
Depression in the Workplace Melinda Adams, SPHR Human Resources Manager [email protected] 1.
date post
19-Dec-2015Category
view
220download
0
Pairwise Sequence Comparison
Dong Xu
Computer Science Department271C Life Sciences Center
E-mail: [email protected]
http://digbio.missouri.edu

Lecture Outline
Introduction
Scoring function
Dynamic programming
Confidence Assessment
Heuristic alignment

Introduction (1)
Biological sequence comparison DNA-DNA, RNA-RNA
Protein-protein
DNA-protein
Sequence comparison is the in most important and fundamental operation bioinformatics
Key to understand evolution of an organism or a gene

Introduction (2)
Applications in most bioinformatics problems Sequence assembly
Gene finding
Protein structure prediction
Phylogenic tree analysis
THE most popular tool: BLAST Foundation of sequence database search
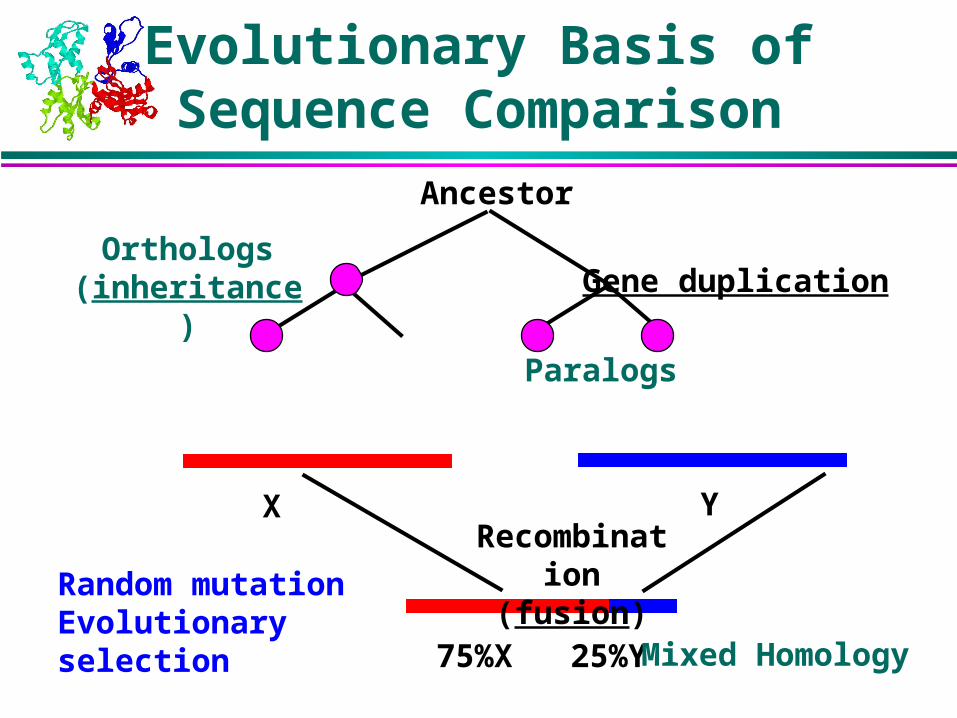
Ancestor
Gene duplication
X YRecombination
(fusion)
75%X 25%Y
Paralogs
Mixed Homology
Orthologs(inheritance)
Evolutionary Basis of Sequence Comparison
Random mutationEvolutionary selection

An Example of Sequence
Comparison
TT....TGTGTGCATTTAAGGGTGATAGTGTATTTGCTCTTTAAGAGCTG || || || | | ||| | |||| ||||| ||| ||| TTGACAGGTACCCAACTGTGTGTGCTGATGTA.TTGCTGGCCAAGGACTG
AGTGTTTGAGCCTCTGTTTGTGTGTAATTGAGTGTGCATGTGTGGGAGTG | | | | |||||| | |||| | || | | AAGGATC.............TCAGTAATTAATCATGCACCTATGTGGCGG
AAATTGTGGAATGTGTATGCTCATAGCACTGAGTGAAAATAAAAGATTGT ||| | ||| || || ||| | ||||||||| || |||||| | AAA.TATGGGATATGCATGTCGA...CACTGAGTG..AAGGCAAGATTAT

Alignement (1)
a correspondence between elements of two sequences with order (topology) kept
pairwise alignment: 2 sequences aligned multiple alignment: alignment of 3 or more
sequences
FSEYTTHRGHR: ::::: ::FESYTTHRPHR
FESYTTHRGHR:::::::: ::FESYTTHRPHR

Similar to ”longest common subsequence” (LCS) problem for strings, (Robinson, 1938)
LCS: define a set of operations (e.g. substitution, insertion or deletion) that transform the aligned elements of one sequence into the corresponding elements of the other and associate with each operation a cost or a score.
Optimal alignment: the alignment that is associated with the lowest cost (or highest score).
Between two sequences several optimal alignments can be constructed with the same optimal score.
Alignement (2)
FSEY-THRGHR: : ::: ::FESYTTHRPHR
FSEYT-HRGHR: :: :: ::FESYTTHRPHR

Components of Sequence Alignment
FDSK-THRGHR:.: :: :::FESYWTH-GHR
Match (:) Mismatch(substitution)
Insertion Deletion{Indel
(1) Scoring function: a measure of similarity between elements (nucleotides, amino acids, gaps);
(2) An algorithm for alignment;
(3) Confidence assessment of alignment result.

Lecture Outline
Introduction
Scoring function
Dynamic programming
Confidence Assessment
Heuristic alignment

Edit Distance(Hamming Distance)
Introduced by Levenshtein in 1966 Binary: match 1 / mismatch 0 (Identity Matrix) Definition: Minimum number of edit
operations to transform one string to another Can be used for DNA/RNA Possible edit operations
Symbol insertionSymbol deletionSymbol substitution

amino acid substitution matrices (20X20) account for probability of one amino acid being substituted for another:
frequency of substitution - genetic codetolerance for changes - natural selection
penalize residues pairs with a low probability of mutation in evolution and rewards pairs with a high probability
empirically derived from observed amino acid substitutions that occur between aligned residues in homologous sequences
Scoring Matrix

Physical Bases of Mutation Matrix
Geometry nature
Physical nature
(charged or hydrophobic)
Chemical nature
Frequencies of amino acids
physical property matrices

PAM
The first substitution matrices derived by Dayhoff et al. (1978)
PAM (point accepted mutation) distance: Two sequences are defined to have diverged by one PAM unit if they show in average one accepted point mutation (i.e. one amino acid change) per hundred amino acids.
Derived from the pairwise alignment of sequences less than 15% divergent.

BLOSUM
Block substitution matrices (Henikoff & Henikoff 1992)
Blocks: highly conserved regions in a set of aligned protein sequences (local multiple alignment)
Number of BLOSUM matrix (e.g. BLOSUM 62) indicates the cutoff of percent identity that defines the clusters - lower cutoffs allow more diverse sequences
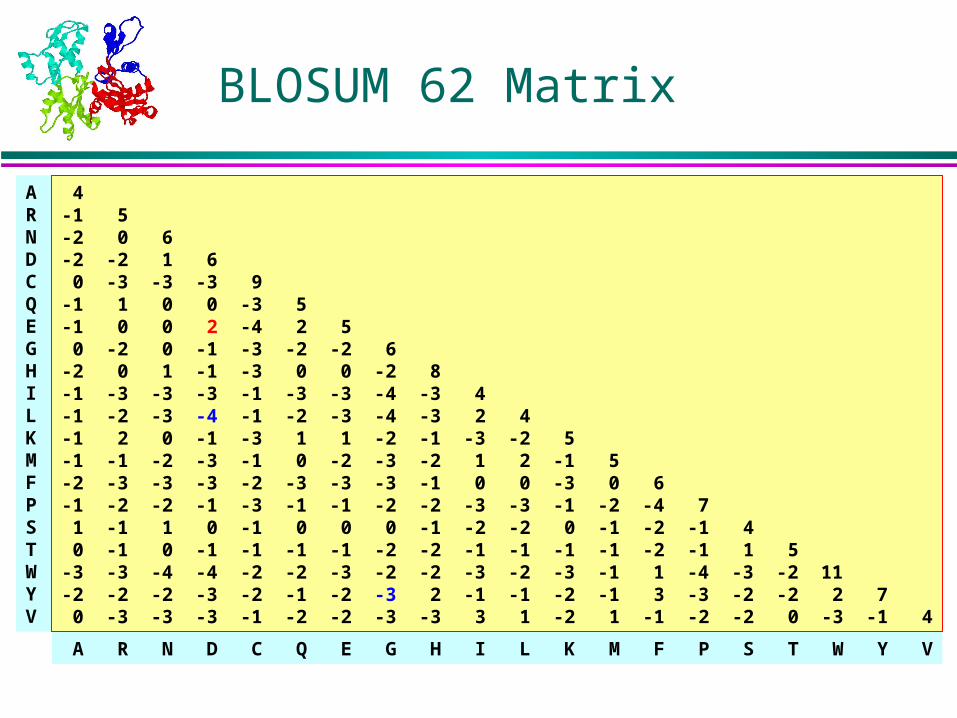
A R N D C Q E G H I L K M F P S T W Y V
ARNDCQEGHILKMFPSTWYV
4-1 5-2 0 6-2 -2 1 6 0 -3 -3 -3 9-1 1 0 0 -3 5-1 0 0 2 -4 2 5 0 -2 0 -1 -3 -2 -2 6-2 0 1 -1 -3 0 0 -2 8 -1 -3 -3 -3 -1 -3 -3 -4 -3 4 -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4
BLOSUM 62 Matrix

Close homolog: high cutoffs for BLOSUM (up to BLOSUM 90) or lower PAM values
BLAST default: BLOSUM 62
Remote homolog: lower cutoffs for BLOSUM (down to BLOSUM 10) or high PAM values (PAM 200 or PAM 250)
A best performer in structure prediction:PAM 250
What Matrices to Use

Gap Penalty Functions
Corresponding to insertion/deletion in evolution
Typically linear gap penalties Easy to implement in algorithmsSatisfactory performance in alignment
Can be derived from alignmentKnown alignmentsPerformance-based (sequence comparison)

Affine Gap Penalty
Most commonly used model w(k) = h + gk , k 1 ,with w(0) = 0.
h: gap opening penalty; g: gap extension penalty
h > g > 0 (e.g., for PAM250, 10.8 + 0.6k)
Non-linear form: h + g log (k)
FDS-T-HRGHR:.: : :::::FESYTTHRGHR
FDS--THRGHR:.: ::::::FESYTTHRGHR

Lecture Outline
Introduction
Scoring function
Dynamic programming
Confidence Assessment
Heuristic alignment

Global alignment: the alignment of complete sequences Good for comparing members of same protein familyNeedleman & Wunsch 1970 J Mol Biol 48:443
Local alignment: the alignment of segments of sequences ignore areas that show little similaritySmith & Waterman 1981, J Mol Biol, 147:195 modified from Needelman-Wunsh algorithmcan be done with heuristics (FASTA and BLAST)
Global vs. Local Alignment

Dot Matrix and Alignment
A A C G G T A T G CA 1 1 1T 1 1C 1 1G 1 1 1G 1 1 1G 1 1 1T 1 1T 1 1G 1 1 1C 1 1
AACGATCG
-GGTGT
A-TGCTGC
Dot matrix:Score between cross-elements
path:Mapping toan alignment

1. Assign scores between elements in dot matrix
2. For each cell in the dot matrix, check all possible pathways back to the beginning of the sequence (allowing insertions and deletions) and give that cell the value of the maximum scoring pathway
3. Construct an alignment (pathway) back from the last cell in the dot matrix (or the highest scoring) cell to give the highest scoring alignment
Dynamic Programming Steps

Dynamic programming
Foundation: any partial subpath ending at a point along the true optimal path must itself be an optimal path leading up to that point. So the optimal path can be found by incremental extensions of optimal subpaths
One of the most fundemental algorithms in bioinfomatics
Applications: sequence comparison, gene finding, mass-spec data analysis, ...

Needleman-WunschAlgorithm (1)
Global alignment
Elementary operations:Single insertion/deletions (s(ai,-) or s(-,bj))
Substitution (s(ai,bj))
Easy case: h=0 for gap penalty (h+gk)

21
0
11
0
00
1),(
1),(
0
ljbsS
liasS
S
j
kkj
i
kki
21
1,
1,1
,1
1,1
),(
,
),(
max ljli
bsS
basS
asS
S
jji
jiji
iji
ij
Needleman-WunschAlgorithm (2)
The optimal score ending at i & j

Calculate S(i,j) in three ways:•By adding a score s(ai,bj) to the score diagonally upwards, i.e. S(i-1,j-1);
•By adding a score s (-,bj) (which represents the introduction of a gap into the alignment) to the score vertically above (i.e. S(i,j-1);
•Or by adding s(ai,-) to the score horizontally to the left (i.e. S(i-1,j)
i A A A
j S(i-1,j-1) S(i,j-1)
A S(i-1,j) S(i,j)
T
C
Needleman-WunschAlgorithm (3)

Alignment Construction (1)
A A C . . .
0 -1 -2 -3 . . .
A -1
T -2
C -3
. . . . . .
-- (AAC)AT (C)
Initialization:
S(0,0) = 0
the outside row and column are given incrementally decreasing values

Alignment Construction (2)
A A C . . .
0 -1 -2 -3 . . .
A -1
T -2
C -3
. . . . . .
1
A(AC)A(TC)
A-(AC)-A(TC)
-A(AC)A-(TC)
S(1,1) : one of three values:
(1) ai = bj, s = 1S(i-1,j-1) + s(ai,bj) = 0+1 = 1
(2) add s(-,bj) to S(i,j-1)s(i,j-1) - s(-,bj) = -2
(3) add s(ai,-) to S(i-1,j) s(i-1,j) - s(ai,-) = -2
choose highest 1 in the cell.

A A C . . .
0 -1 -2 -3 . . .
A -1
T -2
C -3
. . . . . .
1 0
Alignment Construction (3)
For the next cell, as ai = bj again, s(ai,bj) = 1 and the three possible scores are: i,j -1 + 1 = 0
i, j-1 -2 - 1 = -3
i-1, j 1 - 1 = 0
Two degenerate paths! (Max=3)

A A C . . .
0 -1 -2 -3. . .
A -1
T -2
C -3
. . . . . .
1 0 -1
Alignment Construction (4)
For the next cell, as ai bj, s(ai,bj) = 0. The three possible scores are: i,j -2 + 0 = -2
i, j-1 -3 - 1 = -4
i-1, j 0 - 1 = -1

A A C . . .
0 -1 -2 -3. . .
A -1
T -2
C -3
. . . . . .
1 0 -1
0 1 0
-1 0 2
Alignment Construction (5)
C C
AC TC
AACATC
Trace back:

Mathematical Representation
0
0
20
10
ljjS
liiS
j
i
Length sequence 1
Length sequence 2
0
for 1),(
with
0,0),(max 21
1,
1,1
,1
else
babas
ljli
1S
basS
1S
S
jiji
ji
jiji
ji
ij
Initialization
Scoring

Computational Complexity Computational Complexity of Dynamic Programmingof Dynamic Programming
Computing time: O(nm), where n and m are sequence lengths).
Retrieval time: O(Max (n,m))
[worst case: n+m; best case: Min(n,m)]
Required memory: O(nm).
Keeping in mind the computational complexity while programming

Smith-Waterman Algorithm
S0, j Si ,0 0for 0i l1 and 0 j l2
max
Si,j-1
Si-1,j-1
Si-1,j
Sij1,1 l2jl1i
),( bjs
,bjais
),(ais
0
Set all values in top row and left column to 0
Set the value of Sij to 0 if it would otherwise be less than 0
Traceback from highest value of Sij, rather than from
bottom right corner. Stop at 0 rather than top left corner.

Smith-Waterman Algorithm
With Affine Gap Penalties
S0,j Si,0 D0,j Di,0 I0, j Ii ,0 0
for 0il1 and 0 j l2
Si 1,j h - g
Di-1,j g
Si 1,j h – g
Sij max
Si 1,j 1 s ai,bj
0for 1il1 and 1 j l2
Ii,j-1 g
Di 1,j 1 s ai,bj
I
i 1,j 1 s ai,bj
Gap penalty: w(k) = h + gk , k 1
Dij max
Iij max
Boundary conditions:

C A G C C U C G C U U A GA 0.0 1.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 1.0 0.0A 0.0 1.0 0.7 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 1.0 0.7U 0.0 0.0 0.8 0.3 0.0 0.0 0.0 0.0 0.0 1.0 1.0 0.0 0.7G 0.0 0.0 1.0 0.3 0.0 0.0 0.7 1.0 0.0 0.0 0.7 0.7 1.0C 1.0 0.0 0.0 2.0 1.3 0.3 1.0 0.3 2.0 0.7 0.3 0.3 0.3C 1.0 0.7 0.0 1.0 3.0 1.7 1.3 1.0 1.3 1.7 0.3 0.0 0.0A 0.0 2.0 0.7 0.3 1.7 2.7 1.3 1.0 0.7 1.0 1.3 1.3 0.0U 0.0 0.7 1.7 0.3 1.3 2.7 2.3 1.0 0.7 1.7 2.0 1.0 1.0U 0.0 0.3 0.3 1.3 1.0 2.3 2.3 2.0 0.7 1.7 2.7 1.7 1.0G 0.0 0.0 1.3 0.0 1.0 1.0 2.0 3.3 2.0 1.7 1.3 2.3 2.7A 0.0 1.0 0.0 1.0 0.3 0.7 0.7 2.0 3.0 1.7 1.3 2.3 2.0C 1.0 0.0 0.7 1.0 2.0 0.7 1.7 1.7 3.0 2.7 1.3 1.0 2.0G 0.0 0.7 1.0 0.3 0.7 1.7 0.3 2.7 1.7 2.7 2.3 1.0 2.0G 0.0 0.0 1.7 0.7 0.3 0.3 1.3 1.3 2.3 1.3 2.3 2.0 2.0
GCC-UCGGCCAUUG
Alignment retrieval for Smith-Waterman
algorithm

Lecture Outline
Introduction
Scoring function
Dynamic programming
Confidence Assessment
Heuristic alignment

Confidence Assessmentof Sequence Alignment
Why confidence assessment is needed
True homology or alignment by chance
Expected probability by chance Statistical models

Why not to use sequenceidentity as confidence
measure

Real but non-homologous sequences
Real sequences that are shuffled to preserve compositional properties
Sequences that are generated randomly based upon a DNA or protein sequence model (analytic statistical results)
What to Compare

The probability that a variate would assume a value greater than or equal to the observed value strictly by chance P(z>zo)
If the P-value found for an alignment is low (<0.001) then alignment is probably biologically meaningful.
Pre-compute the parameters based on a statistical model
P-value or E-value

The maximum scores of a large number of alignments between random sequences of equal length tends to have an extreme value distribution
P(S’<x)=exp[-exp(-x)]
P(S’>=x)= 1- exp[-exp(-x)]
Bit scores:
S’= (S-lnK)/ln2
and K: scaling factors
(depending on composition, mutation matrix used etc.)
Extremal Value Distribution
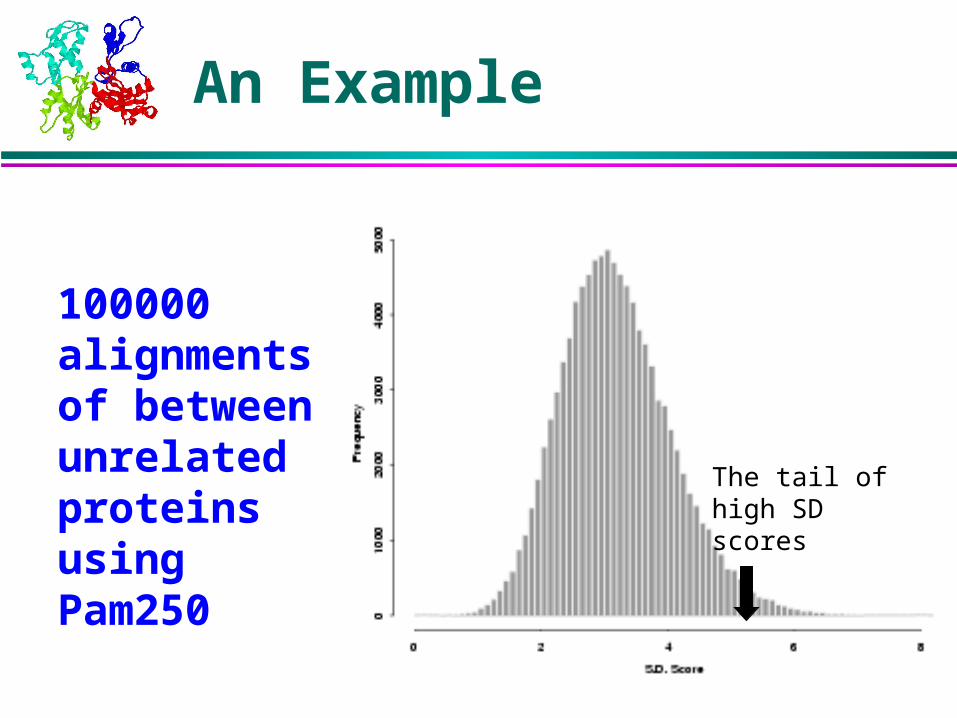
An Example
100000 alignments of between unrelated proteins using Pam250
The tail of high SD scores

Other Issues
Gapless alignment vs. gapped alignment
Low complexity regions Over-estimate or under-estimate of
confidence level

Scalability of Software
The trend of genetic
data growth
Genomes: yeast, human, rice, mouse, fly... Software must be (linearly or close to linearly)
scalable to large datasets.
30 billionin year 2005

Need for Heuristic Alignment
Time complexity for optimal alignment: O(n2) , n -- sequence length
Given the current size of sequence databases, use of optimal algorithms is not practical for database search
Heuristic techniques: BLAST, FASTA, MUMmer, PatternHunter...
20 min (optimal alignment, SSearch) 2min (FASTA) 20 sec (BLAST)

Ideas in Heuristics Search
Indexing and filtering: Google searchGood alignment includes short identical, or
similar fragments
break entire string into substrings, index the substrings
Search for matching short substrings and use as seed for further analysis
extend to entire string and find the most significant local alignment segment

FASTA (1)
Lipman & Pearson, 1985, Science 227, 1435-1441
Four stepsStep 1: Identify regions of the sequences
with the highest density of matches. In this step exact matches of a given length (by default 2 for proteins, 6 for nucleic acids) are determined and regions (fragments of diagonals) with a high number of matches selected.

FASTA (2)

Step 2: Rescan 10 regions with the highest density of identities using the BLOSUM50 matrix. Trim the ends of region to include only those residues contributing to the highest score. Each region is a partial alignment without gaps.
Step 3: If there are several regions with scores greater than a cut off value, try to join these regions. A score for the joined initial regions is calculated given a penalty for each gap.
FASTA (3)

FASTA (4)

Step 4: Select the sequences in the database with the highest score. For each of those sequences construct a Smith-Waterman optimal alignment considering only the positions that lie in a band centred on the best initial region found.
FASTA (5)

A-FTFWSYAIGL--PSSSIVSWKSCHVLHKVLRDGHPNVLHDCQRYRSNI| |||||| || || |||| | | |... | : AIPQFWSYAIERPLNSSWIVVWKSCITTHHLMVYGNERFIQYLAS-RNTL
FASTA (6)
Step 1
Step 2
Step 3 / Step 4

Basic Alignment Search Tool (Altschul et al, 1990, J. Mol. Biol. 215, 403-410)
Uses word matching like FASTA Similarity matching of words (3 aa’s, 11
bases) does not require identical words.
If no words are similar, then no alignmentwon’t find matches for very short sequences
BLAST (1)

Detects alignments with optimal maximal segment pair (MSP) score. Gaps are not allowed.
MSP: Highest scoring pair of segments of identical length. A segment pair is locally maximal if it cannot be improved by extending or shortening the segments.
Homologous sequences will contain a MSP with a high score; others will be filtered out.
BLAST (2)

BLAST (3)

BLAST (4)
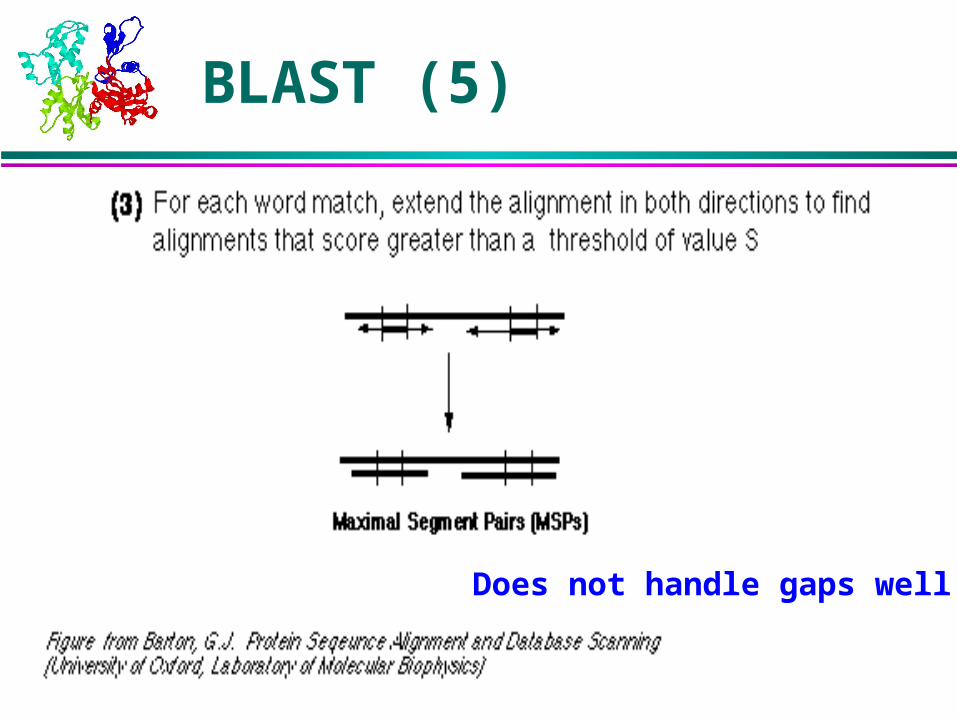
BLAST (5)
Does not handle gaps well

Genome Alignment by PatternHunter(4 seconds)

Comparison with Blastn and MegaBlast
Blastn MegaBlast PatternHunter
E.coli vs H.inf 716s 5s/561M 14s/68M
Arabidopsis 2 vs 4 -- 21720s/1087M 498s/280M
Human 21 vs 22 -- -- 5250s/417M
Human (3G) vs Mouse 20 days

Secret in PatternHunter
Seeds (length of word used) Blastn finds a match of length 11, then
extend from there. MegaBlast in order to increase speed, increases this to 28.
Dilemma: increasing seed size speeds up but loses sensitivity; decreasing seed size gains sensitivity but loses speed.
Spaced Seed: PatternHunter looks for matches of 11 nonconsecutive matches and optimized such seeding scheme.

Super Seeds
ATTTCCGACGCGAGGGGACTTTCAGGAGAG AGGGGACTTTC 11111111111
ATTTCCGACGCGAGGGGACTTTCAGGAGAG GTGATGGAACAATCGAGA 101101101100110011 G*GA*GG*AC**TC**GA

Reading Assignments
Suggested reading: Chapter 6 in “Pavel Pevzner: Computational
Molecular Biology - An Algorithmic Approach. MIT Press, 2000.”
Optional reading: http://www.people.virginia.edu/~wrp/papers/i
smb2000.pdf

Optional Assignment (1)
1. What does 200 mean in PAM 200?
2. What does 62 mean in BLOSUM 62?
3. Prove Needleman-Wunsch algorithm produces optimal global alignment.
4. Prove the computational complexity of Smith-Waterman algorithm.
5. What is the relationship between the alignment score and statistical significance of the alignment?

Explain the difference between PAM40 and PAM250.Why some elements in the matrix have different signs?
Optional Assignment (2)

Construct dynamic program matrix using edit distance and PAM250 distance, respectively.
1. Try different affine gap penalties, h=g or not
2. Try global and local alignment.
M P R C L C TMPCLWCQ
Optional Assignment (3)

Develop a program that can perform optimal global-local alignment for DNA sequences:
1. Fit a short sequence into a long sequence and output ALL optimal alignments.
2. No gap penalty when deleting terminal bases of the long sequence, but with gap penalty for deleting any base of the short sequence.
3. Use edit distance (match 1; otherwise 0) with gap penalty –1 – k (k is gap size).
Project Assignment (1)

4. Use the FASTA format for input of each sequence (see http://www.g2l.bio.uni-goettingen.de/blast/fastades.html).
5. Test on sample sequences, e.g.,
TTTGAGCCTCTGTTTGTGTGTAATTGAT-GTGCATGTGTGGG || |||||| || |||| ................TG-GTAATTAATCATGCAC.......
The above format can be used for your outputs.
Project Assignment (2)


















