







PimE IS A POLYPRENOL-PHOSPHATE-MANNOSE … pime is a polyprenol-phosphate-mannose-dependent...
26
1 PimE IS A POLYPRENOL-PHOSPHATE-MANNOSE-DEPENDENT MANNOSYLTRANSFERASE THAT TRANSFERS THE FIFTH MANNOSE OF PHOSPHATIDYLINOSITOL MANNOSIDE IN MYCOBACTERIA Yasu S. Morita 1 , Chubert B. C. Sena 1 , Ross F. Waller 2 , Ken Kurokawa 3 , M. Fleur Sernee 4 , Fumiki Nakatani 1 , Ruth E. Haites 5 , Helen Billman-Jacobe 5 , Malcolm J. McConville 4 , Yusuke Maeda 1 , and Taroh Kinoshita 1 1 Department of Immunoregulation, Research Institute for Microbial Diseases, Osaka University, Osaka 565-0871, Japan; 2 Plant Cell Biology Research Centre, School of Botany, University of Melbourne, Parkville, Victoria 3010, Australia; 3 Laboratory of Comparative Genomics, Graduate School of Information Science, Nara Institute of Science and Technology, Ikoma, Nara 630-0192, Japan; 4 Department of Biochemistry and Molecular Biology, Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, Victoria 3010, Australia; and 5 Department of Microbiology and Immunology, University of Melbourne, Parkville, Victoria 3010, Australia. Running Title: PimE transfers the fifth mannose in PIM biosynthesis Address correspondence to: Taroh Kinoshita, Department of Immunoregulation, Research Institute for Microbial Diseases, Osaka University, 3-1 Yamada-oka, Suita, Osaka 565-0871, Japan. Tel: +81-6-6879-8328. Fax: +81-6-6875-5233. E-mail: [email protected] Phosphatidylinositol mannosides (PIMs) are a major class of glycolipids in all mycobacteria. AcPIM2, a dimannosyl PIM, is both an end-product and a precursor for polar PIMs, such as hexamannosyl PIM (AcPIM6) and the major cell wall lipoglycan, lipoarabinomannan (LAM). The mannosyltransferases that convert AcPIM2 to AcPIM6 or LAM are dependent on polyprenol-phosphate-mannose (PPM), but have not yet been characterized. Here, we identified a gene, termed pimE that is present in all mycobacteria, and is required for AcPIM6 biosynthesis. PimE was initially identified based on homology with eukaryotic PIG-M mannosyltransferases. PimE-deleted Mycobacterium smegmatis was defective in AcPIM6 synthesis, and accumulated the tetramannosyl PIM, AcPIM4. Loss of PimE had no affect on cell growth or viability, or the biosynthesis of other intracellular and cell wall glycans. However, changes in the cell wall hydrophobicity and plasma membrane organization were detected, suggesting a role for AcPIM6 in the structural integrity of the cell wall and plasma membrane. These defects were corrected by ectopic expression of the pimE gene. Metabolic pulse-chase radiolabeling and cell-free PIM biosynthesis assays indicated that PimE catalyzes the alpha1,2-mannosyl transfer for the AcPIM5 synthesis. Mutation of an Asp residue in PimE that is conserved in and required for the activity of human PIG-M, resulted in loss of PIM-biosynthetic activity, indicating that PimE is the catalytic component. Finally, PimE was localized to a distinct membrane fraction enriched in AcPIM4-6 biosynthesis. Taken together, PimE represents the first PPM-dependent mannosyltransferase shown to be involved in PIM biosynthesis, where it mediates the fifth mannose transfer. Mycobacterium tuberculosis is an etiologic agent of tuberculosis, which causes millions of deaths annually. Other species belonging to the genus also cause important diseases, such as leprosy in humans and Johne’s disease and bovine tuberculosis in livestock. The success of mycobacteria as human and animal pathogens is due, in part, to their unusual cell walls, which are multi-layered structures comprised of peptidoglycan, arabinogalactan, mycolic acids and glycolipids on top of the plasma membrane (1-3). In particular, the outer asymmetric pseudobilayer of mycolic acids and glycolipids represents a hydrophobic permeability barrier against antibiotics and host immune attacks (4,5). http://www.jbc.org/cgi/doi/10.1074/jbc.M604214200 The latest version is at JBC Papers in Press. Published on June 27, 2006 as Manuscript M604214200 Copyright 2006 by The American Society for Biochemistry and Molecular Biology, Inc. by guest on May 27, 2018 http://www.jbc.org/ Downloaded from
Transcript of PimE IS A POLYPRENOL-PHOSPHATE-MANNOSE … pime is a polyprenol-phosphate-mannose-dependent...

1
PimE IS A POLYPRENOL-PHOSPHATE-MANNOSE-DEPENDENT MANNOSYLTRANSFERASE THAT TRANSFERS THE FIFTH MANNOSE OF
PHOSPHATIDYLINOSITOL MANNOSIDE IN MYCOBACTERIA
Yasu S. Morita1, Chubert B. C. Sena1, Ross F. Waller2, Ken Kurokawa3, M. Fleur Sernee4, Fumiki Nakatani1, Ruth E. Haites5, Helen Billman-Jacobe5,
Malcolm J. McConville4, Yusuke Maeda1, and Taroh Kinoshita1
1Department of Immunoregulation, Research Institute for Microbial Diseases, Osaka University,
Osaka 565-0871, Japan; 2Plant Cell Biology Research Centre, School of Botany, University of Melbourne, Parkville, Victoria 3010, Australia; 3Laboratory of Comparative Genomics,
Graduate School of Information Science, Nara Institute of Science and Technology, Ikoma, Nara 630-0192, Japan; 4Department of Biochemistry and Molecular Biology, Bio21 Molecular
Science and Biotechnology Institute, University of Melbourne, Parkville, Victoria 3010, Australia; and 5Department of Microbiology and Immunology, University of Melbourne,
Parkville, Victoria 3010, Australia.
Running Title: PimE transfers the fifth mannose in PIM biosynthesis
Address correspondence to: Taroh Kinoshita, Department of Immunoregulation, Research Institute for Microbial Diseases, Osaka University,
3-1 Yamada-oka, Suita, Osaka 565-0871, Japan. Tel: +81-6-6879-8328. Fax: +81-6-6875-5233. E-mail: [email protected]
Phosphatidylinositol mannosides (PIMs) are a major class of glycolipids in all mycobacteria. AcPIM2, a dimannosyl PIM, is both an end-product and a precursor for polar PIMs, such as hexamannosyl PIM (AcPIM6) and the major cell wall lipoglycan, lipoarabinomannan (LAM). The mannosyltransferases that convert AcPIM2 to AcPIM6 or LAM are dependent on polyprenol-phosphate-mannose (PPM), but have not yet been characterized. Here, we identified a gene, termed pimE that is present in all mycobacteria, and is required for AcPIM6 biosynthesis. PimE was initially identified based on homology with eukaryotic PIG-M mannosyltransferases. PimE-deleted Mycobacterium smegmatis was defective in AcPIM6 synthesis, and accumulated the tetramannosyl PIM, AcPIM4. Loss of PimE had no affect on cell growth or viability, or the biosynthesis of other intracellular and cell wall glycans. However, changes in the cell wall hydrophobicity and plasma membrane organization were detected, suggesting a role for AcPIM6 in the structural integrity of the cell wall and plasma membrane. These defects were corrected by ectopic expression of the pimE gene. Metabolic pulse-chase radiolabeling and cell-free PIM biosynthesis assays indicated that PimE catalyzes the alpha1,2-mannosyl transfer for the AcPIM5
synthesis. Mutation of an Asp residue in PimE that is conserved in and required for the activity of human PIG-M, resulted in loss of PIM-biosynthetic activity, indicating that PimE is the catalytic component. Finally, PimE was localized to a distinct membrane fraction enriched in AcPIM4-6 biosynthesis. Taken together, PimE represents the first PPM-dependent mannosyltransferase shown to be involved in PIM biosynthesis, where it mediates the fifth mannose transfer. Mycobacterium tuberculosis is an etiologic agent of tuberculosis, which causes millions of deaths annually. Other species belonging to the genus also cause important diseases, such as leprosy in humans and Johne’s disease and bovine tuberculosis in livestock. The success of mycobacteria as human and animal pathogens is due, in part, to their unusual cell walls, which are multi-layered structures comprised of peptidoglycan, arabinogalactan, mycolic acids and glycolipids on top of the plasma membrane (1-3). In particular, the outer asymmetric pseudobilayer of mycolic acids and glycolipids represents a hydrophobic permeability barrier against antibiotics and host immune attacks (4,5).
http://www.jbc.org/cgi/doi/10.1074/jbc.M604214200The latest version is at JBC Papers in Press. Published on June 27, 2006 as Manuscript M604214200
Copyright 2006 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

2
Phosphatidylinositol (PI 1 ) is an abundant phospholipid of the mycobacterial plasma membrane, and accounts for about 25% of the total phospholipids (6). In addition to being a structural membrane component, PI also acts as a biosynthetic precursor of the glycosylphosphatidylinositols (GPIs) known as phosphatidylinositol mannosides (PIMs), lipomannan (LM) and lipoarabinomannan (LAM) (7-11). The major PIM species are PI-dimannosides (AcPIM2 and Ac2PIM2) and PI-hexamannosides (AcPIM6 and Ac2PIM6). AcPIM2 and AcPIM6 contain one fatty acid linked to the mannose residue in addition to a diacylglycerol moiety, while Ac2PIM2 and Ac2PIM6 contain an additional fatty acid linked to the inositol residue. Purified PIM species have various immunomodulatory activities. For example, they may activate macrophages to induce TNF-α secretion through TLR2 (12), and can induce granuloma formation by recruiting natural killer T cells (13). More recently, PIMs were identified as natural antigens of CD1d-restricted or CD1b-restricted T cells (14,15). Interestingly, partially digested PIMs, rather than the mature molecules, are more efficiently recognized and presented, and CD1e is involved in facilitating the processing of PIM molecules mediated by lysosomal α-mannosidases (15). On the other hand, LAM is a potent anti-inflammatory molecule, which is thought to be involved in immune suppression (2,16-18) and play a role in blocking phagosomal maturation during macrophage invasion of M. tuberculosis (19).
1 Abbreviations used: ASAM, Aspergillus saitoi α-mannosidase; CL, cardiolipin; DPM, dolichol-phosphate-mannose; DXO, double crossover; GPI, glycosylphosphatidylinositol; GPL, glycopeptidolipid; HPTLC, high performance thin layer chromatography; LAM, lipoarabinomannan; LM, lipomannan; MALDI-TOF-MS, matrix-assisted laser desorption ionization time-of-flight mass spectrometer; MMP, methylmannose polysaccharide; PE, phosphatidylethanolamine; PI, phosphatidylinositol; PIM, phosphatidylinositol mannoside; PPM, polyprenol-phosphate-mannose; SXO, single crossover.
The major end-products, namely PIMs, LM and LAM, all contain a common core structure, suggesting that the early steps in their biosynthesis are the same (20). Recent studies have suggested that the biosynthesis of AcPIM6, LM and LAM may bifurcate after the synthesis of the intermediate AcPIM4. Specifically, in vivo metabolic labeling experiments (21) and assays reconstituting PIM biosynthesis in vitro (8) have indicated that AcPIM4 is the direct precursor of AcPIM6, which is capped with two α1,2-linked mannose residues not found in the mannan backbone of LM and LAM (Fig. 1). Moreover, we recently identified an Mycobacterium smegmatis mutant with a defect in the lpqW gene that accumulates AcPIM4 in response to alterations in both AcPIM6 and LM/LAM biosynthesis (22). However, to date, little is known about the enzymes that direct AcPIM4 toward AcPIM6 or LM/LAM biosynthesis. The first committed step of the PIM biosynthetic pathway is mediated by PimA, a GDP-mannose-dependent mannosyltransferase that transfers mannose to the 2-OH of the inositol moiety of PI (23). The next step is mediated by an acyltransferase that substitutes the 6-OH of the mannose residue with a fatty acyl chain (24). The gene encoding this acyltransferase appears to form an operon with pimA as well as pgsA, which encodes PI synthase (6). The second mannosyltransferase, PimB, transfers mannose from GDP-mannose to the 6-OH of the inositol residue of AcPIM1 (25). The order of the reactions mediated by the acyltransferase and PimB may be reversed, leading to the synthesis of PIM2 rather than AcPIM1 as an intermediate (8-10,23). From AcPIM2 onwards, the enzymes involved in the biosynthesis of AcPIM6 and LM/LAM are poorly defined. PimC has been identified in M. tuberculosis as the third mannosyltransferase, but the gene is only present in some clinical strains of M. tuberculosis and absent from M. smegmatis, suggesting that there must be another gene involved in this step (26). PimF was reported as a gene involved in LM synthesis (27), but later redefined as a glycosyltransferase involved in the synthesis of lipooligosaccharide (28). The only other gene known to be involved in mycobacterial GPI biosynthesis is EmbC, which is an arabinosyltransferase involved in the synthesis of the arabinan side chain of LAM
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

3
(29). Polyprenol-phosphate-mannose (PPM), rather than GDP-mannose, is the mannose donor for the mannosyltransferases involved in the post-AcPIM2 segment of AcPIM6 synthesis and the LM/LAM pathway (7,8). Glycosyltransferases that utilize polyprenol-phosphate-sugars as donors are distinct from those that utilize nucleotide sugars as donors. In eukaryotes, dolichol-phosphate-mannose (DPM) is the bacterial PPM equivalent and DPM-dependent mannosyltransferases have been well described in eukaryotic GPI biosynthetic pathways. We and others have identified four genes encoding mannosyltransferases involved in the GPI glycan biosynthesis, namely PIG-M, PIG-V, PIG-B and Smp3 involved in the first, second, third and fourth mannose transfers, respectively (30-36). Since the mannosyltransferases involved in PIM and eukaryotic GPI biosyntheses must recognize a prenol-based sugar donor and PI-based sugar acceptor, we considered the possibility that these enzymes may be evolutionarily conserved and therefore identifiable through amino acid sequence homology. Here, we describe the identification of PimE as a protein distantly related to eukaryotic PIG-M, and show that it mediates the fifth mannose transfer in the AcPIM6 biosynthetic pathway. EXPERIMENTAL PROCEDURES Genome-wide survey of PIG-M homologs — All the genes identified in the Mycobacterium leprae genome (37) were subjected to SOSUI (38) to predict membrane proteins. All the candidate membrane proteins were then subjected to PSI-BLAST (39) with 6 iterations. Proteins homologous to eukaryotic PIG-M, Rv0051, Rv1159, Rv2181 and Rv2673 were identified by searching the corresponding GI numbers. Mycobacterial strains and culture growth — M. smegmatis strain mc2155 was used throughout this study (40). Unless otherwise indicated, the cells were grown on Middlebrook 7H10 agar plates (Difco) supplemented with 0.2% (w/v) glucose, 0.2% (v/v) glycerol and 15 mM NaCl. For liquid culture, the cells were grown in Middlebrook 7H9 broth (Difco) supplemented
with 0.2% (w/v) glucose, 0.2% (v/v) glycerol, 15 mM NaCl and 0.05% (v/v) Tween-80. Where appropriate, streptomycin and kanamycin were used at 20 µg/ml. Viability, as indicated by colony-forming units, was measured by plating an aliquot of the cells grown in Middlebrook 7H9 broth onto Middlebrook 7H10 agar plates, and counting the number of colonies that had grown after 3 days at 37°C. Disruption of the MSMEG5136 gene — DNA manipulation was performed according to standard protocols (41). Approximately 1 kb fragments on the 5’ and 3’ sides of the MSMEG5136 gene were amplified from genomic DNA by PCR using the following primer pairs (in which SpeI, HindIII, EcoRI and NotI restriction enzyme sites, respectively, are underlined): 5’- GGTCACTAGTTCGGCATACCGGGCACCTG and 5’- GTCGAAGCTTATTGACCTGGCCGTAGTCGAATGTG; 5’- GCAGGAATTCGCCGGTACTTCACCGAACTGCTG and 5’- CGACCGTGCGGCCGCTTTCCGCCAGGAAGTTCCCACAG. A kanamycin-resistance cassette was excised from pHBJ395 (42) using HindIII and EcoRI, ligated with the PCR fragments prepared above and inserted into the vector pHBJ428 (42) at the SpeI and NotI sites. The PCR primers were designed so that the central 213 bases in the coding region of MSMEG5136 were deleted and replaced with the kanamycin-resistance cassette in the final construct. A high-purity plasmid of the resultant construct, designated pYAB28, was prepared using QIAGEN Plasmid Midi Kits and transfected into M. smegmatis competent cells according to a published protocol (43), except that a BTX T820 ElectroSquarePorator was used at 2.8 kV, 99 µsec, and 10 pulses. A single-crossover (SXO) mutant, designated SXO28-6, was selected by its resistance to kanamycin and streptomycin and its inability to grow on Middlebrook 7H10 agar containing 10% (w/v) sucrose due to sacB gene. Double-crossover (DXO) was induced by growing SXO28-6 in PPLO broth (50 g/l brain heart infusion, 10 g/l peptone and 5 g/l NaCl) supplemented with 0.05% Tween-80 in the absence of antibiotics selection. DXO mutants were selected by their ability to grow on sucrose-containing PPLO agar (1.5%),
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

4
sensitivity to streptomycin and resistance to kanamycin. Three clones, designated DXO28-3, DXO28-4 and DXO28-6, were identified and subjected to further analysis. Southern blot analysis — Genomic DNA was extracted as described previously (42). A 10 µg aliquot of SacI-digested DNA was resolved by electrophoresis in a 0.8% agarose gel in 1x TAE buffer, and blotted onto a Hybond N+ nylon membrane (Amersham Biosciences). Probe hybridization and signal detection were performed using a Gene Images AlkPhos Direct Labeling and Detection System (Amersham Biosciences), according to the manufacturer’s guidelines. The probe was prepared by PCR-amplification of a DNA fragment from M. smegmatis genomic DNA using the primer pair 5’-ATCTGAACTGTCCCTGCGGAGAAGC and 5’-CCAGACGGCCTACGTGTTCACC, and covalently cross-linking thermostable alkaline phosphatase following the manufacturer’s protocol. Gene cloning and complementation — The MSMEG5136 gene was amplified by PCR from the M. smegmatis genomic DNA (primers: 5'- CGGTCAACGGTTACCGGGGCG and 5'- CGGAATTCGAGGAGATCAACCGGATCC) and cloned directionally into the MscI and EcoRI sites at the multiple cloning sites of the expression vector pHBJ334 (44), a derivative of pMV261 containing a streptomycin-resistance cassette instead of a kanamycin-resistance cassette. The resultant MSMEG5136 gene expression vector was designated pYAB34. Rv1159 was amplified by PCR from the M. tuberculosis H37Rv genomic DNA (gift from Dr. Takeshi Yamada, Nagasaki University, Japan) (primers: 5'-CCCGCACCCTGATCGACGGCCC and 5'-CGGAATTCTAATTGGCCATGCGCCGCG) and cloned into pHBJ334, resulting in an Rv1159 gene-expression vector designated pYAB35. These constructs were transfected into M. smegmatis DXO mutant cells by electroporation as described above. Expression of epitope-tagged MSMEG5136 protein — A FLAG tag was introduced by PCR amplification of a part of the MSMEG5136 gene using the primers 5'-TGTCGTGGCTCGTGGTGGGT and 5'-TTCGAATTCATTTGTCGTCGTCGTCT
TTGTAGTCAATATTACCGGATCCTTTCCGACTGA (EcoRI and SspI restriction enzyme sites are underlined). The bold characters indicate the sequence encoding the FLAG tag. A part of pYAB34 was replaced with the amplified fragment, resulting in pYAB52, an expression vector for MSMEG5136 protein containing a C-terminal FLAG epitope tag. Site-directed mutagenesis — A QuikChange Site-Directed Mutagenesis Kit (Stratagene) was used according to the manufacturer’s recommended protocol. The primers used to create the D58A mutation were 5’- CGCCAACTTCGTCGCCCTGCACGTCTACG and 5’- CGTAGACGTGCAGGGCGACGAAGTTGGCG, with pYAB52 as the template. Lipid extraction and analysis — To purify PIMs, phospholipids and glycopeptidolipids (GPLs), cell pellets were sequentially extracted in 20 volumes of chloroform/methanol (2:1, v/v), 10 volumes of chloroform/methanol (2:1, v/v) and 10 volumes of chloroform/methanol/water (1:2:0.8, v/v/v). Each extraction involved a brief water bath sonication to resuspend the pellet, followed by a 2 h incubation at room temperature. The combined organic solvents were dried under a nitrogen stream, and the lipids were further purified by biphasic partitioning in 1-butanol/water (2:1, v/v). The 1-butanol phase extracts were dried and applied to aluminium-backed high performance thin layer chromatography (HPTLC) silica gel 60 sheets (Merck). PIMs and phospholipids were resolved in chloroform/methanol/13 M ammonia/1 M ammonium acetate/water (180:140:9:9:23, v/v/v/v/v). GPLs were treated with a base (0.2 M methanolic NaOH at 40°C for 2 h) after the 1-butanol/water partition, purified again by a second round of 1-butanol/water phase partitioning and then analyzed by HPTLC in chloroform/methanol (9:1, v/v). PIMs and GPLs were visualized using an orcinol-H2SO4 spray reagent, while phospholipids visualized using a molybdenum blue spray reagent. For matrix-assisted laser desorption ionization time-of-flight mass spectrometer (MALDI-TOF-MS) analysis, the 1-butanol extract described above was mixed with an equal volume of saturated α-cyano-4-hydroxycinnamic acid and analyzed in a Voyager Elite XL (Perkin-Elmer
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

5
Biosystems) MALDI-TOF-MS in linear negative-ion mode. An accelerating voltage of 20000 V was used with grid and guide wire voltages of 97.5 and 0.04%, respectively. LAM extraction and analysis — The delipidated cell pellets were resuspended in 5 volumes of 50% ethanol in water, and refluxed at 100°C for 2 h. The extraction was repeated three times, and the combined extract was further fractionated by octyl-Sepharose column chromatography. The bound fraction was freeze-dried, separated by SDS-PAGE (10-20% gradient gel) and visualized using a carbohydrate staining kit (ProQ Emerald 488; Molecular Probes) and a fluoroimage analyzer (FLA-5000; Fujifilm). Methylmannose polysaccharide extraction and analysis — The aqueous phase obtained after the 1-butanol/water partitioning (described above) was desalted on a mixed-bed column of AG50-X12(H+) over AG3-X8(OH-) (Bio-Rad). After methanolysis in 0.5 M methanolic HCl, the released monosaccharide methyl esters were converted to trimethylsilyl derivatives for GC-MS analysis. The characteristic 3-O-methylmannose peak was used to calculate the methylmannose polysaccharide (MMP) contents (21). Hexadecane partitioning — A published protocol was followed (45), with minor modifications. Briefly, cells were grown to an OD600 of 1.0-1.5. An aliquot (1.5 ml) of the cells was then transferred to a borosilicate tube and mixed with 0-50 µl of hexadecane. The tube was vortexed for 2 min and the contents were transferred to a cuvette to allow separation of the two phases. After 15 min, the OD600 of the lower aqueous phase was measured. Electron microscopy — Cells were grown to mid-log phase, fixed and embedded in Spurrs resin as previously described (21). Ultrathin sections prepared using a Leica Ultracut R microtome were stained with uranyl acetate for 20 min and Reynold’s lead citrate stain for 4 min and then observed using a Philips CM120 BioTWIN transmission electron microscope. Metabolic labeling — For [3H]inositol labeling, cells at mid-log phase were harvested and resuspended in Middlebrook 7H9 broth (without
supplements) at 0.2 g wet pellet/ml. Next, myo-[2-3H]inositol (20 Ci/mmol; American Radiolabeled Chemicals) was added to a final concentration of 50 µCi/ml and incubated at 37°C for 5 min. The cell suspension was diluted 25-fold with fully supplemented Middlebrook 7H9 medium that also contained 5 mM inositol, and chased for 5 h. For [3H]mannose labeling, mid-log phase cells were harvested and resuspended in Middlebrook 7H9 broth at 0.8 g wet pellet/ml. The cells were then incubated with 50 µCi/ml [2-3H]mannose (20 Ci/mmol; American Radiolabeled Chemicals) for 15 min as a pulse, washed with cold 7H9 medium to remove unincorporated radiolabel and resuspended at 1.6 µg wet pellet/ml in 7H9 medium supplemented with 1 mM mannose for the chase. Lipids were extracted and analyzed as described above. To visualize the radioactive signals, HPTLC plates were either exposed to an imaging plate for the Fujifilm BAS autoradiography system or sprayed with En3Hance and exposed to an X-ray film for fluorography. Cell-free PIM biosynthesis — Log phase cells were harvested, washed twice with 50 mM HEPES-NaOH (pH 7.4) and resuspended at 200 mg pellet weight/ml in lysis buffer containing 25 mM HEPES-NaOH (pH 7.4), 10% (w/v) sucrose, 2 mM EGTA and a protease inhibitor cocktail (Roche). Acid-washed glass beads (106 µm diameter) were added at a concentration of 800 mg/ml of cell suspension, and the cells were lysed by bead-beating using a MicroSmash MS100R (TOMY SEIKO) at 5000 rpm for 30 s. The process was repeated 6 times with cooling after each run. After removing cell debris and aggregates, the cell lysate was supplemented with 5 mM MgCl2 and pre-incubated for 5 min at 37°C. PIM biosynthesis was initiated by adding GDP-[2-3H]mannose (20 Ci/mmol; American Radiolabeled Chemicals) to a final concentration of 10 µCi/ml. The reaction was terminated by adding chloroform/methanol to achieve a final ratio of chloroform/methanol/water of 10:10:3 (v/v/v). The lipids were extracted, purified by 1-butanol/water partitioning, separated by HPTLC and visualized by autoradiography or fluorography as described above. Mannosidase treatment — Dried lipid extracts were resuspended in a buffer containing 0.1 M
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

6
sodium acetate (pH 5.0) and 0.1% (w/v) taurodeoxycholate. Digestion was initiated by adding Aspergillus saitoi α1,2-mannosidase (ASAM; Prozyme) to a final concentration of 20 mU/ml in a 10 µl aliquot and continued for 17 h at 37°C. A second aliquot (80 µU) of fresh enzyme was then added, and the digestion was continued for a further 24 h. Lipids were extracted by 1-butanol/water partitioning and analyzed by HPTLC. Subcellular fractionation — The MSMEG5136-deletion mutant (DXO28-3) complemented with pYAB52 was harvested at mid-log phase, washed with 50 mM HEPES-NaOH (pH 7.4) and resuspended at 0.2 g wet pellet/ml in 25 mM HEPES-NaOH (pH 7.4), 10% (w/v) sucrose, 2 mM EGTA and a protease inhibitor cocktail. The cells were then disrupted by nitrogen cavitation (1,850 psi, 3x 30 min) and cell debris was removed by centrifugation (2,380x g). Next, the crude lysate (1.2 ml) was fractionated by sucrose-gradient sedimentation at 218,000x g for 6 h at 4°C. A sucrose gradient of 10-60% (w/v) was created using a Gradient Master (BioComp Instruments) using the default settings. After centrifugation, 1 ml fractions were collected from the top of the tube using a Piston Gradient Fractionator (BioComp Instruments). The sucrose density of each fraction was measured using a refractometer. GDP-[3H]mannose labeling was performed as described above. The protein concentrations were measured in duplicate using the BCA Protein Assay Reagent (Pierce). The sucrose concentrations were adjusted to 32% prior to radiolabeling or protein concentration assays. SDS-PAGE and western blotting — Following a standard protocol, proteins were fractionated by SDS-PAGE (12.5% gel or 10-20% gradient gel) under reducing conditions. After transferring the proteins to a PVDF membrane, the membrane was blocked with 5% skim milk and sequentially incubated with a primary antibody (mouse anti-FLAG M2 monoclonal antibody; 1:2000 dilution; Sigma) for 1 h and a secondary antibody (horseradish peroxidase-conjugated sheep anti-mouse IgG antibody; 1:2000 dilution; Amersham) for 1 h. The bound probe was visualized by enhanced chemiluminescence (Perkin-Elmer).
RESULTS Candidates for PPM-dependent mannosyltransferases — To identify candidates for PPM-dependent mannosyltransferases involved in the PIM biosynthetic pathway, we conducted BLASTP homology searches of an M. tuberculosis genome (http://genolist.pasteur.fr/TubercuList/) using the amino acid sequences of eukaryotic GPI mannosyltransferases as queries. We identified several mycobacterial proteins sharing some similarities to PIG-M. Because the bit scores were not statistically significant using the standard BLASTP searches (less than 50), we subjected the identified mycobacterial amino acid sequences to PSI-BLAST of a non-redundant database at the NCBI website (http://www.ncbi.nlm.nih.gov/BLAST/). Four proteins were predicted to be homologous to eukaryotic PIG-M proteins after several iterations, resulting in high bit scores ranging from 159 to 230. The four genes encoding these proteins in the genome of M. tuberculosis H37Rv strain were Rv0051, Rv1159, Rv2181 and Rv2673. We did not identify any other mycobacterial proteins showing significant homologies to other GPI mannosyltransferases such as PIG-V, PIG-B, and Smp3 through this strategy. These four genes were also identified using a second bioinformatics approach. Considering the fact that eukaryotic DPM-dependent mannosyltransferases are all polytopic integral membrane proteins, we hypothesized that the mycobacterial PPM-dependent mannosyltransferases are likely to be membrane proteins. We further hypothesized that the PPM-dependent mannosyltransferases should be present in the minimized genome of M. leprae (37), since M. leprae is known to synthesize PIMs and LAM (46,47). Thus, we screened the entire 1605 genes found in the M. leprae genome using SOSUI, a secondary structure prediction software for membrane proteins, and found 415 predicted membrane proteins. We then subjected these 415 genes to PSI-BLAST with 6 iterations. The output data were searched for M. leprae proteins homologous to eukaryotic PIG-M proteins, Rv0051, Rv1159, Rv2181 and Rv2673. As a result, we found two additional M. leprae proteins encoded by ML0591c and ML0899c (homologous to Rv1459c and Rv2174
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

7
in M. tuberculosis H37Rv, respectively). Thus, we identified a total of six candidate genes for PIM mannosyltransferases. All six gene products were most homologous to PIG-M and no significant homologies to other GPI mannosyltransferases were indicated by PSI-BLAST analyses. Consistent with the ubiquitous prevalence of PIMs and LAM in mycobacteria, these six genes were conserved in all pathogenic Mycobacterium species examined, including Mycobacterium bovis, Mycobacterium avium and M. leprae. Since the six genes were also found in the saprophytic M. smegmatis, we decided to create deletion mutants of these genes in this experimentally tractable model organism in order to identify their precise functions. Since Rv0051, Rv1159 and Rv2181 are predicted to be non-essential genes in M. tuberculosis (48), the involvement of these genes in PIM biosynthesis was initially examined. Studies on the other three potentially essential genes (Rv1459c, Rv2174 and Rv2673) are currently underway in our laboratory. We previously deleted the homolog of Rv0051 (locus designated MSMEG6861 in the annotation list at TIGR-CMR, http://cmr.tigr.org/tigr-scripts/CMR/CmrHomePage.cgi) in M. smegmatis mc2155 (49) as part of a study on ponA, which is located directly upstream of MSMEG6861 in the M. smegmatis genome. We reexamined PIM biosynthesis in this mutant, and found that all the major PIM species were synthesized, suggesting that MSMEG6861 is not essential for PIM biosynthesis (not shown). We then deleted the homolog of Rv2181 (MSMEG4250) in M. smegmatis and found that PIM biosynthesis was not defective in this mutant either (not shown). When we deleted the homolog of Rv1159 (MSMEG5136) in M. smegmatis, we found a striking defect in PIM biosynthesis. MSMEG5136 is a multiple membrane spanning protein with 410 amino acid residues (predicted molecular weight, 44.5 kDa), and is highly conserved among Mycobacterium species (Fig. 2A). Disruption of the gene was confirmed by Southern blot analysis using a probe as indicated in Fig. 2B. Bands of the expected sizes were identified in the wild-type (mc2155) strain, a parental SXO strain and three independent DXO
clones (Fig. 2C), confirming the generation of MSMEG5136 deletion mutants. We confirmed the gene deletion with additional probes directed against a sequence within the integrated vector as well as a sequence downstream of the vector integration site, and all the results were consistent with successful deletion of MSMEG5136 (not shown). We further confirmed the targeted disruption of the gene by PCR using primers designed to amplify the integration sites upstream and downstream of the inserted construct (not shown). MSMEG5136 deletion mutant is defective in PIM6 synthesis — Lipids were extracted and purified according to our standard procedure, analyzed by HPTLC and visualized by chemical carbohydrate staining. The lipid profiles of two independent MSMEG5136 deletion mutants (DXO28-3 and DXO28-4) differed from that of the wild-type strain in completely lacking AcPIM6 and Ac2PIM6, while accumulating two novel glycolipids with faster HPTLC mobilities than AcPIM6 species (Fig. 3A, lanes 4 and 7; see also Fig. 4A). These species had similar HPTLC mobilities as AcPIM4 and Ac2PIM4, which were previously identified in an M. smegmatis lpqW mutant (22). We subjected lipid extracts from wild-type and DXO28-3 to MALDI-TOF-MS analysis, and identified molecular ions with the expected m/z corresponding to AcPIM2 and Ac2PIM2 from both wild-type and DXO28-3 lipid extracts (Table 1). While we identified molecular ions corresponding to AcPIM6 and Ac2PIM6 from wild-type extract, these molecular ions were missing in the DXO28-3 extracts. Instead, we identified molecular ions corresponding to AcPIM4 and Ac2PIM4 from the DXO28-3 lipid extracts (Table 1). To confirm the identity further, we purified major PIM species identified in the DXO28-3 lipid extract from HPTLC plates, and reanalyzed by MALDI-TOF-MS. As shown in Figs. 3B and 3C, molecular ions with expected m/z were detected from purified AcPIM2 and AcPIM4, confirming their identities. The open reading frames upstream and downstream of MSMEG5136 are oriented in the reverse direction compared to MSMEG5136 (Fig. 2B), suggesting that MSMEG5136 is not part of an operon. Therefore, it is unlikely that the polar effect of inserting the MSMEG5136
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

8
deletion construct influenced the functions of downstream genes. We confirmed that the PIM6-deficient phenotypes were due to the disruption of MSMEG5136 by introducing an episomal copy of the gene under the control of a constitutive expression promoter in a plasmid derived from pMV261. As a negative control, a vector without the MSMEG5136 gene did not affect the PIM profile (Fig. 3A, lanes 4 and 7). In contrast, a vector carrying the MSMEG5136 gene restored the mutant phenotype to the wild-type phenotype (Fig. 3A, lanes 5 and 8). We confirmed the identity of AcPIM6 from the complemented mutant by purifying this species from an HPTLC plate and detecting an expected molecular ion by MALDI-TOF-MS (not shown). These results indicated that the disruption of MSMEG5136 is responsible for the mutant phenotypes. We also transfected the mutant with the M. tuberculosis homolog Rv1159, and found that the ectopic copy of Rv1159 reversed the PIM6 biosynthetic defect in a similar way to the M. smegmatis gene (Fig. 3A, lanes 6 and 9). This result demonstrates that Rv1159 is a functional homolog of MSMEG5136. When these genes were transfected into wild-type cells, they did not affect the levels of PIM species (Fig. 3A, lanes 1-3). Effect of MSMEG5136 deletion on cell wall structure, in vitro growth and viability — Next, we examined the general characteristics of one of the mutant clones in detail during axenic culture growth. First, we examined various components of the cell wall and plasma membrane. As described above, the mutants themselves (DXO) and those transfected with an empty vector (DXO-V) were devoid of PIM6 species, and instead accumulated PIM4 species (Fig. 4A). As previously observed (50), diacyl species such as Ac2PIM2 and Ac2PIM6 accumulated in stationary phase cultures. The phospholipid profiles were examined by HPTLC and molybdenum blue visualization (Fig. 4B), and consistent with the previous report (50), phosphatidylethanolamine (PE) and PI became less abundant in the stationary phase. Importantly, there were no obvious defects in the synthesis of any of the major phospholipids, including PI, PE and cardiolipin (CL), in the mutant cells. The steady-state levels of a second class of mycobacterial glycolipids, the
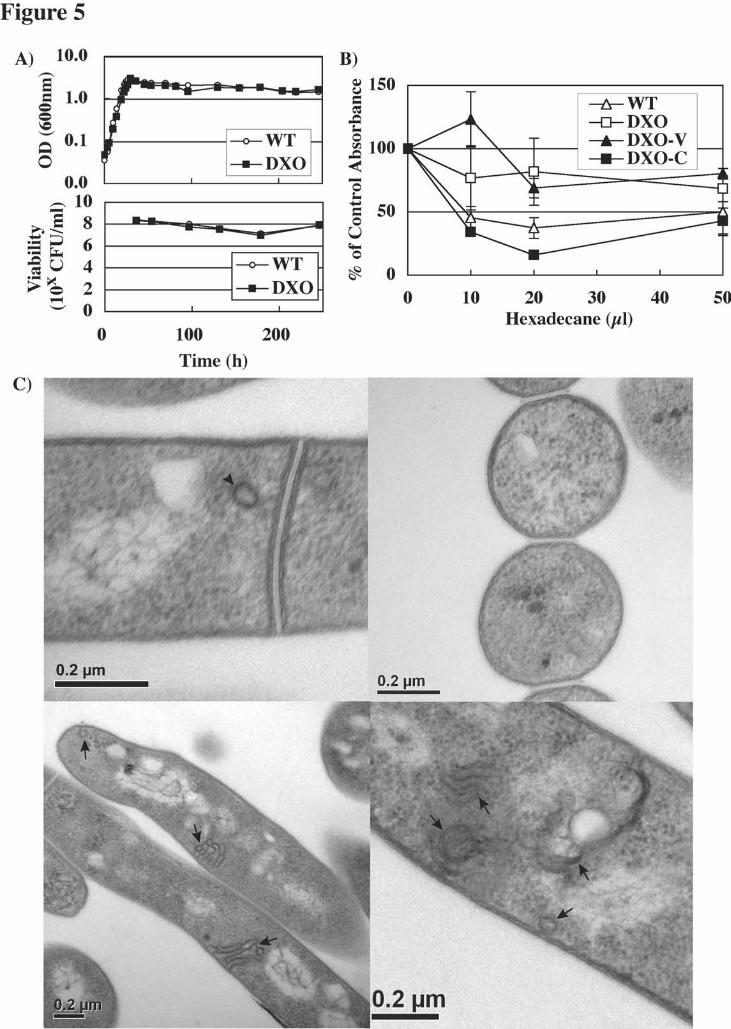
glycopeptidolipids (GPLs) were also identical in both wild-type and mutant strains (Fig. 4C). Significantly, the PimE deletion mutant synthesized wild-type levels of both LM and LAM, confirming that synthesis of AcPIM6 is not required for LM/LAM biosynthesis (Fig. 4D). Interestingly, dramatic and previously unreported reductions in the steady-state concentrations of LM and LAM were observed as both the wild-type and mutant strains entered stationary phase. Finally, GC-MS analysis of the polar metabolite fraction indicated that the mutants synthesized similar levels of an intracellular mannose-containing glycoconjugate, MMP, as the wild-type cells (Fig. 4E). Next, we examined the growth rate of the mutant, and found that it grew at essentially the same rate as the wild-type strain (Fig. 5A, upper panel). We considered the possibility that the viability of the mutant may be compromised during the stationary phase, but it proved able to sustain a similar viability to the wild-type strain during the course of a ~250 h incubation, as evaluated by colony-forming units (Fig. 5A, lower panel). The integrity of the cell wall in the mutant was examined using two approaches. In the first approach, the tendencies of wild-type and mutant cells to partition into a hexadecane phase, a measure of their surface hydrophobicity, were evaluated (45). The mutant showed a lower tendency to partition into hexadecane phase than wild-type or complemented strain (Fig. 5B), suggesting that its surface was less hydrophobic. In the second approach, the cell wall structure was examined directly by transmission electron microscopy. In the mutant cells, there was no obvious ultrastructural defect in the cell wall (Fig. 5C). Instead, we observed a frequent occurrence of internal membrane structures in the mutant cell cytoplasm (Fig. 5C, lower panels, arrows). These membrane structures appear as stacks or vesicle clusters and are often located near plasma membrane. Occasional profiles indicated that they can be contiguous with the plasma membrane. In wild-type cells, internal membrane structures were very rare though simple vesicular structures were occasionally observed (Fig. 5C, upper left panel, arrowhead). Upon complementation of the mutant cells following introduction of an ectopic
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

9
MSMEG5136 gene, the internal membrane structures disappeared, and the overall appearance became indistinguishable from that of wild-type cells (not shown). Taken together, MSMEG5136 gene deletion resulted in the disappearance of PIM6 species and concomitant increases in PIM4 species. This alteration in the PIM composition within the plasma membrane-cell wall complex had minimum effects in terms of the overall cell wall structure and viability, although some defects, especially in the plasma membrane homeostasis, were clearly evident. MSMEG5136 encodes a PPM-dependent mannosyltransferase involved in the fifth mannose transfer — The accumulation of AcPIM4 and Ac2PIM4 suggested that the MSMEG5136 gene product is involved in the fifth mannose transfer of the PIM biosynthesis pathway, which is thought to be mediated by a PPM-dependent mannosyltransferase (8). To demonstrate the specific defects of the mutant more directly, we examined the PIM biosynthesis of the mutant by two different methods. First, we conducted a pulse-chase metabolic labeling experiment using [3H]mannose or [3H]inositol. We labeled logarithmically growing cells with [3H]mannose for 15 min, and then chased in the presence of excess non-radioactive mannose for up to 6 h. The results demonstrated initial synthesis of the mannose donor PPM as well as apolar PIM intermediates, such as Ac2PIM1 and AcPIM2, in both wild-type and mutant cells (Fig. 6A). In the wild-type cells, the radiolabeled PPM decreased during the chase, with a concomitant increase in radiolabeled polar PIM species, such as AcPIM4, AcPIM5 and AcPIM6. After 6 h, the radiolabel was predominantly found in AcPIM2 and AcPIM6. In the mutant cells, radiolabel was chased from PPM into AcPIM4, with no label appearing in AcPIM5 or AcPIM6 (Fig. 6A). Radiolabel was chased into the LM/LAM fraction in both wild-type and mutant cells (Fig. 6B), confirming that the LM/LAM pathway was unaffected by the MSMEG5136 gene deletion. In a parallel experiment, the de novo synthesis of PIMs was followed using [3H]inositol. After a 5 min pulse and 5 h chase, the mutant cells only accumulated AcPIM4, whereas the wild-type cells AcPIM6 (Fig. 6C), again suggesting that the transfer of the fifth mannose is defective in
the deletion mutant. In the second approach, we examined PIM biosynthesis using a previously established cell-free system (8). A cell lysate was incubated with GDP-[3H]mannose for up to 2 h in a continuous labeling experiment. As shown in Fig. 6D, complemented mutant cells that carry the authentic gene episomally (DXO-C) were able to synthesize polar PIM species, such as AcPIM5 and AcPIM6. In contrast, mutant cells transfected with an empty vector (DXO-V) were unable to synthesize AcPIM5 and AcPIM6, but did synthesize apolar PIM intermediates up to AcPIM4. Interestingly, these mutants also synthesized novel polar intermediates that migrated slightly faster than authentic AcPIM5 and AcPIM6 (Fig. 6D, AcPIM5-like and AcPIM6-like). Because terminal mannose residues of authentic AcPIM5 and AcPIM6 are α1,2-linked, we wanted to examine if these novel intermediates carried terminal α1,2-linked mannoses. Total lipid extracts (prepared as in Fig. 6D) were digested with α1,2-specific mannosidase (ASAM) and the sensitivity of these intermediates to ASAM monitored by HPTLC. As a control, AcPIM6 was sensitive to ASAM treatment (>30% digestion) (not shown). In contrast, the two novel intermediates synthesized in the empty-vector-transfected mutant cell lysate were essentially resistant to ASAM treatment (<5% digestion) (not shown). These data suggest that AcPIM5-like and AcPIM6-like species synthesized in vitro do not carry terminal α1,2-linked mannose residues, being consistent with the possibility that PimE is an α1,2-mannosyltransferase. A conserved aspartic acid residue is critical for the enzyme function — In a study of human PIG-M mannosyltransferase, an aspartic acid residue located in the hydrophilic loop between the first and second transmembrane domains was found to be essential for the function of the enzyme (31). We conducted a multiple sequence alignment with the T-COFFEE software (51) using MSMEG5136 homologs from Mycobacterium species together with human PIG-M protein. Remarkably, the aspartic acid residue known to be essential for the function of the human enzyme was conserved in the mycobacterial enzymes (position 58 for M. smegmatis; Fig. 7A). To examine whether this aspartic acid residue is important for the
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

10
function of the mycobacterial enzyme, we conducted site-directed mutagenesis and created a D58A mutant, in which the aspartic acid residue at position 58 was replaced with alanine. We found additional 5 aspartic acid residues in the hydrophilic loop between the first and second transmembrane domains, and therefore created 5 further mutants, each of which carried a site-directed mutation at one of these 5 aspartic acids (D67A, D70A, D77A, D82A and D86A). To confirm that the mutant proteins were expressed, we placed a FLAG epitope tag at the C-terminus of each protein. As shown in Figs. 7B and 7C, the D58A mutant could not restore the mutant phenotype, despite efficient expression of the protein (lanes 3-6). In contrast, FLAG-tagged wild-type protein corrected the mutant phenotype, indicating that the epitope tag did not interfere with the enzyme function (lane 2). The mutations at the other aspartic acid residues did not interfere with the enzyme activity, and these mutant proteins fully restored AcPIM6/Ac2PIM6 synthesis (not shown). Subcellular localization of MSMEG5136 — Previously, we demonstrated that PIM biosynthesis is compartmentalized into two distinct plasma membrane fractions, termed PMf and PM-CW (50). The initial steps in PIM biosynthesis were predominantly localized to the PMf fraction, while the PPM-dependent mannosyltransferase reactions in the later part of the pathway were restricted to the PM-CW fraction. We therefore predicted that MSMEG5136, which mediates the PPM-dependent mannose transfer, should be localized in the PM-CW fraction in our subcellular fractionation assay (Fig. 8A). Consistent with previous studies, PIM1 synthesis was enriched in the PMf fraction (Fig. 8B, fractions 5-7). The synthesis of polar PIM species, such as AcPIM5 and AcPIM6, was enriched in the PM-CW fraction (fractions 8-10). PPM synthetic activities were found in both PMf and PM-CW, as previously described (50). Some PPM synthetic activities were also found in the top fractions of the gradient (fractions 1-2), but the significance of this activity was not pursued in the present study. Consistent with our prediction, FLAG-tagged MSMEG5136 was predominantly localized in the PM-CW fraction (Fig. 8C), and essentially no protein was detected in the PMf fraction.
DISCUSSION We have identified a PPM-dependent α1,2-mannosyltransferase that is required for biosynthesis of a major PIM species. According to the nomenclature for mannosyltransferases involved in PIM biosynthesis, we have designated this enzyme PimE. Previously, PimF was reported as a mannosyltransferase involved in the later stage of PIM/LAM biosynthesis (27). However, a recent study suggests that this enzyme is involved in the biosynthesis of glycosylated acyltrehalose lipooligosaccharides rather than PIM (28). Thus, PimE represents the first example of a PPM-dependent mannosyltransferase involved in PIM/LAM biosynthesis in mycobacteria. Several lines of evidence strongly support our conclusion that PimE is a PPM-dependent mannosyltransferase itself, rather than an auxiliary or regulatory protein. First, PimE is predicted to be a polytopic membrane protein homologous to human PIG-M, a DPM-dependent mannosyltransferase involved in the biosynthesis of eukaryotic GPIs. Second, we identified a functionally important aspartic acid residue in a hydrophilic loop between the first and second transmembrane domains. This aspartic acid residue is conserved in eukaryotic PIG-M, and is important for the function of human PIG-M (31), suggesting that PimE is a mannosyltransferase with a similar catalytic mechanism to PIG-M mannosyltransferases. Third, in support of the bioinformatics prediction that PimE is an integral plasma membrane protein, epitope-tagged PimE was specifically localized in a distinct plasma membrane domain, termed PM-CW, where PPM-dependent mannosyltransferase reactions are enriched (50). Taken together, PimE is most likely to be a PPM-dependent mannosyltransferase with an evolutionary relationship to the eukaryotic GPI mannosyltransferase PIG-M. Considering the evolutionary distance between metazoans and bacteria, it is remarkable that PIG-M and PimE can be identified as homologs by PSI-BLAST searches. It is interesting that PimE, being an α1,2-mannosyltransferase, is most closely related to the α1,4-mannosyltransferase PIG-M, rather than α1,2-mannosyltransferases such as PIG-B and
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

11
Smp3. PIG-M is the first mannosyltransferase of GPI biosynthetic pathway, and it is possible that PIG-M represents the prototype enzyme, which is most closely related to the ancestral protein common to PimE and eukaryotic GPI mannosyltransferases. PIG-V, PIG-B and Smp3 may have evolved from PIG-M in the eukaryotic lineage for the elaboration of GPI core glycan, and thus more divergent from the bacterial mannosyltransferase PimE. The evolutionary conservation may be due to two structural requirements common to PIG-M and PimE enzymes, namely that the donor substrates are polyprenol-based molecules and the acceptor substrates are PI-based molecules. Presumably, a common ancestral enzyme had structural features that were able to fulfill a prototype glycolipid biosynthetic reaction using a PI-based acceptor and polyprenol-phosphate-based mannose donor. These structural requirements were perhaps sufficient to allow conservation of the primary sequence of the protein during the evolution of mannosyltransferases in each lineage. Whether the ancestral enzyme was present before the divergence of bacteria and the archea/eukarya lineages or acquired by lateral gene transfer later during evolution remains to be evaluated. The deletion mutant lacking a functional pimE gene accumulated AcPIM4 and Ac2PIM4. These findings suggest that PimE is an α1,2-mannosyltransferase that transfers mannose from PPM to PIM4 intermediates. This possibility was supported by both metabolic and cell-free radiolabeling studies. In the metabolic labeling experiments, accumulation of AcPIM4 was evident in the mutant cells, and no further reactions toward AcPIM6 synthesis were detected, strongly supporting that PimE mediates the mannosyltransferase reaction from AcPIM4 to AcPIM5. Similarly, in the mutant cell-free system, accumulation of AcPIM4 was observed, and no synthesis of AcPIM5 or AcPIM6 could be detected. In contrast to the metabolic labeling, however, some species more polar than AcPIM4 were identified. These species, designated AcPIM5-like and AcPIM6-like, were resistant to α1,2-specific ASAM treatment, suggesting that they are not involved in the synthesis of AcPIM6. These novel intermediates may be involved in the synthesis of the α1,6 linear chains of LM and LAM, which are still made by the mutant.
The phenotype of pimE deletion mutant is reminiscent of another mutant we recently characterized, which has a defect in the gene lpqW, and lacks PIM6 species but accumulates PIM4 species (22). Similar to pimE deletion mutants, lpqW deletion mutants are viable and have an intact cell wall. These findings are in striking contrast to an Ino1-deleted inositol auxotroph (44), which ceases growth upon inositol starvation and manifests pleiotropic loss of the major cell wall components. A detailed analysis of the inositol auxotroph suggested that the loss of viability of this mutant is associated with the loss of PIM6 species. Both pimE and lpqW deletion mutants still accumulate PIM4 species, suggesting that PIM4 species can functionally replace PIM6 species. Despite the viability of pimE deletion mutants, a significant alteration in the cell surface hydrophobicity was observed by a hexadecane partitioning assay. More striking, the mutant cells accumulated internal membrane structures, indicating that the accumulated PIM4 species do not fully replace the physiological functions of PIM6 species. The exact functions of PIM6 species in mycobacterial cell physiology remain to be determined, but they appear to be important components for the structural integrity of the plasma membrane-cell wall complex. PimE is not involved in LM/LAM synthesis, as evidenced by the fact that LM and LAM were synthesized at comparable levels in the mutant and wild-type cells. Metabolic [3H]mannose labeling also demonstrated that the radioactive precursors were chased into the LM/LAM fraction, while AcPIM5-6 synthesis was absent. These data are consistent with the possibility that the LM/LAM pathway diverges from the PIM pathway before the synthesis of AcPIM5, suggesting that AcPIM4 is the most likely intermediate at the branch point of the PIM and LM/LAM pathways as previously proposed (8,22). Interestingly, LM and LAM were reduced to barely detectable levels in the stationary phase. These reductions in the LM and LAM levels may be associated with shutdown of the LM/LAM biosynthetic pathway and/or enhancement of the AcPIM6 biosynthetic pathway in combination with degradation and/or shedding of the existing LM and LAM. The precise mechanisms of the metabolic control are largely unknown, and it will be interesting to
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

12
delineate the roles that PimE may play in this metabolic regulation. It remains possible that PimE also mediates the sixth mannose transfer, which represents the final step of AcPIM6 synthesis. We previously reported that the two mannosyl transfer steps from AcPIM4 to AcPIM6 are efficient in a cell-free system, which may suggest that these steps are mediated by a processive enzyme (8). We considered the possibility that PimE forms a dimer to mediate the two successive α1,2 mannosyl transfers without releasing the substrate. However, expression of the PimE D58A in wild-type cells does not result in a dominant-negative effect on polar PIM biosynthesis, indicating that PimE does not form oligomers (unpublished data). Ideally, the biochemical function of PimE would be best studied in reconstitution assays using the purified enzyme and synthetic substrates. However, such systems have been proven to be difficult for polytopic membrane proteins, and in the case of PPM- or DPM-dependent mannosyltransferases involved in glycolipid biosynthesis, partial success has only been achieved in the case of human PIG-M activity (31). Reconstitution of PimE enzymatic activities remains a challenge for future studies, and will provide important insights into the
biosynthesis of PIMs and LAM. M. tuberculosis Rv1159 was found to be a functional homolog of MSMEG5136. It will be interesting to examine whether a similar AcPIM6 deficiency in M. tuberculosis affects the integrity of the plasma membrane and cell wall, and whether such defects affect the survival of mycobacteria in macrophages or during infection in animal models. Although pimA is essential for M. smegmatis growth (23), whether or not PIMs and LAM are essential for M. tuberculosis infection remains to be examined. M. tuberculosis pimE deletion mutant will be a useful tool for investigations along these lines. Acknowledgements — We thank Keiko Kinoshita and Fumiko Mori for technical assistance, Dr. Hisashi Ashida for discussion, and Dr. Toshifumi Takao for the MALDI-TOF-MS. This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan and the Core Research for Evolutional Science and Technology, Japan Science and Technology Agency. Y.S.M. was supported by an International Human Frontier Science Program postdoctoral fellowship and the Uehara Memorial Foundation.
REFERENCES
1. Brennan, P. J., and Nikaido, H. (1995) Annu Rev Biochem 64, 29-63 2. Brennan, P. J. (2003) Tuberculosis (Edinb) 83, 91-97 3. Dmitriev, B. A., Ehlers, S., Rietschel, E. T., and Brennan, P. J. (2000) Int J Med Microbiol
290, 251-258 4. Jarlier, V., and Nikaido, H. (1994) FEMS Microbiol Lett 123, 11-18 5. Daffe, M., and Draper, P. (1998) Adv Microb Physiol 39, 131-203 6. Jackson, M., Crick, D. C., and Brennan, P. J. (2000) J Biol Chem 275, 30092-30099 7. Besra, G. S., Morehouse, C. B., Rittner, C. M., Waechter, C. J., and Brennan, P. J. (1997) J
Biol Chem 272, 18460-18466 8. Morita, Y. S., Patterson, J. H., Billman-Jacobe, H., and McConville, M. J. (2004) Biochem J
378, 589-597 9. Brennan, P., and Ballou, C. E. (1967) J Biol Chem 242, 3046-3056 10. Brennan, P., and Ballou, C. E. (1968) J Biol Chem 243, 2975-2984 11. Hill, D. L., and Ballou, C. E. (1966) J Biol Chem 241, 895-902 12. Gilleron, M., Quesniaux, V. F., and Puzo, G. (2003) J Biol Chem 278, 29880-29889 13. Apostolou, I., Takahama, Y., Belmant, C., Kawano, T., Huerre, M., Marchal, G., Cui, J.,
Taniguchi, M., Nakauchi, H., Fournie, J. J., Kourilsky, P., and Gachelin, G. (1999) Proc Natl Acad Sci U S A 96, 5141-5146
14. Fischer, K., Scotet, E., Niemeyer, M., Koebernick, H., Zerrahn, J., Maillet, S., Hurwitz, R., Kursar, M., Bonneville, M., Kaufmann, S. H., and Schaible, U. E. (2004) Proc Natl Acad Sci U S A 101, 10685-10690
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

13
15. de la Salle, H., Mariotti, S., Angenieux, C., Gilleron, M., Garcia-Alles, L. F., Malm, D., Berg, T., Paoletti, S., Maitre, B., Mourey, L., Salamero, J., Cazenave, J. P., Hanau, D., Mori, L., Puzo, G., and De Libero, G. (2005) Science 310, 1321-1324
16. Briken, V., Porcelli, S. A., Besra, G. S., and Kremer, L. (2004) Mol Microbiol 53, 391-403 17. Nigou, J., Gilleron, M., Rojas, M., Garcia, L. F., Thurnher, M., and Puzo, G. (2002) Microbes
Infect 4, 945-953 18. Chatterjee, D., and Khoo, K. H. (1998) Glycobiology 8, 113-120 19. Chua, J., Vergne, I., Master, S., and Deretic, V. (2004) Curr Opin Microbiol 7, 71-77 20. Chatterjee, D., Hunter, S. W., McNeil, M., and Brennan, P. J. (1992) J Biol Chem 267,
6228-6233 21. Patterson, J. H., Waller, R. F., Jeevarajah, D., Billman-Jacobe, H., and McConville, M. J.
(2003) Biochem J 372, 77-86 22. Kovacevic, S., Anderson, D., Morita, Y. S., Patterson, J., Haites, R., McMillan, B. N., Coppel,
R., McConville, M. J., and Billman-Jacobe, H. (2006) J Biol Chem 281, 9011-9017 23. Kordulakova, J., Gilleron, M., Mikusova, K., Puzo, G., Brennan, P. J., Gicquel, B., and
Jackson, M. (2002) J Biol Chem 277, 31335-31344 24. Kordulakova, J., Gilleron, M., Puzo, G., Brennan, P. J., Gicquel, B., Mikusova, K., and
Jackson, M. (2003) J Biol Chem 278, 36285-36295 25. Schaeffer, M. L., Khoo, K. H., Besra, G. S., Chatterjee, D., Brennan, P. J., Belisle, J. T., and
Inamine, J. M. (1999) J Biol Chem 274, 31625-31631 26. Kremer, L., Gurcha, S. S., Bifani, P., Hitchen, P. G., Baulard, A., Morris, H. R., Dell, A.,
Brennan, P. J., and Besra, G. S. (2002) Biochem J 363, 437-447 27. Alexander, D. C., Jones, J. R., Tan, T., Chen, J. M., and Liu, J. (2004) J Biol Chem 279,
18824-18833 28. Burguiere, A., Hitchen, P. G., Dover, L. G., Kremer, L., Ridell, M., Alexander, D. C., Liu, J.,
Morris, H. R., Minnikin, D. E., Dell, A., and Besra, G. S. (2005) J Biol Chem 280, 42124-42133
29. Zhang, N., Torrelles, J. B., McNeil, M. R., Escuyer, V. E., Khoo, K. H., Brennan, P. J., and Chatterjee, D. (2003) Mol Microbiol 50, 69-76
30. Kang, J. Y., Hong, Y., Ashida, H., Shishioh, N., Murakami, Y., Morita, Y. S., Maeda, Y., and Kinoshita, T. (2005) J Biol Chem 280, 9489-9497
31. Maeda, Y., Watanabe, R., Harris, C. L., Hong, Y., Ohishi, K., Kinoshita, K., and Kinoshita, T. (2001) EMBO J 20, 250-261
32. Taron, B. W., Colussi, P. A., Grimme, J. M., Orlean, P., and Taron, C. H. (2004) J Biol Chem 279, 36083-36092
33. Takahashi, M., Inoue, N., Ohishi, K., Maeda, Y., Nakamura, N., Endo, Y., Fujita, T., Takeda, J., and Kinoshita, T. (1996) EMBO J 15, 4254-4261
34. Canivenc-Gansel, E., Imhof, I., Reggiori, F., Burda, P., Conzelmann, A., and Benachour, A. (1998) Glycobiology 8, 761-770
35. Benghezal, M., Lipke, P. N., and Conzelmann, A. (1995) J Cell Biol 130, 1333-1344 36. Fabre, A. L., Orlean, P., and Taron, C. H. (2005) FEBS J 272, 1160-1168 37. Cole, S. T., Eiglmeier, K., Parkhill, J., James, K. D., Thomson, N. R., Wheeler, P. R., Honore,
N., Garnier, T., Churcher, C., Harris, D., Mungall, K., Basham, D., Brown, D., Chillingworth, T., Connor, R., Davies, R. M., Devlin, K., Duthoy, S., Feltwell, T., Fraser, A., Hamlin, N., Holroyd, S., Hornsby, T., Jagels, K., Lacroix, C., Maclean, J., Moule, S., Murphy, L., Oliver, K., Quail, M. A., Rajandream, M. A., Rutherford, K. M., Rutter, S., Seeger, K., Simon, S., Simmonds, M., Skelton, J., Squares, R., Squares, S., Stevens, K., Taylor, K., Whitehead, S., Woodward, J. R., and Barrell, B. G. (2001) Nature 409, 1007-1011
38. Hirokawa, T., Boon-Chieng, S., and Mitaku, S. (1998) Bioinformatics 14, 378-379 39. Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman,
D. J. (1997) Nucleic Acids Res 25, 3389-3402 40. Snapper, S. B., Melton, R. E., Mustafa, S., Kieser, T., and Jacobs, W. R., Jr. (1990) Mol
Microbiol 4, 1911-1919 41. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) Molecular cloning: a laboratory manual,
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

14
2nd ed. Ed. 42. Jeevarajah, D., Patterson, J. H., Taig, E., Sargeant, T., McConville, M. J., and Billman-Jacobe,
H. (2004) J Bacteriol 186, 6792-6799 43. Jacobs, W. R., Jr., Kalpana, G. V., Cirillo, J. D., Pascopella, L., Snapper, S. B., Udani, R. A.,
Jones, W., Barletta, R. G., and Bloom, B. R. (1991) Methods Enzymol 204, 537-555 44. Haites, R. E., Morita, Y. S., McConville, M. J., and Billman-Jacobe, H. (2005) J Biol Chem
280, 10981-10987 45. Rosenberg, M., Gutnick, D., and Rosenberg, E. (1980) FEMS Microbiol Lett 9, 29-33 46. Hunter, S. W., Gaylord, H., and Brennan, P. J. (1986) J Biol Chem 261, 12345-12351 47. Marques, M. A., Chitale, S., Brennan, P. J., and Pessolani, M. C. (1998) Infect Immun 66,
2625-2631 48. Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003) Mol Microbiol 48, 77-84 49. Billman-Jacobe, H., Haites, R. E., and Coppel, R. L. (1999) Antimicrob Agents Chemother 43,
3011-3013 50. Morita, Y. S., Velasquez, R., Taig, E., Waller, R. F., Patterson, J. H., Tull, D., Williams, S. J.,
Billman-Jacobe, H., and McConville, M. J. (2005) J Biol Chem 280, 21645-21652 51. Notredame, C., Higgins, D. G., and Heringa, J. (2000) J Mol Biol 302, 205-217 52. Schafer, A., Tauch, A., Jager, W., Kalinowski, J., Thierbach, G., and Puhler, A. (1994) Gene
145, 69-73 53. Patterson, J. H., McConville, M. J., Haites, R. E., Coppel, R. L., and Billman-Jacobe, H.
(2000) J Biol Chem 275, 24900-24906
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

15
FIGURE LEGENDS Figure 1. PIM biosynthetic pathway. The order of the reactions from PIM1 to AcPIM2 may be flexible, with PIM2 as an intermediate instead of AcPIM1. The terminal two mannoses of AcPIM6 are α1,2-linked. LM and LAM contain a long α1,6 mannose chain with modification by single α1,2 mannose residues (not shown). The exact mannose residue modified by the arabinan side chain has not been determined. Figure 2. MSMEG5136 protein homologs and construction of MSMEG5136 gene-deletion mutants. (A) CLUSTALW alignment of MSMEG5136 protein homologs from various mycobacteria, including M. tuberculosis (MT), M. bovis (MB), M. avium paratuberculosis (MAP), M. leprae (ML) and M. smegmatis (MS). The underlined regions indicate the transmembrane domains of M. smegmatis protein predicted by TMpred. Other algorithms, such as SOSUI, gave similar results (not shown). (B) Strategies for disrupting the MSMEG5136 gene. Black rectangles: MSMEG5136 and other open reading frames in the same orientation as MSMEG5136. Gray rectangles: Open reading frames in the reverse orientation to MSMEG5136. Arrows indicate the orientation of the genes. S: SacI sites. SacB: a genetically modified sacB gene (52), which confers sucrose sensitivity to mycobacteria. Sm: streptomycin-resistance cassette. Km: kanamycin-resistance cassette. Gray bar: region of DNA recognized by the Southern blot probe used in panel C. (C) Southern blot using SacI-digested genomic DNA from wild-type (lane 1), SXO28-6 (lane 2), DXO28-3 (lane 3), DXO28-4 (lane 4) and DXO28-6 (lane 5) cells. Figure 3. PIM profiles in deletion mutants. (A) Wild-type cells and two mutant strains (DXO28-3 and DXO28-4) were transfected with an expression vector carrying no gene (V), MSMEG5136 (MS) or M. tuberculosis Rv1159 (MT) downstream of the promoter. The cells were collected from Middlebrook 7H10 agar plates and the purified lipids were analyzed by HPTLC followed by orcinol staining. Loading was adjusted for equal cell pellet weights. Only part of the HPTLC plate is shown. (B-C) MALDI-TOF mass spectra of putative AcPIM2 (B) or AcPIM4 (C) species derived from mutant cells. Glycolipid bands were scraped from HPTLC sheets, extracted with chloroform/methanol/water (10:10:3, v/v/v) and further purified by 1-butanol/water biphasic partitioning, prior to MALDI-TOF-MS analysis. Figure 4. Compositional analysis of deletion mutants during axenic culture growth. Wild-type cells (WT), DXO28-3 cells (DXO), DXO28-3 cells transfected with an empty expression vector (DXO-V) and DXO28-3 cells complemented with an MSMEG5136 expression vector (DXO-C) were harvested at specific time points (14, 30 and 83 h, representing the log, early stationary and stationary phases, respectively) in a growth curve in Middlebrook 7H9 broth. Loading was adjusted for equal cell pellet weights. Only the relevant parts of the HPTLC plates (A-C) and SDS-PAGE gel (D) are shown. All analyses were repeated twice or more, and representative data are shown. PIMs (A) and GPLs (C) were detected after orcinol staining, while phospholipids (B) were detected with molybdenum blue. Lipid identities were assigned based on previous studies (6,50,53). (D) LM/LAM species were visualized by ProQ Emerald Carbohydrate staining (20,22). (E) Levels of methyl mannose from MMP were quantified by GC-MS. Figure 5. Characterization of mutant phenotypes by the growth rate, cell wall hydrophobicity and electron microscopy. (A) Upper panel: Growth rate in Middlebrook 7H9 broth as determined by the OD600. Lower panel: Viability as determined by colony forming units (CFUs). Each data point represents an average of two measurements. (B) Hexadecane partitioning to determine the cell surface hydrophobicities of wild-type cells (WT), DXO28-3 cells (DXO), DXO28-3 cells transfected with an empty vector (DXO-V) and DXO28-3 complemented with an expression vector carrying MSMEG5136 (DXO-C). The experiment was performed in triplicate, and the average values are plotted with standard errors of the mean. (C) Electron micrographs of wild-type cells (upper panels) and DXO28-3 cells (lower panels). The arrowhead indicates an example of the occasional membrane vesicles in wild-type cells that appear morphologically simple. Arrows indicate the intricate
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

16
membrane structures frequently found in mutant cells. Figure 6. PIM biosynthesis in wild-type and deletion mutant (DXO28-3) cells. (A) Cells in mid-log phase were pulse-labeled with [3H]mannose, and then suspended in medium containing unlabeled mannose. The cells were harvested at the indicated chase times, the labeled glycolipids analyzed by HPTLC, and detected by fluorography. (B) [3H]Mannose-labeled LM and LAM from the pulse-chase labeling experiment (panel A) were extracted from the delipidated pellets and purified by octyl-Sepharose chromatography. The ratios of the scintillation counts of the LM/LAM fractions to the total lipid fractions (combining the butanol-purified PPM/PIM fraction and octyl-Sepharose-purified LM/LAM fraction) over the chase period are shown. (C) Cells in mid-log phase were pulse-labeled with [3H]inositol, and then suspended in medium containing unlabeled inositol for 5 h. Labeled inositol lipids were extracted in chloroform/methanol/water, analyzed by HPTLC and detected by fluorography. (D) Cell lysates were prepared from DXO28-3 cells transfected with an empty vector (DXO-V) or an expression vector carrying MSMEG5136 (DXO-C) and incubated with GDP-[3H]mannose for the indicated times. Labeled lipids were analyzed by HPTLC and detected by autoradiography. Only part of the HPTLC plate is shown. The identity of all labeled lipids were assigned based on previous studies (8,21,44). Figure 7. (A) T-COFFEE alignment of mycobacterial MSMEG5136 homologs and human PIG-M mannosyltransferase. The positions of aspartic acid residues in the M. smegmatis MSMEG5136 sequence are indicated by black dots and numbers. MT, M. tuberculosis; MB, M. bovis; ML, M. leprae; MAP, M. avium paratuberculosis; MS, M. smegmatis; HS, Homo sapiens. (B) Left panel: Western blotting using an anti-FLAG antibody. The ~40 kDa band represents MSMEG5136-FLAG. The reason for the slightly faster migration of MSMEG5136-FLAG than the predicted molecular weight (45.5 kDa) is unknown, but may reflect the highly hydrophobic nature of the protein (~50% frequency of hydrophobic amino acid residues), leading to more avid binding of SDS. The band at ~60 kDa is non-specific. Some faint bands are observed in the high molecular weight region (80-200 kDa), but their physiological relevance is currently unclear. Right panel: Ponceau S staining of a western blot membrane confirming equal loading of the proteins. Lane 1: DXO28-3 cells transfected with an empty vector; lane 2: DXO28-3 cells transfected with an expression vector carrying a gene encoding wild-type MSMEG5136; lanes 3-6: four independent clones of DXO28-3 cells transfected with an expression vector carrying a gene encoding the D58A site-directed mutant of MSMEG5136. (C) Orcinol staining of PIM species extracted and analyzed by HPTLC. The lanes are arranged in the same manner as those in panel B. Figure 8. Subcellular fractionation of PIM biosynthesis by sucrose gradient sedimentation. DXO28-3 cells complemented by an expression vector carrying a gene encoding wild-type MSMEG5136-FLAG was lysed and fractionated by 10-60% sucrose gradient. (A) Sucrose gradient and protein concentration profiles. (B) PIM biosynthetic activity determined by GDP-[3H]mannose labeling. Only part of the HPTLC plate is shown. (C) Localization of FLAG-tagged MSMEG5136 determined by western blotting using an anti-FLAG antibody.
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

17
Table 1 MALDI-TOF-MS analysis of M. smegmatis PIMs Only the major peaks are shown. The fatty acid composition was deduced based on previous analysis (8). N.D., not detected.
[M-H]- (m/z) PIM Species
WT DXO28-3
Predicted [M-H]- (m/z)
Predicted Fatty Acid Composition
AcPIM2 1415.5 1415.9 1414.8 C16:0/C19:0/C16:0 Ac2PIM2 1680.0 1680.4 1681.3 C16:0/C19:0/C16:0/C18:0 AcPIM4 N.D. 1739.6 1739.1 C16:0/C19:0/C16:0 Ac2PIM4 N.D. 2004.0 2005.5 C16:0/C19:0/C16:0/C18:0 AcPIM6 2063.9 N.D. 2063.4 C16:0/C19:0/C16:0 Ac2PIM6 2328.3 N.D. 2329.8 C16:0/C19:0/C16:0/C18:0
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Yusuke Maeda and Taroh KinoshitaFumiki Nakatani, Ruth E. Haites, Helen Billman-Jacobe, Malcolm J. McConville,
Yasu S. Morita, Chubert B. C. Sena, Ross F. Waller, Ken Kurokawa, M. Fleur Sernee,transfers the fifth mannose of phosphatidylinositol mannoside in mycobacteriaPimE is a polyprenol-phosphate-mannose-dependent mannosyltransferase that
published online June 27, 2006J. Biol. Chem.
10.1074/jbc.M604214200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from


















