Part 2 of RNA-seq for DE analysis: Investigating raw data
56
This presentation is available under the Creative Commons Attribution-ShareAlike 3.0 Unported License. Please refer to http://www.bits.vib.be/ if you use this presentation or parts hereof. RNA-seq for DE analysis training Raw data investigation Joachim Jacob 22 and 24 april 2014
-
Upload
joachim-jacob -
Category
Science
-
view
396 -
download
1
description
Second part of the training session 'RNA-seq for Differential expression' analysis. We explain the characteristics of RNA-seq data that allow us to detect differential expression. Interested in following this session? Please contact http://www.jakonix.be/contact.html
Transcript of Part 2 of RNA-seq for DE analysis: Investigating raw data

This presentation is available under the Creative Commons Attribution-ShareAlike 3.0 Unported License. Please refer to http://www.bits.vib.be/ if you use this presentation or parts hereof.
RNA-seq for DE analysis training
Raw data investigation
Joachim Jacob22 and 24 april 2014

2 of 56
Previous section: experimental setup
We have decided on:● how many samples per condition● how deep
This determines how reliable the statistics will be, using experience, and tools like Scotty. A wrong experimental design cannot be fixed. Best approach: pilot data (3 samples per condition, 10M)
But we have other sequencing options to choose!

3 of 56
PE versus SE Illumina● Single end (SE): from each cDNA fragment only
one end is read.
● Paired end (PE): the cDNA fragment is read from both ends: 2 reads per fragment, a given distance apart.
Purify and fragment,..
PE
SE
Part that is read

4 of 56
PE versus SE Illumina
Single end (SE):
● Gene level differential expression
Paired end (PE):
● Novel splice junction detection
● De novo assembly of transcriptome
● Helps with correctly positioning reads on the reference genome sequence.
Note: PE not the same as mate pairs.

5 of 56
Strandedness
● Naive protocols obtain reads from cDNA fragments. BUT the link with the sense or antisense strand is broken.
● Stranded protocols generate reads from one strand, corresponding to the sense or antisense strand (depending on the protocol).

6 of 56
Strandedness
Not strandedStranded

7 of 56
Example of a stranded protocol
● dUTP protocol to generate stranded reads.

8 of 56
Importance of strandedness
● Strandedness can bias the read counts compared to non-stranded protocols.
● Depends on the genome whether you should apply it, e.g. in case genes overlap, the improved benefit of assigning reads to correct genes can outweigh technical variation.

9 of 56
Required length of the reads
● Does not matter so much (when we want to quantify aligning to a reference sequence): 50 bp will do.
● The most important point is to be able to accurately position the read on the reference genome sequence, to assign it to the correct gene.
● Length can become important, if you want to assemble the transcriptome.

10 of 56
For DE on the gene level
The 'cheapest' protocol for high-throughput sequencing suffices to achieve DE detection:● SE● 50bp● Option: strandedness.
Use the money you have left over for increasing the number of biological replicates.

11 of 56
First mRNA is extracted
sdf

12 of 56
After sequencing, you get raw data
The data you get arrives as...
barcode
experiment
Compressed, usually with gzip

13 of 56
Raw Illumina data
@HWI-ST571:202:D1B86ACXX:2:1102:1146:2155 1:N:0:ACAGTG
CCAACATCGAGGTCGCAATCTTTTTNANCGATATGAACTCTCCAAAAAAA
+
@@@FFFDFHHDG?FFHIIJJJJJIJ#1#1:BFFIGJJJJJIJJGIJJJJA
@HWI-ST571:202:D1B86ACXX:2:1102:1073:2240 1:N:0:ACAGTG
CGGAGCTGAAGGAGAAACTGAAATCCCTGCAATGTGAATTGTACGTTCTT
+
CCCFFFFFGGHHHIJJJJJJJIJFHIJIIIJJJJGIIIIIEFGHIFCHJI
@HWI-ST571:202:D1B86ACXX:2:1102:1385:2192 1:N:0:ACAGTG
GTTGGCAGCCCTGGAGCCCTGCCTCGGTGGTTTAGCCAGTACTAGGGGAT
+
CCCFFFFFHHHHHJJJIJJJJJJGIJJCGHFHIGIHJJJBDHGHHJJJIE
@HWI-ST571:202:D1B86ACXX:2:1102:1352:2244 1:N:0:ACAGTG
ATTTCCTCTTATTTACGTTGCTTTAAAGCGAGACTTCAACGCCATTTGAC
+
@@CFFFFFHHFHDFGHIJIIJGIJGGEHGGJB>??FHHGFFFGHIGIECF
@HWI-ST571:202:D1B86ACXX:2:1102:1981:2152 1:N:0:ACAGTG
CATCGAAGCAAAGCATATAAAGTTANTNNTNNCTGAGTTGTACATATTGC
+
??;;D?DB6CDB+<EFE>:AFA443#2##1##11)0:0?9**0??DAGI4
@HWI-ST571:202:D1B86ACXX:2:1102:1877:2165 1:N:0:ACAGTG
GAAGTGCCCCGCTGGCAGCACACAAGGAGCAGCCCGCTGCCGGACCACTC
+
?@@DDDADFFAA:CEGHBFGAHGD?F@BE9BFF?D@F;'-8AG<B92=;;
One read (minimum 4 lines)
http://wiki.bits.vib.be/index.php/.fastq
sequence
certainty reading this base at this position ('quality')
(this one: 87196924 lines)

14 of 56
Exploring the raw data
1) check whether the Fastq file is consistent-
2) Make graphs of some metrics of the raw data
http://wiki.bits.vib.be/index.php/.fastq
http://wiki.bits.vib.be/index.php/RNAseq_toolbox#Quality_control_and_visualization_of_raw_reads

15 of 56
FastQC – graphical exploration
http://www.bioinformatics.babraham.ac.uk/projects/fastqc/

16 of 56
FastQC – perfect example
Reads have good quality!

17 of 56
FastQC – perfect example
Anna Karenina principle: “There is only one way to be good, but there are many ways to be wrong.”
We will start by showing a good sample. Afterwards we will discuss a less good sample.
http://en.wikipedia.org/wiki/Anna_Karenina_principle

18 of 56
FastQC – perfect example
Smooth histogram/ density line towards the right,

19 of 56
FastQC – perfect example
steady nucleotide distribution.
Bias typical for illumina

20 of 56
Not strongly fluctuating GC content
Bias typical for illumina
FastQC – perfect example

21 of 56
GC-content nicely bell shaped
FastQC – perfect example

22 of 56
No N's! (should ring something)
FastQC – perfect example

23 of 56
All reads have length 50bp,
FastQC – perfect example

24 of 56
Reads are nicely duplicated: some amount of duplication is to be expected in RNA-seq data.
FastQC – perfect example

25 of 56
Reads are nicely duplicated: some amount of duplication is to be expected in RNA-seq data.
FastQC – perfect example

26 of 56
Kmers are short sequence stretches. Sometimes they are overrepresented. But in RNA-seq this is not so important (duplication).
FastQC – perfect example

27 of 56
FastQC – less good RNA-seq sample
A relatively large Portion of the reads have mistakes at the 3' end of the read.

28 of 56
FastQC – less good RNA-seq sample
There is an over- representation of reads
with a low mean quality score

29 of 56
FastQC – less good RNA-seq sample
Not a steady levelof different nucleotide
fractions

30 of 56
FastQC – less good RNA-seq sample
Fluctuates

31 of 56
FastQC – less good RNA-seq sample
Heavily skewed versusAT rich reads

32 of 56
FastQC – less good RNA-seq sample
Apparently a mixture of two sets of reads
with different lengths

33 of 56
FastQC – less good RNA-seq sample
Duplication seems abit on the low side
(reported figures are from 60 -75%)

34 of 56
FastQC – less good RNA-seq sample
Very highly skewed read number.Often the sequence of Truseq adaptor, or multiplex identifierscan be found here. BLAST can revealmore information!

35 of 56
FastQC – less good RNA-seq sample
Specific patterns of Specific kmers.
Note: A and T rich

36 of 56
Quality control of raw data
Proceed? Or rerun?
This QC can guide you to which preprocessing steps you need to apply for sure. The extra time and money needed to correct the biases (and still obtain less then optimal results...) can sometimes justify a rerun of the experiment.
This QC shows which preprocessing steps have already been made by the sequencing provider.

37 of 56
Preprocessing: what does it takes
Removing unwanted sequences or parts hereof ('contamination') so it helps as much as possible with reaching our goal: defining differentially expressed genes.
1) removing contamination of technical sources● Low quality read parts● Technical sequences: adaptors● PhiX internal control sequences
2) removing contamination with biological sources● polyA-tails● rRNA sequences● mtDNA sequences
After this, run FastQC again!

38 of 56
Focus on technical contamination
We need to assign reads with a high confidence to the correct genomic location.
We remove low quality read parts: they have a higher chance to contain errors. Errors can lead to wrong alignment, hence noise in our read counts.

39 of 56
Remove low quality read: tools (1)
Our goal is to define DE expression, for this we need to assign reads with a high confidence to the correct genomic location.
Removal of low quality read parts: they have a higher chance to contain errors, and cause noise in our read counts.

40 of 56
Remove low quality read: tools (2)

41 of 56
Technical contamination
We need to assign reads with a high confidence to the correct genomic location.
Removal of adaptor sequences (and other technical sequences, such as multiplex) as they cannot be mapped to the reference genome.

42 of 56
Technical contamination
Our goal is to define DE expression, for this we need to assign reads with a high confidence to the correct genomic location.
Removal of adaptor sequences (and other technical sequences, such as multiplex) as they cannot be mapped to the reference genome.
List of technical sequences
Advised by FastqMcf to use defaults
http://code.google.com/p/ea-utils/wiki/FastqMcf

43 of 56
Fastq-mcf output
http://code.google.com/p/ea-utils/wiki/FastqMcf

44 of 56
Duplicates are not contamination
● Never remove duplicate reads! Highly expressed genes can have genuine duplicate reads, which are not due to the PCR amplification step in the protocol.
● PhiX sequences: the DNA of Phi X bacteriophage is spiked in to monitor and optimize sequencing on Illumina machines. Your sequencing provider should filter out those sequences before delivery. If they are still present, you can filter them out by aligning your reads to the PhiX genome.
http://en.wikipedia.org/wiki/Phi_X_174

45 of 56
Biological contamination: what's in it
Mitochondria containrRNA, mRNA and mtDNA
cell
rRNA and non-coding (95% of RNA)
mRNA (5% of RNA)
nucleus

46 of 56
Biological contamination (1)
mRNAs are captured with oligo-dT coated beads.
Occasionally, non-protein coding sequences are also captured (especially since mtRNA and rRNA can be relatively rich in AT).
We can remove them via homology searching (BLAST) with known non-protein coding sequences.
Mitochondrial
mRNA (5% of RNA)
rRNA and nc

47 of 56
Biological contamination (2)
mRNAs are post-transcrip- tionally modified: e.g. the addition of a poly-A tail. If our goal is to map the reads to a reference genome sequence, the polyA tails should be removed. This can be viewed as some source of 'biological contamination' in our sequences.
AAAAAAAAAAAAA

48 of 56
● Get the non-protein coding sequences via Biomart, and also mitochondrial genome sequence.
Biological contamination: howto

49 of 56
Biological contamination

50 of 56
Biological contamination

51 of 56
Filter the biological contamination
Your reads
The biological readsImported via Biomart
We are interested in the reads that don't map!

52 of 56
Doing this in Galaxy
PRO TIP: take a sample of your reads with the tools: fastq-to-tabular, select random lines, tabular-to-fastq
1. create a new history and load the sample data in3. Run fastqMcf to remove technical sequences4. Run bowtie to match against biological sequence databases you just fetched, and keep reads that don't match.5. Summarize: fastqc
→ create a workflow of this, and run it on all your samples in parallel

53 of 56
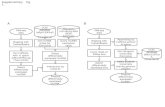
Summary preprocessing
Your reads
…...Format consistent? Errors in quality?
Your groomed reads
…....…... Trends in raw data? QC report
Your groomed reads without technical contamination
….... ... Get biological contaminants- ….- ….
Your groomed reads without technical and biological contamination
…... How does your data look now? QC
... Get technical contaminants- ….

54 of 56
KeywordsPaired end
Stranded reads
gzip
fastq
Biological contamination
Technical contamination
Adapter sequence
Write in your own words what the terms mean

55 of 56
Exercise
→ investigating and preprocessing raw RNA-seq data

56 of 56
Break