
CJBONLINE.NL pc-hulp Organisatie Computer & Windows (Uitgangspunt is Windows-XP SP2/feb2006)
Upload
hoangkhanhCategory
view
221download
0
CA-Sept12-Doc.6.2.a -Final
PA&MRFG-Sept12-Doc.5 – Final
Rev. 2 (28/01/2013)
EUROPEAN COMMISSIONDIRECTORATE-GENERALENVIRONMENTDirectorate D - Water, Marine Environment & ChemicalsENV.D.3 - Chemicals, Biocides and Nanomaterials
Technical Notes for Guidance
Notes for guidance to applicants for product authorisation and mutual recognition
Update of contents:
Revision number Content Page(s) DateRev.1 Annex II - Poland 65-69 08/10/2012Rev.2 Annex II - Luxembourg 57-58 28/01/2013
Commission européenne, B-1049 Bruxelles / Europese Commissie, B-1049 Brussel - Belgium. Telephone: (32-2) 299 11 11.E-mail: [email protected]

NOTES FOR GUIDANCE TO APPLICANTS FOR PRODUCT AUTHORISATION AND MUTUAL RECOGNITION
1. General Principles.......................................................................................5
1.1. Who can apply for authorisation?........................................................5
1.2. When to apply for authorisation?.........................................................5
1.2.1. Existing biocidal products:.....................................................5
1.2.2. New Products.........................................................................6
1.3. Issues to consider before preparing application....................................7
1.4. Structure of application and related dossier.........................................9
1.4.1. General requirements for documentation................................9
1.4.2. Practical details on submission of the application.................11
1.4.3. General principles on the submission of experimental studies11
1.4.4. Risk assessment to be prepared by the applicant...................122. The mutual recognition process ..............................................................14
2.1. Role of the Reference Member State.................................................14
2.2. Involvement of the Concerned Member States.....................................14
2.3. Negative conclusions............................................................................14
2.4. Procedural steps in the MR procedure..................................................153. Phasing-out................................................................................................ 174. Frame formulations..................................................................................17Annex I. Deadline for the submission of applications for product
authorisation or for mutual recognition..................................................18Annex II. Further details on what information is expected and when, as well
as explanations on specific national provisions are provided hereafter. 19Annex III. Common approach on the issue of provisional authorisation......86Annex IV. Standard format to facilitate the comparison between the original
protected data of reference dossier and the new data of the alternative supplier dossier.......................................................................................... 88
Annex V. Applicability of Good Laboratory Practice (GLP).........................89Annex VI. Frame formulations under Directive 98/8/EC...............................97Annex VII. Letters of access...........................................................................106Annex VIII. Products belonging to several product types............................112
2

Introduction
Article 16(3) of the Directive requires the Member States shall ensure that, following the decision by the Commission to include or not to include an active substance into Annex I or IA, authorisations or, where relevant, registrations for biocidal products containing the active substances and complying with the provisions of the Directive are granted, modified or cancelled as appropriate.
The article does not give the Commission the power to adopt legal measures that would oblige the Member States to require submission of dossiers for product authorisation or to withdraw products for which no dossiers have been submitted by a given date. Any such actions must come from the Member States themselves.
During the discussions at the 19th and 20th CA meetings, Member States however agreed that they should all strive to act in a harmonised way to set deadlines by which:
i. applications for authorisation should be received,
ii. unauthorised products be removed from the market,
iii. authorisations should be granted.
The key principle is that the application for the authorisation of a product already placed on the market should be submitted before the formal inclusion in Annex I or IA of the active substance it contains. Otherwise products should be removed from the market.
Applicants are given a period of two years to prepare their dossiers for product authorisation and Member States then have another period of two years to assess these dossiers and come to a decision. That second two-year period is again subdivided in a first period of 15 months during which the Reference Member State shall assess the dossier and another period of 9 months for the process of mutual recognition (see diagram below).
3

The purpose of these notes for guidance is to provide up-to-date advice to applicant on the procedures and administrative details of applications for product authorisation and mutual recognition. For that purpose, Members States are invited to report to the Commission any relevant information to be updated in this document.
These notes are the fruit of discussions within the product authorisation and mutual recognition group (PA&MRFG) and are based on a series of agreement between the Member States, to which they might have subscribed to in full or in part.
Applicants should therefore also consult the different annexes of the document for more concrete information and possible deviations in the different Member States. For more specific questions, they can also request additional information to the relevant contact person in each Member State1.
1 An updated list of the PA&MRFG members is available on CIRCABC.4

1. General Principles
1.1. Who can apply for authorisation?
Application for authorisation shall be made by, or on behalf of, the natural person or legal entity responsible for the first placing of the product on the market of the Member State concerned. The applicant may be either native or foreign but shall have a permanent office within the European Union.
The Applicant may be
o the future authorisation holder or
o person/entity handling practical issues related to the application procedure on behalf of the future authorisation holder.
The Authorisation holder is the person/entity to whom the decision on authorisation is issued to. Responsibility for the placing on the product on the market, classification and labelling etc. always lies on the authorisation holder.
1.2. When to apply for authorisation?
1.2.1. Existing biocidal products:
Existing Biocidal products are those products which will have already been placed on the market of the relevant Member State (as opposed to the EU market as a whole) at the date of inclusion of the substance into Annex I or IA. The concept of existing biocidal product is therefore seen at Member State level.
Either notification numbers, registration or authorisation numbers are considered sufficient evidence that a given product is an existing biocidal product in a Member State territory.
Where the product in question is not covered by a notification, registration or authorisation scheme in the territory of a Member State then, some other form of proof (e.g. sales invoice) is likely to be required to demonstrate that the product in question is an existing biocidal product at time of formal Annex I or IA inclusion.
For existing biocidal products, applicants shall submit:
either an application for authorisation complying with the requirements of Article 8 of the Directive;
or an application for mutual recognition in accordance with Article 4 of the Directive2;
by the date of inclusion of the active substance contained in that product in Annex I or IA . 2 Alternatively, some Member States, such as AT, PT or SK, accept a letter of intention that an
application for mutual recognition will be submitted within 2 months of obtaining a first authorisation in the Reference Member State.
5

If a product contains more than one active substance the deadline for the product application shall be the latest of the inclusion dates relating to its active substances
The inclusion dates can be found in the inclusion directives available on the Commission website.
Please note that for products belonging to several product types, the approach is different.
Where a product belongs to several product types, authorisation for each product type shall be applied for separately and before the deadline given for the relevant product type.3
Those products, for which an application for either authorisation or mutual recognition has been made in due time, can then remain on the market in accordance with existing provisions in the Member States, while the applications are evaluated.
If no application is received by the date of the Annex I or IA inclusion, the product shall be phased out (see below).
1.2.2. New Products4
1.2.2.1. Containing new active substances
For biocidal product containing a new active substance, which is not yet included in Annex I, applications for authorisations can be submitted at any time.
However, it is only after the inclusion in Annex I or IA of the new active substance, that the product can be authorised and placed on the market.
A provisional authorisation5 can nonetheless be granted in accordance with Article 15(2) of the Directive, which would enable the authorisation holder to place its product on the market before the formal inclusion of the active substance in Annex I or IA. Otherwise, the placing the product on the market is not allowed before authorisation.
Once the first authorisation is obtained, companies can ask for mutual recognition in all other Member States where they wish to place the product on the market.
1.2.2.2. Containing existing active substances
Products containing existing active substances for a product type not covered by the review programme of existing active substances will be treated in the same way as products containing new active substances.
3 Further information on biocidal products belonging to several product-types is available from Annex VIII.
4 Those products which will not have already been placed on the market of the relevant Member State at the date of inclusion of the active substance into Annex I or IA for the relevant product type. For products with more than one active substance, those products which will not have already been placed on the market of the relevant Member State at the date of inclusion of its last active substance into Annex I or IA for the product type.
5 See Annex III.6

The active substance will have to be included in Annex I or IA for the relevant product type before the product can be authorised and placed on the market.
For these products it is however not possible to grant a provisional authorisation.
1.3. Issues to consider before preparing application
There are a number of important elements to consider before preparing an application for a biocidal product:
Applicants – if they are not owners of the data submitted for the purpose of the inclusion in Annex I or IA of the active substance – will be required to provide information to demonstrate access to the required data on the active substance. This may be achieved in a number of ways:
o By reference to information previously submitted on the active substance to support inclusion in Annex I or IA, which is not protected in accordance with Article 12 of Directive 98/8/EC.
o By providing evidence of access6 to the information submitted to support inclusion in Annex I or IA, which is protected in accordance with Article 12 of Directive 98/8/EC7.
o By providing alternative and equivalent studies, including published studies, to those protected. In this case, a complete dossier ("alternative supplier" substance dossier) satisfying the requirements of Annex IIA and IIIA has to be submitted.
o By providing a case justifying why certain data are not relevant to the uses which are claimed to be supported.
If applicants are the owner of an alternative supplier dossier, they are invited to contact the future evaluating Competent Authority to verify whether it would like to receive this dossier in advance of the application for product authorisation – in order to ensure that the dossier complies with the requirements of Article 8(2)(b) and prevents unnecessary delays during the product evaluation.
A standard format to facilitate the comparison between the original and the new data is given at Annex IV and shall be used by applicants submitting an 'alternative supplier' dossier on the active substance.
Additional information related to alternative supplier's dossiers is available on CIRCABC in the Note for Guidance to Member States on Compliance check of applications submitted for the authorisation of biocidal products.
Applicants should be aware that the active substance contained in the product will have to be technically equivalent to the one assessed for the purpose of the Annex I or IA inclusion. This means that the active substance has to be similar in terms
6 More details on evidence to be provided and on letters of access are provided in Annex VII.7 Applicants can find information on which data are protected in the reference list of studies provided
in annex of the assessment report of the active substance.7

of its impurity profile to the one assessed for the purpose of the Annex I or IA inclusion.
The evaluating Competent Authority will make the final decision on the technical equivalence based on the data submitted by the applicant.
If technical equivalence is established, the conclusions reached at the time of the Annex I or IA inclusion can be extrapolated to the active substance and, in particular, the endpoints re-used for preparing the product application.
If the active substance is not technically equivalent to the one included into Annex I or IA, then applicants should explore the possibility to refine the production/manufacturing process of the active substance in order to reduce impurities to an acceptable level.
If this option is not feasible or the evaluating CA considers that this refinement does not meet the required standards, the applicant would have to submit a new application dossier for Annex I or IA inclusion of the active substance not being technically equivalent.
More detailed g uidance on technical equivalence is available from the Commission website.
Applicants should make sure that they consider the right product type with respect to the use purpose and pattern of the biocidal product. Further information about scope of the BPD is compiled by the European Commission in the Manual of Decisions (MoD).
In case of any doubts as to whether a product or active substance is falling within the scope of the Directive or to which product type it belongs, applicants are invited to contact the future evaluating Competent Authority.
Applicants should consider carefully the use purpose and pattern of the product and see whether the risk and efficacy assessment conducted with the representative product for the purpose of the Annex I or IA inclusion is valid or relevant for their product.
As in many cases this will not be the case, applicants will have to provide new data to support their own product and carry out a new risk and efficacy assessment.
In order to facilitate the evaluation, it would be preferable for the applicant to request the first product authorisation in the Member State, which was rapporteur for the active substance contained in the product, if, of course, the product is already placed or is intended to be placed on the market of that Member State.
Applicants are however free to choose the Member State where they want to apply to obtain a first authorisation and on the basis of which they can then apply for mutual recognition in other Member States.
8

1.4. Structure of application and related dossier
1.4.1. General requirements for documentation
Application forms
The application form for authorisation of a biocidal product is created via the Register for Biocidal Products (R4BP). The register is maintained by the European Commission. The application form is available in all of the EU official languages.
In the register the applicant fills in the details on the applicant and the product. The applicant has to indicate in which Member States he applies for the first product authorisation and in which EU/EEA countries mutual recognition. Furthermore, the applicant is requested to indicate in which Member States the product is already on the market.
Note! Some Member States8 – where mutual recognition will be applied for – require that the product can only stay on their market beyond the deadline for the submission of the product application and until the final decision is reached, if it is indicated in the application form submitted for the first product authorisation that the product is placed on their market.
When filling in the application form, applicants will be requested to provide a summary of the product characteristics (SPC). This SPC shall be provided in all the official languages of the Member State where the application is to be submitted.
Dossiers
The Member States and the European Commission have agreed that the information included in dossiers for product authorisation should be submitted in a standard format. The format is the same in whichever Member State the dossier is submitted. This will make it easier for the applicants to know exactly what must be done and what a dossier must contain.
The application consists of application forms and the dossier. The applicant is responsible for providing the required information and for including the study reports and other documents needed. The evaluation of the data by the applicant will form the basis of the evaluation by the Competent Authority.
A dossier for product authorisation consists of data and documents as presented in Figure 1.
The application for mutual recognition should include all required data and information other than those parts that can only be submitted following the authorisation/registration of the product in the first Member State (for instance the copy of the first authorisation).
Thus, the following information shall be submitted in a first step:
A covering letter confirming that an application is made for mutual recognition of authorisation for the product;
A signed paper copy of the application form generated via the Register for Biocidal Products (R4BP);
8 PT for instance.9

One electronic copy of the summary dossier for the product as submitted to the first member state;
Letters of access.
The remaining items will have to be submitted within 2 months of the decision being taken on the application for a first authorisation/registration of the product in a first Member State (or “Reference Member State”).
Given the difficulties encountered by the first applicants to produce certified copies of the first authorisation within two months and the fact that this information is available in the R4BP, most Member States9 agreed to accept the dissemination of copies of authorisations by Member States via the R4BP to be equivalent to the submission of certified copies by the applicants; thereby agreeing that the requirement of Article 4 of the Directive would be fulfilled and that it would no longer be necessary for applicants to submit the information themselves.
Most Member States will accept a copy of the first authorisation in English. Those Member States who require a copy of the authorisation in their own language are outlined in the footer 9.
9 Member States requiring a certified copy of the authorisation: BU (until the adoption of new national legislation), HU (until the adoption of new national legislation), LT (until the adoption of new national legislation). Some MS also require a translation of the first authorisation in their own language: DE, FR, PL.
10

Figure 1. Structure of the dossier required for an application for product authorisation.
1.4.2. Practical details on submission of the application
Further details on what information is expected and when, as well as explanations on specific national provisions are provided in Annex II.
1.4.3. General principles on the submission of experimental studies
The applicant shall submit to the Competent Authority all data on physical-chemical properties, toxicological, environmental fate and ecotoxicological effects, efficacy and other properties of the chemical that is necessary for the assessment of the conditions for issue an authorisation. The Technical Notes for Guidance (TNsG) on Data Requirements describes the data needed which was originally set by Annexes II and III of the Directive.
11
Doc. IV-A or LoA*: Test and Study Reports
a.s.(s)
Doc. IV-B or LoA*: Test and Study Reports b.p.**
Doc II-B or LoA*
- Effects Assess.**
- Exposure Assess.
- Efficacy Assess.
for Biocidal Prod.2)
Doc II-A or LoA*
Effects and exposure Ass.
Active Subst.
(s)2)
Doc. II-C Risk Characterisation
for Biocidal Product
Doc. II Risk and Efficacy Assess.
Doc. IOverall
Summary and Assessment1)
Document III-A or LoA*
Study SummariesActive Substance(s)2)
Document III-B or LoA*:
Study SummariesBiocidal Product2)
1) To append: List of end points 2) To append: Reference lists
List of abbreviationsCheck for completeness
Summary DossierComplete Dossier
* LoA = Letter of access** In the case of applications for registration of low-risk products, the effects assessment
is confined to data on the active substance(s) only. In general, the data to be provided in Doc. IV-B and III-B are limited.

The original study reports shall be attached to the application. However, the original study reports (Doc IV) and the study summaries (Doc III) are not required if the applicant has a written proof of his right to refer to them in his application (Letter of Access, LoA) and these documents have already been submitted either for the evaluation of the active substance for Annex I or in another application for product authorisation in the Member State where the application is to be submitted.
Studies must be conducted and reported either according to the methods mentioned in the Council Regulation (EC) N°440/2008 on test methods or according to the OECD (Organisation for Economic Co-operation and Development) guidelines for testing of chemicals. The main rule is that studies must also comply with the principles of Good Laboratory Practice (GLP) and the study report shall contain a certificate of this. Further guidance on GLP is given in the TNsG on Data Requirements, Chapter 6 as well as in Annex V of these notes for guidance.
If it is not technically possible or scientifically justifiable to submit the required information or carry out the required studies, or if the studies are not conducted accor-ding to the guidelines referred above, then the reasoning must be given in the application. If such justifications are not given, or if the application is otherwise insufficient, the Competent Authority will ask the applicant to submit the missing information and studies. The processing of the application will continue after the supplementary data has been presented.
The BPD encourages limiting the duplication of testing on vertebrate animals, whenever possible. According to this principle, before starting a new test, literature searches should be conducted and the other owners of the required documentation should be consulted in order to find out, whether the available information is sufficient for the reliable evaluation of the possible hazards of the chemical. In order to receive the contact details of other data owners the applicants are invited to contact the Competent Authority.
1.4.4. Risk assessment to be prepared by the applicant
The applicant shall carry out a preliminary risk assessment for the product. This risk assessment shall address the following elements:
the assessment of the effects of all active substances and of substances of concern contained in the product;
the assessment of the effects of the product; the assessment of physical-chemical properties of the product; the assessment of the exposure to the product; the assessment of unacceptable effects of the product; the risk characterisation for the product; the assessment of the efficacy of the product.
In most cases, no reassessment of the human health, environmental effects and physical-chemical properties should be carried out for active substances already included in Annex I or IA. Hence, the documents (Doc.II-A) provided with such applications should be used as basis for the effects assessment for the product.
Applicants should nonetheless check in the assessment report of the active substance if there were issues which were not entirely addressed at the time of the substance evaluation and for which additional or complementary data might have been requested before product authorisation can be granted.
12

Product-specific data as required by Annex IIB and IIIB of the Directive have to be provided, summarised and evaluated by the applicant.
More guidance is available on TNsG for Preparation of Dossiers and Study Evaluation and TNsG on Product Evaluation. Detailed advice on risk assessment is given in Technical Guidance Document on Risk Assessment (TGD) and Emission/Exposure Scenario Documents available at JRC website.
13

2. The mutual recognition process 10
2.1. Role of the Reference Member State
Within 3 months of having received the application, the Reference Member State will verify that the submitted dossiers are complete and that the active substance is the same or technically equivalent to the one included into Annex I or IA.
Once a dossier has been considered to be complete, the Reference Member State will start the evaluation of the dossier and, within 12 months, decide whether to authorise the biocidal product or not. This will be done in accordance with the common principles as established in Annex VI to the Directive. The date of the product authorisation, the authorisation, the summary of the product characteristics and the product assessment report will be made available to the other Member States and the Commission via the Register for Biocidal Products (R4BP).
2.2. Involvement of the Concerned Member States
Within two months of having obtained a first authorisation in the Reference Member State, companies shall submit a certified copy of the first authorisation granted in the Reference Member State in accordance with Article 4 of Directive 98/8/EC. Concerned Member States shall evaluate and decide on the mutual recognition of the first authorisation within 120 days from the date of receipt of the certified copy.
Companies should be advised to submit the certified copies of the first authorisation at the same time so that the applications for mutual recognition can be processed in parallel in the Concerned Member States and that arising issues can be discussed within the same 120-day period.
In most Member States, if within the 2 months following the granting of a first authorisation, the certified copy of this authorisation has not been submitted to the Competent Authority, for a product for which an application for mutual recognition was made, the product concerned will be removed from their market within the next 6 months11.
2.3. Negative conclusions
In the case, where, for a product for an application for mutual recognition was made, a Reference Member State comes to the conclusion that a dossier submitted is not complete or, after evaluation, that an authorisation cannot be granted, for the sake of harmonisation the RMS should contact other RMS (assessing similar products at the same time) and Concerned Member States before taking a final decision. The Reference Member State(s) and the Concerned Member States could discuss their concerns before a final decision is taken.
In the event that the Reference Member State decides to refuse to grant an authorisation, companies which had submitted an application for mutual recognition will then not be able to send the copy of the authorisation within 2 months of the decision on their application being taken. Consequently, in the absence of the different elements requested
10 The actions and timelines set in this section shall apply mutatis mutandis to registration of low-risk biocidal products, except that the time for registering a product already registered in another Member State shall be 60 days.
11 See further details in Annex II.14

in Article 4(1) of the Directive, the applications for mutual recognition should be declared incomplete, rejected and the products for, which the applications were made, removed from the market.
2.4. Procedural steps in the MR procedure
Step Timing12 Legal basis
Step 1: Informal discussions between the Concerned Member State and the applicant about submitting an application for mutual recognition.
Step 2: Applicant enters details of application in R4BP.
For new biocidal products
Step 3: Applicant sends application for mutual recognition and the assessment report from the Reference Member State to the Concerned Member State.
Step 4: Concerned Member State receives the application.
Step 5: Concerned Member State confirms receipt of application (directly to applicant and via the R4BP) and checks validity of application (copy of authorisation, summary dossier and fee) within 15 days.
Alternative path for existing biocidal products (Post-Annex I process)
Step 3: Applicant sends: A covering letter confirming that an application is made for mutual
recognition of authorisation for the product A filled in paper copy of the application form generated via the R4BP One electronic copy of the summary dossier for the product as
submitted to the first member state.Step 4: Concerned Member State confirms receipt of application (directly to
applicant and via the R4BP), declares it incomplete, stops the clock and indicates the date by which the missing data are expected to be submitted.
Step 4a: Applicant send copy of the first authorisation and of the assessment report from the Reference Member State to the Concerned Member State.
Step 5: Concerned Member State confirms receipt of application (directly to applicant and via the R4BP) and checks validity of application (copy of authorisation, summary dossier and fee) within 15 days.
Common path
Step 6: Concerned Member State confirms receipt of a valid application (directly to applicant and via the R4BP) – the clock starts.
Day 0
Step 7: Evaluation of the application on the basis of the summary dossier and of the certified copy and of the assessment report of the first authorisation granted.
Step 8: Concerns on or divergence of opinions with the conclusions of the assessment of the Reference Member State and the summary of the
Day 90
12 These indicative milestones are only recommended and not binding on Member States. Only the 120 for the mutual recognition of authorisations is binding.
15

product characteristics to be communicated to the Reference Member State.
Concerned Member State to request applicant that certain conditions referred to in Article 20(3)(e), (f), (h), (j) and (l) of the Directive be adjusted to the different circumstances. 13
Step 9: Reference Member State to respond to concerns or divergence of opinions.
Day 100
Step 10: Applicant to respond to request for adjustment to local circumstances to Concerned Member State.
Day 100
Step 11: If no concerns remain and adjustments are accepted, Concerned Member State to issue the authorisation with a national number and to update the R4BP accordingly.
Day 120 Art. 4.1§1
Start of Formal Conflict Resolution Process
Step 12: Concerned Member State officially notifies the Commission, other Member States and the applicant of any remaining concerns or unaccepted adjustments and updates the R4BP.
Concerned Member State sends the explanatory document containing the name of the product and its specification and setting out the grounds on which it proposes to refuse or restrict the authorisation to the Commission, other Member States and the applicant
Day 0 Art. 4.4§1
Step 13: The Commission allows a period of 90 days during which the Member States and the applicant can submit comments in writing
Day 90 Art. 27.1.b
Step 14: The Commission prepare a proposal for a draft decision Art. 27.2
Step 15: The concerns and the comments are discussed in the CA/PA&MRFG meeting
Step 16: The proposal is voted on in the Standing Committee meeting
Step 17: Commission adopts decision
Step 18: In case the decision confirms the refusal/restriction of mutual recognition, the Reference Member State shall take this refusal/restriction into consideration and withdraw/amend the original authorisation, within 3 months of the decision coming into force.
If the decision is in favour of the mutual recognition, the Concerned Member State must recognise and grant the authorisation, within 3 months of the decision coming into force
Step 19: Member States to issue the authorisation with a national number and to
13 If, in accordance with Article 4, the Concerned Member State establishes that:
(a) the target species is not present in harmful quantities,(b) unacceptable tolerance or resistance of the target organism to the biocidal product is demonstrated, or(c) the relevant circumstances of use, such as climate or breeding period of the target species, differ significantly from those in the Member State where the biocidal product was first authorised, and an unchanged authorisation may therefore present unacceptable risks to humans or the environment, the Concerned Member State may request that certain conditions referred to in Article 20(3)(e), (f), (h), (j) and (l) be adjusted to the different circumstances, so that conditions for issue of an authorisation laid down in Article 5 are satisfied.
16

update the R4BP accordingly.
3. Phasing-out
In most Member States, when no dossier is submitted by the deadline set to apply for product authorisation, existing products shall no longer be placed on the market within 6 months of the deadline to apply for product authorisation or mutual recognition.
Similarly, when a decision is made that a dossier is not complete or that an authorisation cannot be granted, the product shall no longer be placed on their market within 6 months of the decision being made.
The 6-month deadline for the phasing out of products not supported refers to the first placing on the market. For subsequent storage and stocks, Member State may grant a period of grace in accordance with the provisions of Article 7(3) of the Directive.
More information is available on the phasing-out regimen of the different Member States in Annex II.
4. Frame formulations
Specific guidance has been developed for frame formulations14. This guidance gives an overview of the approach to handling frame formulations and authorisation of products covered by a frame formulation. This practical approach was developed in light of the first applications received but some evolution has taken place since its publication.
Note!: The new version of the R4BP allows applicants to create the reference products as well as all additional products within the frame formulation, so it is no longer necessary to send the lists of products within a frame formulation to the Commission for upload in R4BP. The R4BP User Guide for Industry15 provides guidance on frame formulation applications (pages 24-30).
14 For more details see Annex VI.
15 This guidance is available through the R4BP website under the item "More information"17

Annex I
Post-Annex I procedure
Deadline for the submission of applications for product authorisation or for mutual recognition16
2 years after date of adoption of decision
2 years after date of publication of decision
2 years after date of coming into force of decision
Date of inclusion other
(e.g. 20.12.2008 for sulfuryl fluorid)
(e.g. 30.12.2008 for sulfuryl fluorid)
(e.g. 19.1.2009 for sulfuryl fluorid)
(e.g. 1.1.2009 for sulfuryl fluorid)
AT XBE XBUCH XCY XCZ See Annex IIDE XDK XEE XEL XES XFI XFR XHU XIE XISIT XLTLU XLV See Annex IIMT XNO XNL XPL XPT XROSE XSI XSK XUK X
16 In grey, Competent Authorities which have provided no information.18

Annex II
Post-Annex I procedure
Further details on what information is expected and when, as well as explanations on specific national provisions are provided hereafter.
Austria............................................................................................................... 20
Belgium.............................................................................................................. 25
Bulgaria............................................................................................................. 30
Cyprus............................................................................................................... 31
Czech Republic.................................................................................................. 32
Denmark............................................................................................................ 33
Estonia............................................................................................................... 35
Finland............................................................................................................... 37
France................................................................................................................ 39
Germany............................................................................................................ 42
Greece................................................................................................................ 45
Hungary............................................................................................................. 47
Ireland............................................................................................................... 49
Italy.................................................................................................................... 54
Latvia................................................................................................................. 56
Luxembourg...................................................................................................... 57
Malta.................................................................................................................. 59
Netherlands....................................................................................................... 60
Norway.............................................................................................................. 63
Poland................................................................................................................ 65
Portugal............................................................................................................. 69
Slovakia............................................................................................................. 73
Slovenia............................................................................................................. 75
Spain.................................................................................................................. 78
Sweden............................................................................................................... 81
Switzerland........................................................................................................ 83
United Kingdom................................................................................................ 84
19

Austria
In Austria, the relevant national laws for product authorisation and mutual recognition are:
• BiozidG-Altwirkstoffverordnung (BiozidG-Altwirkstoffverordnung, BGBl II Nr. 353/2008)• Biozid-Produkte-Gesetz (BiozidG, BGBl. I Nr. 105/2000)• BiozidG-GebührentarifV I, BGBl. III Nr. 251/2002• BiozidG-GebührentarifV II, BGBl. II Nr. 331/2003, and amendments BGBl. II Nr.
392/2005, BGBl. II Nr. 352/2008, BGBl. II Nr. 209/2009
(First) authorisation/registration:
An application for authorisation/registration (A/R) has to consist of:
Application form, part I (R4BP extract in German; on paper)
Application form, part II (form in German; download from http://www.biozide.at)
Complete dossier on the biocidal product (in electronic format), i.e.
o Document level I (draft PAR)
o Document level II and III as well as studies relating to the biocidal product (document level IV) (reduced data requirements for registrations: see Section 9 (2) of the BP Act)
o Document level II and III as well as active substance studies (document level IV) if deviating from the Competent Authority Report(s) (CAR/s) accompanying the inclusion of the active substance in Annex I/IA; in other cases a reference to the CAR(s) will be sufficient.
Letter(s) of Access relating to the active substance and/or product data insofar as relevant (on paper and in electronic format)
For existing biocidal products containing existing active substances: proof of placing the product on the market before inclusion of the active substance in Annex I/IA of the BPD 98/8/EC (e.g. invoice; on paper and in electronic format)
Current Safety Data Sheet for the biocidal product according to Section 25 of the BP Act in German (in electronic format)
Product label according to Section 24 (5) of the BP Act: label in German, technical leaflet if required (on paper and in electronic format)
In case of an existing product, please submit a copy of the current label as well as a copy of the future label, if different
Proof of payment of the fees (basic fee + fee for checking the completeness of data and dossiers; when completeness has been confirmed: evaluation fee) to the Austrian authority.
Mutual recognition of authorisation/registration:
A written communication announcing a subsequent application for mutual recognition of authorisation/registration, with an authorised representative signature, is a necessary requirement for the admissibility of the marketing of an existing biocidal product (BP) in
20

AT while the corresponding application procedure is underway in the country of the first authorization/registration. A written communication has to consist of:
Letter of intent to announce that an application for authorisation or registration will be submitted for the purpose of mutual recognition (on paper and in electronic format)
Application form, part I (R4BP extract in German; on paper)
Summary dossier on the biocidal product - as submitted in the country of the first authorisation (Reference Member State; in electronic format), i.e.
o Document level I
o Document level II and III for the biocidal product (reduced data requirements for registrations: see section 9 (2) of the Biocidal Products Act)
o Document level II and III for the active substances as far as deviating from the Competent Authority Report(s) (CAR/s) accompanying the inclusion of the active substance in Annex I/IA; in other cases a reference to the CAR(s) will be sufficient.
Letter(s) of Access relating to the active substance and/or the biocidal product, if applicable (on paper and in electronic format)
For existing biocidal products containing existing active substances: proof of placing the product on the market before inclusion of the active substance in Annex I/IA of the BPD 98/8/EC (e.g. invoice; on paper and in electronic format)
Current Safety Data Sheet for the biocidal product according to section 25 of the BP Act in German (in electronic format)
Product label according to section 24 (5) of the BP Act: label in German, technical leaflet if required (on paper and in electronic format)
in case of an existing product, please submit a copy of the current label as well as a copy of the future label, if different.
An application for mutual recognition of the authorisation/registration of a biocidal product includes the following documents, if these have not been submitted already as part of the written communication. If principle changes have to be made as a result of the first A/R procedure (e.g. on the SDS, label etc.), the updated versions have to be submitted as well.
Application form, part I (R4BP extract in German; on paper)
Application form, part II (form in German, in paper; download from http://www.biozide.at)
Official notice of A/R by the country where the first A/R has been granted (on R4BP or notarised in German or English; if in English: a German translation which does not need to have been translated by a juridically certified translator)
Final dossier on the biocidal product after evaluation performed by the authority granting the first A/R (in electronic format), i.e.
o PAR Product Assessment Report
o SPC Summary of Product Characteristics
21

o Document levels II and III as well as studies relating to the biocidal product (document level IV) (reduced data requirements for registrations: see Section 9 (2) of the BP Act
o Document level II and III as well as active substance studies (document level IV) if deviating from the Competent Authority Report(s) (CAR/s) accompanying the inclusion of the active substance in Annex I/IA; in other cases a reference to the CAR(s) will be sufficient
Letter(s) of Access relating to the active substance and/or product data insofar as relevant (on paper and in electronic format)
For existing biocidal products containing existing active substances: proof of placing the product on the market before inclusion of the active substance in Annex I/IA of the BPD 98/8/EC (e.g. invoice; on paper and in electronic format)
Current Safety Data Sheet for the biocidal product according to Section 25 of the BP Act in German (in electronic format)
Proposed product label according to Section 24 (5) of the BP Act: label in German, technical leaflet if required (on paper and in electronic format)
in case of an existing product, please submit a copy of the current label as well as a copy of the future label, if different)
Proof of payment of the fees (basic fee + fee for checking the completeness of data and dossiers; when completeness has been confirmed: evaluation fee) to the Austrian authority.
Deadlines for submission:
(First) application for authorisation or registration - biocidal product containing existing active substances:
An application has to be submitted within three months before the date of inclusion in Annex I/IA (specified in column 4 of the Annex to the Biocides Act-Ordinance on Existing Active Substances), or at the latest on the day when the active substance is included in Annex I/IA.
Special case: If a biocidal product contains several existing active substances of the same active substance/product type combination and not all of these substances have been included in Annex I of the BPD, the last Annex I inclusion date is the reference date for the submission of a first application.
(First) application for authorisation or registration of a biocidal product containing new active substances:
The application for authorisation (or registration) of the biocidal product may be submitted together with the application for inclusion of the active substance in Annex I/IA of the Biocidal Products Directive 98/8/EC, along with all required data and dossiers. The placing on the market of the biocidal product is not allowed until authorisation or registration has been granted. It is, however, possible to apply for provisional authorisation or registration pursuant to section 12 of the Biocidal Products Act. Such provisional authorisation or registration is only valid in the country of the first application. The Biocidal Products Directive 98/8/EC does not provide for the possibility of a mutual recognition of a provisional authorisation or registration.
22

Application for mutual recognition of authorisation/registration:
A written communication has to be submitted within three months before the date of inclusion in Annex I/IA (specified in column 4 of the Annex to the Biocides Act-Ordinance on Existing Active Substances), or at the latest on the day of the inclusion of the active substance in Annex I/IA.
Special case: If a biocidal product contains several existing active substances of the same active substance/product type combination and not all of these have been included in Annex I/IA of the BPD, the last Annex I/IA inclusion date is the reference date for the submission of an application for mutual recognition.
An application for mutual recognition must be submitted not later than four months (authorisation) or two months (registration) before the deadline for compliance (column 5 of the Annex of the Biocides Act-Ordinance on Existing Active Substances) has to be met. According to EU agreement, the application must be submitted within two months after the first application has been granted.
Contact & submission:
The dossier, study reports together with all the necessary information and documentation, and the relevant application form(s) have to be submitted to the following authority:
Bundesministerium für Land- und Forstwirtschaft, Umwelt und Wasserwirtschaft (Federal Ministry of Agriculture, Forestry, Environment and Water Management), attention: Dr. Edmund PlattnerAbteilung V/3, Stubenbastei 5, 1010 Vienna, Austria E-mail: [email protected]; CC: [email protected]
Payment:
Payment of the fees to the following bank account:
Österr. Postsparkasse (ÖPSK)Bank account number: 5060 904;Account holder: BMLFUW-UmweltBank sorting code number: 60000BIC (Swift code): OPSKATWWIBAN: AT77600000005060904
The bank transfer should mention as reference “name of the biocidal product” as well as the relevant product type number.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Austria (1) 1 (1) 1
(For details, please see above.)
23

Sell out period for non-supported biocidal products
If no application is submitted until the deadline for submission, the relevant biocidal products may be placed on the market for no longer than six months from the date of inclusion of the (last) active substance in Annex I/IA. Stocks of the relevant biocidal products may be sold off for another six months, provided they have been in existence in Austria before the expiration of six months from the date specified in column 4 for the relevant active substance/product type combination..
24

Belgium
I. General RemarksAn applicant who has this permanent office in Belgium and who is a legal person has to make use of the Dutch, French our German language depending on the location of his permanent office, at least for the form A and for the R4BP:
- applicant established in Flemish region : Dutch language required - applicant established in Walloon region : French language required- applicant established in Brussels region : Dutch or French language required- applicant established in German community : German language required
A natural person can use his own language, Dutch, French or German, independently of the location of his permanent office in Belgium.
The labelling proposal and the proposal of the directions for first aid as well as the safety data sheet for the product have to be in Dutch and in French. For the other information not given in the form A or R4BP we accept an English version.
An applicant established in another member state of the EC can use one of our national languages for the A form and for the R4BP form and apply the same rules as above for the information not specified in the A- and R4BP form.
In case of mutual recognition, a translation of the SPC, as established by the Reference Member State in English, has to be introduced in Dutch, French or German, depending on the language required for the application.
The applicant should be the same (legal or natural person) as the future authorisation holder and will be responsible for the first placing on the market of the biocide. In case of a consortium, each legal entity that is responsible for the first placing on the market, needs his own authorisation.
In case of products covered by the same frame formulation or in case of identical products, a ‘1-to-1’-approach will be applied : each single product with its unique name will be covered by a separate authorisation with separate SPC and separate authorisation number.
The data (paper and data on electronic support) should be send to the following address :
Federale Overheidsdienst Volksgezondheid, Veiligheid van de Voedselketen en Leefmilieu,Directoraat-generaal Leefmilieu /DG5BiocidenVictor Hortaplein 40, bus 10B-1060 Brussel
The electronic data can also be send by e-mail to : [email protected]
In case of questions, an e-mail can be send to [email protected]
Fees should only be paid after reception of an electronic confirmation that the dossier has been received. This e-mail will mention the exact amount to be paid as well as a structured message which should be used for the payment.
25

During the treatment of the application the applicant or his contact person will be informed by e-mail about the progress of the application. In case additional data are necessary, these will be requested by e-mail. In case an authorisation can be granted, the electronically signed authorisation and the SPC will be send by e-mail.
II. Requirements for applications for existing biocidal products For an existing biocidal product i.e. for which an authorisation has been granted for placing on the Belgian market before the date of inclusion of the active ingredient, the national authorisation expires on the date of inclusion of the active ingredient in Annex I/IA of Directive 98/8, or in case of several active ingredients and/or producttypes, on the last date of inclusion for the first producttype.
The actual authorisation holder has to inform the Belgian competent authority about his intention to introduce an application in Belgium for first authorisation or for mutual recognition, this within 6 months after the coming into force of the decision to include the active substance present in the biocidal product in Annex I/IA of Directive 98/8.
The application for first authorisation or for mutual recognition according to the BPD has to be made by the actual authorisation holder of the existing product who will become the future authorisation holder. Thus Form A should be signed by the actual authorisation holder of the existing product. Only in that case the Minister can grant a prolongation of the existing authorisation for the period necessary for the treatment of the application but no longer than the deadline for compliance with article 16.3 of the BPD, as mentioned in the annex of the inclusion directive.
The application should be introduced at the latest at the date of inclusion of the active ingredient in Annex I, IA of Directive 98/8.
In case the future authorisation holder will differ from the actual authorisation holder, it is necessary to apply for a transfer of the existing authorisation from the actual authorisation holder to the future authorisation holder. It is advised to do this in due time ; the treatment of a transfer takes at least 45 working days. Please contact our service for the requirements by sending an e-mail to [email protected].
In case the product for which a post-annex I authorisation is aimed, differs in composition form the existing product, it is necessary to apply in due time for a change of composition. The treatment of a change in composition takes approximately 6 months. Please contact our service for the requirements by sending an e-mail to [email protected].
III. Application for first authorisation/registrationAn application for first authorisation / registration should include the following documents:
1) a covering letter, form A , as well as form B1, part 2 (Ministerial decree of 3 september 2008, published on 08-09-2009). For Form A and B1 see on our website www.biocide.be; législation / wetgeving. The original signed form A should be sent by post to the address, mentioned on this form.
Note: Form B1, part 1 is not necessary since it has been replaced by the R4BP-form
26

2) the application form generated via the "Register For Biocidal Products (R4BP)", a tool set up by the European Commission. The R4BP is available at: https://webgate.ec.europa.eu/env/r4bp/ . Once you filled in the R4BP, you have to make a print-out of the form, sign it and send the original signed form by post to the address, mentioned on Form A.
3) one electronic copy of the dossier according to the requirements of Directive 98/8/CE. These requirements are more detailed in the Technical Notes for Guidance on Data Requirements (see http://ecb.jrc.ec.europa.eu/biocides/). We would recommend the use of IUCLID 5 for the study summaries.
In addition a proposal for the label in French and Dutch, a safety data sheet for the product in French and Dutch and an instruction leaflet for first aid in French and Dutch are required. This leaflet has to be in accordance with the requirements of the Poison Control Center (see www.biocide.be ; toelatingsaanvraag; Instructies voor het vermelden van eerste hulp maatregelen (.WORD) and Instructies eerste hulp (.EXCEL) or in the French version : demande d'autorisation ; Instructions pour la rédaction des premier soins (.WORD) et Notice premiers soins (.EXCEL))
4) a Letter of Access from the owner of the data which has been used for the annex I/IA inclusion if the applicant doesn’t own the data himself. This LOA should authorise the Belgian authority to use the data for the purpose of granting to the applicant an authorisation/registration of the biocidal product. The original signed Letter of Access should be send by post to the address mentioned on form A.
5) The requested fee should only be paid after reception of an electronic confirmation that the dossier has been received. This e-mail will mention the exact amount to be paid as well as a structured message which should be used for the payment.
IV. Application for mutual recognition of an authorisation/registration An application for mutual recognition has to include the following documents:
In a first step, in case of an existing product at the latest at the date of inclusion of the active ingredient in Annex I,IA of Directive 98/8:
1) a covering letter, (= form A) , as well as form B1, part 2 (Ministerial decree of 3 september 2008, published on 08-09-2009). For Form A and B1 see on our website www.biocide.be; législation / wetgeving. The original signed form A should be sent by post to the address, mentioned on this form.
Note : Form B1, part 1 is not necessary since it has been replaced by the R4BP-form2) the application form generated via the "Register For Biocidal Products (R4BP)", a tool set up by the European Commission. The R4BP is available at: https://webgate.ec.europa.eu/env/r4bp/ . Once you filled in the R4BP, you have to make a print-out of the form, sign it and send the original signed form by post to the address, mentioned on Form A.
3) In case of an application for mutual recognition of an authorisation one electronic copy of the summary dossier as submitted to the reference member state. In default of the summary dossier, one electronic copy of the dossier according to the requirements of Directive 98/8/CE.
27

In case of an application for mutual recognition of a registration, the same data as for a normal registration according to Directive 98/8/CE, article 8, except that only a summary of the efficacy data is required.
4) a Letter of Access from the owner of the data which has been used for the annex I/IA inclusion if the applicant doesn’t own the data himself. This LOA should authorise the Belgian authority to use the data for the purpose of granting to the applicant a mutual recognition of the authorisation/registration for the biocidal product for which he did hold an authorisation before the annex I/IA inclusion of the active substance concerned. The original signed Letter of Access should be sent to the address mentioned on form A.
Once the first authorisation/registration has been granted, in case of an existing product w ithin 2 months :
5) a copy of the authorisation/registration act, certified by the Competent Authority of the Reference Member State. If the Reference Member State has uploaded the first authorisation in R4BP, the certification by the Competent Authority of the Reference Member State is not necessary.
6) the SPC, as approved by the reference Member State, together with a translation of the SPC in French, Dutch or German, depending on the language required for the application.(see part I of this document ‘General Remarks’)
7) One electronic copy of the product assessment report, made by the Reference Member State. We accept assessment reports in Dutch, French or English.
8) A proposal for the label, a safety data sheet for the product and an instruction leaflet for first aid are required ; for all these documents a French and a Dutch version are required. The instruction leaflet for first aid has to be in accordance with the requirements of the Poison Control Center (see www.biocide.be ; toelatingsaanvraag ; Instructies voor het vermelden van eerste hulp maatregelen (.WORD) and Instructies eerste hulp (.EXCEL) or in the French version : demande d'autorisation ; Instructions pour la rédaction des premier soins (.WORD) et Notice premiers soins (.EXCEL))
The copy of the first authorisation has to be sent BY POST to the above mentioned address! After confirmation of receipt of all the abovementioned data, the 60/120 days period, as foreseen in the Directive for treatment of the application for mutual recognition of a registration/authorisation, starts.
28

Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Belgium 1 1 1 1
Payment
Fees should only be paid after reception of an electronic confirmation that the dossier has been received. This e-mail will mention the exact amount to be paid as well as a structured message which should be used for the payment.
Fees should be paid to the following bank account:
Service Public Fédéral Santé publique, Sécurité de la Chaîne alimentaire et Environnement – Redevances & côtisations produits
Place Victor Horta 40, boîte 101060 BruxellesIBAN: BE65 6792 0059 5996BIC: PCHQBEBB
Sell out period for non-supported biocidal products
All existing authorisations expire on the date of the latest Annex I inclusion of one of its active substances for the first producttype. If an acceptable application has been introduced for the product according to Dir 98/8, a prolongation of the authorisation can be granted for the minimal period necessary to evaluate the dossier. If the application can’t be accepted or if no application is made, a 6 months period can be attributed to eliminate, to store or to put on the market existing stocks; 18 months is foreseen to use these stocks. The 6 months and the 18 months starts from the date of declaration that the application can’t be accepted.
Sell-out period for supported Biocidal products following Authorisation/Registration
In case of an existing product for which a prolongation of the existing authorisation has been granted after the date of inclusion in Annex I and for which subsequently an authorization/registration has been granted according to the BPD, a 6 months period will be accepted to eliminate, to store or to put on the market existing stocks ; 18 months will be accepted to use these stocks. The 6 months and the 18 months start from the date on which de first authorization according to the BPD has been granted.
29

Bulgaria
We will be accepting applications in Bulgarian language. 3 electronic copies and 3 paper copy are needed.
30

Cyprus
Our service, accepts application forms in English language.
Payment
The cost for the registration of a biocide under the national scheme or under the Mutual recognizing process is 500 euros.
•The payments should be made net in EURO free of any charges.
• Proof of payment of the fees should be enclosed to the application.
The payment of the registration fees for biocidal products could be done in either of the following ways:
A. For easier and faster payment settlements, it is suggested to proceed with the payment at the Department of Agriculture by your local distributor. The cheque (of a bank Based in Cyprus) must be issued to “Director Department of Agriculture”.
B. For direct payments from abroad, payment could be done via transfer with the following details:
EUR THROUGH TARGET 2SWIFT CODE: CBCYCY2NFBUCENTRAL BANK OF CYPRUSCY 1395, NICOSIA
Please remit to the Central bank of Cyprus (SWIFT CODE: CBCYCY2NFBU) the amount of EUR …………………… through TARGET 2 for the credit of the Government General Account, A/C No. CY 16001000010000000006001010
In favour of the Department of Agriculture, Plant Health Service, Biocide Registration. Product Name ……………………………..
Sell out period for non-supported biocidal products
6 months to phase out the product from the market and supply chain.
31

Czech Republic
In the Czech legislation, products - on the basis of national notifications - can be placed on the market until the end of the transition period (14 May 2010 to be extended to 14 May 2014) or, in the case of products containing substances included in Annex I until the date of compliance with Article 16(3), as stipulated in the Annex I inclusion..
We however recommend via our web pages that applicants submit applications for a first authorisation by the date of inclusion of the active substance in Annex I (e.g. 1 January 2009 for wood preservatives containing sulfuryl fluoride). In case of application for mutual recognition, we recommend the applicants to submit the application no later than 120 days before the date by which the product notified in accordance with the national provisions is to be removed from the market.
Regarding mutual recognition, a complete data set containing a covering letter, the application form generated via the R4BP (1 copies in print and 4 electronic copies), proof of the payment of the fee, 1 electronic copy of the summary dossier and 1 copy of authorization issued by the Reference Member State (we need certified translation of the authorization into Czech) is to be submitted. MSDS and proposed labelling is required in Czech.
Once a dossier has been considered to be complete, it shall be evaluated within 9 months.
If the documentation is incomplete, the clock can be stopped and applicant asked to provide the remaining data.
Payment
Fees should be paid to.
Name of bank: Česká národní banka / Czech National BankAddress: Na příkopě 28, 115 03 Praha1, Czech RepublicIBAN: CZ4507100037110002528001BIC/SWIFT: CNBACZPP
Sell out period for non-supported biocidal products
Non-supported existing products can stay on the market according to national conditions till the date of compliance with article 16 (3) of the BPD. Till this date, it is also possible to notify and subsequently place on the market a „non-existing“ product according to national conditions. No sell out period is possible after this date.
32

Denmark
We at present will require the following - in a first step - for this first application for mutual recognition:
A covering letter confirming that an application is made for mutual recognition of authorization for the product.
A filled in paper copy of the application form generated via the R4BP One electronic copy of the summary dossier for the product as submitted to the first
member state.
Additional information will be required in a second step where the full application for mutual recognition will be submitted. These will, for instance, include: Fees for handling the application to be paid A summary dossier, where the first member state has filled in the evaluation boxes, in
English Other part of application form covered by R4BP (not yet available, in English or
Danish) A certified copy of the first authorization (in Danish or English) and an assessment
report as foreseen being made by the first member state in English Label proposal and instruction leaflets in Danish
In addition we may request for all studies submitted by the applicant in an electronic form according to Article 8 of the BPD. We want 3 electronic copies (CD) and one paper copy.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Denmark 1 1 1 1
Payment
Denmark issues an invoice upon reception of any application.
Sell-out period for supported Biocidal products following Authorisation/Registration
Under the national regulation, the Danish CA grants authorisations for marketing of biocidal products for a period of ten years, in some case only for five years.
In our authorisation letter to the authorisation holder we state that this authorisation will expire within 10/5 years, at a specific date. If the authorisation holder wishes to apply for a renewal of the authorisation, the company must apply at least one year in advance of the expiry date.
33

This means that the authorisation holder has been warned, that he must react at least one year in advance of the expiry of his right to import and sell a specific biocidal product with reference to his authorisation. If the company applies in due time, under normal conditions the company will receive the new authorisation before the original authorisation expires.
When an authorisation is renewed, changed or denied, we state that the right to import and sell biocidal products according to the original authorisation has terminated on receipt of this letter.
However existing stocks may be sold out until three months from the date of the letter. In special cases, on request the Danish EPA can expand the sell-out period for a longer period, if the company has special reasons and can inform us of the size of the stocks and the expected sales period.
Under the national regulation, the Danish EPA has not in normal cases regulated the termination of the sales and usage of biocidal products in the downstream sales channels or at the end users, only the authorisation holder’s rights. The reasoning has been that the products disappear from the market place when the supply dries out.
This will change under the BPD, where the downstream sales channels and end users only will be able to sell and use products with old labels until the deadline for compliance with article 16(3).
In connection with this change, the Danish EPA has made it possible in our legislation to set a slightly earlier date of termination of the right to import or sell the biocidal product than the ultimate deadline stated in the inclusion directive. This is to prevent a situation where a downstream sales channel or an end user may buy a product one day and the next day it will be illegal to use the product. Therefore we have made it possible to impose differentiated deadlines for sales and usage of products with old labels on the different categories in the sales chain and for end users.
Another reason for the differentiated deadlines is the wish to prevent manufactures from building up excessive stocks with old labels very close to the deadline in the inclusion directive.
34

Estonia
“Existing biocidal product” in Estonia refers in the context of this document to those products which have already been placed on the market of the Estonia in accordance with transitional provisions of Chapter 9 of the Biocides Act transposing Directive (EC) 98/8/EC. As a basic requirement, these products are:
must bear a valid registration number on the product label at the date of inclusion of the active substance into Annexes I or IA
and at the same time they must be listed in the Biocides Register.
For a product already placed on the Estonian market for a given Product Type (PT), the deadline for submission of an application is the date of inclusion of the active substance or the last active substance.
Application for authorisation/registration the following is required:
Covering letter stating that the applicant would like to apply for product authorisation/registration.
Copy of the dossier according to the requirements of Directive 98/8/EC, Annex IIB-IIIB-IVB. Dossier is accepted in Estonian or English. We would recommend the use of IUCLID 5 for the study summaries.
Fees for handling the application to be paid
One paper copy and one electronic copy (CD or DVD) are required.
Application for mutual recognition of authorisation/registration the following is required: In a first step the following is required:
A filled in and signed paper copy of the application form generated via the R4BP in Estonian or English.
In a second step the full application for mutual recognition will be submitted within two months of the first authorisation granted. These will, for instance, include:
Fees for handling the application to be paid Copy of the official authorisation granted by the reference Member State (in Estonia
or English) Copy of the Product Assessment Report by the reference Member State, preferably in
English Copy of R4BP form (part 2). Summary dossier (Doc I-III; on CD) Copy of the proposed label, SDS and instruction leaflets, all in Estonian.
One paper copy and one electronic copy (CD or DVD) are required.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Estonia 1 1 1 1
35

Payment
The payment should be made before the application so that the proof of payment can be joined to the application or in parallel to the submission of the application. We don’t issue invoices.
Bank details:
Beneficiary customer: RAHANDUSMINISTEERIUMBeneficiary's account: IBAN EE89 1010220034796011Beneficiary's Bank: SEB, Tallinn, EstoniaBIC: EEUHEE2X
On cross-border payment order, details of payment, must have:
Terviseamet - 2900082087, name of biocide
Sell out period for non-supported biocidal products
From the date of Annex I inclusion 1 year to phase out the product from the supply chain and market (don’t have legal basis for this).
36

Finland
For a product already placed on the Finnish market for a given Product Type (PT), the deadline for submission of an application is the date of inclusion of the last active substance. Procedures after Annex I inclusion and related deadlines are implemented by the Ministry of the Environment Decree 20/2008 as amended.
The application, in accordance with Chemicals Act 1998/1999 and related Decrees, consists of:
- an application for authorisation/registration or - an application for mutual recognition of authorisation/registration.
The content of the dossier (studies, risk assessment etc.) depends on the kind of application (authorisation, registration, mutual recognition etc.) and is specified in the Ministry of The Environment Decree 518/2000 as amended. Application guideline will be available at www.tukes.fi/biocides
This dossier shall be accompanied by:
The application form generated from the Register for Biocidal Products (R4BP) of the EU Commission (part 1 of the application form)
A specific FI application form (part 2 of the application form). It is send upon request. Please contact: [email protected]
A proposal for the Classification and Labelling, the Safety Data Sheet, and the copy of the label of the product in Finnish and Swedish and proposal for the Summary of the Product Characteristics (SPC).
Application for mutual recognition of authorisation/registration takes place in two steps: notice of intention and formal application.
For the notice of intention following shall be submitted by the deadline of application at latest:
A letter indicating that an application for product authorisation is being made in another Member State (Reference Member State, RMS) and mutual recognition of that product is sought in FI
Copy of the R4BP form, printed and signed (part 1) Electronic copy of the summary dossier.
For the formal application for mutual recognition following is submitted within two months after authorisation of the product by the RMS:
Summary dossier as accepted by the RMS Copy of the proposed label and Safety Data Sheet and Summary of the Product
Characteristics.(Copies of the first authorisation or Product Assessment Report by the RMS are not requested provided that the RMS has uploaded them in the R4BP and that the Product Assessment Report is in English).
Additional studies and information considered essential to the assessment may be requested.
37

All documents shall be submitted either in Finnish, in Swedish or in English. However, proposal for the Safety Data Sheet and the proposed label must always be available in Finnish and in Swedish and SPC in Finnish. One paper copy and two electronic copies (CD, DVD or USB-stick) are required.
Fees are according to the Decree 1302/2010. The applicant will be invoiced by the CA.
Applications shall be submitted to the designated CA:
Finnish Safety and Chemicals Agency (Tukes)P.O. Box 66, FI 00521 Helsinkiregister office: [email protected]: [email protected]/biocides
In case no application is submitted before the deadline, the first placing on the market has to be stopped 6 months after the deadline and subsequent storage and use after an additional 12 months.
The same deadlines will apply in case where an application is rejected due to incompleteness of the dossier.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Finland 1 2 1 2
Payment
Tukes will send an invoice indicating banking details is to the applicant. Part of the fees is charged after receipt of on application and handling of an application does not start until the fee is paid. Part of the fees is invoiced at the end of the evaluation if the initial fee does not cover the costs.
Further information is available at: www.tukes.fi/biocides
Sell out period for non-supported biocidal products
6 months sell-out period + 12 months for use or disposal.
Sell-out period for supported Biocidal products following Authorisation/Registration
In FI sell-out period for supported product would normally be 6 months for the first placing on the market and an additional 12 months for resale (total of 18 months) and use i.e. the same as normal phase-out periods. We wish to keep the system simple and timelines similar in different cases as much as possible. However, in case of significant changes in the instructions for use or new restrictions compared to national authorisation, sell-out periods may be shorter. Timelines would then depend on the seriousness of the case
38

France
For a product already placed on the French market (ie “existing product”) for a given Product Type (PT), the deadline for submission of an application is the date of inclusion of the last active substance. For instance, for a wood preservative product PT08 put on the French market with 2 active substances (AS1 and AS2, decision of inclusion of AS1 is taken before AS2), the submission of the dossier can be made from the date of transposition of the inclusion directive of AS2 in France, until the date of inclusion of AS2, at the latest.This application consists in:- either an application to demand the Authorization - or, if a demand of authorization is submitted in another Member State where the product is also put on the market (called “Reference Member State”), an application to demand an intention of Mutual recognition. In such case, the assessment of the dossier would be put on hold until a decision is taken by the Reference Member State and the applicant confirms its intention of mutual recognition.
The content of the “technical” dossier (studies, risk assessment etc.) depends on the kind of demand (authorization, registration, mutual recognition etc.).
This “technical” dossier shall be accompanied by:1) A letter from the applicant written in French, to explain the purpose of the
submission (ie demand for authorization, demand for mutual recognition, registration of a low risk product etc.).
2) The application form generated from the Register for Biocidal Products (R4BP) of the EU Commission, in French version.
3) A proposal for the Classification and Labeling, the Safety Data Sheet, and the copy of the label of the product also have to be submitted in French. A copy of the letter of access in French shall also be submitted.
Application for biocidal product authorisation shall be sent via the French website SIMMBAD (www.simmbad.fr). SIMMBAD facilitates and strengthens exchanges between applicants and administration. SIMMBAD allows companies to fill online an application form, and submit directly their whole electronic dossiers (by upload). This website aims to limit the use of “physical sendings” (paper, CDs etc.). SIMMBAD doesn’t substitute to the application made through the R4BP (https://webgate.ec.europa.eu/env/r4bp/), but is a complimentary tool to complete the process of submission of an application for product authorization in France.
SIMMBAD also provides to applicants an user account, that allows them to have a real-time monitoring of the steps of treatment of their dossier, as well as a platform of dialogue with administration during the treatment of the application. This website is only available in French for the moment, but an English version as well as a German version are under development and should be available at the end of 2011 (translation of user guidance, masks and list of selection in the system).
Companies are strongly encouraged to use SIMMBAD to make their application in France, as this tool is especially designed for the management of application of product authorization in France. In exceptional circumstances, if a company do not succeed to send an application via SIMMBAD, it shall contact ANSES [email protected].
39

For any question related to technical question on the use of SIMMBAD and technical assistance (electronic certificate, problems with the interface etc….), e-mails can be sent to: [email protected]
A specific Helpdesk on Biocides had been developed to explain procedure of submission in France, as well as information on how to build a dossier. http://www.helpdesk-biocides.fr/. An English version is to be provided soon.
Fees are fixed in the Decree of 24 June 2004 modified («Arrêté du 24 juin 2004 modifié fixant le montant de la rémunération due au titre de l’autorisation de mise sur le marché des substances et produits biocides »), and shall be paid directly to ANSES :
- In case of an application to demand the Authorization, 2.500 euros shall be paid at the submission for the completeness check of the dossier. If the dossier is declared as complete, the dossier will be assessed and the remaining part of the fee will have to be paid later (according to the provisions of last paragraph of article 1 of the Decree of 24 June 2004 modified). Several fees are defined in function of the amount of work needed, from 16.000 to 37.000 euros, with the “basic” fee of 32.500 euros.
- In case of an application for Mutual recognition, the fee will have to be paid when the Reference Member State would have taken a decision on the dossier. The fee is 4.000 euros for a “standard product”, 1.800 euros for a “low risk product”.
Further information on the fees is available: http://www.helpdesk-biocides.fr/
Administrative fees must be paid to: ANSESBank references:T.G. VAL DE MARNE1, place du Général Billotte94 040 CRETEIL CEDEXIBAN: FR76 1007 1940 0000 0010 0043 619 BIC: TRPUFRP1The bank transfer to the ANSES should be carried out before sending the application and should mention: French R4BP number / product name.A copy/scan of the bank transfer sheet should be added to the dossier.
Sell out period for biocidal productsIn case no dossier is submitted for a product before the deadline of submission, all placing on the market will be forbidden 6 months after the deadline for submission of the dossier, and their use 12 months after the deadline for submission
In case the dossier is declared uncompleted and rejected, all placing on the market will be forbidden 6 months after the decision, and use forbidden 12 months after the decision, unless otherwise specified.
Sell-out period for supported Biocidal products following Authorisation/Registration
Under the current process, the deadlines implemented in France are shown below. However, these deadlines may be changed depending on EU and national discussions.
40

Summary table of deadlines for compliance with a decision
ScenarioTimelines to withdrawal of
the market the biocidal product
End of use of the biocidal product
Dossier of authorization not submitted at the inclusion
date of the active substance in the annex I of the directive
98/8/CE
6 months after the inclusion date of the active substance
12 months after the inclusion date of the active substance
Dossier of authorization submitted at the inclusion
date of the active substance in the annex I of the directive
98/8/CE but judged inadmissible
6 months after the date of the decision, unless contrary provision in the decision
12 months after the date of the decision, unless contrary
provision in the decision
Dossier of authorization submitted at the inclusion
date of the active substance in the annex I of the directive 98/8/CE but rejected at the
end of the assessment OR
Changes in the authorization of the biocidal product with
risk for health and environment.
Deadlines fixed case by case in the decision, according the
reason of the rejection
Deadlines fixed case by case in the decision, according the
reason of the rejection
First authorization according the directive 98/8/CE after the
transitional periodOR
Changes in the authorization of the biocidal product without
risk for health and environment.
6 months after the date of the decision, unless contrary provision in the decision
12 months after the date of the decision, unless contrary
provision in the decision
Detailed information on the implementation of the biocidal products directive in France, procedures for submission of dossiers etc… can be found on the following websites:http://www.developpement-durable.gouv.fr/-Biocides-.html http://www.helpdesk-biocides.fr/http://www.simmbad.fr
41

Germany
Article 28 (8) of the German Chemicals Act (Chemikaliengesetz, ChemG) provides rules on the interim measures for both new and existing biocidal-products, which contain existing active substances for which the decision of Annex I inclusion was reached. These rules lay down the prerequisites under which such products may be continued to be placed on the market for the time of product authorisation process, registration process or the procedure of mutual recognition (at the latest until the deadline according to Article 16 paragraph 3 of the Directive 98/8/EC, which is given in the Inclusion Directive for the active substance). According to this provision the interim measures only apply if an application for authorisation, registration or mutual recognition has been filed at the latest 24 months after the date the Inclusion Directive has been published in the Official Journal.
For a product containing more than one active substance of a given Product Type (PT), the deadline for submission of an application is 24 months after the date of publication of the Inclusion Directive of the last active substance.
The application for mutual recognition in DE has to be in line with the requirements of Article 4 paragraph 1 of Directive 98/8/EC. Requirements include the following documents/ formats:
Step 1:
1. Hardcopy of the R4BP (register of biocidal products) application form in German language with date and signature (please provide the originally signed document and not a copy).
2. Second part (additional form in Germany) of the application form in German language
3. Statement, that the paper copy is identical to the electronic format (please provide the originally signed document and not a copy).
4. Authorisation for communication during the procedure (if necessary please provide the original document and not a copy; e.g. for a consultant)
5. Letter of Access (if necessary)6. "Summary Dossier" (Doc. Sections I-III), which has also been submitted to the
member state, that has received the application for authorisation (Reference Member State, RMS)
7. A proposal for classification and labelling: please provide the safety data sheet and a copy of the label of the biocidal product in German language
Step 2:
8. A certified copy of the first authorisationo The certified copy of the first authorisation of the Reference Member State
does not have to be provided in line with the application (step 1). This document needs to be provided as soon as it is issued by the Reference Member State (step 2).
o The first authorisation according to Article 4 §1(2) of Directive 98/8/EC does not have to be provided by a specific German authority. It is acceptable, if it is carried out according to your national law by your local administration.
42

9. A German translation of the first authorisation and a statement of the translator, that both documents have the same content (an official certification is not required).
10. The Assessment Report of the Reference Member State (in German or English language)
MS agreed that the RMS should upload the Assessment Report and authorisation in the R4BP and that CMS will generally accept these as “certified”. Upon the submission of the German translation you have to make a reference to these uploaded documents in your cover letter, if you do not submit them yourself.
An application for authorisation in DE has to be in line with the requirements of Article 8 (2) of Directive 98/8/EC and comprises the application form and documents as listed above (points 1. to 7.). In addition a dossier is required, that has to fulfil the following requirements according to Art. 8(2) of Directive 98/8/EC:
a dossier or a letter of access for the biocidal product satisfying, in the light of current scientific and technical knowledge, the requirements set out in Annex IIB and, where specified, the relevant parts of Annex IIIB, and
for each active substance in the biocidal product, a dossier or a letter of access satisfying, in the light of current scientific and technical knowledge, the requirements set out in Annex IIA and, where specified, the relevant parts of Annex IIIA.
Please find the documents under the following link: http://www.baua.de/nn_8584/de/Chemikaliengesetz-Biozidverfahren/Dokumente/Biozid-Formulare.zip
Please note that these forms are frequently updated. Therefore you should check for updated versions before application submission.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Germany 1 1 1 1
Sell-out period for non-supported biocidal products
According to interim measures of Article 28 (8) of the German Chemical Act (Chemikaliengesetz, ChemG), application for authorisation, registration or mutual recognition of an existing biocidal product needs to be submitted within 24 months after publication of the Inclusion Directive of the composing active substance. In case a biocidal product contains several active substances, product dossiers must be submitted by the date which is 24 months after the publication of the inclusion directive for the last active substance. If the applicant fails to submit a complete application within the given deadline the product will no longer be marketable. Germany does not grant a further sell-out or phase-out period. In any case, products may remain on the market latest until the deadline of compliance stated in Article 16(3) of the Directive 98/8/EC.
43

Fees
For the authorisation or the mutual recognition of biocidal products a fee will be charged. For more information please refer to Article 1 (1) of the German “Chemikalien-Kostenverordnung”.
PLEASE DO NOT transfer any money to Germany’s Competent Authority prior the submission of your application. You will receive an invoice from the Competent Authority listing the amount due and all the necessary banking details.
44

Greece
1. For the 1st Authorisation in Greece, are required the followings:
1) The administrative part in Greek, containing a covering letter in paper to explain the purpose of the submission and the application form in paper (generated via the R4BP).
2) The Assessment Report (a synthesis of Doc.I and Doc.II of the biocidal product dossier) can be in Greek or in English. In case where the Assessment Report is in Greek language an English version will be needed for a mutual recognition and we agree to review the draft translation provided by the applicant.
3) A summary of the main product characteristics (SPC)
4) A list, as Annex to the Assessment Report, of the studies reviewed.
5) A proposal for the classification and labelling, the safety data sheet, and the copy of the label of the product also have to be submitted in Greek
6) Fees for handling the application to be paid
The MS will make the Decision and grant the authorisation to the applicant.
2. For the mutual recognition:
An application for mutual recognition would not be rejected if the Member State was to receive until the date of Inclusion of the a.s. in a first step the following information:
A covering letter in paper in Greek or maybe we can accept also an English version confirming that an application is made for mutual recognition of authorisation for the product.
A filled in paper copy of the application form in Greek generated via the R4BP One electronic copy and one paper copy of the summary dossier for the product, as
requested in Article 4 of the Directive and already submitted to the Reference Member State.
The rest of the information would be submitted in a second step, once the first authorisation would have been granted.
A certified copy of the authorisation granted in whatever MS and an official translation in Greek.
An electronic copy and one paper copy of the Assessment Report of the Reference member state.
A proposal for the classification and labelling, the safety data sheet, and the copy of the label of the product also have to be submitted in Greek.
A Safety data Sheet for the product as well as for all inserts in Greek. Fees for handling the application to be paid
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Greece 2 1 All required documentation except for product dossier in
45

paper, while product dossier only in electronic format.
Payment
Regarding biocidal products belonging to PT 8, 9, 10, 12, 13, 14, 15, 16, 17, 18, 19, 21 and 23 for which the responsible Competent Authority is the Hellenic Ministry of Rural Development and Food, fee payment should be facilitated by the means of Bank check (not a check from the Company’s Carne) issued by any bank of applicant’s choice on the grounds that the bank has an operating branch situated in Athens, Greece. The payment order on this check should be issued either to a) “ELLINIKO DHMOSIO”17 or b) “A’ DOY KALLITHEAS”18.
Payment of the fees by bank check stating “Hellenic Ministry of Rural Development and Food” as the one receiving the payment order, is not possible
The bank checks must be posted to the Greek CA. Alternatively, the bank checks can be submitted directly to the Greek CA through applicant’s local representatives.
Regarding biocidal products belonging to PT 8, 9, 10, 12, 13, 14, 15, 16, 17, 18, 19, 21 and 23 for which the responsible Competent Authority is the Hellenic Ministry of Rural Development and Food, placing of non-supported biocidal products (i.e. existing products for which an application has not been submitted, or products that a decision is made that the dossier is not complete or that an authorization cannot be granted) on the market will be ceased from the date of inclusion of their active substances to ANNEX I IA IB of 98/8 EU directive. Existing stock of non-supported biocidal products on the market on the inclusion date may remain on the market for a period not longer than 12 months after the inclusion date.
Sell out period for non-supported biocidal products
In the case of non-supported biocidal products containing mixtures of active substances, the abovementioned “inclusion date”, refers to the inclusion date of the last included active substance.
Sell-out period for supported Biocidal products following Authorisation/Registration
The authorisation of the supported product (so called 'new' product) will be published in replacement of the old authorisation that had been granted following the national rules (so called 'old' product). From the date the 'new' authorisation has been published, the authorisation holder will not be allowed to continue to place on the market products according to the 'old' authorisation. In addition, a 12-month sell out period will be given for the marketing and use of the existing stocks of the 'old' product on the market.
The above applies to biocidal products belonging to PT 8, 9, 10, 12, 13, 14, 15, 16, 17, 18, 19, 21 and 23 for which the responsible Competent Authority is the Hellenic Ministry of Rural Development and Food.
17 “ELLINIKO DHMOSIO” means Hellenic Public Sector or Greek Public Sector.18 “A’ DOY KALLITHEAS” means “A’ tax office” in “Kallithea” region, which is also the region the
corresponding CA service is located to. 46

Hungary
For the first authorisation the following is required:
o Covering letter stating that the applicant would like to apply for product authorisation.
o One electronic and one paper copy of the dossier as stated in the BPD, accepted in Hungarian or English.
o Fee for product authorisation has to be paid according to our fee regulation 1/2009. (I.30.) EüM rendelet.
Mutual recognition of the first authorisation
1. In the first step the following is required:
o A covering letter confirming that an application is made for mutual recognition of authorisation for the product
o A filled in paper copy of the application form generated via R4BP. At the moment English version is only available, but in the future the Hungarian is preferred.
o One electronic copy of the summary dossier for the product as submitted to the first Member State.
2. In the second step of mutual recognition the following is required:
o Covering letter that the first authorisation has been granted.o Certified copy of the first authorisation granted in the Reference Member State.
Certified Hungarian translation is required if it is not in English.o Fee for mutual recognition has to be paid according to the fee regulation 1/2009
(I.30.) EüM rendelet.o One electronic and one paper copy of the Assessment Report.o One paper copy of the summary dossier for the product as submitted to the first
Member State.
Label proposal, Safety Data Sheet and using instructions only accepted in Hungarian.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Hungary 1 1 1 1
Payment
Fees should be paid before sending the application to the main bank of the
Országos Tisztifőorvosi Hivatal (Office of the Chief Medical Officer of State) which is the following:
Magyar Államkincstár (Hungarian State Treasury)Address: 1139 Budapest, Váci út 71.
47

IBAN: HU81 1003 2000 0028 1519 0000 0000BIC: MANEHUHB
The treasury transfer code 311 and the name of the product have to be specified in the comments of the bank transfer.
Sell out period for non-supported biocidal products
There is registration expiry date for product on the on-line registry and this is final date for placing on the market, if product is not supported for product authorisation. Then enforced for disposal.
48

Ireland
Upon publication of a Decision Directive for Inclusion/Non-inclusion:
The Irish CA informs notification holders, of the relevant biocidal product(s) that contain the active substance and associated product-type (PT) combination, that products will need to be either supported for Product Authorisation/Registration or Mutual Recognition as per an inclusion decision, or that the products are revoked from the market as per a non-inclusion decision.
For support of a product based on an inclusion decision, the PCS distinguishes between products containing a single active/PT combination, multiple actives/single PT combination and multiple actives/PT combinations.
For revocation of a product based on a non-inclusion decision, the PCS distinguishes between a full product revocation and a product-type revocation. Following entry into force of the non-inclusion the notification holder of the relevant product has a period of 1-year to use existing stocks of the product.
From 2 months of the date of the above letter informing of an inclusion decision, the PCS requires a letter of intent (that can be emailed) from the authorisation holder of the relevant biocidal products as to whether they will support the product(s) authorisation or mutual recognition.
Within the period between entry into force of an inclusion Directive and the date of inclusion into Annex I of Directive 98/8/EC the PCS are willing to liaise (via email or face-to-face) with applicants regarding specifics of their product application/mutual recognition.
The deadline for submission of an application for Product Authorisation (PA) or Mutual Recognition (MR):
Date of Inclusion (as stated in the specific Decision Directive), e.g. Sulfuryl Fluoride deadline = 01/01/09.
49

General data requirements for Product Authorisation (PA) or Mutual Recognition (MR):
For MR of authorisation:
By the date of inclusion of the active substance, the PCS requires submission of:
Export file from the R4BP (indicating that mutual recognition is sought).
Following authorisation by the reference RMS, the PCS requires:
Copy of Summary dossier (1x electronic copy, 1x paper copy), which includes:
- R4BP application form- Annex IIA/IIIA sections (either equivalent data or LoA)- Annex IIB/IIIB sections (either equivalent data or LoA)- Confidential section
Draft Irish label (1x electronic copy, 1x paper copy)
SDSs for the active substance and co-formulants
For product authorisation:
Required by all applicants:
Fee for product authorisation (€5000.0019 as of 01/01/09)
Completed application form (1x electronic copy, 1x paper copy), which includes:
- Export file of the R4BP
19 There is an additional re-occurring annual fee of €300.00 that comes into effect from 01/09.50

- Similar information, necessary for national authorisations, that is not present in R4BP (incl. SDS and draft Irish label). NB: The PCS is still developing an application form in line with the PA&MRFG discussions on this issue.
- Full composition- Checklist information on active substance and biocidal product- Reference list- Relevant LoA
Required by the Applicant that secured Annex I Inclusion:
Summary dossier (Electronic 1, Paper 1), which include:
- Annex IIA/IIIA updated sections - Annex IIB/IIIB updated sections - Confidential updated section
Required by Applicants that did not support Annex I Inclusion:
Summary dossier (Electronic 1, Paper 1), which include:
- An equivalent Annex IIA/IIIA dossier satisfying requirements of the EU review.- An equivalent Annex IIB/IIIB dossier satisfying requirements of the EU review.- Or a notarised LoA from the primary data holder for the data pertaining to the
active substance and/or biocidal product.
* Please note that for authorisation/mutual recognition of a biocidal product containing more than 1 active substance, the product will be evaluated and subsequently authorised following inclusion in Annex I of the last active substance contained in the product. However, at the inclusion date of the preceding active substances the applicant is required to provide either:
- A notarised LoA, for either the active substance and/or product data, from the primary data holder.
- Equivalent data for the active substance/biocidal product (reference list, study protocols, provisional reports, final reports).
For product registration:
Required by all applicants:
Fee for product registration (€2000.0020 as of 01/01/09)
Completed application form (1x electronic copy, 1x paper copy), which includes:
- Export file of the R4BP- Similar information, necessary for national authorisations, that is not present in
R4BP (incl. SDS and draft Irish label). NB: The PCS is still developing an application form in line with the PA&MRFG discussions on this issue.
- Full composition- Checklist information on active substance and biocidal product- Reference list- Relevant LoA
20 There is an additional re-occurring annual fee of €300.00 that comes into effect from 01/09.51

Required by the Applicant that secured Annex I Inclusion:
Summary dossier (Electronic 1, Paper 1), which include:
- Annex IIA/IIIA updated sections - Annex IIB/IIIB updated sections (as per Article 8 (3) of Directive 98/8/EC)- Confidential updated section
Required by Applicants that did not support Annex I Inclusion:
Summary dossier (Electronic 1, Paper 1), which include:
- An equivalent Annex IIA/IIIA dossier satisfying requirements of the EU review.- An equivalent Annex IIB/IIIB dossier satisfying requirements of the EU review
(as per Article 8 (3) of Directive 98/8/EC).- Or a notarised LoA from the primary data holder for the data pertaining to the
active substance and/or biocidal product.
Additional Information:
It is hoped that by the end of the year the PCS will have available online:
- A full database of all biocidal products authorised in Ireland- Application information for national authorisation and EU dossier evaluation.- Pay structure information- Annex I Inclusion/non-inclusion information- Contact information
Payment
Ireland will invoice companies following receipt of the application. The details for EFT are as follows:
The name of the bank to which fees should be paid: Bank of Ireland The address of the bank: Main Street, Cavan, Co. Cavan, IRELAND IBAN: IE21 BOFI90329317183210 BIC code: BOFIIE2D
However, please note and include in the document: Payment by a Company to the Department of Agriculture, Fisheries and Food should only proceed following receipt of an invoice generated following an application received by the PCS.
Sell out period for non-supported biocidal products
6 months to phase-out the biocidal product from the market and supply chain.
Sell-out period for supported Biocidal products following Authorisation/Registration
Firstly, the Irish CA considers that authorisation holders of products also have a responsibility to manage the supply of existing products/labels into the supply chain following applications made to Competent Authorities for authorisation of the formulation.
52

In relation to the phase-out of products:
1. From the date of a new product authorisation, any new stocks of the authorised product being placed on the market must be in compliance with the conditions of the authorisation.
2. Existing stocks already in the supply chain may continue to be sold and used for a total period of 18 months.
3. However, existing stocks already at the primary distributor level from the date of a new product authorisation can no longer be sold after a period of 6-months.
4. Existing stocks already at the retail/end-user level may continue to be sold and used for a further 12-months following the removal of existing stocks from the primary distributor level.
5. The Irish CA reserves the right to enforce an immediate and full market withdrawal of a product, where appropriate.
53

Italy
Since March 2010 Italy has changed the national procedure, according to the harmonised procedures, regarding the transposition of the inclusion directives and now:
The application for a biocidal product, in order to maintain on the Italian market an existing product, can be submitted only by the inclusion date of the active substance;
There is a 6 months sell-out period (regarding the validity of the authorisation) + 6 months for use and disposal will be applied for non supported products;
If there is a request of authorisation of a new product containing substances included in Annex I (also according mutual recognition), we can accept the submission at any time and we can grant an authorisation from the date of inclusion of the active substances in Annex I.
In any case, we accept the application only in Italian. Technical documentation can be submitted also in English.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Italy - 2 - 2
Payment
The payments have to be done, before the application, on the giro account n. 60413812 in name of the Tesoreria Provinciale di Viterbo.
The bank details for transfers and foreign payments are the following:
Bic BPPIITRRXXX IBAN IT02F0760114500000060413812.
In the “Reason for Payment” section, the applicant should indicate the type of application required, the name of the active substance and of the biocidal product which the request refers to.
The proof of the payment (hard or electronic copy) has to be joined to the application. We don’t issue invoices.
For the amount of the fees, please see the Italian DM 16/4/2004 and our web-site:
http://www.salute.gov.it/biocidi/paginaInternaBiocidi.jsp?id=40&menu=instruments&lingua=english
Sell out period for non-supported biocidal products
Since March 2010 a new procedure applies according to the majority of the Member States: 6-months sell out period (validity of the authorisation) + 6 months for use and disposal.
54

Sell-out period for supported Biocidal products following Authorisation/Registration
Concerning the sell-out period for supported products, we accept that a product can remain on the market after the authorisation of the biocidal product for 12 months, in any case we can consider the possibility of an extension (it depends on the position of the majority of the MSs).
We believe that the old product has the authorization revoked (if authorized) since the entry in force of the decree and it can remain on the market (as disposal) for additional 12 months.
However, in order to apply this procedure, the product already on the market must have the same composition of the new biocidal product (only slight modifications on the composition can be accepted).
If the biocidal product will have a different composition in comparison to the old product, a major modification must be applied for the old product before the submission of the application of the new biocidal product.
55

Latvia
In Latvia we cannot accept the application for product authorisation and mutual recognition before official date of inclusion of active substance in Annex I of Biocidal Directive 98/8/EC. Therefore, Latvian Competent Authority can start accept the documentation after the date of inclusion, in case of sulfuryl fluorid after 1.1.2009.
In Latvia we have the legal requirement to submit product dossier in national language for Latvian companies. If the applicant is international company the dossier can be submitted in other language. Competent Authority, however, in this case may request a translation with a notarial certificate approving it. Usually we do not require that for English and Russian languages, i.e. languages for which we have sufficient expertise and experience.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Latvia 2 2 - 2
Sell-out period for supported Biocidal products following Authorisation/Registration
For the biocidal product for which authorisation or mutual recognition is or not granted, the retail shops (not the producers from their storehouses) can sell out the biocidal products which are bought before the deadline for compliance with Article 16(3) for additional 6 month. The time of buying of biocidal product must be proved by official documents, for example, invoice.
56
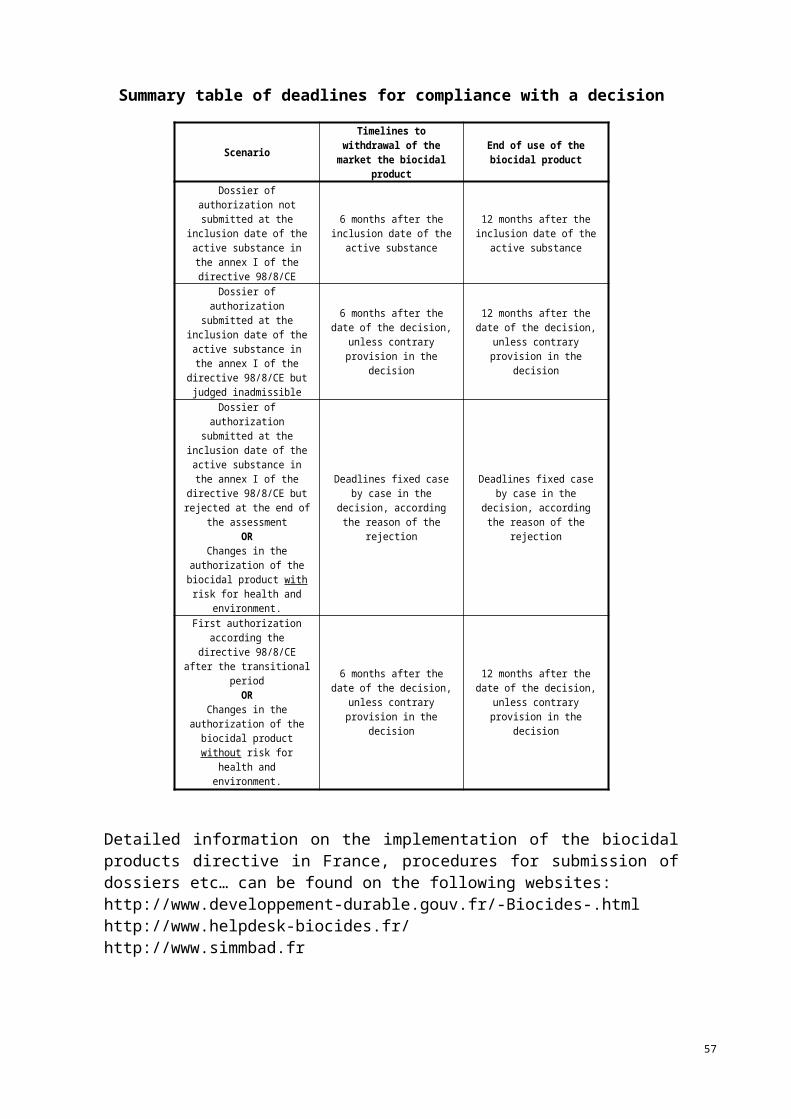
Luxembourg
Mail address:
Direction de la Santé -Division de la Pharmacie et des Médicaments
-BIOCIDES-
Allée Marconi – Villa LouvignyL-2120 Luxembourg
E-mail : [email protected]
For existing (notified) products, the deadline for submission of mutual recognition applications (Step 1) is the date of inclusion of the (last) active substance at the latest.
Please note that:
- Applicants are required to have a permanent office in the EU.- Labeling, instructions for use and safety data sheets must be in at least one of the
following languages: German; French.
❶ Preliminary requirements for MR are: A cover letter (paper copy) confirming that an application for mutual recognition is
made. (French, German preferred - English accepted). PLEASE SPECIFY if the product is already placed on the market + proof thereof (i.e. transitional period notification).
A signed paper copy of the application form generated via the online R4BP (Register for Biocidal Products, Link: https://webgate.ec.europa.eu/env/r4bp/) (French, German - English also accepted).See also: Link: http://ec.europa.eu/environment/biocides/prod_authorisation.htm .
❷An application can only be accepted as complete if the following information is submitted in a 2nd sending , once the first authorisation has been granted by the Reference Member State:
One (1) updated electronic copy of the summary dossier for the product. This dossier consists in a summary of the dossier as required in Article 8(2)(a) and Annex II B, Section X of directive 98/8/EC (Doc, I, I, III – in French or German or English). The updated summary dossier shall also contain any additional studies or information submitted to the Reference MS during the evaluation.
And/orLetter(s) of Access for active substance data / product data, valid for the MR procedure in Luxembourg.
Copy of the first authorisation: A translation into French (or German ), together with the original authorisation, is required.
57

If applicable, details regarding the Frame Formulation:
- composition of the Frame Formulation as established by the Reference MS + products covered by the FF,
- REF MS FF-certificate,
- composition of all products under the FF to be mutually recognized.
o Acknowledgment of receipt or copy of the bank transfer order*. Amount of Fees are provided in the modified “Règlement grand-ducal du 7 juin 2007” –
- For Mutual Recognition of SINGLE BIOCIDAL PRODUCTS the fee amounts to 400 € for both, registrations and authorizations.
- For Mutual Recognition of MULTIPLE PRODUCTS, all of which enter into a FF as established by the Reference Member-State, the fee amounts to 100 €/product (shade).
LINK: http://www.legilux.public.lu/leg/a/archives/2007/0092/2007A1784B.html
Summary of Product Characteristics (SPC), as established by the Reference Member State
Copy of the Assessment Report
Label (1 per user category) + instruction leaflet
Safety Data Sheet (in FR or DE)
Additional Application Form LU (available on demand)
Please submit all documents in electronic format (including a copy of those documents submitted on paper, such as the cover letter etc.).
__________________________.
* Fees for marketing authorization and registration applications of biocidal products in Luxembourg
The fees corresponding to the amount specified in the modified grand-ducal regulation of 7 June 2007 published in the Memorial A N° 92 du 14 June 2007 must be paid using bank transfer to one of the bank accounts stated below.
The communication relating to this bank transfer must contain the keyword “Biocide”+ the name(s) of the product(s).
1) Code Swift : CCPLLULL(IBAN) LU65 1111 0002 4753 0000 (Chèques Postaux, L-1010 Luxembourg)Recipient: Enregistrement, Bureau des Domaines à Luxembourg 1-3, avenue Guillaume L-1651 Luxembourg
2) Code Swift: BCEELULL(IBAN) LU72 0019 1002 0033 8000 (Banque et Caisse d’Epargne de l’Etat, Place de Metz, L-2954 Luxembourg)Recipient:Enregistrement, Bureau des Domaines à Luxembourg
58

1-3, avenue Guillaume L-1651 Luxembourg
59

Malta
We will be accepting applications in English as well as in Maltese since both languages are official languages in Malta.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Malta - 1 - 1
Payment:
Authority’s address: Malta Standards Authority2nd floor, Evans BuildingMerchants Street,Valletta VLT 1179Malta
Bank Details
Bank’s Name: Bank of Valletta plcBank’s Address: Santa Venera Branch229, Fleur De Lys RoadSanta Venera, BKR 09Malta
Account No: 15806183019Swift Code: VALLMTMTSort Code: 22 585IBAN Code: MT74VALL22013000000015806183019Bank’s BIC: VALLMTMT
We would like payments to be submitted in parallel to the submission of the application. Fees can be found on our website with the following link: http://www.msa.org.mt/rad/MSA_Provision_Of_Services_Fees.pdf
60

Netherlands
Following a decision on inclusion of an active substance into Annex I, authorisation holders of existing biocidal products on the Dutch market have to submit either an application for first authorisation to Ctgb, or a notification for mutual recognition. In case Ctgb does not receive such an application nor notification, we assume that the authorisation holder is not planning to keep this product on the Dutch market.
The data requirements are also valid for new biocidal products on the Dutch market.
Applicants need to send the following documents for First authorisation in the Netherlands
- A covering letter confirming that an application is made for first authorisation- A signed copy of the application form generated via the Register for Biocidal
Products (R4BP) available at https://webgate.ec.europa.eu/env/r4bp/- Letter of Access for the active substance(s) dossier(s) - Fee for first authorisation according to the Dutch tariff decree (www.ctgb.nl)
In electronic and paper copies:
- One electronic copy of the summary dossier (Doc I-III) according to the requirements of Directive 98/8/EC.
- Our national application form (Application form B, see our website www.ctgb.nl)- Appendix A (Active substance, see our website www.ctgb.nl)- Appendix B (Biocidal product, see our website www.ctgb.nl)- Appendix Composition (see our website www.ctgb.nl)- Appendix PGB/PUB (Practical Use Biocides, see our website)- A legal use and directions for use in Dutch (so called WG/GA, see our pesticide
database for examples www.ctgb.nl) - MSDS’s of the biocidal product, the active substance(s) and the formulants (not
older than 5 years)- List of references
Applicants need to send the following documents for Mutual recognition of the first authorisation
3. The first step (notification) of mutual recognition.
Ctgb requires the following documents (paper copies):
- A covering letter confirming your notification for mutual recognition of authorisation for the product
- A signed copy of the application form generated via the Register for Biocidal Products (R4BP) available at https://webgate.ec.europa.eu/env/r4bp/
4. The second step (application) of mutual recognition.
Applications for the mutual recognition of existing biocidal products on the NL market (re-registrations) have to be submitted to Ctgb within 2 months after authorisation of the product by the Reference Member State.
61

Applications for the mutual recognition of new products to the NL market can be submitted at any time.
The applications shall include the following:
- A covering letter confirming your application for mutual recognition of authorisation for the product
- Letter of Access (when relevant)- An electronic copy of the summary dossier (Doc I-III) for the product as
submitted to the first member state- Fee for mutual recognition according to the Dutch tariff decree (www.ctgb.nl)
In electronic and paper copies:
- The Product Assessment Report by the Reference Member State - A summary of the product characteristics (SPC) - The authorization document (letter) uploaded by the RMS in R4BP (or
alternatively, a certified copy of the first authorization). - Our national application form (Application form B, see our website www.ctgb.nl)- Composition of the product (Appendix composition, see our website
www.ctgb.nl)- MSDS’s of the biocidal product, the active substance(s) and the formulants (not
older than 5 years)- A legal use and directions for use in Dutch (so called WG/GA, see our pesticide
database for examples www.ctgb.nl) - PGB/PUB (Practical Use Biocides, see our website)
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Netherlands 1 1 1 1
Payment
Fees should be paid before sending the application to the following bank account:
CtgbRabobank Nederland,Bankno. 397076053PO Box 216710 BA EdeIBAN: NL27 RABO 0397076053BIC: RABONL2U
Sell out period for non-supported biocidal products
According to the proposed procedures the decision on the inclusion of an active substance contains three important dates:
1. Directive comes into force (0)
62

2. Inclusion comes into force (+ 2 year).
3. Authorisation to be granted, modified or cancelled (+ 2 year)
If these are the set dates in the decision the national authorisation will be cancelled 2 years after the inclusion comes into force when no application and dossier for authorisation of the biocide is submitted (= date authorisation to be granted, modified or cancelled). If the expire date of national authorisation lies before that date, the national authorisation will expire on the set national date. No sell-out of phase-out period will be decided on; the dates are set and knowable so that the parties concerned can make their preparations on the coming situation that the biocide may not be put into the market or used.
Sell-out period for supported Biocidal products following Authorisation or Registration
In the Netherlands, the judge has ruled that we have to allow for the period of grace that is necessary, which information is to be provided by the applicant. Ctgb is to check the reasonableness of the information, and has to provide a motivation in their Decision. The standard periods are ½ year for sales, 1½ year to use up the product. During that period of grace, two different labels can co-exist.
In conclusion: if the applicant has convincing information why the period of grace should be 24 months, Ctgb may take that up in their Decision.
63

Norway
At present, NO will require the following to be submitted:
A covering letter confirming that an application is made for mutual recognition of authorization for the product. This should preferably be signed by the one presently responsible for placing on the Norwegian market.
A filled in paper copy of the application form generated via the R4BP (in future no printout is needed)
One electronic copy of the summary dossier for the product as submitted to the Reference Member State.
We will at this point of time not require:
Other part of application form not covered by R4B (not yet available) Label proposal and instruction leaflets and SDS in Norwegian (if already available
they could be submitted electronically or on paper copies now). Fees for handling the application to be paid.
This have to be submitted/fulfilled together with a copy of the first authorization and an assessment report as foreseen being made by the Reference Member State. This will have to be submitted at the latest 8 weeks after the first authorization has been given.
In general we accept all part of a product dossier to be submitted in either English or Norwegian (except label proposals, proposal for instruction of use and a copy of the SDS. These documents all have to be in Norwegian).
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Norway - 1 - 1
Payment:
The Norwegian Authority will send an invoice upon receipt of the application.
The details from Norway:
Name of the bank: Nordea Bank Norge ASAAddress of the bank: 1166 Sentrum
0107 OsloNorway
IBAN: NO4663450502997BIC: NDEANOKK
64

Sell out period for non-supported biocidal products
6 months sell-out period + 6 months for use or disposal.
Sell-out period for supported Biocidal products following Authorisation/Registration
12 month (one year) is the standard time frame for new package, labels etc.
However the applicant should be allowed to ask for longer sell out period in cases were e.g. multilingual pre-printed label on the product package are used. For such product subject to mutual recognition the time lag between the date of reference member state authorisation and the concerned member state authorization could also be more than 6 months if the concept of “authorisation in parallel” is not used. Another aspect is also the timing of the authorisation for product with mainly seasonal use (e.g. antifouling in Nordic countries).
However there could also be cases were the product have been authorised under BPD with more restricted use and stricter risk mitigation measures to be applied than under present existing legislation (e.g. rodenticides). In such cases it could be warranted to require a shorter sell out period for products not complying with new labels, use instruction or packaging requirements.
65

Poland
1. Requirements for first authorization of biocidal product:
Confirmation of payment concerning submission of the application. The fee for a demand of biocidal product authorisation has been fixed to 1000 PLN in first step and 24 000 PLN after the completeness check is accepted (the Regulation of Minister of Health of January, 12 2007 concerning charging activities related to authorization for the placing of biocidal product on the market (Official Journal No. 8, item.62). The payment should include in the title ‘payment concerning the placement of the biocidal product (name of product) on the market’;
Application form according to the Regulation of Minister of Health of May, 28, 2008 concerning the model application form for biocidal product registration, authorisation, provisional authorisation, presentation and documentation requirements, which should correspond to documentation necessary for the active substance and biocidal product assessment. The application form is available (in Polish) on:http://www.urpl.gov.pl/system/files/PL/Produkty-biobojcze/formularz_wzoru_wniosku_z_Rozporzadzenia.doc
Application form shall be signed by the future authorisation holder;
Application form generated via R4BP, in Polish, signed by the applicant;
Dossier, in Polish or English;
Safety Data Sheet, in Polish;
Proposed labeling and instruction leaflet (if needed), in Polish;
Proposal for Summary of Product Characteristics, in English;
Document certifying the legal status of the applicant (and of the future authorisation holder, if different);- not older than 6 months- indicating the person authorized to represent it - sworn translated into Polish;
If several applications are filed by the same applicant simultaneously or during 6 months, it is sufficient to submit one original in the documentation and for the rest just a copy and declaration that: ‘the original document is located with the application in the matter of the biocidal product named…’.
Power of Attorney- Applicant may act in person or by attorney;- Power of attorney from abroad should be sworn translated into Polish;- Power of attorney should be original and signed by the person authorized to
represent the company, as indicated in the above mentioned document certifying the legal status of the applicant;
- Document confirming granting of authority to the attorney is subject to the revenue duty. The level of revenue duty is given in the scale of charge in the Appendix to the Act of November 16, 2006 on Revenue Duty (Official Journal No. 225, item 1635) and amounts 17 PLN. If several applications are
66

filed by the same applicant simultaneously, it is sufficient to submit one original document granting of authority in the documentation and for the rest just a copy and declaration that ‘the original document is located with the application in the matter of the biocidal product named…’;Revenue duty of 17 PLN should be paid for submitting both original and copy.
The payment should include in the title: ‘payment concerning power of attorney (names of products shall be listed as well)’.
2. Requirements for MRP:
General remark: According to polish law act on biocides only the company, which is the authorisation holder in RMS can apply for mutual recognition of this authorisation in Poland (act as an applicant). Applicant can indicate any other company as a future holder of authorisation in Poland.
In the first step of MRP we require:
A letter of intention for mutual recognition, in Polish;
Application form generated via R4BP, in Polish, signed by the applicant;
Summary dossier, in Polish or English.
In the second step we will require:
Confirmation of payment concerning submission of the application. The fee for a demand of MRP has been fixed to 6250 PLN for authorization of biocidal product (the Regulation of Minister of Health of January 12, 2007 concerning charging activities related to authorization for the placing of biocidal product on the market (Official Journal No. 8, item.62);
The payment should include in the title ‘payment concerning the placement of a biocidal product (name of product) on the market’.
Application form according to the Regulation of Minister of Health of May, 28 2008 concerning the model application form for biocidal product registration, authorisation, provisional authorisation, presentation and documentation requirements, which should correspond to documentation necessary for the active substance and biocidal product assessment. The application form is available (in Polish) on:http://www.urpl.gov.pl/system/files/PL/Produkty-biobojcze/formularz_wzoru_wniosku_z_Rozporzadzenia.doc
Application form shall be signed by the future authorisation holder.
Print out copy of authorization granted by Reference Member State, from R4BP and its sworn translation into Polish;
Copy of Product Assessment Report of Reference Member State;
Summary of Product Characteristics, in English;
Proposed labeling and instruction leaflet (if needed), in Polish;
Safety data Sheet, in Polish;67

Document certifying the legal status of the applicant (and of the future authorisation holder, if different):
- not older than 6 months;
- indicating the person authorized to represent it;
- sworn translated into Polish.
If several applications are filed by the same applicant simultaneously or during 6 months, it is sufficient to submit one original in the documentation and for the rest just a copy and declaration that ‘the original document is located with the application in the matter of the biocidal product named…’
Power of Attorney
- Applicant may act in person or by attorney;
- Power of attorney from abroad should be sworn translated into Polish;
- Power of attorney should be original and signed by the person authorized to represent the company as indicated in the above mentioned document certifying the legal status of the applicant;
- Document confirming granting of authority to the attorney is subject to the revenue duty. The level of revenue duty is given in the scale of charge in the Appendix to the Act of November 16, 2006 on Revenue Duty (Official Journal No. 225, item 1635) and amounts 17 PLN. If several applications are filed by the same applicant simultaneously, it is sufficient to submit one original document granting of authority in the documentation and for the rest just a copy and declaration that ‘the original document is located with the application in the matter of the biocidal product named…’.
Revenue duty of 17 PLN should be paid for submitting both original and copy. The payment should include in the title: ‘payment concerning power of attorney (names of products shall be listed as well)’.
The deadline for submission of an application for an existing product is at latest the date of inclusion of the active substance into Annex I/IA.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Poland 1 2 1 2
68

Payment:
Payment of the fees for authorization/registration:
Fees should be paid before sending the application to the following bank account:
Urząd Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych,
Ząbkowska 41, 03-736 WarszawaNarodowy Bank Polski
00-950 WarszawaPlac Powstańców Warszawy 4
Bank account number (foreign payments):
PL30 1010 1010 0094 1022 3100 0000Kod BIC NBP - NBPLPLPW
Bank account number (national payments):
NBP O/O Warszawa: 30 1010 1010 0094 1022 3100 0000
Fees should be paid separately for each application
Payment of the fee concerning power of attorney:
Payment shall be transmitted to the bank account of :
Urząd m. st. Warszawy Dzielnica Praga Północ Bank Handlowy w Warszawie S.A.
Bank account number:
PL 96 1030 1508 0000 0005 5002 6074Kod SWIFT- CITIPEPX
Sell out period for non-supported biocidal products
The national authorisation is cancelled after 6 months after the date of Annex I/IA inclusion. Biocidal product can stay on the market no longer than 18 months after the date of Annex I/IA inclusion.
69

Portugal
A - Biocial Products PT8/Wood Preservatives
Application for Mutual Recognition in Portugal, following a decision on inclusion of an existing active substance into Annex I or I-A of the Decree Law nº 121/2002 from 3rd May
Documents to submit to the Portuguese Competent Authority Direcção-Geral de Agricultura e Desenvolvimento Rural (DGADR):
1. Note of intention21:
Within 2 years from entry into force the inclusion directive22(2), the Company that wants to continue to place the existing biocidal product on the market has to submit in a first step the documents below (1.1 to 1-3)
1.1-A cover letter in Portuguese language, signed by a person responsible in the Company, confirming that an application is made for mutual recognition of authorisation for the product.1.2-Print out of the first part of the application form fulfilled in the site of the registration for the Biocidal Products (R4BP): https://webgate.ec.europa.eu/env/r4bp/ 1.3-One electronic copy of the summary dossier (Doc I-III) of the product as submitted in the Reference Member State1.4-Letter of access for the active substance(s) dossier(s)
2. Formal application:
Within two month of having obtained the first authorization for the product in the Reference Member State (RMS) the Applicant shall submit to the Portuguese Competent Authority (DGADR) the documents below (2.1 to 2.6)
2.1-one certified copy of the granted authorization in the Reference Member State (RMS)2.2-The assessment report from the RMS in English or Portuguese language2.3-Print out of the second part of the application form fulfilled in the common register for biocidal products (R4BP) in the site https://webgate.ec.europa.eu/env/r4bp/Dated and signed by a person responsible in the Company2.4-Safety data sheet in Portuguese 2.5-Copy of the proposed label, in Portuguese2.6-Evidence of the payment of the fee23 (Portaria nº702/2006 de 13 de Julho)
B - Direcção Geral de Veterinária (DGV) Portuguese Competent Authority to Veterinary Biocidal Products for the application will require:
1. For the notice of intention (only for products already in the market at the date of inclusion):
21 For products with more than one active substance, the deadline shall be the one set out in the last inclusion decision relating to its active substances
22 Until the date of inclusion of the active substance in the Annex I or I-A23 1500 to 2100 euros depending on the work
70

- A cover letter indicating:
(1) that an application for the product authorisation is being made in another member state;
(2) the intention of submit a mutual recognition for that product in Portugal.
- Copy of the R4BP.
2. For the formal application:
- Application form in Portuguese;- Certified copy of the official authorisation granted by the reference member state;- Certified copy of the reference member state authorisation assessment report;- Proof of payment (the applicant has until 10 day after the application form);- 4 electronic copies and 1 paper copy of the summary dossier of the product;- If necessary we may request 4 electronic copies and 1 paper copy of all the studies
submitted by the applicant;- Label proposal and instruction leaflets in Portuguese;- Safety Data Sheet in Portuguese
C - General Directorate of Health (DGS) is the Competent Authority for all biocidal product types, excluding PT 8 and Veterinary Biocidal uses.
In case we do not receive a submission related to a product already placed on the Portuguese market, DGS will conclude that the product authorisation holder does not intend to continue with this product on the market and will inform him that, in accordance with the provisions of Article 7(3) of the Directive (in Portugal, article 14.º of Decreto-Lei n.º 121/2002), a period of grace of 6 months after the date of inclusion of the active substance will be granted, for storage and stocks phasing-out. After this deadline the products are considered as removed from the market.
For a product, already placed on the Portuguese market, containing more than one active substance for a given Product Type (PT), the deadline to submit an application is the date of inclusion of the last active substance in the product to be included in the BPD Annex (I or IA).
If a biocidal product containing all active substances included in one of the BPD Annexes already exists on the Portuguese market, to be able to remain on the market while applications are evaluated, the companies have to submit till the deadline mentioned above, the following:
1 - For the application to demand the mutual recognition of an authorisation, the following elements must be submitted, on electronic form (1 CD or DVD) and 1 paper copy, to Director General of Health - General Directorate of Health – Alameda D. Afonso Henriques, 45 – 1049-005 Lisboa:
- A covering letter from the Applicant, written in Portuguese language, dated and signed.- Print out of the R4BP application form (available from https://webgate.ec.europa.eu/env/r4bp/), dated and signed (can be in Portuguese or English).- The summary dossier, as described in article 4 paragraph 1 of Directive 98/8/CE (in Portugal, article 22. º, paragraph 1 and 2 of Decreto-Lei n. º 121/2002, de 3 de Maio) or a letter of access, dated and signed (if this is the case).
71

- A certified copy of the first authorisation and the assessment report from the Reference Member State.
The proposal for the Classification and Labelling, the Material Safety Data Sheet, and the copy of the label of the product have to be submitted in Portuguese. Other information in this dossier can be accepted in Portuguese or English.
The fee related to a demand of mutual recognition of an authorisation has been fixed to 1.539 EUR, at the moment, by point 6, table I of the annex of rectified Portaria n.º 702/2006, of 13 July 2006, and must be paid in advance, after the request from our Financial Department
Applications for mutual recognition of an authorisation will be put on hold until the certified copy of the first authorisation, to be granted by the Reference Member State, is received by DGS. The application will only then be considered as complete.
2 - For the application to demand the mutual recognition of a registration, the following elements must be submitted, on electronic form (1 CD or DVD) and 1 paper copy, to Director General of Health - General Directorate of Health – Alameda D. Afonso Henriques, 45 – 1049-005 Lisboa:
- A covering letter from the Applicant, written in Portuguese language, dated and signed.- Print out of the R4BP application form, dated and signed (can be in Portuguese or English).- Information, as described in article 4, paragraph 1 and article 8, paragraph 3 of Directive 98/8/CE (in Portugal, paragraph1, article 16.º and paragraph 3, article 22.ºof Decreto-Lei n. º 121/2002, de 3 de Maio) or a letter of access, dated and signed (if this is the case).- A certified copy of the first registration and the assessment report from the Reference Member State.
The proposal for the Classification and Labelling, the Material Safety Data Sheet, and the copy of the label of the product have to be submitted in Portuguese. Other information in this dossier can be accepted in Portuguese or English.
The fee related to a demand of mutual recognition of a registration has been fixed to 1.539 EUR, at the moment, by point 6, table I of the annex of rectified Portaria n.º 702/2006, of 13 July 2006, and must be paid in advance, after the request from our Financial Department.
Applications for mutual recognition of a registration will be put on hold until the certified copy of the first registration, to be granted by the Reference Member State, is received. The application will only then be considered as complete.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Portugal 1 (4 for products for veterinary use)
1 1 (4 for products for veterinary use)
1
72

Payment:
Portugal has 3 Competent Authorities for the implementation of Directive 98/8/EC, concerning the placing of biocidal products on the market:
1 - Direcção-Geral da Saúde - DGS – Receive applications for authorisations of all biocidal product types excluding wood preservative products and veterinary use products.
The payment should be made upon request of the Competent Authority - which would then send an invoice.
Caixa Geral de DepósitosAv. Almirante Reis, 227-A-B1000-049 LISBOAIBAN: PT50003500630008709343031BIC: CGDIPTPL
2 - Direcção-Geral de Agricultura e Desenvolvimento Rural – DGADR - Receive applications for authorisation of wood preservative products.
The payment should be made upon request of the Competent Authority, by invoice from Direcção de Serviços de Informação, Gestão e Administração.
Instituto de Gestão da Tesouraria e do Crédito Público, IPAv. da República n.º 57-6.º1050-189 LISBOAIBAN: PT50078101120112001248048BIC: IGCPPTPL
3 - Direcção-Geral de Veterinária – DGV – Receive applications for authorisation of veterinary use products.
The payment should be made upon request of the Competent Authority, by invoice from Direcção de Serviços de Informação, Gestão e Administração.
Instituto de Gestão da Tesouraria e do Crédito Público, IPIBAN: PT50 0781 0112 00000007784 96BIC — IGCPPTPL
Sell out period for non-supported biocidal products
6 months sell-out for the biocide manufacturer from the date of Annex I inclusion. No further time is granted for use or disposal.
Sell-out period for supported Biocidal products following Authorisation/Registration
The sell-out period for supported product (this means, the period that an authorised biocidal product can remain on the market with the old labelling after the approval of the product under BPD) would be 6 months.
73

Slovakia
“Existing biocidal product” in Slovakia refers in the context of this document to those products which have already been placed on the market of the Slovak Republic in accordance with transitional provisions of Article 33, paragraph 2 of the Act No 217/2003 Coll. transposing Directive (EC) 98/8/EC. As a basic requirement, these products:
must bear a valid registration number on the product label at the date of inclusion of the active substance into Annexes I or IA
and at the same time they must be listed in the SK On-line Registry of Biocidal Products.
If any applicant wishes to continue to place an existing biocidal product on the market of the Slovak Republic, the SK CA recommends to submit by the inclusion date at the latest (except for products containing more than one active substance, for which the deadline shall be the one set out in the last inclusion decision relating to its active substances):
either an application for authorisation complying with the requirements of Article 4 of the Act No 217/2003 Coll. (Article 8 of the Directive 98/8/EC) to one of the competent authorities for implementation of Directive (EC) 98/8/EC in the European Union
or a letter of intention24 for placing an existing biocidal product on the market of the Slovak Republic based on the procedure of mutual recognition according to Article 10 of the Act No 217/2003 Coll. (Article 4 of the Directive 98/8/EC) to the Centre for Chemical Substances and Preparations. The applicants are recommended to use a standardized form for letter of intention which is placed on the CCSP web page.
The CCSP accepts applications in SK or EN languages. The applicants are also advised to use the European Register for Biocidal Products (R4BP).
Starting March 2009, detailed instructions in SK and EN languages with all necessary links to the related EU web pages are placed on the CCSP web page: http://www.ccsp.sk/biocides/pages/englishbio.html
For more specific information the applicants are advised to use the CCSP electronic Help-Desk: http://helpdesk.ccsp.sk/ .
24 In accordance with Article 4(1) BPD the applicant shall submit a certified copy of the first authorisation granted. Since this Article does not contain any specific provisions related to handling of incomplete applications, the CCSP is obliged to follow the deadlines in accordance with the national Act on State Administrative Procedures. As a consequence, the CCSP cannot wait until the first authorisation in one of the EU MS is granted and hold the incomplete application for more than 60 days. This will result in the decision on rejection of the application for mutual recognition. Due to this fact, the CCSP proposes to amend the existing version of the document on the post-Annex I procedure by adding the words “or a letter of intention” to the second bullet point of Article 2.1.1 Submission of application. This will avoid possible misunderstandings in the future and at the same time it will enable the CCSP to meet the harmonized time frames as proposed by the above mentioned document.
74

Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Slovakia 1 5 - 5
Payment:
The SK CA will invoice companies following receipt of the application. The actual bank details are available on the CCSP website:
http://www.cchlp.sk/, section BIOCIDES, chapter "How to place BP on the market" under point No 4 PAYMENT INSTRUCTIONS.
Sell out period for non-supported biocidal products
There is registration expiry date for product on the on-line registry and this is final date for placing on the market, if product is not supported for product authorisation. Then enforced for disposal.
Sell-out period for supported Biocidal products following Authorisation/Registration
Sell-out period for BP placed on the market under transitional measures is in SK stipulated by Article 33a (7) on Biocides Act No. 217/2003 Coll.
The CCSP will grant the sell-out period for products with labels according Article 33 of the Act on biocides (national registrations) up to 12 months as a maximum.
75

Slovenia
If the product is already placed on the Slovenian market companies have to submit, in order to prolong the stay on the market, before the date of inclusion of the active substance in Annex I:
- Application for the authorisation /registration in accordance with the Biocidal Products Act (OJ RS No 61/06) or- A letter of intention for mutual recognition.
Authorisation / registration:
- a letter in the Slovene language - completed and signed hard copy of the application form, printed from the application R4BP in the Slovene language - one electronic copy of dossier for biocidal product and each active substance in the biocidal product or Letter of Access for biocidal product or for each active substance in the biocidal product; in the English language, if possible in accordance with article 3 of Rules on regular permissions for biocidal products (OJ RS, No. 62/07, 41/08)- proposed label and safety data sheet in the Slovene language stated date of payment of administrative fee on the letter or certificate of its payment - stated date of payment of the administrative fee and cost of procedure fee on the letter or certificate of its payment- authorisation to market dangerous chemicals if the biocidal product is classified as dangerous.
Mutual recognition
In the first step the letter on intention covers:
- A covering letter confirming that an application is made for mutual recognition of authorization for the product in Slovenian language. Other documentation can be in Slovenian or English.- A filled in paper copy of the application form generated via the R4BP- One electronic copy of the summary dossier for the product as submitted to the first member state.- The administrative fee to be paid.
Additional request: a declaration stating that the applicant agrees with the withdrawal of the biocidal product from the market when fails to submit the application for the issue of the mutual recognition of the authorisation within two months of the first issuance of the authorisation.
In the second step we will require:
- The summary dossier in accordance with BPD- Certified copy of the first authorisation and the assessment report- Label proposal, SDS and instruction leaflets, all in Slovenian- Fee (cost of procedure) to be paid.
If a biocidal product is classified as dangerous, the applicant needs to obtain a Licence for trade in hazardous chemicals in Slovenia as well. To get this licence the applicant has
76

to fulfil requirements from the Article 44 and 45 of an Act on Chemicals. http://www.uk.gov.si/en/areas_of_work/authorization_for_carrying_out_activities/
More information regarding the data requirements for the procedures and fees are available at: http://www.uk.gov.si/en/areas_of_work/biocidal_products/
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Slovenia NO 1 NO 1
Payment:
Fee for authorisation is laid down by Article 2 of the Decree on the costs of procedures of placing biocidal products on the market (Official Gazette of the Republic of Slovenia no. 9/09). Fee is paid in two parts, namely:
- upon the submission of the application 1500 EUR shall be charged for review and processing of the application;
- when the Chemicals Office has confirmed that the application and the requested documentation are complete, the Applicant shall be asked to pay the second part of the fee amounting to 4300 – 7200 EUR (for one type a product type of use). Prior to the issue of authorisation (decision), the Chemicals Office can ask the Applicant to pay any additional costs incurred.
Fee for mutual recognition of authorisation permit is laid down by Article 2 of the Decree on the costs of procedures of placing biocidal products on the market (Official Gazette of the Republic of Slovenia no. 9/09). Fee is paid in the second part, amounting to 2400 EUR.
Additionally, the application is subject to the payment of administrative fee per Tariff no. 1 of the Administrative Fees Act (Official Gazette of the RS, no. 42/07-UPB3, 126/07 and 88/10) in the amount of:
- 3.88 EUR from 20.11.2010 till 30.6.2011- 4.21 EUR from 1.7.2011 till 31.12.2011- 4.54 EUR from 1.1.2012The administrative fee should be paid when the application is given (at the beginning) to the following bank account:
Banka SlovenijaSlovenska 35SI-1000 LjubljanaIBAN: SI56011001000315637BIC: BSLJSI2X
The bank transfer should be mention as reference number '27154-7111002-18415000' as well as the name of the biocidal product.
77

Sell out period for non-supported biocidal products
If no new application for authorisation/registration or mutual recognition of authorisation has been filed for the biocidal product until the date of inclusion of the active substance in the Annex I, IA of Directive 98/8/EC the biocidal product will be recalled from the market and supply chain within six months after the date of inclusion.
If the application for authorisation/registration or mutual recognition of authorisation is incomplete biocidal product will be recalled from the market and supply chain within six months after the decision on incomplete application.
Sell-out period for supported Biocidal products following Authorisation/Registration
We can be flexible regarding sell-out period of supported products.
78

Spain
First authorisation
Applicants for authorization of biocidal products have to submit the following documents:
- EU-harmonised application form (R4BP) (*), enclosed by:o Full composition.o Checklist (Active substance/product).o Reference list.o Letters of Access.
- Application form (Second part) with relevant information for Spanish Authority. For this part we have a provisional bilingual form.
- Signed declaration in which applicant confirm that this is the true composition of the product placed on the market (*).
- Certificate of analysis of active substances (Laboratory at least certified to be in conformance with ISO 9001) (*).
- Safety Data Sheet and labelling (in Spanish).- Justification of paid fees.- Full dossier in compliance with Article 8 DPB (1 paper copy, and 2 electronic
copies) (*). Section X of Annex IIB, in Spanish and English.- Certified Spanish translation if pointed documents (*) are not in English.
Mutual recognition
Holders of registered biocides in Spain which active substances have been included in annex I or IA of Directive, who wish follow on marketing their products on the Spanish market after the date of inclusion, they have to submit the following documents:
First step
- EU-harmonised application form (R4BP) (*) , enclosed by:o Full composition, adding a signed declaration in which applicant confirm
that this is the true composition of the product placed on the market.o Checklist (Active substance/product)o Reference listo Letters of Access
- Application form (Second part) with relevant information for Spanish Authority.
For this part we have a provisional bilingual form.
- Safety Data Sheet and labelling (in Spanish).- Justification of paid fees.- Summary of the dossier, as stipulated in Article 4 DPB (1 paper copy, and 2
electronic copies) (*). Section X of Annex IIB, in Spanish and English.- Covering letter confirming that an application has been made in the Reference
Member State (*).
Second step:
- Certified copy of the first granted authorization (*).
79

- Certified copy of the Reference Member State Authorization Assessment Report (*).
And (first and second step):
- Certified Spanish translation if pointed documents (*) are not in English.
Deadline for submission is the date of inclusion of the substance in Annex I or IA.
If the product is not on the Spanish market, the documents are:
- EU-harmonised application form (R4BP) (*) , enclosed by:o Full composition, adding a signed declaration in which applicant confirm
that this is the true composition of the product placed on the market.o Checklist (Active substance/product)o Reference listo Letters of Access
- Application form (Second part) with relevant information for Spanish Authority. For this part we have a provisional bilingual form.
- Safety Data Sheet and labelling (in Spanish).- Justification of paid fees.- Summary of the dossier, as stipulated in Article 4 DPB (1 paper copy, and 2
electronic copies) (*).- Certified copy of the first granted authorization (*).- Certified copy of the Reference Member State Authorization Assessment
Report(*).- Certified Spanish translation if pointed documents (*) are not in English.
Payment:
Bank account:
Name: Ministerio de Sanidad y Política Social Account number: 0182 2370 41 0200000822 IBAN: ES48 0182 2370 4102 00000822BIC: BBVAESMMXXX Bank: Banco Bilbao Vizcaya Argentaria, SA. Address: Pº de Recoletos 10, ALA SUR – PL.1 City (Postal Code) 28001 Madrid Country: SPAIN
Any expenses and bank commissions caused by the transfer will be paid by the person making the transfer.
One of the copies of the bank transfer must be joined to the Application form when it is submitted.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
80

Spain 1 3 1 3
Sell out period for non-supported biocidal products
Concerning the manufacturing process, the deadline to stop the manufacture of the product is the Annex I inclusion date. Regarding the elimination of the remaining stocks, the deadline will be the first happening between the expiry date of the resolution under the existing national register and the deadline for compliance of the inclusion directive. Spain always informs accordingly to the applicant, once the inclusion directive is published.
Sell-out period for supported Biocidal products following Authorisation/Registration
Concerning the period that a biocidal product can remain on the market with the old labelling after the approval of the product under the Directive 98/8, we think that, if the product under the national legislations has a different composition, name or holder than the product evaluated under the Directive, the deadline of compliance must be fulfilled. If the product is the same, and it is only an issue of labelling, 12 months is a period appropriate to eliminate the old product.
81

Sweden
Applications for authorisation and mutual recognition should be submitted at the latest on the day for the inclusion in Annex I to 98/8/EC of the last active substance in the biocidal products. Application forms, guidance and information regarding the data requirements and fees are available at www.kemi.se.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
Sweden25 NO 1 NO 1
Payment:
When the Swedish Chemicals Agency (KemI) has received an application, a confirmation of received application will be sent, together with details for payment and a reference number that should be indicated when paying the fee.
Sell out period for non-supported biocidal products
Biocidal products already authorised in Sweden according to national legislation can stay on the Swedish market until the authorisations expire, but no longer than until the last day of compliance with article 16(3), i.e. 2 years after date of inclusion. Any additional sell-out period for others than the authorisation holder (after the expiry of the authorisation) is decided on a case-by-case basis, but it is usually a period of one year. The timelines in the gentlemen’s agreement have not been implemented in the Swedish legislation due to the lack of interim deadlines in inclusion directives. Therefore phase out before the last date of compliance due to non-submission of an application is not possible for products where the national authorisations are valid until the date of compliance or for products that do not require authorisation according to previous national rules. In both these cases applicants could come in with applications up until the last day and we would be obliged to accept them. However, if an application is submitted after the date of inclusion, we cannot guarantee that the product will be authorised according to the BPD by the last date of compliance. In these cases the products may not be marketed between the expiry of the national authorisation or the compliance date and the date of the decision on an authorisation in accordance with the Directive. Therefore, applicants are advised to submit by the date of inclusion. The harmonised timelines will be followed with regards to evaluation and mutual recognition.
Sell-out period for supported Biocidal products following Authorisation/Registration
Current practice in Sweden for changes of authorisations according to older national practice (not BPD).
If a decision for change of an authorisation leads to a change in information to be included in the labelling of a biocidal product, the product first placed on the market by the authorization holder should have the new label from the date the change enters into
25 However, the application form, letters of access and letters of attorney must be submitted to the Swedish Chemicals Agency in original paper copies (or certified paper copies).
82

force. The date for the decision to enter into force is set dependant on the type of change and if the authorisation holder has expressed a wish regarding that timepoint.
If the change of an authorisation is being decided upon in connection with a renewal of an authorisation, the change will enter into force at the same time as the renewal. The renewal and the change will normally enter into force the day after the expiration date for the old authorization. The decision is normally made some time before it enters into force, allowing for adaption to the change.
In case a decision is made shortly before the change enters into force, the Swedish Chemicals Agency can grant the authorisation holder the right to place the product on the market with the old label for a certain period of time after the change has entered into force.
Currently, only adaption to the change by the authorisation holder for the first placing on the market has been considered. The downstream sellers and users have been able to sell and use the product with the old label (for as long as the product may be sold and used).
In cases where the change of a product cannot be considered a major change due to a greater risk/exposure with the old label, i.e. a more narrow area of use, the product is given a new authorisation number and the product with the older authorization number is phased out. A phase out means that the authorisation holder is not allowed to place the product on the market after the authorisation has expired. Others than the authorisation holder are normally granted the right to sell the product for one more year, and use is normally permitted for two years after the expiry date of the authorisation (for consumer products no last date for use is normally set).
Please note that this is the practice for products authorized in Sweden. We are currently discussing what will be the practice for products not yet authorized according to the older national practice.
83

Switzerland
We ask for a notification of intention the latest on the date of inclusion of the active substance. This is a form to be downloaded from internet. The company must indicate among others in which country it will apply for the first authorization (or registration) and promises with its signature to apply for a mutual recognition the latest two months after the first authorization has been granted in an European or EEA country. We do not require other documents, though most of the companies have sent us a copy of the R4BP form.
A dossier for a mutual recognition required in Switzerland is the one foreseen in the BPD. Though, as we do not have access to not published data, we may ask in some cases for a copy of the documents IIA and IIIA related to the active substances.
We accept dossiers in one of the three national languages (German, French and Italian) or in English.
Payment
An invoice with all payment references is sent after reception of the application.
Sell out period for non-supported biocidal products
Phasing-out period is 12 months after inclusion date.
84

United Kingdom
The Biocidal Products Directive is implemented in the UK in the Biocidal Products Regulation (BPR).
For products already on the market which contain existing active substances, the BPR (Schedule 13) defines transitional arrangements. The applicant must request a Certificate of Exemption (CoE), which exempts their product from the requirements of BPR, in order to remain on the UK market while the evaluation of their product authorisation takes place. The CoE lasts until a decision on the authorisation is made (but not exceeding 3 years).
Prior to an active entering Annex I the company should inform us of their intention to make an application for mutual recognition within 2 months of the first authorisation being granted (a “notice of intention”). Once we have this they are issued with a CoE.
For the notice of intention we would expect to receive:
A cover letter indicating that an application for product authorisation is being made in another Member State and mutual recognition of that product will be sought in the UK
Copy of the R4BP form (part 1)
When the formal application for mutual recognition is made we expect to receive:
Copy of the official authorisation granted by the reference Member State (we receive this just as evidence that the authorisation is given. We do not anticipate expecting a version in English as all relevant information will be in the Product Assessment Report).
Copy of the Product Assessment Report by the reference Member State Copy of R4BP form (part 2). Until R4BP is fully operational we have our own
application form for the applicants to use. Summary dossier (Doc I-III; on CD) Copy of the proposed label (and Safety Data Sheet)
Additional studies and information considered essential to the assessment may be requested on an ad-hoc basis. The applicant will be invoiced at some stage during the evaluation.
We prefer to receive all documentation in electronic form (including e-mail where appropriate) wherever possible, and documents should be in English.
Requests for dossiers for product authorisation and for summary dossiers for mutual recognition
Product authorisation Mutual RecognitionPaper Electronic Paper Electronic
UK 1 1 - 1
85

Payment:
When we receive an application we send out an invoice for the minimum fee (we would indicate banking details with this). The evaluation of the application does not start until this fee is paid. At the end of evaluation we may charge a "top-up" fee if necessary i.e. if the evaluation was more extensive than we initially expected. An authorisation is not issued until all required payments have been made.
Sell out period for non-supported biocidal products
We implement a staged phase-out of products over an 18 month period. From the date of Annex 1 inclusion, we allow 6 months to cease first placing on the market (i.e. by the manufacturer / formulator), a further 6 months to cease retail sale (i.e. by distributors and retail outlets), and a final 6 months to cease all use (amateur and professional) and for storage and disposal.
86

EUROPEAN COMMISSIONDIRECTORATE-GENERALENVIRONMENTDirectorate B - Protecting the Natural EnvironmentENV.B.3 - Biotechnology, Pesticides and Health
Annex III
31st meeting of representatives of Members States Competent Authorities for the implementation of Directive 98/8/EC concerning the placing of biocidal products on the market
Common approach on the issue of provisional authorisation
As explained in detail in the document submitted to the Member States at May CA meeting (CA-May08-Doc.11.4), there are several unclear points with regard to the implementation of Article 15(2) of the Biocides Directive regarding the provisional authorisation of biocidal products containing new active substance(s)26.
The main issues concern:
a) whether all Member States may grant a provisional authorisation, or only the Member State in charge of evaluating the new active substance; and
b) when may a provisional authorisation be granted.
After consideration of practices in similar regulatory frameworks (Directive 91/414/EEC), the interests of the industry, the views of the Member States and the need to ensure a high level of protection for human health and the environment, the following was agreed to be adopted as common practice by the Member States, at least until the revised Directive enters into force27.
1. Any Member State may grant a provisional authorisation, after receiving an application to that effect.
2. Member States other than the one in charge of evaluating the new active substance dossier ('rapporteur') may grant a provisional authorisation to a biocidal product containing that new active substance only after the rapporteur has submitted the CA report with a recommendation to include the said active substance in Annex I to the Directive.
Clarifications to the above two propositions:
1. "Any Member State may grant a provisional authorisation" does not mean that all Member States are obliged to grant provisional authorisations when requested. Only when and if they are individually convinced that the active substance satisfies the requirements of Article 10 and the biocidal product will satisfy the conditions of Article 5(1) b) c) and d).
2. Provisional authorisation is not subject to mutual recognition (Article 4). The Directive does not expressly provide for that and the obligation to mutually recognise is not something that can be presumed.
26 i.e. active substances that were not used in biocidal products before 14 May 2000.27 There is a possibility that the evaluation of new active substances and the authorisation of products containing new active
substances be handled at Community level by an agency. This might render the provisional authorisation provision as it stands today redundant.
87

3. The submission of the favourable CA report is the earliest date after which the Member States agree that they may grant provisional authorisations.
4. After the inclusion of the new active substance in Annex I, the Member States who have granted provisional authorisations should request that an application for a regular authorisation be submitted. The provisional authorisation will cover the placing on the market of the product until the regular authorisation is granted. Please note that there is a maximum time limit for product authorisations: 3 + 1 years, so the evaluation of the authorisation application should not exceed that limit.
5. Provisional authorisations can be granted to products containing a new active substance as well as existing active substances. Such provisional authorisations could be maintained until the inclusion of the last existing active substance in Annex I.
88

Annex IV
Standard format to facilitate the comparison between the original protected data of reference dossier and the new data of the alternative
supplier dossier
Reference dossier Alternative supplier dossier
Annex point /
reference number
Title of study or case for which data protection has
been claimed
Year Title of alternative study or case referenced
Year Reason for equivalence / justification for non-
provision
MS Opinion
89

Annex V
Applicability of Good Laboratory Practice (GLP)
90

EUROPEAN COMMISSIONDIRECTORATE-GENERALENVIRONMENTDirectorate D - Water, Chemicals & BiotechnologyENV.D.3 - Chemicals & Nanomaterials
Members States Competent Authorities for the implementation of Directive 98/8/EC concerning the placing of biocidal products on the market
Product Authorisation and Mutual Recognition Facilitation Group
Guidance regarding the Applicability of Good Laboratory Practice (GLP) to Data Requirements according to
Annex IIB/IIIB of Directive 98/8/EC
According to Article 8 (8) of Directive 98/8/EC, [experimental] tests must be conducted – where appropriate – in accordance with Council Directive 87/18/EEC of 18 December 1986 on the harmonisation of laws, regulations and administrative provisions relating to the application of the principles of good laboratory practice and the verification of their applications for tests on chemical substances.
This provision should not apply to [experimental] tests that have generated test data before 15 May 2000.
The list attached shall give guidance to the Member States in their requirements to applicants performing [experimental] studies for biocidal products required under Directive 98/8/EC.
91

ANNEX IIB
I. APPLICANT
1.1. Name and address
1.2. Formulator of the biocidal product and the active substance(s) (name, addresses, includling location of plant(s))
II. IDENTITY
2.1. Trade name or proposed trade name, and manufacturer’s development code number of the preparation, if appropriate
2.2. Detailed quantitative and qualitative information on the composition of the biocidal product
2.3. Physical state and nature of the biocidal product
III. PHYSYICAL; CHEMICAL AND TECHNICAL PROPERTIES
3.1. Appearance (physical state, colour)
GLP 3.2. Explosive properties
GLP 3.3. Oxidising properties
GLP 3.4. Flash-point and other indications of flammability or spontaneous ignition
GLP 3.5. Acidity/alkalinity and if necessary pH value (1% in water)
GLP 3.6. Relative density
3.7. Storage stability – stability and shelf-life. Effects of light, temperature and humidity on technical characteristics of the biocidal product; reactivity towards container material
3.8. Technical characteristics of the biocidal product
3.9. Physical and chemical compatibility with other products including other biocidal products with which its use is to be authorised
IV. METHODS OF IDENTIFICATION AND ANALYSIS
4.1. Analytical method for determining the concentration of the active substance(s) in the biocidal product
4.2. In so far as not covered by Annex IIA, paragraph 4.2, analytical methods including recovery rates and the limits of determination for toxicologically and ecotoxicologically relevant components of the biocidal product and/or residues thereof, where relevant in or on the following:
92

(a) Soil(b) Air(c) Water (including drinking water)(d) Animal and human body fluids and tissues(e) Treated food or feedingstuffs
V. INTENDED USES AND EFFICACY
5.1. Product type and field of use envisaged
5.2. Method of application including description of system used
5.3. Application rate and if appropriate, the final concentration of the biocidal product and active substance in the system in which the preparation is to be used
5.4. Number and timing of applications, and where relevant, any particular information relating to geographical variations, climatic variations, or necessary waiting periods to protect man and animals
5.5. Function
5.6. Pest organism(s) to be controlled and products, organisms or objects to be protected
5.7. Effects on target organisms
5.8. Mode of action (including time delay) in so far as not covered by Annex IIA, paragraph 5.4
5.9. User: industrial, professional, general public (non-profissional)
Efficacy data
5.10. The proposed label claims for the product and efficacy data to support these claims, including any available standard protocols used, laboratory tests, or field trials, where appropriate
5.11. Any other known limitations on efficacy including resistance
VI. TOXICOLOGICAL STUDIES
6.1. Acute toxicity
For studies 6.1.1 to 6.1.3, biocidal products other than gases shall be administered via at least two routes, one of which should be the oral route. The choice of the second route will depend on the nature of the product and the likely route of human exposure. Gases and volatile liquids should be administered by the inhalation route
GLP 6.1.1. Oral
93

GLP 6.1.2. Dermal
GLP 6.1.3. Inhalation
GLP 6.1.4. For biocidal products that are intended to be authorised for use with other biocidal products, the mixture of products, where possible, shall be tested for acute dermal toxicity and skin and eye irritation, as appropriate
GLP 6.2. Skin and eye irritation (1)
GLP 6.3. Skin sensitisation
GLP 6.4. Information on dermal absorption
GLP 6.5. Available toxicological data relating to toxicologically relevant non-active substances
6.6. Information related to the exposure of the biocidal product to man and the operatorWhere necessary, the test(s) described in Annex IIA, shall be required for the toxicologically relevant non-active substances of the preparation
VII. ECOTOXICOLOGIDAL STUDIES
GLP 7.1. Foreseeable routes of entry into the environment on the basis of the use envisaged
GLP 7.2. Information on the ecotoxicology of the active substance in the product, where this cannot be extrapolated from the information on the active substance itself
GLP 7.3. Available ecotoxicological information relating to exotoxicological relevant non-active substances
VIII. MEASURES TO BE ADOPTED TO PROTECT MAN, ANIMALS AND THE ENVIRONMENT
8.1. Recommended methods and precautions concerning handling, use, storage, transport or fire
8.2. Specific treatment in case of an accident
8.3. Procedures, if any, for cleaning application equipment
8.4. Identity of relevant combustion products in cases of fire
8.5. Procedures for waste management of the biocidal product and its packaging for industry, professional users and the general public (non-professional users)
94

8.6. Possibility of destruction or decontamination following release in or on the following:(a) Air(b) Water, including drinking water(c) Soil
8.7. Observations on undesirable or unintended side-effects
8.8. Specify any repellents or poison control measures included in the preparation that are present to prevent action against non-target organisms
IX. CLASSIFICATION, PACKAGING AND LABELLING
- Proposals for packaging and labelling
- Proposals for safety-data sheets, where appropriate
- Justification for the classification and labelling according to the principles of Article 20 of this Directive- Hazard symbol(s)- Indications of danger- Risk phrases- Safety phrases- Packaging (type, materials, size, etc.), compatibility of the prepara- tion with proposed packaging materials to be included
X. SUMMARY AND EVALUATION OF SECTIONS II TO IX
Notes
(1) Eye-irritation test shall not be necessary where the biocidal product has been shown to have potential corrosive properties.
95

ANNEX IIIB
XI. FURTHER HUMAN HEALTH-RELATED STUDIES
1. Food and feedingstuffs studies
GLP 1.1. If residues of the biocidal product remain on feedingstuffs for a significant period of time, then feeding and metabolism studies in livestock shall be required to permit evaluation of residues in food of animal origin
GLP 1.2. Effects of industrial processing and/or domestic preparation on the nature and magnitude of residues of the biocidal product
2. Other test(s) related to the exposure to humansSuitable test(s) and a reasoned case will be required for the biocidal product
XII. FURTHER STUDIES ON FATE AND BEHAVIOUR IN THE ENVIRONMENT
GLP 1. Where relevant all the information required in Annex IIIA, Section XII
GLP
GLP
GLP
2. Testing for distribution and dissipation in the following:(a) Soil(b) Water(c) AirTest requirements 1 and 2 above are applicable only to ecotoxicologically relevant components of the biocidal product
XIII. FURTHER ECOTOXICOLOGICAL STUDIES
1. Effects on birds
GLP 1.1. Acute oral toxicity, if not already done in accordance with Annex IIB, Section VII
2. Effects on aquatic organisms
2.1. In case of application on, in, or near to surface waters
GLP 2.1.1. Particular studies with fish and other aquatic organisms
GLP 2.1.2. Residue data in fish concerning the active substance and including toxicologically relevant metabolites
GLP 2.1.3. The studies referred to in Annex IIIA, Section XIII, parts 2.1, 2.2, 2.3 and 2.4 may be required for relevant components of the biocidal product
96

2.2. If the biocidal product is to be sprayed near to surface waters then an overspray study may be required to assess risks to aquatic organisms under field conditions
3. Effects on other non-target organisms
GLP 3.1. Toxicity to terrestrial vertebrates other than birds
GLP 3.2. Acute toxicity to honeybees
GLP 3.3. Effects on beneficial arthropods other than bees
GLP 3.4. Effects on earthworms and other soil non-target macro-organisms, believed to be at risk
GLP 3.5. Effects on soil non-target micro-organisms
GLP 3.6. Effects on any other specific, non-target organisms (flora and fauna) believed to be at risk
3.7. If the biocidal product is in the form of bait or granules
GLP 3.7.1. Supervised trials to assess risks to non-target organisms under field conditions
3.7.2. Studies on acceptance by ingestion of the biocidal product by any non-target organisms thought to be at risk
GLP 4. Summary and evaluation of parts 1, 2 and 3
97

Annex VI
Frame formulations under Directive 98/8/EC

CA-Sept10-Doc.
EUROPEAN COMMISSIONDIRECTORATE-GENERALENVIRONMENTDirectorate D - Water, Chemicals & BiotechnologyENV.D.3 - Chemicals & Nanomaterials
Technical Notes for Guidance
Frame formulations
These Technical Notes for Guidance were endorsed during the 40th CA meeting and will apply from 1 June 2011 for application for product authorisation.
Note!: The new version of the R4BP allows applicants to create the reference products as well as all additional products within the frame formulation, so it is no longer necessary to send the lists of products within a frame formulation to the Commission for upload in R4BP. The R4BP User Guide for Industry28 provides guidance on frame formulation applications (pages 24-30).
28 This guidance is available through the R4BP website under the item "More information"
99

CA-Feb11-Doc.6.1.a
EUROPEAN COMMISSIONDIRECTORATE-GENERALENVIRONMENTDirectorate D - Water, Chemicals & BiotechnologyENV.D.3 - Chemicals & Nanomaterials
Members States Competent Authorities for the implementation of Directive 98/8/EC concerning the placing of biocidal products on the market
Notes for Guidance
Frame formulations
This paper gives an overview of the approach to handling frame formulations and authorisation of products covered by a frame formulation. This practical approach has been developed in light of the first applications received so far but is also likely to evolve with experience.
The frame formulation concept
The concept of frame formulation as laid down in Directive 98/8/EC (the Directive) is provided to underpin and allow for a streamlined and simplified procedure for granting authorisations to very similar products which only vary in terms of relatively minor differences in composition and have the same use and user type. To this end a frame formulation is essentially just a generic specification which encapsulates a group of highly related biocidal products.
The primary use of frame formulations is to expedite the evaluation and authorisation of such related products. In the first instance a frame formulation is established by carrying out an assessment of the risks to health and the environment and of the efficacy and by concluding that there are no unacceptable risks and that efficacy is demonstrated. Once established, any real product falling within the frame formulation can be taken to be acceptable without any formal evaluation necessary.
Frame formulations are considered to be an important tool in facilitating the efficient operation of product authorisation and are particularly useful for certain types of product that are often produced in subtle variations (e.g. in different colours/shades).
Commission européenne, B-1049 Bruxelles / Europese Commissie, B-1049 Brussel - Belgium. Telephone: (32-2) 299 11 11.Office: BU9 6/163. Telephone: direct line (32-2) 2986933. Fax: (32-2) 2998558.
E-mail: [email protected]

Formal definition of a frame formulation and practical interpretation
The formal definition of a frame formulation provides the general indication for the use and applicability of frame formulations.
Specifications for a group of biocidal products having the same use and user type
This group of products must contain the same active substances of the same specifications, and their compositions must present only variations from a previously authorised biocidal product, which do not affect the level of risk associated with them and their efficacy.
In this context, a variation is the allowance of a reduction in the percentage of the active substance and/or an alteration in percentage composition of one or more non-active substances and/or the replacement of one or more pigments, dyes, perfumes by others presenting the same or lower risk, and which do not decrease the efficacy.
The interpretation of this definition is that a frame formulation is a generic specification of a series of products for a given use and user type. The specification of the products is in terms of the active- and non-active substances present and the allowable range in the concentration at which each may be present.
A frame formulation has to be established in connection to an authorisation, either at the same time or later. According to the definition, the products must only present variations from a previously authorised biocidal product. In most cases it will however make more sense in practice to establish the frame formulation in the first instance, and then to assess individual products falling within the frame formulation for authorisation.
The frame formulation is based on data owned by a company and may not be used by others unless they have a letter of access.
The definition does not give much indication on the magnitude of the allowable variations in concentration, other than they should not affect the level of risk or decrease efficacy. However, although it is not explicitly stated in the Directive, one would expect other properties, such as the type of formulation, the instructions for use, not to be affected by the variations in concentration.
The definition also indicates that pigments, dyes and perfumes can be replaced by alternatives, with the implication that the alternatives could be present at different concentrations.
The specification of a frame formulation is presented graphically in Figure 1. Each component is specified with its allowable concentration range (see also the annex). What is an acceptable concentration range will depend on the nature of the substance and its properties and how its concentration may affect the product overall. The allowable range will be smaller for components with hazardous properties as changing the concentration significantly will potentially impact on the risk level. The classification of any product within the frame formulation should normally be the same.
101

Figure 1. Composition of a frame formulation.
Evaluating and establishing frame formulations
A frame formulation will be assessed according to the same criteria as for a single product, namely in relation to risks to health and the environment and efficacy. In general terms the assessment of the frame formulation will be conducted to assess the maximum risks to health and environment and the minimum level of efficacy over the whole potential range of composition of the frame formulation as specified.
For risks to health and the environment the maximum risk will usually be where the active substance(s) and any other components with hazardous properties are present at their highest allowable concentrations. Products with lower levels of these substances present will necessarily be of equal or lower risk. Similarly for efficacy, it will be necessary to demonstrate that a product containing the lowest allowable concentration of the active substance(s) is sufficient to achieve the label claims. Products with higher levels of the active substance(s) would be of at least equal efficacy. To help with this assessment it should be clearly indicated what the function (e.g. surfactant, emulsifier, stabiliser) of each component in the product is.
There are no definitive data requirements for establishing a frame formulation. The assessment is conducted using available data on the component substances and possibly products within the frame formulations, utilising to a large extent expert judgement, to make an informed judgement on the frame formulation as a whole. Requiring a considerable amount of extra testing to establish the frame formulation defeats the intention of frame formulations to expedite authorisation of related products. Where additional testing is needed, it may be useful to test products which are at the “low end” and at the “high end” of the frame
102
Concentration(% w/w)
Components
Active A B C D
100
0
Concentration(% w/w)
Components
Active A B C D
100
0

formulation so that data are available that fully cover the range of product compositions allowed in the frame formulation.
The evaluation of the frame formulation would be documented in an assessment report, and, by definition, this assessment would be applicable to any individual product within that frame formulation.
Authorisation of products within frame formulations
A typical application will include a justification for establishing a frame formulation and application for authorisation of one or more products (with all the associated data, Letters of Access etc). The exact composition of each product in the application needs to be specified along with its name. Once the frame formulation has been evaluated and judged to be acceptable each individual product specified in the application can be authorised.
They could be different approaches for authorising a series of products covered by the same frame formulation: a first one, whereby the authorisation covers a single product (1-to-1 relation), resulting in several separate authorisations to authorise the different products covered by the frame formulation; a second one, whereby a single authorisation covers the series of products (1-to-many relation), as exemplified in the figure below.
Figure 2. Possible approaches to product authorisation
For the purpose of the first product authorisation and of mutual recognition, these different approaches would be compatible.
The authorisation and the summary of the product characteristics would include the name of the product, the name of the active substance and of any substance of concern. Where the authorisation covers several products, as many summary of product characteristics would need to be included in the authorisation.
103
1-to-1
Product X
Authorisation number MS/09/1000-01: Product X - oak Authorisation number MS/09/1000-02: Product X - chestnutAuthorisation number MS/09/1000-03: Product X – pine
Authorisation
1-to-many
Product X
Authorisation number MS/09/1001: Product X - oak Authorisation number MS/09/1002: Product X - chestnutAuthorisation number MS/09/1003: Product X - pine
Product X
Authorisation number MS/09/1000: Product X - oak Authorisation number MS/09/1000: Product X - chestnutAuthorisation number MS/09/1000: Product X - pine

The detailed composition of the frame formulation would be available in the assessment report of the frame formulation as well as in the R4BP and that of the individual products in the R4BP.
If the authorisation holder wishes to place on the market additional products and if these are covered by an established frame formulation, the authorisation holder will be required to make a notification to the relevant competent authorities. This notification should be a simple process, but would require, as a minimum, the authorisation holder to indicate the name and the detailed composition of the product.
This is considered acceptable because the frame formulation is established on the basis that any product falling within it is safe and efficacious and therefore any new product within the frame formulation can implicitly be taken to be acceptable without the need for a formal assessment. This approach is intended to give authorisation holders a considerable degree of freedom in managing their product lines without any unnecessary burden of submitting additional applications.
Similarly, if the composition of existing products are modified, but still covered by the frame formulation, then the authorisation would be updated accordingly after notification of the new composition of the product by the authorisation holder.
Lastly, if some products are no longer placed on the market, then the authorisation will be revoked (1-to-1 approach) or amended (1-to-many approach) as the case may be.
Mutual recognition of frame formulations
Where the authorisation holder intends to seek mutual recognition of the authorisation to place one of their products on the market, they will however need to ensure that the product is formally authorised by the Reference Member State.
In the approach outlined above, all of the products covered by a frame formulation would be authorised either through individual decisions or through a single decision. There would be an assessment report on the frame formulation. The detailed composition of the frame formulation would be available in the assessment report and in the R4BP and that of the individual products in the R4BP.
This should allow other Member States to consider the frame formulation and establish it for themselves, if they wish, and to consider the authorisation - through mutual recognition - of the product(s) within that frame.
As a requirement of mutual recognition is that the product is authorised in the Reference Member State, any product for which the authorisation holder intends to seek mutual recognition must be formally authorised by the Reference Member State. It does however not matter if a particular product is only formally authorised with the Reference Member State in order to facilitate mutual recognition by another Member State and is not actually placed on the market of the Reference Member State.
As an alternative, the applicant may prefer to apply for mutual recognition of products to be placed on the markets of both the Reference and Concerned Member States, to request the establishment of the same frame formulation by the Concerned Member States and then to
104

notify the relevant Concerned Member States of additional products to be placed on their market and covered by the frame formulation.
Fees to be charged for the establishment of a frame formulation
The future rules on fees could include a fee for the establishment of a frame formulation as well as fees for the authorisations of individual products covered by a frame formulation. As the main part of the work will relate to the establishment of the frame formulation, the fee to cover the product authorisation would rather be a “top up” fee to cover the additional work involved in processing the application to completion.
In most cases an applicant will apply for authorisation of a product(s) and at the same time seek to establish a frame formulation which includes that product(s). The fee structure described above could however be applied. Thus, there could be a single fee for such an application which would cover both the work involved in establishing the frame formulation and in processing each individual product.
This should be considerably less than the fee for seeking the equivalent number of product authorisations in individual applications.
Any subsequent applications for authorisation of products within that frame formulation would attract an individual fee per product as some processing will be required for each application. This fee would however be similar to the 'top up' fee described above.
Alternative approaches to frame formulations
Frame formulations are of most use for groups of highly related products, for instance where a product is available in a range of colours. However, in cases where few products are involved, or where frame formulations may not be applicable, it may be more appropriate to use a read-across / bridging approach. Here, data principally on one product is used in the assessment of another similar product.
This could apply for instance to rodenticide baits containing the same active and non-active substances in different matrices (e.g. wheat, pasta, wax blocks etc), which can not be formally grouped within a frame formulation. Given that the risks to health and the environment of these products are expected to be more or less the same a read-across / bridging approach using data on one product to assess another product in a different matrix would be appropriate.
In practice, the read-across / bridging and frame formulation approach are very similar and achieve the same aim of expediting the authorisation of similar products with the minimum of extra testing and evaluation.
The future fee regulation should also include the possibility of a reduced fee whenever a read-across approach is used.
105

Annex
Composition of the frame formulation
NB: This information is confidential and should not be disclosed to third parties
Active substance(s) Contents
Minimum Maximum
Common name29 IUPAC name CAS number EC number Concentration
Unit30 w/w (%)
Concentration
Unit w/w (%) minimum purity (% w/w)
Same source as for Annex I inclusion
yes no31
Add rows as necessary
Co-formulants Contents
Minimum Maximum
Common name IUPAC name Function32 CAS number EC number Concentration Unit w/w (%) Concentration Unit w/w (%) Classification Substance of concern
yes noAdd rows as necessary
29 Indicate the common name. Trade names alone are not accepted. For biological products, indicate the scientific name, strain/serovar, as appropriate.30 e.g. g/l, g/kg, other. For biological products, the concentration should state the number of activity units/units of potency (as appropriate) per defined unit of formulation (e.g.
per gramme or per litre).31 If the source is different, information should be provided on the technical equivalence of the two sources. For further guidance, see note for guidance on the assessment of
technical equivalence (CA-May08-Doc.6.7) available at: http://ec.europa.eu/environment/biocides/index.htm .32 e.g.: antioxidant, emetic, dispersing agent, other.
106

Annex VII
Letters of access
107

EUROPEAN COMMISSIONDIRECTORATE-GENERALENVIRONMENTDirectorate D - Water, Marine Environment & ChemicalsENV.D.3 - Chemicals, Biocides and Nanomaterials
Representatives of Members States Competent Authorities for the implementation of Directive 98/8/EC concerning the placing of biocidal products on the market
41st CA meeting
Guidance on the use of letters of access
This document is an attempt to provide guidance in the interest of consistency, and has been agreed between the Commission services responsible for biocidal products and a majority of the Member States' Competent Authorities for biocidal products. Please note, however, that Member States are not legally obliged to follow the approach set out in this document, since only the Court of Justice of the European Union can give authoritative interpretations on the
contents of Union law.
(1) A letter of access ('LoA') is normally a unilateral declaration of the data owner or his representative which states that a competent authority ('CA') can use his data for the benefit of a third party. As such, it cannot impose direct or indirect obligations for CAs.
Contracts accompanying LoAs
(2) In reality, the LoA is often accompanied by a commercial contract between the data owner, or his representative, and the beneficiary. A contract cannot stipulate obligations for third parties which are not parties to the contract and have not agreed to take upon obligations under the contract, such as a CA responsible for the authorisation of biocidal products. The CA cannot be asked to enforce an obligation of a beneficiary, and any commercial agreements between the contractual parties should be kept separate from the LoA.
(3) In case a LoA is nevertheless mixed with content intended to bind the beneficiary to a commercial commitment, this can only be enforced between the parties of the LoA in the framework of civil proceedings under national law. In
108

such cases, the question arises whether illegality of the commercial commitment would affect the validity of the LoA. This is also a matter that would ultimately have to be decided based on national law. Competent authorities can avoid this problem by requiring any commercial agreements between the contractual parties to be kept separate from the LoA.
(4) Tying the provision of a LoA to the supply of an active substance could raise serious concerns with respect to its compatibility with competition rules. A fair competition on the market of both active substances and biocidal products has the advantages, amongst others, to guarantee a wider choice of products for consumers, lead to lower prices, and promote innovation.
(5) In any event, a commercial contract has to be compatible with Articles 101 and 102 of the Treaty on the Functioning of the European Union. If it is not compatible, it is automatically considered void (Article 101) or declared void by the Commission or relevant competent authorities (Article 102).
Conditions in LoAs
(6) Any condition in a LoA for the CA's right to use the data for the benefit of another applicant must be compatible with the Biocidal Products Directive 98/8/EC ('BPD'). The conditions for authorisation of biocidal products are listed in Article 5(1) of the BPD, and terms in a LoA cannot further impose a condition for an authorisation. Terms intended to impose further conditions for an authorisation are consequently not binding upon CAs. In particular, a LoA cannot oblige a CA to authorise only biocidal products containing active substances of a certain origin or from a certain supplier or for certain uses.
(7) Any conditions in a LoA should also comply with the legislation of the Member State receiving it ('the receiving Member State'). This can for example concern an authorising CA's right (or duty) under national law to communicate essential facts in the documentation to the product authorisation applicant, in which case the data owner will have to require the applicant to treat any confidential information as such.
(8) If a LoA stipulates conditions for its validity which are incompatible with the legislation of the receiving Member State, the question whether the receiving CA shall accept or reject the letter is a matter for national law. In case a condition in a LoA renders the letter invalid, it is recommended that the CA makes the data owner aware of this and gives it the possibility to either amend the letter or confirm his withdrawal of the condition in question.
Content and verification of LoAs
(9) A CA receiving a LoA is recommended to check that the letter contains the date of issue, the names of the data owner and of the beneficiary, the list of protected data to which access is granted (or – if the CA accepts it – an unambiguous reference to such a list, such as the list of studies in the given version of a CA report), the statement that the data may be used by the CA for the purpose of granting an authorisation, and the signature of the data owner (or his representative).
109

(10) There is nothing in the BPD obliging a CA to examine ex officio whether the person who signed a LoA effectively has the right to give access to the protected data (which, however, does not prevent the CA from checking this in obvious cases of doubt, or from refusing a letter in case the doubts are confirmed).
(11) In case a LoA is explicitly only addressed to the CAs of certain Member States, the question arises whether other CAs can rely on it for the purpose of product authorisation or mutual recognition. The wording of the definition of a LoA in BPD – a document stating that data may be used by "the competent authority" – suggests that only those CAs to which a LoA is addressed can rely on the letter. For the purpose of a first product authorisation, an applicant who cannot submit the active substance dossier therefore needs to submit a LoA to that data addressed to the CA granting the authorisation.
(12) For mutual recognition, Article 4(1) of the BPD requires the CA to have the summary of the product dossier. In consequence, a CA cannot grant mutual recognition of an authorisation unless the applicant submits this data or a LoA to this data addressed to that CA. On the other hand, the BPD does not require the submission of other data, such as the active substance dossier, for mutual recognition. The question whether, or in what situations, to request a LoA to the active substance dossier from an applicant for mutual recognition has not yet been resolved between CAs..
(13) In case a task force has been created for the purpose of Annex I inclusion of a substance, CAs and the Commission should recommend task force members to entitle each of the other members to give access, for the purpose of product authorisation, to the full protected data package that allowed the Annex I inclusion. More precisely, task forces should be encouraged to adopt the following strategy:
o The task force should generate one general LoA (signed by all members) to the active substance dossier, which lists all the relevant studies, identifies the owners of the individual studies, and confirms that each of the members have access to the dossier and to all studies;
o One original of such letter should be sent to all 27 Member States;
o For the purpose of authorisation of biocidal products, each task force member can issue a 1 page LoA, which refers to the general LoA but does not have to contain the complete list of studies relating to the active substance.
If this strategy or an equivalent one is followed, CAs should accept the 1 page LoAs mentioned in the third bullet point above as a valid LoA to all the data relating to the active substance.
(14) If task forces have not followed this strategy, or in other cases where data referred to in a LoA has several owners but is only signed on behalf of some of them, the receiving CA should require that additional LoAs are submitted on behalf of the rest of the data owners.
110

(15) Nothing prevents a CA from also requesting or accepting that the LoA specifies the name of the active substance or the name of the product for which the LoA is valid. This facilitates the CA's linking a LoA to the relevant product authorisation application, if the two are submitted separately. On the other hand, it may create an additional burden on applicants choosing different product names in different countries. There is also nothing preventing a CA from requiring each individual product authorisation application to contain evidence of the right to make use of the information referred to in the application.
CAs' access to the actual data
(16) A situation could in theory arise where an applicant for product authorisation submits a LoA to data which is not held by the receiving CA, but by another CA ('the data holding CA'). In such cases, the applicant has a strong interest in securing that the receiving CA has access to the actual data, either through submission by the applicant itself or by the data owner.
(17) Should this not happen, it follows from Article 8(10) BPD that the data holding CA is obliged to transmit the data to the receiving CA upon request in case the data has been received by the data holding CA in the course of an application for product authorisation. In the case described, i.e. where the applicant has a LoA, the data can thus be used for the application. In case the data has been submitted in another context (e.g. for the purpose of Annex I inclusion), no such obligation for the data holding CA can be derived directly from Article 8(10) BPD, but it is possible that the Article should be applied by analogy also to data which a data holding CA has received for the purpose of Annex I inclusion. In any event, it follows from the same Article that the receiving CA can, as a last resort, request that the applicant submits a copy of the dossier.
Duration of a LoA
(18) Nothing in BPD prevents the data owner from granting a LoA which is limited in time. Article 12 BPD does not specify the points in time at which a LoA needs to be valid in order to allow a CA to rely on it for the purpose of granting an authorisation of certain duration, leaving CAs a certain margin of discretion to apply the rule in the way they find most appropriate in this respect, provided that the application confirms with national law.
(19) Since a LoA is the expression of a data owner's consent to allow a CA to use his data for the benefit of another person, it appears both fair and pragmatic to require that the LoA is valid when the CA starts using the data in question, i.e. in most cases at the time of submission of an application, and only then. In consequence, a CA cannot rely on a LoA which has expired for the purpose of granting a new authorisation. On the other hand, if it is accepted that a valid LoA is only required at the time of submission of an application for product authorisation, nothing prevents the CA from granting an authorisation which exceeds the validity of the LoA.
(20) Nor is the CA responsible for a modification of an authorisation required to check or request that the LoA submitted for that authorisation is still valid at the time of the modification, insofar as the data does not need to be used by the CA
111

for the purpose of the checking that the modification can be granted in accordance with Article 7(8) BPD (typically the active substance dossier in a case where the source of the substance is not changed).
(21) On the other hand, Articles 12 and 7(8) BPD prevents the CA from unauthorised use of data that needs to be assessed for the purpose of granting the modification, should it be in the possession of such data. In such cases, the LoA should also be valid at the time when the CA starts using it, i.e. at the time of submission of the request for modification.
(22) In line with the interpretation defended above in paragraph 19, nothing in the BPD prevents a CA from relying on a LoA which was valid at the time of submission of the application even if the LoA is later revoked during the authorisation procedure. If the sudden withdrawal of a LoA in the course of the authorisation procedure were to prevent a CA from relying on the letter for the purpose of granting the authorisation, CAs could be faced with a situation where the fee is paid and the evaluation is done, but the authorisation can nevertheless not be granted.
(23) Concerning the question whether the expiry or revocation of a LoA has any impact on the validity of the authorisation, the following comments can be made.
o The only grounds for reviewing an authorisation under BPD are indications that any of the conditions referred to in Article 5 are no longer satisfied. Article 5 does not stipulate that the authorisation holder has to have a dossier or a LoA to a dossier for each active substance in the biocidal product during the validity period of the authorisation.
o Article 7 of Directive 98/8/EC lists situations in which an authorisation has to be cancelled. It does not require that an authorisation is cancelled if a LoA is revoked or expires during the validity period of the authorisation.
(24) As a consequence, if a LoA is limited in time and the authorisation is granted beyond the expiry date, or if the LoA is revoked after its submission, the data owner may have a claim on the beneficiary. However, such an issue would have to be settled between the data owner and the beneficiary in the framework of civil proceedings, and does not limit the scope of action of the CA responsible for the authorisation of biocidal products.
112

Annex VIII
Products belonging to several product types*
* This annex can be updated according to new guidance available in this field.
113

Products belonging to several product types
Question 1:
If a decision for non-inclusion of an existing active substance in a certain product type is made (e.g. product type 8), can a product still be placed on the market for that product type if it contains that substance as a non-active substance or as an active substance for another product type (e.g. product type 10)? The product shall, of course, also contain an active substance included in Annex I or still under review for the first product type (in this case product type 8).
Example: Disodium octaborate tetrahydrate (CAS 12280-03-4) and DDAC (CAS 7173-51-5) were notified in both product type 8 (wood preservatives) and product type 10 (masonry preservatives). They are both included as active substances in products claiming to be efficacious in both product types 8 and 10. A non-inclusion decision has been taken for disodium octaborate tetrahydrate in product type 10 (Commission decision 2010/72/EU, products are to be phased out by 11-02-2011). May such a product still be marketed with claims to be efficacious in both product type 8 and 10, if it is stated that disodium octaborate tetrahydrate is only efficacious as an active substance in product type 8?
Answer (agreed December 2011):
According to Article 4.2 of Commission Regulation (EC) 1451/2007, products containing existing active substances for which a decision was taken to not include these active substances for certain or all of their notified product types in Annex I or IA to Directive 98/8/EC, shall no longer be placed on the market for the product types concerned. However, products containing the substance can be marketed if it is shown that the substance is not efficacious as an active substance for the concerned product type. In that case efficacy data must be submitted to demonstrate that the non-included active substance is not contributing in an essential or significant way to the efficacy for the product type to which the non-inclusion decision applies. If the substance has the function of an active substance in another product type then data must be submitted, as usual, to demonstrate sufficient efficacy in this second product type in accordance with the requirements of Article 5 and Annex VI.
In the case of the specific example, a biocidal product can no longer be placed on the market containing disodium octaborate tetrahydrate as an active substance for product type 10, although it can be placed on the market with disodium octaborate tetrahydrate as an active substance for product type 8. For the product in the example to be authorised for use in both product type 8 and 10, it must be shown that the disodium octaborate tetrahydrate is not efficacious in product type 10 (in addition to all other requirements to fulfill Article 5 and Annex VI).
Question 2:
May a substance be included in a product marketed for a product type for which that substance was never notified (it must, of course, include an active substance which is being defended for that product type), e.g. as an active substance for another product type?
114

Example: A product contains the following existing active substances: X (notified and still under review for pt 2, 8, 10) and Y (notified and still under review for pt 8 and 10, never notified in pt 2). Can this product be placed on the market for product type 2, 8 and 10?
The difference in this case is that one of the substances was never notified for one of the product types as opposed to the non-included product type in the previous example.
Answer (agreed December 2011):
There seems to be no real reason to treat this case differently than Question 1. According to chapter 5.2 in the MoD, such a product would be possible. In the example, it still would have to be made clear that the substance Y is not efficacious in relation to product type 2.
115


















