
Panagiotis Katsaros [email protected] - delab.csd.auth.gr/~katsaros
Upload
allegra-yatesCategory
view
29download
1description

Panagiotis Papapetrou*, Gary Benson** and George Kollios*
*Department of Computer Science**Departments of Biology and Computer Science
Boston University
Discovering Frequent Poly-Regions in DNA Sequences

Introduction and Motivation (1/3) In cells, DNA forms up long chains made up of four chemical units, known as
nucleotides.
A number of important regions (known functional regions) at both large and
small scales, contain a high occurrence of one or more nucleotides (called poly-
regions).
Example:
Poly-A: a region “rich” in nucleotide A.
Poly-(C,T): a region “rich” in nucleotides C and T.
Many methods have addressed the problem. However, they only focus on
specific types of poly-regions.

Introduction and Motivation (2/3) Isochores:
Multi-megabase regions of genomic sequence.
Specifically, GC-rich or GC-poor.
CpG islands:
Regions of several hundreds of nucleotides rich in the dinucleotide
CpG.
The level of methylation of the cystine (C) is associated with gene
expression in nearby genes.
Protein binding regions: Tens of nucleotides long.
DNA flexibility: dinucleotide, base-step composition.
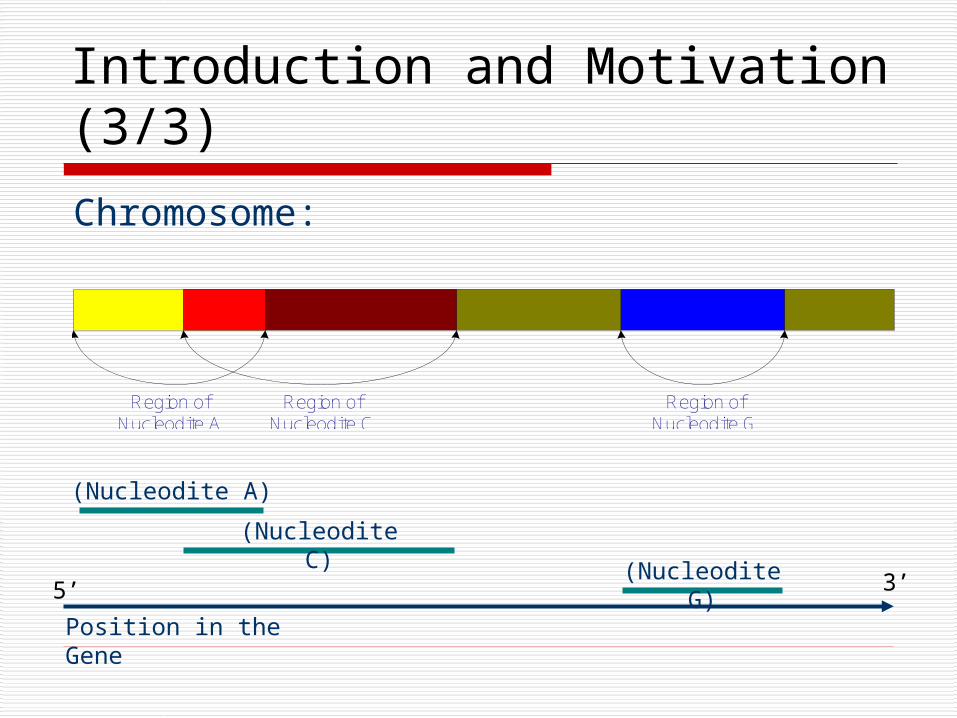
Chromosome:
Region ofNucleodite A
Region ofNucleodite G
Region ofNucleodite C
(Nucleodite C)
(Nucleodite G)
(Nucleodite A)
Position in the Gene
Introduction and Motivation (3/3)
5’ 3’

Related Work Statistical Methods:
Maximum Likelihood Estimation (MLE) of segments
(Auger et. al. 1989, Bement et. al 1977, Fu et. al.1990).
Hidden Markov Chain Model (Churchill et. al. 1992).
Walking Markov Model (Ficket et. al. 1992).
Change-points:
(Carlstein et. al. 1994, Braun et. al. 1998, et. al. 2000).
Hierarchical Segmentation
(Grosse et. al. 2002, Galvan et. al. 2002 Zhang et. al. 2005).

Main Contributions Formal definition of the problem of detecting poly-regions in a
sequence.
Application of an existing recursive segmentation technique to solve
the problem.
Development of an efficient algorithm based on multiple sliding
windows.
Application of an efficient arrangement mining algorithm to extract
the complete set of frequent arrangements of these regions.
Extensive experimental evaluation of our algorithms on the dog
gemone.

Preliminaries (1/4)
SequenceSequence: SS = { = {ss11, , ss22, …, , …, ssmm}, an ordered set of items, }, an ordered set of items,
defined over an alphabet.defined over an alphabet.
In our case, In our case, ssii corresponds to a nucleotide.
k Poly-Region: Hd,k = {I, pstart, pend}
k: number of items in the region.
d: density of the region.
starts and end with one of the k items.
each of the k items has at least (d/k)% frequencuy in the region.

Preliminaries (2/4)
Example of a k Poly-Region:
AACAAGAAAA AACAATCGCC
5 14 20 29
(1) (2)
(1) poly-A A: 8/10
(2) poly-(A,C) A: 4/10
C: 4/10

Preliminaries (3/4)
Different types of relations1 can occur among Poly-Regions.
A[tstart, tend] B[tstart, tend]
(a) Meet of A and B
A[tstart, tend]
B[tstart, tend]
(d)
A[tstart, tend] B[tstart, tend]
(g)
Contain of A and B
Follow of A and B
+/- e
A[tstart, tend]
B[tstart, tend]
(e) Left Contain of A and B
A[tstart, tend]
B[tstart, tend]
(f) Right Contain of A and B
+/- e +/- e
A[tstart, tend]
B[tstart, tend]
(c) Overlap of A and B
A[tstart, tend]
B[tstart, tend]
(b) Match of A and B
+/- e+/- e
1. J. F. Allen and G. Ferguson. “Actions and events in interval temporal logic”. Technical Report 521, The University of Rochester, July 1994”.

Preliminaries (4/4) k-Arrangement:k-Arrangement: a set of kk temporally correlated events in an
e-sequence, denoted as A = {EE , R}, where: E E : the set of labels of the event intervals in the arrangement.
R R : the set of temporal relations between the events in E.
)}E ,(Er ... ),E ,(Er ..., ),E ,(Er ),E ,(Er ..., ),E ,(Er ),E ,(E{r R n1-nn232n13121
}, |, ||, { )E ,(Er ji A
B
C
} }|, |, { C},B,{A, {
E1 Ei Ei+1 EnEi+3Ei+2Ei-1
where is the temporal relation between EEii and EEjj.

Problem Statement1. Given:
A sequence database S.S.
A minimum density constraint d.d.
A range [min, max].
Find the complete set of maximal poly-regions HH of size [min, max], and
density of at least d d %.
2. Given:
The set of maximal poly-regions H. H.
A minimum support threshold min_sup.
Extract the complete set of frequent arrangements of poly-regions in H.H.

Recursive Segmentation (1/2)
Recursively segment the sequence
Homogeneity difference between each segment in
maximized with respect to a measure λ.
In our case we use the Jensen-Shannon Entropy (JSE).
Split point is chosen, where JSE is maximized.
Segmentation of a subsequence is stopped when
minimum poly-region size is reached.
Expand each segment to define poly-regions.

Recursive Segmentation (2/2)
To improve the efficiency of the segmentation:
When looking for H-regions of two nucleotides replace the
rest of the nucleotides with a single literal.
Example:
S = AAACCCAGGTAGCT
Looking for poly-(A,C):
Snew = AACCCAXXXAXCX

Sliding Windows (1/3) Define a set of sliding windows W = {w1, w2, …, wN} over the
sequence
# of windows: N = max – min + 1.
size of window i : min + i -1.
Each window keeps statistics of a segment:
For each nucleotide: the # of occurrences in the segment.
Each window w = {C, Start, End}
C: set of statistics.
Start: pointer to the start point of the segment in the sequence.
End: pointer to the end point of the segment in the sequence.
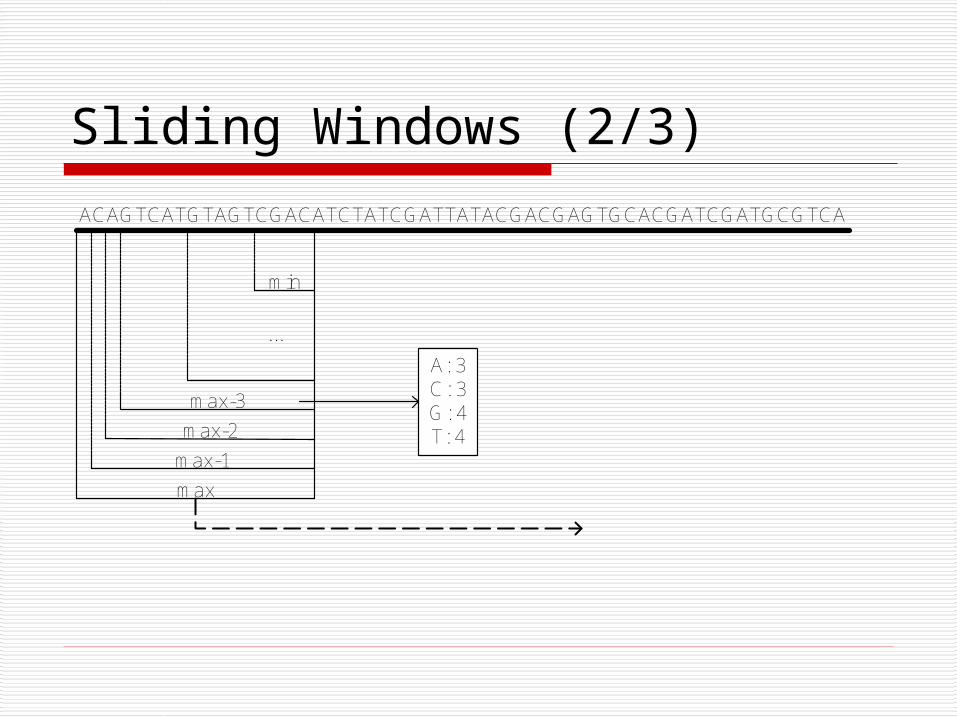
Sliding Windows (2/3)
...
max
max-1
max-2
max-3
min
ACAGTCATGTAGTCGACATCTATCGATTATACGACGAGTGCACGATCGATGCGTCA
A: 3C: 3G: 4T: 4

Sliding Windows (3/3) Heuristic:
NCi = # of items of type C in window i.
C is dense in wi if NCi / |wi| >= d.
Observe: the maximum size of the window where items of type C
can fit and fulfill the density constraint is NCi / d.
This indicates which windows of the lower level should be
searched for a candidate poly-region.
Start with the window of size max, and for each literal apply the
heuristic.
Move to the lower levels.

Frequent Arrangement Mining Algorithm
Use a sliding window W of size M>>max.
At each position of W find the set of arrangements in W.
Keep a global frequency of all arrangements.
Update after each slide.
For the enumeration, the arrangement enumeration tree
is used.

Experimental Setup
39 Chromosomes.
Organism: Canis Familiaris (Dog).
Two phases: Extract poly-regions.
Discover frequent arrangements of poly-regions in the
DNA sequences.
Density constraint d varied between 40-80%.
H-region size varied between 8-64 nucleotides.

Performance Analysis
Recursive Segmentation: Has accuracy of 85-90%.
Performs better in smaller sequences.
Sliding Window: Extracts the complete set of poly-regions.
Faster in terms of run time.
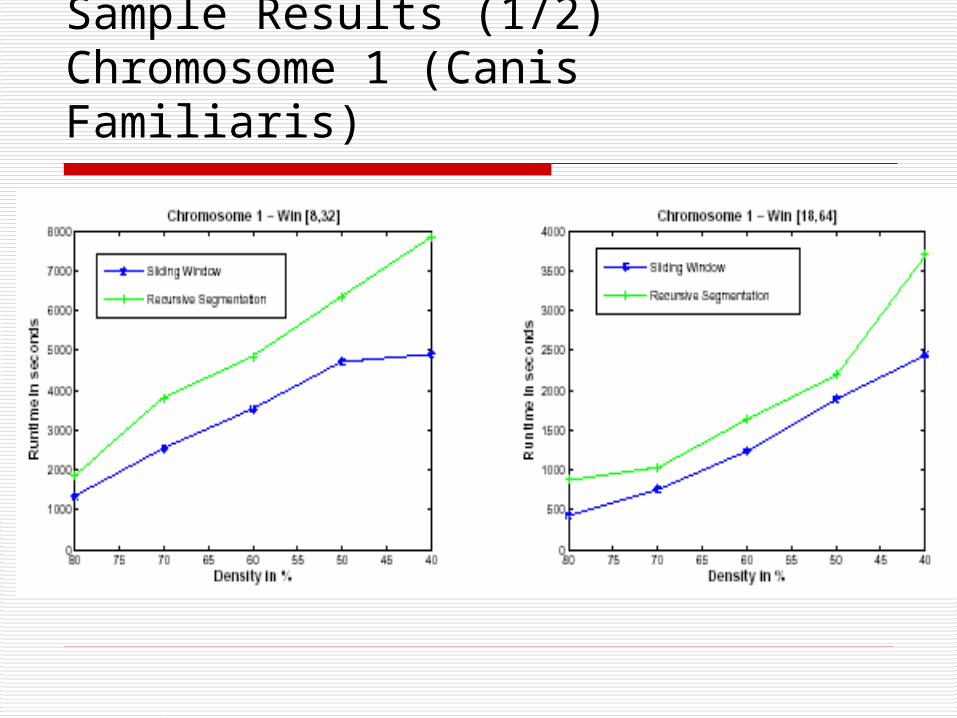
Sample Results (1/2) Chromosome 1 (Canis Familiaris)
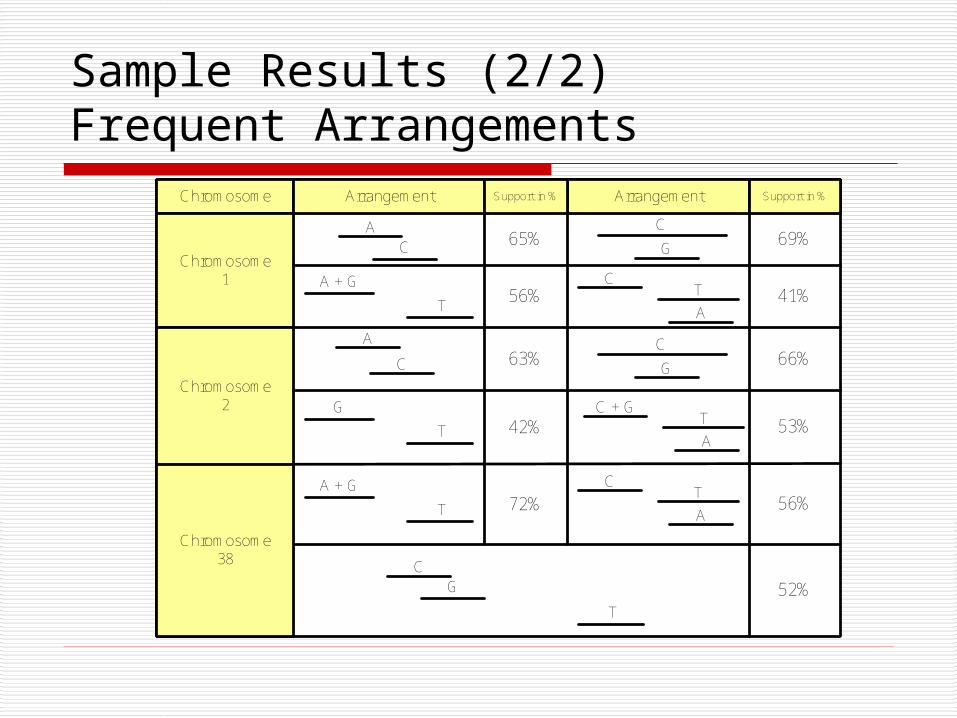
Sample Results (2/2) Frequent Arrangements
Chromosome Arrangement ArrangementSupport in % Support in %
Chromosome1
Chromosome2
Chromosome38
A
A + G
T
C
A
C
G
T
A + G
T
C
G
C
A
T
C
G
C + G
A
T
C
A
T
CG
T
65%
56%
63%
42%
72%
69%
41%
66%
53%
56%
52%

Conclusions The problem of discovering poly-regions and their
frequent arrangements in DNA sequences has been introduced.
Two efficient methods for solving the problem have been discussed. Recursive Segmentation: approximate. Sliding Windows: exact.
An efficient algorithm for mining frequent arrangements of intervals has been applied to the extracted poly-regions.

Future Work
Generalize the definition of a poly-region.
Poly-regions of dinucleotides/trinucleotides.
Poly-patterns.
Detect arrangements of poly-regions that occur
frequently over:
Coding regions (Genes).
Nucleosomes.


















