Prenatal diagnosis of an 8p23.1 deletion in a fetus with a diaphragmatic hernia and review of the...
6
Prenat. Diagn. 18: 1055–1060 (1998) PRENATAL DIAGNOSIS OF AN 8p23.1 DELETION IN A FETUS WITH A DIAPHRAGMATIC HERNIA AND REVIEW OF THE LITERATURE . 1 *, . - 1 , . 1 , . 2 , . 1 , . 3 , . . 3 , . 1 , . 1 , . 1 . 1 1 De ´partement de Ge ´ne ´tique, Ho ˆpital Necker Enfants Malades, Paris, France 2 Service d’Anatomopathologie, Ho ˆpital Port-Royal, Paris, France 3 Centre de Me ´decine Foetale, Ho ˆpital Port-Royal, Paris, France Received 12 January 1988 Accepted 7 March 1998 SUMMARY The prenatal diagnosis of an 8p23.1 deletion is reported. The diagnosis was ascertained at 22 weeks of gestation because of the discovery of a diaphragmatic hernia at ultrasound. Following cytogenetic studies and counselling, the pregnancy was terminated. An autopsy confirmed the presence of a diaphragmatic hernia and revealed also the existence of an atrio-ventricular canal (AVC) and an atrial septal defect (ASD). The clinical features of this antenatally diagnosed case are compared with those observed in 16 previously reported cases with an identical deletion of the short arm of chromosome 8. This suggests that a deletion 8p23.1 should be considered whenever a diaphragmatic hernia and/or an AVC is detected on ultrasound. ? 1998 John Wiley & Sons, Ltd. : deletion of short arm chromosome 8; diaphragmatic hernia; atrio-ventricular canal; prenatal diagnosis INTRODUCTION Since the initial observation of a patient with deletion of the short arm of chromosome 8 (del(8)(p23.1)) (Fagan et al., 1988), 15 further cases have been reported (Fryns et al., 1989; Blennow and Bro ¨ ndum-Nielsen, 1990; Pecile et al., 1990; Pettenati et al., 1992; Hutchinson et al., 1992; Devriendt et al., 1995; Wu et al., 1996). All these cases were detected after birth. The features of this common and recently described deletion include retardation, dysmorphic facies, mental retardation and congenital heart defect. Here we report the first case of a 8p23.1 deletion ascertained prenatally. CASE REPORT A 30-year-old Portuguese mother was referred to our centre at 22 weeks of gestation. Her past medical history was unremarkable. She had three brothers. Her 31-year-old husband is Portuguese and the couple is not consanguineous. They have a healthy three-year-old girl. No spontaneous abortion is reported. A second-trimester ultra- sound examination showed a posterior left dia- phragmatic hernia, with intra-thoracic stomach and spleen. A cardiac fetal ultrasound was reported as normal. All parameters of fetal bi- ometry were between the third and the fifth percentile. A cytogenetic study performed on amniotic fluid cells showed a terminal deletion of the short arm of chromosome 8 (del(8)(p23.1)). Parental chromosomes were normal. After coun- selling, the pregnancy was terminated at 26 weeks of gestation. *Correspondence to: L. Faivre, Service de Ge ´ne ´tique Me ´dicale Ho ˆ pital Necker Enfants Malades, 149 rue de Se `vres, 75015 Paris, France. CCC 0197–3851/98/101055–06 $17.50 ? 1998 John Wiley & Sons, Ltd.
Transcript of Prenatal diagnosis of an 8p23.1 deletion in a fetus with a diaphragmatic hernia and review of the...

Prenat. Diagn. 18: 1055–1060 (1998)
PRENATAL DIAGNOSIS OF AN 8p23.1 DELETIONIN A FETUS WITH A DIAPHRAGMATIC HERNIA
AND REVIEW OF THE LITERATURE
. 1*, . -1, . 1, . 2, . 1, . 3, . . 3,. 1, . 1, . 1 . 1
1Departement de Genetique, Hopital Necker Enfants Malades, Paris, France2Service d’Anatomopathologie, Hopital Port-Royal, Paris, France3Centre de Medecine Foetale, Hopital Port-Royal, Paris, France
Received 12 January 1988Accepted 7 March 1998
SUMMARY
The prenatal diagnosis of an 8p23.1 deletion is reported. The diagnosis was ascertained at 22 weeks ofgestation because of the discovery of a diaphragmatic hernia at ultrasound. Following cytogenetic studies andcounselling, the pregnancy was terminated. An autopsy confirmed the presence of a diaphragmatic hernia andrevealed also the existence of an atrio-ventricular canal (AVC) and an atrial septal defect (ASD). The clinicalfeatures of this antenatally diagnosed case are compared with those observed in 16 previously reported caseswith an identical deletion of the short arm of chromosome 8. This suggests that a deletion 8p23.1 should beconsidered whenever a diaphragmatic hernia and/or an AVC is detected on ultrasound. ? 1998 John Wiley &Sons, Ltd.
: deletion of short arm chromosome 8; diaphragmatic hernia; atrio-ventricular canal; prenataldiagnosis
INTRODUCTION
Since the initial observation of a patient withdeletion of the short arm of chromosome 8(del(8)(p23.1)) (Fagan et al., 1988), 15 further caseshave been reported (Fryns et al., 1989; Blennowand Brondum-Nielsen, 1990; Pecile et al., 1990;Pettenati et al., 1992; Hutchinson et al., 1992;Devriendt et al., 1995; Wu et al., 1996). All thesecases were detected after birth. The features of thiscommon and recently described deletion includeretardation, dysmorphic facies, mental retardationand congenital heart defect.
Here we report the first case of a 8p23.1 deletionascertained prenatally.
*Correspondence to: L. Faivre, Service de GenetiqueMedicale Hopital Necker Enfants Malades, 149 rue de Sevres,
75015 Paris, France.CCC 0197–3851/98/101055–06 $17.50? 1998 John Wiley & Sons, Ltd.
CASE REPORT
A 30-year-old Portuguese mother was referredto our centre at 22 weeks of gestation. Her pastmedical history was unremarkable. She had threebrothers. Her 31-year-old husband is Portugueseand the couple is not consanguineous. They havea healthy three-year-old girl. No spontaneousabortion is reported. A second-trimester ultra-sound examination showed a posterior left dia-phragmatic hernia, with intra-thoracic stomachand spleen. A cardiac fetal ultrasound wasreported as normal. All parameters of fetal bi-ometry were between the third and the fifthpercentile. A cytogenetic study performed onamniotic fluid cells showed a terminal deletion ofthe short arm of chromosome 8 (del(8)(p23.1)).Parental chromosomes were normal. After coun-selling, the pregnancy was terminated at 26 weeks
of gestation.
1056 . .
External examination revealed a female fetuswith facial dysmorphism, including doli-chocephaly and eyelids oedema (Fig. 1).Measurements (weight 970 g; vertex–talus length35 cm; vertex–coccyx length 26 cm; OFC 25 cm;foot 5·2 cm) were compatible with term. No otherexternal malformations were found. X-ray studiesof the fetus were normal. At necropsy, the leftdiaphragmatic hernia was confirmed. The leftlobe of the liver, the stomach, the spleen and partof the small intestine were found in the thorax.The left lung was hypoplastic and pushed to theright. Cardiac examination showed an atrio-ventricular canal (AVC) and an atrial septaldefect (ASD). Examination of other organs,including a neuropathological examination, wasnormal.
Fig. 1—Picture of the fetus after termination at 22 weeksof gestation. Note the facial dysmorphism includingdolichocephaly and eyelid oedema
? 1998 John Wiley & Sons, Ltd.
Fig. 2—Partial karyotype of the propositus showing the RHG-(left) and GTG-banded (right) chromosomes 8. The normalchromosome 8 is on the left, and the deleted chromosome 8 onthe right
CYTOGENETIC AND MOLECULARANALYSIS
Chromosome analysis of the fetus was per-formed on amniotic fluid cells using solidGiemsa, R and G banding techniques. A distaldeletion of the short arm of one chromosome 8was seen in all examined cells, with a breakpointin 8p23.1 (Fig. 2). Chromosome analyses ofthe parents performed on blood samples werenormal.
Fluorescence in situ hybridization (FISH)analysis with a chromosome 8 specific sub-telomeric probe (YAC 931B2, locus D8S264provided by Dr T. Haaf) was performed on thefetus and his parents. These studies confirmedthe terminal deletion of the short arm ofchromosome 8 on the fetus and indicated that noother rearrangement, such as small undetect-able translocation, was involved (data notshown).
A molecular analysis was done in this familyto determine the parental origin of the deletion(Fig. 3). Total cellular DNA was prepared frompulmonary cell culture of the fetus and fromperipheral blood lymphocytes of the parents.Polymorphic markers D8S261, D8S265, D8S550,D8S277 and D8S264 located in chromosome8p23.1 (centromere to telomere) were used. Toestablish which polymorphic loci were deleted,alleles were compared between the fetus and theparents. The results showed that the deletion wasof maternal origin and extended to the regionbetween the D8S261 and D8S265 markers.
Prenat. Diagn. 18: 1055–1060 (1998)

1057 8p
Fig. 3—Polymorphic marker analysis for 8p23 deletion. Fullyinformative analysis of an 8p23 marker is shown for the fetusand his parents. The fetus demonstrates inheritance of only oneallele (from his father) for marker D8S550 and therefore has adeletion for this marker on the maternally-derived chromosome
DISCUSSION
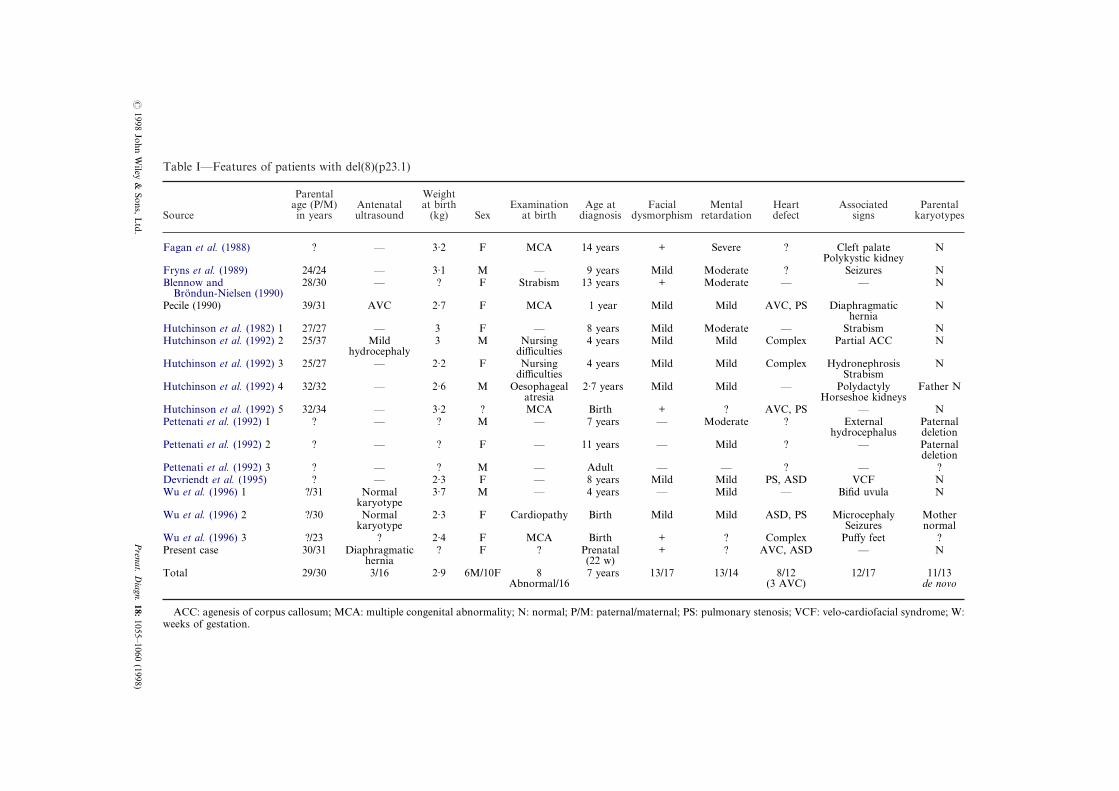
Here, we describe the prenatal diagnosis of afetus with partial deletion of 8p. Molecular cyto-genetic analysis of the fetus confirmed the terminaldeletion and molecular studies showed that thedeletion was maternal in origin. In all 16 previouscases, diagnosis was made during infancy, usuallyaround seven years old in a child with pre and/orpostnatal growth retardation (mean birth weight,2·9 kg), slight facial dysmorphism, moderatemental retardation and congenital heart defect(Table I). Usually, the deletion occurs de novoalthough it can be inherited (Pettenati et al., 1992).Maternal and paternal age, as well as sex, does notappear to be associated with the occurrence of thedeletion. Retrospectively, only two patients pre-sented as abnormal on an antenatal ultrasoundexamination. One observation (Pecile et al., 1990)describes the presence of an AVC in utero wherethe cytogenetic analysis performed on amnioticfluid cells was interpreted as normal. The secondcase (Hutchinson et al., 1992) was ascertainedbecause of a mild dilatation of cerebral ventriclesat 36 weeks of amenorrhea, but no further cyto-genetic study was performed. Two further cases
illustrate the difficulty of prenatal identification of? 1998 John Wiley & Sons, Ltd.
such a deletion. Amniocentesis was performedbecause of low maternal serum alpha-fetoproteinin the first case and because of maternal anxiety inthe second case. Both results were interpreted asnormal (Wu et al., 1996).
Congenital cardiac malformation, in particularpulmonary stenosis and/or AVC is present in 6/11reported cases. Therefore, search of an 8p23deletion appears to be indicated whenever thisfeature is present. In addition, 4/18 cases had adiaphragmatic hernia (Thorpe-Beeston et al., 1989;Hutchinson et al., 1992; Howe et al., 1996; thepresent case). Other malformations, such as cleftpalate (1/14), kidney anomalies (polycystic 1/14;horseshoe 1/14; hydronephrosis 1/14), agenesis ofcorpus callosum (1/14), and polydactyly (1/14), canalso be present.
Diaphragmatic hernia (CHD) is a commoncongenital malformation (David and Illingworth,1976). It occurs in 0·45 per 1000 total birthsand appears to be aetiologically, as well asanatomically, heterogeneous (Philip et al., 1991).For example, CHD is associated with chromosomeanomalies in 3–40 per cent of the cases accordingto different studies. The vast majority of theseanomalies are changes in chromosome number,especially trisomy 18, but more recent studiesreport a higher incidence of structural chromo-some abnormalities, such as tetrasomy 12p or 4p,and 8p deletions (David and Illingworth, 1976;Thorpe-Beeston et al., 1989; Scott Adzick et al.,1989; Philip et al., 1991; Kalousek et al., 1992;Bollman et al., 1995; Dommergues et al., 1996;Howe et al., 1996).
AVC results from failure of the normal fusionprocess of the superior and inferior endocardialcushions. It occurs in 0·36 per 1000 total births,and make up two per cent of cases of congenitalheart disease. Down syndrome accounts for 60 percent of the cases of AVC (Romero et al., 1988).Two reports (Marino et al., 1992; Diligio et al.,1993) already showed the high prevalence of AVCin del(8p) syndrome and suggested a non-randomassociation of the two conditions. Interestingly,with our report, 3/11 cases with del(8)(p23.1)(Table I) and 5/13 cases with del(8)(p21) or (p22)are associated with AVC (Reiss et al., 1979;Dobyns et al., 1985; Brocker-Vriends et al., 1986;Marino et al., 1992; Digilio et al., 1993).
Comparison of the clinical features of 17patients with del(8)(p23) (Table I) and those withdel(8)(p21) or (p22) deletions shows that moreproximal deletions appear to be associated with
Prenat. Diagn. 18: 1055–1060 (1998)
Facialmorphism
Mentalretardation
Heartdefect
Associatedsigns
Parentalkaryotypes
+ Severe ? Cleft palatePolykystic kidney
N
Mild Moderate ? Seizures N+ Moderate — — N
Mild Mild AVC, PS Diaphragmatichernia
N
Mild Moderate — Strabism NMild Mild Complex Partial ACC N
Mild Mild Complex HydronephrosisStrabism
N
Mild Mild — PolydactylyHorseshoe kidneys
Father N
+ ? AVC, PS — N— Moderate ? External
hydrocephalusPaternaldeletion
— Mild ? — Paternaldeletion
— — ? — ?Mild Mild PS, ASD VCF N
— Mild — Bifid uvula N
Mild Mild ASD, PS MicrocephalySeizures
Mothernormal
+ ? Complex Puffy feet ?+ ? AVC, ASD — N
13/17 13/14 8/12(3 AVC)
12/17 11/13de novo
ternal; PS: pulmonary stenosis; VCF: velo-cardiofacial syndrome; W:
?1998
JohnW
iley&
Sons,L
td.P
renat.D
iagn.18:
1055–1060(1998)
Table I—Features of patients with del(8)(p23.1)
Source
Parentalage (P/M)in years
Antenatalultrasound
Weightat birth
(kg) SexExamination
at birthAge at
diagnosis dys
Fagan et al. (1988) ? — 3·2 F MCA 14 years
Fryns et al. (1989) 24/24 — 3·1 M — 9 yearsBlennow and
Brondun-Nielsen (1990)28/30 — ? F Strabism 13 years
Pecile (1990) 39/31 AVC 2·7 F MCA 1 year
Hutchinson et al. (1982) 1 27/27 — 3 F — 8 yearsHutchinson et al. (1992) 2 25/37 Mild
hydrocephaly3 M Nursing
difficulties4 years
Hutchinson et al. (1992) 3 25/27 — 2·2 F Nursingdifficulties
4 years
Hutchinson et al. (1992) 4 32/32 — 2·6 M Oesophagealatresia
2·7 years
Hutchinson et al. (1992) 5 32/34 — 3·2 ? MCA BirthPettenati et al. (1992) 1 ? — ? M — 7 years
Pettenati et al. (1992) 2 ? — ? F — 11 years
Pettenati et al. (1992) 3 ? — ? M — AdultDevriendt et al. (1995) ? — 2·3 F — 8 yearsWu et al. (1996) 1 ?/31 Normal
karyotype3·7 M — 4 years
Wu et al. (1996) 2 ?/30 Normalkaryotype
2·3 F Cardiopathy Birth
Wu et al. (1996) 3 ?/23 ? 2·4 F MCA BirthPresent case 30/31 Diaphragmatic
hernia? F ? Prenatal
(22 w)Total 29/30 3/16 2·9 6M/10F 8
Abnormal/167 years
ACC: agenesis of corpus callosum; MCA: multiple congenital abnormality; N: normal; P/M: paternal/maweeks of gestation.

1059 8p
more severe manifestations, suggesting the pres-ence of a relationship between the size of thedeletion and the importance of the clinical signs(Plomp et al., 1995). For example, Pettenati et al.(1992) described a seven-year-old child referred fora chromosome study because of moderate mentalretardation. Interestingly, the deletion was alsoobserved in the father and the 11-year-old sisterwho were clinically normal and mildly retarded,respectively, and whole chromosome painting witha chromosome 8 specific DNA library was used toexclude the presence of a familial chromosometranslocation.
The mechanism of 8p deletions has recently beenproposed (Floridia et al., 1996). Briefly, abnormalpairing of chromosomes 8 at maternal meiosis Ifollowed by abnormal recombination betweenmispaired copies of repeated and inverselyorientated sequences could generate a dicentricchromosome. Then, the dicentric chromosomebreaks at anaphase I, generating, respectively, an8p duplication and an 8p deletion in each daughtercell. Thus, the size of the deletion (or the dupli-cation) is dependent on the site of the breakpointon the dicentric chromosome. Apparently, this siteis not related to the presence of a fragile site. Also,the broken end of the deleted chromosome couldheal by the addition of telomeric repeats (Wilkieet al., 1990).
In conclusion, we describe a case of deletion ofthe short arm of chromosome 8 ascertained pre-natally through the discovery of a diaphragmatichernia on ultrasound. Following counselling, thepregnancy was terminated and a fetal autopsyperformed, which revealed in addition the presenceof an atrio-ventricular canal. We conclude that adeletion 8p23 should be excluded whenever a dia-phragmatic hernia and/or an atrio-ventricularcanal is detected on ultrasound.
REFERENCES
Blennow, E., Brondum-Nielsen, K. (1992). Partialmonosomy 8p with minimal dysmorphic signs, J.Med. Genet., 27, 327–329.
Bollmann, R., Kalache, K., Mau, H., Chaoui, R.,Tennstedt, C. (1995). Associated malformations andchromosomal defects in congenital diaphragmatichernia, Fetal Diagn. Ther., 10, 52–59.
Brocker-Vriends, A.H.J.T., Mooij, P.D., Van Bel, F.,Beverstocks, G.C., Van de kamp, J.J.P. (1986). Mono-somy 8p: an easily overlooked syndrome, J. Med.
Genet., 23, 153–154.? 1998 John Wiley & Sons, Ltd.
David, T.J., Illingworth, C.A. (1976). Diaphragmatichernia in the south-west of England, J. Med. Genet.,13, 253–262.
Devriendt, K., De Mars, K., De Cock, P., Gewillig, M.,Fryns, J.P. (1995). Terminal deletion in chromosomeregion 8p23.1–8pter in a child with features of velo-cardio-facial syndrome, Ann. Genet., 38, 228–230.
Digilio, M.C., Giannotti, A., Marino, B., Dallapiccola,B. (1993). Atrioventricular canal and 8p- syndrome,Am. J. Med. Genet., 47, 437–438.
Dobyns, W.B., Dewald, G.W., Carlson, R.O., Mair,D.D., Michels, V.V. (1985). Deficiency of chromo-some 8p21.1]8pter: case report and review of theliterature, Am. J. Med. Genet., 22, 125–134.
Dommergues, M., Louis-Sylvestre, C., Mandelbrot, L.,Oury, J.F., Herlicoviez, M., Body, G., Gamerre, M.,Sarramon, M.F., Favre, R., Dumez, Y. (1996).Hernies de la coupole diaphragmatique: valeurpronostique de l’echographie prenatale, MedecineFoetale et Echographie en Gynecologie, 27, 30–34.
Fagan, K., Wilkinson, I., Allen, M., Brownlea, S.(1988). The coagulation factor is located on 8p23.1,Hum. Genet., 79, 365–367.
Floridia, G., Piantanida, M., Minelli, A., Dellavecchia,C., Bonaglia, C., Rossi, E., Gimelli, G., Croci, G.,Franchi, F., Gilgenkrantz, S., Grammatico, P.,Dalpra, L., Wood, S., Danesino, C., Zuffardi, O.(1996). The same molecular mechanism at thematernal meiosis I produces mono- and dicentric 8pduplications, Am. J. Hum. Genet., 58, 785–796.
Fryns, J.P., Kleczkowska, A., Vogels, A., Van denBerghe, H. (1989). Normal phenotype and slightmental retardation in de novo distal 8p deletion(8pter]8p23.1), Ann. Genet., 32, 171–173.
Howe, T., Kilby, D., Sirry, H., Barker, G.M., Roberts,E., Davidson, E.V., McHugo, J., Whittle, M.J. (1996).Structural chromosome anomalies in congenitaldiaphragmatic hernia, Prenat. Diagn., 16, 1003–1009.
Hutchinson, R., Wilson, M., Voullaire, L. (1992). Distal8p deletion (8p23.1]8pter): a commun deletion?,J. Med. Genet., 29, 407–411.
Marino, B., Reale, A., Giannoti, A., Digilio, M.C.,Dallapiccola, B. (1992). Nonrandom association ofatrioventricular canal and del (8p) syndrome, Am. J.Med. Genet., 42, 424–427.
Pecile, V., Petroni, N.G., Fertz, N.C., Filippi, G. (1990).Deficiency of distal 8p-: report of two cases andreview of the literature, Clin. Genet., 37, 271–278.
Pettenati, M.J., Rao, N., Johnson, C., Hayworth, R.,Crandall, K., Huff, O., Thomas, I.T. (1992).Molecular cytogenetic analysis of a familial 8p23.1deletion associated with minimal dysmorphic features,seizures, and mild mental retardation, Hum. Genet.,89, 602–606.
Philip, N., Gambarelli, D., Guys, J.M., Camboulives, J.,Ayme, S. (1991). Epidemiological study of congeni-tal diaphragmatic defects with special reference to
aetiology, Eur. J. Pediatr., 150, 726–729.Prenat. Diagn. 18: 1055–1060 (1998)

1060 . .
Plomp, A.S., Schrander-Stumpel, C.T.R.M., Engelen,J.J.M., Sijstermans, J.M.J., Loneus, W.H., Fryns, J.P.(1995). Interstitial deletion of the short arm ofchromosome 8: report of a patient and review of theliterature, Genet. Counsel., 6, 55–60.
Reiss, J.A., Brenes, P.M., Chamberlin, J., Magenis,R.E., Lovrien, E.W. (1979). The 8p- syndrome, Hum.Genet., 47, 135–140.
Romero, R., Pilu, G., Jeanty, P., Caidini, A., Hobbins,J.C. (1988). The heart. In: Romero, R., Pilu, G.,Jeanty, P., Caidini, A., Hobbins, J.C. (Eds). PrenatalDiagnosis of Congenital Anomalies, Appleton andLange, 144–145.
Scott Adzick, N., Vacanti, J.P., Lillehei, C.W., PearlO’Rourke, P., Crone, R.K., Wilson, J.M. (1989).Fetal diaphragmatic hernia: ultrasound diagnosis andclinical outcome in 38 cases, J. Pediatr. Surg., 24,654–658.
? 1998 John Wiley & Sons, Ltd.
Thorpe-Beeston, J.G., Gosden, C.M., Nicolaides, K.H.(1989). Prenatal diaphragmatic hernia: associatedmalformations and chromosomal defects, Fetal Ther.,4, 21–26.
Wilkie, A.O.M., Lamb, J., Harris, P.C., Finney, R.D.,Higgs, D.R. (1990). A truncated human chromosome16 associated with á thalassaemia is stabilized byaddition of telomeric repeat (TTAGGG)n, Nature,346, 868–871.
Wu, B.L., Schneider, G.H., Sabatino, D.E., Bozovic,L.Z., Cao, B., Korf, B.R. (1996). Distal 8p deletion(8)(p23.1): an easily missed chromosomal abnormalitythat may be associated with congenital heart defectand mental retardation, Am. J. Med. Genet., 62,77–83.
Prenat. Diagn. 18: 1055–1060 (1998)