
Paul Hanna DVM, MSc, DACVP General Pathology (VPM 152...
20
“All organ injuries start with structural or molecular alterations in cells” concept began by Virchow in 1800's. present day study of disease attempts to understand how cells react to injury, often at the subcellular / molecular level, and how this is manifested in the whole animal. NORMAL CELLS differentiated eukaryotic cells vary from one another, depending on their specialized function (eg hepatocyte, neuron, keratinocyte, etc), however all share the basic organelles for the synthesis of proteins, lipids, carbohydrates; energy production; and for transport of ions and other substances. to understand pathology you first need to understand the normal; review normal structure and function of cells. Plasma membrane: phospholipid bilayer with embedded proteins / glycoproteins / glycolipids (eg ion pumps, receptors, adhesion molecules, etc) semipermeable membrane with pumps for ionic / osmotic homeostasis Cellular Pathology Paul Hanna DVM, MSc, DACVP General Pathology (VPM 152) January 2018
Transcript of Paul Hanna DVM, MSc, DACVP General Pathology (VPM 152...

“All organ injuries start with structural or molecular alterations in cells” concept began by Virchow in 1800's.
present day study of disease attempts to understand how cells react to injury, often at the subcellular / molecular
level, and how this is manifested in the whole animal.
NORMAL CELLS
differentiated eukaryotic cells vary from one another, depending on their specialized function (eg hepatocyte,
neuron, keratinocyte, etc), however all share the basic organelles for the synthesis of proteins, lipids,
carbohydrates; energy production; and for transport of ions and other substances.
to understand pathology you first need to understand the normal; review normal structure and function of cells.
Plasma membrane: phospholipid bilayer with embedded proteins / glycoproteins / glycolipids
(eg ion pumps, receptors, adhesion molecules, etc)
semipermeable membrane with pumps for ionic / osmotic homeostasis
Cellular Pathology
Paul Hanna DVM, MSc, DACVP General Pathology (VPM 152) January 2018

Gen Path (VPM 152) Cell Pathology 2
Nucleus: chromatin (euchromatin vs heterochromatin)
nucleolus (synthesis of ribosomal RNA / subunits)
transcription of genes
Mitochondria: inner & outer membranes, cristae
intermembranous and inner matrix compartments
oxidative phosphorylation (main source of ATP)
Endoplasmic reticulum (ER), Ribosomes & Golgi Apparatus:
RER & Golgi - synthesis and packaging of proteins for export, membranes, lysosomes
SER - lipid biosynthesis (eg membranes, steroids)
- detoxification of harmful compounds (via P450’s)
- sequestration of Ca2+
ions
Chaperones &
Proteasomes: chaperones assist proper folding of proteins and transport across organelle membranes.
proteasomes degrade both excess proteins and incorrectly folded (misfolded) proteins.
Lysosomes: enzymatic digestion (acid hydrolases) of materials in the cell
primary vs secondary lysosomes; residual bodies
autophagy vs heterophagy / endocytosis
phagocytosis / phagosome; pinocytosis / pinocytotic vesicle; receptor-mediated endocytosis
Cytoskeleton: structure and movement of cells / organelles / granules / surface molecules / phagocytosis
Microfilaments: actin in various forms cell shape & movement
Microtubules: polymers of tubulin organelle movement / flagella / cilia / mitotic spindle
Intermediate filaments: cytokeratins, vimentin, desmin, GFAP, neurofilament proteins
Peroxisomes: enzymes (eg catalase, oxidases) metabolism of hydrogen peroxide & fatty acids.
CELL ADAPTATION, CELL INJURY and CELL DEATH
I. DEFINITIONS AND TERMINOLOGY
1) Homeostasis
cells are able to maintain normal structure and function (eg ion balance, pH, energy metabolism) in response to
normal physiologic demands.
2) Cellular Adaptation
as cells encounter some stresses (eg excessive physiologic demand or some mild pathologic stimuli) they may
make functional or structural adaptations to maintain viability / homeostasis.
cells may respond to these stimuli by either increasing or decreasing their content of specific organelles.
adaptive processes: atrophy, hypertrophy, hyperplasia and metaplasia are forms of adaptation.
3) Cell Injury
if the limits of adaptive response are exceeded, or in certain instances when adaptation is not possible (eg with
severe injurious stimulus), a sequence of events called cell injury occurs.

Gen Path (VPM 152) Cell Pathology 3
a) Reversible Cell Injury
removal of stress / injurious stimulus results in complete restoration of structural and functional integrity.
b) Irreversible Cell Injury / Cell Death
if stimulus persists (or severe enough from the start) the cell will suffer irreversible cell injury and death.
cell death is one of the most crucial events in pathology and can affect any type of cell.
two principle morphologic patterns that are indicative of cell death:
Necrosis: type of cell death characterized by severe membrane injury and enzymatic degradation;
always a pathologic process.
Apoptosis: regulated form of cell death; can be a physiologic or pathologic process.
II. CELLULAR ADAPTATIONS OF GROWTH AND DIFFERENTATION
major adaptive responses: atrophy, hypertrophy, *hyperplasia, *aplasia,*hypoplasia,*metaplasia,*dysplasia.
(*to be discussed in more detail in neoplasia section)
1) Atrophy
definition: decrease in the amount of a tissue or organ after normal growth has been attained.
adaptive response whereby a tissue or organ undergoes a reduction in mass (size), due to a decrease in the size
and/or number of cells.
a) Etiology
can be physiologic, eg postpartum uterine & mammary gland involution.
can be pathologic, eg=s: decreased workload (disuse atrophy), loss of innervation (denervation atrophy),
loss of hormonal (trophic) stimulation, reduced blood supply / hypoxia, inadequate nutrition, compression
(by tumors, etc) persistent cell injury, aging (senile atrophy).
atrophic cells are not dead or necessarily badly injured but they have a reduced functional capacity (note,
may progress to cell injury / death if stimulus persists or worsens).
retain ability to control their internal environment and produce enough energy to survive.
given "enough time" and removing the reason for cellular atrophy, the cells can return to 'normal'.
b) Mechanisms and Biochemistry
decreased amount of substance produced within cell, ie catabolic processes exceed anabolic processes.
eg: with muscle atrophy (due to denervation atrophy or disuse atrophy) : myofilaments, mitochondria,
endoplasmic reticulum, metabolic activity.
organelles removed by autophagocytosis and decreased proteins by ubiquitin-proteosome pathway.
cell shrinks in volume and shuts downs its differentiated functions decreases its energy requirements.
c) Gross Appearance tissue/organ is decreased in size.
d) Microscopic Appearance cells are smaller than normal
eg’s
endocrine gland, following decreased trophic stimulation
- physiologic: uterus/breast atrophy with decreased hormonal stimulus following pregnancy / lactation
- pathologic: loss of trophic hormone production due to pituitary destruction or reduced trophic hormone
production (eg ACTH) due to medical supplementation of a hormone (eg corticosteroid)

Gen Path (VPM 152) Cell Pathology 4
starvation, results in atrophy of fat (eg serous atrophy of fat) and various other tissues (eg muscle, liver).
- there is a definite sequence in which body proteins are broken down during starvation in order to preserve
the blood glucose level (via glucogenic amino acids): first digestive enzymes of the gastrointestinal tract
and pancreas & various hepatic enzymes that normally process incoming nutrients from the intestine; later,
muscle proteins.
- there is a sequential use (& loss) of muscle mass as an endogenous sources of protein and energy; postural
muscles (eg supraspinatus) are preserved at the expense of other muscles (eg longissimus dorsi) that are
used for locomotion.
- there is also a sequential mobilization of fat depots; in most terrestrial mammals, subcutaneous fat is used
first, visceral fat is used next and bone marrow fat is used last.
2) Hypertrophy
definition: organs are increased in size due to an increase in cell size without cellular proliferation.
in organs / tissues which have minimal proliferative capacity (cardiac and skeletal muscle) see only
hypertrophy, whereas in tissues / organs with cells capable of division see both hypertrophy and hyperplasia.
a) Etiology
a response to increased work load:
physiologic: eg with exercise see increase in muscle cell size (grossly muscles increase in size).
pathologic: eg heart failure see enlargement of myocardial fibers (grossly heart increases in size).
a response to trophic signals:
physiologic hypertrophy (& hyperplasia), eg uterus and mammary gland during pregnancy / lactation.
pathological hypertrophy, eg myocardial hypertrophy in hyperthyroid cats.
response to certain drugs or toxins: increased SER in liver (organelle hypertrophy) with phenobarbital.
b) Mechanisms and Biochemistry
anabolic processes exceed catabolic ones (ie, increased synthesis of cellular constituents).
increase in organelles / total cellular proteins: mitochondria, endoplasmic reticulum, myofibrils.
may not always be advantageous (eg myocardial hypertrophy can exceed the limits of vascular supply).
c) Gross Appearance: tissue / organ increased in size / weight.
d) Microscopic Appearance
cellular enlargement due to a proportional increase in the number and size of organelles.
must be distinguished from cellular swelling, which is due to an increased intake of fluid by the cell.
3) *Hyperplasia
increase in organ size or tissue mass caused by an increase in the number of constituent cells.
hypertrophy and hyperplasia are not mutually exclusive and are often seen together in structures which can
undergo division (esp reproductive and endocrine organs).
[*to be discussed in more detail in Disturbances of Growth / Neoplasia section of course]

Gen Path (VPM 152) Cell Pathology 5
III. CAUSES OF CELL INJURY
1) Hypoxia (Oxygen Deficiency)
one of the most important and common causes of cell injury and cell death.
hypoxia causes impairment of oxidative respiration, ie it interferes with energy production.
occurs with:
a) Deficient blood supply
ischemia = deficiency of blood supply from impeded arterial flow or reduced venous drainage = hypoxia +
delivery of nutrients and removal of metabolites.
cells may adapt to mild ischemia (eg muscle atrophy) or die with severe ischemia.
infarction = localized area of ischemic necrosis.
b) Reduced oxygen-carrying capacity of the blood
due to anemia = reduction in numbers or volume of erythrocytes or quantity of hemoglobin (Hb).
due to Hb dysfunction, eg methaemoglobinemia - nitrate / nitrite poisoning,
carboxyhaemoglobinemia - carbon monoxide poisoning.
c) Interference with respiratory chain / oxidative phosphorylation
eg, cyanide poisoning inactivates cytochrome oxidase in mitochondria blocks oxidative phosphorylation.
2) Physical agents
severity of a physical injury may be increased by tissue hypoxia due to associated local vascular injury.
a) Direct mechanical trauma - lacerations or crush injuries.
b) Temperature extremes - heat (thermal burn), cold (frostbite).
c) Radiation - radioactive isotope emissions or electromagnetic radiation (eg UV light, x-rays).
d) Electrocution - pets chewing electric cords, faulty wiring in barns, lightning strike, etc.
e) Sudden changes in atmospheric pressure - marine mammals have mechanisms to mostly avoid the “bends”
3) Chemicals, Drugs & Toxins a) Inorganic poisons - eg lead, copper, arsenic, selenium, mercury, etc.
b) Organic poisons - eg nitrate/nitrite, oxalate, hydrocyanic acid, etc.
c) Manufactured chemicals - eg drugs (overdose / idiosyncratic), pesticides, herbicides, rodenticides, etc.
d) Physiologic compounds - eg salt, glucose, oxygen, etc.
e) Plant toxins - eg ragwort, sweet clover, braken fern, etc.
f) Animal toxins - eg snake or spider venom, tick toxin, etc.
g) Bacterial toxins / Mycotoxins - eg botulinum toxin, aflatoxin, ergot, etc.
4) Infectious agents (such important causes of disease, you have individual courses to study them in detail!)
a) Viruses
b) Bacteria / rickettsiae / chlamydia
c) Fungi
d) Protozoa
e) Metazoan parasites
5) Immunologic Reactions a) Immune response - eg cells damaged as “innocent bystanders” in immune / inflammatory response.
b) Hypersensitivity (allergic) reactions - eg anaphylactic reaction to a foreign protein or drug.
c) Autoimmune diseases - reactions to self-antigens.

Gen Path (VPM 152) Cell Pathology 6
6) Genetic abnormalities a) Cytogenetic disorders / chromosomal aberrations - one cause of congenital anomalies.
b) Mendelian disorders (mutant genes)
enzyme defects, eg lysosomal storage disease.
structural / transport protein defects - eg collagen dysplasia, cystic fibrosis, sickle cell anemia, etc.
c) Multifactorial inheritance - combined effects of environmental factors and 2 or more mutated genes
(eg neoplasia, hypertension, coronary artery disease, etc).
7) Nutritional Imbalances a) Deficiencies - deficiencies of protein-calories (starvation), vitamins (A to E), minerals (eg copper).
b) Overnutrition - eg excess lipids / calories obesity, diabetes, atherosclerosis, etc.
8) Workload Imbalances a) Overworked cells - cell injury occurs if stimulus prolonged and/or exceeds ability to adapt.
b) Underworked cells - prolonged lack of stimulation (eg disuse, denervation, lack of trophic hormones)
can lead to atrophy and eventually the loss of cells.
9) Cell Aging
the cumulative effects of a life time of cell damage (chemical, infectious, nutrition, etc) leads to a diminished
capacity of aged cells / tissues to maintain homeostasis and adapt to harmful stimuli.
Mnemonic acronym for agents of disease = “double MINT”
Malformation (genetics, teratogens, etc)
Miscellaneous (metabolic, aging, hypoxia, etc)
Infectious (viruses, bacteria, fungi, etc)
Immune (immune mediated, hypersensitivity, autoimmune, etc)
Nutritional (protein-caloric intake, vitamins, minerals, etc)
Neoplastic (genetic, viral, chemical, radiation, etc)
Trauma (mechanical, temperature, radiation, etc)
Toxicity (inorangic, chemicals / drugs, plant toxins, etc)

Gen Path (VPM 152) Cell Pathology 7
IV. MECHANISMS OF CELL INJURY
1) General Considerations a) the cellular response to injurious stimuli is dependant on the type of injury, its duration and its severity.
eg, low doses of toxins or brief durations of ischemia may lead to reversible cell injury, whereas larger toxin
doses or longer ischemic intervals may result in irreversible injury / cell death.
b) consequences of an injurious stimulus are dependent on the type of cell injured and its current status, ie
nutritional, hormonal, metabolic, oxygen requirement, etc.
c) 4 intracellular systems are particularly vulnerable to injury.
cell membranes - especially ionic / osmotic homeostasis.
mitochondria - oxidative phosphorylation / ATP production.
protein synthesis, folding and packaging - structural and functional proteins.
genetic apparatus - DNA / RNA.
d) interrelationship of structural and biochemical elements means that damage at one focus leads to wide-
ranging secondary effects.
eg, impairment of aerobic respiration (eg O2 or mitochondrial damage) disruption of the energy-
dependent membrane sodium pump ionic and fluid imbalance cell swelling.
2) Biochemical Mechanisms
several molecular / biochemical sites are commonly damaged by a variety of inciting causes.
a) ATP depletion
ATP depletion and ATP synthesis are common consequences of both ischemic and toxic injury.
AMP activates phosphofructokinase & phosphorylase anaerobic glycolysis depletion of glycogen
stores lactic acid & [Pi] intracellular pH impaired cell enzyme activity.
ATP required for membrane transport (Na+/K
+ & Ca
2+ pumps) / osmotic balance, protein synthesis, protein
stability (proper folding), lipogenesis, etc.
cells with greater glycolytic capacity (eg liver cells) have an advantage over cells which are more reliant on
oxidative phosphorylation (eg neurons).
Tissue sensitivity to hypoxia - dependant upon energy demands of the cell and/or its ability to utilize
anaerobic glycolysis as a source of energy SUSCEPTIBILITY CELL TYPE TIME (to irreversible cell injury) HIGH Neurons ~ 3 to 5 minutes INTERMEDIATE Cardiac myocytes ~ 30 minutes to 1 hour Hepatocytes " Renal epithelium " LOW Fibroblasts many hours Keratinocytes " Skeletal myocytes "
HIGH Neurons ~ 3 to 5 minutes
INTERMEDIATE Cardiac myocytes ~ 30 minutes to 1 hour Hepatocytes " Renal epithelium "
LOW Fibroblasts many hours
Keratinocytes " Skeletal myocytes "

Gen Path (VPM 152) Cell Pathology 8
b) Free radical induced injury (Oxidative Stress)
free radicals are chemical species with a single unpaired electron in outer orbit (donate or steal electrons,
extremely unstable); readily react with organic or inorganic chemicals, avidly attack/degrade membranes,
proteins & nuclei acids.
free radical-induced injury, particularly that induced by activated oxygen species, is an important mechanism
of cell damage in many disease processes (chemical, radiation, O2 toxicity, inflammation, reperfusion, etc).
cell injury occurs when the free radical generation overwhelms antioxidant defense mechanisms.
Generation of free radicals
Cellular metabolism - small amounts produced from cell redox reactions, eg normal oxidative
phosphorylation (leakage from mitochondria), other intracellular oxidases (eg peroxisomes), PMN’s in
inflammation, excess O2, altered metabolism in cell stress (eg reperfusion injury)
Enzymatic metabolism of exogenous chemicals - some intermediary metabolites of chemical / drugs are
highly reactive free radicals.
Ionizing radiation - hydrolyzes water into hydroxyl (•OH) and hydrogen (H•) free radicals.
Divalent metals - the transition metals (copper and iron), accept or donate free electrons during certain
intracellular reactions, ie catalyze free radical formation.
Important reactants
superoxide anion (O2.-), hydroxyl radical (•OH), hydrogen peroxide (H2O2) and peroxynitrite (ONOO
•).
H2O2 (not a free radical) is a frequent by-product of oxidative metabolism that can generate hydroxyl
radicals from reactions with copper or ferrous ions (eg Fenton reaction = Fe2+
+ H2O2 •OH + OH- + Fe
3+)
most intracellular stored iron is in the ferric (Fe3+
) state and must be reduced to the ferrous (Fe2+
) state to act
in the Fenton reaction (often reduced by O2.-); iron & O2
.- required for maximal cell damage.
Main sites of damage
Damage of Membranes (lipid peroxidation)
free radicals (esp •OH) are highly reactive & unstable (don’t last long or travel far) “steal” single
electrons from the hydrogen next to a double bond in unsaturated fatty acids in cell membranes form
lipid peroxides (which themselves are reactive & unstable) autocatalytic chain reaction (self-
propagating) can cause rapid widespread membrane / organelle damage.
Damage of Proteins
free radicals cause fragmentation and cross-linkage between proteins damaged structural proteins / loss
of enzymatic activity increased degradation by proteosomes.
Damage to DNA
free radicals damage nuclear & mitochondrial DNA, producing strand breaks & DNA-protein adducts
(short-term apoptosis; long-term low level damage implicated in cell aging & neoplasia).
Protective Mechanisms of the Cell
Storage and transport proteins:
iron & copper can catalyze formation of reactive oxygen forms; they are minimized by being bound to
storage and transport proteins (eg ceruloplasmin, transferrin, lactoferrin, apoferritin/ferritin) and kept in an
oxidized state.
Antioxidants - either block the formation of free radicals or inactivate / scavenge them:
Vitamins A&E (lipid soluble, found in cell membranes), Vitamin C (aqueous-phase antioxidant) and
glutathione (reduced form [GSH] reacts with H2O2 or •OH oxidised glutathione [GSSG] + H2O).
Enzymes which are involved in neutralizing free radicals:
Glutathione peroxidase - a selenium-containing enzyme which catalyzes GSH to GSSG.
- this enzyme also catalyses the reduction of lipid peroxides by glutathione,
preventing propagation of lipid peroxidation reactions.
Superoxide dismutase (SOD) - catalyzes the conversion of O2.- to H2O2.
Catalase - catalyzes the breaks down H2O2 to O2 + H2O.
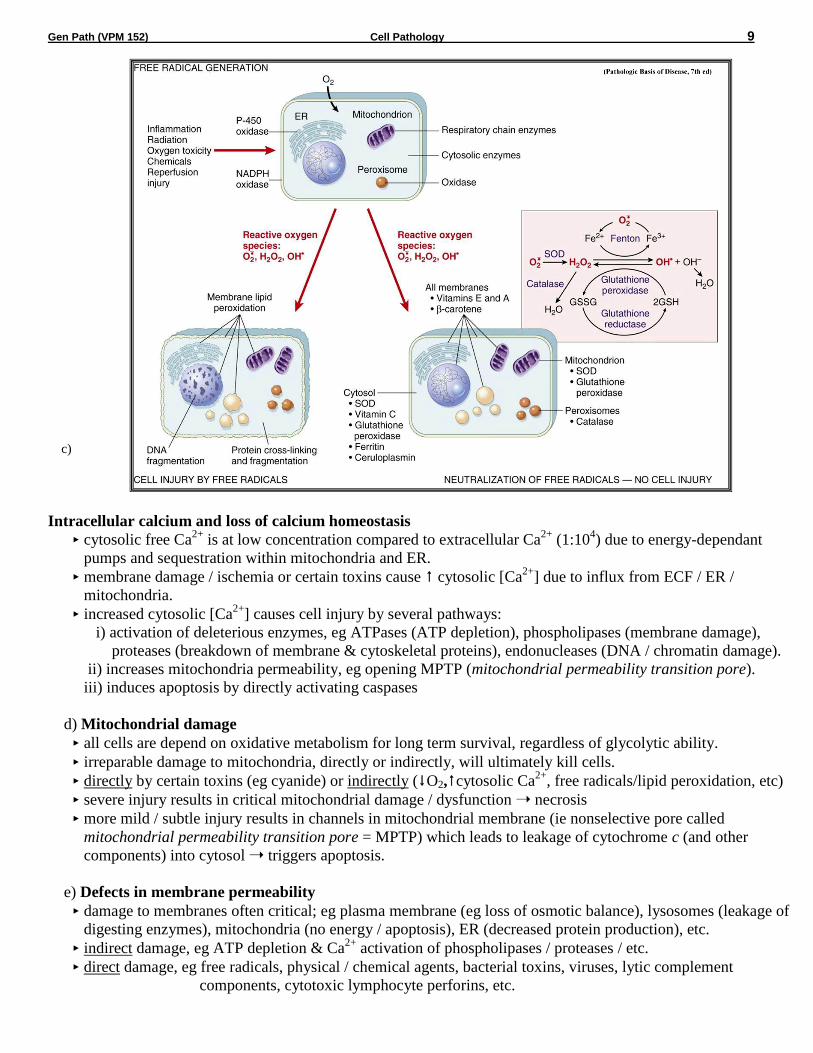
Gen Path (VPM 152) Cell Pathology 9
c)
Intracellular calcium and loss of calcium homeostasis
cytosolic free Ca2+
is at low concentration compared to extracellular Ca2+
(1:104) due to energy-dependant
pumps and sequestration within mitochondria and ER.
membrane damage / ischemia or certain toxins cause cytosolic [Ca2+
] due to influx from ECF / ER /
mitochondria.
increased cytosolic [Ca2+
] causes cell injury by several pathways:
i) activation of deleterious enzymes, eg ATPases (ATP depletion), phospholipases (membrane damage),
proteases (breakdown of membrane & cytoskeletal proteins), endonucleases (DNA / chromatin damage).
ii) increases mitochondria permeability, eg opening MPTP (mitochondrial permeability transition pore).
iii) induces apoptosis by directly activating caspases
d) Mitochondrial damage
all cells are depend on oxidative metabolism for long term survival, regardless of glycolytic ability.
irreparable damage to mitochondria, directly or indirectly, will ultimately kill cells.
directly by certain toxins (eg cyanide) or indirectly (O2,cytosolic Ca2+
, free radicals/lipid peroxidation, etc)
severe injury results in critical mitochondrial damage / dysfunction necrosis
more mild / subtle injury results in channels in mitochondrial membrane (ie nonselective pore called
mitochondrial permeability transition pore = MPTP) which leads to leakage of cytochrome c (and other
components) into cytosol triggers apoptosis.
e) Defects in membrane permeability
damage to membranes often critical; eg plasma membrane (eg loss of osmotic balance), lysosomes (leakage of
digesting enzymes), mitochondria (no energy / apoptosis), ER (decreased protein production), etc.
indirect damage, eg ATP depletion & Ca2+
activation of phospholipases / proteases / etc.
direct damage, eg free radicals, physical / chemical agents, bacterial toxins, viruses, lytic complement
components, cytotoxic lymphocyte perforins, etc.

Gen Path (VPM 152) Cell Pathology 10
3) Chemical (Toxic) Injury
chemicals and certain drugs / toxins produce damage in one of two general ways.
a) Direct interaction
some chemicals act directly by damaging particular organelles or critical cell molecules.
- eg cyanide damages mitochondrial cytochrome oxidase blocks oxidative phosphorylation.
- eg fluoroacetate converted to fluorocitrate prevents citrate from being used in the citric acid cycle.
b) Conversion to reactive toxic metabolites
toxic metabolites usually produced by cytochrome P-450 mixed function oxidase (MFO) in the SER of liver.
toxic metabolites can be in the form of reactive free radicals (see previous discussion) or adducts (ie
chemicals that form strong covalent bonds with cell molecules, thus damaging them).
since liver is a major site of chemical modification of drugs and toxins, it is therefore particularly susceptible
to drug / toxin-induced injury.
Acetaminophen (Tylenol) Toxicity
in humans & dogs most acetaminophen is detoxified in liver to glucuronide and sulfate conjugates which are
then excreted in the urine; only small amounts converted to highly reactive metabolite NAPQI by P450 MFO.
cats are relatively deficient in glucuronyl transferase and a large portion of acetaminophen is metabolized to
NAPQI, which is electrophilic binds to molecules in hepatocytes (ie adduct formation) and causes necrosis.
additionally cats & dogs (compared to humans) produce relatively more of the metabolite para-aminophenol,
which is released from the liver cells and results in oxidative damage to hemoglobin (ie methemoglobinemia).
Carbon Tetrachloride (CCl4) Toxicity
CCl4 metabolized by P450 MFO enzyme system on the SER of the hepatocyte (CCl4 + e CCl3• + Cl
- ).
CCl3• is highly reactive and causes lipid peroxidation (autocatyzing) see severe and rapid membrane
destruction protein synthesis (30 min); ER swelling & ribosomal dissociation (2 hrs).
V. MORPHOLOGY OF CELL INJURY AND NECROSIS
gross morphologic changes usually require hours to develop, ie no sign of myocardial necrosis if an animal dies
immediately from a myocardial infarct (however a thrombus would be evident in a coronary artery!)
in general:
i) cell swelling (reversible injury)
- can start within minutes of the initial insult.
- note, loss of function is also rapid, eg heart muscle stops contracting with 60 sec of coronary occlusion.
ii) cell death (irreversible injury)
- eg can occur in the myocardium within 20 to 60 min of coronary artery occlusion.
- biochemical changes (eg creatine kinase, troponins) within minutes & EM changes within few hrs.
- LM changes in 4 to 12 hrs and gross changes not apparent for 12 to 24 hrs after initial insult.
the critical transition point to irreversible injury is not known; however two features consistently characterize
irreversibility (ie “point of no return” or “lethal hit”):
inability to reverse mitochondrial dysfunction.
profound disturbances of membrane function.
I. REVERSIBLE CELL INJURY
two main types of reversible cell injury are recognized by LM cellular swelling and fatty change.

Gen Path (VPM 152) Cell Pathology 11
1) Cellular Swelling a) Etiology / Pathogenesis of Cellular Swelling (see prior discussion)
early and almost universal manifestation of cell injury.
due to loss of ion and fluid homeostasis (esp Na+ pump) net [intracellular H2O].
b) Gross Appearance of Cellular Swelling
organ swollen with rounded edges, tissue may show slight palor.
cut surface: tissue bulges and can not be easily put in correct apposition, heavy.
c) Histologic Appearance of Cellular Swelling
affected cells appear enlarged, with pale cytoplasm, nucleus in normal position.
staining characteristics are altered:
- if mild, may have cloudy appearance = "cloudy swelling".
- as the process continues many vacuoles of variable size appear in the cytoplasm; called “hydropic or
vacuolar degeneration” or when severe (esp viral infected cells) called “ballooning degeneration”.
d) Ultrastructural Changes of Cellular Swelling (especially ischemia)
plasma membrane: blunting / loss of microvilli, blebbing, +/- myelin figures (membrane fragments).
endoplasmic reticulum: swelling of cisternae (and cell in general), detachment of ribosomes.
mitochondria: swelling, appearance of small densities.
nucleus: clumping of chromatin.
e) Prognosis of Cellular Swelling
cell swelling is reversible, ie if stimulus / stress is removed the cell returns to its normal state.
2) Fatty Change a) Etiology / Pathogenesis of Fatty Change
occurs in various forms of injury (esp hypoxic, toxic, metabolic).
see abnormal accumulation of lipids within the cell (ie: intracellular).
mainly in cells dependant on fat metabolism, esp liver, less in renal tubular epithelium and myocardium.
seen in abnormalities of uptake, utilization (metabolism) and/or mobilization (export) of fat.
may be an expression of cell injury or a stage of injury in cells that are destined to die.
may be preceded or accompanied by cell swelling.
Lipid metabolism and storage in the liver:
lipids enter hepatocytes as free fatty acids (FFA); note,
fatty acids can also be synthesized from acetate.
lipids are metabolized by hepatocytes
- most are esterified to form triglycerides (TG).
- some synthesized to cholesterol esters or phospholipids.
- some oxidized for energy & produce ketone bodies.
lipids are exported from hepatocyte
- TG’s must be complexed with a lipid-acceptor protein
(apoprotein) to form lipoproteins to be exported.
- accumulation of hepatocellular intracellular lipid can occur due to abnormalities in any of these steps; esp
important is the synthesis of lipid-acceptor protein (apoprotein) to form lipoproteins.
- eg: excess dietary fat (eg foie gras); starvation / prolonged fasting & diabetes (FA mobilization & protein
synthesis); hypoxia (inhibits fatty acid oxidation & protein synthesis); some toxins (apoprotein synthesis)

Gen Path (VPM 152) Cell Pathology 12
b) Gross Appearance of Fatty Change
in liver: - diffusely yellow if all cells affected or reticular pattern if zones of hepatocytes are affected.
- edges rounded and will bulge on cut section, cuts easily and has a greasy texture.
- if severe, liver may float in fixative / water.
c) Histologic Appearance of Fatty Change presence of well delineated, lipid-filled vacuoles in the cytoplasm.
may be single, large vacuoles (macrovesicular) or multiple, small vacuoles (microvesicular).
may need to confirm with special stain, since not all clear vacuoles are lipid (ie could be water or
glycogen); use frozen sections with an oil-soluble dye called "oil red O" (if it is lipid, it will stain red).
II. IRREVERSIBLE CELL INJURY
1) Necrosis
term used to describe the range of morphologic changes that occur following cell death in living tissue.
the morphologic appearance is due to 2 concurrent processes:
denaturation of proteins (ie nonproteolytic structural alteration in 2o or 3
o structure).
enzymatic digestion of the cell:
- endogenous enzymes derived from the lysosomes of the dying cells = autolysis (self digestion).
- lysosomes of immigrant leukocytes = heterolysis.
note, the term autolysis is also often used to describe the changes that occur in all of the cells after an animal
has died; however the proper term is postmortem autolysis or postmortem decomposition.
a mass of necrotic tissue may exhibit distinctive morphologic patterns depending on whether enzyme
catabolism or protein denaturation predominates.
a) Coagulation (coagulative) Necrosis
is the most common manifestation of cell death.
is characteristic of hypoxic / ischemic death of cells in all tissues (except brain).
basic outline of coagulated cell persists at least for a few days; protein denaturation predominates over
enzymatic digestion (presumably some of the lysosomal enzymes are denatured as well).
Gross appearance
architecture of necrotic tissue resembles normal tissue, but color and texture may be visibly different.
lighter in color (pale); due to coagulation of cytoplasmic proteins and decreased blood flow, eg infarcts.
usually firm texture.
may be swollen (due to swelling of individual cells) or shrunken (due to cell loss).
may see a local reaction to necrotic tissue (surrounding red zone of hyperemia or white layer of
inflammatory cells).
Microscopic appearance
original cell shape and tissue architecture preserved, ie dead cells resemble an eosinophilic "shadow" of
the original cells.
cytoplasm:
- increased eosinophilia (on H&E stain) due to binding of eosin to altered proteins & loss of ribosomes.
- cytoplasm is usually hyalinized (homogeneous glassy appearance) due to the loss of glycogen particles.
- dead cells / cell debris may also be mineralized (“calcified”).
- eventually necrotic cells are removed by white blood cell proteolysis and phagocytosis.
nucleus (see 3 patterns of nuclear changes):
- karyolysis = dissolution / fading of the nucleus (ie fading of chromatin 2N to DNAases).
- pyknosis = shrunken and densely basophilic nuclei (also seen in apoptosis).
- karyorrhexis = nuclear fragmentation.
- even with these latter two types, after 1-2 days following the death of the cells, the nuclei disappear.

Gen Path (VPM 152) Cell Pathology 13
Ultrastructural appearance (especially ischemia)
myelin figures and disrupted plasma & organelle membranes loss of proteins (eg CK from muscle, ALT
from liver), essential coenzymes & RNA from leaky membranes and marked influx of Ca2+
/
macromolecules from the ECF.
severe swelling of mitochondria with large densities in the matrix unable to restore ATP production.
initial swelling of lysosomes further injury results in leakage of lysosomal enzymes into cytoplasm
acid hydrolases are activated in the pH degradation of cytoplasmic and nuclear components.
dense pyknotic nuclei.
cell debris either removed by phagocytosis or broken down into fatty acid residues ( calcification).
b) Liquefactive Necrosis
occurs when enzymatic digestion of necrotic cells predominates over protein denaturation.
see in many bacterial infections, due to attraction of neutrophils which contain potent hydrolases which are
capable of digesting dead cells.
also, for unknown reasons, hypoxic damage of the brain/spinal cord, often causes liquefactive necrosis.
Gross appearance
affected tissue is liquefied; becomes a soft to viscous fluid.
if process was initiated by inflammation, the liquid is often mostly dead neutrophils (ie called pus).
Microscopic appearance
may see degenerate neutrophils and amorphous material (or nothing if necrotic tissue has flowed out).
c) Caseous Necrosis
typical lesion seen with specific bacterial diseases, eg tuberculosis, caseous lymphadenitis.
common in birds since heterophils don’t have the potent hydrodrolytic enzymes to liquefy cells.
Gross appearance
grey-white and dry with friable (ie crumbly) to pasty texture; note, caseous = cheese like.
Microscopic appearance
necrotic areas consist of dead cells persisting as amorphous, coarsely, granular debris; often mineralized.
necrotic cells do not retain cellular outline as seen with coagulation necrosis.
necrotic cells do not undergo complete dissolution as seen in liquefactive necrosis.
d) Gangrenous Necrosis [= Gangrene]
definition: necrosis (usually ischemic) of extremities, eg distal limbs / digits or tips of ears.
dry gangrene = coagulation necrosis of an extremity.
wet gangrene = when the coagulative necrosis of dry gangrene is modified by the liquefactive action of
invading saprophytic / putrefactive bacteria.
gas gangrene = production of gas bubbles in the necrotic tissue by invading bacteria (esp Clostridia).
e) Fat Necrosis
type of necrosis distinguished by its location within body fat stores, esp abdominal or subcutaneous fat.
etiology: with recurrent pancreatic necrosis, Vit E deficiency, trauma, unknown causes (idiopathic).
eg, pancreatic necrosis - digestive enzymes activated extracellularly adipose & pancreatic tissues are
digested fatty acids combine with Ca2+
precipitated as insoluble Ca2+
soaps ("saponification of fat")
Gross appearance
firm to hard, white / chalky, gritty areas (often adjacent to normal fat).
Microscopic appearance
areas of coagulative to liquefactive necrosis of fat tissue often with cholesterol clefts, basophilic calcium
deposits and often surrounded by inflammatory cells.

Gen Path (VPM 152) Cell Pathology 14
2) Apoptosis a) Definition (derived from the Greek = “falling off”)
death of single cells as a result of activation of a genetically programmed "suicide" pathway.
differentiating from necrosis is important, in that necrosis indicates widespread tissue injury due to severe
pathologic stimuli, while apoptosis indicates selective elimination of cells, due to either physiologic or specific
pathologic stimuli.
apoptosis involves death of single cells (or small clusters) with intact cell membranes and rapid removal by
phagocytosis with little inflammation, while necrosis involves locally extensive areas with loss of cell
membrane integrity, enzymatic digestion and an inflammatory response
note, intermediate / hybrid forms of cell death exist that share aspects of necrosis & apoptosis (necroptosis).
b) Causes
seen in many physiologic / adaptive situations or pathologic events (when cells have irreparable damage):
Physiologic
Cells undergoing programmed cell death during embryologic development (eg sculpting of the digits).
Cells undergoing normal turnover
- hormone-dependent involution (eg endometrium, mammary, prostate).
- cell depletion in proliferating population (eg unused neutrophils, balancing #’s of enterocytes in crypts)
Immune System
- depletion of autoreactive T cells in thymus.
- immune regulation (eg lymphocytes at the end of an immune response)
Pathologic
DNA damage - when DNA is damaged beyond repair, eg radiation, toxins, hypoxia.
Misfolded proteins - excess accumulation in ER, beyond ability to adapt, leads to apoptosis.
Specific infectious agents - esp viruses; induced by either direct viral damage or by T-cell response.
Specific immune responses - apoptosis of altered cells by cytotoxic T cells (eg viral infected cells, mutated
tumor cells, rejection of transplant cells).
Pathologic atrophy of organs after duct obstruction (eg pancreas, kidney, salivary gland).
c) Morphologic Features
because the events are rapid, considerable apoptosis may occur in tissues before it evident on histology.
Cell shrinkage - cytoplasm has packed organelles
Chromatin condensation - dense aggregates of chromatin fragmentation
Formation of cytoplasmic blebs then apoptotic bodies with intact membranes ( nuclear fragments)
Phagocytosis of apoptotic cells / cell bodies - usually by macrophages with no inflammatory response
d) Biochemical Mechanisms Signaling Pathways that Initiate Apoptosis - stimulate targets on surface (eg TNF / TNFR) or in the cell.
Control & Integration - balance of pro- & anti-apoptotic molecules determines outcome of affected cell.
Common Execution Phase - actual death program accomplished by proteases (esp caspase family).
Removal of Dead Cells - apoptotic cells have ligands for phagocyte receptors; efficient, no inflammation.
e) Consequence of “too much” or “too little” Apoptosis Disorders associated with defective apoptosis (increased cell survival)
- increased survival of abnormal cells with neoplasia (eg p53 mutation) or increased survival of autoreactive
lymphocytes causing autoimmune disorder.
Disorders associated with increased apoptosis (excessive cell death)
- increased loss of cells in neurodegenerative diseases, ischemic injured cells or viral infected cells.

Gen Path (VPM 152) Cell Pathology 15
PIGMENTS AND OTHER TISSUE DEPOSITS
many pathologic processes are accompanied by accumulations of material either within the cell (intracellular) or
within the extracellular space.
1) Lipid Accumulation
a) Intracellular
types of lipids which can accumulate:
Triglycerides - (see previous discussion of fatty change in reversible cell injury)
Inherited Storage Diseases (Lipid storage disorders)
Cholesterol accumulations
eg: - inflammation and necrosis: foamy macrophages at sites of cell injury and inflammation, ie phagocytosis
of lipid material from dead cells.
- atherosclerosis: accumulation of lipids in smooth muscle cells and macrophages in walls of arteries /
arterioles (in vet med, in some dogs with hypothyroidism / hypercholesterolemia).
- eg xanthomas: tumor like masses in skin formed by clusters of foamy macrophages; seen with inherited
or acquired hyperlipidemic states.
b) Adipose (Fatty) Tissue Infiltration
occasionally seen in skeletal muscle and myocardium (sometimes called muscle steatosis).
microscopically see normal fat tissue, which has replaced lost (congenital or acquired) muscle fibers.
usually not associated with functional disturbance.
2) Glycogen Accumulation
excessive intracellular deposits of glycogen, ie seen with abnormalities of glucose or glycogen metabolism.
eg’s, in renal tubular epithelium with diabetes mellitus, in hepatocytes of dogs with excess corticosteroids (so-
called “steroid hepatopathy”), in various cells with glycogen storage diseases.
microscopically, see poorly delineated vacuoles within cell; to confirm use PAS stain (ie rule-out fat or H20).
3) Protein Accumulation
terminology:
hyaline - is the name given to any substance, intracellular or extracellular, which has a homogeneous, glassy,
eosinophilic appearance; the substance is often protein in nature (eg’s amyloid; Ag-Ab complexes
causing thickened BM’s; protein droplets in renal tubular epithelium).
fibrinoid - is a nonspecific term for hyaline material within an arterial wall.
- the presence of plasma proteins / Ag-Ab / complement within a damaged vascular wall causes
intense eosinophilic staining (often called “fibrinoid necrosis”).
eg’s
reabsorption droplets in renal tubular epithelium: often with glomerular damage protein is lost in the urine,
some of this protein can be reabsorbed by proximal tubular cells; see eosinophilic droplets in the cytoplasm.
"Russell bodies" of plasma cells - a result of immunoglobulin accumulating in the cisternae of the RER.
defects in protein folding can cause intracellular or extracellular accumulations of protein:
a) ER stress / “unfolded protein response”- is induced by accumulation of unfolded & misfolded proteins.
b) aggregates of misfolded proteins (genetic / acquired) - eg amyloid, prions in TSE’s, Alzheimer’s, etc.

Gen Path (VPM 152) Cell Pathology 16
4) Amyloid and Amyloidosis amyloid = a pathologic proteinaceous substance (95% amyloid fibrils) which is resistant to proteolysis.
- amyloid fibrils are insoluble aggregates that result from the self-assembly of abnormally folded proteins
(typically the abnormal folded protein has excess sheet conformational change).
- almost all types of misfolded proteins are degraded intracellularly (proteosomes) or extracellularly
(macrophages); however a few types of protein which misfold with excessive and/or externalized sheet
structures can form amyloid.
- ultrastructurally see masses of linear, nonbranching fibrils 7.5-10 nm in diameter.
amyloidosis = a disorder of protein folding in which normally soluble proteins are deposited as abnormal,
insoluble fibrils that disrupt tissue structure and function.
amyloid can be formed in a variety of pathologic conditions (eg neoplasia, inflammation, genetic) from a
variety of proteins (derived from about 20 unrelated proteins in humans).
in vet medicine, mostly see reactive systemic amyloidosis secondary to chronic inflammatory disease.
in humans most commonly associated with plasma cell neoplasia (less common in vet med).
chemical composition of most common forms of amyloid:
Protein AA - derived from an acute phase protein called serum amyloid A (SAA) in chronic inflammation.
Protein AL - derived from immunoglobulin light chains with plasma cell neoplasia.
Familial amyloid - in humans, Shar pei dogs, Abyssinian cats.
Endocrine amyloid - derived from polypeptide hormones or prohormones in neoplastic or degenerative
conditions, eg islet amyloidosis in cats & humans with type 2 diabetes mellitus.
Other amyloids - eg’s, misfolded prion protein (PrPsc
) in TSE’s; amyloid plaques in Alzheimer disease.
it can be deposited locally or systemically; once deposited is difficult to digest / remove.
grossly: organ involved may be normal or enlarged, and is often firm.
amyloid stains brown/black when Lugol's iodine is applied to fresh tissue.
microscopically:
on H&E staining see lightly eosinophilic, amorphous, hyaline material that is deposited extracelluarly.
on congo red staining amyloid is orange-red & under polarized light shows apple green birefringence.
sites and significance:
renal amyloid in glomerular BM impairs glomerular filtration, causing protein losing glomerulopathy.
amyloid in medullary interstitium impairs medullary function.
other organs (eg liver, pancreas, spleen, vessel walls, skin)
can enlarge, distort and/or compress organ.
can interfere with function (eg blocking space of Disse in liver, obliterating pancreatic islets).
5) Endogenous Pigments
endogenous pigments (colored substances) are those that originate in the animal.
a) Lipofuscin
also known as "wear-and-tear" or “aging” pigment.
origin: breakdown products of lipids, usually derived from cell membranes (esp lipid peroxidation).
sites: aged cells, eg myocardial cells and neurons of old animals, or in chronically injured cells.
grossly: when severe can give yellow-brown discoloration to tissue.
microscopically: golden brown, finely granular, intracellular pigment.
significance: does not injure cell, but is a sign of aging or excess free radical damage.
Lipofuscinosis - disorders characterized by the excess storage of lipofuscin.
eg “brown gut syndrome” of dogs with vitamin E/selenium deficiency increased free radical damage.
grossly the intestine has a distinct yellow-brown discoloration (intestinal lipofuscinosis);
microscopically see increased amount of lipofuscin in smooth muscle cells.

Gen Path (VPM 152) Cell Pathology 17
b) Ceroid
variant of lipofuscin which is acid-fast positive and autofluorescent.
c) Melanin
insoluble, intracellular, brown-black pigment derived from tyrosine.
origin: normal pigment found in epidermis & some cells of the eye.
- in the skin, melanocytes (derived from neural crest cells) produce melanin granules which they inject
into keratinocytes for skin / hair coloration & protection from UV light.
sites: melanin can also occur incidentally at other sites and when in excess called melanosis,
eg leptomeninges (esp ruminants), intestine, kidney, lung, base of aorta, etc.
grossly: melanosis will appear as a dark (pigmented) area of otherwise normal tissue.
microscopically: see finely granular, brown, intracellular pigment.
d) Copper
an essential trace element; the liver is the major organ involved in the regulation of copper levels, and
homeostasis is maintained by the balance of dietary intake and copper excretion via the bile
copper toxicity - common in sheep because of the reduced biliary excretion of copper in this species
- some dog breeds predisposed to inherited copper storage disease.
significance: storage of large amounts of copper is toxic to the hepatocyte and may produce a sudden
onset of acute hepatocellular degeneration (via catalyzing lipid peroxidation).
e) Hemosiderin
origin: represents stored iron (ferric form = Fe3+
), recovered from the hemoglobin of destroyed rbc’s.
- in cells, Fe3+
bound to apoferritin forming ferritin; excess ferritin forms hemosiderin granules.
sites: normal in macrophages of spleen and bone marrow.
- also anywhere there is excessive breakdown of erythrocytes or excessive accumulations of iron, eg,
in macrophages at sites of congestion / hemorrhage (eg bruise)
splenic macrophages and Kupffer cells in hemolytic anemia
alveolar macrophages in congestive heart failure (“heart failure cells”).
grossly: may give a light brown color to the tissues.
microscopically: yellow-brown, granular, intracellular pigment; stains positive with Perl’s Prussian blue.
significance: a mild or even moderate accumulation, usually causes no damage, called hemosiderosis.
- called hemochromatosis when a massive accumulation causes cell damage; common in humans,
rare in other species (eg idiopathic hemochromatosis seen in mynahs and some other tropical birds)
f) Bilirubin
bilirubin is end product of heme degradation (no iron); mostly from senescent rbc’s via macrophages.
origin: heme biliverdin (by heme oxygenase) uncongugated bilirubin (by biliverdin reductase)
binds albumin & transported to the liver conjugated to its glucuronide (water soluble)
excreted in bile.
occurrence:
prehepatic jaundice primarily with increased breakdown of erythrocytes, eg hemolytic disease.
hepatic jaundice with failure of conjugation or excretion.
post hepatic jaundice obstruction of bile duct flow.
sites: in hepatocytes (intracytoplasmic or in canaliculi); in renal tubular epithelium (can be toxic).
grossly: in blood and tissues produces a yellow discoloration, called jaundice or icterus.
microscopically: green-brown to yellow-brown granular pigment in cytoplasm of hepatocytes or
distending bile canaliculi.

Gen Path (VPM 152) Cell Pathology 18
g) Hematoidin
bright yellow-brown homogenous pigment occasionally seen at sites of previous hemorrhage.
believed to be locally precipitated bilirubin (stains negative for iron).
h) Acid-Hematin
blood + acid = acid hematin (black color).
imparts black color to blood originating in stomach (eg gastric ulcers), ie blood + stomach HCl = melena.
is also an artifact in histologic tissue, ie black precipitate on slide, due to improperly buffered (acidic)
formalin reacting with blood from tissues (called “formalin pigment”).
6) Pathologic Calcification (Mineralization)
a) Dystrophic Calcification
calcification of injured cells (no hypercalcemia or other disturbances of calcium homeostasis).
occurrence: - in areas of necrosis (either coagulative, liquefactive or caseous), small membrane
fragments produced during cell necrosis act as a nidus for Ca2+
/PO4 precipitation.
- in cells injured by many mechanisms, eg vascular, toxic, metabolic, inflammation.
- most prominent when there is a good blood supply in the injured tissue.
grossly: white, irregular areas, sometimes dry and gritty, (may not be visible).
microscopically: - intracellular or extracellular, amorphous, basophilic granular deposits.
- stains black with a Von Kossa special stain.
eg’s “white muscle disease” (nutritional myopathy) associated with Vit E / selenium deficiency in farm
animals, tuberculosis nodules, abdominal fat necrosis, etc.
b) Metastatic Calcification
when apparently normal tissue undergoes calcification with hypercalcemia / altered Ca2+
metabolism.
occurrence: - primary hyperparathyroidism
- renal failure / secondary hyperparathyroidism
- vitamin D toxicosis (iatrogenic or plant analogues in cattle; rodenticides in dogs & cats)
- paraneoplastic syndrome (with certain neoplastic diseases); eg, canine lymphosarcoma.
sites: many sites can be affected, especially gastric and intestinal mucosa, interstitium of blood vessel
walls, basement membranes of many tissues (esp lung and kidney).
grossly: if visible, often as white plaques or streaks.
histo: deposits may be irregular / clumped (eg in necrotic tissue) or smooth / linear (eg in BM’s and
elastic fibers), stains basophilic with H&E stain or black with Von Kossa stain.
7) Crystals a) Calcium Oxalate
origin: plants with high levels of oxalic acid, eg halogeton, rhubarb, greasewood.
- antifreeze contains ethylene glycol (via hepatic alcohol dehydrogenase) glycoaldehyde glycolic
acid glyoxcylic acid oxalic acid calcium oxalate (precipitated in lumina of renal tubules).
- many of the chemical intermediates of ethylene glycol metabolism are toxic to renal tubular epithelium
and/or nervous tissue.
occurrence: - in cats and dogs due to ingestion of antifreeze.
- in sheep due to ingestion of poisonous plants.
grossly: kidneys often appear normal or may be slightly swollen.
microscopically: degeneration & necrosis of tubular epithelium and often see refractile, translucent,
yellow crystals (ie calcium oxalate) in renal tubular lumina / interstitium.
significance: severe renal dysfunction & electrolyte imbalances (eg hyperkalemia, acidosis) death.

Gen Path (VPM 152) Cell Pathology 19
b) Urates and Uric Acid
gout: humans: disorder of purine metabolism with hyperuricemia and deposition of urates in tissues.
birds/reptiles: uric acid is the end product of nitrogen metabolism, so any significant renal
dysfunction (including dehydration) can lead to abnormal accumulations/deposits of urates.
occurrence: birds, reptiles and humans.
sites: - in and around the joints, called articular gout.
- on serosal surfaces or in tissues, called visceral gout.
grossly: chalky white masses in tissues (called "tophi") or “frosting” of crystals on serosal membranes.
microscopically: variably sized clear crystals, frequently associated with macrophages.
c) Cholesterol Clefts
occurrence: any tissue following severe necrosis or hemorrhage (from cell membranes), esp fat necrosis;
also see in metabolic disorders of cholesterol accumulation (eg atherosclerosis, xanthomas).
microscopically: see elongate, needle-like crystals, often in picket fence-type arrangement.
8) Exogenous Pigments exogenous pigments are those that originate outside the body, ie from external environment.
eg, carbon, soot, dusts, silica, asbestos, tattoo pigments, etc.
a) Anthracosis
deposition of carbon particles (esp from air pollution), into lungs / lymph nodes.
gives tissue a black discoloration to tissue; see in macrophages in lungs and draining lymph nodes.
relatively harmless unless present in large quantities (eg chronic lung injury in coal miners).
b) Silicosis
deposition of silica dust in the lungs, is a special problem for miners; causes granulomatous pneumonia.
c) Asbestosis
deposition of asbestos into lung, associated with mesotheliomas, and chronic lung injury.
9) Parasite Pigments
several parasites are commonly associated with pigmented material, eg’s,
Pneumonyssus simicola - brown to black pigment in the airways of monkeys with lung mites.
Plasmodia - malarial pigment from excretion of catabolized hemoglobin (large deposits in
macrophages of spleen and liver).
Fascioloides magna - liver fluke of ruminants; see black discoloration of tissue around bile ducts due
to regurgitated iron-porphyrin pigment (“fluke puke”).

Gen Path (VPM 152) Cell Pathology 20
POST MORTEM ARTIFACTS [for information only - see also PM artifacts slideshow on website]
“Once you know the normal and the artifacts, all the rest is pathology!!” Dr. T Van Winkle
1) Postmortem Scavenging vs Predation
postmortem scavenging = removal of organs (esp eyes, tongue, rectum) of carcass by carrion eating animals
(eg dogs, coyotes, ravens, vultures, etc).
2) Rigor Mortis
contraction of muscles after death.
usually within 1-6 hrs after death (depending on glycogen stores, ambient temperature, etc) & lasts 1-2 days
after death, circulation of blood ceases muscle cells resort to anaerobic glycolysis when glycogen stores
run out & ATP becomes depleted (needed for muscle relaxation) Ca2+
floods into muscle cells causing
myofilaments to contract (note: all muscles affected, flexors / extensors, causing rigidity of joints) rigor
mortis gradually dissipates with autolysis of structural and functional muscle proteins.
3) Algor Mortis
gradual cooling of the cadaver to ambient temperature (in humans 1.5oF per hr).
4) Livor Mortis (hypostatic congestion)
gravitational pooling of blood to the dependant regions ("down side") of the body.
5) Postmortem Clotting
occurs in heart and vessels.
rbc’s may separate from plasma (esp in animals with high fibrinogen levels, eg horses) = “chicken fat clot”
6) Hemoglobin Imbibition
HgB released by rbc breakdown (after death) staining tissues.
especially lining of heart and blood vessels; also common in tissues of aborted fetuses and frozen tissues.
7) Bile Imbibition
leakage of bile from gall bladder and major bile ducts which stains adjacent tissues green to yellow.
8) Pseudomelanosis
term used to describe an artifactual black discoloration of tissues (similar in an appearance to melanosis).
due to saprophytic/putrefactive bacterial production of hydrogen sulfide (H2S) + iron iron sulfide (FeS).
9) Postmortem autolysis / decomposition
after death, decomposition of tissues begins by progressive release of endogenous enzymes (autolyis) and more
gradually by the action of saprophytic bacteria (especially from gut).
rate of progression dependent upon several factors, eg body temperature at time of death; ambient temperature;
size of body; amount of fat/hair/wool; cause of death (eg bacterial infection, hyperthermia, etc).
grossly the tissues become progressively more pale (often mottled) and soft.
10) Putrefaction and Postmortem Emphysema (bloating)
putrefaction (rotting), refers to the enzymatic decomposition of organic material (tissue) with production of
foul-smelling compounds (eg H2S, NH3, mercaptans), especially by saprophytic / putrefactive bacteria.
postmortem emphysema occurs when saprophytic bacteria produce gas, causing gaseous distention of G-I tract,
organs and body cavities.
11) Postmortem Ruptures and Organ Displacements
with swelling of viscera (due to postmortem emphysema) rectal prolapse, visceral dislocation, gastric
rupture (horses), diaphragmatic hernia.
12) Other Abnormalities
rib indentations on pulmonary pleura.
pale discolored compressions on capsular surface of liver from distended intestine loops and/or rib imprints.
euthanasia with barbiturate splenomegaly, accumulation of fluid and blood in lung, froth in trachea.
lens opacity in frozen carcasses.


















