidad
asumirá plenamente la EMEA, sin la intervención de un ponente y un ponente adjunto. El concepto de reuniones anuales con los titulares de las autorizaciones de comercialización para
planificar la estrategia posterior a la autorización de cada producto, que se inició como un proyecto piloto en 2002, se implantará en 2003.
Seguirá mejorándose la política de transparencia de la EMEA, con la actualización procedimental y científica periódica de los informes públicos europeos de evaluación (EPAR) para todos los medicamentos autorizados por el procedimiento centralizado (cerca de 250 productos en 2003), así como la publicación de resúmenes de dictámenes para algunas actividades posteriores a la autorización.
Objetivos: Cumplimiento de los plazos de revisión por parte del CPMP Publicación de resúmenes de dictámenes en el momento de su adopción por el CPMP para algunas
actividades posteriores a la autorización que repercuten significativamente en el uso del medicamento.
Actualización periódica de los aspectos procedimentales y científicos del EPAR en la fase posterior a la autorización.
2.3 Actividades de farmacovigilancia y mantenimiento Aquí se incluyen actividades relacionadas con la farmacovigilancia (informes sobre reacciones adversas a medicamentos (RAM) e informes periódicos actualizados de seguridad (PSUR), medidas de seguimiento, obligaciones específicas, reevaluaciones anuales y renovaciones de las autorizaciones de comercialización.
Informes recibidos/esperados sobre reacciones adversas a medicamentos
dentro y fuera de la UE (Entrada)
20000
27800
3260015300
14808
14334
0 5000 10000 15000 20000 25000 30000 35000
2001
2002
2003
RAM en la UE RAM fuera de la UE

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 14/58
Informes periódicos actualizados de seguridad, medidas de seguimiento y obligaciones
específicas (Entrada)
654
738
700
180
223
264
0 200 400 600 800
2001
2002
2003
PSUR FUM y SO
Tendencias: Aumento continuado de las actividades de mantenimiento en comparación con 2002.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Mantener, actualizar y mejorar la base de datos Eudravigilance y la red de procesamiento de datos.
Las actividades previstas están relacionadas con la estrecha vigilancia del progreso en la implantación del sistema de transmisión de informes de seguridad de casos individuales (ICSR), incluidos el aseguramiento de la calidad, la gestión del trabajo atrasado con estos informes, la preparación y coordinación de un programa de formación para la industria farmacéutica y un mayor desarrollo del sistema, con la incorporación de herramientas de transmisión electrónica para pequeñas y medianas empresas.
Hacer frente, como parte de la aplicación de la estrategia de gestión de riesgos de la EMEA, a un aumento importante del número de reuniones del Grupo de Trabajo “Farmacovigilancia”, así como a la contribución de los expertos en la fase previa y posterior a la autorización.
Contribuir, junto con los responsables de las agencias nacionales, a la formulación de una estrategia europea de gestión de riesgos, realizando la contribución necesaria a los distintos aspectos de esa estrategia.
2.4 Asesoramiento científico y asistencia de protocolo Este ámbito se refiere a la prestación de asesoramiento científico y asistencia de protocolo a los promotores durante la investigación y el desarrollo de medicamentos. El asesoramiento científico constituye un área prioritaria para la EMEA y se ofrece para cualquier aspecto de la investigación y el desarrollo que tenga relación con la calidad, la seguridad o la eficacia de los medicamentos. Además, la Agencia ofrece asesoramiento a los promotores de los medicamentos designados huérfanos a través de la asistencia de protocolo, que puede incluir también asesoramiento sobre los beneficios más significativos de su producto.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 15/58
Solicitudes de asesoramiento científico y asistencia de protocolo (Entrada)
59
75
5
25
56
15
0 20 40 60 80
2001
2002
2003
Solicitudes de asistencia de protocolo y seguimientoSolicitudes de asesoramiento científico y seguimiento
Asesoramiento científico y asistencia de protocolo concluidos (Salida)
65
65
68
4
13
18
0 20 40 60 80
2001
2002
2003
Asistencia de protocolo y seguimiento concluidosAsesoramiento científico y seguimiento concluidos
Tendencias: Aumento continuado del número de solicitudes de asesoramiento científico y seguimiento. Aumento significativo del número de solicitudes de asistencia de protocolo. Aumento general de la carga de trabajo del 20%.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: El asesoramiento científico y la asistencia de protocolo constituyen un área prioritaria del trabajo
de la EMEA. El asesoramiento científico se prestará en 2003 a través del Grupo de trabajo sobre asesoramiento científico.
La situación de este grupo ha cambiado al haberse convertido en un grupo de trabajo del CPMP. El grupo se reunirá 11 veces en 2003, durando las reuniones 2 días enteros y celebrándose todos los meses separadas del CPMP. De esta forma se dejará más tiempo para el diálogo entre el grupo y las empresas que solicitan asesoramiento y para una mejor preparación de las conclusiones del asesoramiento científico antes de las reuniones del CPMP, pudiendo ofrecer un asesoramiento y asistencia más rápidos a los promotores.
Mayor participación de los expertos externos en cuestiones relacionadas con enfermedades comunes y poco comunes.
Se vigilará y analizará la repercusión del asesoramiento científico en el resultado de las solicitudes de autorización de comercialización como parte de la memoria científica y las bases de datos sobre asesoramiento científico.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 16/58
Aumento de la comunicación y la interacción con las partes interesadas, incluidas las instituciones académicas.
Objetivos: Vigilancia de la aplicación del nuevo procedimiento en términos de plazos, reuniones con los
promotores y participación de expertos externos. Vigilancia continua de la repercusión de los procedimientos de asesoramiento científico y
asistencia de protocolo en las futuras solicitudes de autorización de comercialización.
2.5 Arbitraje y remisiones comunitarias Los procedimientos de arbitraje (conforme al artículo 29 de la Directiva 2001/83/CE o el apartado
5 del artículo 7 del Reglamento (CE) nº 542/95 de la Comisión) se inician por discrepancia entre los Estados miembros en el marco del procedimiento de reconocimiento mutuo.
Las remisiones conforme al artículo 30 se inician principalmente para conseguir la armonización en la Comunidad de las condiciones de autorización de productos ya autorizados en los Estados miembros.
Los procedimientos de remisión conforme a los artículos 31 y 36 se inician principalmente en caso de interés comunitario y por cuestiones relacionadas con la seguridad.
Arbitraje y remisiones comunitarias a la EMEA (Entrada)
6
23
24
0 5 10 15 20 25 30
2001
2002
2003
Arbitraje y remisiones comunitarias
Tendencias: En 2003 se esperan un total de 24 arbitrajes y remisiones comunitarias. Se espera que el número de remisiones relacionadas con cuestiones de farmacovigilancia se
mantenga al mismo alto nivel que en 2002. Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: La carga de trabajo generada por las remisiones de armonización aumentará dependiendo de las
primeras experiencias con el proyecto europeo para armonizar los resúmenes de las características del producto de algunos medicamentos en las principales áreas terapéuticas.
La carga de trabajo generada por las remisiones es mayor que la generada por las solicitudes de autorizaciones de comercialización. Las remisiones suelen implicar a muchas autorizaciones de comercialización y titulares de autorizaciones de comercialización.
La transparencia del arbitraje y las remisiones comunitarias mejorarán con la publicación puntual de información explicando los motivos en los que se basan las recomendaciones del CPMP, las condiciones y, en su caso, los cambios efectuados en la información sobre el producto.
Objetivos: Hacer frente a la carga de trabajo relacionada con remisiones y arbitrajes. Cumplir los plazos establecidos para el arbitraje y las remisiones comunitarias. Publicar puntualmente información sobre los procedimientos de remisión y arbitraje.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 17/58
Distribución paralela Una autorización de comercialización comunitaria es válida en toda la UE y un medicamento autorizado por el procedimiento centralizado es, por definición, idéntico en todos los Estados miembros. Los productos lanzados al mercado en un Estado miembro pueden ser comercializados en cualquier otra parte de la Comunidad por un distribuidor («distribuidor paralelo») independiente del titular de la autorización de comercialización. Esto suele hacerse para beneficiarse de las diferencias de precios. La EMEA comprueba que los productos distribuidos en paralelo cumplen las condiciones establecidas en la autorización de comercialización comunitaria.
Notificaciones recibidas (Entrada)
366
418
300
0 100 200 300 400 500
2001
*2002
2003
Notificaciones de distribución paralela
* 294 de las 418 notificaciones recibidas en 2002 fueron válidas Tendencias: En 2003 se espera que el número de notificaciones iniciales válidas de distribución paralela se
mantenga relativamente estable, mientras que el número de notificaciones de cambios seguirá aumentando debido a la actualización del etiquetado de los productos.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Aumentará la presión para mantener o mejorar los tiempos de procesamiento, pero se necesitará
una revisión importante del proceso dependiendo de los recursos disponibles. Objetivos: Simplificar el procedimiento actual teniendo en cuenta la experiencia adquirida hasta la fecha,
especialmente mediante el diseño de una solución pragmática para poder disponer con rapidez de las actualizaciones del etiquetado de los productos.
2.7 Actividades internacionales Tendencias: Se espera que el nivel de actividades internacionales se mantenga elevado, tanto en términos de
los compromisos de la Agencia con sus colaboradores internacionales como del interés en el trabajo de la Agencia por parte de las autoridades reguladoras no comunitarias.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Participación activa en el PERF III tanto para la evaluación previa como posterior a la
autorización de medicamentos de uso humano en 2003. Apoyo a los observadores de los países candidatos cuando empiecen a participar en el trabajo de
los comités científicos y los grupos de trabajo de la Agencia. Relaciones con la FDA de Estados Unidos para seguir reforzando la cooperación en áreas tales
como medicamentos huérfanos, asesoramiento científico, nuevas solicitudes y farmacovigilancia, y con intercambio de personal en prácticas.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 18/58
Las relaciones con Canadá y Japón continuarán con el programa de estancias de expertos destacados en la EMEA.
Aumentarán las actividades de la ICH, sobre todo en la preparación de la VI Conferencia ICH que se celebrará en otoño de 2003 en Japón.
Colaboración y participación en las reuniones científicas y las actividades de formación, conjuntamente o a petición de la Organización Mundial de la Salud (OMS).
Contribución a las actividades del Observatorio Europeo de la Droga y las Toxicomanías (EMCDDA) con sede en Lisboa por medio de la contribución a las Acciones Conjuntas de la UE y el proyecto Trend.
Contribución a las actividades de la Dirección General de Investigación de la Comisión Europea relacionadas con los medicamentos destinados a los países en desarrollo.
2.8 Medicamentos huérfanos Los medicamentos huérfanos se desarrollan para el diagnóstico, la prevención o el tratamiento de enfermedades que ponen en peligro la vida o producen debilitamiento crónico y que afectan a no más de cinco por 10.000 personas en la Comunidad Europea, o cuando por razones económicas esos medicamentos no se desarrollarían sin incentivos. El Comité de Medicamentos Huérfanos (COMP) tiene la responsabilidad de recomendar a la Comisión Europea la designación de medicamentos huérfanos para enfermedades poco comunes. Es responsable de asesorar a la Comisión Europea sobre la creación de una política de medicamentos huérfanos, y de colaborar en la relación con los socios y organizaciones de pacientes internacionales a este respecto. La contribución especial de la Comunidad («contribución para medicamentos huérfanos») debe servir para apoyar las solicitudes nuevas y la asistencia de protocolo, además de las actividades posteriores a la autorización, ante el creciente número de medicamentos huérfanos con autorizaciones de comercialización comunitarias. Se espera que la contribución para medicamentos huérfanos aprobada por las autoridades presupuestarias de la Unión Europea en 2003 ascienda a 3.300.000 euros. Para satisfacer las expectativas de los promotores y las organizaciones de pacientes y tener en cuenta la cuantía del fondo para los medicamentos huérfanos en 2003, se propone que el nivel de reducción de las tasas cubra: el 100% de las tasas para la asistencia de protocolo y el 50% para las solicitudes iniciales y las
inspecciones relacionadas el 50 % de las tasas para las solicitudes posteriores a la autorización, dando prioridad a los
medicamentos huérfanos el primer año de su autorización.
Solicitudes de designación (Entrada)
84
79
75
0 20 40 60 80 100
2001
2002
2003
Solicitudes de designación

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 19/58
Dictámenes sobre la designación de medicamentos huérfanos (Salida)
62
44
50
0 20 40 60 80 100
2001
2002
2003
Dictámenes emitidos sobre la designación
Tendencias: Tras el gran número inicial de solicitudes de designación de medicamentos huérfanos que se
recibieron tras la adopción la política comunitaria sobre los medicamentos huérfanos, se espera que ese número se mantenga estable en relación con 2002, con 75 solicitudes.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Con 134 medicamentos designados como huérfanos a finales de 2002, se espera que la carga de
trabajo posterior a la designación aumente en 30% en 2003. Procesamiento y seguimiento de unos 100 informes anuales que se presentarán en 2003 para
medicamentos designados como huérfanos. Incremento de las actividades de seguimiento y evaluación de los criterios de designación de
medicamentos huérfanos en el momento de la autorización de comercialización, al aumentar las solicitudes de autorización para medicamentos huérfanos.
Se espera que las solicitudes de designación de medicamentos huérfanos incluyan terapéuticas nuevas más complejas.
Nuevas iniciativas para mejorar la transparencia en el procedimiento de designación de medicamentos huérfanos.
Mayor cooperación con las autoridades reguladoras internacionales y las instituciones de la Unión Europea.
Objetivos: Cumplimiento de los plazos establecidos para las solicitudes de designación de medicamentos
huérfanos. Publicación de resúmenes de dictámenes cuando la Comisión Europea decida sobre su
designación. Gestión y organización del COMP El COMP celebrará 11 reuniones en 2003, reuniéndose 2 días cada mes. El mandato del Comité actual concluye en 2003 y en abril de ese año se designará a un nuevo Comité. El nuevo Comité elegirá también a su presidente y vicepresidente en esa misma reunión de abril.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 20/58
Reuniones del COMP en 2003
9-10 de enero 29-30 de julio
6-7 de febrero No se celebra reunión en agosto
18-19 de marzo 9-10 de septiembre
14-15 de abril 9-10 de octubre
7-8 de mayo 6-7 de noviembre
12-13 de junio 4-5 de diciembre
2.9 Grupos de trabajo y grupos ad hoc Los grupos de trabajo de los comités científicos de la EMEA responsables de medicamentos de uso humano participan en la redacción y revisión de directrices, la formulación de recomendaciones y el asesoramiento sobre medicamentos para los que se solicita la designación de huérfanos, el asesoramiento científico, la ayuda a los protocolos y las actividades de autorización de comercialización o posteriores a la autorización, en función del área concreta de responsabilidad de cada comité. Su labor incluye asesoramiento y recomendaciones sobre cuestiones generales de salud pública relacionadas con los medicamentos. Esta actividad también respalda el trabajo de las autoridades nacionales competentes en el funcionamiento del procedimiento de reconocimiento mutuo. Muchas de las directrices adoptadas por el CPMP se derivan del trabajo de la Agencia como parte de la Conferencia internacional trilateral UE-Japón-Estados Unidos para la armonización de los requisitos técnicos de los medicamentos de uso humano (ICH). El proceso ICH cuenta con la participación de las autoridades reguladoras y la industria farmacéutica en la elaboración de directrices. Este proceso realiza una importante contribución a la armonización de los requisitos técnicos y reglamentarios para la investigación y el desarrollo de medicamentos. Tendencias: Está previsto que se adopten o publiquen para consulta 60 proyectos de directrices o directrices
definitivas del CPMP. Está previsto que se adopten o publiquen para consulta 5 directrices ICH-CPMP.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Los grupos de trabajo del CPMP simplificarán sus procesos y criterios en materia de transparencia
y eficacia. Los grupos ad hoc de nuevas terapias emergentes, nuevas tecnologías y reproducibilidad de
medicamentos biotecnológicos seguirán reuniéndose en 2003. Esto será importante mientras la Agencia se prepara para recibir tales solicitudes en el futuro y contribuye también a las iniciativas internacionales de regulación que forman parte del proceso ICH.
El trabajo que se realizará en 2003 para preparar la legislación comunitaria sobre los medicamentos de uso pediátrico prevista en 2004 incluirá apoyo a la Comisión Europa en la elaboración de sus propuestas. El Grupo de expertos en pediatría se reunirá con varias empresas para discutir el desarrollo de formulación pediátricas y trabajar en relación con la disponibilidad de información sobre medicamentos de uso pediátrico.
Están previstas también nuevas actividades relacionadas con ficheros maestros de plasma, ficheros maestros de antígenos para vacunas, equipos médicos que contienen productos de biotecnología y hemoderivados.
Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 21/58
Trabajo y conocimientos necesarios para ayudar al CPMP a asesorar a la Comisión Europea sobre terrorismo químico en 2003 y seguimiento del trabajo iniciado en 2002 sobre amenazas bioterroristas.
El Grupo ad hoc de biotecnología del COMP se reunirá todas las veces que sea necesario para apoyar el proceso de designación para terapias nuevas, y el grupo del COMP con las partes interesadas seguirá reuniéndose una vez cada tres meses.
Se prestará apoyo a los seminarios organizados por iniciativa del COMP y el CPMP sobre nuevos aspectos científicos y metodológicos, así como a las actividades relacionadas con la formación de auditores nacionales que se acuerden con los comités científicos de la EMEA y las autoridades nacionales competentes de la UE.
Los recursos que demandarán la elaboración de directrices nuevas y la revisión de directrices serán vigilados de cerca por el CPMP y los grupos de trabajo cuando planifiquen sus actividades. Se crearán nuevas herramientas para conocer los recursos disponibles en colaboración con las autoridades nacionales competentes, simplificando y mejorando el proceso actual. Grupos de trabajo y grupos ad hoc del CPMP
Número de reuniones
Nuevas directrices
Directrices actuales
Grupo de trabajo “Farmacovigilancia” 11 9 17 Grupo de trabajo “Biotecnología” 9 6 13 Grupo de trabajo conjunto CPMP/CVMP “Calidad”
4 7 9
Grupo de trabajo "Hemoderivados" 3 4 12 Grupo de trabajo “Eficacia” 4 2 28 Grupo de trabajo “Seguridad” 3 3 10 Grupo de trabajo sobre medicamentos de origen vegetal
3 18 28
Grupo de expertos en pediatría 4 -- -- Grupo de expertos en vacunas 4 3 4 En el anexo 3 se facilitan detalles sobre los documentos orientadores que está previsto publicar para consulta o concluir en 2003.
2.10 Grupo de facilitación del reconocimiento mutuo Algunos sitios web útiles: Responsables de las agencias de medicamentos humanos http://heads.medagencies.org Secretaría de la EMEA/MRFG (e-mail) email: [email protected] Índice europeo de productos http://mri.medagencies.com/prodidx El Grupo de Facilitación del Reconocimiento Mutuo (MRFG) seguirá recibiendo el apoyo de la EMEA en sus reuniones mensuales, que se celebrarán el día anterior al inicio de las reuniones del CPMP.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 22/58
3. Medicamentos veterinarios Prioridades para medicamentos de uso veterinario en 2003: Avanzar el trabajo de definición de las normas de la UE para la transmisión electrónica con el fin
de implantar el programa EudraVigilance, ya retrasado en el sector veterinario durante el año. Ese retraso se debe a que el proceso VICH para concluir la directriz sobre los datos para la transmisión electrónica de presuntas reacciones adversas a medicamentos veterinarios no ha progresado.
De acuerdo con su directriz sobre la extrapolación de límites máximos de residuos (MRL) a otras
especies, el CVMP seguirá evaluando formas de obtener los datos necesarios para esa extrapolación con nuevas iniciativas que faciliten la mayor disponibilidad de medicamentos para especies menores.
Apoyándose en las conclusiones del seminario conjunto EMEA/FEDESA/FVE sobre
farmacovigilancia veterinaria en mayo de 2002, la EMEA tratará de poner en práctica algunas de las recomendaciones adoptadas en dicho seminario para promover una mayor aceptación y eficiencia de la notificación de acontecimientos adversos para medicamentos veterinarios en toda la UE.
Continuando con los buenos resultados conseguidos por el PERF II en el sector veterinario, la
EMEA, apoyada por el CVMP y sus expertos, seguirá abordando cuestiones reglamentarias pendientes en el contexto de la asistencia continuada a los países candidatos que preparan su ingreso en la Unión Europea por medio de una serie de seminarios y una mini-conferencia como parte del programa PERF III.
El CVMP publicará para consulta una directriz sobre la seguridad de los manipuladores de
medicamentos veterinarios en la Unión Europea. El CVMP adoptará un protocolo de acciones necesarias tras la detección de contaminación de
medicamentos veterinarios por partículas del virus de la diarrea vírica bovina (DVB) por medio de la técnica PCR y elaborará una directriz sobre la necesidad de realizar pruebas de seguridad de lotes en los animales destinatarios de medicamentos veterinarios inmunológicos con objeto de reducir los ensayos en animales.
Tras el aumento de las solicitudes de asesoramiento científico recibidas por el CVMP en 2002, la
Agencia seguirá instando a los posibles solicitantes a que utilicen este servicio y que apliquen plenamente el nuevo procedimiento de trabajo y las directrices ahora disponibles en la Agencia.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 23/58
3.1 Evaluación inicial
Nuevas solicitudes para medicamentos y límites máximos de residuos (Entrada)
9
3
10
7
3
2
0 2 4 6 8 10 12
2001
2002
2003
Solicitudes para nuevos límites máximos de residuosSolicitudes para nuevos medicamentos
Tendencias: Se espera un aumento del número de solicitudes para la evaluación inicial de medicamentos como
consecuencia de los retrasos en la presentación de solicitudes ocurridos en 2002. La disminución progresiva del número de solicitudes de establecimiento de MRL para sustancias
nuevas refleja el hecho de que se estén desarrollando menos moléculas nuevas para medicamentos veterinarios dirigidos a animales destinados al consumo humano.
Objetivos: Asegurar el procesamiento de todas las solicitudes por el procedimiento centralizado y de MRL
dentro de los plazos establecidos. Considerar otras oportunidades para aumentar la transparencia de la información sobre el
funcionamiento del Comité de Medicamentos Veterinarios (CVMP) y sus grupos de trabajo a las partes interesadas.
Publicar informes EPAR en menos de cinco días desde la notificación de la decisión de la Comisión Europea de conceder una autorización de comercialización.
Publicar los informes completos de evaluación del CVMP para las solicitudes de nuevos MRL. Gestión y organización del CVMP El CVMP se reunirá 11 veces en 2003. El grupo de planificación estratégica se reunirá 4 veces, presidido por el vicepresidente del CVMP. Se crearán grupos ad hoc de expertos sobre la disponibilidad de medicamentos, la extrapolación de MRL, la preparación para el Codex Alimentarius y la finalización de la contribución de la UE al VICH sobre las pruebas de toxicidad medioambiental para los medicamentos veterinarios.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 24/58
Reuniones del CVMP en 2003
14-16 de enero 22-24 de julio
11-13 de febrero 19-21 de agosto
11-13 de marzo 16-18 de septiembre
8-10 de abril 14-16 de octubre
13-15 de mayo 11-13 de noviembre
17-19 de junio 9-11 de diciembre
* El CVMP sólo se reunirá en Agosto si lo considera necesario.
3.2 Establecimiento de límites máximos de residuos para sustancias antiguas Se avanzará el trabajo relacionado con las últimas 8 sustancias que quedan con MRL provisionales en el Anexo III del Reglamento (CEE) nº 2377/90 del Consejo. Estas sustancias son:
Alfa-cipermetrina Altrenogest Cipermetrina Deltametina Acetato de flugestona Kanamicina Metamizol Morantel
Dictámenes emitidos sobre límites máximos de residuos para sustancias antiguas (Salida)
25
10
8
0 5 10 15 20 25 30
2001
2002
2003
Dictámenes sobre MRL para sustancias antiguas
Tendencia: La carga de trabajo aumentará cuando se establezcan MRL definitivos para el resto de las
sustancias antiguas contenidas en el Anexo III.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 25/58
3.3 Actividades posteriores a la autorización
Solicitudes recibidas (Entrada)
18
12
10
25
15
20
10
6
8
0 5 10 15 20 25 30
2001
2002
2003
Variaciones de tipo II Variaciones de tipo I Ampliaciones
Solicitudes recibidas (Entrada)
4
6
8
0 2 4 6 8 10
2001
2002
2003
Solicitudes de ampliación/modificación de MRL
Tendencias: Se espera que en 2003 sigan aumentando las actividades posteriores a la autorización, mediante el
incremento del número de medicamentos autorizados por el procedimiento centralizado. Se espera que en 2003 siga aumentando el número de solicitudes para ampliar y modificar los
MRL, en parte como respuesta a las iniciativas del CVMP para promover la extrapolación de MRL a especies menores.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 26/58
3.4 Actividades de farmacovigilancia y mantenimiento
Informes periódicos actualizados de seguridad recibidos (Entrada)
49
46
43
0 10 20 30 40 50 60
2001
2002
2003
Informes periódicos actualizados de seguridad
Tendencias: Se elaborarán informes anuales para 27 productos. Se esperan 4 solicitudes de renovación de autorizaciones de comercialización. El CVMP evaluará 43 informes periódicos actualizados de seguridad.
Nuevas actividades que se acometerán en 2003 y que repercutirán en la carga de trabajo: Continuarán las actividades de la Conferencia Veterinaria Internacional sobre Armonización
(VICH) para finalizar las directrices en el ámbito de la farmacovigilancia y establecer normas para la transmisión electrónica, teniendo en cuenta los requisitos europeos.
Entre tanto, el CVMP elaborará normas comunitarias para la transmisión electrónica de reacciones adversas como parte de la implantación del sistema EudraVigilance para la farmacovigilancia de los medicamentos veterinarios.
Las recomendaciones adoptadas por el CVMP en el seminario conjunto EMEA/FEDESA/FVE sobre farmacovigilancia celebrado en 2002 tendrán como resultado las siguientes actividades en 2003: Publicación anual por la EMEA de un boletín sobre la farmacovigilancia de los productos
autorizados por el procedimiento centralizado. Armonización de un formato común para la notificación de reacciones adversas en toda la
Comunidad. Medidas para promover desde los estamentos educativos unas buenas prácticas de
farmacovigilancia destinadas a los veterinarios y los propietarios de animales.
3.5 Asesoramiento científico Tendencias: Se espera que el número de solicitudes de asesoramiento científico aumente progresivamente
respecto al año anterior, con 5 solicitudes previstas. Los solicitantes están empezando a conocer mejor el procedimiento y las ventajas que puede ofrecer a la hora de preparar la presentación de una solicitud de autorización de comercialización.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 27/58
3.6 Arbitraje y remisiones comunitarias
Arbitraje y remisiones comunitarias recibidas/esperadas (Entrada)
2
3
6
0 2 4 6 8
2001
2002
2003
Procedimientos de arbitraje y remisiones comunitarias
Tendencias: En 2003 se espera un aumento significativo de la carga de trabajo en esta área. Se esperan 3 remisiones como resultado de cuestiones de seguridad relacionadas con
medicamentos veterinarios. Se esperan 3 remisiones de arbitraje remitidas al CVMP.
3.7 Partes interesadas Aprovechando las excelentes relaciones establecidas en el pasado, proseguirán los esfuerzos para aprovechar otras oportunidades con las principales partes interesadas, como la Federación de Veterinarios en Europa, FEDESA, la Federación de Veterinarios en la Industria y grupos de consumidores. Seguirán celebrándose reuniones bilaterales con FEDESA, se organizará una serie de Jornadas Informativas y se aplicará el nuevo concepto de grupos representativos desarrollado en 2001, sobre todo para comunicar con más antelación los programas a los grupos de trabajo del CVMP.
3.8 Actividades internacionales Como siempre, la EMEA y el CVMP seguirán participando en una serie de actividades internacionales. Plena coordinación de la contribución de la UE a la VICH y participación en grupos de trabajo de
expertos, en dos Comités Rectores y posiblemente también en grupos de trabajo sobre la resistencia a los antibióticos.
Participación de expertos científicos en las reuniones del Codex Alimentarius y la OMS. Actividades con las instituciones europeas, como los Comités Permanentes y de Productos
Farmacéuticos Veterinarios de la Comisión Europea, así como con los responsables de las agencias nacionales de medicamentos veterinarios (HEVRA)
El PERF III seguirá demandando apoyo técnico, logístico y científico para completar el programa antes de la adhesión de los países candidatos.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 28/58
3.9 Grupos de trabajo y grupos ad hoc Tendencias: Se espera que el CVMP remita una serie de temas y cuestiones importantes a los grupos de trabajo
para recibir de ellos asesoramiento y recomendaciones en 2003. Por consiguiente, se espera que aumente el número de reuniones.
El mayor número de actividades de farmacovigilancia exigirá 6 reuniones al año del Grupo de trabajo “Farmacovigilancia” del CVMP.
Grupos de trabajo y grupos ad hoc del CPMP
Número de reuniones
Nuevas directrices
Directrices actuales
Grupo de trabajo “Medicamentos veterinarios inmunológicos”
4 5 4
Grupo de trabajo “Eficacia” 2 2 2 Grupo de trabajo “Farmacovigilancia” 6 1 3 Grupo de trabajo “Seguridad” 4 1 2 Grupo de trabajo conjunto CPMP/CVMP “Calidad”
4 5 4
Grupo ad hoc sobre evaluación del riesgo medioambiental
2 3 --
Grupo ad hoc sobre resistencia antimicrobiana
3 -- --
En el Anexo 3 pueden encontrarse más detalles sobre los documentos orientadores que se prevé publicar para consulta o finalizar en 2003.
3.10 Grupo de facilitación del reconocimiento mutuo Algunos sitios web útiles: Responsables de la agencias de medicamentos veterinarios http://www.hevra.org La Agencia seguirá prestando apoyo para hacer frente a la creciente carga de trabajo del VMRFG en 2003, con la ayuda de un experto nacional del Irish Medicines Board destacado en la EMEA.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 29/58
4. Inspecciones Prioridades de las inspecciones en 2003: Concluir con éxito la fase de preparación del acuerdo de reconocimiento mutuo (MRA) con Japón
y finalizar la entrada en vigor del acuerdo de la UE con Canadá. En 2003 seguirán vigilándose los otros acuerdos operativos.
Participar activamente en las actividades exigidas por la Directiva sobre ensayos clínicos con medicamentos de uso humano y, en particular, finalizar las directrices y crear las bases de datos relacionadas con ensayos clínicos.
Prestar un apoyo eficaz a los países candidatos que preparan su adhesión a la UE a través del PERF III con la organización de inspecciones observadas y dos seminarios técnicos de buenas prácticas de fabricación (BPF).
Coordinar y gestionar eficazmente las solicitudes de inspecciones de BPF, buenas prácticas clínicas (BPC) y buenas prácticas de laboratorio (BPL) relacionadas con la autorización de productos por el procedimiento centralizado en los plazos contemplados por la legislación comunitaria y conforme a las normas establecidas en el sistema de gestión de calidad de la Agencia.
Utilizar el sistema de gestión de crisis en el caso de que aparezcan problemas de calidad y defectos en productos autorizados por el procedimiento centralizado y coordinar junto con los Estados miembros la retirada eficaz de los productos cuando sea necesario.
Poner en práctica el nuevo acuerdo con el EDQM para los próximos cinco años sobre los procedimientos de muestreo y ensayo establecidos previamente, para garantizar una vigilancia eficaz y adecuada de la calidad de los medicamentos autorizados por el procedimiento centralizado que se comercializan en la UE.
Realizar un esfuerzo concertado para armonizar las actividades de inspección de BPF y BPC, en particular con los países candidatos que se preparan para su adhesión a la UE en 2004.
4.1 Inspecciones
Inspecciones realizadas (Salida)
64
63
62
8
12
16
0
0
2
0 10 20 30 40 50 60 70
2001
2002
2003
Inspecciones de BPF realizadas Inspecciones de BPC realizadasInspecciones de BPL realizadas

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 30/58
En 2003 se espera un aumento del número de inspecciones de BPC, al adquirir más relevancia las cuestiones relacionadas con las BPC como resultado de la preparación de los Estados miembros para la transposición de la Directiva sobre ensayos clínicos. También se espera un aumento de las inspecciones de las actividades de conformidad en farmacovigilancia. Las solicitudes de inspección de BPF no aumentarán significativamente en 2003, puesto que muchos de los centros de producción mencionados en las nuevas solicitudes han sido ya inspeccionados en relación con productos anteriores. Además, el acuerdo de MRA con Suiza, que entró en vigor en junio de 2002, hará innecesaria la inspección de los centros de producción suizos. Aunque en 2002 no se recibió ninguna solicitud de inspección de BPL, se han adoptado las disposiciones oportunas por si ha de realizarse un pequeño número de inspecciones de BPL. El Grupo ad hoc de servicios de inspección de BPF se reunirá cuatro veces en 2003. Su trabajo se centrará en la armonización gradual de los procedimientos de inspección, con la adopción de un enfoque común para la implantación de los sistemas de calidad y la coordinación de actividades de seguimiento de las inspecciones de BPF, así como el apoyo a las actividades continuas relacionadas con los MRA. Una de esas reuniones se celebrará con el Grupo de trabajo conjunto CPMP/CVMP “Calidad”. El grupo ad hoc de servicios de inspección de BPC se reunirá cinco veces en 2003. Además del trabajo de armonización, la atención se centrará en la elaboración y consolidación de directrices y procedimientos como preparación para la transposición de la Directiva sobre ensayos clínicos en 2004. Tendencias: Se espera que el número de solicitudes de inspección de BPF se mantenga estable en 2003, puesto
que muchos de los centros de producción citados en las nuevas solicitudes fueron inspeccionados recientemente en relación con medicamentos ya autorizados y se han empezado a sentir los efectos de los MRA con Suiza y Canadá. Esta tendencia se verá compensada por la creciente necesidad de volver a inspeccionar los centros autorizados.
Se espera que las solicitudes de inspección de BPF en los centros de producción de medicamentos huérfanos constituyan una parte importante de las solicitudes de BPF recibidas en 2003.
Se espera que siga aumentando la carga de trabajo relacionada con defectos de los productos, al ser cada vez mayor el número de productos autorizados.
Los grupos ad hoc de inspectores de BPF y BPC seguirán reuniéndose 4 y 5 veces al año, respectivamente, mientras continúen con las actividades de armonización en la UE.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Los servicios de inspección de los países candidatos exigirán un trabajo adicional antes de su
adhesión. Se ha previsto un programa de 8 inspecciones observadas en los países candidatos y 2 seminarios como parte de las actividades relacionadas con BPF del programa PERF III.
Preparación para la aplicación de la directiva relativa a los ensayos clínicos
4.2 Acuerdos de reconocimiento mutuo Tendencias: MRA CE-Suiza: 2003 será el primer año completo de la fase operativa de este MRA. MRA CE-Canadá: Está previsto que entre en vigor a principios de 2003. MRA CE-Japón: Se tratará de concluir la fase de preparación de este MRA con una serie de
visitas y reuniones asociadas con representantes de la UE.
4.3 Muestreos y ensayos Tendencias: En 2003 continuará el programa de muestreos y ensayos de productos autorizados por el
procedimiento centralizado. A principios de 2003 entrará en vigor un nuevo acuerdo con el EDQM.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 31/58
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Por primera vez, observadores de los países candidatos participarán en el programa de muestreos y
ensayos. Se iniciará un procedimiento piloto para la vigilancia de los resultados de los ensayos. En 2003 se organizará un seminario para auditores, inspectores y representantes de la red de
Laboratorios Oficiales de Control de Medicamentos para controlar y revisar el programa.
4.4 Certificados
Solicitudes de certificados de medicamentos (Entrada)
12517
13135
16000
0 5000 10000 15000 20000
2001
2002
2003
Solicitudes de certificados
Tendencias: Se prevé un aumento de 20% debido al mayor número de productos autorizados.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Seguirán manteniéndose y simplificándose los procedimientos. El aumento de la demanda influirá en los recursos disponibles y posiblemente obligue a revisar la
estructura de tasas para este servicio.
4.5 Transposición de la Directiva relativa a los ensayos clínicos La fecha límite para la transposición de la Directiva 2001/20/CE del Consejo relativa a la realización de ensayos clínicos de medicamentos de uso humano es el 1 de mayo de 2004 (DO L 121, 1.5.2001, p. 34). La EMEA dirige la elaboración de varios documentos orientadores necesarios para la transposición de la Directiva. Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Se hará un esfuerzo importante para diseñar, construir y desarrollar la base de datos de ensayos
clínicos y la base de datos para la notificación de presuntas reacciones adversas inesperadas en ensayos clínicos.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 32/58
5. Comunicaciones y redes Las redes de telecomunicaciones e información de la Agencia son fundamentales para mantener la relación entre la EMEA y las 27 autoridades nacionales competentes diferentes que existen en la UE y en los Estados miembros del EEE-AELC, la Comisión Europea y los ciudadanos. La Unidad de comunicaciones y redes se ocupa del funcionamiento y el mantenimiento de estas redes.
5.1 Aplicación de la estrategia comunitaria de telemática Tendencias: Creciente uso de las herramientas de transmisión electrónica y almacenamiento de datos en la
industria farmacéutica. Demanda cada vez mayor de información fiable sobre los productos por las partes interesadas. Necesidad de un sistema más fiable y específico de detección de señales en farmacovigilancia a
escala paneuropea. Necesidad de unos procedimientos de evaluación más eficientes.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Transferencia de responsabilidades de los proyectos de TI en el sector farmacéutico que forman
parte de la familia ‘Eudra’ (excluido el sistema de seguimiento EudraTrack de los Estados miembros) del Centro de Investigación Común de la Comisión Europea (JRC) a la EMEA. Esta transferencia se hará previa consulta de la Comisión Europea con las autoridades de los Estados miembros.
La EMEA espera que la aplicación de la estrategia completa de telemática dure 4 años, con un presupuesto calculado de aproximadamente 39 millones de euros. La autoridad presupuestaria de la UE decidió financiar con 7 millones de euros esta fase inicial. La estrategia de telemática tiene como finalidad: apoyar y facilitar el uso de los procedimientos según se establece en la legislación crear y mejorar la transparencia proporcionar herramientas eficaces para divulgar información aumentar la eficiencia y hacer el mejor uso posible de los recursos disponibles.
La aplicación de esta estrategia se basa en cinco proyectos básicos, que se articulan en torno a una infraestructura común y que se pondrán en marcha al mismo tiempo: Iniciativa Actividades EudraNet EudraNet se creó para proporcionar una red segura de telecomunicaciones entre las
autoridades y los servicios relacionados que incluya un medio seguro de intercambiar archivos con colaboradores internos y externos. Los principales objetivos para 2003 son: Transferencia de responsabilidades del Centro de Investigación Común de la
Comisión Europea a la EMEA el 1 de enero de 2003. Puesta en marcha de EudraSafe II en enero de 2003. Implantación de una banda ancha mayor. Puesta en marcha de EudraWorkSpace en junio de 2003 Implantación de la infraestructura PKI antes de diciembre de 2003 Implantación de la infraestructura de IP/VPN antes de diciembre de 2003
Base de datos EuroPharm
EuroPharm pretende ser una base de datos europea con información sobre todos los medicamentos comercializados en la Unión Europea. Su revisión ha traído consigo un aumento del tipo de información que la Agencia recogerá y mantendrá, lo cual repercutirá en el desarrollo de la base de datos, cuyos objetivos principales para 2003

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 33/58
son: Acuerdo paneuropeo sobre el modelo de datos de referencia antes de abril de
2003. Acuerdo sobre la norma de intercambio entre agencias para la transmisión de
datos antes de julio de 2003. Especificaciones funcionales para la primera fase piloto disponibles antes de
septiembre de 2003. Conclusión del procedimiento de licitación para la selección de contratistas en la
primera fase piloto. EudraVigilance EudraVigilance es una base de datos que contiene información sobre la seguridad de
todos los productos que se venden en el mercado de la UE, actualizada por medios electrónicos y a disposición de todas las autoridades reguladoras de la UE para su análisis. Los principales objetivos para 2003 son: Crear e instalar infraestructura adicional antes de febrero de 2003. Implantar almacenes de datos e inteligencia operativa antes de finales de 2003.
Transmisión electrónica
La finalidad de este proyecto es establecer normas de intercambio e implantar aplicaciones que permitan el envío de datos para la evaluación de medicamentos a la EMEA y su procesamiento electrónico. Se está trabajando en dos subproyectos, la implantación del documento técnico común electrónico (eCTD – una estructura definida y un conjunto de formatos para el envío de información por los solicitantes a las autoridades competentes en apoyo de una solicitud de autorización de comercialización) y el proyecto de gestión de información sobre productos (PIM – una estructura definida para el intercambio de Resúmenes de Características del Producto, prospectos y etiquetados entre el solicitante y las autoridades competentes). Los principales objetivos para 2003 son: eCTD: Proceso para recibir, almacenar y acceder a los eCTD enviados utilizando el
procedimiento centralizado acordado en abril de 2003. Puesta en marcha del Sistema Básico Europeo de Revisión (EURES) en junio de
2003. Revisión de los requisitos basándose en la experiencia de los eCTD recibidos en
diciembre de 2003.
PIM: Decisión del modelo de financiación antes de febrero de 2003. Informe sobre la prueba del concepto y la documentación asociada en marzo de
2003. Requisitos para el sistema de la Agencia antes de junio de 2003.
Base de datos de ensayos clínicos
Este proyecto se ha puesto en marcha para crear una base de datos europea que registre todos los ensayos clínicos solicitados en la UE, así como las supuestas reacciones adversas graves inesperadas que ocurran durante esos ensayos. Los principales objetivos para 2003 son: Preparar las especificaciones de los requisitos (antes de marzo de 2003). Convocar una licitación para el desarrollo del sistema y el contrato posterior
(antes de junio de 2003). Infraestructura Los proyectos de tecnología de la información de Eudra requieren una infraestructura
común y con una arquitectura sólida enraizada en unos procesos operativos claramente definidos y respaldada por personal debidamente cualificado. Esta infraestructura se crea en paralelo a la ejecución de los proyectos descritos antes. Los principales objetivos para 2003 son: Instalar software adecuado para la gestión de sistemas y proyectos. Acompañar a los proyectos individuales del hardware, el software y la
administración de sistemas adecuados. Mejorar los recursos empleados en asistencia técnica.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 34/58
5.2 TI y dirección de proyectos en la EMEA El correcto funcionamiento de los sistemas internos de tecnología de la información (TI) de la EMEA es crítico para que la Agencia pueda realizar sus tareas. El objetivo es prestar unos servicios fiables y sólidos al personal de la EMEA y a los delegados, junto con niveles adecuados de apoyo operativo, introduciendo al tiempo nuevos servicios y mejoras en la infraestructura según las necesidades de la Agencia y los usuarios. El apoyo de la tecnología de información corporativa de la Agencia requiere el mantenimiento y la mejora de los sistemas, a ser posible sin que los usuarios de esos sistemas sean conscientes del trabajo que se realiza entre bastidores. Los objetivos operativos específicos para 2003 son: Utilizar más de 20 servidores. Utilizar, prestar apoyo y mantener todo el hardware y el software de la Agencia. Mantener unos niveles de servicio help desk superiores al objetivo del 95% de resolución de
problemas. Introducir nuevas formas de almacenamiento, acceso y recuperación de información. Implantar los requisitos de TI solicitados por la Agencia. Asegurar una disponibilidad mínima del sistema del 99,5 % de los servicios TI durante las horas
de trabajo de la EMEA. Asegurarse de que el entorno TI esté correctamente dimensionado para las necesidades de la
EMEA. A medida que evoluciona la carga de trabajo de la Agencia, se desarrollan e implantan requisitos nuevos o revisados de apoyo a la tecnología de la información. Estos requisitos se definen mediante un procedimiento normalizado de trabajo y se introducen ya sea mejorando los sistemas existentes o añadiendo otros nuevos. En 2003, los objetivos en este terreno son: Desarrollar las aplicaciones básicas de la EMEA (SIAMED, SI2, ActiTrak, y la base de datos del
personal). Implantar el servicio de retransmisión a través de Internet (videostreaming). Ampliar los servicios de videoconferencia de sobremesa. Continuar con la implantación del sistema informatizado de gestión de documentos conjuntamente
con el Sector de gestión y publicación de documentos.
5.3 Organización de reuniones y conferencias
Reuniones celebradas/previstas (Salida)
629
564
516
5319
3326
3434
0 1000 2000 3000 4000 5000 6000
2001
2002
2003
Número de días de reunión Número de delegados

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 35/58
Este Sector es responsable de asegurar un apoyo eficiente a las reuniones de la EMEA, poniendo a su disposición las mejores instalaciones y servicios posibles y mejorando constantemente los recursos disponibles, así como ayudando a los delegados con servicios logísticos y administrativos. Su labor incluye la organización de reuniones, viajes y alojamiento en hoteles para delegados y anfitriones, el recibimiento de los visitantes, así como la organización del reembolso de los gastos de desplazamiento (en colaboración con el Sector de Contabilidad) y la preparación y vigilancia del acondicionamiento de las salas de reunión. Tendencias: El número de reuniones y delegados aumentará en comparación con 2002, debido en parte a las
actividades relacionadas con reuniones aplazadas de 2002 a 2003 y en parte a las reuniones relacionadas con EudraVigilance y la implantación de la estrategia de telemática de la UE.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Implantación del sistema informatizado de gestión de reuniones. Participación de observadores de los países candidatos en reuniones de la EMEA. Puesta en marcha de un proyecto piloto para la retransmisión en directo de las reuniones a través
de la web. Con esto se pretende fomentar la participación de expertos externos en las reuniones del CPMP.
Creación de un centro de visitantes en el sitio web de la EMEA. Participación en la implantación del PERF III. Aumento del número de videoconferencias a cerca de 40 en 2003.
5.4 Gestión y publicación de documentos Este Sector es responsable de la publicación, catalogación, distribución y conservación de los documentos de la EMEA. Sus actividades abarcan la gestión de la calidad (especialmente en el área de las traducciones, la información sobre los productos y el control de la calidad y la coherencia de los documentos normativos) y la logística. Abarcan también el funcionamiento y la gestión de la biblioteca de la EMEA, así como los archivos electrónicos. Tendencias: Aumento del número de documentos publicados en el sitio web, entre ellos publicaciones
multilingües. Aumento del número de solicitudes de información.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Continuar con el desarrollo y la implantación del sistema informatizado de gestión de documentos
(EDMS). Continuar con el desarrollo del sitio web de la EMEA y la organización de la gestión de la web,
con creación de varios servicios nuevos que respondan al compromiso de la Agencia de aumentar la transparencia y la comunicación.
Revisar continuamente las plantillas de los dictámenes y la información sobre los productos, teniendo en cuenta la legibilidad de los prospectos.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 36/58
6. Administración La Unidad de Administración consta de tres sectores que son responsables de la administración de los ingresos, los gastos y las cuentas de la Agencia conforme a las reglas y normas vigentes; la contratación, gestión y administración del personal y los expertos destacados, y la prestación y organización de los servicios logísticos necesarios para el buen funcionamiento de la Agencia.
6.1 Personal y presupuesto Los principales objetivos del Sector de personal y presupuesto son el desarrollo y la gestión puntual y certera de los recursos financieros y humanos de la Agencia. Tendencias: La contratación de personal se mantendrá en niveles moderados, pero las actividades de
administración del personal aumentarán proporcionalmente más, como resultado de la rotación del personal, la movilidad interna y la formación.
Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Seguir mejorando la gestión de los recursos humanos en la EMEA, con la adopción de normas
relativas al trabajo a tiempo parcial, la revisión del sistema de evaluación del rendimiento y la adopción de normas relativas a la transferencia interna de personal.
Finalizar la implantación del sistema informatizado de gestión y datos del personal. Poner en práctica una política de formación avanzada para seguir desarrollando competencias. Prepararse para la entrada en vigor del futuro Reglamento del personal de la UE y adaptar las
reglas de aplicación internas. Implantar el nuevo Reglamento financiero de la UE y la EMEA.
6.2 Servicios de infraestructura El desarrollo técnico general y la diversificación de las actividades de la EMEA requieren la ejecución de una serie de proyectos para acomodar al personal de la Agencia y también para crear un lugar de trabajo adecuado y unas instalaciones apropiadas para el personal y los visitantes. Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Desarrollo y mantenimiento de un plan de continuidad operativa para la EMEA. Formación continua del equipo de apoyo técnico para las salas de reuniones y las oficinas. Mantenimiento de las oficinas y las zonas públicas. Revisión de la estrategia de acondicionamiento de la EMEA, incluidas las necesidades de espacio
para oficinas, salas de reuniones y equipos técnicos, especialmente ante la futura ampliación de la UE.
Evaluación de la adopción de medidas de salud y seguridad, así como de planes contra incendios y emergencias.
La ampliación del ámbito de la actividades de la EMEA y su crecimiento obligarán a ampliar de nuevo el espacio de las oficinas, con la creación de nuevas oficinas y salas de reunión para las delegaciones.
Mejora de las instalaciones de los archivos externos para que puedan albergar el creciente número de expedientes que conserva la EMEA, junto con un mejor sistema para la gestión de los registros.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 37/58
6.3 Contabilidad
Número de pagos recibidos y efectuados
1775
1752
1642
10399
12955
14393
0 5000 10000 15000 20000
2001
2002
2003
Pagos recibidos Pagos efectuados
Tendencias: Se espera que la carga de trabajo total del Sector de Contabilidad aumente en 21% en 2003, en
comparación con 2001. Las mejoras conseguidas en la productividad no serán suficientes para compensar los nuevos
aumentos. Las tendencias en el número de solicitudes centralizadas tendrán sólo un efecto limitado en la
carga de trabajo del Sector. El mayor número de delegados y reuniones en 2003 determinará la necesidad de realizar un mayor
número de transacciones. Nuevas actividades que se acometerán en 2003 y repercutirán en la carga de trabajo: Revisión del Reglamento financiero de la EMEA y normas de aplicación tras la reforma del
Reglamento financiero de la UE. Implantación de mejores sistemas para facilitar el reembolso de los gastos de las reuniones y el
seguimiento de las cuestiones relacionadas con el pago de tasas con los solicitantes y los titulares de autorizaciones de comercialización.
Mejora del sistema de contabilidad SI2. Mejora continua del sistema ActiTrak y mejora de los sistemas de cálculo de los costes de las
actividades (incluidos los costes de los ponentes). Participación en la revisión del sistema de tasas de la EMEA.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 38/58
Anexos 1. Plan de recursos humanos de la EMEA 2001 -2003
2. Resúmenes de los presupuestos de la EMEA 2001 -2003
3. Documentos orientadores de la EMEA en 2003
4. Puntos de contacto y documentos de referencia de la EMEA
5. Perfil de los directivos de la EMEA

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 39/58
Anexo 1 Plan de recursos humanos de la EMEA 2001 -2003
2001 2002 2003 Categoría y
Grado Ocupado hasta
31.12.2001 Autorizados para
2002 Ocupado hasta
31.12.2002 Autorizados para
2003 A1 -- -- -- -- A2 1 1 1 1 A3 4 5 5 5 A4 26 29 28 32 A5 24 28 26 32 A6 24 29 24 34 A7 24 30 30 32 A8 -- -- -- --
TOTAL A 103 122 114 136
B1 4 4 2 6 B2 8 9 8 10 B3 9 11 10 12 B4 5 9 8 12 B5 5 8 6 9
TOTAL B 31 41 34 49
C1 14 15 15 19 C2 13 19 19 23 C3 42 44 43 47 C4 -- 4 4 6 C5 -- -- -- --
TOTAL C 69 82 81 95
D1 1 1 1 2 D2 4 5 5 5 D3 -- -- -- -- D4 -- -- -- --
TOTAL D 5 6 6 7
TOTAL PUESTOS
208 251 235 287
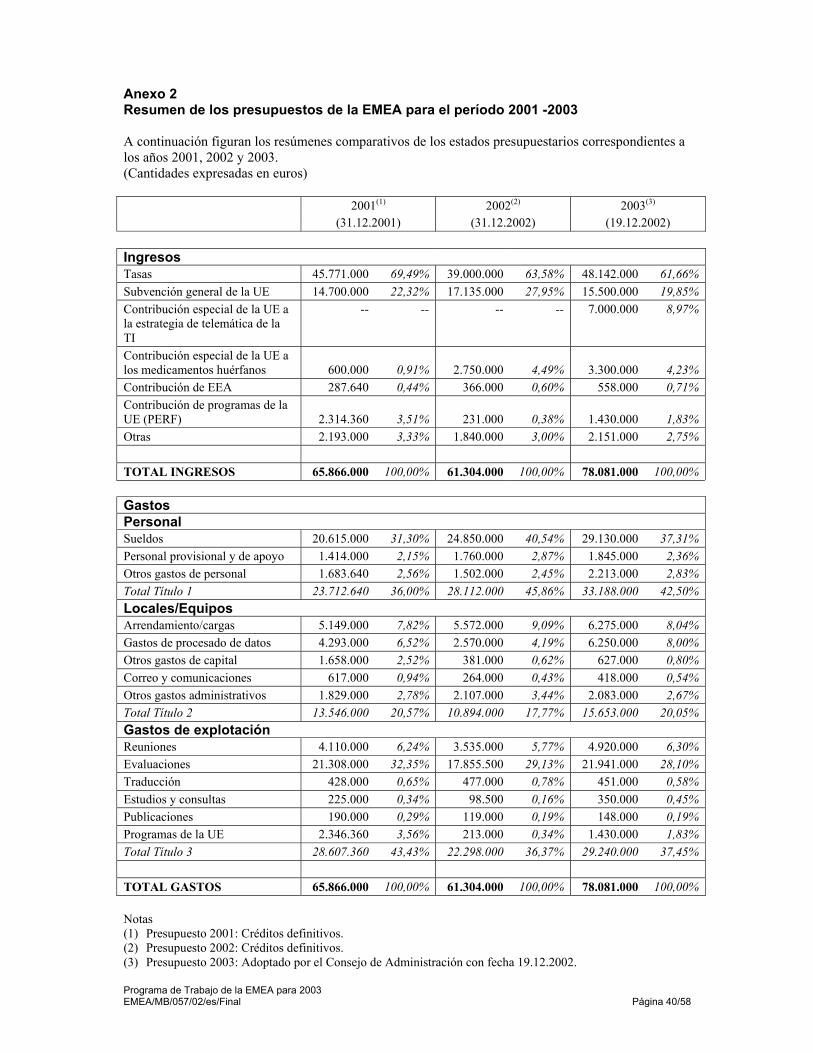
Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 40/58
Anexo 2 Resumen de los presupuestos de la EMEA para el período 2001 -2003 A continuación figuran los resúmenes comparativos de los estados presupuestarios correspondientes a los años 2001, 2002 y 2003. (Cantidades expresadas en euros)
2001(1) 2002(2) 2003(3) (31.12.2001) (31.12.2002) (19.12.2002)
Ingresos Tasas 45.771.000 69,49% 39.000.000 63,58% 48.142.000 61,66% Subvención general de la UE 14.700.000 22,32% 17.135.000 27,95% 15.500.000 19,85% Contribución especial de la UE a la estrategia de telemática de la TI
-- -- -- -- 7.000.000 8,97%
Contribución especial de la UE a los medicamentos huérfanos 600.000 0,91% 2.750.000 4,49%
3.300.000 4,23%
Contribución de EEA 287.640 0,44% 366.000 0,60% 558.000 0,71% Contribución de programas de la UE (PERF) 2.314.360 3,51% 231.000 0,38%
1.430.000 1,83%
Otras 2.193.000 3,33% 1.840.000 3,00% 2.151.000 2,75% TOTAL INGRESOS 65.866.000 100,00% 61.304.000 100,00% 78.081.000 100,00% Gastos Personal Sueldos 20.615.000 31,30% 24.850.000 40,54% 29.130.000 37,31% Personal provisional y de apoyo 1.414.000 2,15% 1.760.000 2,87% 1.845.000 2,36% Otros gastos de personal 1.683.640 2,56% 1.502.000 2,45% 2.213.000 2,83% Total Título 1 23.712.640 36,00% 28.112.000 45,86% 33.188.000 42,50% Locales/Equipos Arrendamiento/cargas 5.149.000 7,82% 5.572.000 9,09% 6.275.000 8,04% Gastos de procesado de datos 4.293.000 6,52% 2.570.000 4,19% 6.250.000 8,00% Otros gastos de capital 1.658.000 2,52% 381.000 0,62% 627.000 0,80% Correo y comunicaciones 617.000 0,94% 264.000 0,43% 418.000 0,54% Otros gastos administrativos 1.829.000 2,78% 2.107.000 3,44% 2.083.000 2,67% Total Título 2 13.546.000 20,57% 10.894.000 17,77% 15.653.000 20,05% Gastos de explotación Reuniones 4.110.000 6,24% 3.535.000 5,77% 4.920.000 6,30% Evaluaciones 21.308.000 32,35% 17.855.500 29,13% 21.941.000 28,10% Traducción 428.000 0,65% 477.000 0,78% 451.000 0,58% Estudios y consultas 225.000 0,34% 98.500 0,16% 350.000 0,45% Publicaciones 190.000 0,29% 119.000 0,19% 148.000 0,19% Programas de la UE 2.346.360 3,56% 213.000 0,34% 1.430.000 1,83% Total Título 3 28.607.360 43,43% 22.298.000 36,37% 29.240.000 37,45% TOTAL GASTOS 65.866.000 100,00% 61.304.000 100,00% 78.081.000 100,00% Notas (1) Presupuesto 2001: Créditos definitivos. (2) Presupuesto 2002: Créditos definitivos. (3) Presupuesto 2003: Adoptado por el Consejo de Administración con fecha 19.12.2002.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 41/58
Anexo 3 Documentos orientadores de la EMEA en 2003 En 2003 se pretende finalizar o publicar para consulta los siguientes documentos: Grupo de trabajo del CPMP “Biotecnología” Título del documento Note for Guidance on the Use of Bovine Serum in the manufacture of Human Biological Medicinal Products Points to Consider on Quality Aspects of Medicinal Products containing Active Substances Produced by Stable Transgene Expression in Higher Plants Revision of Note for guidance on minimising the risks of TSE transmission via medicinal products Points to Consider on the Development of Live Attenuated Influenza Vaccines Grupo de trabajo del CPMP “Hemoderivados” Título del documento Note for Guidance on the Clinical investigation of plasma derived fibrin sealants Note for guidance on the Clinical investigation of von Willebrand factor Warning on transmissible agents for patient leaflets and SPCs Core SPC for Plasma derived fibrin sealants
Grupo de trabajo del CPMP “Eficacia” Título del documento Points to consider on the clinical development of fibrinolytic medicinal products in the treatment of patients with ST segment elevation acute myocardial infarction (STEMI) Note for Guidance on the clinical development of medicinal products for the treatment of HIV infection. Note for Guidance on clinical investigation of medicinal products for the treatment of migraine. Note for Guidance on evaluation of anticancer medicinal products in man (CPMP/EWP/205/95 rev. 2) – Addendum on paediatric oncology. Points to consider on the evaluation of new anti-fungal agents for invasive fungal infections. Points to consider on clinical investigation of medicinal products for treatment of rheumatoid arthritis. Points to consider on the evaluation of medicinal products for the treatment of irritable bowel syndrome. Appendix to the Note for Guidance on the Clinical investigation of medicinal products in the treatment of schizophrenia (CPMP/EWP/559/95) – methodology of clinical trials concerning the development of depot preparations of approved medicinal products in schizophrenia. Points to consider on the requirements for clinical documentation for metered dose inhalers (MDI) Addendum on neuropathic pain to the Note for Guidance on clinical investigation of medicinal products for nociceptive Pain treatment Note for Guidance on Clinical Investigation of Medical Products in the Treatment of Generalised Anxiety Disorder Note for Guidance on Clinical Investigation of Medical Products in the Treatment of Panic Disorder. Note for Guidance on Clinical Investigation of Medical Products in the Treatment of Obsessive-compulsive Disorder Addendum on acute cardiac failure to the CPMP Note for Guidance on clinical investigation of medicinal products in the treatment of acute cardiac failure Note for guidance on the evaluation of medicinal products for the treatment of dyslipoproteinaemia Note for Guidance on Clinical investigation of steroid contraceptives in women Revision of Note for Guidance on evaluation of new anti-bacterial medicinal products (CPMP/EWP/558/95) and Note for Guidance on the pharmacodynamic section of the summary of product characteristics for antibacterial medicinal products

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 42/58
Título del documento Points to consider on Biostatistical/methodological issues arising from CPMP discussion on licensing applications: Choice of Non-inferiority margin Points to consider on Biostatistical/methodological issues arising from CPMP discussion on licensing applications: Adjustment for baseline covariates Points to consider on the use of statistical methods for flexible design and analysis of confirmatory clinical trials Points to consider on clinical pharmacokinetic investigation of the pharmacokinetics of peptides and proteins Note for guidance on the evaluation of the pharmacokinetics of medicinal products in patients with impaired renal function Points to consider on the evaluation of the pharmacokinetics of medicinal products in the paediatric population Note for guidance on the evaluation of the pharmacokinetics of medicinal products in patients with hepatic impairment Note for guidance on clinical investigation of medicinal products for the treatment of psoriasis Points to consider on allergic rhino-conjuctivitis Addendum to the Note For Guidance on Modified Release Oral and Transdermal Dosage Forms: Section II (PharmacoKinetic and Clinical Evaluation) on the clinical requirements of modified release products submitted as a line-extension of an existing marketing authorisation Points to consider on Xenogenic Cell Therapy Note for Guidance on Comparability of Medicinal Products containing biotechnology- derived proteins as active substance Note for Guidance on the use of medicinal products during pregnancy Note for Guidance on risk assessment of medicinal products on human reproductive and development toxicities: from data to labelling Grupo de trabajo del CPMP “Seguridad” Título del documento Position Paper on the Non-clinical safety studies to support clinical trials with a single low dose of a compound Note for Guidance on specification limits for residues of metal catalysts Note for Guidance on Comparability of Medicinal Products Containing Biotechnology-derived Proteins as Drug Substance: Annex on Non-Clinical and Clinical Considerations Note for Guidance on the Non-Clinical Documentation of Medicinal Products with Well-Established Use Position Paper on the limits of genotoxic impurities Note for Guidance on the Need for Pre-clinical Testing of Human Pharmaceuticals in Juvenile Animals Note for Guidance on Environmental Risk Assessments for Pharmaceuticals Grupo de trabajo del CPMP “Farmacovigilancia” Título del documento Note for Guidance on Criteria for Recall and Repackaging Following Urgent Safety Restriction and Variation Procedures CPMP Points-to-Consider Document on Xenogeneic Cell Therapy (CPMP/1199/02) ICH-V1: Clinical Safety Data Management: Periodic Safety Update Reports for Marketed Drugs – Addendum to ICH-E2C (CPMP/ICH/4679/02) ICH-V2: Post-Approval Safety Management: Definitions and Standards for Expedited Reporting and Good Case Management Practices ICH-V3: Prospective Planning of Pharmacovigilance Notice to Applicants – Guideline on the Summary of Product Characteristics (EC December 1999 – Revised Version)

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 43/58
Grupo de trabajo del CPMP “Medicamentos de origen vegetal” Título del documento Note for guidance on non-clinical testing of herbal drug preparations with long-term marketing experience - guidance to facilitate mutual recognition and use of bibliographic data Concept paper for a Note for Guidance on the investigation of biopharmaceutical characterisation and bioavailability/bioequivalence of herbal drugs/preparations Position paper on the risk associated with the use of herbal products containing estragole Position paper on the risk associated with the use of herbal products containing methyleugenol Position paper on the risk associated with the use of herbal products containing á- and β-asarone Position paper on the use of Sassafras albidum as active substance or ingredient in herbal medicinal products Concept paper on the levels of scientific evidence required for the authorisation of well-established use and traditional herbal medicinal products Core-data on Urticae radix Core-data on Lini semen Core-data on Rosmarini folium cum flore Core-data on Primulae Radix Grupo de trabajo del CVMP “Eficacia” Título del documento Antimicrobials for general use in target animal species Summary of product characteristics for antimicrobial products Efficacy requirements for ectoparasiticides for cattle Fluid therapy Grupo de trabajo del CVMP “Medicamentos inmunológicos veterinarios” Título del documento Requirements and controls applied to bovine serum used in the production of immunological veterinary medicinal products EU requirements for batches with maximum and minimum titre or batch potency for developmental safety and efficacy studies Requirements for live recombinant vectored vaccines Requirements for compatibility statements for veterinary vaccines Harmonisation of requirements for potency and batch consistency of vaccines VICH: Testing for the detection of mycoplasma contamination VICH: Biologicals: testing of residual fomaldehyde VICH: Target animal safety for veterinary biological products VICH: Tests on the presence of extraneous viruses in veterinary viral vaccines Grupo de trabajo del CVMP “Farmacovigilancia” Título del documento VICH: Pharmacovigilance of veterinary medicinal products: Management of adverse event reports VICH: Pharmacovigilance of veterinary medicinal products: Management of periodic summary update reports VICH: Pharmacovigilance of veterinary medicinal products: Controlled list of terms VICH: Pharmacovigilance of veterinary medicinal products: Electronic standards for transfer of data
Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 44/58
Grupo de trabajo del CVMP “Seguridad” Título del documento VICH: Pre-approval information for registration of new medicinal products for food producing animals with respect to antimicrobial resistance Injection site residues User safety Estimation of predicted environmental concentrations, including harmonisation of default values and development of a harmonised computer model Toxicity of substances to dung fauna Degradation of substances in manure Grupo de trabajo conjunto CPMP/CVMP “Calidad” Título del documento Use of near infrared spectroscopy by the pharmaceutical industry Modified release oral and transdermal dosage forms Revision: European Drug Master File Declaration of storage conditions for pharmaceutical veterinary medicinal products in the product particulars and active substances: Annex: Stability testing of new active substances and medicinal products Annex: Stability testing of existing active substances and related finished products Summary of requirements for active substances Process validation (update) Quality aspects of veterinary medicinal products administered via drinking water

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 45/58
Anexo 4 Puntos de contacto con la EMEA Farmacovigilancia e informes sobre productos defectuosos La vigilancia constante de la seguridad de los medicamentos una vez concedida la autorización de comercialización (“farmacovigilancia”) constituye una parte importante del trabajo de las autoridades nacionales competentes y de la EMEA. La EMEA recibe informes sobre seguridad de dentro y fuera de la UE con respecto a los medicamentos autorizados por el procedimiento centralizado y coordina las actividades referentes a la seguridad y calidad de los medicamentos. Para asuntos relativos a la farmacovigilancia de Panos TSINTIS medicamentos de uso humano Teléfono directo (44-20) 75 23 71 08 E-mail: [email protected] Para asuntos relativos a la farmacovigilancia de Medicamentos de uso veterinario Medicamentos de uso veterinario Teléfono directo (44-20) 74 18 85 81 E-mail: [email protected] Para asuntos relativos a productos defectuosos y E-mail: [email protected] otros asuntos sobre la calidad de los productos Fax: (44-20) 74 18 85 90 Out of hours telephone: (44-7880) 55 06 97 Certificados de medicamentos La EMEA emite certificados de medicamentos de conformidad con las disposiciones establecidas por la Organización Mundial de la Salud. Estos certificados pueden ser utilizados en apoyo de las solicitudes de autorizaciones de comercialización dentro y fuera de la UE. Para preguntas relativas a los certificados de Jonna SUNELL-HUET medicamentos autorizados para uso humano Teléfono directo (44-20) 74 18 84 65 E-mail: [email protected] Servicios de documentación La EMEA ha publicado un gran número de documentos, entre ellos notas de prensa, documentos con información general, informes anuales y programas de trabajo. El acceso a estos y otros documentos puede realizarse a través de Internet en http://www.eudra.org/emea.html o dirigiéndose por escrito a: Servicio de Documentación de la EMEA Agencia Europea para la Evaluación de Medicamentos 7 Westferry Circus Canary Wharf RU – Londres E14 4H Puede obtener más información en la E-mail: [email protected] anterior dirección o contactando con Fax: (44-20) 74 18 86 70 Los documentos de información general Amanda BOSWORTH deben solicitarse a Teléfono directo (44-20) 74 18 84 08 E-mail: [email protected]

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 46/58
Lista de expertos europeos La EMEA utiliza unos 3.000 expertos en su trabajo de evaluación científica. La lista de estos expertos europeos está disponible para su consulta previa petición a las oficinas de la EMEA. Las peticiones deben enviarse por escrito a la EMEA o a la siguiente dirección de correo electrónico: E-mail: [email protected] Gestión integrada de la calidad Asesora GIC Marijke KORTEWEG Teléfono directo (44-20) 74 18 85 56 E-mail: [email protected] Oficina de prensa Encargado de la oficina de prensa Martin HARVEY-ALLCHURCH Teléfono directo (44-20) 74 18 84 27 E-mail: [email protected]

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 47/58
Anexo 5 Perfil de las personalidades de la EMEA Keith Jones, Presidente del Consejo de Administración, nacido el 14 Octubre de 1937, nacionalidad británica Estudios: El Dr. Jones es doctor en medicina y ha trabajado en medicina clínica e investigación para varios hospitales clínicos del Reino Unido. Posteriormente trabajó como toxicólogo en la industria agroquímica. Trayectoria profesional hasta la fecha: El Dr. Jones trabajó durante 22 años en la industria como Jefe del Departamento Médico de Fisons Agrochemical Divisions, Jefe de Evaluación de la Seguridad y Farmacología Clínica en Beecham Pharmaceuticals y Director Ejecutivo de Asuntos Médicos en Merck Sharp and Dohme en Estados Unidos. En 1991, fue nombrado Director Ejecutivo de la Agencia de Control de Medicamentos británica y actualmente es el delegado británico en el Comité Farmacéutico y el Comité Regulador Permanente de la UE, y miembro del Comité de Asesoramiento Científico de la UE perteneciente a la Dirección General de Sanidad y Protección de los Consumidores de la Comisión Europea. En la actualidad también es profesor visitante de Farmacología en la Facultad de Farmacia de la Universidad de Londres y ha publicado numerosos trabajos. El Dr. Jones se incorporó al Consejo de Administración de la EMEA en 1995, en 2001 fue nombrado presidente del Consejo y reelegido en 2003. Philippe Duneton, Vicepresidente del Consejo de Administración, nacido el 15 de septiembre de 1961, nacionalidad francesa Estudios: El Dr. Duneton se doctoró en medicina en la Universidad de Paris VI, Facultad de Lariboisière Saint Louis. Fue médico de familia y jefe de sección adjunto en los hospitales de París. Trayectoria profesional hasta la fecha: De 1992 a 1993, el Dr. Duneton fue asesor técnico de salud pública en el gabinete del ministerio francés responsable de salud y ayuda humanitaria. De 1993 a 1995 fue coordinador del C-Clin Paris-Nord (centro interregional para infecciones hospitalarias). Fue nombrado jefe de grupo “SIDA y Drogadicción” de los hospitales públicos de París. Trabajó en el Gabinete del Secretario de Estado francés para la Salud, en calidad de asesor sanitario de 1997 a 1998, antes de ser nombrado Secretario General de la Agencia Francesa del Medicamento en 1998. En 1999 fue nombrado Director General de la nueva Agence Française de Sécurité Sanitaire des Produits de Santé (AFSSAPS). En 1999 ingresó en el Consejo de Administración de la EMEA y fue elegido vicepresidente del mismo en 2003. Thomas Lönngren, Director Ejecutivo, nacido el 16 de diciembre de 1950, nacionalidad sueca Estudios: Doctor en farmacia por la Facultad de Farmacia de la Universidad de Uppsala. Máster en farmacia social y legal. Estudios de posgrado en administración y economía sanitaria. Trayectoria profesional hasta la fecha: De 1976 a 1978 fue profesor en la Universidad de Uppsala. Perteneció al Consejo Nacional de Salud y Bienestar de Suecia desde 1978 hasta 1990, tiempo durante el cual fue responsable de medicamentos de origen vegetal, cosméticos, equipos médicos, narcóticos y anticonceptivos. Fue consultor farmacéutico jefe para el programa sueco de cooperación sanitaria con Vietnam de 1982 a 1994. Se incorporó a la agencia sueca de medicamentos en 1990, primero como Director de Operaciones y posteriormente como Subdirector General. Es Director Ejecutivo de la EMEA desde enero de 2001.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 48/58
Comités científicos de la EMEA Daniel Brasseur, Presidente del CPMP, nacido el 7 de junio de 1951, nacionalidad belga Estudios: Doctor en medicina por la Universidad Libre de Bruselas. Estudios de posgrado en pediatría y doctorado en nutrición. Trayectoria profesional hasta la fecha: De 1976 a 1986, el Dr. Brasseur trabajó como pediatra en el Hospital Universitario Sint Pieter de Bruselas. En 1986 y 1987 trabajó un corto período de tiempo para la industria farmacéutica, antes de volver al trabajo clínico en el Hospital Universitario Infantil Reina Fabiola de Bruselas como jefe de la unidad de nutrición y farmacodinámica, un puesto que sigue ocupando en la actualidad. Se incorporó al Cuerpo de Inspectores Farmacéuticos del Ministerio de Salud Pública belga como jefe de asesores médicos en 1997. Fue nombrado miembro del CPMP en 1997. El Dr. Brasseur ha ocupado diversos puestos académicos y es actualmente profesor de nutrición y enfermedades asociadas en la Universidad Libre de Bruselas. Eric Abadie, Vicepresidente del CPMP, nacido el 14 de julio de 1950, nacionalidad francesa Estudios: Doctor en medicina por la Universidad de París. Estudios de posgrado en medicina interna, endocrinología, diabetología y cardiología. También es máster en administración de empresas. Trayectoria profesional hasta la fecha: De 1981 a 1983, el Dr. Abadie ocupó diferentes puestos clínicos y de laboratorio, antes de entrar en la industria farmacéutica en 1983. Fue director de asuntos médicos de la asociación francesa del sector farmacéutico de 1985 a 1993 y regresó a la industria hasta 1994. Se incorporó a la agencia francesa de medicamentos en 1994 como director de evaluación farmacoterapéutica, un puesto que sigue ocupando actualmente. El Dr. Abadie es consultor en cardiología y diabetología desde 1984. Gérard Moulin, Presidente del CVMP, nacido el 18 de octubre de 1958, nacionalidad francesa Estudios: Doctor en microbiología por la Universidad de Lyon. Trayectoria profesional hasta la fecha: De 1981 a 1984, el Dr. Moulin trabajó en el Laboratorio de Patología Bovina de Lyon. En 1984 se incorporó al Laboratorio de Medicina Veterinaria de Fougères como asesor y ponente para dosieres de autorización de comercialización. También fue responsable de una unidad del laboratorio. En 1997, fue nombrado Jefe de la unidad de evaluación de productos farmacéuticos de la agencia veterinaria francesa (AFSSA-ANMV). Johannes Hoogland, Vicepresidente del CVMP, nacido el 22 de febrero de 1956, nacionalidad holandesa Estudios: Licenciado en química analítica por la Universidad de Amsterdam en 1984, y doctor en bioquímica por la Universidad de Amsterdam en 1988. Trayectoria profesional hasta la fecha: Trabajó para la industria alimentaria (1976-1977) y para el laboratorio biológico de la Universidad Pública de Amsterdam (1977-1978). Contratado por el Ministerio de Agricultura, Naturaleza y Pesca desde 1988; entre 1988 y 1998 trabajó para el Instituto Estatal de Control de la Calidad de los Productos Agrícolas (RIKILT-DLO) como asesor de medicamentos veterinarios y aditivos de los piensos, investigación sobre el desarrollo de métodos analíticos y desarrollo de sistemas de calidad para la producción agrícola. Desde 1998 hasta la actualidad trabaja en el Bureau Registratie Diergeneesmiddelen (BRD). Es miembro del CVMP desde 1998 y presidente del Grupo ad hoc sobre evaluación del riesgo medioambiental del CVMP. Fue nombrado vicepresidente del CVMP en enero de 2002.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 49/58
Josep Torrent i Farnell, Presidente del COMP, nacido el 2 de mayo de 1954, nacionalidad española Estudios: Licenciado en farmacia y doctor en medicina y cirugía por la Universidad de Barcelona, además de cursar estudios de posgrado en farmacología y toxicología, salud pública e instituciones europeas. Especialista en medicina interna y farmacología clínica. Doctor en farmacología clínica por la Universidad Autónoma de Barcelona (UAB). Trayectoria profesional hasta la fecha: De 1977 a 1990, el Prof. Torrent i Farnell trabajó en medicina interna y farmacología clínica en España y fue Profesor Adjunto de Farmacología en la UAB. De 1990 a 1994, fue consejero técnico de Evaluación Clínica y Farmacología del Ministerio de Sanidad español y miembro del Grupo de trabajo “Eficacia”, además de participar en el grupo “Eficacia” de la ICH. En 1992 pasó a ser profesor de Farmacología Clínica y Terapéutica y director del curso de máster/diplomatura sobre Registro Europeo de Medicamentos (UAB). Se incorporó a la EMEA en 1995 como administrador científico principal y de 1996 a 1998 fue jefe del Sector de nuevas sustancias químicas. En 1998 fue nombrado director coordinador para la creación de la agencia española de medicamentos y su director ejecutivo de 1999 a 2000. Fue nombrado presidente del Comité de Medicamentos Huérfanos en mayo de 2000. En noviembre de 2000 fue nombrado director general del Centro Avanzado de Servicios y Formación para la Salud y la Vida, Fundación Dr. Robert (UAB). Yann Le Cam, Vicepresidente del COMP, nacido el 15 de julio de 1961, nacionalidad francesa Estudios: Es licenciado en administración de empresas por el Institut Supérieur de Gestion de París. Obtuvo también un MBA por el Centre de Perfectionnement aux Affaires, Groupe HEC-CPA, 2000, Jouy-en-Josas, Francia. Trayectoria profesional hasta la fecha: El Sr. Le Cam tiene 15 años de experiencia profesional y compromiso personal con organizaciones no gubernamentales de salud e investigación médica en Francia, Europa y Estados Unidos, en los campos del sida y las enfermedades genéticas. Fue Director General de la AIDES Fédération Nationale de 1992 a 1998. Posteriormente se incorporó a la Asociación Francesa de Enfermedades Neuromusculares (AFM) como asesor especial para promover políticas de salud pública sobre enfermedades poco comunes, para crear la Alliance Maladies Rares francesa, una agrupación nacional de 70 asociaciones de pacientes, y para asesorar a la Organización Europea de Enfermedades poco comunes (Eurordis), con sede en París. Es también vicepresidente de la Alianza Internacional de Organizaciones de Pacientes (IAPO) con sede en Londres. El Sr. Le Cam tiene tres hijas, la mayor de las cuales tiene fibrosis quística.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 50/58
Unidad para la evaluación previa a la autorización de medicamentos de uso humano Patrick Le Courtois, Jefe de Unidad, nacido el 9 de agosto de 1950, nacionalidad francesa Estudios: Doctor en medicina por la Universidad de París. Doctor en salud pública por la Universidad de Burdeos. Otros diplomas de posgrado en medicina tropical, investigación clínica y epidemiología. Trayectoria profesional hasta la fecha: De 1977 a 1986, el Dr. Le Courtois trabajó como médico general y como director de un centro médico en París. En 1986 se incorporó a la Universidad de Burdeos y participó en distintas áreas de investigación de la salud pública, como epidemiología, investigación clínica, farmacovigilancia, enfermedades infecciosas y tropicales, economía sanitaria y educación sanitaria. En 1990 se incorporó a la Dirección de Farmacia del Ministerio de Sanidad francés y, en 1993, a la agencia francesa de medicamentos como miembro del CPMP. Se incorporó a la EMEA en septiembre de 1997, fue nombrado Jefe del Sector de nuevas sustancias químias en junio de 1998 y Jefe del Sector de medicamentos huérfanos y asesoramiento científico en enero de 2001. Agnès Saint Raymond, Jefa del Sector de medicamentos huérfanos y asesoramiento científico, nacida el 7 de septiembre de 1956, de nacionalidad francesa Estudios: Doctora en medicina por la Universidad de París. Diplomas de posgrado en pediatría y metodología. Trayectoria profesional hasta la fecha: La Dra. Saint Raymond ocupó la plaza de pediatra en un hospital pediátrico universitario en París, seguido por años de trabajo para laboratorios farmacéuticos. En 1995 se incorporó a la Agencia Francesa del Medicamento como Directora de la Unidad de evaluación fármaco-tóxico-clínica. Se incorporó a la EMEA en enero de 2000 y fue nombrada Jefe del Sector de asesoramiento científico y fármacos huérfanos en diciembre de 2001. También se ocupa de temas relacionados con medicamentos pediátricos. John Purves, Jefe de Sector de calidad de los medicamentos, nacido el 22 de abril de 1945, nacionalidad británica Estudios: Licenciado en farmacia por la Universidad de Heriot-Watt, Edimburgo. Doctor en filosofía, titulado en microbiología farmacéutica por la Universidad de Strathclyde, Glasgow. Trayectoria profesional hasta la fecha: De 1972 a 1974, el Dr. Purves trabajó en la industria farmacéutica. Entre 1974 y 1996 ocupó diversos cargos en la División de Medicamentos y en la Agencia de Control de Medicamentos del Reino Unido, incluido el de inspector de fabricación farmacéutica, revisor de documentación y director de la Unidad de Biotecnología y Biología. Representó al Reino Unido en el grupo de trabajo “Biotecnología” y participó en la formulación de numerosas directrices relativas a biotecnologías y productos biológicos. Se incorporó a la EMEA en agosto de 1996 como jefe del Sector de biotecnología y productos biológicos. Fue nombrado jefe de Sector de calidad de medicamentos en enero de 2001. Isabelle Moulon, Jefa del Sector de seguridad y eficacia de los medicamentos, nacida el 9 de marzo de 1958, nacionalidad francesa Estudios: Doctora en medicina por la Universidad de Grenoble, Francia. Especialista en endocrinología. Estudios de posgrado en estadística, metodología y nutrición. Trayectoria profesional hasta la fecha: Trabajó como endocrinóloga clínica en un hospital francés hasta 1987, cuando se incorporó a la Dirección de Farmacia del Ministerio de Sanidad francés. De 1992 a 1995 trabajó para la industria farmacéutica, antes de incorporarse a la EMEA en julio de 1995. Fue nombrada Jefa del Sector de seguridad y eficacia de los medicamentos en enero de 2001.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 51/58
Marisa Papaluca Amati, Jefa Adjunta del Sector de seguridad y eficacia de los medicamentos, nacida el 12 de octubre de 1954, nacionalidad italiana Estudios: Licenciada en medicina y cirugía por la Universidad de Roma. Especialista en medicina interna. Estudios de posgrado en cardiología y endocrinología. Trayectoria profesional hasta la fecha: De 1978 a 1983, la Dra. Papaluca trabajó en el Tercer Departamento de Medicina Interna de la Universidad de Roma, ocupándose de proyectos de investigación en el área de la inmunología clínica y la inmunología celular. Entre 1984 y 1994 fue directora médica del Departamento Farmacéutico del Ministerio de Sanidad italiano. Fue miembro del antiguo Comité de Especialidades Farmacéuticas en representación de su país, ponente para un tema sobre eficacia de la ICH y miembro de los Grupos de Trabajo Internacionales CIOMS I y II sobre farmacovigilancia. Se incorporó a la EMEA en octubre de 1994. Fue nombrada jefa adjunta de Sector de seguridad y eficacia de los medicamentos en enero de 2001.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 52/58
Unidad de Evaluación de Medicamentos de Uso Humano después de su Autorización Noël Wathion, Jefe de Unidad, nacido el 11 de septiembre de 1956, nacionalidad belga Estudios: Licenciado en farmacia por la Universidad Pública de Bruselas. Trayectoria profesional hasta la fecha: El Sr. Wathion inició su trayectoria profesional como farmacéutico en una farmacia minorista. Posteriormente, fue designado inspector jefe del Servicio de Inspección Farmacéutica de Bruselas (Ministerio de Asuntos Sociales y Sanidad Pública), actuando como secretario de la Comisión Belga de Medicamentos. Ha sido miembro belga del CPMP (Comité de Especialidades Farmacéuticas) y del CVMP (Comité de Medicamentos Veterinarios), y representante en el Comité Farmacéutico. Se incorporó a la EMEA en agosto de 1996 como Jefe del Sector de asuntos reglamentarios y farmacovigilancia y fue nombrado Jefe de la Unidad para la evaluación de medicamentos en septiembre de 2000. Después de la reestructuración de la Unidad de evaluación de medicamentos de uso humano en 2001, fue nombrado Jefe de la Unidad para la evaluación de medicamentos de uso humano después de su autorización. Tony Humphreys, Jefe del Sector de asuntos reglamentarios y servicios de apoyo, nacido el 12 de diciembre de 1961, nacionalidad irlandesa Estudios: Licenciado en Farmacia y máster en ciencias farmacéuticas en el área de investigación de la microencapsulación por el Trinity College de Dublín. Trayectoria profesional hasta la fecha: Desde que terminó sus estudios en 1983, el Sr. Humphreys ha trabajado en el área del desarrollo farmacéutico para un fabricante nacional de genéricos de marca y una empresa internacional de investigación y desarrollo. En 1991 se incorporó a la División Internacional de Asuntos Reglamentarios de Glaxo Group Research Limited, donde fue responsable del desarrollo y la presentación de una serie de solicitudes de registro internacionales en diferentes áreas terapéuticas. Se incorporó a la EMEA en mayo de 1996 y fue nombrado Jefe del Sector de asuntos reglamentarios y servicios de apoyo en enero de 2001. Panos Tsintis, Jefe del Sector de farmacovigilancia y seguridad y eficacia de medicamentos ya autorizados, nacido el 18 de septiembre de 1956, nacionalidad británica. Estudios: Licenciado en medicina por la Universidad de Sheffield en 1983. Estudios de posgrado en medicina interna (FRCP) y medicina farmacéutica (FFPM). Trayectoria profesional hasta la fecha: Seis años de experiencia clínica en hospitales del Reino Unido, 5 años como Director de Farmacovigilancia y Asuntos Reglamentarios para Astra Pharmaceuticals en el Reino Unido y un total de 7 años en la Agencia de Control de Medicamentos del Reino Unido. Antes de ser nombrado Jefe de la Unidad de Farmacovigilancia, ocupó diversos puestos en las áreas de actividades previas y posteriores a la autorización de medicamentos y fue también delegado británico en el Grupo de trabajo del CPMP “Farmacovigilancia”. El Dr. Tsintis se incorporó a la EMEA como Jefe del Sector de Farmacovigilancia y seguridad y eficacia de medicamentos ya autorizados en marzo de 2002. Sabine Brosch, Jefa Adjunta del Sector de farmacovigilancia, seguridad y eficacia de medicamentos ya autorizados, nacida el 17 de agosto de 1963, nacionalidad austriaca Estudios: Máster en farmacia y Doctora en farmacología por la Universidad de Viena. Estudios de posgrado en farmacología por la Universidad de Melbourne y Auckland. Trayectoria profesional hasta la fecha: De 1988 a 1992, la Dra. Brosch trabajó como profesora adjunta del Departamento de Farmacología y Toxicología de la Universidad de Viena, donde se especializó en electrofisiología. En 1992 se trasladó al Departamento de Farmacovigilancia del Ministerio de Salud austriaco y en 1995 completó un programa de formación de seis meses en la Unidad de Productos Farmacéuticos de la Comisión Europea. Se incorporó a la EMEA en noviembre de 1996 y fue nombrada Jefa Adjunta del Sector de farmacovigilancia, seguridad y eficacia de medicamentos ya autorizados en enero de 2001.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 53/58
Unidad de medicamentos e inspecciones en Veterinaria Peter Jones, Jefe de Unidad, nacido el 9 de agosto de 1947, nacionalidad británica Estudios: Licenciado por la Facultad de Ciencias Veterinarias de la Universidad de Liverpool y miembro del Real Colegio de Veterinarios del Reino Unido. Trayectoria profesional hasta la fecha: Después de trabajar durante varios años como veterinario general en el Reino Unido y Canadá, el Dr. Jones se incorporó a la industria farmacéutica en el sector de la sanidad animal. Ha ocupado diversos puestos en los campos de la investigación y los asuntos reglamentarios en empresas multinacionales y, más recientemente, fue nombrado director de Asuntos Reglamentarios Internacionales de Productos de Sanidad Animal de Merck Sharp & Dohme en Nueva Jersey, Estados Unidos. Se incorporó a la EMEA en junio de 1995 y fue nombrado jefe de la Unidad Veterinaria en diciembre de ese mismo año. Es coordinador de la UE en la VICH. Jill Ashley-Smith, Jefa del Sector de procedimientos para la autorización de comercialización de medicamentos veterinarios, nacida el 18 de diciembre de 1962, nacionalidad británica Estudios: Licenciada en farmacología por el Kings College de la Universidad de Londres. Licenciada en cirugía veterinaria por el Royal Veterinary College de la misma universidad. Trayectoria profesional hasta la fecha: De 1987 a 1994, la Sra. Ashley-Smith trabajó en la industria farmacéutica veterinaria, primero como asesora técnica y posteriormente como gestora de registros. En 1994 se incorporó a la Dirección de Medicamentos Veterinarios como asesora veterinaria en el equipo de productos farmacéuticos y aditivos alimentarios. Participó como miembro del Reino Unido en el CVMP desde 1996, hasta su incorporación a la EMEA en julio de 1997. Melanie Leivers, Asistente del Jefe de Sector de procedimientos para la autorización de comercialización de medicamentos veterinarios, nacida el 1 de diciembre de 1958, nacionalidad británica Estudios: Licenciada en bioquímica y farmacología por la Universidad de Leeds. Diploma de posgrado en legislación de la Comunidad Europea otorgado por King’s College, Londres. Trayectoria profesional hasta la fecha: La Sra. Leivers colaboró con el Milk Marketing Board para Inglaterra y Gales (MMB) como Liaison Chemist 5 años antes de ser nombrada Directora Adjunta del MMB/oficina de la Federación de Cooperativas Agrícolas en Bruselas, en representación de todos los sectores de cooperación agrícola con las instituciones europeas. A continuación tuvo un contrato de corta duración con la Comisión Europea (DG XI) y luego se incorporó a Pfizer (anteriormente SmithKline Beecham Animal Health) como directora de asuntos administrativos. La Sra. Leivers se incorporó a la EMEA en febrero de 1996 y fue nombrada asistente del Jefe de Sector en junio de 2001. Kornelia Grein, Jefa de Sector de seguridad de medicamentos veterinarios, nacida el 24 de julio de 1952, nacionalidad alemana Estudios: Licenciada en química y farmacia por la Universidad Libre de Berlín. Doctora en química orgánica por la misma Universidad. Trayectoria profesional hasta la fecha: De 1976 a 1987, la Sra. Grein trabajó como asesora científica en la Universidad Libre de Berlín y como farmacéutica. En 1987 se incorporó a la agencia alemana del medio ambiente como administradora científica. Destacada a la Comisión Europea en 1992, regresó a Alemania al Ministerio de Medio Ambiente en 1995. Ha participado en la elaboración del sistema de clasificación y etiquetado de la UE y en la armonización de los procedimientos de valoración de riesgos y los requisitos de información sobre sustancias químicas relativos a la salud humana y el medio ambiente, tanto en la Unión Europea como en la OCDE. Se incorporó a la EMEA en abril de 1996.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 54/58
Emer Cooke, Jefa del Sector de inspecciones, nacida el 9 de abril de 1961, nacionalidad irlandesa Estudios: Licenciada en farmacia y Master en química farmacéutica y dirección de empresas (MBA) por el Trinity College de Dublín. Miembro de la Sociedad de Farmacéuticos de Irlanda. Trayectoria profesional hasta la fecha: La Sra. Cooke trabajó en varias industrias farmacéuticas irlandesas antes de incorporarse a la Irish Medicines Board como asesora farmacéutica en 1988. Después de obtener un MBA en 1991, se incorporó a la EFPIA, la asociación de industrias farmacéuticas europeas, como Directora de Asuntos Científicos y Reglamentarios. Sus responsabilidades consistían en coordinar los aspectos reglamentarios de los procedimientos europeos, así como las actividades relacionadas con la Conferencia Internacional de Armonización (ICH). Después de una estancia de tres años en Praga (República Checa), donde trabajó como consultora en asuntos farmacéuticos europeos además de proseguir su trabajo con la EFPIA, se incorporó a la Unidad de Productos Farmacéuticos de la Comisión Europea en septiembre de 1998. Sus responsabilidades en este puesto consistían en coordinación de las actividades de la ICH, relaciones con la FDA, aspectos farmacéuticos de los acuerdos de reconocimiento mutuo, asuntos relacionados con BPF e inspecciones, medicamentos huérfanos, preparación de un Reglamento sobre medicamentos de uso pediátrico y cuestiones relacionadas con la ampliación de la UE. Se incorporó a la EMEA como Jefa del Sector de Inspecciones en julio de 2002.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 55/58
Unidad de comunicaciones y redes Hans-Georg Wagner, Jefe de Unidad, nacido el 29 de noviembre de 1948, nacionalidad alemana Estudios: Doctor en ciencias naturales (física aplicada y ciencias de los materiales) por la Universidad de Saarbruecken, diplomado en física por la Universidad de Tuebingen, Master of Arts (matemáticas) por la Universidad de Cambridge, Reino Unido. Trayectoria profesional hasta la fecha: El Dr. Wagner fue investigador y profesor adjunto en la Universidad de Saarbruecken entre 1976 y 1981. Más tarde fue profesor y profesor titular en esa misma universidad, hasta que se incorporó a Comisión Europea de Luxemburgo en enero de 1986. Allí fue responsable de varios grupos de la división de apoyo técnico de la Dirección de Salvaguardias de Euratom. El Dr. Wagner fue nombrado Jefe del Sector de TI en el mismo servicio en 1993. Se incorporó a la EMEA el 1 de mayo de 2002. Beatrice Fayl, Jefa del Sector de gestión y publicación de documentos, nacida el 9 de octubre de 1959, nacionalidad danesa Estudios: Idiomas y lingüística en la Universidad de East Anglia y título de posgrado en biblioteconomía y ciencias de la información en la Universidad de Gales. Trayectoria profesional hasta la fecha: Ha ocupado diversos cargos como documentalista en varios países europeos, siendo su puesto más reciente el ocupado de 1988 a 1995 en el Servicio de Documentación de la Delegación de la Comisión Europea en Noruega. La Sra. Fayl se incorporó a la EMEA en abril de 1995. Sylvie Bénéfice, Jefa de Sector de organización de reuniones y conferencias, nacida el 28 de diciembre de 1954, nacionalidad francesa Estudios: Doctora en ciencias físicas; estudios de gestión de la investigación; título de química orgánica física; master en química orgánica física; licenciada en bioquímica. Trayectoria profesional hasta la fecha: De 1982 a 1986, la Dra. Bénéfice fue investigadora de la Universidad de Montpellier, Francia. En 1986 se incorporó al Centro Nacional de Investigaciones Científicas de Francia como Chargé de Recherche 1ère Classe y fue designada “directora para Asuntos Europeos” en 1991. De 1993 a 1997 trabajó en comisión de servicios en la Comisión Europea (DG XII) como secretaria científica para las acciones COST en el campo de la química, desempeñando tareas de coordinación de redes de investigación y de organización de conferencias y seminarios científicos en Europa. Se incorporó a la EMEA en septiembre de 1997. Tim Buxton, Jefe de Sector de dirección de proyectos, nacido el 27 de febrero de 1959, nacionalidad británica Estudios: Licenciado en Derecho por la Universidad de Birmingham, miembro del Institute of Chartered Accountants en Inglaterra y Gales. Trayectoria profesional hasta la fecha: Tim Buxton publicó artículos con Touche Ross & Co en Londres en 1987. Tras un año de trabajo en banca fue director financiero de una empresa privada de 1988 a 1995. Trabajó de asesor de dirección para proyectos a largo plazo hasta enero de 1997, fecha en que se incorporó a la EMEA. Fue nombrado Jefe de Sector el 1 de mayo de 2002.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 56/58
Michael Zouridakis, Jefe de Sector de tecnología de la información, nacido el 8 de febrero de 1958, nacionalidad sueca Estudios: Magister en informática y Licenciado en administración de empresas y economía por la Universidad de Gotemburgo. Trayectoria profesional hasta la fecha: De 1985 a 1989, el Sr. Zouridakis ocupó diversos puestos en el campo de las tecnologías de la información como programador, analista de sistemas y gestor de proyectos, y trabajó como consultor de 1990 a 1992. En 1993 pasó a desempeñar las tareas de Director de Sistemas de Información/Tecnología de la Información en Astra AB en Grecia. Se incorporó a la EMEA en abril de 1998. David Drakeford, Subjefe del Sector de tecnología de la información, nacido el 4 de diciembre de 1957, nacionalidad irlandesa Estudios: Licenciado en física experimental y máster en ingeniería electrónica por el Trinity College de Dublín. Trayectoria profesional hasta la fecha: David Drakeford trabajó en Telecom Eireann, donde dirigió la instalación de una red nacional de comunicación de datos. En 1987, entró a trabajar en Coopers & Lybrand, donde fue consultor jefe especialista en la gestión y el control financiero de grandes proyectos, principalmente relacionados con la TI. Participó también en numerosos proyectos multinacionales, como la instalación de un sistema mundial de gestión de la información sobre ensayos clínicos por encargo de una empresa farmacéutica con sede en Suiza. Se incorporó a la EMEA en febrero de 1997.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 57/58
Unidad de Administración Andreas Pott, Jefe de la Unidad de Administración, nacido el 14 de abril de 1949, nacionalidad alemana Estudios: Máster en ciencias políticas, historia e inglés por la Universidad de Hamburgo. Certificat de Hautes Etudes Européennes por la Escuela Universitaria Europea, Brujas. Trayectoria profesional hasta la fecha: De 1972 a 1989, el Sr. Pott ocupó una serie de puestos docentes y de investigación, entre ellos una beca de investigación en el Instituto de Investigación para la Paz y Política de Seguridad de la Universidad de Hamburgo. El Sr. Pott entró en la Secretaría del Parlamento Europeo en 1989, en calidad de secretario de la Comisión de Investigación, Desarrollo Tecnológico y Energía, de la Comisión de Presupuestos y, más tarde, de la Oficina del Parlamento y la Conferencia de Presidentes. Se incorporó al Centro de Traducción para los Organismos de la Unión Europea en 1999 como jefe del Departamento de Organización y Cooperación Interinstitucional. Entró en la EMEA en mayo de 2000. Frances Nuttall, Jefe del Sector de personal y presupuestos, nacida el 11 de noviembre de 1958, nacionalidad irlandesa Estudios: Licenciada en administración pública y máster en economía por el Trinity College de Dublín. Trayectoria profesional hasta la fecha: Ha ocupado diversos cargos en la administración pública de Irlanda, prestando sus servicios en los Departamentos de Sanidad y Hacienda y la Oficina de Obras Públicas. La Sra. Nuttall trabajó posteriormente en la Organización de las Naciones Unidas para la Agricultura y la Alimentación durante cinco años, hasta su incorporación a la EMEA en mayo de 1995. Sara Mendosa, Jefa del Sector de servicios de infraestructura, nacida el 23 de enero de 1950, nacionalidad británica Estudios: Estudios de negocios e idiomas en la Politécnica Loughborough Trayectoria profesional hasta la fecha: De 1975 a 1990 la Sra. Mendosa ocupó algunos puestos en la Comisión Europea en Luxemburgo, como el servicio de conferencias, la oficina de publicaciones oficiales y el departamento de estadística. En 1991 la Sra. Mendosa fue trasladada a la oficina de Londres de la representación de la Comisión Europea en el Reino Unido. Se incorporó a la EMEA en noviembre de 1994 y fue nombrada Jefa de Sector en noviembre de 2002. Gerard O’Malley, Jefe del Sector de contabilidad, nacido el 14 de octubre de 1950, nacionalidad irlandesa Estudios: Licenciado en comercio por el University College de Dublín. Miembro del Institute of Chartered Accountants de Irlanda. Censor jurado de cuentas y miembro del Registro Oficial de Auditores de Cuentas de España. Trayectoria profesional hasta la fecha: De 1971 a 1974, el Sr. O’Malley realizó su período de prácticas en Stokes Kennedy Crowley. De 1974 a 1985 ocupó el puesto de director de auditoría en España de Ernst and Young, y, de 1985 a 1995 fue interventor financiero en Johnson Wax Española. Se incorporó a la EMEA en abril de 1995.

Programa de Trabajo de la EMEA para 2003 EMEA/MB/057/02/es/Final Página 58/58
Oficina de prensa Martin Harvey Allchurch, responsable de la oficina de prensa, nacido el 20 de octubre de 1966, nacionalidad británica Estudios: Licenciado en Derecho por la Universidad de Dundee del Reino Unido. Master en legislación europea e internacional por la Universidad Libre de Bruselas, Bélgica. Trayectoria profesional hasta la fecha: Tras un período de formación en la Comisión Europea en 1991-92, Martin Harvey trabajó de asesor para asuntos europeos en Bruselas, de 1992 a 1995. En este período también trabajó de editor colaborador para una publicación sobre asuntos europeos y corresponsal en Bruselas para una publicación farmacéutica estadounidense. Se incorporó a la EMEA en septiembre de 1995, trabajando en la oficina del Director Ejecutivo. Fue nombrado responsable de la oficina de prensa en septiembre de 2001.


















