![Advances in Porous Organic Polymers for Efficient Water Capture · 2019-10-31 · Advances in Porous Organic Polymers for Efficient Water Capture Yearin [a]Byun+, ... Summary of textural](https://static.fdocuments.in/doc/165x107/5f49bd1f748e9e12703af3d1/advances-in-porous-organic-polymers-for-efficient-water-capture-2019-10-31-advances.jpg)
Porous aluminum-based DUT metal-organic frameworks for the ... · Metal−organic framework (MOF)...
10
Contents lists available at ScienceDirect Catalysis Today journal homepage: www.elsevier.com/locate/cattod Porous aluminum-based DUT metal-organic frameworks for the transformation of CO 2 into cyclic carbonates: A computationally supported study Jintu Francis Kurisingal a , Yixin Li a , Yryskul Sagynbayeva a , Ramesh Kumar Chitumalla b , Srimai Vuppala b , Yadagiri Rachuri a , Yunjang Gu a , Joonkyung Jang b , Dae-Won Park a, * a Division of Chemical and Biomolecular Engineering, Pusan National University, Busan, 46241, Republic of Korea b Department of Nanoenergy Engineering, Pusan National University, Busan, 46241, Republic of Korea ARTICLE INFO Keywords: DUT CO 2 cyclic carbonates DFT ABSTRACT Two aluminum-based DUT (Dresden University of Technology) metal organic frameworks were synthesized, and their structural and chemical properties were exploited for the cycloaddition reaction of CO 2 and epoxide under solvent-free condition. Both the catalysts have the same supramolecular architecture but different ligands. DUT- 5 (Al(OH)(bpdc), bpdc = 4,4′-biphenyldicarboxylate) has a higher number of acidic and basic sites and larger BET surface area and specific pore volume compared with DUT-4 (Al(OH)(ndc), ndc = 2,6-naphthalenedi- carboxylate). The highly porous DUT-5 exhibited better catalytic conversion of epichlorohydrin (ECH) than DUT-4, with > 99 % selectivity in the presence of tetrabutylammonium bromide (TBAB). Short linkered MIL-53 (MIL = Materials Institute Lavoisier) (Al(OH)(bdc), bdc = 1,4-benzenedicarboxylate) exhibited lower ECH conversion compared with DUT-4 and DUT-5, even though all the catalysts possessed the same crystal structure. DUT catalyst could be recycled at least five times without any noticeable loss in the ECH conversion. In addition, the mechanism for the DUT-5/TBAB mediated cycloaddition of CO 2 to ECH, affording the five‐membered ECH carbonate, has been investigated in detail using the density functional theory. The energy barrier for the ECH ring opening in the presence of DUT-5/TBAB (15.14 kcal/mol) is significantly lower than those of un-catalyzed (61.96 kcal/mol) and TBAB-catalyzed (39.60 kcal/mol) reactions, clearly showing the vital role of the Al 3+ /Br − bifunctional catalyst system. 1. Introduction Over the past few decades, the rapid increase in the concentration of greenhouse gases, especially carbon dioxide (CO 2 ), in the earth’s at- mosphere is of great concern for the scientific community [1,2]. This global issue should be addressed, and immediate action must be taken to control the atmospheric CO 2 level. CO 2 is not only a typically harmful gas but also an attractive C1 feed stock, because it is non-toxic, abundant, and easily available [3–5]. Various valuable chemicals, such as urea, formic acid, cyclic carbonates, methanol, polycarbonates, and sodium 2-hydroxybenzoate, are commercially synthesized from CO 2 [6–11]. The synthesis of cyclic carbonates through epoxide addition is generally accepted as a green and 100 % atom economic reaction among the CO 2 transformation reactions [12–18]. The coupling of CO 2 with epoxide is attractive because of the wide application of cyclic carbonates as aprotic solvents, electrolytes of batteries, pharmaceutical intermediates, and polymer precursors [19–23]. To date, many cata- lysts have been reported for the cycloaddition reactions, but their en- vironmental benignity and economic performance still face challenges. Metal − organic framework (MOF) is a class of tailorable crystalline porous organic-inorganic material and have attracted significant re- search interest over the last few decades owing to their characteristic features such as high pore volume, tunable porosity, low framework density, and large surface area [24–32]. The connectivity of the organic linkers to the metal clusters plays a crucial role in determining the structural topology of the MOFs. The pore size of MOFs can be easily tuned by varying the length of the linkers. Due to these varied prop- erties, MOFs have potential applications in the field of gas storage, separation and purification, catalysis, light harvesting, chemical sen- sing, and drug delivery [33–40]. Owing to their high CO 2 adsorption capacity, large surface areas, and suitable acidic and basic sites, MOFs possess an advantage in the CO 2 cycloaddition reaction with epoxides. https://doi.org/10.1016/j.cattod.2019.12.038 Received 30 June 2019; Received in revised form 13 November 2019; Accepted 26 December 2019 ⁎ Corresponding author. E-mail address: [email protected] (D.-W. Park). Catalysis Today 352 (2020) 227–236 Available online 31 December 2019 0920-5861/ © 2020 Elsevier B.V. All rights reserved. T
Transcript of Porous aluminum-based DUT metal-organic frameworks for the ... · Metal−organic framework (MOF)...

Contents lists available at ScienceDirect
Catalysis Today
journal homepage: www.elsevier.com/locate/cattod
Porous aluminum-based DUT metal-organic frameworks for thetransformation of CO2 into cyclic carbonates: A computationally supportedstudy
Jintu Francis Kurisingala, Yixin Lia, Yryskul Sagynbayevaa, Ramesh Kumar Chitumallab,Srimai Vuppalab, Yadagiri Rachuria, Yunjang Gua, Joonkyung Jangb, Dae-Won Parka,*a Division of Chemical and Biomolecular Engineering, Pusan National University, Busan, 46241, Republic of KoreabDepartment of Nanoenergy Engineering, Pusan National University, Busan, 46241, Republic of Korea
A R T I C L E I N F O
Keywords:DUTCO2cyclic carbonatesDFT
A B S T R A C T
Two aluminum-based DUT (Dresden University of Technology) metal organic frameworks were synthesized, andtheir structural and chemical properties were exploited for the cycloaddition reaction of CO2 and epoxide undersolvent-free condition. Both the catalysts have the same supramolecular architecture but different ligands. DUT-5 (Al(OH)(bpdc), bpdc= 4,4′-biphenyldicarboxylate) has a higher number of acidic and basic sites and largerBET surface area and specific pore volume compared with DUT-4 (Al(OH)(ndc), ndc=2,6-naphthalenedi-carboxylate). The highly porous DUT-5 exhibited better catalytic conversion of epichlorohydrin (ECH) thanDUT-4, with> 99 % selectivity in the presence of tetrabutylammonium bromide (TBAB). Short linkered MIL-53(MIL=Materials Institute Lavoisier) (Al(OH)(bdc), bdc= 1,4-benzenedicarboxylate) exhibited lower ECHconversion compared with DUT-4 and DUT-5, even though all the catalysts possessed the same crystal structure.DUT catalyst could be recycled at least five times without any noticeable loss in the ECH conversion. In addition,the mechanism for the DUT-5/TBAB mediated cycloaddition of CO2 to ECH, affording the five‐membered ECHcarbonate, has been investigated in detail using the density functional theory. The energy barrier for the ECHring opening in the presence of DUT-5/TBAB (15.14 kcal/mol) is significantly lower than those of un-catalyzed(61.96 kcal/mol) and TBAB-catalyzed (39.60 kcal/mol) reactions, clearly showing the vital role of the Al3+/Br−
bifunctional catalyst system.
1. Introduction
Over the past few decades, the rapid increase in the concentration ofgreenhouse gases, especially carbon dioxide (CO2), in the earth’s at-mosphere is of great concern for the scientific community [1,2]. Thisglobal issue should be addressed, and immediate action must be takento control the atmospheric CO2 level. CO2 is not only a typicallyharmful gas but also an attractive C1 feed stock, because it is non-toxic,abundant, and easily available [3–5]. Various valuable chemicals, suchas urea, formic acid, cyclic carbonates, methanol, polycarbonates, andsodium 2-hydroxybenzoate, are commercially synthesized from CO2
[6–11]. The synthesis of cyclic carbonates through epoxide addition isgenerally accepted as a green and 100 % atom economic reactionamong the CO2 transformation reactions [12–18]. The coupling of CO2
with epoxide is attractive because of the wide application of cycliccarbonates as aprotic solvents, electrolytes of batteries, pharmaceutical
intermediates, and polymer precursors [19–23]. To date, many cata-lysts have been reported for the cycloaddition reactions, but their en-vironmental benignity and economic performance still face challenges.
Metal− organic framework (MOF) is a class of tailorable crystallineporous organic-inorganic material and have attracted significant re-search interest over the last few decades owing to their characteristicfeatures such as high pore volume, tunable porosity, low frameworkdensity, and large surface area [24–32]. The connectivity of the organiclinkers to the metal clusters plays a crucial role in determining thestructural topology of the MOFs. The pore size of MOFs can be easilytuned by varying the length of the linkers. Due to these varied prop-erties, MOFs have potential applications in the field of gas storage,separation and purification, catalysis, light harvesting, chemical sen-sing, and drug delivery [33–40]. Owing to their high CO2 adsorptioncapacity, large surface areas, and suitable acidic and basic sites, MOFspossess an advantage in the CO2 cycloaddition reaction with epoxides.
https://doi.org/10.1016/j.cattod.2019.12.038Received 30 June 2019; Received in revised form 13 November 2019; Accepted 26 December 2019
⁎ Corresponding author.E-mail address: [email protected] (D.-W. Park).
Catalysis Today 352 (2020) 227–236
Available online 31 December 20190920-5861/ © 2020 Elsevier B.V. All rights reserved.
T

Recently, MOFs received great attention in the field of cyclic carbonatesynthesis, as compared with other catalysts, because of their highthermal and chemical stabilities and heterogeneous nature [41–46].
In this work, we synthesized two aluminum-based DUT catalystsand investigated their catalytic potential for CO2–epoxide cycloaddition(Scheme 1). A limited number of aluminum-based MOFs have beenreported for the synthesis of cyclic carbonates. Aluminum-based MOFsare especially attractive because of their superior chemical and thermalstability, octahedral coordination, flexible nature, and structural tun-ability. Till now, only a few types of linker units (mainly bdc units) havebeen reported for the synthesis of aluminum-based MOFs. Herein, weused 2,6-naphthalenedicarboxylate (ndc) and 4,4′-biphenyldicarbox-ylate (bpdc) to prepare aluminum-based DUT-4 [C48H28O20Al4] andDUT-5 [C56H36O20Al4] catalysts, respectively [47]. To study the influ-ence of the length of the ligand, we synthesized aluminum-based MIL-53 [Al(OH)(bdc)(H2bdc)0.7], which contains BDC (benzene-1,4-di-carboxylic acid) as the linker unit [48]. The mechanism of the cy-cloaddition reaction catalyzed by DUT catalyst was proposed using thedensity functional theory (DFT). The cycloaddition reaction was in-vestigated using theoretical calculations under three conditions, viz.,un-catalyzed reaction, tetrabutylammonium bromide (TBAB)-catalyzedreaction, and DUT/TBAB catalyzed reaction. This was done in order tostudy the key role of the Al3+/TBAB synergism in the CO2–epoxidecycloaddition reaction.
2. Results and discussion
2.1. Characterization of catalysts
In DUT-4, the aluminum atom is connected to six oxygen atoms in adistorted octahedral geometry and has an orthorhombic crystal struc-ture with the space group Pnna. The axial positions of the octahedra areoccupied by hydroxyl groups forming –Al−OH– chains. The ndc linkersbridge the –Al−OH– chains to form a three dimensional framework.When a longer rigid ligand (bpdc) was used instead of the ndc ligand forDUT-5, the same supramolecular architecture with the space groupImma was obtained with slightly larger pores (Scheme 2). The co-ordination environments generated by the ndc and bpdc ligands aroundthe Al3+ centers in DUT-4 and DUT-5 and their packing diagrams areshown in Scheme 2. Al-O(ligand) bond distance in DUT-4 and DUT-5 are1.78 and 1.80 Å, respectively, while the distance between the two ad-jacent aluminum atoms (connected through O atom) in DUT-4 andDUT-5 are 3.4 and 3.3 Å, respectively.
Initially, the textural features of the DUT catalysts were investigatedby field emission scanning electron microscopy (FE-SEM). Fully growncrystals of DUT-4 and DUT-5 were observed in the FE-SEM images(Fig. 1(a) and (c)). The crystallinity and structural integrity of bothDUT-4 and DUT-5 were characterized by powder X-ray diffraction(PXRD) (Fig. 1(b) and (d)). The positions and relative intensities ofmain peaks in the PXRD patterns obtained experimentally was inagreement with the simulated patterns in the crystallographic
information file, thereby confirming the successful syntheses of thecatalysts. The similarities in composition and chemical integrity of theDUT MOFs were further studied by Fourier transform infrared (FTIR)spectroscopy (Fig. 2(a)). The peaks observed at 3452 cm−1 correspondsto the OeH stretching vibration. The bands in the range1608–1570 cm−1 represents the asymmetric stretching vibrations of thecarboxylate group (νas(CeO)), while those at 1430 and 1364 cm−1 canbe ascribed to the symmetric vibrations of the carboxylic group(νs(CeO)). The band at 478 cm−1 corresponds to the stretching vibra-tions of the Al–O bonds.
The thermal stability and robustness of the DUT catalysts (driedcatalysts) were estimated using thermogravimetric analyses (TGA)(Fig. 2(b)). Both the catalysts were thermally stable up to ∼500 °C, butthe DUT framework started to collapse beyond this temperature. Thethermal stability of these catalysts (∼500 °C) is much higher than thoseof other MOFs (usually ∼300 °C), and hence, are expected to be sui-table for the cycloaddition at moderate temperatures (80–100 °C). DUTcatalysts also attract attention for high temperature applications be-cause of their tremendous thermal stability. X-ray photoelectron spec-troscopy data confirmed the oxidation state of aluminum and the co-ordination of the ligands to the aluminum atom (Fig. 3(a) and (b). TheAl 2p3 scan showed the characteristic peak for Al(III) at a binding en-ergy of 74.1 eV (Fig. 3(c) and (d)). In order to determine the elementalcomposition of the DUT catalysts, inductively coupled plasma–opticalemission spectrometry analysis and elemental analysis were performed,and the results are shown in Table S1. The calculated elemental com-positions of both the DUT catalysts almost matched with the observedelemental compositions.
In order to perform the cycloaddition reaction, the knowledge aboutthe acidic and basic strength of the catalyst is vital. Thus, we performedCO2 and NH3 temperature‐programmed desorption analysis of the DUTcatalysts (Fig. S1 and Table S2). The total amounts of CO2 and NH3
desorbed for DUT-5 were higher than DUT-4. The larger pores in DUT-5compared with DUT-4 was advantageous, since larger pores make ea-sier diffusion of the gas molecules to the active sites. The N2 ad-sorption–desorption measurements for the thermally activated DUTcatalysts were carried out at 77 K and revealed reversible type I iso-therms, which are characteristic of microporous materials (Fig. 4(a)).The estimated Brunauer–Emmett–Teller (BET) surface area and porevolume were 968.6m2 g−1 and 0.36 cm3 g−1 for DUT-4 and1697.0 m2 g−1 and 0.63 cm3 g−1 for DUT-5. CO2 sorption analysis forthe catalysts was conducted at 298 K, and a type I behavior was ob-served in the range of 0–850mmHg absolute pressure (Fig. 4(b) andTable S3). The CO2 adsorption–desorption profile of DUT-5 revealedslightly higher adsorption capacity than DUT-4. The CO2 uptake capa-cities of DUT-4 and DUT-5 were 32.62 and 33.06 cm3 g−1, respectively.The excellent thermal stability, good CO2 adsorption capacity, andlarge BET surface area and pore volume make them promising candi-dates in the catalytic transformation of CO2 into cyclic carbonates.
Scheme 1. Schematic presentation of the synthesis of cyclic carbonates from epoxides and CO2.
J.F. Kurisingal, et al. Catalysis Today 352 (2020) 227–236
228

2.2. Cycloaddition reaction of epichlorohydrin with CO2
In order to explore the catalytic potential of the DUT catalysts, theCO2 cycloaddition reaction was carried out using epichlorohydrin(ECH) as the model substrate. The reaction was conducted under sol-vent-free conditions for 6 h at 80 °C and 1.2MPa CO2 pressure(Table 1). A blank reaction was performed in the absence of any
catalyst. No product formation was observed (Table 1, entry 1) in thiscase, indicating the importance of the catalyst in the conversion of CO2
into cyclic carbonates. The homogeneous precursors of the DUT cata-lysts were examined for their individual catalytic potential but did notshow any noticeable ECH conversion (Table 1, entries 2–4). Under theemployed reaction conditions, both DUT-4 and DUT-5 showed ECHconversion of 25 % and 32 %, respectively (Table 1, entries 5 and 6).
Scheme 2. Structural view and pore size details of MIL-53, DUT-4 and DUT-5 catalyst.
Fig. 1. (a) and (c) FE-SEM images and (b) and (d) PXRD patterns of DUT-4 and DUT-5 catalysts.
J.F. Kurisingal, et al. Catalysis Today 352 (2020) 227–236
229

The importance of nucleophiles (especially halide ions) in the initiationof cycloaddition reaction through the synergistic role with the metalcenter is well known. When the reaction was carried out only in thepresence of co-catalyst, TBAB yielded 68 % ECH conversion (Table 1,entry 7). A tremendous increase in the ECH conversion was observedupon combining DUT catalysts and TBAB, which served as a binarycatalyst system under the same reaction conditions. In the presence ofTBAB, DUT-4 showed an ECH conversion of 93 %, with excellent se-lectivity towards ECH carbonate (Table 1, entry 9). The replacement ofthe shorter NDC ligand (9.2 Å) in DUT-4 (Fig. 5) with the longer rigidligand, BPDC (11.4 Å), resulted in DUT-5, which had a better porevolume. The porous structure of DUT-5 compared with DUT-4 offersfacile accessibility of the substrates to its inner active sites, and the
well-defined catalytic environment enables the activation of the ECHwith the Al3+ Lewis acidic centers [49–51]. The combination of TBABand DUT-5 showed the best catalytic activity, giving 99 % yieldand> 99 % selectivity for ECH carbonate (Table 1, entry 10). In orderto study the influence of the length of the ligand, we synthesized alu-minum-based MIL-53, which contained BDC as the linker unit. MIL-53has less pore volume and porosity as compared with DUT catalystsbecause of the shorter ligand (Fig. 5). Both DUT-4 and DUT-5 providedbetter ECH conversion than the short lingered MIL-53 catalyst owing tothe permanent porosity and larger pore volume in the former, whichfacilitated the easy approach of reactant molecules toward the activesites (Table 1, entry 8).
The effects of reaction parameters such as the amount of catalyst,
Fig. 2. (a) FT-IR spectra and (b) TGA curves.
Fig. 3. (a) and (b) XPS survey spectra and (c) and (d) Al 2p spectra of DUT-4 and DUT-5.
J.F. Kurisingal, et al. Catalysis Today 352 (2020) 227–236
230

reaction temperature, reaction time, and CO2 pressure were optimizedusing DUT-5 (Fig. 6). Fig. 6a shows that the ECH conversion increasedlinearly with reaction temperature, suggesting that it has a significantrole in influencing the activation energy of the cycloaddition reaction.The maximum ECH conversion (99 %) and ECH carbonate selectivity(> 99 %) were observed at 80 °C. The dependence of the cycloadditionreaction on reaction time is illustrated in Fig. 6b. A steep increase in theECH conversion was observed with increasing reaction time, andmaximum ECH conversion was attained at 6 h. As expected, ECH con-version improved linearly with an increase in CO2 pressure, reaching 99% ECH conversion at 1.2MPa (Fig. 6c). A stable increase in ECH con-version was observed as the amount of DUT-5 was increased from 0.1 to0.5 mol%, reaching a maximum ECH conversion of 99 % and>99 %selectivity toward ECH carbonate (Fig. 6d).
2.3. Catalyst activity with different epoxides and reusability
The potential of DUT as a heterogeneous catalyst in the cycloaddi-tion reaction of CO2 and epoxide was further evaluated using various
Fig. 4. a) N2 adsorption isotherms at 77 K and b) CO2 adsorption isotherms at 298 K.
Table 1Performance of DUT catalysts in the cycloaddition of CO2 with epichlorohydrin.
Entry Catalyst Conversion (%) Selectivity (%)
1 None – –2 Al(NO3)3.9H2O 5 >993 H2NDC 9 >994 H2BPDC 11 >995 DUT-4 25 >996 DUT-5 32 >997 TBAB 68 >998 MIL-53 /TBAB 84 >999 DUT-4/TBAB 93 >9910 DUT-5 /TBAB 99 >99
Reaction Conditions: Epichlorohydrin (ECH) =25mmol, Catalyst =0.5 mol%,TBAB =0.5mol%, Pressure =1.2MPa CO2, Temperature= 80 °C, Time =6 h,semi batch reaction.
Fig. 5. Structural details of catalyst.
J.F. Kurisingal, et al. Catalysis Today 352 (2020) 227–236
231

epoxide substrates (Fig. 7). An efficient conversion of the terminalaliphatic epoxides such as epichlorohydrin, propylene oxide, and allylglycidyl ether was observed with> 99 % selectivity of the corre-sponding cyclic carbonates. The bulky aromatic epoxides such asstyrene oxide and internal epoxide cyclohexene gave comparativelylower conversions than the aliphatic epoxides, mainly because of thesterically hindered structure [52–54]. 1H nuclear magnetic resonancespectra of the synthesized cyclic carbonates are given in the SupportingInformation. Recyclability was examined to verify the heterogeneous
nature of the DUT-5 catalysts. Heterogeneous nature of the catalysts iscrucial in industrial processes in order to minimize waste streams andpower utilization. DUT-5 was separated easily from the reaction mix-ture by simple centrifugation and could be used for five reaction cyclesin the cycloaddition of ECH and CO2 under the optimized reactionconditions. DUT-5 maintained its ECH conversion and ECH carbonateselectivity in all the recycles (Fig. 8). PXRD pattern, FTIR spectrum,TGA curve and N2 adsorption isotherms of the reused DUT-5 were si-milar to those of the freshly synthesized DUT-5, indicating that thechemical structure and textual properties of the catalyst were well
Fig. 6. Effect of reaction parameters for DUT-5 on the cycloaddition of ECH and CO2: (a) reaction temperature (0.5mol%, 6 h, 1.2MPa PCO2), (b) reaction time(0.5 mol%, 80 °C, 1.2MPa PCO2), (c) pressure (0.5 mol%, 80 °C, 6 h) and (d) catalyst amount (80 °C, 6 h, 1.2MPa PCO2).
Fig. 7. Synthesis of cyclic carbonates from various epoxides. Reaction condi-tions: Epoxide =25mmol, Temperature =80 °C, Time =6 h, Pressure=1.2MPa, TBAB =0.5mol%, Catalyst amount =0.5mol%.
Fig. 8. Catalyst reusability at 80 °C, 6 h, 1.2MPa CO2 pressure, 0.5 mol% cat-alyst/co-catalyst using 25mmol of epichlorohydrin (> 99 % selectivity towardsthe ECH carbonate).
J.F. Kurisingal, et al. Catalysis Today 352 (2020) 227–236
232

preserved even after recycling (Fig. S2–S5).
2.4. Density functional theory study
We have employed a computational study based on density func-tional studies (DFT) for the investigation of the cycloaddition reactionmechanism. The theoretical calculations were performed under threeconditions, viz., un-catalyzed reaction, tetrabutylammonium bromide(TBAB)-catalyzed reaction, and DUT-5/TBAB catalyzed reaction.Similar to previous investigations, [53,55] we used the meta-hybridGGA functional M06 level of theory using the Gaussian 16 program[56]. A combination of “‘double– ξ”’ quality basis set consisting of Hayand Wadt’s effective core potentials (LanL2DZ ECP) [57,58] were ap-plied for the heavy atom Br whereas the basis set 6–31 G(d) was usedfor the rest of atoms Al, H, C, N, O, and Cl. The geometric optimizationwas followed by vibrational frequency analysis, and all the reactants,intermediates (Int), and products were confirmed with no imaginaryfrequencies, whereas the transition states (TS) were confirmed withonly one imaginary frequency. We investigated the reaction mechanismof the un-catalyzed cycloaddition of CO2 with ECH through a single stepsimulation leading to the formation of epichlorohydrin carbonate(ECH). The un-catalyzed cycloaddition reaction has been proceedingthrough a nucleophilic attack from an oxygen atom of CO2 to the leastsubstituted carbon atom of epichlorohydrin. The energy barrier for therate-determining step (RDS) in the uncatalyzed reaction, i.e., epi-chlorohydrin ring opening, was 61.96 kcal/mol (Fig. 9), which is ingood agreement with the earlier reports from Zhang and Bo(55–63 kcal/mol) [59,60].
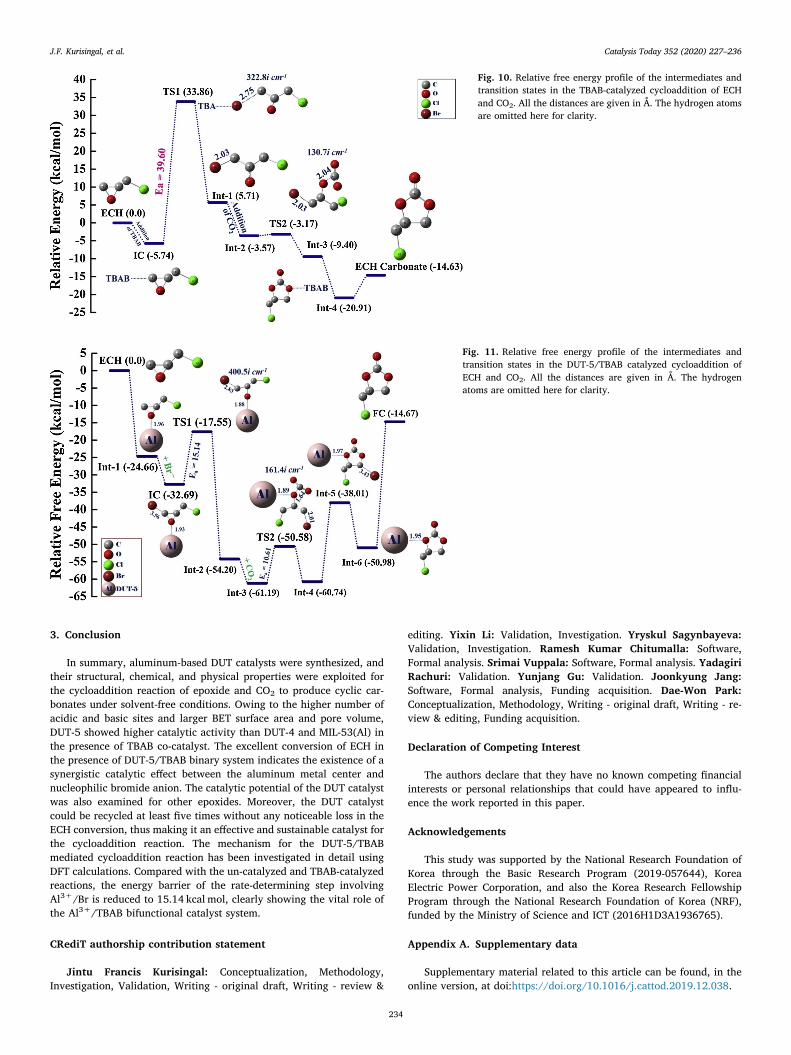
Similar to the uncatalyzed reaction, the cycloaddition reaction ofECH with CO2 catalyzed by the co-catalyst TBAB was carried out andthe energy profile diagram is depicted in Fig. 10. The sum of the relativeenergies of ECH, CO2 and bromide ion was set to 0.00 kcal/mol. Theinitial step is the introduction and interaction of bromide ion fromTBAB to the oxygen atom of epichlorohydrin to form the initial complex(IC) having the energy of –5.74 kcal/mol. In the next step, IC followedby a concerted ring-opening of ECH by the nucleophilic attack of bro-mide ion to the least substituted carbon atom of epichlorohydrin re-sulted in the formation of TS-1. Here, the distance between bromide ionand the beta carbon of ECH (βC) was 2.75 Å and the TS-1 have theenergy of 33.86 kcal/mol. The energy barrier for the rate-determiningstep (RDS) in the TBAB-catalyzed reaction was 39.60 kcal/mol. Theaddition of a CO2 molecule into the reaction medium caused the for-mation of new intermediate (Int-2) with the energy -3.57 kcal/mol. Inthe later step, the negatively charged oxygen atom of the ring openedECH attacked the CO2 to form the second transition state (TS-2) withthe energy of -3.17 kcal/mol. Thereafter, CO2 insertion (Int–3) processtook place in between Al and alkoxide group with a total energy of-9.40 kcal/mol. Upon ring closure (Int–4), the total relative energy
decreased to -20.91 kcal/mol, and the final product, epichlorohydrincarbonate was formed with the detachment of bromide ion and have arelative energy of -14.63 kcal/mol.
In the DUT-5/TBAB catalyzed cycloaddition reaction, we simplifiedthe DUT-5 catalyst structure with 65 atoms (Fig. S6) and used Br− ionto represent the TBAB for computational calculations. Similarly, withprevious simulations, we set the sum of the relative energy of DUT-5,ECH, CO2 and bromide ion as 0.00 kcal/mol. The energy profile dia-gram along with the optimized structures of the DUT-5 catalyzed re-actions are shown in Fig. 11. The studies suggested that the interactionof Al metal atom with the oxygen atom of ECH caused the formation ofInt-1. In Int-1, the Al is at a distance of 1.96 Å from the oxygen atom ofECH and have the total energy of -24.66 kcal/mol. The approach ofbromide ion into the beta carbon of ECH (βC) resulted in a further dropin the relative energy to -32.69 kcal/mol, termed as initial complex(IC). Here, the distance between bromide ion and βC of ECH is 3.56 Å,while the distance between Al and O(ECH) become closer (1.93 Å). In thering-opening step of ECH, bromide ion makes a nucleophilic attacktowards the βC of ECH to form an alkoxide complex (TS-1) with relativeenergy of -17.55 kcal/mol. The TS-1 was characterized by the breakingof the βC(ECH)-O(ECH) and the simultaneous formation of a βC(ECH)-Brbond as shown in the relative imaginary frequencies at 400.5i cm-1. Thebond length of the newly formed bond was 2.83 Å and the distance fromAl to the O(ECH) became closer to 1.88 Å. The ring opening step wasconsidered as the rate-determining step (RDS) in the CO2 cycloadditionreactions [13,52,53,55,59]. Herein, the energy barrier for this crucialstep required the energy of 15.14 kcal/mol. Interestingly, the energybarrier for the ECH ring opening in the presence of DUT-5/TBAB issignificantly lower than those of un-catalyzed (61.96 kcal/mol) andTBAB-catalyzed (39.60 kcal/mol) reactions [55].
In the later step, TS-1 was transformed into an intermediate Int-2 bythe introduction of CO2 with relative energy of –61.19 kcal/mol. TheCO2 reacted with the negatively charged O(ECH) of the Int-2 caused theformation of Int-3. In the subsequent step, another transition state (TS-2) was developed with the formation of new C(CO2)-O(ECH) bond havingthe length of 1.64 Å, with an energy barrier of 10.61 kcal/mol as shownin the relative imaginary frequencies at 161.4i cm−1. TS-2 undergoes anintramolecular ring-closing by the attack of negatively charged O(CO2)
to the βC(ECH) leading to the formation of Int-4 with an energy value of-60.74 kcal/mol. Later, the formation of new C(CO2)-O(ECH) bond and thedetachment of C(ECH)-Br bond caused the Int-5 with the energy of-38.01 kcal/mol. In Int-5, the distance between C(ECH) and Br increasedfrom 2.01–3.43 Å. Finally, the epichlorohydrin carbonate (FC) isformed with relative energy of -14.67 kcal/mol. Thus, these results arein good agreement with the experimental data obtained for the DUT-5/TBAB system and also showed the importance of the binary catalystreaction in the cycloaddition reaction as compared to the un-catalyzedand TBAB catalyzed reactions.
A reasonable reaction mechanism was proposed based on the ex-perimental and theoretical studies (Scheme 3). The coordinatively un-saturated aluminum centers of DUT-5 acts as Lewis acidic sites thatbind to the oxygen atom of the epoxide by weak interactions. The nu-cleophilic anion, Br−, of TBAB attacks the activated carbon atom of theepoxide and results in the ring opening of the epoxide, forming an in-termediate. Then, the anion of the intermediate (O−) attacks thecarbon atom of CO2 to form a carbonate complex (CO2 insertion). Fi-nally, intramolecular cyclization of the carbonate complex and si-multaneous elimination of Br− produce the cyclic carbonate, and theregenerated DUT-5 catalyst can be reused for the next cycloadditionreaction.
A comparison study was conducted with the other reported catalystsfor cycloaddition reaction of CO2 in order to evaluate the catalyticpotential of DUT-5 (Table 2). The results showed that the DUT-5 cat-alyst here stands alongside of or a step ahead of the MOFs reportedearlier in the cycloaddition reaction of epoxide with CO2.Fig. 9. Relative free energy diagram of the un-catalyzed cycloaddition reaction
of ECH and CO2. The distances are given in Å.
J.F. Kurisingal, et al. Catalysis Today 352 (2020) 227–236
233
3. Conclusion
In summary, aluminum-based DUT catalysts were synthesized, andtheir structural, chemical, and physical properties were exploited forthe cycloaddition reaction of epoxide and CO2 to produce cyclic car-bonates under solvent-free conditions. Owing to the higher number ofacidic and basic sites and larger BET surface area and pore volume,DUT-5 showed higher catalytic activity than DUT-4 and MIL-53(Al) inthe presence of TBAB co-catalyst. The excellent conversion of ECH inthe presence of DUT-5/TBAB binary system indicates the existence of asynergistic catalytic effect between the aluminum metal center andnucleophilic bromide anion. The catalytic potential of the DUT catalystwas also examined for other epoxides. Moreover, the DUT catalystcould be recycled at least five times without any noticeable loss in theECH conversion, thus making it an effective and sustainable catalyst forthe cycloaddition reaction. The mechanism for the DUT-5/TBABmediated cycloaddition reaction has been investigated in detail usingDFT calculations. Compared with the un-catalyzed and TBAB-catalyzedreactions, the energy barrier of the rate-determining step involvingAl3+/Br is reduced to 15.14 kcal mol, clearly showing the vital role ofthe Al3+/TBAB bifunctional catalyst system.
CRediT authorship contribution statement
Jintu Francis Kurisingal: Conceptualization, Methodology,Investigation, Validation, Writing - original draft, Writing - review &
editing. Yixin Li: Validation, Investigation. Yryskul Sagynbayeva:Validation, Investigation. Ramesh Kumar Chitumalla: Software,Formal analysis. Srimai Vuppala: Software, Formal analysis. YadagiriRachuri: Validation. Yunjang Gu: Validation. Joonkyung Jang:Software, Formal analysis, Funding acquisition. Dae-Won Park:Conceptualization, Methodology, Writing - original draft, Writing - re-view & editing, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financialinterests or personal relationships that could have appeared to influ-ence the work reported in this paper.
Acknowledgements
This study was supported by the National Research Foundation ofKorea through the Basic Research Program (2019-057644), KoreaElectric Power Corporation, and also the Korea Research FellowshipProgram through the National Research Foundation of Korea (NRF),funded by the Ministry of Science and ICT (2016H1D3A1936765).
Appendix A. Supplementary data
Supplementary material related to this article can be found, in theonline version, at doi:https://doi.org/10.1016/j.cattod.2019.12.038.
Fig. 10. Relative free energy profile of the intermediates andtransition states in the TBAB-catalyzed cycloaddition of ECHand CO2. All the distances are given in Å. The hydrogen atomsare omitted here for clarity.
Fig. 11. Relative free energy profile of the intermediates andtransition states in the DUT-5/TBAB catalyzed cycloaddition ofECH and CO2. All the distances are given in Å. The hydrogenatoms are omitted here for clarity.
J.F. Kurisingal, et al. Catalysis Today 352 (2020) 227–236
234

References
[1] M. Aresta, A. Dibenedetto, A. Angelini, Chem. Rev. 114 (2014) 1709–1742.[2] J.D. Shakun, P.U. Clark, F. He, S.A. Marcott, A.C. Mix, Z. Liu, B. Otto-Bliesner,
A. Schmittner, E. Bard, Nature 484 (2012) 49–54.[3] J. Vaitla, Y. Guttormsen, J.K. Mannisto, A. Nova, T. Repo, A. Bayer, K.H. Hopmann,
ACS Catal. 7 (2017) 7231–7244.[4] Y. Zhao, X. Liu, K.X. Yao, L. Zhao, Y. Han, Chem. Mater. 24 (2012) 4725–4734.[5] A. Kumar, D.G. Madden, M. Lusi, K.-J. Chen, E.A. Daniels, T. Curtin, J.J. Perry IV,
M.J. Zaworotko, Angew. Chem., Int. Ed. 53 (2014) 7527–7533.[6] Z. Chen, N. Hadjichristidis, X. Feng, Y. Gnanou, Macromolecules 50 (2017)
2320–2328.[7] H. He, J.A. Perman, G. Zhu, S. Ma, Small 12 (2016) 6309–6324.[8] C.-L. Chiang, K.-S. Lin, H.-W. Chuang, J. Cleaner Prod. 172 (2018) 1957–1977.[9] J.F. Kurisingal, Y. Rachuri, Y. Gu, Y. Choe, D.-W. Park, Inorg. Chem. Front. (2019),
https://doi.org/10.1039/C9QI01163C.[10] S. Ravi, P. Puthiaraj, W.-S. Ahn, ACS Sustainable Chem. Eng. 6 (2018) 9324–9332.[11] G.-H. Kim, J.F. Kurisingal, Y. Gu, M.-H. Lee, Y. Choe, D.-W. Park, J. Nanosci.
Nanotechnol. 20 (2020) 752–759.[12] Z. Zhang, F. Fan, H. Xing, Q. Yang, Z. Bao, Q. Ren, ACS Sustainable Chem. Eng. 5
(2017) 2841–2846.[13] J.F. Kurisingal, Y. Rachuri, R.S. Pillai, Y. Gu, Y. Choe, D.-W. Park, ChemSusChem 12
(2019) 1033–1042.
[14] Y. Rachuri, J.F. Kurisingal, R.K. Chitumalla, S. Vuppala, Y. Gu, J. Jang, Y. Choe,E. Suresh, D.-W. Park, Inorg. Chem. 58 (2019) 11389–11403.
[15] L. Longwitz, J. Steinbauer, A. Spannenberg, T. Werner, ACS Catal. 8 (2018)665–672.
[16] J.F. Kurisingal, R. Babu, S.-H. Kim, Y.X. Li, J.-S. Chang, S.J. Cho, D.-W. Park, Catal.Sci. Technol. 8 (2018) 591–600.
[17] G. Bresciani, M. Bortoluzzi, F. Marchetti, G. Pampaloni, ChemSusChem 11 (2018)2737–2743.
[18] R. Babu, J.F. Kurisingal, J.-S. Chang, D.-W. Park, ChemSusChem 11 (2018)924–932.
[19] B. Scrosati, J. Hassoun, Y.-K. Sun, Energy Environ. Sci. 4 (2011) 3287–3295.[20] V. Besse, F. Camara, C. Voirin, R. Auvergne, S. Caillol, B. Boutevin, Polym. Chem. 4
(2013) 4545–4561.[21] B. Schäffner, F. Schäffner, S.P. Verevkin, A. Bçrner, Chem.Rev. 110 (2010)
4554–4581.[22] M. Bahr, R. Mulhaupt, GreenChem. 14 (2012) 483–489.[23] Nvd. Assen, A. Sternberg, A. Kätelhön, A. Bardow, Faraday Discuss. 183 (2015)
291–307.[24] H.-J. Kim, J.F. Kurisingal, D.-W. Park, Reac. Kinet. Mech. Catal. 124 (2018)
335–346.[25] A.H. Valekar, K.-H. Cho, S.K. Chitale, D.-Y. Hong, G.-Y. Cha, U.-H. Lee,
D.W. Hwang, C. Serre, J.-S. Chang, Y.K. Hwang, Green Chem. 18 (2016) 4542.[26] Y. Rachuri, B. Parmar, K.K. Bisht, E. Suresh, Dalton Trans. 47 (2018) 898–908.[27] G. Liu, A. Cadiau, Y. Liu, K. Adil, V. Chernikova, I.-D. Carja, Y. Belmabkhout,
Scheme 3. Plausible reaction mechanism of CO2–epoxide cycloaddition reaction catalyzed by DUT-5 catalyst.
Table 2Comparison of the catalytic potential of the DUT-5 catalyst with previously reported MOFs.
No Catalysts Epoxide T ( ˚C) P CO2 (MPa) T (h) Catalyst amount (mol%) Yield (%) Ref.
MOF catalysts1 ZnMOF-1-NH2 ECH 80 0.8 18 1.0 89 [60]2 {[Zn(CHDC)(L)].H2O}n ECH 80 1.0 18 1.8 91 [61]3 {[Cd(CHDC)(L)].H2O}n ECH 80 1.0 8 1.8 89 [61]4 DUT-52(Zr) ECH 80 1.2 6 0.5 96 [62]5 UiO-66/Cu-BTC ECH 60 1.2 8 0.16 91 [63]6 DUT-5 ECH 80 1.2 6 0.5 99 This work7 (Me2NH2) [In(SBA)(BDC NH2)] PO 80 2 24 0.15 92 [64]8 (NH4)3[In3Cl2(BPDC)5] PO 80 2 24 0.15 95 [64]9 DUT-5 PO 80 1.2 6 0.5 98 This work
J.F. Kurisingal, et al. Catalysis Today 352 (2020) 227–236
235

M. Karunakaran, O. Shekhah, C. Zhang, A.K. Itta, S. Yi, M. Eddaoudi, W.J. Koros,Angew. Chem. Int. Ed. 57 (2018) 14811–14816.
[28] G.-G. Choi, J.F. Kurisingal, Y.G. Chung, D.-W. Park, Korean J. Chem. Eng. 35 (2018)1373–1379.
[29] H. Furukawa, K.E. Cordova, M. O’Keeffe, O.M. Yaghi, Science 341 (2013) 1230444.[30] Y. Rachuri, B. Parmar, K.K. Bisht, E. Suresh, Dalton Trans. 46 (2017) 3623–3630.[31] J. Zhu, P.M. Usov, W. Xu, P.J. Celis-Salazar, S. Lin, M.C. Kessinger, C. Landaverde-
Alvarado, M. Cai, A.M. May, C. Slebodnick, D. Zhu, S.D. Senanayake, A.J. Morris, J.Am. Chem. Soc. 140 (2018) 993–1003.
[32] D.-H. Nam, O.S. Bushuyev, J. Li, P. De Luna, A. Seifitokaldani, C.-T. Dinh,F.P. García de Arquer, Y. Wang, Z. Liang, A.H. Proppe, C.S. Tan, P. Todorović,O. Shekhah, C.M. Gabardo, J.W. Jo, J. Choi, M.-J. Choi, S.-W. Baek, J. Kim,D. Sinton, S.O. Kelley, M. Eddaoudi, E.H. Sargent, J. Am. Chem. Soc. 140 (2018)11378–11386.
[33] Y. Rachuri, B. Parmar, E. Suresh, Cryst. Growth Des. 18 (2018) 3062–3072.[34] A.C. Kathalikkattil, K.K. Bisht, N. Aliaga-Alcalde, E. Suresh, Cryst. Growth Des. 11
(2011) 1631–1641.[35] S. Senthilkumar, R. Goswami, N.L. Obasi, S. Neogi, ACS Sustainable Chem. Eng. 5
(2017) 11307–11315.[36] M. Ding, H.-L. Jiang, ACS Catal. 8 (2018) 3194–3201.[37] Z.-J. Lin, H.-Q. Zheng, J. Chen, W.-E. Zhuang, Y.-X. Lin, J.-W. Sun, Y.-B. Huang,
R. Cao, Inorg. Chem. 57 (2018) 13009–13019.[38] Q. Han, Y.-L. Wang, M. Sun, C.-Y. Sun, S.-S. Zhu, X.-L. Wang, Z.-M. Su, Chem. Eur. J.
24 (2018) 15089–15095.[39] M. Chen, H. Zhao, C.-S. Liu, X. Wang, H.-Z. Shi, M. Du, Chem. Commun. 51 (2015)
6014–6017.[40] B. Parmar, P. Patel, R.I. Kureshy, N.-U.H. Khan, E. Suresh, Chem. Eur. J. 24 (2018)
15831–15839.[41] J.F. Kurisingal, Y. Rachuri, Y. Gu, Y. Choe, D.-W. Park, Chem. Eng. J. (2019),
https://doi.org/10.1016/j.cej.2019.05.061.[42] P. Patel, B. Parmar, R.I. Kureshy, N.-u. Khan, E. Suresh, ChemCatChem 10 (2018)
2401–2408.[43] X.-H. Ji, N.-N. Zhu, J.-G. Ma, P. Cheng, Dalton Trans. 47 (2018) 1768–1771.[44] R. Babu, S.-H. Kim, J.F. Kurisingal, H.-J. Kim, G.-G. Choi, D.-W. Park, J. CO2 Util. 25
(2018) 6–13.[45] W. Clegg, R.W. Harrington, M. North, R. Pasquale, Chem. Eur. J. 16 (2010)
6828–6843.[46] B. Parmar, P. Patel, R.S. Pillai, R.I. Kureshy, N.-u.H. Khan, E. Suresh, J. Mater.
Chem. A 7 (2019) 2884–2894.[47] I. Senkovska, F. Hoffmann, M. Fröba, J. Getzschmann, W. Böhlmann, S. Kaskel,
Micropor. Mesopor. Mater. 122 (2009) 93–98.[48] J. Yan, S. Jiang, S. Ji, D. Shi, H. Cheng, Sci. China Chem. 58 (2015) 1544–1552.[49] R. Babu, A.C. Kathalikkattil, R. Roshan, J. Tharun, D.-W. Kim, D.-W. Park, Green
Chem. 18 (2016) 232–242.[50] S.S. Dhankhar, C.M. Nagaraja, New J.Chem. 43 (2019) 2163–2170.[51] J. Yin, T. Zhang, E. Schulman, D. Liu, J. Meng, J. Mater. Chem. A 6 (2018)
8441–8448.[52] F. Castro-Gómez, G. Salassa, A.W. Kleij, C. Bo, Chem. Eur. J. 19 (2013) 6289–6298.[53] A.C. Kathalikkattil, R. Babu, R.K. Roshan, H. Lee, H. Kim, J. Tharun, E. Suresh, D.-
W. Park, J. Mater. Chem. A 3 (2015) 22636–22647.[54] A.C. Kathalikkattil, R. Roshan, J. Tharun, R. Babu, G.-S. Jeong, D.-W. Kim, S.J. Cho,
D.-W. Park, Chem. Commun. 52 (2016) 280–283.[55] R. Babu, R. Roshan, Y. Gim, Y.H. Jang, J.F. Kurisingal, D.-W. Kim, D.-W. Park, J, .
Mater. Chem. A 5 (2017) 15961–15969.[56] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,
G. Scalmani, V. Barone, G.A. Petersson, H. Nakatsuji, X. Li, M. Caricato,A.V. Marenich, J. Bloino, B.G. Janesko, R. Gomperts, B. Mennucci, H.P. Hratchian,J.V. Ortiz, A.F. Izmaylov, J.L. Sonnenberg, D. Williams–Young, F. Ding, F. Lipparini,F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe,V.G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara,K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao,H. Nakai, T. Vreven, K. Throssell, J.A. Montgomery Jr., J.E. Peralta, F. Ogliaro,M.J. Bearpark, J.J. Heyd, E.N. Brothers, K.N. Kudin, V.N. Staroverov, T.A. Keith,R. Kobayashi, J. Normand, K. Raghavachari, A.P. Rendell, J.C. Burant, S.S. Iyengar,J. Tomasi, M. Cossi, J.M. Millam, M. Klene, C. Adamo, R. Cammi, J.W. Ochterski,R.L. Martin, K. Morokuma, O. Farkas, J.B. Foresman, D.J. Fox, Gaussian, Inc.:Wallingford, CT, 2016.
[57] P.J. Hay, W.R. Wadt, J. Chem. Phys. 82 (1985) 270–283.[58] W.R. Wadt, P.J. Hay, J. Chem. Phys. 82 (1985) 284–298.[59] H. Sun, D. Zhang, J. Phys. Chem. A 111 (2007) 8036–8043.[60] P. Patel, B. Parmar, R.I. Kureshy, N.H. Khan, E. Suresh, Dalton Trans. 47 (2018)
8041–8051.[61] B. Parmar, P. Patel, R.I. Kureshy, N.-u.H. Khan, S. Eringathodi, Chem. Eur. J. 24
(2018) 15831–15839.[62] J.F. Kurisingal, Y. Rachuri, Y. Gu, Y. Choe, D.-W. Park, ACS Appl. Mater. Interfaces
44 (2019) 41458–41471.[63] J.F. Kurisingal, Y. Rachuri, Y. Gu, G.-H. Kim, D.-W. Park, Appl. Catal. A Gen. 571
(2019) 1–11.[64] Y.-H. Li, S.-L. Wang, Y.-C. Su, B.-T. Ko, C.-Y. Tsai, C.-H. Lin, Dalton Trans. 47 (2018)
9474–9481.
J.F. Kurisingal, et al. Catalysis Today 352 (2020) 227–236
236


















