
Design and Synthesis of Inhibitors Targeting the Hepatitis C Virus ...

Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology 952
Peptide-Based Inhibitors ofHepatitis C Virus NS3 SerineProtease: Kinetic Aspects and
Inhibitor Design
BY
ANTON POLIAKOV
ACTA UNIVERSITATIS UPSALIENSISUPPSALA 2004


Papers included in the thesis
I. Poliakov A, Hubatsch I, Shuman CF, Stenberg G, Danielson UH. Expression and purification of recombinant full-length NS3 protease-helicase from a new variant of Hepatitis C virus. Protein Expr Purif. 2002 Aug;25(3):363-71.
II. Johansson A, Poliakov A, Åkerblom E, Lindeberg G., Winiwarter S, Samuelsson B, Danielson UH, Hallberg A. Tetrapeptides as potent protease inhibitors of Hepatitis C Virus full-length NS3 (protease-helicase/NTPase). Bioorg Med Chem. 2002 Dec;10(12):3915-22.
III. Johansson A, Poliakov A, Åkerblom E, Wiklund K, Lindeberg G, Winiwarter S, Danielson UH, Samuelsson B, Hallberg A. Acyl sulfonamides as potent protease inhibitors of the hepatitis C virus full-Length NS3 (protease-helicase/NTPase): a comparative study of different C-terminals. Bioorg Med Chem. 2003 Jun 12;11(12):2551-68.
IV. Oscarsson K, Poliakov A, Oscarson S, Danielson UH, Hallberg A, Samuelsson B. Peptide-based inhibitors of hepatitis C virus full-length NS3 (protease-helicase/NTPase): model compounds towards small molecule inhibitors. Bioorg Med Chem. 2003 Jul 3;11(13):2955-63.
V. Poliakov A, Johansson A, Åkerblom E, Oscarsson K, Samuelsson B, Hallberg A, and Danielson UH. Structure-activity relationships for the selectivity of hepatitis C virus NS3 protease inhibitors. Biochimica et Biophysica Acta, in press.
Reprints of papers I – IV were made with permissions from the publishers

Contents
Introduction.....................................................................................................1Origins of virology .....................................................................................1
Fighting against infections (historical perspectives) .............................1History of discovery of viruses in a nutshell .........................................2
The hepatitis C virus ..................................................................................3History of hepatitis C virus discovery ...................................................3Hepatitis C virus ....................................................................................4
Taxonomy .........................................................................................4Genome structure ..............................................................................5
5’-untranslated region (5’UTR)....................................................53’-untranslated region (3’UTR)....................................................6Open reading frames (ORF) .........................................................7
HCV structural proteins ....................................................................7Core protein..................................................................................7E1 and E2 .....................................................................................9p7..................................................................................................9
Non-structural proteins .....................................................................9NS2...............................................................................................9NS3 and NS4A ...........................................................................10NS4B ..........................................................................................15NS5A..........................................................................................15NS5B ..........................................................................................15Replicase complex......................................................................16
xORF, protein F. .............................................................................17HCV virion structure.......................................................................17
HCV epidemiology..............................................................................17Infected population .........................................................................17

HCV transmission routes ................................................................18HCV pathogenesis and symptoms .......................................................19How to kill a virus ...............................................................................21
Modulation of host antiviral defence ..............................................21Viral life-cycle intervention ............................................................21
How to inhibit the NS3 protease ..............................................................23Classifications of serine protease inhibitors ........................................23
Non-covalent serine protease inhibitors..........................................23Protein non-covalent serine protease inhibitors..........................23Peptide non-covalent inhibitors..................................................25Non-peptidic, non-covalent inhibitors........................................26
Covalent serine protease inhibitors. ................................................26Protein covalent serine protease inhibitors.................................27Peptide covalent inhibitors. ........................................................28Non-peptidic covalent inhibitors ...............................................29
Inhibitor optimization strategy. ...........................................................29
Present study .................................................................................................31Aims of the study .....................................................................................31
Obtaining the target enzyme................................................................31Establishment of assay for NS3 protease (construct V). .....................33
Substrate..........................................................................................33Inner filter effect (IFE)....................................................................34NS4A cofactor.................................................................................34Buffer conditions.............................................................................35Substrate inhibition .........................................................................36Kinetic constants of NS3 protease ..................................................36Selectivity measurements................................................................36Slow-binding inhibition ..................................................................36
Optimization of peptide-based inhibitors of the NS3 protease (inhibitory potency and selectivity) .....................................................38
Inhibitor design strategy..................................................................38N-terminal capping groups..............................................................39P5 and P6 residues. .........................................................................39C-terminal residues .........................................................................40P1 side chains..................................................................................40P2, P3, P4 side chains (Paper II) .....................................................42
Structure-activity relationships of electrophilic inhibitors with cyclopropane side chain residue ..........................................................44

Influence of cyclopropane P1 residue on the activity of electrophilic inhibitors .........................................................................................44High selectivity of ACPC-based electrophilic keto acid inhibitor ..45Indirect evidence for non-covalent interaction of peptidyl electrophilic inhibitors with NS3 protease. .....................................45
Conclusions...................................................................................................50
Acknowledgements.......................................................................................52
References.....................................................................................................54

Abbreviations
ACPC 1-aminocyclopropane-1-carboxylic acid BPC Bovine pancreatic chymotrypsin DTT Dithiothreitol HCV Hepatitis C virus HLE Human leucocyte elastase IFE Inner filter effect IMAC Immobilized metal affinity chromatography IRES Internal ribosome entry site Nva Norvaline PPE Porcine pancreatic elastase RET Resonance energy transfer SAR Structure/activity relationship


1
Introduction
Origins of virology
‘’Virus (Latin - poison) - any of a group of submicroscopic entities consisting of a single nucleic acid surrounded by a protein coat and capable of replication only within the cells of animals and plants: many are pathogenic’’. http://www.wordreference.com
‘’Enzyme (from Medieval Greek enzumos, leavened, from Greek EN-2 + zume leaven) - any of a group of […] proteins that are produced by living cells and act as catalysts in specific biochemical reactions’’. http://www.wordreference.com
Fighting against infections (historical perspectives)
It would not be an exaggeration to say that infectious diseases have shaped the history of mankind. Bacterial diseases like plague and cholera, viral diseases caused by smallpox and poliovirus have been known since ancient times and have often devastated local communities and whole continents, thus slowing down the development of civilization. The history of combat against infections is probably as ancient as the infections themselves. It is known that ancient Greeks treated infected wounds with heat. Many plants, such as garlic and onion, have been used for treatment of different infections in folk medicine. Moreover, vaccination against smallpox (also called variolation) was widespread in India and China already by the 11th century.
The discovery of anthrax vaccine by Pasteur in 1881 and antitoxins against diphtheria and of tetanus by Von Behring in 1890 was the beginning of the end of the reign of bacterial diseases. It was later helped by the discovery of sulphanilamides and penicillin, highly effective, broad-range antibacterial substances.

2
The development of antiviral vaccines was also successful: the introduction of cowpox virus vaccine by Edward Jenner and the revolutionary efforts of Louis Pasteur with rabies vaccine were later followed by discoveries of vaccines against other viral infections. This eventually resulted in complete eradication of smallpox and effective control of many deadly viruses. Many, but far from all. Unfortunately, the development of powerful and specific antiviral chemicals did not follow. Without an effective vaccine at hand, physicians were often left with only a handful of palliative tools for helping patients. However the quality of antiviral treatment had satisfied expectations of society for some time until the balance was tipped over by the arrival of AIDS, understanding of the connection between viruses and cancer and discovery of the viral nature of many previously known diseases. All that necessitated active search for specific antiviral drugs. But before that, a drug target had to be found. Viral enzymes, like polynucleotide polymerases and proteases, had become prime targets in this search. The successful launching of drugs against Herpes Simplex Virus (DNA polymerase inhibitor) and Human Immunodeficiency Virus (reverse transcriptase inhibitor and protease inhibitors) told us that this approach can be fruitful and should be applied to other viral infections.
History of discovery of viruses in a nutshell
Not visible in a light microscope, unable to grow on any kind of media by themselves, viruses were elusive for a long time after the discovery of bacteria. The first virus to be discovered was not from humans. In 1887, the Russian scientist Dimitri Ivanovski, investigating a case of tobacco mosaic disease in tobacco plants in Crimea, provided the first operational definition of viruses by showing that ‘’the sap of [tobacco] leaves infected with tobacco mosaic disease retains its infectious properties even after filtration through Chamberland filter candles’’. Defined in this way, viruses were called ‘’filterable agents’’ for several decades after Ivanovski’s discovery. Another important step was taken by the Dutch microbiologist Martinus Beijerinck in 1898. He showed that the filterable agent of tobacco mosaic disease regained its infectious ‘’strength’’ after dilution only in the presence of tobacco plant cells. Viruses thus became defined as filterable infectious agents that were invisible in a microscope and only multiplied in living cells. Guided by this definition, the finding of other viruses was only a matter of time and

3
the first human filterable agent, yellow fever virus, was soon (1901) discovered by Walter Reed and co-workers. Invention of the plaque assay, culturing of viruses in living animals and cell cultures, nucleotide sequencing, etc. had resulted in the accumulation of an enormous amount of knowledge about viruses by the 20th century. The discovery of PCR paved the way for the identification of the virus that is the main subject of the present thesis: in 1989 hepatitis C virus became the first virus to be discovered by molecular cloning (Choo et al. 1989).
The hepatitis C virus
History of hepatitis C virus discovery
By the middle of the seventies of the 20th century it was already known that hepatitis A and B viruses were not the only causative agents of viral hepatitis in humans (Barker et al. 1976, Feinstone et al. 1975). A number of patients with mild hepatitis did not fit into the clinical picture of hepatitis A or B infections and their blood serum was negative for immunological markers of the two known hepatitis viruses; this disease was called as what it was not: non-A non-B hepatitis (NANBH). By the end of the seventies, the clinical picture of the disease was well established (Wyke et al. 1980), but the etiological factor was still unknown and laboratory tests for identification of the disease were not available. No specific antibody was found in the blood of diseased individuals and infectivity was only shown with chimpanzees (this was the first direct proof of existence of NANBH), which, of course, was not suitable for broad screening. However, it was possible to observe virus-like particles in liver tissue from both infected humans and chimpanzees. The morphology of the particles raised speculations about the existence of more than one virus (Maugh TH 2nd, 1980). In the eighties, the number of research centres that were dealing with NANBH and the number of publications on the matter had exploded. But quantity had not grown into quality and at the end of the eighties no etiological agent had been identified and no reliable diagnostic test had been discovered (Choksi et al. 1989). Amidst growing anxiety, a long-awaited discovery was made by Choo et al. (Choo et al. 1989) at

4
Chiron Corporation in 1989. Using an extensive set of random primers, they were able to pick up a polynucleotide sequence from the plasma of HCV-infected chimpanzees that did not belong to humans or chimpanzees. Peptides encoded by this sequence did bind antibodies from the plasma of individuals infected with NANBH, but not of healthy individuals. Hepatitis C virus (HCV) was thus discovered. The procedure used in the latter experiment was immediately recognised as a ready-to-use diagnostic tool (Kuo et al. 1989). As blood screenings began, the extent of the hepatitis C problem was soon unveiled. In the years to follow, HCV was extensively studied: it was found to contain at least four enzymes besides structural proteins: two proteases (NS2-3 and NS3), NS3 helicase and RNA-dependent RNA polymerase. These enzymes have become prime targets in the search for anti-HCV chemicals, and one of them (NS3 protease inhibitor) may soon make its way to drug stores (Lamarre et al. 2003).
Hepatitis C virus
Taxonomy
HCV belongs to the Hepacivirus genus of the Flaviviridae
family. It is an enveloped, single-strand positive-sense RNA-containing virus. Like other Flaviviruses, the RNA of HCV propagates by using an RNA-dependent RNA polymerase (RdRp), which lacks a proofreading mechanism. This gives rise to great heterogeneity in the HCV genome. Based on sequence analysis of the 5’-non-coding region (Bukh et al. 1992), genes coding for different structural and non-structural proteins (Bukh et al. 1993, Bukh et al. 1994, Simmonds et al. 1993) or the whole open reading frame (ORF) (Bukh et al. 1995), six major genotypes (I-VI) and 12 subtypes of HCV were originally identified. Despite difficulties in classifying some HCV isolates into any of the six major genotypes, the classification is still in use. Due to its high mutational rate, HCV circulates as a population of related, but slightly different genomes, the so-called quasispecies (Martell et al. 1992) and identification of a viral strain infecting a particular patient may be quite complicated by that fact.

5
Genome structure
The HCV genome is about 9.5 kbp long and consists of a major open reading frame flanked by two non-coding, highly conserved regions (Fig.1) (Pozzetto et al. 1996, Plagemann 1991); an additional smaller reading frame was also proposed (Ina et al. 1994).
Figure 1. HCV genome and polyprotein processing. The genome is ≈9.5 kb long, the polyprotein is ≈3010 aa long. Cleavage of different junctions of the polyprotein by responsible proteases is indicated by arrows. UTR – untranslated region, IRES – internal ribosome entry site, polyU-tract – poly uracil stretch of RNA.
5’-untranslated region (5’UTR)
The 5’-UTR is approximately 340 nt long and is considered to contain regulatory elements important for viral gene expression and replication. Devoid of cap, HCV RNA contains an internal ribosome entry site (IRES), which attaches itself to a ribosome and initiates viral RNA translation (Tsukiyama-Kohara et al. 1992). The IRES spans the 5’-UTR from 40 nt to 370 nt, which overlaps the ORF (starts at 342 nt) (Reynolds et al. 1995). The 5’-UTR has a complex secondary structure, which is important for proper functioning of the IRES (Brown et al. 1992, Le et al. 1995, Rijnbrand et al. 1995). Secondary structural elements may play regulatory roles by interacting with cellular factors (Ali et al. 2000, Spangberg et al. 1999). The 5’-UTR

6
can also play a role in the viral RNA replication, as was shown by deletion mutagenesis (Kim et al. 2002).
3’-untranslated region (3’UTR)
The 3’UTR is approximately 200 to 235 nt long and consists of several regions: 1) a short (~40 nt) variable sequence; 2) a poly(U) region followed by U(C)n-stretch and 3) an approximately 98 nt long sequence (98nt stretch) that is highly conserved among different HCV strains (Kolykhalov et al. 1996, Yamada et al. 1996). The 3’-UTR is predicted to form several loop structures (Fig. 2).
Figure 2. 3’-untranslated region (3’UTR) of the HCV genome.
Removal of loops in the variable region results in impaired RNA replication, whereas removal of the poly(U) stretch or 98nt stretch completely abolishes replication (Friebe et al. 2002). The length of the poly(U) stretch is an important determinant of its functionality – only truncation to fewer than 26 nt results in loss of RNA replication. Changes in the 98nt-stretch are much less tolerated: removal of any of the three loop structures present in 98nt abolishes RNA replication, but do not affect RNA stability or translation. This indicates that the 3’-UTR is of major importance for HCV RNA replication (Friebe et al. 2002). This is supported by the fact that blocking of the 3’ end of the 3’-UTR inhibits initiation of RNA replication (Reigadas et al. 2003).

7
Finally, removal of both the poly(U) and the 98nt stretches results in loss of HCV infectivity in chimpanzees (Kolykhalov et al. 2000). Taken together, these facts point out the importance of the 3’-UTR for viral proliferation and explain the high conservation of this region among different HCV strains.
Open reading frames (ORF)
An interesting feature of the flavivirus genome is the presence of more than one start codon in the 5’-UTR. In the case of HCV, the ORF starts at the fourth AUG codon (Tsukiyama-Kohara et al. 1992). The main ORF (mORF) is about 9 kb long and is translated into a single polyprotein of about 3000 amino acids. Based on the functions of the proteins encoded by the mORF, it can be divided into two regions: a) structural proteins, which are clustered on the 5’-end and b) non-structural proteins, which are clustered on the 3’-end (Takamizawa et al. 1991, Choo et al. 1991, Houghton et al. 1994) (Fig. 1).
Processing of the polyprotein into separate functional proteins is carried out both by cellular and viral proteases: cellular proteases cleave between structural proteins and between the p7 and NS2 proteins, whereas cleavage between the rest of the proteins is carried out by two viral proteases encoded by the NS2-3 and NS3 genes (Lohmann et al. 1996).
The presence of another ORF (xORF), which overlaps the Core gene, was predicted by computer-assisted analysis of the HCV genome (Ina et al. 1994, Smith et al. 1997). The xORF lacks a start codon and is supposed to be expressed via a ribosomal frameshift (see 4.2.5.). Several reports suggest that xORF is functional (Varaklioti et al. 2002, Xu et al. 2001), but the function of the encoded protein is not known.
HCV structural proteins
Core protein
The sequence of the Core protein is highly conserved among different HCV strains (Bukh et al. 1994). The Core protein is released from the polyprotein by proteolytic cleavage at residue 191 (Fig. 1) and between residues 174 and 191, most probably by a signal peptidase complex on the lumenal site of the ER (McLauchlan et al. 2000). Core protein variants of 21 kDa, 19 kDa and 16 kDa have been

8
described (Lo et al. 1995). The direct function of the Core protein, as implied by its name, is to form the capsid shell of the viral particle (Fig. 3). The Core protein can be homo-oligomerized to form a viral capsid. The binding of RNA and the formation of the nucleocapsid seem to stabilize the whole assembly (Shimoike et al. 1999, Kunkel et al. 2002). Very little is known about the structure of the HCV capsid and the virion in general, since HCV does not proliferate in cell cultures and very few viral particles are generated upon infection in living organisms.
A B
Figure 3. HCV virion structure. A. Image of a HCV virion particle in an electron microscope. B. Hypothetical structure of a HCV particle.
Viral genomes are limited in size, especially in RNA viruses, and whatever information is encoded has to be used with maximum efficiency. Viruses have developed multiple ways of maximizing the information stored in the genome without physically increasing the genome size. One such way is to have multiple functions associated with one gene product. The nucleotide sequence coding for the Core protein appears to be involved in the regulation of HCV translation (Zhao et al. 1999). The presence of a DNA-binding motif, nuclear localization signals, the localization of the protein in the cytoplasm, nucleus and mitochondria and its binding to a broad range of host proteins suggests that the Core protein has a regulatory function besides being a structural protein (McLauchlan et al. 2000, Ray et al. 2001). It has been shown that the Core protein might be involved in different cellular processes, such as apoptosis (Kountouras et al. 2003), cell growth/transformation (Schulze zur Wiesch et al. 2003),

9
lipid metabolism (Petit et al. 2003) and gene expression (McLauchlan et al. 2000). It also plays an important role in the evasion of the immune system by the virus (Moorman et al. 2001). In general, it appears that the Core protein plays an important role in the pathogenesis of HCV infection.
E1 and E2
E1 (35 kDa) and E2 (70 kDa) are two highly glycosylated proteins that are presumed to be present on the surface of the viral particle (Dubuisson et al. 2000) (Fig. 3). The lipid envelope of HCV originates from the ER membrane with E1 and E2 already integrated into it via their C-terminally localized transmembrane regions (Cocquerel et al. 2000, Cocquerel et al. 1999, Blanchard et al. 2002). It is likely that viral cell entry is mediated by the interaction of both envelope proteins with CD81 (Pileri et al. 1998, Cocquerel et al. 2003) and low-density lipoprotein (Monazahian et al. 1999, Agnello et al. 1999) receptors and, possibly, with some other cellular proteins (Pandya et al. 2002). The N-terminal end of the E2 protein contains two hypervariable regions (Kurosaki et al. 1994), which are proposed to play an important role in the evasion of the immune system by the virus (Dubuisson et al. 2000, Chang 2003).
p7
p7 is a small (7 kDa) protein, which has been shown to be essential for viral infectivity (Sakai et al. 2003). Recent reports suggest that p7 forms an ion channel (Pavlovic et al. 2003, Griffin et al. 2003), but its exact role in the HCV life cycle is not yet clear.
Non-structural proteins
NS2
NS2 (23 kDa) is a highly hydrophobic, transmembrane protein (Yamaga et al. 2002) with a still unknown function. It has been shown that NS2 inhibits apoptosis by interaction with the liver-specific pro-apoptotic CIDE-B protein, which suggests involvement of NS2 in the pathogenesis of HCV infection (Erdtmann et al. 2003).
The NS2-3 protease spans the C-terminal half of NS2 and the N-terminal part of NS3 proteins (Hijikata et al. 1993) and is responsible for cleaving itself at the NS2/NS3 junction (Thibeault et al. 2001, Grakoui et al. 1993, a). The question of which family of proteases the NS2-3 protease belongs to is still unresolved. The NS2-3 protease has no sequence homology to other known proteases, but

10
mutagenesis studies have revealed the importance of His-952 and Cys-993 for NS2-3 autocleavage (Hijikata et al. 1993, Grakoui et al. 1993), which indicates that NS2-3 is a cysteine protease. On the other hand, it was discovered that NS2-3 activity is stimulated by Zn and inhibited by EDTA, which led to the suggestion that NS2-3 might be metalloprotease (Thibeault et al. 2001, Hijikata et al. 1993). Inhibition studies have revealed that the NS2-3 protease is inhibited by serine, cysteine and metalloprotease inhibitors (Thibeault et al. 2001), which has also contributed to the confusion about classification of the protease.
NS3 and NS4A
The NS3 protein is a 631-amino acid long, multifunctional protein with protease, helicase and NTPase activities. It consists of two distinct domains: protease and helicase/NTPase.
NS3 protease
The N-terminal part of the NS3 protein of flaviviruses had been predicted to contain a serine protease (Gorbalenya et al. 1989a,Bazan et al. 1989) before the discovery of HCV. This was later confirmed by mutagenesis studies where substitution of proposed catalytic residues led to the lack of polyprotein processing (Bartenschlager et al. 1993, Eckart et al. 1993, Tomei et al. 1993, Grakoui et al. 1993b), by biochemical studies with purified NS3 protease (Shoji et al. 1995) and by structure determination (Love et al. 1996, Kim et al. 1997, Yan et al. 1998, Love et al. 1998).
The NS3 protease spans 180 N-terminal amino acids of the NS3 protein and is responsible for separation of non-structural proteins from each other (Fig. 1). It belongs to the chymotrypsin family of serine proteases and consists of two canonical Greek-key β-barrels with the active site placed between the domains (Fig. 4). The NS3 protease lacks the long surface loops that feature the substrate-binding site of other chymotrypsin-like serine proteases. The NS3 protease substrate-binding site is unusually flat, pocketless and solvent-exposed (Love et al. 1996).

11
The C-terminal β-barrel of the NS3 protease contains a structural Zn2+, which is coordinated by Cys-97, Cys-99, Cys-145 and His-149 via a water molecule (Love et al. 1996, Kim et al. 1997, De Francesco et al. 1996). Refolding of NS3 without Zn2+ results in misfolding and aggregation of the protein (Urbani et al. 1998). Zn2+
thus seems to stabilize structure of the protease. The Zn2+-binding site in NS3 is equivalent to that in another chymotrypsin-like protease, the 2A protease of Picornaviridae (Voss et al. 1995). In both proteases, the Zn2+-binding site seems to correspond to a disulfide bond found in a similar position in other chymotrypsin-like proteases. Like other intracellular proteins, NS3 is devoid of S-S bonds, since they would not survive in the reducing intracellular milieu; Zn2+ may play the role of the S-S bond without being reduced (De Francesco et al. 2000).
Figure 4. Structure of the protease domain of HCV NS3 (coordinates are from PDB file 1A1R). The structure is colored according to the secondary structure: β-strands in blue, α-helices in red. The N-terminal β-barrel with the NS4A cofactor (magenta) is on the right, the C-terminal β-barrel with the structural Zn2+ (green) is on the left. The catalytic triad, showed in ball and stick, is in the interdomain cleft: Ser-139 (on the C-terminal β-barrel), His-57 and Asp-81 (behind His-57) (on the N-terminal β-barrel).
Another interesting feature of the NS3 protease is its dependence on the NS4A protein, which activates the NS3 protease and is absolutely necessary for cleavages at the NS3-NS4A, NS4A-NS4B and NS4B-NS5A junctions of the viral polyprotein (Failla et al. 1994, Bartenschlager et al. 1994). NS4A is a 54 amino acid long

12
protein with a highly hydrophobic N-terminus. The central part of NS4A has been shown to interact with the NS3 protease by forming an antiparallel β-strand with its N-terminus (Fig. 4). NS3 in complex with NS4A is less susceptible to proteolysis and is more stable both invitro and in vivo (Urbani et al. 1999, Tanji et al. 1995). Several theories were proposed to explain the mechanism of activation of the NS3 protease by NS4A cofactor, including proper alignment of catalytic residues (Archer et al. 2002, Yan et al. 1998) and influence on the prime side of the substrate binding site (Kim et al. 1997, Landro et al. 1997).
Sequence at the junction Junctions between non-structural proteins
P6 P5 P4 P3 P2 P1 - P1’P2’P3’ P4’ NS3-NS4A D L E V V T - S T W VNS4A-NS4B D E M E E C - A S H LNS4B-NS5A D C S T P C - S G S WNS5A-NS5B E D V V C C - S M S Y Consensus sequence D X X X X C - S X X X
Figure 5. Natural cleavage sites of HCV NS3 protease. Sequences at the junctions are shown using the nomenclature of Schechter and Berger (1967). Prime residues (P’) lie on the right of the scissile bond (marked with a dash), non-prime residues (P) lie on the left. NS3/NS4A junction is the only junction with non-Cys P1. P6 is invariably occupied by an amino acid with acidic side chain – Asp or Glu. P1’ is occupied by an amino acid with a small, hydrophobic side chain. Most of the remaining prime residues are occupied by hydrophobic amino acids.
The substrate specificity of the NS3 protease is rather unusual (Fig. 5), with P1 being Cys in all natural cleavage sites except for the NS3-NS4A junction, where Thr is found (Grakoui et al. 1993b, Pizzi et al. 1994, Zhang et al. 1997). The S1 pocket is a rather shallow cavity that is lined by the side chains of Ala-157 and Leu-135 and by the aromatic ring of Phe-154 at the bottom (Love et al. 1996, Kim et al. 1997, Yan et al. 1998). Apparently, only amino acids with non-bulky side chains (like cysteine, homocysteine, allylglycine, 2-aminobutyric acid, norvaline and valine) are accepted in the P1 position of the NS3 substrate. There are no other well-defined pockets on either the prime or non-prime parts of the substrate-binding site (Love et al. 1996). Another important interaction also resides in the non-prime side. The P6 residue, which is invariably Asp or Glu in all natural NS3 substrates (Fig. 5), was shown to interact with a

13
positively charged area on the surface of the protease. Removal of P6 or its substitution by a non-charged amino acid results in great loss of substrate affinity (Steinkuhler et al. 1996, Komoda et al. 1994). Prime side interactions (P1’-P4’) have also been shown to be important for substrate recognition, but again – no single interaction was especially important for the substrate affinity (Steinkuhler et al. 1996). The interaction of NS3 protease with its substrate is mediated by multiple weak interactions spread along the substrate-binding site, with most of the interaction energy coming from the non-prime side. As a consequence, the NS3 protease effectively cleaves only long peptides (minimum decapeptides spanning P6-P4’) (Steinkuhler et al. 1996).
It has been noticed that the N-terminal hexapeptide product of NS3 protease substrate is a rather potent competitive inhibitor of the enzyme (Steinkuhler et al. 1998, Llinas-Bruneta et al. 1998) (Fig. 13). Its optimization led to the creation of hexapeptide inhibitors with Ki
values in the low nanomolar range (Ingallinella et al. 1998). Besides interactions observed with the non-prime side of the substrate, the additional interaction between the C-terminal carboxylate created upon proteolytic cleavage and the active site is present in the product inhibitor (Fig. 13) (Tsantrizos et al. 2003).
NS3 helicase
The NS3 helicase/NTPase comprises the C-terminal two-thirds of the NS3 protein. It is capable of unwinding RNA, DNA and heteroduplexes in a 3’ to 5’ direction (Hong et al. 1996, Morgenstern et al. 1997) and its main function appears to be to unwind the RNA duplex during RNA replication.
The NS3 helicase belongs to super family II of helicases (DEAD-box family, DexH subfamily) according to (Gorbalenya et al. 1989b). The NS3 helicase contains the conserved motifs I and II (Walker motifs), shared by all helicases, and motifs III and IV (Kwong et al. 2000). Motif I is responsible for binding of nucleotide triphosphates, motif II binds Mg for NTP hydrolysis, motif III participates in coupling of NTP hydrolysis and RNA unwinding, whereas the role of motif IV is not yet known (Kwong et al. 2000). The structure of NS3 helicase has been resolved and revealed that the protein consists of three domains; a groove between the two first domains and the third domain forms a binding site for the oligonucleotide substrate (Kim et al. 1998, Cho et al. 1998).

14
Full length NS3 protein
Although both the protease and the helicase domains of NS3 had been shown to be active when expressed alone, there is no evidence that they become separated in vivo. This may imply that there must be a reason for two enzymes to be linked. Several reports suggest a mutual influence between the helicase and protease domains in the full-length protein (Morgenstern et al. 1997, Johansson et al. 2001, Gallinari et al. 1998). The crystal structure has revealed that the domains are clustered together although interdomain interactions do not appear to be extensive (Yao et al. 1999). More interesting is the fact that the helicase domain ‘hangs’ over the substrate binding site of the protease (Fig. 6), thus providing a structural rationale for how the helicase domain influences the activity of the protease.
Figure 6. Full length HCV NS3 protein (coordinates are from PDB file 1CU1). The structure is colored according to the secondary structure: β-strands in blue, α-helicesin red. The protease domain is at the bottom of the structure, the helicase domain is at the top. Active site residues of the protease are depicted in ball and stick; the C-terminus of the helicase domain occupies the substrate-binding site of the protease.
Helicase domain
Protease domain

15
NS4B
NS4B is a membrane protein with still unknown function (Lundin et al. 2003). NS4B has been suggested to encode selenium-dependent glutathione peroxidase in an overlapping –1 reading frame (Zhang et al. 1999). Expression of NS4B alone or in fusion with NS4A results in distinct structural rearrangements of the cell secretory apparatus and a reduced rate of protein traffic (Konan et al. 2003, Egger et al. 2002).
NS5A
NS5A (56-58 kDa) is a highly phosphorylated protein with an as yet unknown function (Pawlotsky et al. 1999). Production of p58, the hyperphosphorylated form of p56, may depend on the presence of NS4A (Kaneko et al. 1994). NS5A has been found to be localized in the cytoplasmic membrane surrounding the nucleus, but not in the nucleus, despite the presence of a nuclear localization signal (Pawlotsky et al. 1999). NS5A lacking the 146 N-terminal amino acids, but not a full length NS5A, is a potent transcriptional activator in yeast and mammalian cells (Tanimoto et al. 1997, Kato et al. 1997) but it is not clear whether NS5A can perform this function in vivo.
NS5A binds to PKR (a 68 kDa serine-threonine protein kinase) (Satoh et al. 1998, Gale et al. 1997). Interferon produced in response to viral infection increases PKR expression, which is further activated by binding of viral-encoded or cellular double-strand RNA (activated PKR inactivates eucaryotic initiation factor 2α via phosphorylation). This, in turn, inhibits initiation of mRNA translation and blocks viral replication by inhibiting protein synthesis (Clemens et al. 1997). Inhibition of PKR by NS5A prevents this sequence of events and clears the way for viral replication. NS5A can interfere with the IFN-induced signalling upstream of PKR activation (Heim et al. 1998). It has also been reported that the NS5A protein can interact in vitro with a cellular putative tumour suppressor, which may play a role in HCV carcinogenesis (Herion et al. 1998).
NS5B
NS5B (66 kDa) is an RNA-dependent RNA polymerase (RdRp), which is able to copy homologous as well as heterologous RNA templates (Behrens et al. 1996). Together with the NS3 helicase, NS5B forms the core of the HCV replicase complex. NS5B polymerase can utilize primers for initiation of RNA synthesis (Behrens et al. 1996) but is also able to initiate de novo RNA synthesis

16
on a single-stranded RNA template (Luo et al. 2000). The latter mechanism is probably the one that takes place in vivo, since the template-dependent initiation of transcription of the self-primed 3’-end of the HCV genome (Fig. 2) would result in the loss of genetic information.
NS5B has an overall structure that is typical for most polynucleotide polymerases; it consists of three domains: ‘’fingers’’, ‘’palm’’ and ‘’thumb’’ (Ago et al. 1999, Bressanelli et al. 1999) and contains a GDD motif that is shared by all RdRps. NS5B has a highly hydrophobic C-terminal region which mediates binding of the protein to the ER membrane (Shirota et al. 2002, Ivashkina et al. 2002). This is consistent with the idea that the HCV replicase complex is assembled on the membrane of ER.
Replicase complex
The exact sequence of events that take place at the 3’-UTR during RNA replication is not fully understood, but it is obvious that HCV NS5B (RdRp) must be around to start the synthesis of the negative sense strand. Indeed, NS5B binds to 3’UTR, but the binding is not very strong (Oh et al. 2000) and the question remains of how NS5B, which does not possess helicase activity, can initiate a primer-independent synthesis of RNA which is double stranded on the 3’-UTR end with only one terminal U unpaired (Fig. 2). HCV proteins NS3, NS4B (Piccininni et al. 2002, Banerjee et al. 2001) and NS5A (Shirota et al. 2002) were shown to be involved in this process. The NS3 protein, which possesses helicase activity (see below), produces the most dramatic effect on NS5B activity, increasing primer-independent RNA synthesis (Piccininni et al. 2002), probably via unwinding of the 3’-terminal duplex (Fig. 2).
Replication of the HCV genome has been proposed to take place on the membrane of the endoplasmic reticulum (Hwang et al. 1997), which is supported by the fact that all of the non-structural proteins of HCV associate with the ER membrane (Dimitrova et al. 2003). Complex interplay of interactions between non-structural proteins and the genomic RNA during replication is only possible in an ordered, confined in space structure, where local concentrations of viral factors are high and where sequential, concerted action of all players is permitted. The exact structure of the replicase complex is still not known but it is possible that all of the non-structural and, possibly the structural proteins are involved in the replication of the viral genome (Dimitrova et al. 2003).

17
xORF, protein F.
An additional protein is expressed from the Core coding sequence (xORF) by a –2/+1 ribosomal frameshift in the vicinity of codon 11 (Varaklioti et al. 2002, Xu et al. 2001, Roussel et al. 2003). Protein F (as it has been called by Xu et al. 2001) is a 16 or 17 kDa. In fact, expression of a 16 kDa form of Core protein has been described earlier (Lo et al. 1995). HCV-positive sera specifically recognize protein F, which confirms the in vivo expression of this protein (Varaklioti et al. 2002, Xu et al. 2001, Boulant et al. 2003). The function of protein F is still unknown, but it has been shown to be associated with the ER, like other HCV proteins, which may point to the participation of protein F in HCV replication (Xu et al. 2003).
HCV virion structure
The lack of methods for viral replication in cell culture and the low number of viral particles produced upon infection in living tissue has proved to be a serious obstacle to studying the structure of the HCV virion. However, virus-like particles are visible in the electron microscope (Fig. 3A) in liver tissue or blood serum extracts from infected humans and chimpanzees infected with HCV (Ishida et al. 2001, Li et al. 1995, Kaito et al. 1994, Prince et al. 1996, Shimizu et al. 1996). A number of studies have been performed using artificial virus-like particles (Blanchard et al. 2002, Clayton et al. 2002). Based on the data obtained with virions of other viruses from the Flaviviridae family, a structure of HCV has been deduced: the oligomerized Core protein forms complex with viral RNA and is surrounded by a lipid envelope in which the E1 and E2 proteins are embedded (Fig. 3B). Direct observation of virions has revealed that they are approximately 50 – 70 nm in diameter and that the HCV core has an icosahedron-like structure (Ishida et al. 2001, Li et al. 1995, Prince et al. 1996, Shimizu et al. 1996).
HCV epidemiology
Infected population
Globally, about 200 million persons are estimated to be chronically infected with HCV, and 3 to 4 million persons are newly infected each year (World Health organisation). Thanks to the introduction of HCV tests, the number of new cases has dropped

18
significantly, but their number still remains high, making HCV one of the most common chronic infections in the world.
The prevalence rate of HCV infection differs significantly among different regions of the world (Fig. 7). The lowest prevalence rates are observed in the United Kingdom and Scandinavia (0.01 – 0.1%), followed by South and North America, Western Europe, Australia and South Africa (0.2 – 0.5%), Brazil, Eastern Europe, the Mediterranean, the Mideast and the Indian subcontinent (1 - 5%). The highest rate is observed in Egypt (17 –20%) (Wasley et al. 2000).
Figure 7. Global prevalence of HCV infection (WHO, 1998).
HCV transmission routes
Transmission of HCV occurs via direct blood contact. This can occur in different situations:
Injection drug use. Use of contaminated injection material or needles for injections is the cause of the high prevalence (70 to 90%) of HCV among drug users (Thorpe et al. 2000).
Blood transfusions and use of blood products. Multiple cases of non-A, non-B hepatitis after blood transfusions were the principal motivation of the search for HCV in the 70th. Accurate statistical data on blood-transfusion-associated cases of HCV during that time are not

19
available since the screening for potential contamination of donor blood had begun in the beginning of the 1980s, before discovery of the virus and the availability of a reliable detection method. The risk of HCV infection in connection with blood transfusion became as low as 0.001% per unit of transfused blood by the beginning of the 1990s (Donahue et al. 1992). Before vapor treatment of factors VIII and IX became standard, up to 95% of patients with haemophilia were seropositive for HCV (Blanchette et al. 1991).
Chronic hemodialysis. The current risk of HCV infection after hemodialysis is around 10% per year (Fabrizi et al. 2001).
Organ transplantation.Occupational. Health care workers are obvious risk group for
HCV. In the early 1990s, the prevalence of HCV among health care workers was 3 times higher than in the rest of the population (Lanphear et al. 1994).
Sexual transmission (Fletcher 2003). Vertical transmission (mother to child). Infection of a child
during birth and breast-feeding is possible (Tajiri et al. 2001). Miscellaneous: tattooing, body piercing, etc.
HCV pathogenesis and symptoms
The sequence of events after infection with HCV is shown in Fig. 8. The incubation period (time between exposure and the appearance of the first symptoms) is 6 to 8 weeks. As is clear from the scheme in Fig. 8, most cases of primary HCV infection are asymptomatic and in the rest of cases symptoms are often quite mild, which explains the big reservoir of HCV infection. After 3 to 12 weeks either clearance of the virus or development of chronic infection takes place (Zoulim et al. 2003). In general, about 80-95% of newly infected patients progress to chronic infection (Fig. 8). Cirrhosis develops in about 10% to 20% of persons with chronic infection, and liver cancer develops in 1% to 5% of persons with chronic infection over a period of 20 to 30 years (World Health organisation).
HCV infection is famous for its weak clinical manifestations (Zoulim et al. 2003). Primary infection with symptoms can hardly be described as acute hepatitis. Even less pronounced symptoms are observed in the chronic phase of the infection. Considering the serious outcomes of the disease, it is easy to understand why HCV is often referred to as a ‘’silent killer’’. As in the case of other types of viral

20
hepatitis, all symptoms of HCV infection can be grouped into four major syndromes: a) cytolytic - presence of liver enzymes in the blood due to the cytolysis); b) cholestatic – impairment of gall drainage; c) insufficiency of liver function: lowered protein synthesis and detoxication; d) inflammatory syndrome.
As in the case of Hepatitis B virus, the principal cause of liver destruction in HCV infection appears to be the immune system rather than the virus itself (Nakamoto et al. 2003). The immune response to viral proteins and the ability of the virus to evade the immune system by different means creates a continuous circle of sub-acute inflammation. Regeneration of liver tissue takes place, but in a rather disordered way, leading to the creation of liver tissue devoid of its natural cellular and vascular architecture and with a lot of connective tissue – liver cirrhosis (Nakamoto et al. 2003, Cerny et al. 1999). Continuous chronic inflammation leads to autoimmunization (Pleister et al. 2003, Pawlotsky et al. 1995) and, together with viral factors, to the development of hepatocellular carcinoma (Block et al. 2003).
Figure 8. Course and outcomes of HCV infection. The primary infection occurs 3-12 weeks after exposure to the virus. It takes 20-30 years to develop liver cirrhosis and a further 5-10 years to develop hepatocellular carcinoma.

21
Multiple extra hepatic symptoms are associated with HCV infection. There is a strong correlation between HCV and essential mixed cryoglobulinaemia (Pawlotsky et al. 1995), membranous and membranoproliferative glomerulonephritis (Meyers et al. 2003), porphyria cutanea tarda (Gisbert et al. 2003) and rheumatic manifestations such as Sjögren’s syndrome (Manns et al. 1999). There’s a possible link between HCV and other diseases associated with autoantibody production: systemic vasculitis-polyarteriitis, autoimmune thrombocitopenia and type II diabetes (Manns et al. 1999). Evidence is mounting for the possible involvement of HCV in different types of lymphomas (Weng et al. 2003).
How to kill a virus
The aim of any antiviral therapy is to clear the host organism of virus. This might be achieved either by a) modulation of the host’s antiviral defence mechanisms or b) intervening in the viral life cycle and preventing it from proliferating.
Modulation of host antiviral defense
a) Vaccination is the oldest way of preventing viral infections. Unfortunately, the development of vaccine against HCV has not been successful due to the high genetic variability of the virus.
b) Interferon-α, either alone or in combination with ribavirin (Patel et al. 2003), is the cornerstone of anti-HCV treatment today. Interferon-α has been shown to possess antiviral, antiproliferative and immunomodulatory activities, but the exact mechanism of interferon-α influence in anti-HCV treatment is not known. Although the host immune system is known to be involved in the response to interferon-α therapy (Lau 1998), it is not clear whether the immune system is affected by interferon-α directly.
Viral life-cycle intervention
Interfering with different steps of the viral life cycle could stop viral proliferation: a) Adsorption of the virus to a host cell. In the case of HCV, both
envelope proteins, E1 and E2, take part in the interaction with receptors on the cell surface (see description of E1 and E2). Disruption of this interaction will prevent viral adsorption and

22
will stop the virus from proliferating (VanCompernolle et al. 2003).
b) Penetration of enveloped viruses happens via fusion or endocytosis. The latter is the case with HCV. A number of substances like lysosomotropic agents (ammonium chloride, chloroquine), carboxylic ionophores (monensin and nigericin) and dicyclohexylcarbodiimide can abolish acidification of the viral endosome, thereby preventing entry of the core viral particle into the cell (Iacoangeli et al. 2000). Unfortunately, these substances lack specificity and are toxic to the host.
c) Replication. Viruses adopt different replication strategies. According to The International Committee on Taxonomy of Viruses (http://www.ncbi.nlm.nih.gov/ICTV/) HCV belongs to group IV, (+) sense RNA viruses with no DNA intermediate in their life cycle. Translation of the HCV genome starts right after entry into the cell. The translation product, the polyprotein, needs to be processed, which is achieved by both cellular and HCV NS2-3 and NS3 proteases. After that, the NS5B polymerase, with the help of the NS3 helicase and some other viral and cellular factors, replicates the HCV genome, which, in turn can be used for further translation or be packed into new viral particles. Inactivation of the viral enzymes is lethal to the virus (Kolykhalov et al. 2000) and the design of inhibitors for those enzymes is under way (De Francesco et al. 2003, Kleymann 2003). Inhibition of translation could be achieved by other means than inhibition of enzymes, such as blockage of regions on the viral genome important for its replication (Reigadas et al. 2003).
d) Virus assembly. Inhibition of any stage of assembly of the infectious viral particle will prevent viral proliferation (Tang et al. 2003, Deres et al. 2003, Tan 2002).
e) Viral release. Inhibition of the release of virus from the cell will end the spread of the virus and will stop viral proliferation (Wen et al. 2003). In the present study, we concentrated on trying to find good
inhibitors of the NS3 protease with the aim of obtaining a good lead compound for future anti-HCV drug development.

23
How to inhibit the NS3 protease
Classifications of serine protease inhibitors
Enzyme inhibition (inhibeo (lat) – to hold in, to restrain) means suppression of the enzymatic reaction. An inhibitor is a chemical substance that specifically inhibits an enzyme. There are many ways to do that but, in general, enzyme-inhibitor interaction results in: a) prevention of substrate binding and/or b) disruption of the catalytic machinery of an enzyme. Upon closer inspection, most inhibitors (except purely competitive inhibitors) will influence both substrate binding and the catalytic machinery. This influence can be direct (competing for the substrate or prosthetic group binding site) or indirect (competition for binding of non-catalytic cofactor).
Kinetic classification of inhibitors is the only well established classification that is valid for all types of enzymes (Table 1, I) (Nomenclature Committee of the International Union of Biochemistry, http://www.chem.qmul.ac.uk/iubmb/kinetics/). Any inhibitor can be more or less well described by a kinetic classification, but this does not always tell us about the exact mechanism of the inhibitor-enzyme interaction. Additional mechanistic classification of inhibitors that takes into account the inhibition mechanism (and, possibly, some structural features of inhibitors that influence this mechanism) is needed. Classifications of inhibitors that appear in the literature (like classification II (Sanderson 1999), III (Ripka et al. 1998) and VI (Demuth 1990) in Table 1) do not account for all aspects of the inhibitory mechanisms and structures. Box IV of Table 1 lists different inhibitory types that often appear in the literature, many of which are synonymous to each other. In this work, two classifications (II and V, Table 1) have been adopted for describing different types of inhibitors.
Non-covalent serine protease inhibitors
Protein non-covalent serine protease inhibitors.
A large number of non-covalent protein inhibitors exist in nature. These inhibitors are physiologically important, since they are involved in regulation of serine protease activity in vivo (Roberts et al.

24
Table 1 Different classifications of serine protease inhibitors.
Classification Types of inhibitors I. Kinetic(Nomenclature Committee of the International Union of Biochemistry)
1. a) reversible b) irreversible (catalytic poisons)
2. a) linear b) non-linear
3. a) competitive (specific) b) uncompetitive (catalytic or coupling inhibition) c) mixed and non-competitive
4. a) fast-binding b) slow-binding
5. a) tight-binding b) non-tight-binding
II. Mechanistic (Sanderson E.J., 1999)
a) non-covalent b) covalent
III. Peptidomimetic (Ripka A.S., 1998)
a) Peptidomimetics I: peptides and peptide-like substances
b) Peptidomimetics II: small non-peptidic inhibitors of peptide receptors
c) Peptidomimetics III: non-peptide inhibitors that mimic interaction of enzyme with peptidic inhibitor
IV. Other terminology commonly used in scientific literature
Substrate analogs Substrate based inhibitor Product based inhibitor Alternate substrate inhibitor Suicide substrate inhibitor Suicide inactivator Pseudo-suicide inactivator (paracatalytic inhibitor)
V. Structural classification a) Protein inhibitors b) peptide and peptide-mimicking
inhibitors c) non-peptide inhibitors
VI. Mechanistic classification of covalent serine and cysteine protease inhibitors (Demuth 1990)
1. Affiniy-mediated inhibitors a) affinity labels b) transition-state analogs
2. Mechanism-based inhibitors a) acyl-enzyme inhibitors b) enzyme-activated inhibitors

25
1995, Bode et al. 1992). There are at least 59 different families of protein inhibitors of serine proteases (MEROPS database, http://merops.sanger.ac.uk) and most of them do not form a covalent bond with the protease - like the Kunitz family (inter-α-trypsin inhibitor (Salier 1990), the Kazal family (avian egg ovomucoid inhibitor (Saxena et al. 1997), dipetalin (Schlott et al. 2002), etc.
The need for parenteral administration of protein inhibitors and their inability to pass through cellular membranes limits their medical applications to conditions where excessive activity of extracellular proteases is a problem (bacterial sepsis, acute pancreatitis).
Peptide non-covalent inhibitors
Peptides that interact with a protease in a substrate-like way but are not cleaved become inhibitors. There are two strategies for the design of such inhibitors:
- Non-cleavable substrate. Replacement of the scissile bond in a natural protease substrate by an uncleavable bond turns a substrate into an inhibitor (Ingallinella et al. 2000).
- Product inhibitor. The products of nearly all enzyme-catalyzed reactions behave as inhibitors when they are present in the reaction mixture (Nomenclature Committee of the International Union of Biochemistry, 1981). Product inhibition is a phenomenon that is known to occur with serine proteases too (Edwards et al. 1994, Bru et al. 1991, Knight et al. 1995). Product inhibition is especially pronounced in the case of the HCV NS3 protease (Steinkuhler et al. 1998): very potent non-prime-site (Ingallinella et al. 1998) and less potent prime-site (Ingallinella et al. 2002) based product inhibitors were developed based on the sequence of peptides originated form natural HCV NS3 cleavage sites. Peptides that do not mimic a substrate constitute another class
of non-covalent peptide inhibitors of serine proteases. Exosite thrombin inhibitors (Dennis et al. 2001) and inhibitor of the NS4A cofactor interaction with HCV NS3 protease (Shimizu et al. 1996) are examples. These inhibitors do not interact with the substrate-binding site of the protease.
Peptide inhibitors offer high selectivity due to specific and often unique interactions with a target protease. But, being peptides, they are likely to have poor pharmacokinetic properties and are often viewed as unpromising drug candidates. However, peptide-based inhibitors may serve as templates for optimization toward compounds

26
with higher inhibitory potency, a less peptidic nature and better pharmacokinetic properties. Recent successes with HIV (Dash et al. 2003) and HCV (Lamarre et al. 2003) protease inhibitors, which originated from peptides demonstrate the importance of this class of inhibitors in drug design in the future.
Non-peptidic, non-covalent inhibitors
A number of potent non-peptidic inhibitors of serine proteases have been developed in the last decade, for example, towards thrombin, factor Xa (Sanderson 1999) and HCV NS3 protease (Dymock 2001). These inhibitors are usually specific and have better pharmacokinetic properties than do peptide-based inhibitors. Discovery of such compounds is complicated by the fact that their structures are difficult to deduce a priori. As a consequence, the design of such compounds is often guided by SAR previously obtained with substrate-derived inhibitors, by sheer luck or by a combinatorial approach using known templates.
Covalent serine protease inhibitors.
A large number of serine protease inhibitors form a covalent bond with catalytic residues of proteases, thus inhibiting them. Classification VI in Table 1 summarizes different types of covalent inhibitors. Covalent inhibitors usually exploit catalytic mechanism of enzymes for inhibition.
The general mechanism of action of serine proteases is depicted in (Fig. 9). The charge relay system, Asp, His and Ser (Fersht et al. 1973) results in abstraction of a proton from the catalytic serine and transformation of its hydroxyl group from a poor nucleophile into a good one (Fig. 9a). The next step is a nucleophilic attack on the carbonyl carbon of the scissile bond by the activated serine hydroxyl and the creation of a tetrahedral intermediate that is stabilized by hydrogen bonding of the carbonyl’s oxyanion by the amide protons of Ser-195 and Gly-193 (oxyanion hole) (Fig. 9b). His-57 transfers a proton to the amine of the scissile bond whereby it breaks (Fig. 9c). The remaining part of the substrate is now attached to the catalytic Ser via an ester bond, which is easily hydrolyzed, assisted by general acid catalysis by His-57 (Silverman 2000) (Fig. 9d-f).
Covalent inhibitors act like substrates from the beginning and they proceed through several catalytic steps up to a tetrahedral intermediate (reversible inhibitors) or an acyl intermediate (often

27
irreversible inhibitors) but not further, thus occupying the catalytic center and inhibiting protease.
Figure 9. General catalytic mechanism of serine proteases. Members of the catalytic triad are marked Asp102, His57, Ser195 (numbering is based on chymotrypsin sequence). Backbone amides of Ser195 and Gly193 form an oxyanion hole. A)Activation of the hydroxyl group of Ser195 by proton abstraction. Nucleophilic attack on the peptide bond carbonyl. B) Formation of the tetrahedral intermediate and its stabilization by the oxyanion hole. C) Collapse of the tetrahedral intermediate with peptide bond breakage. Creation of the acyl intermediate. D, E, F) Hydrolysis of the ester bond of the acyl intermediate by water, activated by the catalytic histidine. Release of peptide moiety.
Protein covalent serine protease inhibitors.
The serpins constitute a large family of protein protease inhibitors (Gettins 2002). Interaction of a serpin with protease results in cleavage of the specificity loop of the serpin (Gettins 2002). After formation of an acyl-intermediate (Fig. 9c) the serpin-protease complex undergoes a conformational transformation that results in disruption of the catalytic machinery of the enzyme, stabilization of the serpin-enzyme acyl-intermediate complex and irreversible enzyme

28
inhibition (Gettins 2002). The HCV NS3 protease has also been shown to be inhibited by serpins (Drouet et al. 1999, Richer et al. 2003).
Peptide covalent inhibitors.
Inhibitors of this class consist of two principal parts: a) a peptidic moiety, which is usually derived from a substrate or product (denoted as R in Fig. 10a), and b) an electrophilic moiety (a serine trap or an electrophilic ‘warhead’). The peptidic moiety guides the ‘warhead’ into the active center of the protease and the inhibitor’s electrophilic residue (carbonyl in Fig. 10a) becomes a subject for nucleophilic attack by the catalytic serine with subsequent formation of the covalent tetrahedral intermediate.
Figure 10. Interaction of covalent inhibitors with a serine protease active site. The numbering of catalytic residues is based on the NS3 protease sequence. A.Interaction of the electrophilic moiety of the covalent peptide inhibitor with the catalytic center of serine protease. Formation of the tetrahedral hemiketal structure. R - peptide chain, EWG – electron withdrawing group. B. Interaction of the non-peptide covalent inhibitor, phenylmethylenesulfonylfluoride (PMSF), with a serine protease. Formation of an acyl-intermediate-like structure (sulfonester).
There is a big variety of electrophilic moieties in peptidyl electrophilic inhibitors, including diketones, ketoamides, ketoesters, ketoacids, carbamates, carbazates, peptidy α-keto heterocycles, aldehydes, boronates, phosphonates, chloro- and fluoroketones and inverse substrates (Walker et al. 2001, Edwards et al. 1994). Inhibition of serine proteases by peptidyl chloromethyl ketones involves not only
S Cl
O
O
HO Ser195
-HClS O
O
O
Ser195
B
N
N
His57
H
O
O
Asp102 Ser195
N
OH H
NGly193
H
Nucleophilicattack
Electrophilic inhibitor
N
His57
H
O
O
Asp102
N+ H
Tetrahedralhemiketal
A
R
EWG
O
Ser195
N
O H
R
EWG
O-
NGly193
H

29
formation of a covalent bond with the catalytic serine but also alkylation of the catalytic histidine residue.
Many types of peptide covalent inhibitors have been described for the NS3 protease, including peptidyl aldehydes (Landro et al. 1997, Llinas-Brunetb et al. 1998, Attwood et al. 1999, Fattori et al. 2000), boronates (Archer et al. 2002), trifluoromethyl ketones, pentafluoromethyl ketones, α-ketoamides (Llinas-Brunetb et al. 1998, Yaya et al. 2004) and α-keto acids (Narjes et al. 2000).
Electrophilic peptide inhibitors are usually very potent, but seldom selective (Llinas-Brunetb et al. 1998), inhibiting many serine and even cysteine proteases. The metabolic stability of electrophilic groups is low due to their high reactivity. Another problem is the slow-binding nature of many electrophilic inhibitors (Edwards et al. 1994). Such inhibitors are likely to be useless in cases where the target protease does its business shortly after its activation (like thrombin, Stone 1995) or translation (like NS3 protease). For these reasons, few electrophilic inhibitors proceeded from in vitro inhibition studies into clinical trials (Walker et al. 2001, Edwards et al. 1994, Sanderson 1999) and none (to my knowledge) have yet been approved for use in humans.
Non-peptidic covalent inhibitors
The mechanism of inhibition by this type of compounds is similar to that of electrophilic peptide inhibitors (Fig. 10b). A nucleophilic attack of an electrophilic group in the inhibitor results in the formation of a stable acyl-intermediate (boronic acids, phosphonates, sulfonyl fluorides (Fig. 10b), chloropyrones, benzoxazinones, benzisothiazolines (Edwards et al. 1994)). It sometimes also results in a subsequent alkylation of a close-to-catalytic-center residue (usually a catalytic histidine): halo enol and ynenol lactones, isocoumarins, β-lactams and succinimides (Edwards et al. 1994).
Like covalent peptide inhibitors, non-peptide inhibitors are seldom selective and have not yet produced any medically useful compound for the very same reason.
Inhibitor optimization strategy.
The first step in inhibitor design is the selection of a lead compound, i.e., a compound that is known to inhibit the target protease and, for various reasons, looks promising as a scaffold for

30
future improvements. Guided by the results of different in vitro and invivo tests, different chemical groups of the lead compound should be modified in order to improve the following properties of the compound:
- Inhibitory potency. Guided by in vitro inhibition studies using purified enzyme.
- Selectivity. In vitro inhibition studies using different related enzymes.
- Chemical stability. In vitro tests. - Pharmacokinetic properties (metabolic stability of the
drug, its absorption/excretion, etc.). Many pharmacokinetic properties can be predicted using the wealth of knowledge accumulated in the area of drug design. In vivo tests are needed to verify these predictions.
- Pharmacodynamic properties. In vivo tests. Improvement of four of five parameters can, and should be
done, without in vivo tests.

31
Present study
Aims of the study
1. To obtain and clone the gene corresponding to full length NS3 protein. Express the gene and purify NS3 protein.
2. To develop an assay for the NS3 protease and to study its enzymatic properties.
3. To obtain potent peptide-based inhibitors of the NS3 protease, to study their structure-activity relationships (SAR) and to optimize them according to what SAR revealed.
4. To study the selectivity of peptide based inhibitors of the NS3 protease by measuring the inhibition of other serine and cysteine proteases.
Obtaining the target enzyme
The sequence of the NS3 gene obtained was close to that of H77 strain of type 1a hepatitis C virus. Total RNA was extracted from the serum of a patient infected with HCV and reverse transcribed using HCV-specific primers. NS3 cDNA was then introduced into pGEM-3Z(+) vector (Promega, Madison, WI, USA) (Paper I) and this sequence was used for making different constructs of NS3. All of the constructs of NS3 tested in the present study and the results of their expression and purification are summarized in Table 2. Early experiments with constructs I and III (Table 2) (unpublished data) showed that NS3 is poorly expressed, often with accumulation of a big portion of the protein in an insoluble fraction of the cell. Furthermore, it requires quick handling during purification due to rapid loss of activity after cell lysis. The problem of protein instability is probably

32
Table 2 Different constructs of NS3.
related to the absence of the NS4A cofactor. It has been previously shown that NS3 is quickly degraded in the cell if NS4A is not present (Tanji et al. 1995). In the absence of NS4A, the N-terminal part of NS3 is rather disordered and accessible for degradation by proteolytic enzymes and aggregation (Urbani et al. 1999). The problem of instability of NS3 from constructs I-III and V was solved when we found that 25% glycerol and detergents like CHAPS or n-octyl-β-D-glucoside prevented loss of protease activity during purification, indicating that protein aggregation is the most likely cause of the loss of the protease activity. However, the presence of glycerol and detergent in the buffers has created other problems:
1. Initially, lysis of the host cells expressing NS3 was performed by sonication. The presence of detergent in the lysation buffer led to foaming immediately after the first bursts of ultrasound. Foam is a very poor conductor of sound waves and lysis of cells cannot be achieved if foam is present. The use of a French press for cell lysis solved this problem.
2. Due to the high viscosity of buffers with 25% glycerol, the efficiency of many purification procedures, like gel filtration and ion-exchange chromatography, was dramatically decreased. This, together with the fact that the concentration of soluble protein in the cell lysate was very low, made purification of constructs with no affinity tags (I and III) impossible. Due to the presence of the affinity tag (glutathione transferase) we were able to partially purify construct II; this
Construct Host Expression
Activity Purification
I E.coli − −
II E.coli + ±
III E.coli + −
IV P.pastoris − −
V E.coli + +
VI E.coli + −
NS3 protease
NS3 whole length
GST NS3 protease NS4A
NS3 full-length
His6 Linker, EK site NS3 whole length
NS4A Linker His7 NS3 whole length

33
indicated that use of affinity techniques is required. Successful purification of construct V by means of IMAC and polyU-Sepharose chromatography (Paper I) proved it was right. Whole length NS3 protein fused to a histidine tag (his-tag) via
a linker containing an enterokinase cleavage (EK) site (construct V, Table 2) was successfully purified using immobilized metal affinity chromatography (IMAC) and affinity chromatography using polyU-Sepharose (Paper I). Attempts to cleave the affinity tag were unsuccessful, probably due to inaccessibility of the EK cleavage site in the protein. All of the kinetic and inhibition studies presented in this study were performed using the protein with the tag. We have validated the eligibility of the purified enzyme for the inhibition studies by showing that it cleaves the specific NS3 protease substrate and that it is inhibited by the existing specific NS3 protease inhibitors (Paper I).
Construct VI, with the NS4A peptide cofactor fused to the N-terminus of full-length protein via a linker containing a stretch of seven histidines has been successfully designed and expressed in E.coli (unpublished data). No glycerol or detergent was required for preservation of the protease activity, which supports the idea of the stabilizing effect of the NS4A cofactor on the NS3 protein. Purification of construct VI has not yet been achieved, probably due to the formation of complexes between the NS3 protein and cellular components present in the cell lysate.
Establishment of assay for NS3 protease (construct V).
In order to perform an efficient screening of the inhibitors and to obtain reliable and reproducible results, a robust assay is needed. Important parameters found to influence NS3 protease activity are presented below.
Substrate
The substrate used in this work mimics the natural cleavage sites of the NS3 protease (Taliani et al. 1996). It contains a chromophore and a fluorophore placed on opposite sides of the scissile bond. The detection principle is based on resonance energy transfer (RET), where the quenching effect of the chromophore on the fluorescence of fluorophore is greatly reduced upon cleavage of the molecule and separation of the two groups. The advantages of this method are that it is continuous and more sensitive than absorbance measurement.

34
Inner filter effect (IFE)
IFE is a problem common to all RET substrates. It arises from the fact that the quenching group in a substrate absorbs some light even after separation from the fluorophore. The degree of this absorption is directly proportional to the substrate concentration (Liuet al. 1999) and, thanks to that, a correction coefficient can be determined for each substrate concentration (using substrate and free fluorophore) and used for calculating true fluorescence values. IFE correction was only required in measurements where the substrate concentration was varied (unpublished data).
NS4A cofactor
Since construct V (Table 2) had no NS4A cofactor we were forced to use synthetic peptides mimicking a central part of the NS4A cofactor (Fig. 11) (the central part has been found to be sufficient for modulation of NS3 protease activity (Koch et al. 1996, Tomei et al. 1996)). With the second cofactor (NS4A 2) (Fig. 11) the activity of the NS3 protease was higher and it was therefore used in all kinetic and inhibition studies done thereafter (Papers II-V).
Figure 11. HCV NS4A cofactor peptides used in NS3 protease assay. The sequence of the NS4A protein of HCV strain H77 is given in the upper row. The minimal fragment of NS4A required for activation of NS3 is framed. Peptides mimicking this region are also in frame, aligned to the upper sequence.
The titration curve of NS3 with NS4A peptide has a maximum, suggesting that high cofactor concentrations are detrimental to the protease activity (Fig. 12) (unpublished data). At high ionic strength (150 mM NaCl) the decrease of NS3 activity at higher NS4A concentrations is more pronounced and maximum activity occurs at a lover cofactor concentration (switched to the left, (Fig. 12)), which suggests that hydrophobic forces might be blamed for the observed phenomenon.
NS4A protein STWVLVGGVLAALAAYCLSTG CVVIVGRIVLSGK PAIIPDREVLYQEFDEMEEC
NS4A 1 CVVIVGRVVLSGK
NS4A 2 KKG SVVIVGRIVLSGK
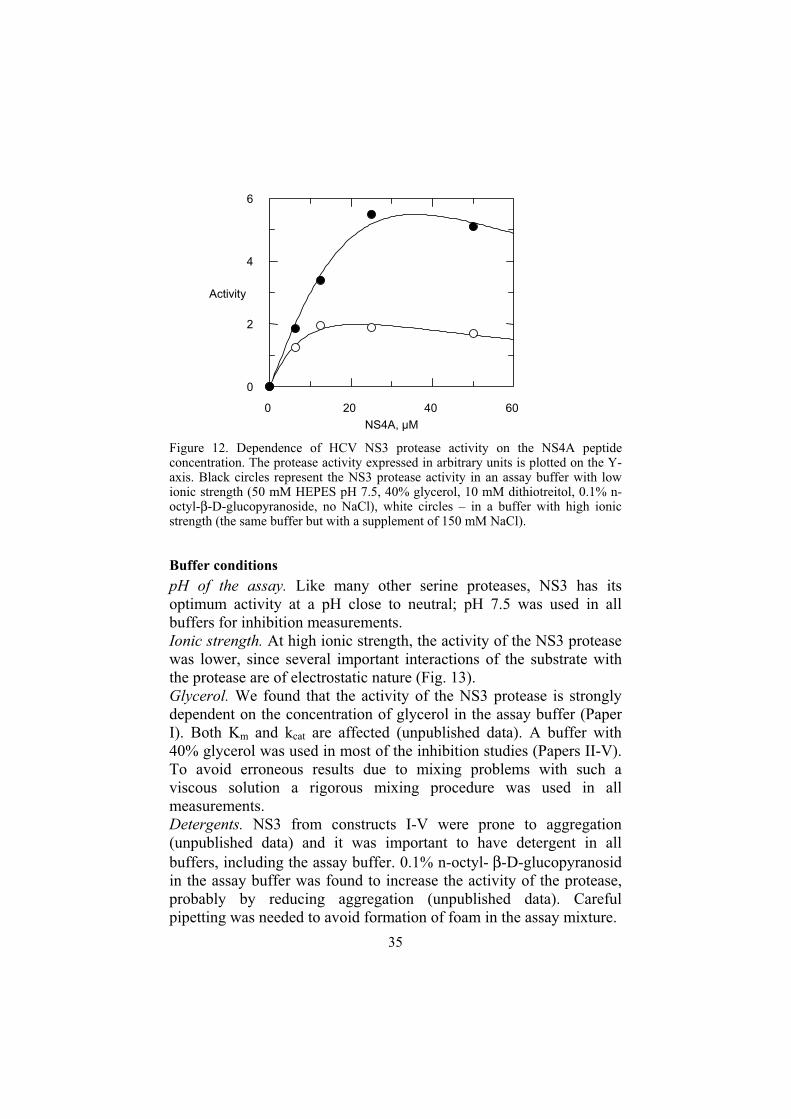
35
Figure 12. Dependence of HCV NS3 protease activity on the NS4A peptide concentration. The protease activity expressed in arbitrary units is plotted on the Y-axis. Black circles represent the NS3 protease activity in an assay buffer with low ionic strength (50 mM HEPES pH 7.5, 40% glycerol, 10 mM dithiotreitol, 0.1% n-octyl-β-D-glucopyranoside, no NaCl), white circles – in a buffer with high ionic strength (the same buffer but with a supplement of 150 mM NaCl).
Buffer conditions
pH of the assay. Like many other serine proteases, NS3 has its optimum activity at a pH close to neutral; pH 7.5 was used in all buffers for inhibition measurements. Ionic strength. At high ionic strength, the activity of the NS3 protease was lower, since several important interactions of the substrate with the protease are of electrostatic nature (Fig. 13). Glycerol. We found that the activity of the NS3 protease is strongly dependent on the concentration of glycerol in the assay buffer (Paper I). Both Km and kcat are affected (unpublished data). A buffer with 40% glycerol was used in most of the inhibition studies (Papers II-V). To avoid erroneous results due to mixing problems with such a viscous solution a rigorous mixing procedure was used in all measurements. Detergents. NS3 from constructs I-V were prone to aggregation (unpublished data) and it was important to have detergent in all buffers, including the assay buffer. 0.1% n-octyl- β-D-glucopyranosidin the assay buffer was found to increase the activity of the protease, probably by reducing aggregation (unpublished data). Careful pipetting was needed to avoid formation of foam in the assay mixture.
NS4A, µM
0 20 40 60
Activity
0
2
4
6

36
Reducing agents. NS3 is an intracellular enzyme and is devoid of disulfide bridges. In order to keep all of NS3’s cysteines reduced, 10 mM dithiothreitol was included in the assay buffer.
Substrate inhibition
We have noticed that at higher substrate concentrations (>10Km), the activity of the protease decreases. This phenomenon cannot be explained by the IFE, since the trend remains even after IFE correction. Therefore it is likely that we are observing substrate inhibition. In order to minimize the substrate inhibition effect on the inhibition data we used a low substrate concentration (3-4 Km) in the inhibition studies.
Kinetic constants of NS3 protease
The kinetic constants of the NS3 protease (construct V, Table 2) measured in 50 mM HEPES pH 7.5, 40% glycerol, 10 mM DTT, 0.1% n-octyl-β-D-glucopyranoside at 30°C were as follows: Km=0.15µM, kcat=0.42 s-1, kcat/Km=2.8 µM-1 s-1 (Paper II). The substrate concentrations used for the measurements were between 0.25 Km and 3Km in order to avoid problems with substrate inhibition.
Selectivity measurements
One of the important properties of any drug is its selectivity, i.e., the compound should interact only with the intended target and thus produce only the desired pharmacological effect. In order to select a lead compound for further optimization, one has to perform a selectivity study to check the activity of the compound on other enzymes related to the target enzyme. We were thus interested to know whether the chosen NS3 protease inhibitors would inhibit other serine and cysteine proteases. Peptidic inhibitors with different C-terminal residues and P1 side chains, but otherwise identical sequence, were tested on the serine proteases HLE, PPE, BPC and the cysteine protease CatB. Selectivity indexes were calculated for each compound with every protease. The results of the selectivity studies are presented in Table 4 and are discussed below.
Slow-binding inhibition
In most inhibition studies, the inhibitor was incubated with enzyme for at least 10 minutes before starting the reaction by the addition of the substrate (Papers II-IV). However, with electrophilic inhibitors we tried to look for signs of slow-binding inhibition (Paper V), since many electrophilic inhibitors of serine and cysteine proteases

37
are slow binders, and it had also been shown to occur with the NS3 protease (Narjes et al. 2000). Slow-binding inhibition is characterized by two-phase association kinetics in non-stop-flow measurements (Morrison et al. 1988):
1. Fast phase – the formation of (usually) a loose non-covalent enzyme-inhibitor complex: steady state is achieved in few seconds. This phase is characterized by Ki (Morrison et al. 1988), which is an inhibition (equilibrium) constant of the complex formed.
2. Slow phase – intramolecular rearrangement of enzyme-inhibitor complex, often with the formation of a covalent complex: this phase may take from few minutes to many hours and is characterized by Ki* (Morrison et al. 1988), which is an inhibition constant of the final enzyme-inhibitor complex. In this work we have always measured only this final Ki*, which we always denote as Ki (Paper V).
Detection of slow-binding inhibition is achieved by continuous measurement of the enzymatic reaction and inspection of the results for signs of the two-phase progress curve. An important determinant of successful detection is the duration of the linear part of the progress curve, which should last for longer than it takes to reach the steady state of the slow phase (hours? days?). This could be achieved by a) high substrate concentration (ideally >10Km) and b) as low enzyme concentration as possible. Another important parameter of slow-binding measurements is protein stability. Some enzymes loose their activity with time and this will complicate analysis of inhibition data, especially when the loss of protease activity is caused by autoproteolysis. The presence of protease inhibitor will slow down autoproteolysis along with cleavage of the substrate, which will make comparison of progress curves of inhibited and uninhibited reactions impossible.
We were able to maintain linearity of the progress curve for most of the enzymes used in our study (Paper V). HLE, however, was a problem, since a loss of activity of uninhibited enzyme was obvious just 10 minutes after the start of the reaction.
The Ki values of slow-binding inhibitors were calculated using the slope of the progress curve after the steady state of the slow phase was achieved.

38
Optimization of peptide-based inhibitors of the NS3 protease (inhibitory potency and selectivity)
Figure 13. Schematic representation of the interaction of the NS3 protease with the N-terminal part of the substrate (product inhibitor). Positively charged subsites 5 and 6 are indicated as S5,6.
Inhibitor design strategy
The choice of the lead compound(s) is the first step. Pronounced inhibition of the NS3 protease by its N-terminal reaction product has been shown (Steinkuhler et al. 1998) and potent N-terminal product based hexapeptide inhibitors were created (Ingallinella et al. 1998) (Fig. 13). These inhibitors belong to non-covalent peptide inhibitors of proteases and looked promising as lead compounds (see 5.1.1.2.) for optimization towards higher inhibitory potency and better pharmacokinetic properties. The initial goals were:
- To find a good substitution for the P1 cysteine residue, which produced the best inhibition results (Paper II and III). The thiol group of cysteine is highly reactive and will be catabolysed very quickly in vivo.
- To shorten hexapeptide inhibitors from the N-terminal part. - To increase inhibitory potency by modifying side chain
residues (Paper II).
CH2
S5,6
S5,6
S5,6
Phe154
Gly137
Hydrophobic interaction
Hydrogen bonding inoxyanion hole
Electrostatic interactions
His57
N
H
CapNH
NHNH
NHNH
SH
O
O
O
O
O
O
R2
R3
R4
R5
R6
NH
O
Ser139
Hydrogen bonding with catalytic histidine
H N
NH

39
- To increase inhibitory potency and selectivity by modifying the inhibitor’s C-terminal residues and P1 side chains (Papers III and IV).
Optimization strategy is depicted in (Fig. 14). Below is a description of the optimization of different groups.
Figure 14. Strategy for optimization of the product inhibitor depicted in Fig.13.
N-terminal capping groups
The interaction of the N-terminal part of the product inhibitor of the NS3 protease is electrostatic in nature (Fig.13), which explains why the presence of the positively charged amino group is detrimental to the enzyme-inhibitor interaction. The negatively charged acidic groups on the N-terminus produce better results. A succinic acid residue invariably produced slightly better inhibitors, since it bears more negative charge than acetic acid residue (Paper II). The effect of the capping group on selectivity was not studied.
P5 and P6 residues.
Removal of P5 and P6 residues from hexapeptide inhibitors resulted in 10 to 25-fold loss of inhibitory potency (Johansson et al. 2001). This must be compared with 600 to 1600-fold loss of inhibitory potency in assays where only the protease domain of NS3 was used (Ingallinell et al. 1998, Cicero et al. 1999).
Modification of P1 and capping
groups
Shortening of the peptides from the
N-terminus
Modification of P2, P3, P4residues
Shortening of the peptides from the
N-terminus

40
C-terminal residues
We have tested several types of C-terminal residues: 1. Free carboxylate (Fig. 15A) (Papers III and IV). This moiety
occurs naturally in NS3 protease product inhibitors. The interaction of carboxylate group with the active site of the protease is schematically depicted in (Fig. 13). Carboxylate group uses unique properties of the NS3 active site structure and produces quite potent and generally selective inhibitors (compounds 15-19, Table 4). But from the pharmacokinetic point of view the carboxylate group is not good, since it is easily metabolized in vivo and its high polarity worsens absorption of the drug administered per os.
2. Carboxylic acid isosteres (Fig. 15B) (Paper III). Bioisosteric replacements are often used to enhance the pharmacokinetic and pharmacodynamic properties of drug candidates (Olesen 2001, Patani et al. 1996). Inhibitors with tetrazole and acyl sulfonamide residues (Fig. 15B) were both potent and very selective (compounds 20-23, Table 4). Molecular modeling had revealed that specific interactions of acyl sulfonamide group, different from free carboxilate containing inhibitors, might be the reason for the high potency and selectivity of this type of compounds (Fig. 16).
3. Electrophilic residues (Fig. 15C) (Paper III and IV). In order to boost inhibitory potency we decided to include an electrophilic group into peptide inhibitors. Introduction of an electrophilic group into inhibitor often increases inhibitory potency by several orders of magnitude (Narjes et al. 2000, Wiley et al. 1995). These inhibitors are electrophilic(covalent) peptide inhibitors. To our surprise, inhibitors with an electrophilic C-terminal residue were equally or less potent than corresponding inhibitors with a free carboxylate (Table 4). The selectivity of electrophilic inhibitors was low. In fact, some compounds were better at inhibiting other proteases than NS3, such as compound 1 on PPE and compounds 11 on HLE (Table 4).
P1 side chains
We found that amino acids with non-bulky, hydrophobic side chain residues are best accepted in the P1 position (cysteine being the best) (Fig. 15A) in full accordance with NS3’s substrate specificity (Paper II and III). Cysteine is not suitable in potential drugs, since it is metabolically instable. The cyclopropane side chain (Fig.15A, compound 52b) is especially promising in non-electrophilic inhibitors,

41
since it is chemically stable and produces potent inhibitors (Paper III). The P1 cyclopropane side chain residue is not well accepted in electrophilic inhibitors (Fig. 15C) (see below).
Figure 15. Structure-activity relationships of peptide-based inhibitors with A: non-electrophilic, B: with electrophilic and C: carboxylic acid bioisostere C-terminal residues. Top – general structure of the peptide inhibitor; bottom – different P1 residues, their numbers as they appear in Paper III (in bold) and Ki-values of inhibitors containing corresponding residues.
We have observed that inhibitors with an amino butyric acid-based P1 side chain residue (compounds 6-14 and 19, Table 4) are usually good inhibitors of HLE, which is in good agreement with HLE preference for small hydrophobic P1 side chains in substrates. This result highlights the importance of the P1 side chain residue for selectivity of both covalent and non-covalent peptide inhibitors and it
SucN
NN
NN
R
H
H
H
H
H O
O
O
O
O
O
OHO
NOH
H O
HS
NOH
H O
NOH
H O
NOH
H O
NOH
H O
50b
Ki=0.028 µM52b
Ki=0.091 µM53b
Ki=0.12 µM51b
Ki=0.22 µM54b
Ki=1.5 µM
HO
46b
Ki=0.0038 µM45b
Ki=0.0136 µM43b
Ki=0.156 µM44b
Ki=0.19 µM
NHNH
S
OO O
NH
N N
NNH
NH
N N
NNH
NHNH
S
OO O
NHC2F5
O
NHC2F5
O
NHC2F5
O
40b
Ki=0.081 µM38b
Ki=0.12 µM39b
Ki=1.8 µM
NH
O
O
OH NH N
N
NHN
O
NH N
N
NHN
O
49b
Ki=0.098 µM41b
Ki=0.3 µM42b
Ki=1.9 µM
A
B
C

42
is in a good agreement with the major role of the P1 side chain residue in the substrate specificity of serine proteases in general (Hedstrom 2002).
Figure 16. Model of the interaction of a peptide inhibitor with a C-terminal acyl sulfonamide group (Suc-D-Glu-Leu-Ile-Cha-Nva-NHSO2Ph, compound 45a in Paper III) in P1 position with full length NS3 protein (Paper III). A – Full length NS3 protein (coordinates are taken from PDB file 1CU1), the helicase domain is colored blue and the protease domain is pink. B and C – two predicted modes of interaction of two P1 and P2 groups of the inhibitor with the active site of HCV NS3 protease and with the helicase. The inhibitor is colored with all carbons in green, oxygens in red, nitrogens in blue and hydrogens in gray. Hydrogen bonds are indicated by a dashed line.
P2, P3, P4 side chains (Paper II)
Using a library of tetrapeptides (Table 3), we revealed that structure-activity relationships (SAR) for P2-P4 side chain residues are rather complex. 2-Nal is best accepted in the P2 position when
B
C
A

43
Figure 17. Model of the interaction of a tetrapeptide inhibitor Suc-Chg-Glu-2-Nal-Cys (compound 9 in Paper II) with the full-length NS3 protein (Paper II). A – Full length NS3 protein (coordinates are taken from PDB file 1CU1), the helicase domain is colored blue and the protease domain is pink. B and C – two predicted modes of interaction of the tetrapeptide inhibitor with the active site of HCV NS3 protease and with the helicase. Inhibitor is colored with all carbons in green, oxygens in red, nitrogens in blue and hydrogens in light blue. Hydrogen bonds are indicated by a dashed lines. Both possible tautomeric forms of His528 are considered: B – protonation at His-N4, C – protonation at His-N2.
Chg occupies P4. Replacement of Chg in P4 by Dif makes Glu rather than 2-Nal preferable at the P2 position. To explain rather high inhibitory potency of the tetrapaptides inhibitors we hypothesized that the helicase domain of NS3 might constitute a part of the substrate/inhibitor binding site. Molecular modeling had revealed a hydrogen bond between His528 of the helicase domain and the
B
C
A
44
carbonyl oxygen of the P4 residue and there is a large S2 pocket separated from the S4 pocket by the side chain of Gln526 (Fig. 17).
This result highlights the importance of using full-length enzyme in inhibition studies. The importance of the P2-P4 residues for inhibitory selectivity was not studied.
Structure-activity relationships of electrophilic inhibitors with cyclopropane side chain residue
Influence of cyclopropane P1 residue on the activity of electrophilic
inhibitors
1-aminocyclopropane-1-carboxylic acid (ACPC)-based inhibitors (2 and 4 in Table 4) were invariably poorer than norvaline
Capping group P4 P3 P2 P1 AcSucGlt
ChgDif Leu
IleGluVal
ChaLeu Glu
2-Nal PheBip
Cys ACPC
Table 3 Building blocks for tetrapeptide inhibitors (Paper II). Ac, acetyl-; Chg, cyclohexylglycine; Cha, β-cyclohexylalanine; Suc, succinyl-/3-carboxypropanoyl-; Dif, 3,3-diphenylalanine; ACPC, 1-aminocyclopropane-1-carboxylic acid; Glt, glutaryl-/4-carboxybutanoyl-; 2-Nal, 2-naphthylalanine; Bip, β-(4-biphenylyl)-alanine.
(Nva)-based inhibitors (1 and 3 in Table 4) on HLE, PPE, BPC and Cathepsin B. In contrast, the ACPC-based non-electrophilic compound 16 was better than a Nva-based non-electrophilic compound 15 in inhibiting the NS3 protease (Table 4), which is in agreement with data from the literature (Llinas-Brunet et al. 2000). The observed poor performance of ACPC-based electrophilic compounds 2 and 4 (Table 4) might depend on: a) unfavorable interaction of the cyclopropane residue with the S1 pocket of the protease; b) instability of the tetrahedral intermediate; c) the electron donating nature of the cyclopropane residue, which might diminish the electrophilicity of the neighboring carbonyl group (Francis et al. fourth edition). The first explanation is not likely, since all of the proteases have different S1 site structures and cannot produce the same trend in the P1 side chain SAR. Calculations have shown that the electron-donating ‘strength’ of cyclopropane is not enough to lower the electrophilicity of the neighboring carbonyl (unpublished data). Therefore b) seems to be the most reasonable explanation for

45
the observed phenomenon. The strain imposed by cyclopropane on the α-carbon of the P1 amino acid should result in instability of tetrahedral hemiketal (Fig. 10a) and should weaken the inhibitor-enzyme interaction.
High selectivity of ACPC-based electrophilic keto acid inhibitor
A ketoacid inhibitor (compound 5 in Table 4) having the suboptimal cyclopropane group as the P1 side-chain was still a relatively good inhibitor of the NS3 protease, but a poor inhibitor of the reference proteases, being the most selective electrophilic inhibitor used in this study. This is a little bit surprising, since ketoacids are generally good inhibitors of serine proteases (Narjes et al. 2000, Tanizawa et al. 1985). The high selectivity of the ACPC-based ketoacid inhibitor may be explained by an insignificant contribution of the electrophilic group to the interaction with the proteases. In fact, the inhibitory potency of the ketoacid on NS3 is equal to that of the non-electrophilic product-based inhibitor with similar structure (compound 16, Table 4), which breaks the pattern observed with other ACPC-based electrophilic inhibitors and raised the question of whether formation of a covalent bond between inhibitor and the NS3 protease takes place at all. Unfortunately, the ketoacid (compound 5, Table 4) does not have a reference substance with Nva in P1 position and cannot be compared with the other ACPC-containing inhibitors. However, from the discussion above, it is likely that an Nva-based ketoacid could be a more potent, but less selective inhibitor of the NS3 protease.
Indirect evidence for non-covalent interaction of peptidyl electrophilic
inhibitors with NS3 protease.
Close inspection and comparison of inhibition data obtained with the NS3 protease and HLE, PPE, BPC and cathepsin B enabled us (Paper III, IV and V) to raise the question about the mode of interaction of electrophilic compounds with the NS3 protease. Although we have not yet obtained direct evidence, we speculate that covalent bond formation (Fig. 10a) between electrophilic compounds used in our study and the NS3 protease does not take place. The possible indications of that are:
1. The introduction of electrophilic functionality into peptide inhibitors was expected to boost their inhibitory potency by exploiting covalent bonding of the inhibitor’s electrophilic group to the enzyme. It turned out that non-ACPC-based electrophilic compounds (compounds 1,3, 6-11, Table 4)

46
were not much better (or even worse, like compounds 3) at inhibiting the NS3 protease than were their non-electrophilic counterparts (compounds 15, 16 and 19, Table 4).
2. Many electrophilic inhibitors of serine and cysteine proteases are characterized by slow binding to their target enzymes (Narjes et al. 2000, Brady et al. 1990, Morrison et al. 1988, Smith et al. 1988) and that is what we observed in the inhibition of HLE, PPE, BPC and cathepsin B by electrophilic inhibitors. None of the electrophilic inhibitors exhibited slow-binding with the NS3 protease in our study (Table 4).

47
Ki (µM), or inhibition (%)
NR
NS3 HLE PPE BPC CatB
1
NC2F5
H O
0.12a 0.35 + 0.2 0.065 + 0.0013c 4.8 + 1.1c0%
at 100 µM
2 NC2F5
H O
1.8a 5.6 + 1.5 0%
at 100 µM
21%
at 100 µM
0%
at 100 µM
3N
H O
NN
NHN0.30a 8.1 + 1.8 7.9 + 1.7c
15%
at 100 µM45 + 8
4 N
H O
NN
NHN
1.9a45%
at 100 µM
0%
at 100 µM
8%
at 100 µM
0%
at 100 µM
5 N
H O
OH
O
0.098a49%
at 100 µMc
0%
at 100 µM
3%
at 100 µM19 + 2c
6 N
H O
S
N
0.32b n.d. 4%
at 100 µM 8.6 + 2.4c 4.7 + 0.5c
CapN
NN
NN
R
H
H
H
H
H O
O
O
O
O
O
OHO
HO

48
7 N
H O
0.20b 16.2 + 1.429%
at 100 µM 35 + 12c 9.1 + 0.7c
8 N
H O
0.39b 0.44 + 0.040%
at 100 µM 32 + 7.7 1.54 + 0.11c
9S
N
N
H O
≈ 1 3.1 + 0.2 9%
at 100 µM 38 + 5 9.3 + 0.8c
10 N
H O
0.65b 1.77 + 0.1833%
at 100 µM 18 + 3c
30%
at 100 µM
11 N
H O
0.46b 0.37 + 0.082%
at 100 µM 30 + 4 15 + 2
12 S
N
N
H OH
2.3b n.d. 8%
at 100 µM 7.7 + 2.4 41 + 3
13 N
H OH
17 31 + 2.5 0%
at 100 µM 44 + 4.7
0%
at 100 µM
14 N
H OH
0.98b 0.12 + 0.030%
at 100 µM 60 + 13 17 + 3
15 N
OH
H O
0.22a19.9%
at 200 µM
0%
at 100 µM
18%
at 200 µM
0%
at 200 µM
16 NOH
H O
0.091a7.5%
at 100 µM
0%
at 100 µM
9%
at 100 µM
0%
at 100 µM

49
17 N
OH
H O
HS
0.028a27.5%
at 200 µM
0%
at 200µM
16%
at 200 µM
20%
at 200 µM
18 N
OH
H O
HS
0.092a40%
at 100 µM
0%
at 100µM
0%
at 100µM
0%
at 100 µM
19 N
OH
H O
1.54b 3.8 + 0.8 0%
at 100 µM 22.8 + 3.9
37%
at 100 µM
20 N
H NN
N
HN 0.16a 3.8 + 1.2
34%
at 200 µM
15%
at 200 µM
0%
at 100 µM
21 N
H NN
N
HN
0.19a 49.5 + 6.60%
at 200 µM
21%
at 200 µM
13%
at 200 µM
22 N
H O
NS
H
OO
0.014a42%
at 200 µM
9%
at 200 µM
0%
at 200 µM
0%
at 100 µM
23 NS
H
OO
N
H O
0.0038a 39.9 + 3.70%
at 200 µM
0%
at 200 µM
0%
at 200 µM
Table 4. Activity of NS3 protease inhibitors on other serine and cysteine proteases. This table is a truncated version of Table 1 in Paper V. On the top is a general inhibitor structure. Cap - capping group was either succinic or acetic acid residue (not shown in the table). cSlow-binding inhibition observed.

50
Conclusions
In this work we successfully obtained and cloned HCV NS3 gene from a HCV infected patient. Despite the low level of the protein expression and high glycerol concentration in all buffers we have successfully purified the full-length NS3 protein by combination of IMAC and polyU-Sepharose chromatography. Assay for NS3 protease was developed and kinetic constants of the enzyme were obtained.
The results obtained in the present work have provided an important insight into structure-activity relationships of peptide-based HCV NS3 protease inhibitors. This information may help in the creation of more potent and selective inhibitors of the NS3 protease and, eventually, may result in the development of a drug candidate for treatment of HCV infection.
Of special interest were compounds with bioisosterically replaced C-terminal residues, which produced the most potent NS3 inhibitors in this work. Molecular modeling has revealed that the high inhibitory potency of bioisosteric inhibitors with an acyl sulfonamide moiety is most likely due to specific interaction of the bioisosteric group with the active center of the NS3 protease.
In order to investigate selectivity of the NS3 protease inhibitors we have measured their inhibitory activity of other serine (human leucocyte elastase, bovine pancreatic chymotrypsin and porcine pancreatic elastase) and cysteine (Cathepsin B) proteases.
NS3 protease inhibitors with an electrophilic C-terminal residue were very non-selective and were not more potent than inhibitors with a C-terminal carboxylate. These results have led to exclusion of electrophilic inhibitors form further efforts towards inhibitor optimization.
Our results have highlighted an important role of the P1 residues of peptide inhibitors in the interaction with serine proteases, especially in selectivity issues.

51
Analysis of inhibition data of electrophilic versus non-electrophilic inhibitors has allowed us to question the occurrence of covalent bond formation between electrophilic inhibitors and the NS3 protease.
The SAR of non-covalent inhibitors and molecular modeling of interactions of these inhibitors with the NS3 protein have revealed the possible involvement of helicase domain of the NS3 in inhibitor binding. This highlights the importance of using a full-length NS3 protein for inhibition studies, since most research groups have earlier used only the protease domain for this purpose.

52
Acknowledgements
I would like to thank a lot of people here in Uppsala and outside it for contributing a lot for the success of this work: Helena Danielson for inviting me to Uppsala, for guiding me through all the years of my work at BMC, for many interesting discussions and for teaching me to take it easy when the bike is stolen. Special thanks for understanding my problem with prepositions. David Eaker for careful reading of my thesis and for being brave enough to face such an overwhelming amount of grammar bugs there. Bengt Mannervik for having examining me before accepting for PhD studentship. Who else could have forced me to read the whole Van Holde’s Biochemistry tree times in just two months? Anja Johansson and Eva Åkerblom for showing me the power of organic chemistry when combined with enzymology. Thank you for very interesting and fruitful collaboration. Discussions with you have always been inspiring. Special thanks for careful reading of my thesis and useful comments. Ina Hubatsch for teaching me a lot of things about molecular biology and protein expression. Cynthia Schumann, Gun Stenberg, Göran Ahlsen, Peo Markgren for starting HCV project and passing it over to me. Göran Dahl and Thomas Gossas for the readiness to continue HCV project and taking it over from me. Gunnar Lindeberg for productive collaboration and help with HPLC equipment. Anders Hallberg for quick and efficient handling of our articles. Matthis Geitmann for showing me the futility of trying to obtain an inhibitor of HCV protease form plants (tannins, you know). Andrei Sanin – nothing is complex enough not to be explained in simple words. Organic chemistry is not an exception. Thanks for showing me that. Alexander Gorbalenya for inviting me to Leiden and for many inspiring conversations. Wasn’t it after meeting with him that I decided to go west?

53
Dmitry Usoskin for sharing with me his vast knowledge of molecular biology and for car-towing assistance at a short notice. Oles Plashkevich for showing me that Bikupan, Grindstugan and Sven Dufva are not the only lunch places in Uppsala. Dan Backman, Maria Lindgren for being good labkamrater. Omar Gutierres Arenas for many interesting enzyme kinetics discussions.Ann-Christin Lindås and Birgitta Tomkinson for being good and friendly neighbours. Ingo Gottschalk – Kålarmoraån journey was unforgettable. Have to borrow his boat again. Per-Axel Lindström for being helpful in many seemingly hopeless situations. Hardware is not as hard to handle as it looks! Elisabet Boija, Malena Andersson, Maryam Hansson Edalat, Ylva Ivarsson, Birgitta Eklund, Lars Emeren, Caroline Engvall, Birgitta Evremar, Lilian Forsberg, Usama Hegazy, Inger Hermanson, Jessica Jacobsson, Sanela Kurtovich, Christine Lagerquist Hägglund, Anna-Karin Larsson, Mikael Nilsson, Birgit Olin, Abeer Shokeer, Lisa Tronstad Elfström, Akos Vegvari, Michael Widersten, Shu-Sheng Zuo for sharing the labspace with me and helping me with different problems in my work. Special thanks to Medivir AB that provided financial support to my activities at BMC.

54
References
Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci U S A. 1999 Oct 26;96(22):12766-71.
Ago H, Adachi T, Yoshida A, Yamamoto M, Habuka N, Yatsunami K, Miyano M. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure Fold Des. 1999 Nov 15;7(11):1417-26.
Ali N, Pruijn GJ, Kenan DJ, Keene JD, Siddiqui A. Human La antigen is required for the hepatitis C virus internal ribosome entry site-mediated translation. J Biol Chem. 2000 Sep 8;275(36):27531-40.
Archer SJ, Camac DM, Wu ZJ, Farrow NA, Domaille PJ, Wasserman ZR, Bukhtiyarova M, Rizzo C, Jagannathan S, Mersinger LJ, Kettner CA. Hepatitis C virus NS3 protease requires its NS4A cofactor peptide for optimal binding of a boronic acid inhibitor as shown by NMR. Chem Biol. 2002 Jan;9(1):79-92.
Archer SJ, Camac DM, Wu ZJ, Farrow NA, Domaille PJ, Wasserman ZR, Bukhtiyarova M, Rizzo C, Jagannathan S, Mersinger LJ, Kettner CA. Hepatitis C virus NS3 protease requires its NS4A cofactor peptide for optimal binding of a boronic acid inhibitor as shown by NMR. Chem Biol. 2002 Jan;9(1):79-92.
Ascenzi P, Bocedi A, Bolognesi M, Spallarossa A, Coletta M, De Cristofaro R, Menegatti E. The bovine basic pancreatic trypsin inhibitor (Kunitz inhibitor): a milestone protein. Curr Protein Pept Sci. 2003 Jun;4(3):231-51.
Attwood MR, Bennett JM, Campbell AD, Canning GG, Carr MG, Conway E, Dunsdon RM, Greening JR, Jones PS, Kay PB, Handa BK, Hurst DN, Jennings NS, Jordan S, Keech E, O'Brien MA, Overton HA, King-Underwood J, Raynham TM, Stenson KP, Wilkinson CS, Wilkinson TC, Wilson FX. The design and synthesis of potent inhibitors of hepatitis C virus NS3-4A proteinase. Antivir Chem Chemother. 1999 Sep;10(5):259-73.
Banerjee R, Dasgupta A. Specific interaction of hepatitis C virus protease/helicase NS3 with the 3'-terminal sequences of viral positive- and negative-strand RNA. J Virol. 2001 Feb;75(4):1708-21.
Barker LF, Gerety RJ. The clinical problem of hepatitis transmission;. Prog Clin Biol Res. 1976;11:163-82.
Bartenschlager R, Ahlborn-Laake L, Mous J, Jacobsen H. Kinetic and structural analyses of hepatitis C virus polyprotein processing. J Virol. 1994 Aug;68(8):5045-55.
Bartenschlager R, Ahlborn-Laake L, Mous J, Jacobsen H. Nonstructural protein 3 of the hepatitis C virus encodes a serine-type proteinase required for cleavage at the NS3/4 and NS4/5 junctions. J Virol. 1993 Jul;67(7):3835-44.
Bazan JF, Fletterick RJ. Detection of a trypsin-like serine protease domain in flaviviruses and pestiviruses. Virology. 1989 Aug;171(2):637-9.
Behrens S-E, Tomei L, De Francesco R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J 1996; 15: 12 22.

55
Blanchard E, Brand D, Trassard S, Goudeau A, Roingeard P. Hepatitis C virus-like particle morphogenesis. J Virol. 2002 Apr;76(8):4073-9.
Blanchette VS, Vorstman E, Shore A, Wang E, Petric M, Jett BW, Alter HJ. Hepatitis C infection in children with hemophilia A and B. Blood. 1991 Jul 15;78(2):285-9.
Block TM, Mehta AS, Fimmel CJ, Jordan R. Molecular viral oncology of hepatocellular carcinoma. Oncogene. 2003 Aug 11;22(33):5093-107.
Bode W, Huber R. Natural protein proteinase inhibitors and their interaction with proteinases. Eur J Biochem. 1992 Mar 1;204(2):433-51.
Boulant S, Becchi M, Penin F, Lavergne JP. Unusual multiple recoding events leading to alternative forms of hepatitis C virus core protein from genotype 1b. J Biol Chem. 2003 Nov 14;278(46):45785-92. Epub 2003 Sep 02.
Brady K, Abeles RH. Inhibition of chymotrypsin by peptidyl trifluoromethyl ketones: determinants of slow-binding kinetics. Biochemistry. 1990 Aug 21;29(33):7608-17.
Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, Rey FA. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc Natl Acad Sci U S A. 1999 Nov 9;96(23):13034-9.
Brown EA, Zhang H, Ping LH, Lemon SM. Secondary structure of the 5' nontranslated regions of hepatitis C virus and pestivirus genomic RNAs. Nucleic Acids Res. 1992 Oct 11;20(19):5041-5.
Bru R, Walde P. Product inhibition of alpha-chymotrypsin in reverse micelles. Eur J Biochem. 1991 Jul 1;199(1):95-103.
Bukh J, Miller RH, Purcell RH. Genetic heterogeneity of hepatitis C virus: quasispecies and genotypes. Semin Liver Dis. 1995 Feb;15(1):41-63.
Bukh J, Purcell RH, Miller RH. At least 12 genotypes of hepatitis C virus predicted by sequence analysis of the putative E1 gene of isolates collected worldwide. Proc Natl Acad Sci U S A. 1993 Sep 1;90(17):8234-8.
Bukh J, Purcell RH, Miller RH. Sequence analysis of the 5' noncoding region of hepatitis C virus. Proc Natl Acad Sci U S A. 1992 Jun 1;89(11):4942-6.
Bukh J, Purcell RH, Miller RH. Sequence analysis of the core gene of 14 hepatitis C virus genotypes. Proc Natl Acad Sci U S A. 1994 Aug 16;91(17):8239-43.
Cerny A, Chisari FV. Pathogenesis of chronic hepatitis C: immunological features of hepatic injury and viral persistence. Hepatology. 1999 Sep;30(3):595-601.
Chang KM. Immunopathogenesis of hepatitis C virus infection. Clin Liver Dis. 2003 Feb;7(1):89-105.
Cho HS, Ha NC, Kang LW, Chung KM, Back SH, Jang SK, Oh BH. Crystal structure of RNA helicase from genotype 1b hepatitis C virus. A feasible mechanism of unwinding duplex RNA. J Biol Chem. 1998 Jun 12;273(24):15045-52.
Choksi AP, Desai HG. Non-A, non-B hepatitis. Biomed Pharmacother. 1989;43(10):743-51.
Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989 Apr 21;244(4902):359-62.
Choo QL, Richman KH, Han JH, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby R, Barr PJ, et al. Genetic organization and diversity of the hepatitis C virus. Proc Natl Acad Sci U S A. 1991 Mar 15;88(6):2451-5.
Cicero DO, Barbato G, Koch U, Ingallinella P, Bianchi E, Nardi MC, Steinkuhler C, Cortese R, Matassa V, De Francesco R, Pessi A, Bazzo R. Structural characterization of the interactions of optimized product inhibitors with the N-

56
terminal proteinase domain of the hepatitis C virus (HCV) NS3 protein by NMR and modelling studies. J Mol Biol. 1999 Jun 4;289(2):385-96.
Clayton RF, Owsianka A, Aitken J, Graham S, Bhella D, Patel AH. Analysis of antigenicity and topology of E2 glycoprotein present on recombinant hepatitis C virus-like particles. J Virol. 2002 Aug;76(15):7672-82. Erratum in: J Virol 2002 Sep;76(18):9562.
Clemens MJ & Elia A. The double-stranded RNA-dependent protein kinase PKR. Structure and function. J Interferon Cytokine Res 1997; 6: 503-524.
Cocquerel L, Duvet S, Meunier JC, Pillez A, Cacan R, Wychowski C, Dubuisson J. The transmembrane domain of hepatitis C virus glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J Virol. 1999 Apr;73(4):2641-9.
Cocquerel L, Kuo CC, Dubuisson J, Levy S. CD81-dependent binding of hepatitis C virus E1E2 heterodimers. J Virol. 2003 Oct;77(19):10677-83.
Cocquerel L, Wychowski C, Minner F, Penin F, Dubuisson J. Charged residues in the transmembrane domains of hepatitis C virus glycoproteins play a major role in the processing, subcellular localization, and assembly of these envelope proteins. J Virol. 2000 Apr;74(8):3623-33.
Dash C, Kulkarni A, Dunn B, Rao M. Aspartic peptidase inhibitors: implications in drug development. Crit Rev Biochem Mol Biol. 2003;38(2):89-119.
De Francesco R, Steinkuhler C. Structure and function of the hepatitis C virus NS3-NS4A serine proteinase. Curr Top Microbiol Immunol. 2000;242:149-69.
De Francesco R, Tomei L, Altamura S, Summa V, Migliaccio G. Approaching a new era for hepatitis C virus therapy: inhibitors of the NS3-4A serine protease and the NS5B RNA-dependent RNA polymerase. Antiviral Res. 2003 Mar;58(1):1-16.
De Francesco R, Urbani A, Nardi MC, Tomei L, Steinkuhler C, Tramontano A. A zinc binding site in viral serine proteinases. Biochemistry. 1996 Oct 15;35(41):13282-7.
Demuth HU. Recent developments in inhibiting cysteine and serine proteases. J Enzyme Inhib. 1990; 3(4): 249-78.
Dennis MS, Roberge M, Quan C, Lazarus RA. Selection and characterization of a new class of peptide exosite inhibitors of coagulation factor VIIa. Biochemistry. 2001 Aug 14;40(32):9513-21.
Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Kramer T, Niewohner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RN, Reimann A, Jaeger R, Gross R, Beckermann B, Schlemmer KH, Haebich D, Rubsamen-Waigmann H. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science. 2003 Feb 7;299(5608):893-6.
Di Marco S, Rizzi M, Volpari C, Walsh MA, Narjes F, Colarusso S, De Francesco R, Matassa VG, Sollazzo M. Inhibition of the hepatitis C virus NS3/4A protease. The crystal structures of two protease-inhibitor complexes. J Biol Chem. 2000 Mar 10;275(10):7152-7.
Dimitrova M, Imbert I, Kieny MP, Schuster C. Protein-protein interactions between hepatitis C virus nonstructural proteins. J Virol. 2003 May;77(9):5401-14.
Donahue JG, Munoz A, Ness PM, Brown DE Jr, Yawn DH, McAllister HA Jr, Reitz BA, Nelson KE. The declining risk of post-transfusion hepatitis C virus infection. N Engl J Med. 1992 Aug 6;327(6):369-73.
Drouet C, Bouillet L, Csopaki F, Colomb MG. Hepatitis C virus NS3 serine protease interacts with the serpin C1 inhibitor. FEBS Lett. 1999 Sep 24;458(3):415-8.
Dubuisson J. Folding, assembly and subcellular localization of hepatitis C virus glycoproteins. Curr Top Microbiol Immunol. 2000;242:135-48.

57
Dymock BW, Emerging therapies for hepatitis C virus. Emerging Drugs (2001) 6(1):13-42.
Eckart MR, Selby M, Masiarz F, Lee C, Berger K, Crawford K, Kuo C, Kuo G, Houghton M, Choo QL. The hepatitis C virus encodes a serine protease involved in processing of the putative nonstructural proteins from the viral polyprotein precursor. Biochem Biophys Res Commun. 1993 Apr 30;192(2):399-406.
Edwards PD, Bernstein PR. Synthetic inhibitors of elastase. Med Res Rev. 1994 Mar;14(2):127-94.
Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol. 2002 Jun;76(12):5974-84.
Erdtmann L, Franck N, Lerat H, Le Seyec J, Gilot D, Cannie I, Gripon P, Hibner U, Guguen-Guillouzo C. The hepatitis C virus NS2 protein is an inhibitor of CIDE-B-induced apoptosis. J Biol Chem. 2003 May 16;278(20):18256-64.
Fabrizi F, Martin P. Hepatitis C virus infection in dialysis: an emerging clinical reality. Int J Artif Organs. 2001 Mar;24(3):123-30.
Failla C, Tomei L, De Francesco R. Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J Virol. 1994 Jun;68(6):3753-60.
Fattori D, Urbani A, Brunetti M, Ingenito R, Pessi A, Prendergast K, Narjes F, Matassa VG, De Francesco R, Steinkuhler C. Probing the active site of the hepatitis C virus serine protease by fluorescence resonance energy transfer. J Biol Chem. 2000 May 19;275(20):15106-13.
Feinstone SM, Kapikian AZ, Purcell RH, Alter HJ, Holland PV. Transfusion-associated hepatitis not due to viral hepatitis type A or B. N Engl J Med. 1975 Apr 10;292(15):767-70.
Fersht AR, Sperling J. The charge relay system in chymotrypsin and chymotrypsinogen. J Mol Biol. 1973 Feb 25;74(2):137-49.
Fletcher S. Sexual transmission of hepatitis C and early intervention. J Assoc Nurses AIDS Care. 2003 Sep-Oct;14(5 Suppl):87S-94S.
Francis A. Carey and Richard J. Sundberg. ‘’Advanced organic chemistry’’. Fourth edition, Part A, p. 284-285.
Friebe P, Bartenschlager R. Genetic analysis of sequences in the 3' nontranslated region of hepatitis C virus that are important for RNA replication. J Virol. 2002 Jun;76(11):5326-38.
Gale MJr, Korth MJ, Tang NMet al.Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 1997; 6: 217-227.
Gallinari P, Brennan D, Nardi C, Brunetti M, Tomei L, Steinkuhler C, De Francesco R. Multiple enzymatic activities associated with recombinant NS3 protein of hepatitis C virus. J Virol. 1998 Aug;72(8):6758-69.
Gettins PG. Serpin structure, mechanism, and function. Chem Rev. 2002 Dec;102(12):4751-804.
Gisbert JP, Garcia-Buey L, Pajares JM, Moreno-Otero R. Prevalence of hepatitis C virus infection in porphyria cutanea tarda: systematic review and meta-analysis. J Hepatol. 2003 Oct;39(4):620-7.
Gorbalenya AE, Donchenko AP, Koonin EV, Blinov VM. N-terminal domains of putative helicases of flavi- and pestiviruses may be serine proteases. Nucleic Acids Res. 1989 May 25;17(10):3889-97.

58
Gorbalenya AE, Koonin EV, Donchenko AP, Blinov VM. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989 Jun 26;17(12):4713-30.
Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM. A second hepatitis C virus-encoded proteinase. Proc Natl Acad Sci U S A. 1993 Nov 15;90(22):10583-7.
Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM. Characterization of the hepatitis C virus-encoded serine proteinase: determination of proteinase-dependent polyprotein cleavage sites. J Virol. 1993 May;67(5):2832-43.
Griffin SD, Beales LP, Clarke DS, Worsfold O, Evans SD, Jaeger J, Harris MP, Rowlands DJ. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 2003 Jan 30;535(1-3):34-8.
Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002 Dec;102(12):4501-24.
Heim MH, Moradpour D, Blum HE. Expression of hepatitis C virus proteins inhibits signal transduction through the Jak-Stat pathway. Hepatology 1998; 6: 321A.
Herion DW, Malayaman N, Hoofnagle JH, Liang TJ. Interaction of hepatitis C virus (HCV) NS5A with a cellular signaling protein involved in receptor tyrosine kinase mitogenic pathway. Proceedings of the Fifth International Meeting on Hepatitis C Virus and Related Viruses. Venice, Italy. June 25-28, 1998.
Hijikata M, Mizushima H, Tanji Y, Komoda Y, Hirowatari Y, Akagi T, Kato N, Kimura K, Shimotohno K. Proteolytic processing and membrane association of putative nonstructural proteins of hepatitis C virus. Proc Natl Acad Sci U S A. 1993 Nov 15;90(22):10773-7.
Hong Z, Ferrari E, Wright-Minogue J, Chase R, Risano C, Seelig G, Lee CG, Kwong AD. Enzymatic characterization of hepatitis C virus NS3/4A complexes expressed in mammalian cells by using the herpes simplex virus amplicon system. J Virol. 1996 Jul;70(7):4261-8.
Houghton M, Selby M, Weiner A, Choo QL. Hepatitis C virus: structure, protein products and processing of the polyprotein precursor. Curr Stud Hematol Blood Transfus. 1994;(61):1-11.
Hwang SB, Park KJ, Kim YS, Sung YC, Lai MM. Hepatitis C virus NS5B protein is a membrane-associated phosphoprotein with a predominantly perinuclear localization. Virology. 1997 Jan 20;227(2):439-46.
Iacoangeli A, Melucci-Vigo G, Risuleo G. The ionophore monensin inhibits mouse polyomavirus DNA replication and destabilizes viral early mRNAs. Biochimie. 2000 Jan;82(1):35-9.
Ina Y, Mizokami M, Ohba K, Gojobori T. Reduction of synonymous substitutions in the core protein gene of hepatitis C virus. J Mol Evol. 1994 Jan;38(1):50-6.
Ingallinella P, Altamura S, Bianchi E, Taliani M, Ingenito R, Cortese R, De Francesco R, Steinkuhler C, Pessi A. Potent peptide inhibitors of human hepatitis C virus NS3 protease are obtained by optimizing the cleavage products. Biochemistry. 1998 Jun 23;37(25):8906-14.
Ingallinella P, Bianchi E, Ingenito R, Koch U, Steinkuhler C, Altamura S, Pessi A. Optimization of the P'-region of peptide inhibitors of hepatitis C virus NS3/4A protease. Biochemistry. 2000 Oct 24;39(42):12898-906.
Ingallinella P, Fattori D, Altamura S, Steinkuhler C, Koch U, Cicero D, Bazzo R, Cortese R, Bianchi E, Pessi A. Prime site binding inhibitors of a serine protease: NS3/4A of hepatitis C virus. Biochemistry. 2002 Apr 30;41(17):5483-92.

59
Ishida S, Kaito M, Kohara M, Tsukiyama-Kohora K, Fujita N, Ikoma J, Adachi Y, Watanabe S. Hepatitis C virus core particle detected by immunoelectron microscopy and optical rotation technique. Hepatol Res. 2001 Jul;20(3):335-347.
Ivashkina N, Wolk B, Lohmann V, Bartenschlager R, Blum HE, Penin F, Moradpour D. The hepatitis C virus RNA-dependent RNA polymerase membrane insertion sequence is a transmembrane segment. J Virol. 2002 Dec;76(24):13088-93.
Johansson A, Hubatsch I, Akerblom E, Lindeberg G, Winiwarter S, Danielson UH, Hallberg A. Inhibition of hepatitis C virus NS3 protease activity by product-based peptides is dependent on helicase domain. Bioorg Med Chem Lett. 2001 Jan 22;11(2):203-6.
Johansson A, Poliakov A, Akerblom E, Lindeberg G, Winiwarter S, Samuelsson B, Danielson UH, Hallberg A. Tetrapeptides as potent protease inhibitors of Hepatitis C Virus full-length NS3 (protease-helicase/NTPase). Bioorg Med Chem. 2002 Dec;10(12):3915-22.
Johansson A, Poliakov A, Akerblom E, Wiklund K, Lindeberg G, Winiwarter S, Danielson UH, Samuelsson B, Hallberg A. Acyl sulfonamides as potent protease inhibitors of the hepatitis C virus full-Length NS3 (Protease-Helicase/NTPase): A comparative study of different C-terminals. Bioorg Med Chem. 2003 Jun 12;11(12):2551-68.
Kaito M, Watanabe S, Tsukiyama-Kohara K, Yamaguchi K, Kobayashi Y, Konishi M, Yokoi M, Ishida S, Suzuki S, Kohara M. Hepatitis C virus particle detected by immunoelectron microscopic study. J Gen Virol. 1994 Jul;75 ( Pt 7):1755-60.
Kaneko T, Tanji Y, Satoh Set al.Production of two phosphoproteins from the NS5A region of the hepatitis C viral genome. Biochem Biophys Res Commun 1994; 6:320-326.
Kato N, Lan KH, Ono-Nita SK, Shiratori Y, Omata M. Hepatitis C virus nonstructural region 5A protein is a potent transcriptional activator. J Virol 1997; 6: 8856-8859.
Kim JL, Morgenstern KA, Griffith JP, Dwyer MD, Thomson JA, Murcko MA, Lin C, Caron PR. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure. 1998 Jan 15;6(1):89-100.
Kim JL, Morgenstern KA, Lin C, Fox T, Dwyer MD, Landro JA, Chambers SP, Markland W, Lepre CA, O'Malley ET, Harbeson SL, Rice CM, Murcko MA, Caron PR, Thomson JA. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell. 1996 Oct 18;87(2):343-55. Erratum in: Cell 1997 Apr 4;89(1):159.
Kim YK, Kim CS, Lee SH, Jang SK. Domains I and II in the 5' nontranslated region of the HCV genome are required for RNA replication. Biochem Biophys Res Commun. 2002 Jan 11;290(1):105-12.
Kleymann G. New antiviral drugs that target herpesvirus helicase primase enzymes. Herpes. 2003 Aug;10(2):46-52.
Knight CG, Dando PM, Barrett AJ. Thimet oligopeptidase specificity: evidence of preferential cleavage near the C-terminus and product inhibition from kinetic analysis of peptide hydrolysis. Biochem J. 1995 May 15;308 ( Pt 1):145-50.
Koch JO, Lohmann V, Herian U, Bartenschlager R. In vitro studies on the activation of the hepatitis C virus NS3 proteinase by the NS4A cofactor. Virology. 1996 Jul 1;221(1):54-66.

60
Kolykhalov AA, Feinstone SM, Rice CM. Identification of a highly conserved sequence element at the 3' terminus of hepatitis C virus genome RNA. J Virol. 1996 Jun;70(6):3363-71.
Kolykhalov AA, Mihalik K, Feinstone SM, Rice CM. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3' nontranslated region are essential for virus replication in vivo. J Virol. 2000 Feb;74(4):2046-51.
Komoda Y, Hijikata M, Sato S, Asabe S, Kimura K, Shimotohno K. Substrate requirements of hepatitis C virus serine proteinase for intermolecular polypeptide cleavage in Escherichia coli. J Virol. 1994 Nov;68(11):7351-7.
Konan KV, Giddings TH Jr, Ikeda M, Li K, Lemon SM, Kirkegaard K. Nonstructural protein precursor NS4A/B from hepatitis C virus alters function and ultrastructure of host secretory apparatus. J Virol. 2003 Jul;77(14):7843-55.
Kountouras J, Zavos C, Chatzopoulos D. Apoptosis in hepatitis C. J Viral Hepat. 2003 Sep;10(5):335-42.
Kunkel M, Watowich SJ. Conformational changes accompanying self-assembly of the hepatitis C virus core protein. Virology. 2002 Mar 15;294(2):239-45.
Kuo G, Choo QL, Alter HJ, Gitnick GL, Redeker AG, Purcell RH, Miyamura T, Dienstag JL, Alter MJ, Stevens CE, et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989 Apr 21;244(4902):362-4.
Kurosaki M, Enomoto N, Marumo F, Sato C. Evolution and selection of hepatitis C virus variants in patients with chronic hepatitis C. Virology. 1994 Nov 15;205(1):161-9.
Kwong AD, Kim JL, Lin C. Structure and function of hepatitis C virus NS3 helicase. Curr Top Microbiol Immunol. 2000;242:171-96.
Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, Bos M, Cameron DR, Cartier M, Cordingley MG, Faucher AM, Goudreau N, Kawai SH, Kukolj G, Lagace L, LaPlante SR, Narjes H, Poupart MA, Rancourt J, Sentjens RE, St George R, Simoneau B, Steinmann G, Thibeault D, Tsantrizos YS, Weldon SM, Yong CL, Llinas-Brunet M. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003 Nov 13;426(6963):186-189.
Landro JA, Raybuck SA, Luong YP, O'Malley ET, Harbeson SL, Morgenstern KA, Rao G, Livingston DJ. Mechanistic role of an NS4A peptide cofactor with the truncated NS3 protease of hepatitis C virus: elucidation of the NS4A stimulatory effect via kinetic analysis and inhibitor mapping. Biochemistry. 1997 Aug 5;36(31):9340-8.
Lanphear BP, Linnemann CC Jr, Cannon CG, DeRonde MM, Pendy L, Kerley LM. Hepatitis C virus infection in healthcare workers: risk of exposure and infection. Infect Control Hosp Epidemiol. 1994 Dec;15(12):745-50.
Lau JY. Mechanisms of hepatic toxicity. IV. Pathogenetic mechanisms involved in hepatitis C virus-inducedliver diseases. Am J Physiol. 1998 Dec;275(6 Pt 1):G1217-20.
Le SY, Sonenberg N, Maizel JV Jr. Unusual folding regions and ribosome landing pad within hepatitis C virus and pestivirus RNAs. Gene. 1995 Mar 10;154(2):137-43.
Li X, Jeffers LJ, Shao L, Reddy KR, de Medina M, Scheffel J, Moore B, Schiff ER. Identification of hepatitis C virus by immunoelectron microscopy. J Viral Hepat. 1995;2(5):227-34.
Liu Y, Kati W, Chen CM, Tripathi R, Molla A, Kohlbrenner W. Use of a fluorescence plate reader for measuring kinetic parameters with inner filter effect correction. Anal Biochem. 1999 Feb 15;267(2):331-5.

61
Llinas-Brunet M, Bailey M, Fazal G, Ghiro E, Gorys V, Goulet S, Halmos T, Maurice R, Poirier M, Poupart MA, Rancourt J, Thibeault D, Wernic D, Lamarre D. Highly potent and selective peptide-based inhibitors of the hepatitis C virus serine protease: towards smaller inhibitors. Bioorg Med Chem Lett. 2000 Oct 16;10(20):2267-70.
Llinas-Bruneta M, Bailey M, Deziel R, Fazal G, Gorys V, Goulet S, Halmos T, Maurice R, Poirier M, Poupart MA, Rancourt J, Thibeault D, Wernic D, Lamarre D. Studies on the C-terminal of hexapeptide inhibitors of the hepatitis C virus serine protease. Bioorg Med Chem Lett. 1998 Oct 6;8(19):2719-24.
Llinas-Brunetb M, Bailey M, Fazal G, Goulet S, Halmos T, Laplante S, Maurice R, Poirier M, Poupart MA, Thibeault D, Wernic D, Lamarre D. Peptide-based inhibitors of the hepatitis C virus serine protease. Bioorg Med Chem Lett. 1998 Jul 7; 8(13): 1713-8.
Lo SY, Masiarz F, Hwang SB, Lai MM, Ou JH. Differential subcellular localization of hepatitis C virus core gene products. Virology. 1995 Nov 10;213(2):455-61.
Lohmann V, Koch JO, Bartenschlager R. Processing pathways of the hepatitis C virus proteins. J Hepatol. 1996;24(2 Suppl):11-9.
Love RA, Parge HE, Wickersham JA, Hostomsky Z, Habuka N, Moomaw EW, Adachi T, Hostomska Z. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell. 1996 Oct 18;87(2):331-42.
Love RA, Parge HE, Wickersham JA, Hostomsky Z, Habuka N, Moomaw EW, Adachi T, Margosiak S, Dagostino E, Hostomska Z. The conformation of hepatitis C virus NS3 proteinase with and without NS4A: a structural basis for the activation of the enzyme by its cofactor. Clin Diagn Virol. 1998 Jul 15;10(2-3):151-6.
Lundin M, Monne M, Widell A, Von Heijne G, Persson MA. Topology of the membrane-associated hepatitis C virus protein NS4B. J Virol. 2003 May;77(9):5428-38.
Luo G, Hamatake RK, Mathis DM, Racela J, Rigat KL, Lemm J, Colonno RJ. De novo initiation of RNA synthesis by the RNA-dependent RNA polymerase (NS5B) of hepatitis C virus. J Virol. 2000 Jan;74(2):851-63.
Manns MP, Rambusch EG. Autoimmunity and extrahepatic manifestations in hepatitis C virus infection. J Hepatol. 1999;31 Suppl 1:39-42.
Martell M, Esteban JI, Quer J, Genesca J, Weiner A, Esteban R, Guardia J, Gomez J. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J Virol. 1992 May;66(5):3225-9.
Maugh TH 2nd. Where is the hepatitis C virus? Science. 1980 Nov 28;210(4473):999-1000.
McLauchlan J. Properties of the hepatitis C virus core protein: a structural protein that modulates cellular processes. J Viral Hepat. 2000 Jan;7(1):2-14.
Meyers CM, Seeff LB, Stehman-Breen CO, Hoofnagle JH. Hepatitis C and renal disease: An update. Am J Kidney Dis. 2003 Oct;42(4):631-57.
Monazahian M, Bohme I, Bonk S, Koch A, Scholz C, Grethe S, Thomssen R. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J Med Virol. 1999 Mar;57(3):223-9.
Moorman JP, Joo M, Hahn YS. Evasion of host immune surveillance by hepatitis C virus: potential roles in viral persistence. Arch Immunol Ther Exp (Warsz). 2001;49(3):189-94.
Morgenstern KA, Landro JA, Hsiao K, Lin C, Gu Y, Su MS, Thomson JA. Polynucleotide modulation of the protease, nucleoside triphosphatase, and

62
helicase activities of a hepatitis C virus NS3-NS4A complex isolated from transfected COS cells. J Virol. 1997 May;71(5):3767-75.
Mori A, Yamada K, Kimura J, Koide T, Yuasa S, Yamada E, Miyamura T. Enzymatic characterization of purified NS3 serine proteinase of hepatitis C virus expressed in Escherichia coli. FEBS Lett. 1996 Jan 2;378(1):37-42.
Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol. 1988;61:201-301.
Nakamoto Y, Kaneko S. Mechanisms of viral hepatitis induced liver injury. Curr Mol Med. 2003 Sep;3(6):537-44.
Narjes F, Brunetti M, Colarusso S, Gerlach B, Koch U, Biasiol G, Fattori D, De Francesco R, Matassa VG, Steinkuhler C. Alpha-ketoacids are potent slow binding inhibitors of the hepatitis C virus NS3 protease. Biochemistry. 2000 Feb 22;39(7):1849-61.
Oh JW, Sheu GT, Lai MM. Template requirement and initiation site selection by hepatitis C virus polymerase on a minimal viral RNA template. J Biol Chem. 2000 Jun 9;275(23):17710-7.
Olesen PH. The use of bioisosteric groups in lead optimization. Curr Opin Drug Discov Devel. 2001 Jul;4(4):471-8.
Oscarsson K, Poliakov A, Oscarson S, Danielson UH, Hallberg A, Samuelsson B. Peptide-based inhibitors of hepatitis C virus full-length NS3 (protease-helicase/NTPase): model compounds towards small molecule inhibitors. Bioorg Med Chem. 2003 Jul 3;11(13):2955-63.
Pandya J, Chakraborti A, Chawla Y, Dilawari JB, Sehgal S, Ganguly NK. Identification of human hepatocyte protein(s), which binds specifically to the recombinant envelope-2/non-structural-1 protein of hepatitis C virus. Virus Res. 2002 Aug;87(2):135-143.
Patani GA, LaVoie EJ. Bioisosterism: A Rational Approach in Drug Design. Chem Rev. 1996 Dec 19;96(8):3147-3176.
Patel K, McHutchison JG. Current therapies for chronic hepatitis C. Drug combination achieves sustained response in more than half of patients. Postgrad Med. 2003 Jul;114(1):48-52, 57-9, 62.
Pavlovic D, Neville DC, Argaud O, Blumberg B, Dwek RA, Fischer WB, Zitzmann N. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc Natl Acad Sci U S A. 2003 May 13;100(10):6104-8. Epub 2003 Apr 28.
Pawlotsky JM, Germanidis G. The non-structural 5A protein of hepatitis C virus. J Viral Hepat. 1999 Sep;6(5):343-56.
Pawlotsky JM, Roudot-Thoraval F, Simmonds P, Mellor J, Ben Yahia MB, Andre C, Voisin MC, Intrator L, Zafrani ES, Duval J, et al. Extrahepatic immunologic manifestations in chronic hepatitis C and hepatitis C virus serotypes. Ann Intern Med. 1995 Feb 1;122(3):169-73.
Petit JM, Benichou M, Duvillard L, Jooste V, Bour JB, Minello A, Verges B, Brun JM, Gambert P, Hillon P. Hepatitis C virus-associated hypobetalipoproteinemia is correlated with plasma viral load, steatosis, and liver fibrosis. Am J Gastroenterol. 2003 May;98(5):1150-4.
Piccininni S, Varaklioti A, Nardelli M, Dave B, Raney KD, McCarthy JE. Modulation of the hepatitis C virus RNA-dependent RNA polymerase activity by the non-structural (NS) 3 helicase and the NS4B membrane protein. J Biol Chem. 2002 Nov 22;277(47):45670-9.
Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. Binding of hepatitis C virus to CD81. Science. 1998 Oct 30;282(5390):938-41.

63
Pizzi E, Tramontano A, Tomei L, La Monica N, Failla C, Sardana M, Wood T, De Francesco R. Molecular model of the specificity pocket of the hepatitis C virus protease: implications for substrate recognition. Proc Natl Acad Sci U S A. 1994 Feb 1;91(3):888-92.
Plagemann PG. Hepatitis C virus. Arch Virol. 1991;120(3-4):165-80. Pleister A, Eckels DD. Cryptic infection and autoimmunity. Autoimmun Rev. 2003
May;2(3):126-32. Poliakov A, Hubatsch I, Shuman CF, Stenberg G, Danielson UH. Expression and
purification of recombinant full-length NS3 protease-helicase from a new variant of Hepatitis C virus. Protein Expr Purif. 2002 Aug;25(3):363-71.
Pozzetto B, Bourlet T, Grattard F, Bonnevial L. Structure, genomic organization, replication and variability of hepatitis C virus. Nephrol Dial Transplant. 1996;11 Suppl 4:2-5.
Prince AM, Huima-Byron T, Parker TS, Levine DM. Visualization of hepatitis C virions and putative defective interfering particles isolated from low-density lipoproteins. J Viral Hepat. 1996 Jan;3(1):11-7.
Ray RB, Ray R. Hepatitis C virus core protein: intriguing properties and functional relevance. FEMS Microbiol Lett. 2001 Aug 21;202(2):149-56.
Reigadas S, Ventura M, Andreola ML, Michel J, Gryaznov S, Tarrago-Litvak L, Litvak S, Astier-Gin T. An oligonucleotide complementary to the SL-B1 domain in the 3'-end of the minus-strand RNA of the hepatitis C virus inhibits in vitro initiation of RNA synthesis by the viral polymerase. Virology. 2003 Sep 15;314(1):206-20.
Reynolds JE, Kaminski A, Kettinen HJ, Grace K, Clarke BE, Carroll AR, Rowlands DJ, Jackson RJ. Unique features of internal initiation of hepatitis C virus RNA translation. EMBO J. 1995 Dec 1;14(23):6010-20.
Richer MJ, Juliano L, Hashimoto C, Jean F. Serpin mechanism of hepatitis C virus NS3 protease inhibition: Induced-fit as a mechanism for narrow specificity. J Biol Chem. 2003 Dec 29 [Epub ahead of print]
Rijnbrand R, Bredenbeek P, van der Straaten T, Whetter L, Inchauspe G, Lemon S, Spaan W. Almost the entire 5' non-translated region of hepatitis C virus is required for cap-independent translation. FEBS Lett. 1995 May 29;365(2-3):115-9.
Ripka AS, Rich DH. Peptidomimetic design. Curr Opin Chem Biol. 1998 Aug;2(4):441-52.
Roberts RM, Mathialagan N, Duffy JY, Smith GW. Regulation and regulatory role of proteinase inhibitors.Crit Rev Eukaryot Gene Expr. 1995;5(3-4):385-436.
Roussel J, Pillez A, Montpellier C, Duverlie G, Cahour A, Dubuisson J, Wychowski C. Characterization of the expression of the hepatitis C virus F protein. J Gen Virol. 2003 Jul;84(Pt 7):1751-9.
Sakai A, Claire MS, Faulk K, Govindarajan S, Emerson SU, Purcell RH, Bukh J. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc Natl Acad Sci U S A. 2003 Sep 30;100(20):11646-51.
Salier JP. Inter-alpha-trypsin inhibitor: emergence of a family within the Kunitz-type protease inhibitor superfamily. Trends Biochem Sci. 1990 Nov;15(11):435-9.
Sanderson PE. Small, noncovalent serine protease inhibitors. Med Res Rev. 1999 Mar;19(2):179-97.
Satoh S, Hirota M, Inudoh Met al. Protein processing of hepatitis C virus and investigation of the effects of viral proteins on cell proliferation. In: Schinazi RF, Sommadossi JP, Thomas HC eds. Therapies for Viral Hepatitis. London: International Medical Press, 1998: 263-269.

64
Saxena I, Tayyab S. Protein proteinase inhibitors from avian egg whites. Cell Mol Life Sci. 1997 Jan;53(1):13-23.
Schlott B, Wohnert J, Icke C, Hartmann M, Ramachandran R, Guhrs KH, Glusa E, Flemming J, Gorlach M, Grosse F, Ohlenschlager O. Interaction of Kazal-type inhibitor domains with serine proteinases: biochemical and structural studies. J Mol Biol. 2002 Apr 26;318(2):533-46.
Schulze zur Wiesch J, Schmitz H, Borowski E, Borowski P. The proteins of the Hepatitis C virus: their features and interactions with intracellular protein phosphorylation. Arch Virol. 2003 Jul;148(7):1247-67.
Selectivity article Shimizu Y, Yamaji K, Masuho Y, Yokota T, Inoue H, Sudo K, Satoh S, Shimotohno
K. Identification of the sequence on NS4A required for enhanced cleavage of the NS5A/5B site by hepatitis C virus NS3 protease. J Virol. 1996 Jan;70(1):127-32.
Shimizu YK, Feinstone SM, Kohara M, Purcell RH, Yoshikura H. Hepatitis C virus: detection of intracellular virus particles by electron microscopy. Hepatology. 1996 Feb;23(2):205-9.
Shimoike T, Mimori S, Tani H, Matsuura Y, Miyamura T. Interaction of hepatitis C virus core protein with viral sense RNA and suppression of its translation. J Virol. 1999 Dec;73(12):9718-25.
Shirota Y, Luo H, Qin W, Kaneko S, Yamashita T, Kobayashi K, Murakami S. Hepatitis C virus (HCV) NS5A binds RNA-dependent RNA polymerase (RdRP) NS5B and modulates RNA-dependent RNA polymerase activity. J Biol Chem. 2002 Mar 29;277(13):11149-55.
Shoji I, Suzuki T, Chieda S, Sato M, Harada T, Chiba T, Matsuura Y, Miyamura T. Proteolytic activity of NS3 serine proteinase of hepatitis C virus efficiently expressed in Escherichia coli. Hepatology. 1995 Dec;22(6):1648-55.
Silverman RB. The organic chemistry of enzyme-catalyzed reactions. Academic press, 2000.
Simmonds P, Holmes EC, Cha TA, Chan SW, McOmish F, Irvine B, Beall E, Yap PL, Kolberg J, Urdea MS. Classification of hepatitis C virus into six major genotypes and a series of subtypes by phylogenetic analysis of the NS-5 region. J Gen Virol. 1993 Nov;74 ( Pt 11):2391-9.
Smith DB, Simmonds P. Characteristics of nucleotide substitution in the hepatitis C virus genome: constraints on sequence change in coding regions at both ends of the genome. J Mol Evol. 1997 Sep;45(3):238-46.
Smith RA, Copp LJ, Donnelly SL, Spencer RW, Krantz A. Inhibition of cathepsin B by peptidyl aldehydes and ketones: slow-binding behavior of a trifluoromethyl ketone. Biochemistry. 1988 Aug 23;27(17):6568-73.
Spangberg K, Schwartz S. Poly(C)-binding protein interacts with the hepatitis C virus 5' untranslated region. J Gen Virol. 1999 Jun;80 ( Pt 6):1371-6.
Steinkuhler C, Biasiol G, Brunetti M, Urbani A, Koch U, Cortese R, Pessi A, De Francesco R. Product inhibition of the hepatitis C virus NS3 protease. Biochemistry. 1998 Jun 23;37(25):8899-905.
Steinkuhler C, Urbani A, Tomei L, Biasiol G, Sardana M, Bianchi E, Pessi A, De Francesco R. Activity of purified hepatitis C virus protease NS3 on peptide substrates. J Virol. 1996 Oct;70(10):6694-700.
Stone SR, Tapparelli C. Thrombin inhibitors as antithrombotic agents: the importance of rapid inhibition. J Enzyme Inhib. 1995;9(1):3-15.
Tajiri H, Miyoshi Y, Funada S, Etani Y, Abe J, Onodera T, Goto M, Funato M, Ida S, Noda C, Nakayama M, Okada S. Prospective study of mother-to-infant transmission of hepatitis C virus. Pediatr Infect Dis J. 2001 Jan;20(1):10-4.

65
Takamizawa A, Mori C, Fuke I, Manabe S, Murakami S, Fujita J, Onishi E, Andoh T, Yoshida I, Okayama H. Structure and organization of the hepatitis C virus genome isolated from human carriers. J Virol. 1991 Mar;65(3):1105-13.
Taliani M, Bianchi E, Narjes F, Fossatelli M, Urbani A, Steinkuhler C, De Francesco R, Pessi A. A continuous assay of hepatitis C virus protease based on resonance energy transfer depsipeptide substrates. Anal Biochem. 1996 Aug 15;240(1):60-7.
Tan WS. Inhibition of hepatitis B virus assembly with synthetic peptides derived from the viral surface and core antigens. J Gen Appl Microbiol. 2002 Apr;48(2):103-7.
Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, Sun Y, Huang M, Summers MF. Antiviral inhibition of the HIV-1 capsid protein. J Mol Biol. 2003 Apr 11;327(5):1013-20.
Tanimoto A, Ide Y, Arima N, Sasaguri Y, Padmanabhan R. The amino terminal deletion mutants of hepatitis C virus nonstructural protein NS5A function as transcriptional activators in yeast. Biochem Biophys Res Commun 1997; 6: 360-364.
Tanizawa K, Kanaoka Y, Wos JD, Lawson WB. Transition-state inhibition of thrombin and trypsin by amidinophenylpyruvates. Biol Chem Hoppe Seyler. 1985 Sep;366(9):871-8.
Tanji Y, Hijikata M, Satoh S, Kaneko T, Shimotohno K. Hepatitis C virus-encoded nonstructural protein NS4A has versatile functions in viral protein processing. J Virol. 1995 Mar;69(3):1575-81.
Thibeault D, Maurice R, Pilote L, Lamarre D, Pause A. In vitro characterization of a purified NS2/3 protease variant of hepatitis C virus. J Biol Chem. 2001 Dec 7;276(49):46678-84.
Thorpe LE, Ouellet LJ, Levy JR, Williams IT, Monterroso ER. Hepatitis C virus infection: prevalence, risk factors, and prevention opportunities among young injection drug users in Chicago, 1997-1999. J Infect Dis. 2000 Dec;182(6):1588-94.
Tomei L, Failla C, Santolini E, De Francesco R, La Monica N. NS3 is a serine protease required for processing of hepatitis C virus polyprotein. J Virol. 1993 Jul;67(7):4017-26.
Tomei L, Failla C, Vitale RL, Bianchi E, De Francesco R. A central hydrophobic domain of the hepatitis C virus NS4A protein is necessary and sufficient for the activation of the NS3 protease. J Gen Virol. 1996 May;77 ( Pt 5):1065-70.
Tsantrizos YS, Bolger G, Bonneau P, Cameron DR, Goudreau N, Kukolj G, LaPlante SR, Llinas-Brunet M, Nar H, Lamarre D. Macrocyclic inhibitors of the NS3 protease as potential therapeutic agents of hepatitis C virus infection. Angew Chem Int Ed Engl. 2003 Mar 28;42(12):1356-60.
Tsukiyama-Kohara K, Iizuka N, Kohara M, Nomoto A. Internal ribosome entry site within hepatitis C virus RNA. J Virol. 1992 Mar;66(3):1476-83.
Urbani A, Bazzo R, Nardi MC, Cicero DO, De Francesco R, Steinkuhler C, Barbato G. The metal binding site of the hepatitis C virus NS3 protease. A spectroscopic investigation. J Biol Chem. 1998 Jul 24;273(30):18760-9.
Urbani A, Biasiol G, Brunetti M, Volpari C, Di Marco S, Sollazzo M, Orru S, Piaz FD, Casbarra A, Pucci P, Nardi C, Gallinari P, De Francesco R, Steinkuhler C. Multiple determinants influence complex formation of the hepatitis C virus NS3 protease domain with its NS4A cofactor peptide. Biochemistry. 1999 Apr 20;38(16):5206-15.

66
VanCompernolle SE, Wiznycia AV, Rush JR, Dhanasekaran M, Baures PW, Todd SC. Small molecule inhibition of hepatitis C virus E2 binding to CD81. Virology. 2003 Sep 15;314(1):371-80.
Varaklioti A, Vassilaki N, Georgopoulou U, Mavromara P. Alternate translation occurs within the core coding region of the hepatitis C viral genome. J Biol Chem. 2002 May 17;277(20):17713-21.
Voss T, Meyer R, Sommergruber W. Spectroscopic characterization of rhinoviral protease 2A: Zn is essential for the structural integrity. Protein Sci. 1995 Dec;4(12):2526-31.
Walker B, Lynas JF. Strategies for the inhibition of serine proteases. Cell Mol Life Sci. 2001 Apr;58(4):596-624.
Wasley A, Alter MJ. Epidemiology of hepatitis C: geographic differences and temporal trends. Semin Liver Dis. 2000;20(1):1-16.
Wen YM, Lin X, Ma ZM. Exploiting new potential targets for anti-hepatitis B virus drugs. Curr Drug Targets Infect Disord. 2003 Sep;3(3):241-6.
Wen YM, Lin X, Ma ZM. Exploiting new potential targets for anti-hepatitis B virus drugs. Curr Drug Targets Infect Disord. 2003 Sep;3(3):241-6.
Weng WK, Levy S. Hepatitis C virus (HCV) and lymphomagenesis. Leuk Lymphoma. 2003 Jul;44(7):1113-20.
Wiley MR, Nickolay Y. Chirgadze, David K. Clawson, Trelia J. Craft, Donetta S. Gifford-Moore, Noel D. Jones, Jennifer L. Olkowski, Aaron L. Schacht, Leonard C. Weir and Gerald F. Smith. Serine protease selectivity of the thrombin inhibitor D-Phe-Pro-Agmatine and its homologs. Bioorg Med Chem Lett, 1995, 5(23) Pages 2835-2840.
World Health organisation Fact Sheet No. 164 Wyke RJ, Williams R. Clinical aspects of non-A, non-B hepatitis infection. J Virol
Methods. 1980 Dec;2(1-2):17-29. Xu Z, Choi J, Lu W, Ou JH. Hepatitis C virus f protein is a short-lived protein
associated with the endoplasmic reticulum. J Virol. 2003 Jan;77(2):1578-83. Xu Z, Choi J, Yen TS, Lu W, Strohecker A, Govindarajan S, Chien D, Selby MJ, Ou
J. Synthesis of a novel hepatitis C virus protein by ribosomal frameshift. EMBO J. 2001 Jul 16;20(14):3840-8.
Yamada N, Tanihara K, Takada A, Yorihuzi T, Tsutsumi M, Shimomura H, Tsuji T, Date T. Genetic organization and diversity of the 3' noncoding region of the hepatitis C virus genome. Virology. 1996 Sep 1;223(1):255-61.
Yamaga AK, Ou JH. Membrane topology of the hepatitis C virus NS2 protein. J Biol Chem. 2002 Sep 6;277(36):33228-34.
Yan Y, Li Y, Munshi S, Sardana V, Cole JL, Sardana M, Steinkuehler C, Tomei L, De Francesco R, Kuo LC, Chen Z. Complex of NS3 protease and NS4A peptide of BK strain hepatitis C virus: a 2.2 A resolution structure in a hexagonal crystal form. Protein Sci. 1998 Apr;7(4):837-47.
Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure Fold Des. 1999 Nov 15;7(11):1353-63.
Yaya Liu, Vincent S. Stoll, Paul L. Richardson, Ayda Saldivar, Jeffrey L. Klaus, Akhteruzzaman Molla, William Kohlbrenner and Warren M. Kati. Hepatitis C NS3 protease inhibition by peptidyl-α-ketoamide inhibitors: kinetic mechanism and structure. Archives of Biochemistry and Biophysics, 2004, 421(2), 207-216
Zhang R, Durkin J, Windsor WT, McNemar C, Ramanathan L, Le HV. Probing the substrate specificity of hepatitis C virus NS3 serine protease by using synthetic peptides. J Virol. 1997 Aug;71(8):6208-13.

67
Zhang W, Cox AG, Taylor EW. Hepatitis C virus encodes a selenium-dependent glutathione peroxidase gene. Implications for oxidative stress as a risk factor in progression to hepatocellular carcinoma. Med Klin (Munich). 1999 Oct 15;94 Suppl 3:2-6.
Zhao WD, Wimmer E, Lahser FC. Poliovirus/Hepatitis C virus (internal ribosomal entry site-core) chimeric viruses: improved growth properties through modification of a proteolytic cleavage site and requirement for core RNA sequences but not for core-related polypeptides. J Virol. 1999 Feb;73(2):1546-54.
Zoulim F, Chevallier M, Maynard M, Trepo C. Clinical consequences of hepatitis C virus infection. Rev Med Virol. 2003 Jan-Feb;13(1):57-68.

Acta Universitatis UpsaliensisComprehensive Summaries of Uppsala Dissertations
from the Faculty of Science and Technology
Editor: The Dean of the Faculty of Science and Technology
Distribution:Uppsala University Library
Box 510, SE-751 20 Uppsala, Swedenwww.uu.se, [email protected]
ISSN 1104-232XISBN 91-554-5905-6
A doctoral dissertation from the Faculty of Science and Technology, UppsalaUniversity, is usually a summary of a number of papers. A few copies of thecomplete dissertation are kept at major Swedish research libraries, while thesummary alone is distributed internationally through the series Comprehensive
Summaries of Uppsala Dissertations from the Faculty of Science and Technology.(Prior to October, 1993, the series was published under the title “ComprehensiveSummaries of Uppsala Dissertations from the Faculty of Science”.)





![ORIGINAL ARTICLE fic Glucose-Induced Control of Insulin ... · cellular sources. Calcineurin inhibitors (FK506, cyclosporin A, and a peptide calcineurin inhibitor [CAIN]) abolished](https://static.fdocuments.in/doc/165x107/5e35e15c9ec8741478479276/original-article-ic-glucose-induced-control-of-insulin-cellular-sources-calcineurin.jpg)












