
Princípios de Farmacocinética Clínica e Farmacocinética...
43
Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada 1 Pós-Graduação a Distância Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada Professor Ms. André Luiz Moura Princípios de Farmacocinética Clínica/ Farmacocinética Clínica Aplicada Professor André Luiz Moura
Transcript of Princípios de Farmacocinética Clínica e Farmacocinética...

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
1
Pó
s-G
rad
uaç
ão a
Dis
tân
cia
Princípios de Farmacocinética Clínica e Farmacocinética Clínica AplicadaProfessor Ms. André Luiz Moura
Princípios de Farmacocinética Clínica/ Farmacocinética Clínica AplicadaProfessor André Luiz Moura

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
2
Introdução 3Farmacocinética X Farmacodinâmica 5Os quatro processos da farmacoterapia. 6Farmacocinética. 8Objetivos da farmacocinética clínica. 8Revisão de farmacocinética básica. 8Passagem de fármacos pela membrana. 9Fatores que afetam a absorção de um fármaco. 11Influencia do pH e pKa: 12Biodisponibilidade: 14Fatores fisiológicos que podem influenciar na absorção. 14Resumo dos fatores que podem modificar a absorção 18Distribuição de fármacos. 18Modelos compartimentais em farmacocinética. 19Modelos não compartimentais 21Fatores que podem influenciar as proteínas plasmáticas 22Concentração de ácidos graxos livres 23Volume aparente de distribuição: 23Principal aplicação do Vd 25Relação entre dose de ataque e dose de manutenção 26Área Sob a Curva (AUC): 26Eliminação e Metabolismo de fármacos (excreção e biotransformação). 29Principais enzimas envolvidas com o processo de fase I. 29Reações de fase II 31Excreção de fármacos 31Clearance ou depuração 34Cálculo do clearance e sua aplicabilidade. 34Administração de medicamentos em doses múltiplas 35Monitoramento farmacocinético. 37Requisitos essenciais para controle terapêutico 38Informações sobre o modo de administração e definição dos tempos de coleta 38Fármacos que devem ser submetidos ao controle terapêutico 41Referências 43
SUMÁRIO

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
3
INTRODUÇÃO No homem, o instinto de sobrevivência geralmente
é guiado pelo pensamento abstrato. Desta forma, a
espécie humana utiliza-se – além das respostas reflexas
de autoproteção comuns aos seres vivos – de formas
elaboradas de preservação e restauração da saúde.
Ao se observar a história da raça humana, verifica-se a
existência de inúmeros registros ações que objetivam o
benefício, entre estas, pode-se destacar as mudanças de
comportamento, ritos religiosos, atos cirúrgicos, utilização
de substâncias que presumivelmente possuem alguma
ação terapêutica, entre outras. Em muitas destas ações
ocorreu alívio do sofrimento por razões diversas de
qualquer efeito intrínseco.
Algumas intervenções terapêuticas apresentaram nítido
efeito intrínseco, como por exemplo, a contenção de uma
hemorragia por meio de uma simples compressão local, o
alinhamento de ossos fraturados, a retirada de tumores e
membros em situação gangrenosa certamente contribuíram
para o alívio de sofrimento e até para o prolongamento
da vida. Alguns dos medicamentos utilizados também
apresentavam poder intrínseco, como opióides e álcool,
mas a maioria dos supostos remédios eram certamente
desprovidos de qualquer ação. Demonstrou-se que grande
parte dos resultados obtidos com tratamentos antigos
provinha de fatores diversos do efeito intrínseco, sendo
muitos deles, deletérios ao homem e animais. Atualmente
descreve-se que a quantificação do efeito intrínseco
de medicamentos é feito por métodos farmacológicos e
farmacológico-clínico.
Um fato importante e que não se pode negar, é que o
início do tratamento de doenças por meio de drogas é de
certa forma muito antigo. As primeiras drogas empregadas
como medicamento foram as de origem natural, extraídas
principalmente de plantas, e destinavam-se à terapia de
doenças infecciosas. Por exemplo, o imperador chinês
Sheng Nung (há mais de 3000 anos), em seus escritos
sobre ervas medicinais, recomendava o uso da planta
Ch’ang shang para o tratamento da malária. Atualmente
sabe-se que esta planta contém o alcaloide febrifugina,
que possui forte ação antimalárica. Índios brasileiros
empregavam a raiz da ipeca para disenteria e diarreia; esta
planta, contem a emetina, que é eficaz para o tratamento
daquelas moléstias. Já os índios peruanos, utilizavam para
o tratamento da malária, cascas da quina, de onde se
extrai a quinina.
Conceitos
- Droga: a palavra droga origina do holandês
antigo droog que significa folha seca, pois, antigamente
quase todos os medicamentos eram feitos à base de
vegetais. Atualmente, droga é definida como sendo
qualquer substância capaz de modificar a função dos
organismos vivos, resultando em mudanças fisiológicas
ou de comportamento. Assim, droga consiste em
qualquer substância química que possui a capacidade
de produzir efeito farmacológico, ou seja, que provoque
alterações funcionais ou somáticas. Se estas alterações
forem benéficas, podemos denominar de fármaco ou
droga-medicamento ou apenas medicamento, e se forem
maléficas denominamos de tóxico ou droga-tóxico.
- Princípio ativo: corresponde à substância (ou
grupo destas), responsável pela ação terapêutica, com
composição química e ação farmacológica conhecida.
- Fármaco: substância química, estruturalmente
definida, utilizada para o fornecimento de elementos
essenciais ao organismo, na prevenção, no tratamento
e no diagnóstico de doenças, infecções ou de situações
de desconforto e na correção de funções orgânicas
desajustadas.
- Medicamento: é o mesmo que fármaco,
especialmente quando este se encontra em uma formulação
farmacêutica. Corresponde ao fármaco na especialidade
farmacêutica (comprimidos, drágeas, cápsulas, soluções,
pomadas, etc.). Em algumas situações, determinado
medicamento é constituído de dois ou mais fármacos, nas
chamadas associações medicamentosas.
-Farmacodinâmica: embora dependa da
Farmacocinética, estuda o local de ação, mecanismo de
ação, e os efeitos que as drogas promovem no organismo.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
4
- Farmacocinética: estuda o movimento da droga
através do organismo, envolvendo os processos de
absorção, distribuição, biotransformação (metabolismo), e
eliminação ou excreção.
- Dose de ataque: consiste na dose única que
é suficiente para elevar rapidamente a concentração
plasmática terapêutica do fármaco no organismo. A palavra
dose é derivada do grego dosis o que significa ação de dar
ou o que pode ser dado.
- Dose eficaz mediana: corresponde a dose
necessária para produzir determinada intensidade de um
efeito em 50% dos indivíduos.
- Dose letal: consiste no efeito observado da morte
dos animais de experiência com drogas.
- Efeito adverso ou indesejado: consiste na ação
provocada pelo fármaco diferente do efeito planejado.
Alguns autores denominam de efeito colateral, e que não
está relacionado à principal ação do medicamento.
- Medicamentos bioequivalentes: são
medicamentos que contém o mesmo fármaco ou princípio
ativo, na mesma quantidade e forma farmacêutica,
podendo ou não conter excipientes idênticos e de mesma
biodisponibilidade. Devem cumprir com as mesmas
especificações atualizadas da Farmacopéia Brasileira e,
na ausência destas, com as de outros códigos autorizados
pela legislação vigente, ou com outros padrões aplicáveis
de qualidade, relacionados à identidade, dosagem,
pureza, potência, uniformidade de conteúdo, tempo de
desintegração e velocidade de dissolução, quando for o
caso.
- Biodisponibilidade: é a fração de um dado princípio
ativo liberada por uma formulação farmacêutica em um
intervalo de tempo, disponível para absorção e que atingiu
a corrente sanguínea sistêmica na forma inalterada (não
biotransformada).
- Biotransformação: processo de alteração na
estrutura química que os fármacos sofrem no organismo,
geralmente por conversão enzimática, com a finalidade de
diminuir sua lipossolubilidade e facilitar sua excreção.
- Compartimento central: compreende ao volume
plasmático junto com o líquido extracelular dos tecidos,
neste compartimento a droga difunde-se instantaneamente
gerando equilíbrio, fazem parte deste compartimento
pulmões, fígado, cérebro.
- Compartimento periférico: compreende os tecidos
de menor perfusão, o que demora a gerar equilíbrio.
Fazem parte deste compartimento músculos, pele, tecido
gorduroso.
- Curva de Concentração Plasmática: determinação
laboratorial da quantidade de um fármaco no sangue,
este parâmetro reflete a dosagem inicial administrada,
a absorção, a biodisponibilidade, a meia-vida e a taxa
de metabolismo e excreção. Na curva de concentração
plasmática é possível determinar-se o pico sérico, ou seja,
o momento em que é atingida a concentração máxima
do fármaco na circulação. Estudando-se a área abaixo da
curva é possível entender a biodisponilidade do fármaco.
Esquema demonstrando a curva de concentração plasmática onde aparece o pico sérico (Fonte: http://www.anvisa.gov.br/servicosaude/controle/rede_rm/cursos/rm_controle/opas_web/modulo1/propriedades2.htm)
- Efeito de primeira passagem: metabolização
hepática de fármacos que acontece antes que o fármaco
seja distribuído pelo corpo, que acontece todas as vezes
que o medicamento é administrado por via oral. Na maioria
das vezes, diminui a concentração que fica disponível na
circulação sistêmica para posterior distribuição aos locais
de ação.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
5
Esquema demonstrando o efeito de primeira passagem(fonte: http://www.medscape.org/viewarticle/421137_1)
- Eficácia ou efetividade: capacidade de um fármaco
produzir o efeito máximo desejado.
- Índice terapêutico: expressa a margem de segurança
que um determinado fármaco apresenta em relação a
seus efeitos tóxicos e terapêuticos. Quanto maior o índice
terapêutico, maior é a segurança do fármaco. É a relação
entre a Dose Letal que mata 50% das cobaias (DL50) e a
Dose Efetiva em 50% das cobaias (DE50).
Esquema demonstrando como é determinado o índice terapêutico (Fonte: http://pharmacologycorner.com/therapeutic-index/ )
FARMACOCINÉTICA X FARMACODINÂMICA
Nos dias atuais, tanto os médicos como os pacientes
reconhecem a importância fundamental do tratamento com
fármacos como um dos meios primordiais para a prevenção
e alívio da doença. O médico tem a tarefa nada invejável
de selecionar a(as) droga(s) de melhor propriedade dentro
de um vasto acervo terapêutico. O grau de dificuldade na
escolha da droga apropriada varia de médico para médico,
dependendo de sua especialidade e da natureza da
população de pacientes. Para tentar amenizar os erros, estes
profissionais dentro de sua especialidade, geralmente usa
um número limitado de drogas, e detém uma experiência
ampla em relação à seleção da droga, posologia e reações
adversas. Ao contrário, médicos que tratam de populações
diversas, e que necessitam de adentrarem em diferentes
especialidades, são continuamente desafiados, pois
necessitam de trabalhar com um acervo terapêutico ainda
maior. Assim, estes profissionais correm um grande risco
de em determinada situação realizar uma prescrição com
medicamentos diferentes que podem interagir entre si, e
desencadear no paciente uma reação adversa.
A farmacologia dentro de um enfoque aplicado
fundamenta o ato de prescrever, permitindo que se
faça uma terapêutica medicamentosa mais científica e
racional. Esta se caracteriza pela seleção do medicamento
adequado para prevenir, reverter ou atenuar um processo
patológico. Entretanto, isto pode não ser o suficiente para
se obter sucesso na terapêutica medicamentosa, pois é
necessário garantir que o medicamento escolhido, atinja
em concentrações adequadas, o órgão ou sistema alvo.
Escolha inadequada do fármaco ou esquemas posológicos
inapropriados podem produzir efeitos indesejáveis. Para
que isto não ocorra, é necessário escolher doses, vias
de administração e intervalo entre doses que garantam
a chegada e a manutenção da concentração plasmática
e concentração no local de ação do fármaco. Esquemas
inadequados podem proporcionar concentrações
insuficientes ou subterapêuticas que falseiam a
interpretação sobre a eficácia do fármaco escolhido, ou
em outra hipótese, pode ocorrer a disponibilização de
concentrações muito acima da concentração tida como
terapêutica, desencadeando inúmeros efeitos adversos
oriundos das ações farmacológicas do medicamento, por
este motivo, deve-se sempre objetivar doses e esquemas
terapêuticos que proporcionem concentrações plasmáticas
tidas como terapêutica (faixa terapêutica; Figura 1).

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
6
A disponibilidade para o sistema biológico de um
determinado fármaco contido em uma forma farmacêutica
é essencial para o propósito do desenvolvimento de
formas de dosagem e possui fundamental importância
para a eficácia do medicamento. Portanto, antes de ser
absorvido por um sistema biológico, o fármaco deverá ser
liberado de sua forma farmacêutica (comprimido, solução,
drágea, etc) ou de um sistema de liberação de fármaco
(adesivo transdérmico, implante) e solubilizar-se nos
fluidos biológicos.
Curvas concentração plasmática em função do tempo demonstrando na primeira figura um perfil normal de um medicamento administrado por via oral e, na segunda figura, curvas que saíram da faixa terapêutica (Lüllmann et al. 2008)
OS QUATRO PROCESSOS DA FARMACOTERAPIA.
De acordo com o disposto anteriormente, para
se alcançar uma farmacoterapia de excelência, além
da escolha do melhor medicamento, da melhor forma
farmacêutica e da via de administração correta, para
que um determinado produto possa ser utilizado como
medicamento com finalidades de cura, diagnóstico,
prevenção, este deverá primeiramente estar de acordo
com as regras farmacológicas de ação, ou seja, deverá
respeitar as quatro fases ou processos farmacológicos,
que são por sua vez classificados em:
a) Fase ou processo farmacêutico: está relacionado
com todos os processo inerentes à formulação farmacêutica
e com a via de administração do fármaco. Neste processo,
não há nenhuma relação com os processos de absorção,
distribuição, metabolismo ou excreção dos fármacos, mas
está relacionado com a biodisponibilidade e bioequivalência
dos fármacos e medicamentos, respectivamente.
b) Fase ou processo farmacocinético: este processo
relaciona-se intimamente com os processos de absorção,
distribuição, metabolismo e excreção de fármacos.
c) Fase ou processo farmacodinâmico: está
relacionado com o efeito farmacológico produzido pelo
fármaco quando este atinge seu local de ação. Não
abrange apenas os efeitos farmacológicos que podem
ser responsáveis por um efeito terapêutico, mas também
aqueles responsáveis por causar reações adversas.
d) Fase ou processo terapêutico: Para que o
paciente seja beneficiado com a farmacoterapia, o efeito
farmacológico produzido pela droga deverá se transformar
em um efeito benéfico clínico, em outras palavras o efeito
produzido pelo medicamento tem significado clínico
desejável? Por exemplo, ao se administrar um diurético
tiazídico a um paciente portador de edema de membros
inferiores, este estará eliminando grandes quantidades de
sódio e consequentemente de água, o que poderá acarretar
em redução do edema, mas devemos lembrar que este tipo
de ação produzida pelo diurético também leva à depleção
de potássio, o que pode acarretar em distúrbios como
hipocalemia severa, assim o efeito terapêutico desejável
acaba produzindo um efeito indesejável.
Cada um dos processos descritos acima é de extrema
importância para o bom desempenho do medicamento.
Na verdade existem questões deverão ser respondidas
para que possamos entender melhor a importância destes
processos:
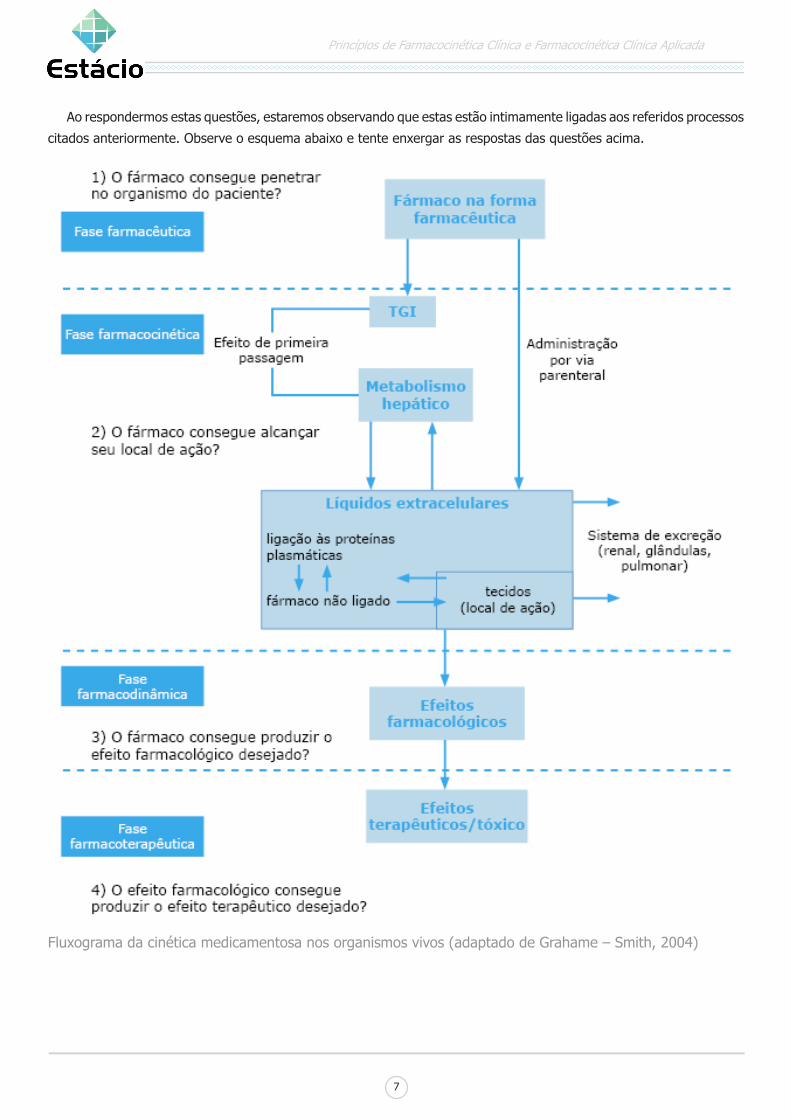
1) O fármaco consegue penetrar no organismo do
paciente?
2) O fármaco consegue alcançar seu local de ação?
3) O fármaco consegue produzir o efeito farmacológico
desejado?
4) O efeito farmacológico consegue produzir o efeito
terapêutico desejado?
Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
7
Ao respondermos estas questões, estaremos observando que estas estão intimamente ligadas aos referidos processos
citados anteriormente. Observe o esquema abaixo e tente enxergar as respostas das questões acima.
Fluxograma da cinética medicamentosa nos organismos vivos (adaptado de Grahame – Smith, 2004)

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
8
FARMACOCINÉTICA.A administração de um determinado fármaco contido
em uma forma farmacêutica, por uma via de administração,
em uma única dose ou em doses múltiplas (repetidas), tem
por finalidade a obtenção de um efeito farmacoterapêutico
à um indivíduo em situação patológica. O efeito
terapêutico desejado, como alívio da dor, diminuição da
ansiedade, regularização da pressão arterial, controle da
taxa glicêmica entre outras, depende da concentração do
princípio ativo no local de ação ,e também da velocidade
com que o princípio ativo consegue chegar no local
(esquema abaixo).
A intensidade da resposta farmacodinâmica, é
geralmente, proporcional à concentração do princípio
ativo no local de ação. Se o principio ativo se difundir
rapidamente do plasma para seu local de ação, a sua
concentração plasmática pode refletir sua concentração
no local de ação.
OBJETIVOS DA FARMACOCINÉTICA CLÍNICA.
A farmacocinética clínica é uma ciência de caráter
multidisciplinar e de grande interesse sanitário, que
possui como principal objetivo na prática assistencial a
individualização posológica, e a otimização do tratamento
farmacoterapêutico, a fim de alcançar a máxima eficácia
terapêutica com a mínima incidência de problemas
relacionados aos medicamentos.
Habitualmente os medicamentos são utilizados
partindo de critérios pré-estabelecidos que contribuam
com a estratégia do “acerto e erro”. Este empirismo está
embasado na resposta clínica ou bioquímica em relação
aos efeitos farmacológicos do medicamento prescrito.
Entretanto, este método não é passível de ser aplicado
a todos os casos, sendo necessárias adequações à
farmacoterapia em função da situação encontrada para
cada paciente.
Portanto, os estudos de farmacocinética clínica
permitem:
- Avaliar o sucesso ou insucesso de uma terapêutica
- Um meio de avaliar a extensão e velocidade de
chegada do fármaco ao seu local de ação
- Previsão e compreensão de efeitos secundários dos
fármacos
- Prever condições não testadas experimentalmente,
tais como dosagens, intervalos de administração, etc.
- Prever níveis teciduais, mesmo sem amostras dos
mesmos
- Comparar resultados entre diferentes indivíduos da
mesma espécie ou entre espécies diferentes.
- Descrever matematicamente o organismo
REVISÃO DE FARMACOCINÉTICA BÁSICA.
Até mesmo a mais promissora das terapias
farmacológicas irá fracassar em estudos clínicos se o
fármaco for incapaz de alcançar o seu órgão-alvo numa
concentração suficiente para exercer um efeito terapêutico.
Muitas das características que tornam o corpo humano
resistente a danos causados por invasores estranhos e
substâncias tóxicas também limitam a capacidade dos
fármacos modernos de combater os processos patológicos
no paciente. O reconhecimento dos numerosos fatores
que afetam a capacidade de um fármaco de atuar em
determinado paciente, bem como da natureza dinâmica
desses fatores com o transcorrer do tempo, é de suma
importância para a prática clínica da medicina.
Vários fatores podem contribuir para a liberação
de um fármaco, estes incluem as características físico-
químicas do princípio ativo (tamanho de suas partículas,
solubilidade em água ou gordura, pKa) e as características
das formas de dosagem ou do sistema de liberação, como a
natureza dos adjuvantes farmacotécnicos empregados na
formulação e o método utilizado para o desenvolvimento
da forma farmacêutica.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
9
O estabelecimento de esquemas posológicos padrões,
e de seus ajustes em presença de situações fisiológicas
(idade, sexo, peso, gestação), hábitos do paciente
(tabagismo, ingestão de álcool) e algumas doenças
(insuficiência renal, hepática, cardíaca) é orientado por
informações provenientes da farmacocinética.
Desta forma, amenos que um determinado fármaco
promova seu efeito topicamente, isto é, no seu local
de administração, o mesmo deverá alcançar a corrente
sanguínea sistêmica e, chegar até seu local de ação. Porém,
o simples fato do fármaco estar na corrente sanguínea não
garante uma resposta farmacológica efetiva. Para que isto
ocorra de forma eficaz, o fármaco deverá sair do plasma e
alcançar o sistema de líquido intersticial (líquido que banha
as células), ou até mesmo o meio intracelular, podendo
ser este simplesmente o citoplasma e/ou o núcleo da
célula. Como visto anteriormente, o efeito farmacológico
é dependente da velocidade com que o fármaco consegue
atingir seu sítio de ação, e este fenômeno é totalmente
dependente de dois processos: absorção e distribuição.
No entanto, para que uma determinada droga possa
ser absorvida e distribuída, a mesma deverá possuir a
capacidade de transpor diversas barreiras biológicas, as
quais são classificadas como se segue:
1. Membranas celulares;
2. Barreira hematoencefálica;
3. Barreira placentária.
Além destes fatores, a característica físico-química
do fármaco (pKa, lipossolubilidade/hidrosolubilidade,
peso molecular, estabilidade química, concentração
no local de administração, forma farmacêutica), e os
fatores relacionados ao paciente (via de administração,
fluxo sanguíneo no local de administração, velocidade
de esvaziamento gástrico, pH do local de administração,
patologias presentes no local de administração), além
de fatores como a administração simultânea de dois ou
mais fármacos (interações físico-químicas no local de
administração) ou ainda a administração de fármacos
simultaneamente com determinados alimentos, são de
grande importância para o processo de absorção. Estes
fatores principalmente estão relacionados com as questões
anteriormente vistas:
1. O fármaco consegue penetrar no organismo do
paciente? (administração, liberação do princípio ativo,
forma farmacêutica, via de administração, característica
físico-química, características do paciente, barreiras
biológicas).
2. O fármaco consegue alcançar seu local de ação?
(absorção, distribuição).
PASSAGEM DE FÁRMACOS PELA MEMBRANA.
O termo absorção significa que a droga saiu do seu
local de administração e chegou até a corrente sanguínea,
ou seja, “é a passagem da droga ou fármaco, do seu local
de administração para a corrente sanguínea e/ou linfática”
(figura ); com exceção da via intravenosa, todas as demais
vias de administração podem propiciar absorção de
fármacos, isto porque no caso daquela via, o fármaco está
sendo administrado diretamente na corrente sanguínea e,
portanto, será pulada a etapa de absorção.
Para que o fármaco possa transpor a membrana celular
que compõem as células epiteliais da pele, do intestino,
dos alvéolos pulmonares, das córneas, das mucosas e até
mesmo dos vasos sanguíneos, será necessário um ou mais
dos seguintes processos:
a. Difusão passiva;
b. Filtração;
c. Fluxo de massa;
d. Transporte ativo;
e. Transporte facilitado;
f. Endocitose;

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
10
Representação de processos utilizados para a absorção
dos medicamentos.
a. Difusão passivaA maioria dos fármacos atravessam as membranas
por difusão passiva, desde que este esteja no seu estado
não ionizado (não carregado eletricamente). A velocidade
deste fenômeno é dependente do coeficiente de partição
lipídio-água, mais do que da lipossolubilidade do fármaco.
EX:
O anticonvulsivante fenobarbital, que se encontra
quase que totalmente na forma não ionizada quando
em pH fisiológico, deveria ser capaz de passar por todas
as membranas de forma facilitada. No entanto, isto não
ocorre na prática, seu coeficiente de partição lipídio-
água é baixo a ponto de dificultar sua passagem pelas
membranas celulares, desta forma o fármaco é mais
lentamente absorvido. Este fator pode explicar o porque
este medicamento possui um tempo de latência (tempo
necessário para que o fármaco atinja a concentração
plasmática mínima necessária para promover o efeito
terapêutico) maior quando comparado a outros
anticonvulsivantes.
b. FiltraçãoA taxa de filtração depende tanto da existência de um
gradiente de pressão como força propulsora quanto do
tamanho da molécula do fármaco em relação ao tamanho
do poro através do qual deverá ser filtrado. Nos sistemas
biológicos, a passagem de solutos hidrossolúveis pequenos
através dos canais aquosos existentes nas membranas
é realizada por filtração. O diâmetro hipotético destes
poros é de aproximadamente 7Å, o que acaba impedindo
a passagem de fármacos que possuam peso molecular
maior que 100.
c. Fluxo de massaEm sua maioria, fármacos lipossolúveis ou não, passam
com maior rapidez pelas paredes dos capilares quando
comparado à sua velocidade de passagem através de
outras membranas. O resultado deste fenômeno é limitado
em maior grau pelo fluxo sanguíneo tecidual, quando
comparado à limitação determinada pela parede do
capilar. Esse fluxo de massa de líquido ocorre através dos
poros intercelulares e constitui o principal mecanismo de
passagem de fármacos através da maioria das membranas
endoteliais que reveste os capilares internamente, com
exceção daquelas no sistema nervoso central.
d. Transporte ativoO transporte ativo ocorre quando o movimento de
fármacos através da membrana celular, de um lado para
o outro, ocorre por meio de transportadores ATPdependentes.
Esse transporte envolve a ligação reversível da molécula
do fármaco a uma estrutura proteica da membrana
(transportador), o qual irá movimentar o fármaco através
da membrana, ou seja, o complexo formado se difunde
através da membrana para o lado oposto, onde o complexo
sofre dissociação, liberando a molécula do fármaco no
compartimento aquoso existe do outro lado. Em seguida,
o transportador pode retornar ao local de início e se
ligar novamente a outra molécula de fármaco repetindo
novamente o trajeto, que ocorre em uma única direção.
É preciso se atentar ao fato de que quando dois ou
mais fármacos são administrados simultaneamente, e que
cujas moléculas se assemelham, uma poderá competir
com a outra pelo mesmo transportador, e desta forma,
a molécula de maior afinidade pelo transportador poderá
prejudicar a absorção da outra.
e. Difusão facilitadaO transporte de fármacos por difusão facilitada
assemelha-se de certa forma ao transporte ativo,
incluindo um sistema de transporte mediado por uma
proteína transportadora, que apresenta saturabilidade

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
11
e seletividade. Porém, s difere do transporte ativo pelo
simples fato de não necessitar de ATP para executar sua
tarefa. Neste sistema de transporte, a movimentação
do fármaco ocorre do meio mais para o meio menos
concentrado, de forma que o gradiente de concentração
possa servir como força propulsora do fenômeno.
f. EndocitoseEste fenômeno envolve a captação celular de moléculas
ou complexos exógenos por meio de vesículas localizadas
na membrana celular. Este processo pode ser dividido em
duas categorias: a) fagocitose e b) pinocitose. O soluto no
interior da vesícula é liberado no meio intracelular.
FATORES QUE AFETAM A ABSORÇÃO DE UM FÁRMACO.
Além do coeficiente de partição lipídio-água,
diversos outros fatores podem influenciar de forma
positiva ou negativa a absorção de um determinado
fármaco. A absorção poderá ser influenciada por fatores
físico-químicos do fármaco, fatores relacionados à forma
farmacêutica e via de administração, bem como por
condições fisiológicas e patológicas do paciente.
Propriedades físico-químicas do fármacoO pH do meio onde o fármaco está sendo administrado,
a solubilidade, o coeficiente de ionização e de dissociação
(pKa), possuem grande influencia sobre a capacidade de
absorção do fármaco. Para que o processo de absorção seja
bem sucedido, o princípio ativo deverá estar solubilizado
no meio, além de estar em seu estado não ionizado.
Portanto, para que a solubilidade de um determinado
fármaco seja elevada, é necessário que o pH do meio onde
este se encontra em solução seja adequado ao mesmo;
isto é, existe um pH ótimo, que permite uma dissolução e
absorção adequadas para cada fármaco.
Estabilidade: A estabilidade química do fármaco deve ser estudada
sob a forma sólida e em solução em diferentes pH. Estes
estudos objetivam encontrar formulações estáveis e vias
de administração adequada a cada fármaco. Assim, um
produto instável em meio ácido não deve ser administrado
por via oral (ex.: a penicilina G quando administrada por
via oral se transforma em ácido penicilânico, que é inativo).
Como solução para este problema da penicilina, o fármaco
teve sua estrutura molecular modificada (penicilina V),
possibilitando sua administração por via oral.
Lipossolubilidade: Para que um determinado fármaco possa passar
facilmente pela membrana celular, este deverá possuir boa
lipossolubilidade, entretanto, um fármaco composto apenas
por lipídio e vice-versa, terá seu coeficiente lipídio-água
diminuído, e consequentemente não conseguirá passar
pela membrana celular; assim, para que um fármaco possa
elevar sua capacidade de se difundir pela membrana, este
deverá possuir em sua estrutura molecular, componentes
lipídicos e componentes aquosos. Quando um fármaco é
totalmente hidrossolúvel ou lipossolúvel, este só consegue
passar pela membrana quando houver um transportador.
Concentração da droga no local de administração: Para alguns modelos cinéticos, quanto maior a
concentração do fármaco no local de administração, maior
será a velocidade e a quantidade absorvida deste, e vice-
versa.
Forma farmacêutica: A forma farmacêutica é a responsável pela veiculação
do fármaco para dentro do organismo, é por meio desta
que o fármaco consegue entrar em contato (cápsulas,
comprimidos, drágeas, soluções, suspensões entre outras)
ou consegue ser mantido em contato com o organismo
(implantes subcutâneos, adesivos transdérmicos, cremes,
pomadas, esmaltes entre outras). Na figura abaixo se
observa diferentes formulações administradas por via
oral, as regiões de cor mais acentuada representa o local
onde há maior liberação de princípio ativo. É observado
na figura que formulações representadas por cápsulas
e comprimidos normais, isto é, sem qualquer tipo de
tratamento, liberam o princípio ativo ainda no estomago,
enquanto que formulações que sofreram tratamento
gastro-resistente, só promovem a liberação do fármaco ou
na primeira porção do duodeno ou no intestino grosso. Na
mesma figura, se observa que formulações de liberação
controlada, a liberação do fármaco ocorre ao longo de todo

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
12
trato-gastrintestinal, e que formulações líquidas (solução)
o fármaco já se encontra disponível para a absorção no
momento em que é administrado.
Propriedades de diferentes formas farmacêuticas relacionadas à sua capacidade em liberar o princípio ativo em diferentes regiões do trato gastrintestinal (Lüllmann et al., 2008).
Difusão através de lipídio: Mecanismo pelo qual os fármacos atravessam
a membrana celular de acordo com a gradiente de
concentração. Para que isto ocorra, o fármaco deve ser
lipossolúvel, ou seja, sua molécula deve ser não-ionizada
e apolar.
Esquema demonstrando a difusão de fármacos lipossolúveis (representados por círculos rosa) através da membrana celular. Fonte: LULLMANN, H; MOHR, K; HEIN, L. Farmacologia: Texto e Atlas. 5ed. Porto Alegre: Artmed, 2008.
INFLUENCIA DO PH E PKA:
A maioria dos fármacos quando em solução são ácidos
ou bases fracas podendo ser encontrados em estado não-
ionizado ou ionizado. A fração não ionizada apresenta
característica lipossolúvel e, portanto, possui a capacidade
de se difundir com maior facilidade pela membrana,
de modo que a fração ionizada é incapaz de penetra a
membrana devido sua baixa lipossolubilidade(veja figura
abaixo).

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
13
Na figura acima, note que quando a molécula
está elétricamente carregada ocorre uma repulção na
membrana. Deve ser lembrado que toda membrana é
eletricamente carregada (em estado de repouso, o lado
externo à célula está positivamente carregado, enquando
que o lado interno está negativamente carregado), desta
forma quando um fármaco está protonado (carga (+))
este sofre repulção pela carga positiva da membrana
impedindo sua passagem.
Portanto, a difusão transmembrana de um eletrólito
fraco é determinada por seu pKa e pelo pH do meio.
Em termos quantitativos, o valor pKa de um fármaco
representa o valor de pH em que metade do fármaco
encontra-se na forma ionizada. A Equação de Henderson-
Hasselbalch (abaixo) descreve a relação entre o valor pKa
de um fármaco ácido (A) ou básico (B) e o pH do meio
biológico contendo este fármaco:
Onde pKa é a constante de ionização do fármaco,
pH é o valor do pH do meio onde o fármaco está sendo
administrado, [AH] é a concentração molar do fármaco
no seu estado não ionizado (fração não ionizada das
moléculas) e [A-] é a concentração molar da droga em
seu estado ionizado (fração ionizada das moléculas),
note que a equação (B) é diferente apenas pela posição
da fração ionizada, esta relação deve ser respeitada para
os cálculos. Para se definir qual das duas equações a se
utilizar, deve-se observar as características do fármaco, se
este tiver característica ácida, a equação de escolha deverá
ser aquela representada por (A) e, quando o fármaco for
de característica básica, a equação representada por (B)
deverá ser a escolhida. Uma observação muito importante
é que, esta escolha deverá ser baseada na característica
do fármaco e não no pH do meio, este não deverá ser o
limitante para a escolha da equação.
EX:
Um paciente deverá receber como medicação o ácido
acetilsalicílico (AAS), entretanto, há uma grande dúvida:
o AAS será melhor absorvido em um meio de pH= 1,5 ou
em um meio de pH tamponado de valor 7,5? Dado: pKa do
AAS é igual a 3,5.
Muito bem, em primeiro lugar lembrar que a equação
a ser utilizada deverá ser escolhida pela característica da
droga e não pelo pH do meio, portanto deverá ser realizado
dois cálculos, um para o meio de pH ácido e outro para
o meio de pH básico, porém para os dois cálculos será
utilizada apenas a equação (A) anteriormente vista, isto
porque a droga é de característica ácida. Veja a resolução
abaixo.
Resolução.
Outro exemplo clássico é o que ocorre com o anestésico
local quando administrado em regiões infeccionadas,
edemasiadas e inflamadas. Ao ser administrado em
tecidos com estas características, a anestesia local não se
instala (“não pega” no popular), isto porque o anéstésico
é um fármaco de característica básica e para promover seu
efeito deverá se difundir pela membrana para o interior
da célula onde promoverá seu efeito, mas para que este
fármaco possa ser estocado de forma segura, o mesmo
está solubilizado em um veículo ácido (pH≈2,8 - 3,4), isto
implaca que o princípio ativo está praticamente quase que
todo no seu estado ionizado; quando este for administrado

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
14
em tecidos com as características normais, o sitema tampão
do organismo irá equilibrar o pH, e o sal irá se desprotonar
voltando ao seu estado molecular, e então conseguirá se
difundir para a membrana celular. Por outro lado, quando
o medicamento for administrado em um tecido inflamado
ou infeccionado, o pH local geralmente é ácido, e o
sistema tampão não funciona adequadamente; portanto,
o fármaco não irá se desprotonar e consequentemente
não passará pela membrana celular e não promoverá seu
efeito (veja o esquema abaixo).
Representação esquemática da influência de carga elétrica presente na estrutura molecular da lidocaína na difusão desta pela membrana celular. Observa que o próton (+) presente na estrutura, impede a difusão do fármaco.
BIODISPONIBILIDADE:
Na farmacologia, o termo biodisponibilidade é usado
para descrever a fração (quantidade ou concentração)
de uma droga que atinge a corrente sanguínea sistêmica
na forma inalterada. É uma das principais propriedades
farmacocinéticas das drogas. Por definição, quando
uma medicação é administrada por via intravenosa, sua
biodisponibilidade é de 100%. Entretanto, quando uma
medicação é administrada por outras vias (como a via oral,
retal intramuscular entre outras), sua biodisponibilidade
pode diminuir (devido à absorção incompleta e ao
metabolismo de primeira passagem), no entanto, devemos
lembrar que muitos fármacos podem ser 100% absorvidos
quando administrados por via oral, intramuscular, retal ou
outra.
A biodisponibilidade é uma das ferramentas essenciais
da farmacocinética, já que seu valor deve ser considerado
quando se calcula as doses para administração de drogas
por vias diferentes da intravenosa. A biodisponibilidade de
um fármaco é expressa em porcentagem ou valor unitário
(100% ou 1 unidade) e este valor é representado pela
letra F, e nada mais é do que um fator de correção.
FATORES FISIOLÓGICOS QUE PODEM INFLUENCIAR NA ABSORÇÃO.
Além dos fatores relacionados às características
físico-químicas dos fármacos, as condições fisiológias do
indivíduo pode ou favorecer ou desfavorecer o processo
de absorção de um fármaco. Asseguir serão relaciodados
alguns destes fatores.
Presença da P-glicoproteína: É um transportado protéico expresso na membrana
celular, e que tem a função de transportar para o meio
extracelular o fármaco que se difundiu para o interior da
célula, ou seja, é um transportador reverso que prejudica
a absorção do fármaco no intestino. Estudos apontam que
este transportador também é resposnsável por impedir
que inúmeros fármacos cheguem ao sitema nervos
central (SNC), onde deacordo com as pesquisas, esta
proteína estaria na membrana celular das células epiteliais
que revestem a barreira hematoencefálica e impediria
portanto, a passangem do fármaco do leito vascular para
o SNC (veja esquema abaixo).

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
15
Vista plana da estrutura de uma p-glicoproteína
(http://www.masterborderskennel.
com/artigos/art_02_mdr1.html).
Observe que o fármaco que está chegando por meio de um capilar, ao se difundir para dentro da célula, é imediatamente transportado de volta para fora pela p-glicoproteína (em verde) (http://www.ff.up.pt/toxicologia/monografias/ano0708/g46_docetaxel/resiste.htm).
A tabela abaixo apresenta alguns fármacos que podem
ser prejudicados pela p-glicoproteína.
Drogas antineoplásicas: Vinca alcalóides ( vincristina, vinblastina)
Antraciclinas (doxorrubicina, daunorrubicina, epirrubicina)
Epipodofilotoxina (etoposide, tenoposide)
Paclitaxel (taxol)
Topotecan
Mitramicina
Outros agentes citotóxicos
Colchicina
Emetina
Puromicina
Inibidores HIV proteaseRitonavir Indinavir Saquinavir
Difusão através canais aquosos: Devido ao tamanho diminuto dos poros ou canais
aquosos presentes nas membranas celulares, este tipo
de passagem através da membrana celular não é muito
importante para absorção de fármacos, pois as moléculas
dos fármacos geralmente são maiores que os poros.
Difusão facilitada: Mecanismo de transporte especializado dependente de
uma molécula transportadora, geralmente uma proteína
transmembrana, que se liga às moléculas dos fármacos,
transportando-as para o outro lado da membrana, que é
realizado de forma passiva, sem gasto de energia e de
acordo com gradiente eletroquímico.
Esquema demonstrando a difusão facilitada de fármacos por meio de uma proteína transportadora através da membrana celular. Fonte: LULLMANN, H; MOHR, K; HEIN, L. Farmacologia: Texto e Atlas. 5ed. Porto Alegre: Artmed, 2008.
Transporte ativo: Mecanismo de transporte especializado dependente de
uma molécula transportadora, geralmente uma proteína
transmembrana, que se liga às moléculas dos fármacos,
transportando-as para o outro lado da membrana, que é
realizado ativamente contra um gradiente eletroquímico
com gasto de energia.
O transporte mediado por transportadores apresenta
como limitação a possibilidade de ocorrer saturação, ou
seja, na presença de altas concentrações do fármaco,
pode ocorrer a ocupação de todas as proteínas
transportadoras, limitando a velocidade do transporte.
Além disso, pode ocorrer inibição competitiva, caso uma
proteína transportadora seja capaz de transportar duas
moléculas diferentes. Este tipo de transporte é importante
principalmente nos túbulos renais, no trato biliar, na
barreira hematoencefálica e no trato gastrointestinal.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
16
Endocitose: Mecanismo de transporte de fármacos que são
moléculas grandes, a endocitose depende da ligação do
fármaco a receptores presentes na membrana celular,
o que gera a formação de uma vesícula que liberará o
fármaco do outro lado da membrana.
Esquema demonstrando a endocitose de fármacos, o que leva ao transporte destes fármacos através da membrana celular. Adaptado de LULLMANN, H; MOHR, K; HEIN, L. Farmacologia: Texto e Atlas. 5ed. Porto Alegre: Artmed, 2008.
Fluxo sanguíneo e vascularização no local de administração:
Quanto mais vascularizado e maior o fluxo sanguíneo
no local da administração do fármaco, mais rápido e em
maior concentração será absorvida deste. Assim, qualquer
alteração no fluxo sanguíneo local poderá interferir positiva
ou negativamente na absorção de um medicamento.
Motilidade gastrintestinal: A absorção dos fármacos ocorre geralmente na
primeira porção do intestino delgado (duodeno), de modo
que a velocidade de esvaziamento gástrico determina
a velocidade em que o fármaco alcança seu local de
absorção, ou seja, quanto maior for a velocidade de
esvaziamento gástrico, mais rápido o fármaco irá conseguir
atingir o duodeno e, consequentemente ser absorvido. Por
exemplo, no caso de pacientes portadores de enxaqueca,
a velocidade de absorção de analgésicos é reduzida, isto
ocorre porque esta condição ativa o centro do arco-reflexo
do vômito (enxaqueca associada com ânsia) e, portanto,
a velocidade de esvaziamento gástrico encontra-se
reduzida, e por isto, é costume do paciente tomar mais de
um comprimido em um intervalo de tempo muito curto.
Porém, quando o fármaco analgésico for administrado
simultaneamente com um agente antiemético, ocorrerá
aumento na velocidade de esvaziamento gástrico, princípio
este utilizado nos medicamentos comerciais Ormigrein® e
Cefalium, os quais possuem substâncias antieméticas.
Patologias gastrintestinais: Quando o paciente possui algum distúrbio patológico
no TGI, este poderá favorecer ou desfavorecer a absorção
de fármacos, por exemplo, a absorção de propranolol,
co-trimoxazol e cefalexina apresenta-se aumentada em
pacientes celíacos, mas a digoxina e a levotiroxina são
menos absorvidas nesta condição ou em outras como
enterite induzida por radiação. O mesmo pode ocorrer
com a vit. B12, que necessita do fator intrinseco produzido
pelas células parietais do estômago para ser absorvida,
em casos de úlcera gástrica e duodenal a produção deste
fator encontra-se diminuída, sendo assim, ocorrerá uma
redução da absorção intestinal de vit. B12.
Volume de líquido (água) ao se administrar uma formulação sólida:
É explicitamente recomendado que todo medicamento
seja administrado apenas com água, há algumas exceções
como, por exemplo, o caso do sulfato ferroso que tem sua
biodisponibilidade melhorada quando este é administrado
com suco de laranja. No mais, todo medicamento deverá
ser administrado com água, pois esta não apresenta
nenhum interferente que possa quelar, aumentar ou
diminuir a secreção gástrica e, consequentemente
prejudicar a absorção do fármaco. A recomendação é
de que deva se utilizar um volume de aproximadamente
100 mL de água, não menos que isto para se administrar
uma formulação sólida. Nos gráficos abaixo se encontra
demonstrado como o volume de água é importante para
o processo de desintegração e liberação do princípio ativo
da formulação e, assim, melhorando a absorção daquele.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
17
Observa-se no gráfico acima que a dois grupos foram administrados comprimidos de eritromicina, sendo que um
dos grupos recebeu a formulação com um volume de 20 mL de água (linha tracejada), enquanto que o segundo grupo
recebeu a mesma formulação com um volume de 250 mL (linha contínua). Verifica-se que a concentração plasmática
máxima obtida nos dois grupos é significativamente diferente, sendo que para o grupo que recebeu apenas 20 mL a
concentração plasmática do fármaco foi bem inferior quando comparada à concentração plasmática do mesmo fármaco
para o grupo que recebeu a formulação com 250 mL de água.
Alimentação: O alimento pode intensificar ou comprometer a taxa de absorção de um fármaco, podendo também afetar a extensão
de sua absorção. Por exemplo, ovos podem comprometer a absorção de ferro, e alimentos ricos em cátions polivalentes
(Ca2+, Fe2+, Mg2+ entre outros) diminui a absorção de fármacos como tetraciclinas, aminoglicosídeos. A tabela a seguir
mostra exemplos de fármacos que podem apresentar interação alimentar.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
18
Metabolismo de primeira passagem: Quando um medicamento é administrado a um paciente,
este poderá sofre metabolismo ou efeito de primeira
passagem. Este metabolismo é muito definido como:
“metabolismo de um determinado fármaco antes que este
alcance a corrente sanguínea sistêmica”. Este metabolismo
ocorre tanto no trato gastrintestinal (luz) e no fígado (logo
após a absorção do fármaco). Outro ponto importante é
que este metabolismo está relacionado à administração do
medicamento pela via oral e retal, quando a formulação
é administrada pelas vias parenterais e sublingual, o
principio ativo não sofre efeito de primeira passagem. A
figura abaixo esquematiza o quanto o efeito de primeira
passagem interfere com a concentração plasmática do
fármaco após este sofrer ação deste fenômeno.
O esquema abaixo demonstra que após a administração
de um comprimido por via oral e, liberação do fármaco no
TGI (A), quando este se difundiu pelo epitélio intestinal
(B), na membrana celular ocorreu o metabolismo de 70%
da concentração do fármaco, que após passar pelo fígado
(C) mais 15% foi metabolizado, resultando na chegada à
circulação sistêmica de apenas 15% deste.
Representação figurativa da relação entre o metabolismo de primeira passagem e o processo de absorção de medicamentos (http://dc217.4shared.com/doc/eAymVhw2/preview.html)
RESUMO DOS FATORES QUE PODEM MODIFICAR A ABSORÇÃO
Os principais fatores que modulam a absorção de
fármacos e seus efeitos encontram-se no quadro abaixo.
Quadro: Fatores que modulam a absorção dos
fármacosFatores Absorção (seta
para cima)Absorção (seta para baixo)
Concetração do fármaco
Maior Menos
Peso molecular do fármaco
Pequeno Grande
Solubilidade do fármaco
Lipossolúvel Hidrossolúvel
Forma farmacêutica
Líquida Sólida
Dissolução sólida Grande Pequena
Área absortiva Grande Pequena
Espessua membrana
Menor Maior
Circulação local Grande Pequena
Condições Patológicas
Inflamação, queimadura
Edema, Choque
DISTRIBUIÇÃO DE FÁRMACOS.
Embora a absorção do fármaco seja considerada um
pré-requisito para atingir níveis plasmáticos adequados
desse fármaco, este também necessita alcançar seu órgão
ou órgãos-alvo em concentrações terapeuticamente ideais
para exercer o efeito desejado.
Entende-se por distribuição o fenômeno de transferência
do fármaco do sistema circulatório (linfa e sangue) para
os diversos tecidos e órgãos do organismo. Esse é um
processo reversível para permitir que o fármaco atinja o
tecido-alvo e promova sua ação terapêutica, e ao mesmo
tempo, permite que haja sua retirada do tecido e que o
mesmo possa ser removido do organismo. As características
do fármaco devem ser amplamente reconhecidas, pois
estas são de grande importância para a determinação da
posologia durante a etapa final dos ensaios clínicos para o
desenvolvimento de um novo medicamento.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
19
A distribuição de um fármaco ocorre através do
sistema circulatório, enquanto o sistema linfático contribui
em menor grau para este processo. Após ser absorvido
ou administrado diretamente na corrente sanguínea,
o fármaco se distribui por todos os sistemas incluído o
líquido intersticial, líquido intracelular, tecido muscular,
tecido ósseo, SNC, tecido adiposo além de unhas e pêlos,
dependendo dos fatores fisiológicos e características físico-
químicas, cada fármaco terá um perfil próprio em termos
de distribuição, ou seja, tecidos mais vascularizados
deverão receber em menor tempo uma concentração maior
quando comparado a tecidos menos ou não vascularizados
como tecido adiposo e pêlos; outro fator relacionado à
circulação sanguínea é o débito cardíaco, pois se compara
a um paciente normal, portadores de insuficiência cardíaca
possuem um prejuízo considerável na velocidade de
distribuição. Ainda, fármacos altamente lipossolúveis
quando comparados a fármacos hidrossolúveis irão se
difundir com maior afinidade para o tecido adiposo. Veja
diagrama abaixo.
Representação esquemática da distribuição de fármacos de acordo com a vascularização dos sitemas (Raffa et al., 2006).
MODELOS COMPARTIMENTAIS EM FARMACOCINÉTICA.
O interesse em correlacionar o tempo e a ação de
um fármaco no organismo, após a administração do
medicamento, justifica-se para o conhecimento de três
fatores:
20 mg
a. Tempo entre a administração do medicamento e o
início da ação farmacológica (tempo de latência), que é
influenciado pela velocidade de absorção, distribuição,
localização do sítio-alvo e, indiretamente pela eliminação;
b. Tempo necessário para desencadear o efeito máximo
(relacionado à concentração máxima), resultante do
balanço entre os processos que levam o fármaco ao sítio-
alvo e aqueles que o retiram deste local;
c. Duração do efeito, que é dependente da velocidade
de eliminação e, em alguns casos, da distribuição.
Na prática correlacionando-se as concentrações
plasmáticas do fármaco com a amplitude dos efeitos em
diferentes períodos após a administração, pressupondo-se
que, depois de atingido o equilíbrio, estas concentrações
refletem as concentrações no sítio de ação do fármaco.
Portanto, os modelos compartimentais na
farmacocinética são úteis para a interpretação dos
processos de transporte e metabolismo dos fármacos.
Nos modelos matemáticos que descrevem os parâmetros
farmacocinéticos o organismo é representado como um
sistema de compartimentos cujo número é representado
arbitrariamente, jé que o corpo pode ser dividido pelo
número de órgãos, sistemas ou tecidos.
Na farmacocinética são propostos modelos matemáticos
para interpretar os dados obtidos após a administração
de um medicamento ao organismo, de modo que podem
ser descritos na literatura como modelos compartimentais
(abordagem clássica) e não compartimentais.
Assim, os modelos compartimentais em farmacocinética
podem ser classificados em Sistema Monocompartimental e
Sistema Multicompartimental; no primeiro caso assume-se
que o organismo funcione como um único compartimento,
ou seja, o fármaco se distribui de forma homogênea
por todo sistema, no segundo caso, adota-se que o
organismo seja constituído por um compartimento central
(sistema sanguíneo, tecidos de alta perfusão: cérebro,
pulmão, coração, rins, fígado) e um compartimento
periférico (diversos sistemas ou compartimentos menos

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
20
vascularizados: tecido adiposo, muscular, pele, unhas,
pêlo) onde o fármaco poderá se distribuir de forma
não homogênea nestes. Desta forma, em um sistema
multicompartimental, o fármaco deixa o compartimento
central e se distribui pelo compartimento periférico de
acordo com suas características físico-químicas e também
fisiológicas do paciente Veja esquema abaixo.
Conforme mostrado acima, após a administração do
fármaco, este se distribui rapidamente por todo sistema e,
em seguida é eliminado do organismo. No segundo caso,
os tecidos mais vascularizados recebem mais rapidamente
maior concentração do fármaco (Silva, 2002).
Na figura acima, observa-se que o fármaco é distribuído
através do sangue para os vários tecidos do organismo
(1). A distribuição pode ser restrita ao compartimento
extracelular, que considera o compartimento intersticial
junto com o volume plasmático (2), ou pode ser estendida
para o compartimento intracelular (3). Alguns fármacos
podem ligar-se fortemente a estruturas teciduais
de tal forma que sua concentração plasmática caia
significativamente mesmo antes que sua eliminação tenha
começado (4).
Ao se avaliar uma curva concentração plasmática
em função do tempo de um fármaco pertencente a um
sistema monocompartimental, nota-se que após atingir
a concentração plasmática máxima, ocorre uma queda
contínua deste, a qual representa o processo de distribuição
e eliminação, isto pode ser observado no gráfico abaixo.
Conforme observado acima, no sistema bicompartimental observa-se uma curva bi-fásica onde a primeira inclinação de caida, representa a primeira fase (distribuição) e a segunda fase dacurva representa o sistema de depuração.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
21
MODELOS NÃO COMPARTIMENTAIS
Os modelos não compartimentais geralmente se aproxi-
mam mais da realidade fisiológica do organismo por des-
crever a disposição do fármaco em cada tecido ou órgão.
Entretanto, perdem a universalidade devido à complexida-
de do tratamento matemático envolvido. Assim, do ponto
de vista da aplicação dos princípios de farmacocinética às
situações clínicas, utiliza-se na maioria dos casos, o mo-
delo monocompartimental e, especialmente, o modelo bi-
-compartimental.
Por meio destes modelos matemáticos, simulam-se os pro-
cessos de absorção, distribuição e eliminação do fármaco
do organismo como uma função do tempo com os seguin-
tes objetivos:
- Predizer as concentrações do fármaco no plasma,
tecidos e urina, com o emprego de determinado esquema
terapêutico.
- Estabelecer o esquema terapêutico ótimo para cada
paciente, individualmente.
- Estimar o possível acumulo de um determinado
fármaco ou até mesmo, de seus metabólitos no organismo.
- Descrever como as alterações fisiológicas ou
enfermidades podem modificar a absorção, distribuição e
a eliminação de um fármaco no organismo.
- Explicar casos de interações medicamentosas.
As análises compartimentais são desenvolvidas
por meio de estudos continuados das concentrações
plasmáticas do fármaco, uma vez que se conhece a
quantidade administrada por via intravascular; porém
também é possível se estimar a fração absorvida no
caso da administração se dar por outra via diferente a
intravascular. O principal método empregado neste análise
é o estudo das curvas obtidas (veja curvas apresentadas
acima).
Ligação às proteínas plasmáticas e tecidos:
Após alcançar a corrente sanguínea, o fármaco irá se
ligar às proteínas transportadoras. A ligação do fármaco a
este carreador será dependente da afinidade estrutural e
da característica físico-química do mesmo. Desta forma,
fármacos de característica ácida terão maior afinidade
por proteínas plasmáticas com característica básica e vice
versa. As principais proteínas plasmáticas transportadoras
de fármacos são: albumina, alfa1-glicoproteína ácida e
globulina.
Os fármacos de característica ácida possuem forte
afinidade pela albumina, enquanto que os fármacos básicos
se ligam preferencialmente à alfa1-glicoproteína ácida e os
hormônios as globulinas; porém, isto não quer dizer que
estes dois últimos não possam se ligar à albumina, pois
esta é encontrada em maior concentração no plasma e, de
certa forma, mesmo que em menor grau, possui afinidade
por inúmeros fármacos.
Perfil de ligação de fármacos a proteínas transportadoras.
Observe que quando a afinidade do fármaco possui
baixa afinidade pela proteína, a distribuição deste para
os tecidos aumenta, e quando sua afinidade é alta pela
proteína transportadora, ocorre menor distribuição tecidual
(Lüllmann, 2008).

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
22
Na figura acima está representado um diagrama de ligação de fármacos às proteínas e seu sequestro pelos sistemas de depuração. (fonte: Golan, 2009)
Um fármaco ligado à albumina ou a outras proteínas
plasmáticas é incapaz de difundir-se do espaço vascular
para os tecidos circundantes. Em (A) os fármacos que não
se ligam às proteínas plasmáticas sofrem visivelmente
uma rápida difusão (mostrada na figura acima na forma
do Fármaco A nos tecidos). Isso resulta em alto nível de
ligação ao local de ação farmacológica (habitualmente
receptores) e numa alta taxa de eliminação (representada
pelo fluxo através de um órgão de depuração). Entre os
exemplos desses fármacos, destacam-se o acetaminofeno,
o aciclovir, a nicotina e a ranitidina. Em (B) ocorre em
contrapartida, para os fármacos que exibem altos níveis
de ligação às proteínas plasmáticas (mostrados aqui na
forma do Fármaco B), é necessária uma concentração
plasmática total mais elevada do fármaco para assegurar
uma concentração adequada do fármaco livre (não-ligado)
na circulação.
Caso contrário, apenas uma pequena fração do
fármaco poderá sofrer difusão no espaço extravascular,
e apenas uma pequena porcentagem dos receptores
estará ocupada. Entre os exemplos desses fármacos,
destacam se a amiodarona, a fluoxetina, o naproxeno e
a varfarina. É preciso ressaltar que a ligação às proteínas
plasmáticas constitui apenas uma das numerosas variáveis
que determinam a distribuição dos fármacos. O tamanho
molecular, a lipofilicidade e a intensidade do metabolismo
de um fármaco são outros parâmetros importantes
que precisam ser considerados quando se estuda a
farmacocinética de determinado fármaco.
FATORES QUE PODEM INFLUENCIAR AS PROTEÍNAS PLASMÁTICAS
Patológicos
Os principais fatores patológicos relacionados à
hipoproteinemia estão relacionados abaixo:
Distúrbios hepáticosEntre os distúrbios hepáticos que podem gerar
hipoproteinemia, os mais importantes são cirrose hepática,
hepatite viral aguda e problemas hepáticos causados pelo
consumo excessivo de álcool, que causam diminuição na
síntese de proteica ou produção de proteínas plasmáticas
com conformação modificada que, portanto, não
conseguem se ligar aos fármacos.
Distúrbios renaisOs distúrbios renais alteram de forma variável a
concentração das proteínas plasmáticas, modificando a
ligação dos fármacos a estas proteínas.
Assim, em pacientes com insuficiência renal, fármacos
ácidos ligam-se pouco às proteínas plasmáticas, ficando
livre na circulação, o que aumenta a ocorrência de efeitos
colaterais; enquanto, em pacientes com transplante renal,
ocorre aumento da ligação dos fármacos às proteínas
plasmáticas, diminuindo os efeitos farmacológicos.
Distúrbios cardíacosApós a ocorrência de um infarto agudo do miocárdio,
há uma modificação na concentração de algumas proteínas
plasmáticas como α1-Glicoproteína Ácida, o que modifica a
fração de fármaco livre para ação farmacológica.
Distúrbios tireoidianosOs distúrbios tireoidianos alteram de forma variável a
concentração das proteínas plasmáticas, modificando a
ligação dos fármacos a estas proteínas.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
23
Assim, no hipertireoidismo, ocorre diminuição da
concentração plasmática de albumina, levando ao aumento
da concentração de fármacos ácidos livre na circulação
sanguínea.
Fatores fisiológicos
Os principais fatores fisiológicos relacionados à
hipoproteinemia estão relacionados abaixo:
IdadeA idade do paciente é capaz de modificar a ligação
dos fármacos a proteínas plasmáticas, modificando a
fração de fármaco livre, isso acontece devido à variação
da concentração de proteínas plasmáticas, à presença de
albumina fetal (que possui baixa afinidade por fármacos),
ao pH sangüíneo (que é mais baixo e modifica a ionização
dos fármacos) e à concentração de ácidos graxos livre
(que maior no adulto).
GestaçãoA concentração de albumina é alterada durante o
primeiro trimestre de gestação, modificando a ligação de
alguns fármacos, que se tornam livres na circulação.
Estado hormonalO uso de medicamentos anovulatórios, que reduzem
a síntese de α1-Glicoproteína Ácida, altera a ligação de
certos fármacos a estas proteínas plasmáticas, o que pode
levar ao aumento dos efeitos colaterais.
CONCENTRAÇÃO DE ÁCIDOS GRAXOS LIVRES
Os ácidos graxos livres, quando presentes em altas
concentrações ligam-se a albumina, diminuindo a ligação
dos fármacos às proteínas plasmáticas; enquanto, que,
quando presentes em baixas concentrações, os ácidos
graxos favorecem a ligação dos fármacos às proteínas
plasmáticas.
Além do fato de que os fármacos estão ligados às
proteínas transportadoras, outra situação que deve ser
levada em consideração é que a fração livre deste irá
se difundir do plasma para os tecidos. Assim, fármacos
de característica hidrossolúvel terão maior afinidade
por tecidos como músculo esquelético, enquanto que
substâncias lipossolúveis se alojarão em maior grau em
tecido adiposo (veja figura abaixo).
Observe na figura acima que em condições normais o
organismo é composto por 70% de água e 30 % de gordura
e, que em pacientes obesos a distribuição é na ordem de
50% para cada parte. Além disto, é possível verificar que
drogas hidrossolúveis apresentam-se mais concentradas
no compartimento hídrico de pessoas obesas, enquanto
que fármacos lipossolúveis estão em concentrações
maiores no compartimento gorduroso (tecido adiposo).
Estes fenômenos descritos acima, todos interferem
no parâmetro farmacocinético conhecido como volume
aparente de distribuição.
VOLUME APARENTE DE DISTRIBUIÇÃO:
É um parâmetro farmacocinético que expressa a
extensão da distribuição além do plasma. Também pode
ser conceituado como o volume no qual o fármaco deve se
dissolver para que sua concentração se iguale a do plasma.
Em termos gerais, o volume de distribuição
também pode ser considerado como uma constante de
proporcionalidade entre duas variáveis: entre a dose
administrada e a concentração plasmática do fármaco.
Este dado está relacionado à concentração plasmática e a
quantidade do fármaco no organismo em um dado tempo.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
24
EX:
Considerando a concentração plasmática de um
dado fármaco como 10 mg/L, quando há 1000 mg deste
dissolvidos no organismo de um paciente de 70 kg, verifica-
se que será necessário cerca de 100 L para solubilizá-lo,
ou seja, o volume aparente de distribuição corresponde
a 100 litros, o que evidentemente não é um volume real,
visto que um paciente de 70 kg, possui no máximo, 50 L
de líquido em seu organismo. Ainda, é necessário lembrar
que muitos fármacos não conseguem deixar a corrente
sanguínea e atingir os tecidos, ou seja, seu volume
aparente de distribuição está relacionado com a quantidade
de sangue que este indivíduo possua (é considerado que
aproximadamente 8% do peso corpóreo de um indivíduo
adulto normal seja constituído por sangue).
O fato é que este parâmetro pode exceder
acentuadamente qualquer volume físico do corpo, visto
que se trata do volume aparente necessário para conter a
quantidade de droga homogeneamente na concentração
encontrada no sangue (plasma ou água). Fármacos que
possuem volume de distribuição muito alto apresentam
concentrações muito mais elevadas em tecidos
extravasculares do que no compartimento vascular, isto é,
apresentam distribuição não homogênea, por outro lado,
fármacos que possuem baixo volume de distribuição, são
encontrados em maior concentração no plasma e menor
nos tecidos. Este parâmetro pode ser determinado pela
seguinte equação:
Onde Vd é o volume de distribuição, dose administrada
(D) é a dose que o paciente está recebendo e concentração
plasmática (Cp; C0; Cp1, Ce; Css) representa a
concentração plasmática após a administração do fármaco
ou, concentração plasmática inicial ou, concentração
plasmática 1 ou, concentração plasmática efetiva, ou
ainda, concentração plasmática em estado de equilíbrio.
Ex.
Determine o volume de distribuição para um dado
fármaco que foi adminitrado a um paciente na dose
igual a 20 mg e, que após dosagem plasmática deste foi
encontrado um valor de 5mg/L de plasma.
Resolução:
Portanto o volume de distribuição é igual a 4 litros, ou
seja o fármaco está contido em um sistema de volume
igual a 4 litros do organismo. Veja o esquema abaixo para
melhor ilustrar.
Observe que após a administração do fármaco, este se
distribui em função do tempo para todos os compartimentos
orgânicos, e com isto, ocorre redução de sua concentração
plasmática e aumento de sua concentração nos tecidos;
no último painel é possível verificar que o fármaco está em
maior concentração em um determinado compartimento
quando comparado aos demais, por este motivo o Vd
expressa o volume “aparente” de distribuição, e pode
exceder qualquer volume físico.
Na tabela abaixo estão relacionados exemplos de
fármacos e seus respectivos volumes de distribuição.
Observe que para alguns fármacos, este parâmetro se
encontra bem acima da massa corpórea do paciente; isto

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
25
é verdade, visto que fármacos como, por exemplo, a cloroquina, se distribui por todas as estruturas do organismo
(plasma, liquido extracelular, líquido intracelular, tecido muscular, tecido adiposo entre outros).
Droga dispon. oral (F) (%)
ligação plasm. (%)
depuração (L/h/70 kg)
vol. de distrib. (L/70 kg)
meia vida (h)
conc. eficaz conc. tóxica
acetaminofen 88 0 21 67 2 10-20 mg/L >0,3 g/L
ac. Salicílico 100 85 0,84 12 13 200 mg/mL > 200 mg/mL
carbamazepina 70 74 5,34 98 15 6 mg/mL > 9 mg/mL
ciclosporina 23 93 24,6 85 5,6 200 ng/mL > 400 ng/L
cloroquina 89 61 45 13000 214 20 ng/mL > 250 ng/mL
digoxina 70 25 7 500 50 1 ng/mL > 2 ng/mL
imipramina 40 90 63 1600 18 0,2 ug/mL > 1 mg/L
indomeacina 98 90 8,4 18 2,4 1 mg/L > 5 mg/L
lidocaína 35 70 38,4 77 1,8 3 mg/L > 6 mg/L
nortriptilina 51 92 30 1200 31 100 ng/mL > 500 ng/mL
Valores de parâmetros farmacocinéticos já estabelecidos e encontrados na literatura básica (adaptado de Hardman et al., 2003).
PRINCIPAL APLICAÇÃO DO VD
Uma das principais aplicações do Vd é a determinação da melhor dose de ataque no início de uma terapia
medicamentosa. Deve ser considerado que a dose de ataque visa alcançar uma concentração plasmática terapêutica
em estado de equilíbrio mais rapidamente quando comparado ao método tradicional de administração. Este cálculo pode
ser determinado pela seguinte equação:
DA = Vd x Cp
Onde: DA é a dose de ataque e Cp é a concentração plasmática desejada (esta deve ser a terapêutica).
A dose de ataque é calculada para preencher o volume de distribuição, de modo a alcançar a concentração
terapêutica rapidamente. Esta concentração terapêutica alcançada deverá ser mantida posteriormente pelas doses de
manutenção. É importante ressaltar que as doses seguintes à dose de ataque, deve ser a dose normalmente utilizada
na terapêutica, ou seja, a dose de ataque não deve ser mantida como dose de manutenção.
EX:
Deseja-se verificar, se para um paciente de 100 kg de peso corpóreo, a dose igual a 1500 mg de paracetamol é
efetiva ou tóxica quando administrada em dose única. Dados: Vd = 67 L/70 kg, Cefetiva = 10 a 20 mg/L e Ctoxica = > 300
mg/L

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
26
Resolução:
No exemplo acima, verificou-se que para um paciente
de 100 kg, a administração de 1500 mg de paracetamol
em dose única ou como dose de ataque, promoveu uma
concentração plasmática efetiva (Ce) de 15,67 mg/L; isto
indica que nesta situação o paciente está seguro, pois
manteve-se a concentração plasmática terapêutica ( que
é entre 10 e 20 mg/L). Porém, é necessário lembrar que
deve ser corrigido sempre o Vd para cada paciente.
RELAÇÃO ENTRE DOSE DE ATAQUE E DOSE DE MANUTENÇÃO
Nos casos em que a meia vida plasmática do fármaco
apresenta um valor muito elevado, não é possível adotar
esquema terapêutico considerado ideal, no qual este
parâmetro corresponda ao intervalo entre a administração
das doses. Porém, nesta situação pode-se optar pela
administração de uma dose inicial (Di) maior, a qual é
conhecida como dose de ataque.
A finalidade desta dose (Di) é promover mais
rapidamente uma concentração plasmática terapêutica
(concentração plasmática de equilíbrio (Css)), mas deve
ser frisado que esta concentração deverá ser mantida
pelas doses de manutenção (Dm), necessitando, portanto,
de se avaliar a relação entre Di e Dm.
Ao se estabelecer um regime posológico, o principal
objetivo é obter níveis plasmáticos considerados
terapêuticos, que normalmente, são atingidos em estado
de equilíbrio. Por isto, os parâmetros farmacocinéticos
envolvidos são aqueles expressos por meio da seguinte
ralação:
Dose = Css x Vd
Onde: Css é a concentração plasmática do fármaco
no estado de equilíbrio, Vd é o volume aparente de
distribuição.
Para analisar a relação Di e Dm descrita acima, deve-se
considerar que a dose inicial administrada corresponde a
uma quantidade do fármaco (X) que será alterada com o
tempo em função de um termo cinético, representado por
e-k.t, ou seja:
X = Di x e-k.t
ÁREA SOB A CURVA (AUC):
Em estudos de farmacocinética e de biodisponibilidade,
é frequente avaliar-se a AUC das concentrações plasmáticas
em função do tempo, a partir do tempo zero, que é o
tempo correspondente à administração da formulação até
o tempo desejado, ou seja, poderá ser determinado a AUC
do tempo zero ao infinito, por exemplo (AUC0 - ∞) ou a um
determinado tempo de escolha.
Este parâmetro pode ser facilmente determinado pelo
método trapezóide (equação abaixo), porém, deverá ser
determinado a AUC para cada um dos intervalos, e a soma
de todas as áreas encontradas em cada um dos intervalos,
representará a AUC total.
Ex:
Determine a AUC da curva concentração plasmática em
função do tempo representada abaixo, no intervalo 2 a 3
h.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
27
Note que este procedimento deverá ser realizado para
CADA UM DOS INTERVALOS, caso seja realizado o
procedimento de se determinar a área total em uma única
equação, haverá a somatória de uma área inexistente.
Veja o exemplo.
Quando realizado o cálculo correto para cada uma
das áreas, e somando todas, o valor da AUCtotal será igual
a 212,5 mg/L . h; e não o valor determinado acima no
exemplo. Veja ainda que no gráfico acima há uma linha
tracejada que demonstra a área inexistente que foi
contabilizada juntamente com a área real sob a curva.
Desta forma, é importante frisar que não se pode utilizar
a equação para se determinar a AUC total com apenas o
primeiro e o último ponto, encontrados no gráfico.
O cálculo da área sob a curva de concentrações
plasmáticas de um fármaco em função do tempo é
importante e aplicável à determinação do Clearence, assim
como para o cálculo do volume aparente de distribuição e
de outros parâmetros farmacocinéticos.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
28
Sua aplicação também envolve a avaliação da biodisponibilidade absoluta no caso de medicamentos inovadores ou
da biodisponibilidade relativa (bioequivalência) para medicamentos genéricos e similares.
No gráfico apresentado abaixo está demonstrado a relação de duas formulações contendo ibuprofeno como principio
ativo, que passaram por este de bioequivalência.
Na figura acima é possível observar que a curva de concentração em função do tempo obtida para a formulação teste
está deslocada à direita da curva obtida para a formulação referência, e que a concentração plasmática máxima observada
para o medicamento teste também está acima deste parâmetro determinado para o medicamento referência. Por estas
observações, seria possível inferir que as formulações não são bioequivalentes. Entretanto, ao se avaliar os parâmetros
corretos, como a área sob a curva (tabela abaixo) é possível verificar que não foi encontrado diferença significativa entre
as formulações teste. Este é um dos indícios de que as formulações podem ser consideradas bioequivalentes.
Parâmetros farmacocinéticos das formulações ibuprofeno referencia e ibuprofeno teste.Não foram encontradas diferenças estatisticamente significativas entre os parâmetros apresentados na tabela acima.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
29
ELIMINAÇÃO E METABOLISMO DE FÁRMACOS (EXCREÇÃO E BIOTRANSFORMAÇÃO).
A menos que se fixe em estruturas orgânicas,
qualquer substância estranha (xenobiótico) introduzida
no organismo será eventualmente eliminada, algumas
mais rapidamente, e outras de uma forma mais lenta. O
mesmo princípio é utilizado para os medicamentos, que
deverão se submeter ao processo de eliminação, que
é constituido pelos processos de biotransformação ou
metabolismo e excreção. Alguns fármacos são excretados
em sua forma inalterada, porém a maioria das substâncias
medicamentosas necessitam de sofrer um processo de
biotransformação para que seja melhor excretado pelo
sistema.
Embora as reações bioquímicas que modificam as
drogas, convertendo-as em formas passíveis de excreção
renal, constituam uma parte essencial do metabolismo dos
fármacos, esse metabolismo abrange mais do que essa
simples função. A biotransformação dos fármacos pode
alterá-los de quatro maneiras importantes:
- Um fármaco ativo pode ser convertido em fármaco
inativo
- Um fármaco ativo pode ser convertido em um
metabólito ativo ou tóxico.
- Um pró-fármaco inativo pode ser convertido em
fármaco ativo.
- Um fármaco não-excretável pode ser convertido em
metabólito passível de excreção (por exemplo, aumentando
a depuração renal ou biliar).
Para melhor ilustrar, veja o quadro abaixo.
A figura mostra exemplos de biotransformação de
fármacos, alguns estão em estado ativo e são convertidos
a substâncias ainda ativas e posteriormente a compostos
inativos, e ainda, exemplos de drogas inativas (pró-droga)
que são metabolizadas a fármacos ativos.
Fases da biotransformação.
O processo de biotransformação pode ser subdividido
em duas fases distintas, sendo elas:
Reações de fase I: São conhecidas também como reações não-sintéticas
ou catabólicas. Esta fase tem por objetivo modificar a
estrutura molecular do fármaco e, envolvem os processos
de oxidação, redução, hidrólise, hidroxilação, desaminação,
ciclização, desciclização entre outras.
Reações de fase II: Também denominada de reação sintética ou anabólica
ou ainda de fase de conjugação. Nesta fase, será conjugada
uma molécula endógena de característica hidrossolúvel à
molécula do fármaco que passou pelo processo de fase
I. As principais moléculas endógenas envolvidas neste
processo são: ácido glicurônico, metil, sulfato, glutationa e
acetato. (veja esquemas a seguir)
Representação geral das fases I e II do processo de biotransformação.
PRINCIPAIS ENZIMAS ENVOLVIDAS COM O PROCESSO DE FASE I.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
30
O principal grupo de enzimas relacionadas com a
metabolização de fase I é o complexo microssomal de
função oxidativa mista Citocromo P-450 ou também
denominada de complexo microssomal CYP. Atualmente
são descritas mais de 95 subtipos de CYP, entretanto,
com função oxidativa para fármacos as subunidades de
principal função são: CYP2 (CYP2D6) e CYP3 (CYP3A4).
Além destas enzimas, outras classes enzimáticas
também possuem grande importância no processo de
biotransformação, entre as diversas outras destacam-
se as: monoxigenase com flavina, monoamina oxidades,
desidrogenases, hidrolases e outras. A figura abaixo
exemplifica a distribuição das principais CYPs.
Hidrólise
Distribuição das principais CYPs envolvidas com o processo de metabolismo de fase I no organismo humano (Raffa et al., 2006).
Exemplos de reações de fase I:
Exemplos de processos de biotransformação de fase I (Lüllmann, 2008).

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
31
Exemplos de processos de biotransformação de fase I (Lüllmann, 2008).
REAÇÕES DE FASE II As principais enzimas envolvidas com os processos
de biotransformação de fase II são: uridinodifosfato
glicuroniltransferase, que responsável pela transferência
do ácido glicurônico; sulfotransferase, transfere radicais
de sulfato; glutationa S-transferase, transfere uma
molécula de glutationa para o radical S do fármaco;
N-acetiltransferase, que transfere radicais de acetato. Nos
esquemas abaixo é possível ter uma ideia de como estes
processos ocorrem.
Exemplos de reações de conjugação de fármacos, esta alteração faz com que o fármaco se torne mais hidrossolúvel e seja mais facilmente excretado pelo sistema renal.
As reações de oxidação/redução e de conjugação/
hidrólise aumentam a hidrofilicidade de um fármaco
hidrofóbico e seus metabólitos, permitindo que esses
fármacos sejam excretados por meio de uma via final
comum. Os fármacos e seus metabólitos são, em sua
maioria, eliminados do corpo através de excreção renal
e biliar. A excreção renal constitui o mecanismo mais
comum de excreção de fármacos e baseia-se na natureza
hidrofílica de um fármaco ou seu metabólito. Apenas um
número relativamente pequeno de fármacos é excretado
primariamente na bile. Muitos fármacos de administração
oral sofrem absorção incompleta pelo trato gastrintestinal
superior, e o fármaco residual é então eliminado por
excreção fecal. De outro modo, os fármacos podem ser
excretados em quantidades mínimas através das vias
respiratória e dérmica.
EXCREÇÃO DE FÁRMACOS A remoção de um fármaco do organismo humano
pode ocorrer através de varias vias: renal, biliar, intestinal,
pulmonar, além do suor, saliva, secreção nasal, e, leite
em mães que amamentam. A principal via de excreção
de fármacos é constituída pelo sistema renal, entretanto
há outras vias de grande importância, como por exemplo,

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
32
a via pulmonar (muito importante para fármacos voláteis
como o álcool e anestésicos gasosos), leite materno,
sistema biliar (é uma via muito importante para fármacos
que dependem do ciclo enterohepático (veja diagrama a
seguir) como os contraceptivos hormonais, cloranfenicol,
tetraciclinas, rifampicina e digoxina), lágrimas, saliva
(atualmente muitos fármacos e substâncias endógenas
tem sido monitorados por meio de sua concentração
salivar), suor, fecal entre outras.
- Ciclo enterohepático: após a ingestão do
fármaco pela via oral, este será absorvido pelo intestino
e transportado pelo sangue portal até o fígado, onde
será biotransformado e conjugado a substâncias mais
hidrossolúveis, como por exemplo, ao ácido glicurônico,
sulfatos ou outro componente endógeno. Este conjugado
por estar carregado com cargas elétricas, pode passar
do hepatócito para a bile, sendo armazenado na vesícula
biliar e posteriormente eliminado junto ao fluido biliar no
intestino. Os metabolitos conjugados ao ácido glicurônico
podem ser clivados no interior do cólon intestinal por
enzimas β-glicoronidases liberando do conjugado a
molécula do fármaco, a qual será absorvida para o
organismo.
Representação do processo enterohepático (Lüllmann, 2000).
Para que um fármaco possa ser excretado pelo
sistema renal, o mesmo poderá passar por um ou mais
dos seguintes processos: filtração glomerular, reabsorção
tubular passiva e, secreção tubular ativa.
- Filtração glomerular: Todos os fármacos são filtrados no glomérulo renal,
porém, apenas as moléculas não ligadas às proteínas
transportadoras é que passam pelos microporos. A
extensão da filtração é diretamente proporcional à taxa
de filtração glomerular e a fração do fármaco não ligado
às proteínas transportadoras. A lipossolubilidade, a
ionização, a não-ionização, inclusive o pH não influenciam
a passagem dos fármacos para o filtrado glomerular. Os
capilares glomerulares permitem a difusão de moléculas
de drogas de peso molecular inferior a 20.000 no filtrado
glomerular. Como a albumina plasmática possui o peso
molecular 68.000, esta é quase totalmente retida,
enquanto os fármacos livres (com peso inferior a 20.000)
atravessam a rede capilar para o espaço de Bowman, como
parte do filtrado glomerular. Cerca de apenas 20% do fluxo
plasmático renal são filtrados através do glomérulo.
Na representação acima pode ser observado que o
sangue contendo o fármaco perfunde todo o interior do
glomérulo, na sequencia, a droga (com peso molecular
inferior a 20 KDa) passa pelos poros de uma membrana
semi-permeável (painel B) para dentro da cápsula de

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
33
Bowmam e em seguida para dentro do túbulo contornado
proximal (painel A); fonte (Lüllmann, 2008).
- Reabsorção tubular passiva: Todo fármaco lipossolúvel e que esteja em sua forma não
ionizado depois de filtrado, poderá ser reabsorvido quando
passar pelo túbulo contornado proximal do néfrom. Assim,
para que um fármaco não sofra este processo no momento
de sua excreção, o mesmo deverá estar na sua forma
hidrossolúvel (para melhor se homogeneizar na urina)
e ionizado, pois uma vez ionizado, sua lipossolubilidade
diminui e a molécula não pode ser reabsorvida. Portanto,
fármacos básicos quando presentes no filtrado renal serão
mais facilmente eliminados, uma vez que a urina possui pH
ácido, por outro lado, fármacos ácidos serão eliminados
com maior dificuldade (Veja esquema abaixo).
Representação esquemática da eliminação e reabsorção
passiva de fármacos e a relação do pH com estes fatores
(Lüllmann, 2000).
- Secreção tubular ativa:
Fármacos que não são filtrados e que também não
conseguem se difundir para a luz tubular, podem ser
secretados ativamente para o túbulo renal. A principal
região responsável pela secreção é o túbulo contornado
proximal. As penicilinas, cefalosporinas, digoxina, diuréticos
tiazídicos são exemplos de fármacos que são secretados
ativamente. Por outro lado, drogas como a probenicida,
são capazes de inibir o sistema de transporte de penicilinas,
cefalosporinas, digoxina e outras, aumentando o tempo de
excreção renal destes fármacos. Ainda, outro ponto muito
importante é que os fármacos descritos anteriormente
competem entre si pelo sistema de secreção, ou seja,
a penicilina e os diuréticos tiazídicos (hidroclorotiazida),
diuréticos de alça (furosemida) podem diminuir de forma
considerável a eliminação de digoxina, podendo ocorrer
intoxicação digitálica.
A figura acima é representativa dos sistemas de
filtração, secreção e reabsorção dos fármacos no rim. Os
fármacos podem ser (1) filtrados no glomérulo renal, (2)
secretados no túbulo proximal, (3) reabsorvidos a partir
da luz tubular e transportados de volta ao sangue, e (4)
excretados na urina. O equilíbrio relativo das taxas de
filtração, secreção e reabsorção é que determina a cinética
de eliminação dos fármacos pelos rins. O aumento do fluxo
sanguíneo, o aumento da taxa de filtração glomerular e
a diminuição da ligação às proteínas plasmáticas causam
uma excreção mais rápida do fármaco, visto que todas
essas alterações resultam em aumento da filtração do

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
34
fármaco no glomérulo. Alguns fármacos, como a penicilina,
são secretados ativamente no túbulo proximal. Embora a
reabsorção possa diminuir a taxa de eliminação de um
fármaco, muitos fármacos sofrem sequestro pelo pH no
túbulo distal e, portanto, são excretados eficientemente na
urina. Para os fármacos que dependem do rim para a sua
eliminação, a presença de comprometimento da função
renal pode resultar em concentrações plasmáticas mais
altas do fármaco, de modo que é preciso modificar a dose
e a frequência de administração desses fármacos.
CLEARANCE OU DEPURAÇÃO O clearance (Cl) total de um fármaco no
organismo descreve a eficiência do processo de eliminação
(irreversível) e pode ser considerado como um parâmetro
que relaciona sua velocidade de eliminação com a
concentração plasmática. Desta forma, o Cl pode ser
determinado pela seguinte equação:
Cl = VE/Cp
Onde VE é a velocidade de eliminação (mg/h); Cl é o
Clearance (L/h) e Cp é a concentração plasmática (mg/L).
O clearance também pode indicar a fração do volume
de distribuição que é depurada completamente na unidade
de tempo, podendo ser calculado pela seguinte equação:
Cl = Vd x Ke
Assim, Cl é o clearance, Vd é o volume aparente de
distribuição da droga e Ke a constante de eliminação.
CÁLCULO DO CLEARANCE E SUA APLICABILIDADE. VIDEO 12: DETERMINAÇÃO DO CLEARANCE E DOSES DE MANUTENÇÃO.
O método clássico para determinar o clearance
renal de uma determinada substância, como por exemplo,
da creatinina, requer a medida de sua velocidade de
eliminação na urina e da concentração plasmática no
mesmo tempo. Para isto, é possível aplicar a seguinte
equação:
ClR = Cu x V/Cp
Onde:
ClR é o clearance; Cu é a concentração da substância
na urina; V é a velocidade do fluxo urinário e Cp é a
concentração da substância no sangue e/ou no plasma.
Portanto, esta equação pode ser utilizada para a
determinação de qualquer substância (fármaco), desde
que seja possível quantifica-la no plasma ou sangue do
paciente. Além da equação anterior, o clearance de drogas
pode ser calculado por meio da relação da porcentagem
do fármaco excretado na forma inalterada na urina (fe) e
clearance total (Cl):
ClR = fe x Cl
Resumindo, o clearance tornou-se um parâmetro
importante em função de seu uso na prática clínica.
Sua determinação é relativamente fácil e seu cálculo é
realizado empregando-se as equações anteriormente
descritas. Por meio deste parâmetro farmacocinético pode-
se estabelecer a dose de manutenção de um determinado
fármaco necessária para atingir a concentração terapêutica
no estado de equilíbrio. Em situação de equilíbrio, a
quantidade de fármaco que é introduzida no organismo
deve ser igual à eliminada (depurada), ou seja, a velocidade
de administração (dose/intervalo entre as doses) é igual a
velocidade de eliminação.
DM = Cl x Css
Portanto, DM é a dose de manutenção, Cl é o clearance
e Css é a concentração em estado de equilíbrio (steady-
state). Portanto o cálculo da dose de manutenção adequada
é o objetivo primário de uma terapia medicamentosa. Para
manter o estado de equilíbrio escolhido, ou a concentração
alvo desejada, a taxa de administração do fármaco deverá
ser ajustada de modo que a quantidade de fármaco
administrado deverá ser igual à taxa de perda (eliminação
ou saída). Para tanto, a equação seguinte pode descrever
esta relação:
Velocidade de infusão = Cpalvo x Cl/F
Caso o clínico já tenha definido a concentração
plasmática alvo (Cpalvo) e conhecer a depuração e

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
35
biodisponibilidade do fármaco prescrito, é possível calcular
a dose e os intervalos entre as doses apropriados.
EX:
Para um paciente portador de ICC internado em uma
unidade hospitalar foi prescrito o medicamento digoxina.
O indivíduo possui 65 Kg e como dose em estado de
equilíbrio, foi estipulado a concentração plasmática de 1,5
ng/mL, com base no fato que a depuração de creatinina
deste paciente é de 80 mL/min, a depuração de digoxina
pode ser estimada com base na seguinte equação abaixo
(estipulada especialmente para a digoxina).
Cl = 0,88 x Clcr + 0,33 mL.min-1.Kg-1
Esta equação é baseada em apontamentos clínicos e
acompanhamento de pacientes com algum grau de ICC.
Logo, ao se desenvolver a situação descrita acima,
temos que:
Cl = 0,88 x (80 mL/min/65 kg) + 0,33 mL.min-1.Kg-1
Cl = 0,88 x (1,23 mL/min/kg) + 0,33 mL.min-1.Kg-1
Cl = 1, 083 mL/min/kg + 0,33 mL.min-1.Kg-1
Cl = 1,41 mL.min-1.Kg-1
Ou corrigido pelo peso total do paciente
Cl = 91, 85 mL.min-1
Cl = 5,5 L.H-1
Com base nestes dados apresentados pode-se
estimar a taxa de administração do medicamento para
este paciente nesta situação em que se encontra. No
entanto, deve ser lembrado que caso o paciente receba o
medicamento por via oral, devemos levar em consideração
a biodisponibilidade da formulação por esta via de
administração (F = 70%)
D = Cp alvo x Cl/F
D = 1,5 ng/mL x 1,41 mL.min-1.kg-1/0,7
D = 1,5 ng/mL x 2,014 mL.min-1.kg-1
D = 3,02 ng. min-1.kg-1
Logo para um paciente de 65 kg:D = 3,02 ng. min-1.kg-1 x 65 kg
D = 196 ng. min-1
Ou em 24 hs
D = (196 ng. min-1 x 60 min) x 24 H
D = 282,76 µg/dia ou 0,282 mg/dia
No entanto, não dispomos de formulações
industrializadas com esta concentração de PA por unidade.
Desta forma, na prática atualmente utilizada, é arredondado
para a dose mais próxima. Assim, a concentração mais
próximas que encontramos disponível no mercado é igual a
0,25 mg. Logo, 1,5 ng/mL x (0,25/0,282), resulta em uma
concentração plasmática de equilíbrio igual a 1,33 ng/mL,
o que está dentro da faixa terapêutica estabelecida para
a digoxina (Veja tabela de parâmetros farmacocinéticos,
apresentada no tópico volume aparente de distribuição).
ADMINISTRAÇÃO DE MEDICAMENTOS EM DOSES MÚLTIPLAS
A administração de drogas em doses múltiplas é
considerada como a modalidade mais frequentemente
utilizada na farmacoterapia, seja para o tratamento de
uma condição clínica aguda ou para uma situação crônica.
Portanto, é fundamental que a concentração plasmática
terapêutica seja mantida em estado de equilíbrio ao longo
de todo o tratamento do paciente.
Desta forma, com base nos achados e condições
clínicas do indivíduo, o clínico (prescritor) estabelece um
esquema terapêutico ou regime de dosagens (posologia)
em conformidade com o medicamento estabelecido,
sua forma farmacêutica e via de administração. Estes
esquemas posológicos geralmente são estabelecidos em
conformidade com as informações constantes na bula
do medicamento, que são estabelecidas com base nos
ensaios clínicos multicêntricos de fase II e III, por meio dos
quais a Indústria Farmacêutica responsável pelo produto
comprovou sua eficácia clínica e segurança.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
36
Quando ocorre a administração de doses semelhantes
em intervalos regulares, o princípio ativo tende a ser
introduzido ao organismo a uma velocidade constante,
o que se assemelha ao processo de infusão contínua de
medicamento; no entanto, as concentrações plasmáticas
estabelecidas por um sistema de administração em doses
múltiplas podem variar em conformidade com o intervalo
de cada administração, ao passo que em um sistema
de administração em infusão contínua, não há alteração
da concentração plasmática, desde que, não ocorram
alterações dos sistemas de metabolismo e excreção
(clearance).
Na figura acima é possível observar que quando o
esquema de administração em sistema de infusão (linha
sólida) é utilizado, a concentração de equilíbrio (steady-
state: Css) é alcançada e mantida. Porém, quando o
sistema de administração de doses múltiplas é escolhido
como sistema terapêutico, observa-se que a cada intervalo
de administração, a concentração plasmática aumenta
logo após a administração do medicamento e, depois de
um intervalo de tempo, esta está em menor concentração
no plasma, indicando que a mesma foi depurada do
organismo.
De um ponto de vista conceitual, em farmacocinética,
é muito importante diferenciar um esquema posológico
de doses múltiplas e doses repetidas. No primeiro caso,
as doses são administradas a determinados intervalos, é
fundamental que haja acumulo do fármaco no organismo,
ou seja, há uma quantidade remanescente da dose no
organismo, antes da administração da próxima dose. Tal
fato é essencial do ponto de vista clínico, uma vez que
se espera que sejam geradas concentrações plasmáticas
dentro das margens terapêuticas de cada fármaco,
quando o estado de equilíbrio é alcançado, concentrações
que ainda não são atingidas em tempos anteriores após o
inicio da administração das doses (figura a seguir).
Em um sistema de administração de doses repetidas
também existe uma dose e um intervalo entre as doses,
mas praticamente todo o fármaco é eliminado antes que
seja administrada a dose seguinte, desta forma, não
há acumulo. Em consequência a este fato, é necessário
alcançar a concentração terapêutica útil, e do ponto de
vista farmacocinético, essa administração deve ser tratada
como dose única.
Na tabela a seguir encontram-se alguns exemplos de
administração de medicamentos segundo terapia de doses
repetidas e múltiplas doses.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
37
Fármaco Tempo de meia vida Intervalo de administração (h)
Esquema de administração
amoxicilina 1,2 8 Doses repetidas
furosemida 1,5 12 Doses repetidas
gentamicina 2 8 a 24 Doses repetidas
digoxina 43 24 Doses múltiplas
clonazepam 27 12 Doses múltiplas
lítio 24 12 Doses múltiplas
Em geral, não são desejadas acentuadas flutuações nas concentrações entre as doses administradas. Se a absorção
e a distribuição fossem instantâneas as flutuações das concentrações de fármacos administrados entre as doses seriam
regidas praticamente pelo tempo de meia vida de eliminação do fármaco.
Se um fármaco for relativamente atóxico, de modo que a concentração muitas vezes maior que a necessária para a
terapia possa ser facilmente tolerada, pode ser utilizada estratégia de dose máxima e o intervalo entre as doses pode
ser muitas vezes, maior que o tempo de eliminação da droga. A meia vida da amoxicilina é de 2 horas, mas geralmente
são administradas dose em intervalos de 12 ou 8 horas em decorrência da elevada dose administrada.
No caso de fármacos com estreita faixa terapêutica, pode ser importante estimar as concentrações máxima e mínima
que irão ocorrer entre os intervalos de administração. Para se determinar a concentração mínima pode-se utilizar a
seguinte equação:
Onde K representa a Ke (constante de eliminação) e é igual a 0,693 dividido pelo tempo de meia vida plasmático, T
é o intervalo entre as doses. O termo exp(-KT) é na verdade a fração da ultima dose (corrigida pela biodisponibilidade)
que permanece no corpo ao final de um intervalo entre as doses.
MONITORAMENTO FARMACOCINÉTICO.As interações dinâmicas entre a absorção, a distribuição, o metabolismo e a excreção de um fármaco determinam a
sua concentração plasmática e estabelecem a capacidade do fármaco de alcançar o seu órgão-alvo numa concentração
efetiva. Com frequência, a duração desejada da terapia farmacológica é maior do que a que pode ser obtida por uma
dose única, tornando necessário o uso de múltiplas doses para proporcionar concentrações plasmáticas relativamente
constantes do fármaco dentro dos limites de sua eficácia e toxicidade.
O planejamento da terapia farmacológica para um paciente, as tomadas de decisões sobre a escolha do fármaco
e sua posologia, são de fundamental importância para a obtenção de uma terapêutica medicamentosa eficaz. Para se
tomar estas decisões, normalmente é objetivado um efeito farmacológico desejado, e a administração de doses do
medicamento escolhido é manipulada de tal forma a se obter o efeito planejado inicialmente. Esta estratégia geralmente
funcional para alguns determinados fármacos, como por exemplo, no controle da pressão arterial de um dado paciente,
que pode ter sua pressão monitorada e a dose do fármaco escolhido é administrada em conformidade com os valores
pressóricos deste. Por outro lado, para fármacos que possuam um índice terapêutico estreito ou quando ocorram

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
38
mudanças repentinas no estado do paciente de forma
que exija alguma alteração posológica do medicamento,
esta manobra poderá ser muito comprometedora para o
paciente.
Na maioria das situações clínicas, é muito importante
manter um efeito terapêutico por um período prolongado.
Isto implica que a concentração plasmática do fármaco
deverá ser mantida pelo tempo necessário. Normalmente,
a concentração plasmática do fármaco é mantida dentro
de um sistema de administração de múltiplas doses ou
dependendo da situação, em um esquema de administração
contínua. Por outro lado, em situações menos complicadas,
é possível realizar uma terapêutica com um sistema de
administração de dose única. O fato importante é que é
possível se realizar o monitoramento cinético do fármaco
em todos os casos. Para isto, se faz necessário controlar os
processos de distribuição, metabolismo e eliminação, de
posse destes resultados é possível se determinar a melhor
dose e o melhor intervalo de tempo para se administrar
esta dose ao paciente.
REQUISITOS ESSENCIAIS PARA CONTROLE TERAPÊUTICO
AnalíticosSão necessários métodos analíticos validados,
infraestrutura, equipamentos (HPLC, CG, LC-MS) e equipe
treinada em boas práticas de laboratório.
Dados do pacienteComo dados primordiais do paciente, é necessário:
- Ficha médica;
- Dados gerais do paciente (nome, sexo, idade, hábitos,
raça);
- Medicamentos a serem monitorados;
- Razão da solicitação;
- Histórico clínico (evolução da patologia e
complicações);
- Função dos órgãos de depuração (rins, fígado e
pulmão).
INFORMAÇÕES SOBRE O MODO DE
ADMINISTRAÇÃO E DEFINIÇÃO DOS TEMPOS DE COLETA
Deve-se ser considerado que caso o tratamento
estipulado envolva o sistema de administração de
múltiplas doses, haverá acumulo de fármaco no organismo
e, portanto, será alcançada a concentração no estado
de equilíbrio (Css). Porém, se o tratamento ocorrer por
meio de uma sucessão de doses, mas o intervalo de
administração for superior ao tempo de meia vida, não
haverá acumulo e o tratamento que se dará aos dados
obtidos será o tratamento de dados referentes ao sistema
de administração dosse única.
Esses modos de administração podem ser resumidos
em:
- Bolus IV em dose única: a concentração máxima
é obtida imediatamente após a administração da dose.
Nesse caso administra-se apenas uma única dose do
fármaco, ou quando o tratamento é longo, a dose
seguinte é administrada quando já não há mais fármaco
remanescente. Portanto, o tempo necessário para que
a concentração plasmática seja mínima é de 5 vezes o
tempo de meia vidada.
- Bolus IV em doses múltiplas: se ocorrer
acúmulo de fármaco por este método, é recomendado o
monitoramento apenas quando o estado de equilíbrio for
atingido, ou seja, depois de transcorrido um período de
administração de 4 a 5 vezes a meia vida de eliminação.
A concentração máxima é alcançada imediatamente
após a administração da dose e a concentração mínima
imediatamente antes à administração de uma nova dose.
- Infusão IV: neste método de administração,
a concentração plasmática máxima será o estado de
equilíbrio, o qual será alcançado após transcorrido um
período de administração de no mínimo, 5 vezes o tempo
de meia vida.
- Infusão IV intermitente em dose única: consistem em administrar infusões por um período curto,
as quais serão repetidas após a eliminação do fármaco,

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
39
uma vez que não existe acúmulo. A concentração máxima
é obtida ao término da infusão.
- Infusão IV intermitente em doses múltiplas: esse modo de administração é caracterizado por
administração de infusões por períodos curtos, mas com
ocorrência de acúmulo. A monitoração deverá ser realizada
quando o período estacionário (equilíbrio) for atingido.
A concentração máxima é a concentração no estado de
equilíbrio, a qual é obtida imediatamente após a infusão
ser finalizada e a concentração mínima é obtida ao final do
intervalo de administração das doses.
- Dose única extravascular: neste sistema de
administração, a concentração máxima é dependente da
velocidade de absorção. Na ausência de dados precisos,
pode-se considerar que a absorção é rápida quando o Tmax
está ao redor de 1 a 1,5 h e mais lenta entre 3 e 4 h.
- Doses múltiplas extravasculares: nesse sistema
de administração, a concentração máxima é dependente
da velocidade de absorção. A concentração mínima é
obtida ao final do intervalo de doses. Caso haja acumulo
do fármaco, deve ser considerado o estado estacionário.
Para se realizar este procedimento, normalmente
amostras de sangue são coletadas do paciente de tempos
em tempos, como por exemplo, 15; 30; 45; 60 min.,
1; 1,5; 2; 3; 4; 5; 6; 8; 10; 12; 24 horas. É possível se
elaborar um protocolo de coletas dentro de um sistema
cientificamente já estabelecido (estes protocolos podem
ser obtidos por meio de pesquisa às bases de busca como
pubmed, medline, lilacs bireme, entre outras).
Após a coleta das amostras, os resultados são
transferidos para um programa de estatística ou podem
ser inseridos em papel milimetrado, obtendo-se assim,
uma curva concentração plasmáticos em função do tempo.
Por meio desta curva, será possível determinar o tempo de
meia vida, a AUC, Cmax, Tmax, a constante de eliminação
do fármaco.
A seguir veremos um exemplo prático de como este recurso poderá ser aplicado na prática clínica.
Exemplo:
Foi administrado a um paciente 200 mg de uma
determinada droga por via endovenosa na forma de bôlus.
Na tabela abaixo, temos as concentrações plasmáticas da
droga em função dos tempos de coleta. O paciente em
questão pesa 70kg.
Tempo (h) Concentrações (mg/L)
0.5 12.6
1.0 10.6
1.5 8.9
2.0 7.5
2.5 6.3
3.0 5.3
3.5 4.5
4.0 3.8
5.0 2.7
6.0 1.9
8.0 0.9
10.0 0.5
12.0 0.2
Com estes dados, é possível elaborar em papel
milimetrado normal (A) ou em escala logarítmica
utilizando papel monolog (B) (exemplos abaixo) a curva
concentração plasmática em função do tempo (abaixo).
(A)
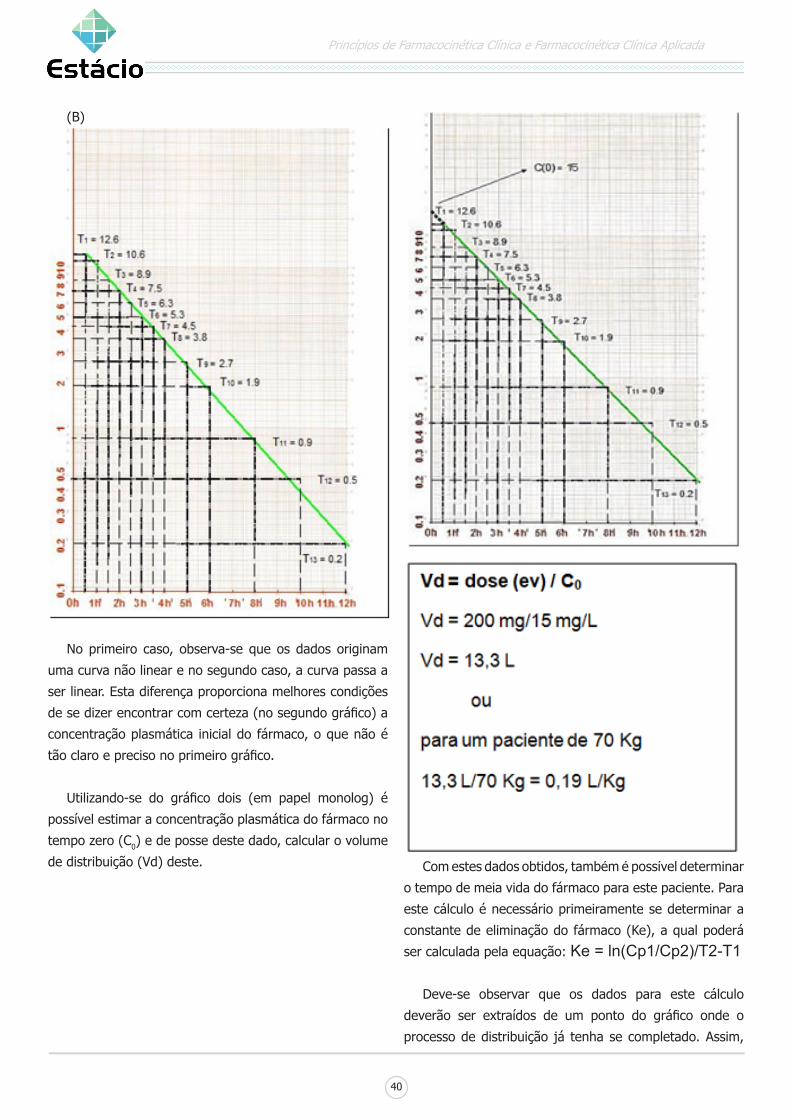
Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
40
(B)
No primeiro caso, observa-se que os dados originam
uma curva não linear e no segundo caso, a curva passa a
ser linear. Esta diferença proporciona melhores condições
de se dizer encontrar com certeza (no segundo gráfico) a
concentração plasmática inicial do fármaco, o que não é
tão claro e preciso no primeiro gráfico.
Utilizando-se do gráfico dois (em papel monolog) é
possível estimar a concentração plasmática do fármaco no
tempo zero (C0) e de posse deste dado, calcular o volume
de distribuição (Vd) deste.
Com estes dados obtidos, também é possível determinar
o tempo de meia vida do fármaco para este paciente. Para
este cálculo é necessário primeiramente se determinar a
constante de eliminação do fármaco (Ke), a qual poderá
ser calculada pela equação: Ke = ln(Cp1/Cp2)/T2-T1
Deve-se observar que os dados para este cálculo
deverão ser extraídos de um ponto do gráfico onde o
processo de distribuição já tenha se completado. Assim,

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
41
adotando-se o intervalo entre os tempos 4 e 6 hs, onde
encontramos as concentrações 3,8 e 1,9, respectivamente,
teremos:
Uma vez determinado a Ke, o tempo de meia vida
poderá ser calculado pela seguinte equação:
T1/2 = 0,693/Ke; onde 0,693 é uma constante do tempo
de meia vida, e Ke é a constante de eliminação que já foi
determinada acima.
Desta forma temos:
T1/2 = 0,693/0,346 logo, T1/2 = 2 h
Caso seja necessário manter uma concentração
plasmática em estado equilíbrio para este medicamento
por infusão intravenosa (contínua) será necessário utilizar
a seguinte equação:
Assim, caso seja necessário manter uma concentração
de equilíbrio (Css) igual a 0,5 mg, será necessário:
Conforme visto até o momento, é possível notar que
com estes parâmetros farmacocinéticos, há diversas
possibilidades de se manipular as concentrações
plasmáticas de fármacos mediante as necessidades de cada
indivíduo. Por isto o acompanhamento farmacocinético é
muito importante quando se trata de medicamentos com
faixa terapêutica estreita.
FÁRMACOS QUE DEVEM SER SUBMETIDOS AO CONTROLE TERAPÊUTICO
Para que um fármaco seja candidato ao
monitoramento farmacoterapêutico, este deverá
apresentar uma ou mais das seguintes características:
- Alta variabilidade interindividual (como por exemplo,
antirretrovirais e cardiotônicos);
- Faixa terapêutica estreita (digitálicos);
- Correlação entre concentração plasmática e efeito
terapêutico (anticonvulsivantes).
Alta variabilidade interindividual está relacionada à
existência de concentrações plasmáticas muito diferentes
quando uma mesma dose de um determinado fármaco
é administrada a diferentes pacientes. Em casos onde o
fármaco possui estreita faixa terapêutica, encontra-se uma
faixa de concentração plasmática de baixa amplitude em
que há maior evidência de eficácia do fármaco e menor
incidência de efeitos adversos.
Outros fatores que podem ser levados em consideração
para o monitoramento farmacocinético do fármaco podem
ser:
- Fármacos que apresentam cinética não linear (ex:
fenitoina);
- Fármaco que apresenta alta variabilidade intraindividual
(respostas diferentes em um mesmo individuo);
- Quando a cinética é modulada por fatores genéticos
(polimorfismo genético) como, por exemplo, diferentes
velocidades de metabolização;
- Dificuldade de avaliar o efeito farmacológico (como os
imunossupressores);
- Quando a toxicidade pode desencadear um risco de
morte para o paciente (insuficiência renal desencadeada
por aminoglicosídeo);
- Quando há diferentes condições fisiológicas ou
patológicas que possam modificar a farmacocinética:
gestação, idades de extremo (neonatos ou idosos),
septicemia, insuficiência renal, transplantados, hepatopatas
ou cardíacos.

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
42
Exemplos de fármacos que necessitam de monitoramento farmacocinético em uma terapia. Classe farmacológica Exemplo Faixa terapêutica
imunossupressores CiclosporinaMicofenolato
Tacrolimo
100 a 300 µg/L1 a 3,5 mg/L5 a 20 g/L
Broncodilatadores Teofilina 10 a 20 µg/L
Antibióticos VancomicinaGentamicina
20 a 40 mg/L8 mg/L
Antidepressivos AmitriptilinaLítio
100 a 250 mg/L0,6 a 1,2 mEq/L
Antiarrítmicos LidocaínaQuinidina
1 a 5 mg/L1 a 4 mg/L
Anticonvulsivantes FenitoínaÁcido valpróicoCarbamazepina
10 a 20 mg/L50 a 100 mg/L4 a 12 mg/L
Fármacos não submetidos ao controle cinético Quando é possível avaliar o efeito farmacológico do fármaco por meio de sinais e sintomas, exames laboratoriais
ou outros procedimentos, não se faz necessário, o monitoramento terapêutico.
Exemplos de fármacos que não são avaliados por meio de controle terapêutico.Fármaco Medida do efeito
Anticoagulantes oraisAnti-hipertensivos
Diuréticoshipoglicemiantes
Tempo de protrombinaPressão arterial
Aumento da diurese/pressão arterialGlicemia

Princípios de Farmacocinética Clínica e Farmacocinética Clínica Aplicada
43
REFERÊNCIAS
CRAIG, C. R.; STITZEL, R. E. Farmacologia Moderna com Aplicações Clínicas. 6 ed. Rio de Janeiro:
Guanabara Koogan. 2005. 832 p.
FUCHS, F. D.; WANNMACHER, L. E FERREIRA, M. B. C..
Farmacologia Clínica: Fundamentos da Terapêutica Racional. 3 ed. Rio de Janeiro: Guanabara Koogan. 2004.
1096 p.
GOLAN, D. E. Princípios de Farmacologia: a Base Fisiopatológica da Farmacoterapia. 2 ed. Rio de
Janeiro: Guanabara Koogan. 2009. 922 p.
GONÇALVES, J. E.; GAI, M. N.; CAMPOS, D. R.;
STORPIRTIS, S. Farmacocinética - Básica e Aplicada. Rio
de Janeiro: Guanabara Koogan. 2011. 240 p.
GRAHAME-SMITH, D. G; ARONSON, J. K. Tratado de Farmacologia Clínica e Farmacoterapia. 3 ed. Rio de
Janeiro: Guanabara Koogan, 2004. 640 p.
HARDMAN, J. G; LIMBIRD, L. E; MOLINOFF, P. B.
GOODMAN & GILMAN: As Bases Farmacológicas da Terapêutica. 10 ed. Rio de Janeiro: Mac Graw Hill, 2003.
1647 p.
KATZUNG, B. G.; Farmacologia Básica & Clínica. 9
ed. Rio de Janeiro: Guanabara Koogan, 2006. 1068 p.
LAUBANE, J.P. Farmacocinética. São Paulo: Editora
Andrei, 1993. 193p.
LEBLANC, P.P. Tratado de Biofarmácia e Farmacocinética. Lisboa: Instituto Piaget, 1997. 419p.
MINNEMAN, K. P.; WECKER, L. Brody: Farmacologia Humana. 4 ed. Rio de Janeiro: Elsevier Editora Ltda,
2005, 724p.
RANG, H. P.; DALE, M. M.; RITTER, J. M.; Farmacologia.
6 ed. Rio de Janeiro: Guanabara Koogan, 2006. 904 p.
SILVA, P.; Farmacologia. 6 ed. Rio de Janeiro:
Guanabara Koogan, 2002. 1400 p.
RAFFA, R. B.; RAWLS, S. M.; BEYZAROV, E. P. Atlas de farmacologia de Netter. Porto Alegre: Artmed editora
SA. 2005. 410p.
LÜLLMANN, H.; MOHR, K.; HEIN, L.; BIEGER, D.
Farmacologia: texto e atlas. 5 ed. Porto Alegre: Artmed
editora SA., 2005. 416 p.


















