
Genomic and comparative genomic analysis BIO520 BioinformaticsJim Lund.
Upload
irene-pierceCategory
view
223download
2
Phylogenetic Inference
Data
Optimality Criteria
Algorithms
Results
Practicalities
BIO520 Bioinformatics Jim Lund
Reading: Ch8

Our Goals
• Infer Phylogeny– Optimality criteria– Algorithm
• Determine the sequence of branching events that reflects the history of a group of organisms.

Phylogenetic Model Assumptions
• No transfer of genetic information by hybridization
• All sequences are homologous (orthologous, really)
• Each position in alignment homologous• Observed variation is valid sample from
included group• Positions evolve independently

Steps in Analysis
1. Data Model (Alignment)– alignment method– “trimming” to a phylogenetic set
2. DNA base substitution model3. Build Trees
– Algorithm based vs Criterion based– Distance based vs Character-based
4. Assess tree quality.

Choice of Input Data
• Data Type– Aligned sequences, RFLP, morphological
data…
• Molecule of interest– rRNA (general purpose)– Mitochondrial DNA– Selected genes
• Number/type of taxa– ingroup and outgroup

rRNA Genes
• Conserved across kingdoms
• Varies within species
• Widely sequenced, easy
• Long, lots of characters

Multiple Alignment Method
• Phylogenetic Assumptions
• Alignment parameters– (substitution matrix, gap cost)
• Aligned features– primary sequence, structure
• Optimization– statistical, non-statistical

Typical Alignment Method
• CLUSTAL, then manual editing– Manual editing for phylogeny– phylogenetic assumption in guide tree– parameters a priori and dynamic– Optimization
• Non-statistical
• Remove poorly aligned regions
• Test several gap penalties

Substitution Models
• G to A, C to T versus N to N
• Amino acid substitution
• Forwards and backwards weights identical?
• Site-to-site variation

Tree-Building Methods
• Distance-based methods– NJ, FM, ME, UPGMA
• Character-based methods– Maximum Parsimony (PAUP)– Maximum Likelihood (PHYLIP)
Algorithm choice is a contested, active research field.

Molecular phylogenetic tree building methods:
Are mathematical and/or statistical methods for inferring the divergence order of taxa, as well as the lengths of the branches that connect them. There are many phylogenetic methods available today, each having strengths and weaknesses. Most can be classified as follows:
COMPUTATIONAL METHODClustering algorithmOptimality criterion
DA
TA
TY
PE
Ch
arac
ters
(b
p, a
a)D
ista
nce
s
PARSIMONY
MAXIMUM LIKELIHOOD
UPGMA
NEIGHBOR-JOINING
MINIMUM EVOLUTION
LEAST SQUARES

Distance Methods
• Measure distance (dissimilarity)• Accurate if distances are all summative
(ultrametric)– NEVER true over large distanceNEVER true over large distance
• Methods– NJ (Neighbor joining)– FM (Fitch-Margoliash)– ME (Minimal Evolution)– UPGMA (Unweighted pair group method with
Arithmetic Mean)

Which Distance Method?
• UPGMA (Unweighted pair group method with Arithmetic Mean)
– Least accurate, still commonly used
• NJ (Neighbor joining)– EXTREMELY RAPID– GIVES ONLY 1 TREE
• ME (Minimal Evolution) and FM (Fitch-Margoliash) seem best
– Minimize tree path lengths

Inferring Trees and Ancestors
CCCAGGCCCAAG->
CCCAAGCCCAAA->
CCCAAACCCAAA->
CCCAAC

Different Criteria
1 CCCAGG2 CCCAAG3 CCCAAA4 CCCAAC
1-2 1
1-3 2
1-4 2
2-3 1
2-4 1
3-4 1
1,2 can be sister taxaAND
3,4 can be sister taxa
Infer ancestor of 1,2 and 3,4
Distance from 1/2, 3/4 equal

Character Methods
• Maximum Parsimony– minimal changes to produce data– can use different substitution models
• Maximum Likelihood– turns problem “inside out”, single most likely tree that
explains data• coin flip analogy
– increasingly popular• Bayesian
– Searches for Best Set of trees that explains data AND fits evolutionary model

Parsimony
CCCAGGCCCAAG->
CCCAAGCCCAAA->
CCCAAACCCAAA->
CCCAAC4 TAXA, 3 changes minimum
Search for shortest tree, the one with the fewest changes.

Likelihood Models
TEAM WIN LOSS
Yanks 100 40
Sox 90 50
Tigers 60 80
Hypothesis 1: All 3 teams are equally good.
Hypothesis 2: The Yankees are the best team.
Hypothesis 3: The Tigers are the worst team

Searching for Trees
# of Taxa # of Trees
3 1
4 3
5 15
10 2 x 106
50 3 x 1074
100 2 x 10182

Tree Search Algorithms
• Exhaustive– VERY
INTENSIVE
• Branch and Bound– Compromise
• Heuristic– FAST (usually
start with NJ)
# of taxa NJ Parsimony ML Bayes
10 0.2s 0.05s 4.1s 0.5 hr
50 .2s .7s 7hr 4hr

Evaluating Trees
• Consensus Tree• Randomized Trees
– Skewness tests
• Randomized Character Data– Permutation tests (permuted by column)
• Bootstrap, Jackknife– resampling techniques– Counts how often each clade appears in test data.– >70% probably correct; 50% overestimates
accuracy

Tree Congruence
• Tree-to-Tree Comparison– 2 different characters/same groups– Important for evaluating biological hypotheses
• Example:
• Did lentiviruses diverge within their current hosts only?
• Or did plant pathogenicity has arisen many times in fungi?

Inferring evolutionary relationships between the taxa requires rooting
the tree: To root a tree mentally, imagine that the tree is made of string. Grab the string at the root and tug on it until the ends of the string (the taxa) fall opposite the root:
A
BC
Root D
A B C D
RootNote that in this rooted tree, taxon A is no more closely related to taxon B than it is to C or D.
Rooted tree
Unrooted tree

Now, try it again with the root at another position:
A
BC
Root
D
Unrooted tree
Note that in this rooted tree, taxon A is most closely related to taxon B, and together they are equally distantly related to taxa C and D.
C D
Root
Rooted tree
A
B

Rooting Trees
• Molecular Clock– Root=midpoint of longest span– Unreliable, often wrong.
• Evidence– select fungus as root for plants, eg
• long branch attraction can be Extrinsic problem
• Paralog rooting– long branch problems

Phylogenetic Software
• PHYLIP– http://evolution.genetics.washington.edu/phylip.html– http://saf.bio.caltech.edu/www/saf_manuals/phylip/phylip.html
• PAUP: Pileup, Lineup, Paupsearch, Paupdisplay– http://paup.csit.fsu.edu/versions.html
• MrBayes– Bayesian trees– http://mrbayes.csit.fsu.edu/
• Treeview – Several programs going by this name have been written.– Draw/format phylogenic trees– Jave TreeView: http://jtreeview.sourceforge.net/

Phylogenetic Stories
• HIV– complete genome accessible– evolution rapid
• selection, neutralism?
• Primate evolution– Which primate is the closest relative to modern
humans?

HIV Genome Diversity
• Error prone (RT) replication
• High rate of replication– 1010 virions/day
• In vivo selection pressure
And In vivo recombination!

HIV tree
Recombinants?
ENV
GAG
AIDS 1996, 10:S13

Subtype E
ENV=A
“Bootscanning”
AIDS 1996, 10:S13
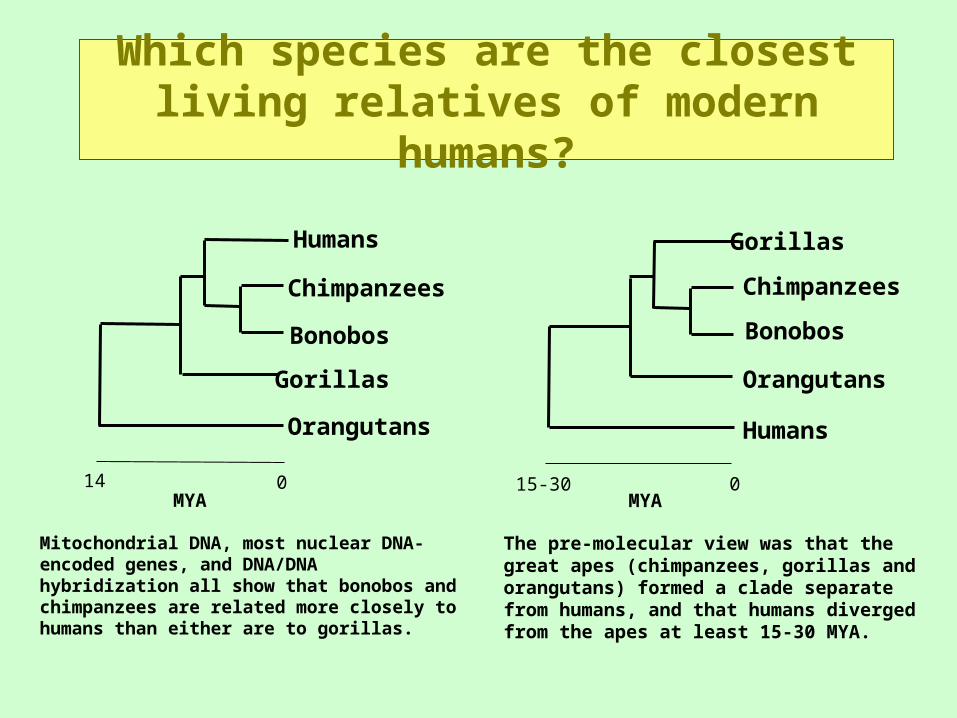
Which species are the closest living relatives of modern humans?
Mitochondrial DNA, most nuclear DNA-encoded genes, and DNA/DNA hybridization all show that bonobos and chimpanzees are related more closely to humans than either are to gorillas.
The pre-molecular view was that the great apes (chimpanzees, gorillas and orangutans) formed a clade separate from humans, and that humans diverged from the apes at least 15-30 MYA.
MYA
Chimpanzees
Orangutans Humans
Bonobos
GorillasHumans
Bonobos
Gorillas Orangutans
Chimpanzees
MYA015-30014

Phylogenetic Resources
• NCBI Taxonomy Browser– http://www.ncbi.nlm.nih.gov/Taxonomy/
• RDP database (Ribosomal Database Project)– http://rdp.cme.msu.edu/index.jsp
• “Tree of Life”– http://tolweb.org/tree/phylogeny.html

Practicalities
• Quality of input alignment critical• Examine data from all possible angles
– distance, parsimony, likelihood, Bayes
• Outgroup taxon critical– problem if outgroup shares a selective
property with a subset of ingroup
• Order of input can be problematic– Jumble them!


















