
Phylogenetic Analysis Unit 16 BIOL221T: Advanced Bioinformatics for Biotechnology Irene Gabashvili,...
61
Phylogenetic Analysis Phylogenetic Analysis Unit 16 Unit 16 BIOL221T BIOL221T : Advanced : Advanced Bioinformatics for Bioinformatics for Biotechnology Biotechnology Irene Gabashvili, PhD
-
Upload
alyson-chambers -
Category
Documents
-
view
214 -
download
0
Transcript of Phylogenetic Analysis Unit 16 BIOL221T: Advanced Bioinformatics for Biotechnology Irene Gabashvili,...

Phylogenetic AnalysisPhylogenetic AnalysisUnit 16Unit 16
BIOL221TBIOL221T: Advanced : Advanced Bioinformatics for Bioinformatics for
BiotechnologyBiotechnologyIrene Gabashvili, PhD

B&O, chapter 14B&O, chapter 14
Bioinformatics analyses should be Bioinformatics analyses should be interpreted in evolutionary contextinterpreted in evolutionary context
Good-quality sequence alignments Good-quality sequence alignments important for evolutionary analysisimportant for evolutionary analysis
Common phylogenetic methods and Common phylogenetic methods and software are different – be cautious software are different – be cautious when using and interpreting your when using and interpreting your resultsresults

Terminology and the Terminology and the BasicsBasics
Phylogenetics is sometimes called Phylogenetics is sometimes called claudisticsclaudistics
Clade – a set of descendants from a Clade – a set of descendants from a single ancestor (greek for branch).single ancestor (greek for branch).
3 basic assumptions:3 basic assumptions: Any group of organism descended from a Any group of organism descended from a
common ancestorcommon ancestor Bifurcating pattern of cladogenesisBifurcating pattern of cladogenesis Change in characteristics occurs in lineages Change in characteristics occurs in lineages
over timeover time

Brief Brief Introduction to Introduction to
the Theory of the Theory of EvolutionEvolution

Classification: LinnaeusClassification: Linnaeus
Carl LinnaeusCarl Linnaeus1707-17781707-1778

Classification: LinnaeusClassification: Linnaeus• Hierarchical systemHierarchical system
– KingdomKingdom (Rige)(Rige)– PhylumPhylum (Række)(Række)– ClassClass (Klasse)(Klasse)– OrderOrder (Orden)(Orden)– FamilyFamily (Familie)(Familie)– GenusGenus (Slægt)(Slægt)– SpeciesSpecies (Art)(Art)

Classification depicted Classification depicted as a treeas a tree

Classification depicted Classification depicted as a treeas a tree
SpeciesSpecies GenusGenus FamilyFamily OrderOrder ClassClass

Theory of evolutionTheory of evolution
Charles DarwinCharles Darwin1809-18821809-1882

Phylogenetic basis of Phylogenetic basis of systematicssystematics
LinnaeusLinnaeus: : Ordering principle is God.Ordering principle is God.
DarwinDarwin: : Ordering principle is shared Ordering principle is shared descent from common ancestors.descent from common ancestors.
Today, systematics is explicitly Today, systematics is explicitly based on phylogeny.based on phylogeny.

Darwin’s four postulatesDarwin’s four postulates
I.I. More young are produced each generation than More young are produced each generation than can survive to reproduce.can survive to reproduce.
II.II. Individuals in a population vary in their Individuals in a population vary in their characteristics.characteristics.
III.III. Some differences among individuals are based Some differences among individuals are based on genetic differences.on genetic differences.
IV.IV. Individuals with favorable characteristics have Individuals with favorable characteristics have higher rates of survival and reproduction.higher rates of survival and reproduction.
Evolution by means of natural selectionEvolution by means of natural selection Presence of ”design-like” features in organisms:Presence of ”design-like” features in organisms:
quite often features are there “for a reason”quite often features are there “for a reason”

Theory of evolution as Theory of evolution as the basis of biological the basis of biological
understandingunderstanding
”Nothing in biology makes sense, except in the light of evolution.
Without that light it becomes a pile of sundry facts - some of them interesting or curious but making no meaningful picture as a whole”
T. Dobzhansky

Phylogenetic Phylogenetic Reconstruction:Reconstruction:Distance Matrix Distance Matrix
MethodsMethods

Trees: terminologyTrees: terminology

TerminologyTerminology
Clades – monophyletic taxonClades – monophyletic taxon Taxons – any named group of Taxons – any named group of
organismorganism Branches – divergence (length may Branches – divergence (length may
indicate the degree)indicate the degree) Nodes – any bifurcating branch pointNodes – any bifurcating branch point

Trees: terminologyTrees: terminology

Trees: representationsTrees: representations
Three different representations of the same tree
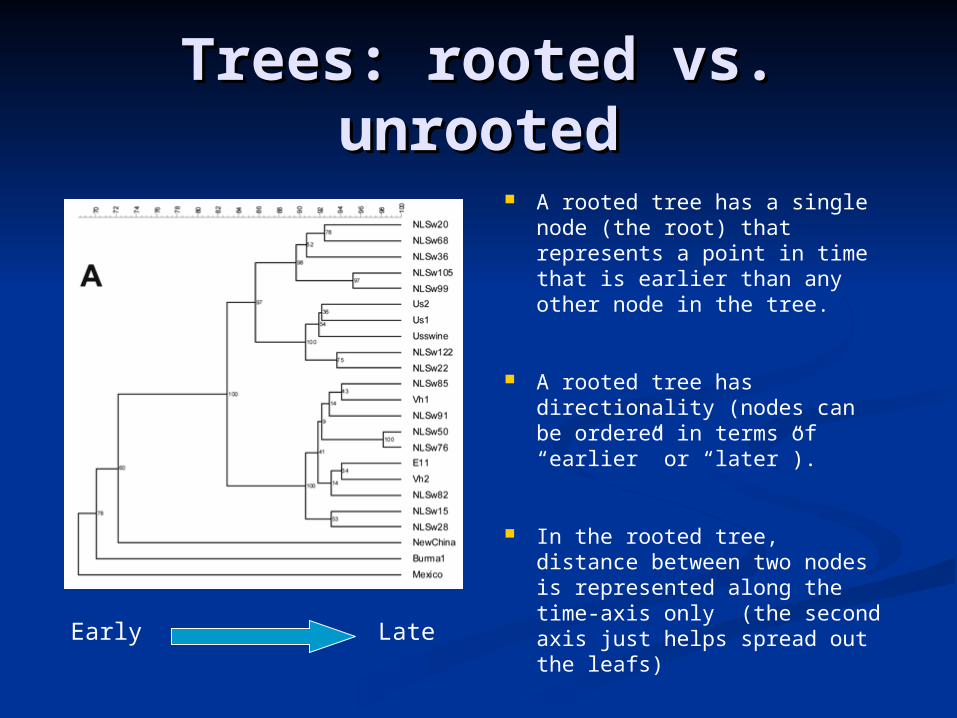
Trees: rooted vs. Trees: rooted vs. unrootedunrooted
A rooted tree has a single node (the root) that represents a point in time that is earlier than any other node in the tree.
A rooted tree has directionality (nodes can be ordered in terms of “earlier” or “later”).
In the rooted tree, distance between two nodes is represented along the time-axis only (the second axis just helps spread out the leafs)
Early Late

Trees: rooted vs. Trees: rooted vs. unrootedunrooted
A rooted tree has a single node (the root) that represents a point in time that is earlier than any other node in the tree.
A rooted tree has directionality (nodes can be ordered in terms of “earlier” or “later”).
In the rooted tree, distance between two nodes is represented along the time-axis only (the second axis just helps spread out the leafs)
Early Late
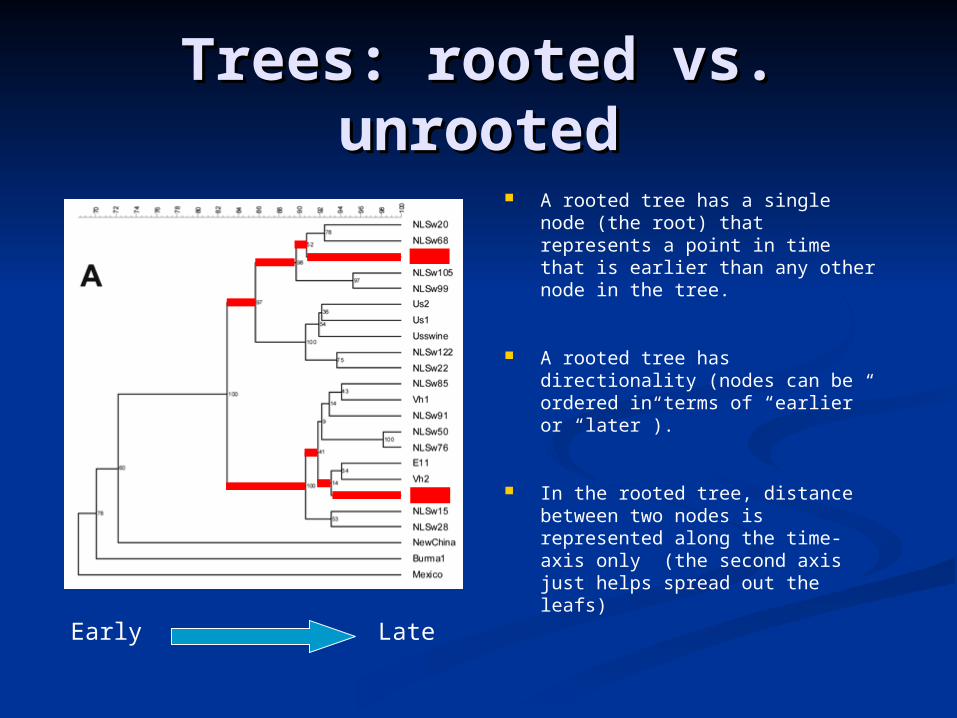
Trees: rooted vs. Trees: rooted vs. unrootedunrooted
A rooted tree has a single node (the root) that represents a point in time that is earlier than any other node in the tree.
A rooted tree has directionality (nodes can be ordered in terms of “earlier” or “later”).
In the rooted tree, distance between two nodes is represented along the time-axis only (the second axis just helps spread out the leafs)
Early Late

Trees: rooted vs. Trees: rooted vs. unrootedunrooted
In unrooted trees there is no directionality: we do not know In unrooted trees there is no directionality: we do not know if a node is earlier or later than another nodeif a node is earlier or later than another node
Distance along branches directly represents node distanceDistance along branches directly represents node distance

Trees: rooted vs. Trees: rooted vs. unrootedunrooted
In unrooted trees there is no directionality: we do not know In unrooted trees there is no directionality: we do not know if a node is earlier or later than another nodeif a node is earlier or later than another node
Distance along branches directly represents node distanceDistance along branches directly represents node distance

Reconstructing a tree Reconstructing a tree using non-using non-
contemporaneous datacontemporaneous data

Reconstructing a tree using present-day Reconstructing a tree using present-day datadata

Data: molecular Data: molecular phylogenyphylogeny
DNA sequencesDNA sequences genomic DNAgenomic DNA mitochondrial DNAmitochondrial DNA chloroplast DNAchloroplast DNA
Protein sequencesProtein sequences
Restriction site polymorphismsRestriction site polymorphisms
DNA/DNA hybridizationDNA/DNA hybridization
Immunological cross-reactionImmunological cross-reaction

Morphology vs. Morphology vs. molecular datamolecular data
African white-backed vulture(old world vulture)
Andean condor (new world vulture)
New and old world vultures seem to be closely related based on morphology.
Molecular data indicates that old world vultures are related to birds of prey (falcons, hawks, etc.) while new world vultures are more closely related to storks
Similar features presumably the result of convergent evolution

Molecular data: single-Molecular data: single-celled organismscelled organisms
Molecular data useful for analyzing single-celled organisms (which have only few prominent morphological features).

Distance Matrix MethodsDistance Matrix Methods
1.1. Construct multiple Construct multiple alignment of sequencesalignment of sequences
2.2. Construct table listing all Construct table listing all pairwise differences pairwise differences (distance matrix)(distance matrix)
3.3. Construct tree from Construct tree from pairwise distancespairwise distances
Gorilla : ACGTCGTAHuman : ACGTTCCTChimpanzee: ACGTTTCG
GoGo HuHu ChCh
GoGo -- 44 44
HuHu -- 22
ChCh --
Go
Hu
Ch
2
11
1

Finding Optimal Branch Finding Optimal Branch LengthsLengths
SS11 SS22 SS33 SS44
SS11 -- DD1212 DD1313 DD1414
SS22 -- DD2323 DD2424
SS33 -- DD3434
SS44 --Observed distance
S1
S3
S2
S4
a
b
c
d e
Distance along tree
D12 d12 = a + b + cD13 d13 = a + dD14 d14 = a + b + eD23 d23 = d + b + cD24 d24 = c + eD34 d34 = d + b + e
Goal:

Optimal Branch Optimal Branch Lengths: Least SquaresLengths: Least Squares
Fit between given tree Fit between given tree and observed distances and observed distances can be expressed as “sum can be expressed as “sum of squared differences”:of squared differences”:
Q = Q = (D(Dijij - d - dijij))22
Find branch lengths that Find branch lengths that minimize Q - this is the minimize Q - this is the optimal set of branch optimal set of branch lengths for this tree.lengths for this tree.
S1
S3
S2
S4
a
b
c
d e
Distance along tree
D12 d12 = a + b + cD13 d13 = a + dD14 d14 = a + b + eD23 d23 = d + b + cD24 d24 = c + eD34 d34 = d + b + e
Goal:
j>i

Least Squares Optimality Least Squares Optimality CriterionCriterion
Search through all (or many) tree topologiesSearch through all (or many) tree topologies
For each investigated tree, find best branch For each investigated tree, find best branch lengths using least squares criterionlengths using least squares criterion
Among all investigated trees, the best tree is Among all investigated trees, the best tree is the one with the smallest sum of squared the one with the smallest sum of squared errors. errors.

Exhaustive search impossible for large Exhaustive search impossible for large data setsdata sets
No. No. taxataxa
No. treesNo. trees
33 11
44 33
55 1515
66 105105
77 945945
88 10,39510,395
99 135,135135,135
1010 2,027,0252,027,025
1111 34,459,42534,459,425
1212 654,729,075654,729,075
1313 13,749,310,57513,749,310,575
1414 316,234,143,225316,234,143,225
1515 7,905,853,580,6257,905,853,580,625

Heuristic searchHeuristic search
1.1. Construct initial tree; determine sum of squaresConstruct initial tree; determine sum of squares
2.2. Construct set of “neighboring trees” by making small Construct set of “neighboring trees” by making small rearrangements of initial tree; determine sum of squares for rearrangements of initial tree; determine sum of squares for each neighboreach neighbor
3.3. If any of the neighboring trees are better than the initial tree, If any of the neighboring trees are better than the initial tree, then select it/them and use as starting point for new round of then select it/them and use as starting point for new round of rearrangements. (Possibly several neighbors are equally good)rearrangements. (Possibly several neighbors are equally good)
4.4. Repeat steps 2+3 until you have found a tree that is better Repeat steps 2+3 until you have found a tree that is better than all of its neighbors.than all of its neighbors.
5.5. This tree is a “local optimum” (not necessarily a global This tree is a “local optimum” (not necessarily a global optimum!) optimum!)

Clustering AlgorithmsClustering Algorithms Starting point: Distance matrixStarting point: Distance matrix
Cluster least different pair of sequences:Cluster least different pair of sequences: Tree: pair connected to common ancestral node, compute branch Tree: pair connected to common ancestral node, compute branch
lengths from ancestral node to both descendantslengths from ancestral node to both descendants
Distance matrix: combine two entries into one. Compute new distance Distance matrix: combine two entries into one. Compute new distance
matrix, by finding distance from new node to all other nodesmatrix, by finding distance from new node to all other nodes
Repeat until all nodes are linkedRepeat until all nodes are linked
Results in only one tree, there is no measure of tree-Results in only one tree, there is no measure of tree-goodness.goodness.
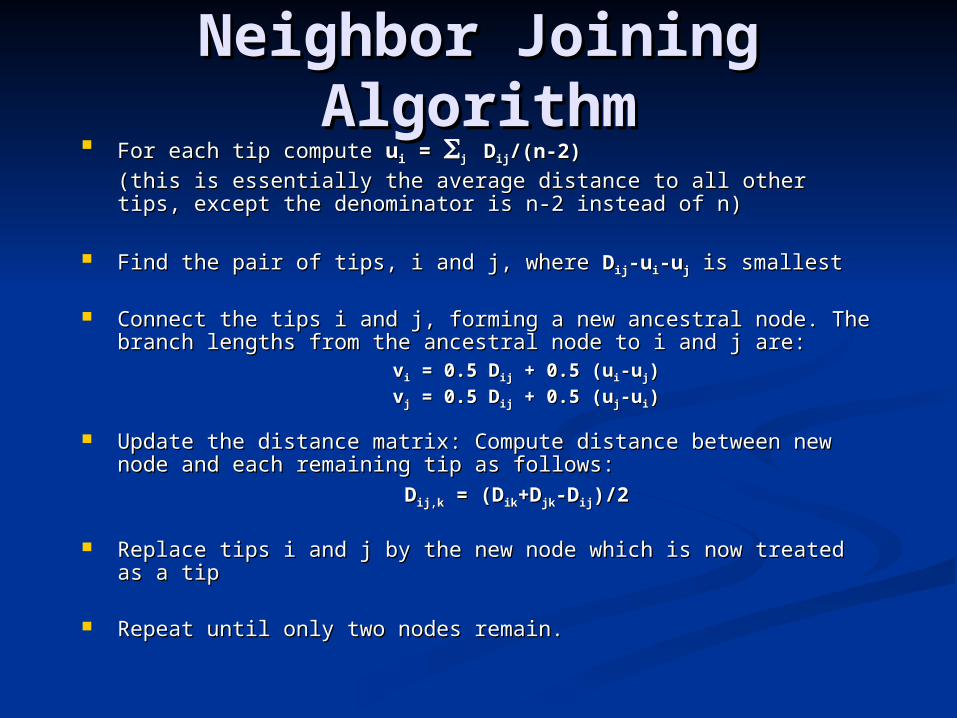
Neighbor Joining Neighbor Joining AlgorithmAlgorithm
For each tip compute For each tip compute uuii = = jj DDijij/(n-2)/(n-2)
(this is essentially the average distance to all other tips, except the (this is essentially the average distance to all other tips, except the denominator is n-2 instead of n)denominator is n-2 instead of n)
Find the pair of tips, i and j, where Find the pair of tips, i and j, where DDijij-u-uii-u-ujj is smallest is smallest
Connect the tips i and j, forming a new ancestral node. The branch Connect the tips i and j, forming a new ancestral node. The branch lengths from the ancestral node to i and j are:lengths from the ancestral node to i and j are:
vvii = 0.5 D = 0.5 Dijij + 0.5 (u + 0.5 (uii-u-ujj))
vvjj = 0.5 D = 0.5 Dijij + 0.5 (u + 0.5 (ujj-u-uii))
Update the distance matrix: Compute distance between new node and Update the distance matrix: Compute distance between new node and each remaining tip as follows:each remaining tip as follows:
DDij,kij,k = (D = (Dikik+D+Djkjk-D-Dijij)/2)/2
Replace tips i and j by the new node which is now treated as a tipReplace tips i and j by the new node which is now treated as a tip
Repeat until only two nodes remain.Repeat until only two nodes remain.

Superimposed Superimposed SubstitutionsSubstitutions
Actual number of Actual number of
evolutionary events:evolutionary events: 55
Observed number of Observed number of
differences:differences: 22
Distance is (almost) always Distance is (almost) always underestimatedunderestimated
ACGGTGC
C T
GCGGTGA

Model-based correction Model-based correction for for
superimposed superimposed substitutionssubstitutions Goal: try to infer the real number of Goal: try to infer the real number of
evolutionary events (the real evolutionary events (the real distance) based ondistance) based on
1.1. Observed data (sequence alignment)Observed data (sequence alignment)
2.2. A model of how evolution occursA model of how evolution occurs

Jukes and Cantor ModelJukes and Cantor Model Four nucleotides assumed to Four nucleotides assumed to
be equally frequent (f=0.25)be equally frequent (f=0.25)
All 12 substitution rates All 12 substitution rates assumed to be equalassumed to be equal
Under this model the Under this model the corrected distance is:corrected distance is:
DDJCJC = -0.75 x ln(1-1.33 x = -0.75 x ln(1-1.33 x DDOBSOBS))
For instance:For instance:
DDOBSOBS=0.43 => D=0.43 => DJCJC=0.64=0.64
A C G T
A -3
C -3
G -3
T -3

Other models of Other models of evolutionevolution

HomologsHomologs
Orthologs - speciationOrthologs - speciation Paralogs - duplicationParalogs - duplication Xenologs – horizontal transferXenologs – horizontal transfer

Clustering AlgorithmsClustering Algorithms
Clustering algorithms use distances to Clustering algorithms use distances to calculate phylogenetic trees. These calculate phylogenetic trees. These trees are based solely on the relative trees are based solely on the relative numbers of similarities and differences numbers of similarities and differences
between a set of sequences.between a set of sequences. Start with a matrix of pairwise distancesStart with a matrix of pairwise distances
Cluster methods construct a tree by linking Cluster methods construct a tree by linking the least distant pairs of taxa, followed by the least distant pairs of taxa, followed by successively more distant taxa. successively more distant taxa.

From Multiple Sequence From Multiple Sequence AlignmentAlignment
Best cluster: {ATCC,ATGC}Best cluster: {ATCC,ATGC}
ATCC ATGC TTCG TCGG
ATCC 0 1 2 4
ATGC 0 3 3
TTCG 0 2
TCGG 0
{ATCC,ATGC} TTCG TCGG
{ATCC,ATGC}
0 ½(2+3)=2.5
½(4+3)=3.5
TTCG 0 2
TCGG 0 Best cluster: {TTCG,TCGG}Best cluster: {TTCG,TCGG}

ExampleExample
{ATCC,ATGC} {TTCG,TCG{TTCG,TCGG}G}
{ATCC,ATGC} 0 ½(2.5+3.5)=3
{TTCG,TCGG} 0
ATCC ATGC TTCG ATCC ATGC TTCG TCGGTCGG
1.5 1.5
0.50.51 1
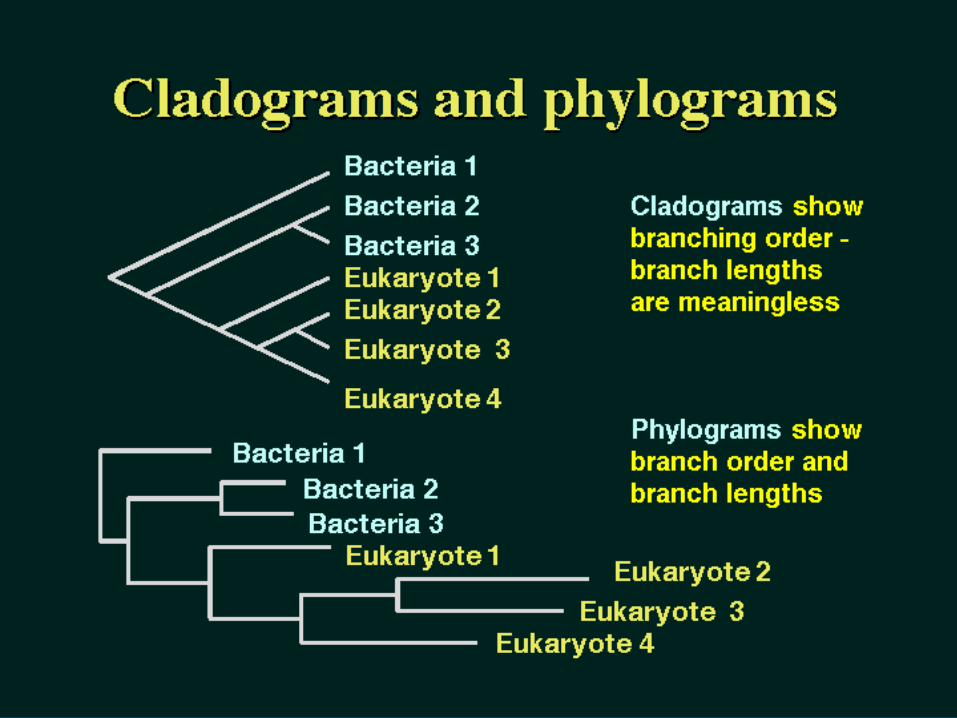
A Cladogram or a Phylogram?

Cladistic MethodsCladistic Methods Evolutionary relationships are documented Evolutionary relationships are documented
by creating a branching structure, termed by creating a branching structure, termed a phylogeny or tree, that illustrates the a phylogeny or tree, that illustrates the relationships between the sequences. relationships between the sequences.
CladisticCladistic methods construct a tree methods construct a tree ((cladogramcladogram) by considering the various ) by considering the various possible pathways of evolution and choose possible pathways of evolution and choose from among these the best possible tree.from among these the best possible tree.
A A phylogramphylogram is a tree with branches that is a tree with branches that are proportional to evolutionary distances.are proportional to evolutionary distances.

Hamming distanceHamming distance
<between two strings of equal <between two strings of equal length>: the number of positions for length>: the number of positions for which the corresponding symbols which the corresponding symbols are different. The number of are different. The number of substitutionssubstitutions required to change one required to change one into the other, or the number of into the other, or the number of errorserrors that transformed one string that transformed one string into the other.into the other.

Hamming distanceHamming distance
The Hamming distance between The Hamming distance between 10111011011101 and and 10010011001001 is 2. is 2.
The Hamming distance between The Hamming distance between 21438962143896 and and 22337962233796 is 3. is 3.
The Hamming distance between The Hamming distance between ""tonedtoned" and "" and "rosesroses" is 3. " is 3.

Levenshtein DistanceLevenshtein Distance
A measure of the similarity between two A measure of the similarity between two strings: number of deletions, insertions, or strings: number of deletions, insertions, or substitutionssubstitutions
For example, For example, If s is "test" and t is "test", then LD(s,t) = If s is "test" and t is "test", then LD(s,t) =
0, 0, If s is "test" and t is "tent", then LD(s,t) = If s is "test" and t is "tent", then LD(s,t) =
1, because one substitution (change "s" to 1, because one substitution (change "s" to "n") is sufficient to transform s into t. "n") is sufficient to transform s into t.
If s os “test” and t is “attempt”, LD(s,t)=4If s os “test” and t is “attempt”, LD(s,t)=4

Levenshtein distanceLevenshtein distance
The Levenshtein distance algorithm The Levenshtein distance algorithm has been used in: has been used in:
Spell checking Spell checking Speech recognition Speech recognition DNA analysis DNA analysis Plagiarism detection Plagiarism detection

DNA DistancesDNA Distances
Distances between pairs of DNA sequences are Distances between pairs of DNA sequences are usually computed as the sum of all base pair usually computed as the sum of all base pair differences between the two sequences. differences between the two sequences. If sequences are similar enough to be alignedIf sequences are similar enough to be aligned
Generally all base changes are considered Generally all base changes are considered equalequal
Insertion/deletions are generally given a larger Insertion/deletions are generally given a larger weight than replacements (gap penalties).weight than replacements (gap penalties).
It is also possible to correct for multiple It is also possible to correct for multiple substitutions at a single site, which is common substitutions at a single site, which is common in distant relationships and for rapidly evolving in distant relationships and for rapidly evolving sites.sites.

Phylogenetic methods (1)
Distance matrix/cluster (UPGMA, NJ):Bacterial taxonomy based on morphological, chemical, biochemical and physiological chacters did not allow natural relationships to be deduced
Numerical taxonomy (Sneath and Sokal, 1963, 1973)
Parsimony (maximum parsomony)The taxonomy of animals shall reflect their natural relatioonships
Phylogenetic Systematics (Willi Hennig 1950, 1966)Without direction (eg. Wiley 1980)

UPGMAUPGMA
The simplest of the distance methods is the The simplest of the distance methods is the UPGMAUPGMA ( (UUnweighted nweighted PPair air GGroup roup MMethod using ethod using AArithmetic averages)rithmetic averages)
The The PHYLIPPHYLIP programs programs DNADISTDNADIST and and PROTDISTPROTDIST calculate absolute pairwise distances between a calculate absolute pairwise distances between a group of sequences. Then the group of sequences. Then the GCGGCG program program GROWTREEGROWTREE uses uses UPGMAUPGMA to build a tree. to build a tree.
Many multiple alignment programs such as Many multiple alignment programs such as PILEUPPILEUP use a variant of use a variant of UPGMAUPGMA to create a to create a dendrogram of DNA sequences which is then used dendrogram of DNA sequences which is then used to guide the multiple alignment algorithm.to guide the multiple alignment algorithm.

Neighbor JoiningNeighbor Joining
The The Neighbor JoiningNeighbor Joining method is the most method is the most popular way to build trees from distance popular way to build trees from distance measurements measurements
(Saitou and Nei 1987, Mol. Biol. Evol. 4:406)(Saitou and Nei 1987, Mol. Biol. Evol. 4:406) Neighbor JoiningNeighbor Joining corrects the UPGMA method for its corrects the UPGMA method for its
(frequently invalid) assumption that the same rate of (frequently invalid) assumption that the same rate of evolution applies to each branch of a tree.evolution applies to each branch of a tree.
The distance matrix is adjusted for differences in the rate of The distance matrix is adjusted for differences in the rate of evolution of each taxon (branch).evolution of each taxon (branch).
Neighbor JoiningNeighbor Joining has given the best results in simulation has given the best results in simulation studies and it is the most computationally efficient of the studies and it is the most computationally efficient of the distance algorithms distance algorithms (N. Saitou and T. Imanishi, Mol. Biol. Evol. 6:514 (N. Saitou and T. Imanishi, Mol. Biol. Evol. 6:514 (1989)(1989)

Cladistic methods Cladistic methods
CladisticCladistic methods are based on the assumption methods are based on the assumption that a set of sequences evolved from a common that a set of sequences evolved from a common ancestor by a process of mutation and selection ancestor by a process of mutation and selection without mixing (hybridization or other horizontal without mixing (hybridization or other horizontal gene transfers).gene transfers).
These methods work best if a specific tree, or at These methods work best if a specific tree, or at least an ancestral sequence, is already known so least an ancestral sequence, is already known so that comparisons can be made between a finite that comparisons can be made between a finite number of alternate trees rather than calculating number of alternate trees rather than calculating all possible trees for a given set of sequences. all possible trees for a given set of sequences.

ParsimonyParsimony
ParsimonyParsimony is the most popular method is the most popular method for reconstructing ancestral for reconstructing ancestral relationships.relationships.
ParsimonyParsimony allows the use of all known allows the use of all known evolutionary information in building a evolutionary information in building a treetree
In contrast, distance methods compress all of In contrast, distance methods compress all of the differences between pairs of sequences the differences between pairs of sequences into a single numberinto a single number

Building Trees with Building Trees with ParsimonyParsimony ParsimonyParsimony involves evaluating all involves evaluating all
possible trees and giving each a score possible trees and giving each a score based on the number of evolutionary based on the number of evolutionary changes that are needed to explain the changes that are needed to explain the observed data. observed data.
The best tree is the one that requires The best tree is the one that requires the fewest base changes for all the fewest base changes for all sequences to derive from a common sequences to derive from a common ancestor.ancestor.

MethodsMethods
Distance-based: UPGMA, NJ, FM, Distance-based: UPGMA, NJ, FM, MEME
Other: Maximum Parsimony, ML, etcOther: Maximum Parsimony, ML, etc
Neighbor JoiningNeighbor Joining methods generally produce just one tree, methods generally produce just one tree, which can help to validate a tree built with the which can help to validate a tree built with the parsimonyparsimony or or maximum likelihoodmaximum likelihood method method

Phylogenetic methods
Maximum likelihood methodsPhylogenies should be formulated in a probalistic framework and statistically testable. Protein and DNA sequence data are extraordinary goodfor phylogenetic interpreation and can ”resist” such treatment.
Cavalli-Sforza and Edwards 1967 (theory)
Felsenstein 1981 first practically useful algorithms.

Phylogenetic analysis.Phylogenetic analysis. Comparison of phylogenetic Comparison of phylogenetic methodsmethods
Consistency:Consistency: a phylogenetic method is a phylogenetic method is consistent for an evolutionary model, if the consistent for an evolutionary model, if the method converges on the corrrect tree as method converges on the corrrect tree as the data becomes infinite. the data becomes infinite.
Efficiency:Efficiency: a phylogenetic method have high a phylogenetic method have high efficiency if it quickly converges on the efficiency if it quickly converges on the correct solution as more data are applied to correct solution as more data are applied to the problem. the problem.
Robustness:Robustness: a phylogenetic method is robust a phylogenetic method is robust if converges on the correct solution with if converges on the correct solution with violations of the assumptions about the violations of the assumptions about the evolutionary model. evolutionary model.
Hillis 1995. Syst. Biol. 44, 3-16. Hillis 1995. Syst. Biol. 44, 3-16.

Phylogenetic analysis. Test of robustness. Bootstrap
Purpose. To show how well supported the nodes are by the data.
Performance. The original data are simulated by drawing columns randomly with replacement 100 or 1000 times. The phylogenetic analysis is repeated and the number of nodes common in all 100 or 1000 trees summarized.
Example. Original data 1 replicate 2 replicateSpecies 1 AGGA AAGA GGAASpecies 2 ACGT AACT CGTTSpecies 3 ACGT AACT CGTTSpecies 4 ACTT AACT CTTTSpecies 5 CCGT CCCT CGTT
linear form (2,3)4)5)1; (2,3)4)5)1; (2,3)5)4)1;

Are there Are there CorrectCorrect trees??trees?? Despite all of these caveats, it is actually quite simple Despite all of these caveats, it is actually quite simple
to use computer programs calculate phylogenetic to use computer programs calculate phylogenetic trees for data sets.trees for data sets.
Provided the data are clean, outgroups are correctly Provided the data are clean, outgroups are correctly specified, appropriate algorithms are chosen, no specified, appropriate algorithms are chosen, no assumptions are violated, etc., assumptions are violated, etc., can the true, correct can the true, correct tree be foundtree be found and proven to be scientifically and proven to be scientifically
valid?valid? Unfortunately, it is impossible to ever conclusively Unfortunately, it is impossible to ever conclusively
state what is the "true" tree for a group of sequences state what is the "true" tree for a group of sequences (or a group of organisms); taxonomy is constantly (or a group of organisms); taxonomy is constantly under revision as new data is gathered (example: 80s under revision as new data is gathered (example: 80s revisionrevision of the seals and sea lions tree) of the seals and sea lions tree)


















