
Osteopontin controls endothelial cell migration in vitro and in excised human valvular tissue from...
11
Osteopontin Controls Endothelial Cell Migration In Vitro and in Excised Human Valvular Tissue from Patients with Calcific Aortic Stenosis and Controls PAOLO POGGIO, 1 JUAN B. GRAU, 1 BENJAMIN C. FIELD, 1 RACHANA SAINGER, 1 WILLIAM F. SEEFRIED, 1 FLAVIO RIZZOLIO, 2,3 AND GIOVANNI FERRARI 1 * 1 Division of Cardiovascular Surgery, Department of Surgery, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania 2 Department of Biology, Sbarro Institute for Cancer Research and Molecular Medicine, Center for Biotechnology, College of Science and Technology, Temple University, Philadelphia, Pennsylvania 3 Human Health Foundation, Terni and Spoleto, PG, Italy Calcific aortic stenosis (CAS) is a pathological condition of the aortic valve characterized by dystrophic calcification of the valve leaflets. Despite the high prevalence and mortality associated with CAS, little is known about its pathogenetic mechanisms. Characterized by progressive dystrophic calcification of the valve leaflets, the early stages of aortic valve degeneration are similar to the active inflammatory process of atherosclerosis including endothelial disruption, inflammatory cell infiltration, lipid deposition, neo-vascularization and calcification. In the vascular system, the endothelium is an important regulator of physiological and pathological conditions; however, the contribution of endothelial dysfunction to valvular degeneration at the cellular and molecular level has received little attention. Endothelial cell (EC) activation and neo-vascularization of the cusps characterizes all stages of aortic valvular degeneration from aortic sclerosis to aortic stenosis. Here we reported the role of osteopontin (OPN) in the regulation of EC activation in vitro and in excised tissue from CAS patients and controls. OPN is an important pro-angiogenic factor in several pathologies. High levels of OPN have been demonstrated in both tissue and plasma of patients with aortic valve sclerosis and stenosis. The characterization of valvular ECs as a cellular target for OPN will help us uncover the pathogenesis of aortic valve degeneration and stenosis, opening new perspectives for the prevention and therapy of this prevalent disease. J. Cell. Physiol. 226: 2139–2149, 2011. ß 2010 Wiley-Liss, Inc. Calcific aortic stenosis (CAS) is a slow but progressive pathological condition of the aortic valve characterized, in its final stage, by dystrophic calcification of the valve leaflets (Freeman and Otto, 2005; Goldbarg et al., 2007; Beckmann et al., 2010). It is the most frequent valvular disease, with a prevalence of 3–9%, and the main cause for valve replacement in the adult population (Bach et al., 2007). The aortic valve degeneration starts with a normal trileaflet aortic valve; initial phases of the disease include mild thickening of the leaflets whereas more advanced stages are associated with impaired leaflet motion and increased resistance to forward blood flow (Freeman and Otto, 2005). These conditions are known as aortic valve sclerosis (AVSc) and aortic valve stenosis (AVS), respectively. Despite the high prevalence and mortality associated with aortic valve calcification, little is known about its pathogenetic mechanisms. As a result, the current treatment of choice for symptomatic AVS is surgical valve replacement (Cowell et al., 2004). Other treatment options, such as percutaneous valve replacement or aortic valvuloplasty, offer some benefits in terms of lower invasiveness and hospitalization time, but are not applicable to all patients (Balmer et al., 2004; Perin et al., 2009). Notably, surgical valve replacement in any of its forms leaves the underlying mechanism that caused the original valvular degeneration, untreated. Degenerative lesions in human valves are characterized by endothelial cell (EC) disruption, increased cellularity and extracellular matrix deposition, accumulation of oxidized lipoproteins, non-foam cell and foam cell macrophages, and occasional T cells within the valve interstitium (O’Brien et al., 1996; Olsson et al., 1999). These histological findings resemble early sclerotic lesions of the vasculature, and together with the shared risk factors, suggest that, as in atherosclerosis, the initiation of aortic valvular sclerosis involves chronic inflammatory and neo-angiogenesis processes potentiated by systemic factors. In advanced calcific valvular lesions, prominent features include mineralized deposits composed of hydroxyapatite, several bone matrix proteins, and mature osteoblasts and osteoclasts (O’Brien et al., 1995; Mohler et al., 2001; Rajamannan et al., 2003) (Fig. S1). Therefore, valvular Additional Supporting Information may be found in the online version of this article. Contract grant sponsor: National Heart, Lung and Blood Institute; Contract grant number: RC1HL100035. Contract grant sponsor: NIH. Contract grant sponsor: University of Pennsylvania School of Medicine (Harrison Memorial Fund). *Correspondence to: Giovanni Ferrari, Research Assistant Professor of Surgery, University of Pennsylvania School of Medicine, 500 S. Ridgeway Ave, Room 257, Glenolden, PA 19036. E-mail: [email protected] Received 25 August 2010; Accepted 9 November 2010 Published online in Wiley Online Library (wileyonlinelibrary.com), 6 December 2010. DOI: 10.1002/jcp.22549 ORIGINAL RESEARCH ARTICLE 2139 Journal of Journal of Cellular Physiology Cellular Physiology ß 2010 WILEY-LISS, INC.
-
Upload
paolo-poggio -
Category
Documents
-
view
212 -
download
0
Transcript of Osteopontin controls endothelial cell migration in vitro and in excised human valvular tissue from...

Osteopontin Controls EndothelialCell Migration In Vitro and inExcised Human Valvular Tissuefrom Patients with Calcific AorticStenosis and ControlsPAOLO POGGIO,1 JUAN B. GRAU,1 BENJAMIN C. FIELD,1 RACHANA SAINGER,1
WILLIAM F. SEEFRIED,1 FLAVIO RIZZOLIO,2,3 AND GIOVANNI FERRARI1*1Division of Cardiovascular Surgery, Department of Surgery, University of Pennsylvania School of Medicine,
Philadelphia, Pennsylvania2Department of Biology, Sbarro Institute for Cancer Research and Molecular Medicine, Center for Biotechnology,
College of Science and Technology, Temple University, Philadelphia, Pennsylvania3Human Health Foundation, Terni and Spoleto, PG, Italy
Calcific aortic stenosis (CAS) is a pathological condition of the aortic valve characterized by dystrophic calcification of the valve leaflets.Despite the high prevalence and mortality associated with CAS, little is known about its pathogenetic mechanisms. Characterized byprogressive dystrophic calcification of the valve leaflets, the early stages of aortic valve degeneration are similar to the active inflammatoryprocess of atherosclerosis including endothelial disruption, inflammatory cell infiltration, lipid deposition, neo-vascularization andcalcification. In the vascular system, the endothelium is an important regulator of physiological and pathological conditions; however, thecontribution of endothelial dysfunction to valvular degeneration at the cellular andmolecular level has received little attention. Endothelialcell (EC) activation and neo-vascularization of the cusps characterizes all stages of aortic valvular degeneration from aortic sclerosis toaortic stenosis. Herewe reported the role of osteopontin (OPN) in the regulation of EC activation in vitro and in excised tissue fromCASpatients and controls. OPN is an important pro-angiogenic factor in several pathologies. High levels of OPN have been demonstrated inboth tissue and plasma of patients with aortic valve sclerosis and stenosis. The characterization of valvular ECs as a cellular target forOPNwill help us uncover the pathogenesis of aortic valve degeneration and stenosis, opening new perspectives for the prevention and therapyof this prevalent disease.J. Cell. Physiol. 226: 2139–2149, 2011. � 2010 Wiley-Liss, Inc.
Calcific aortic stenosis (CAS) is a slow but progressivepathological condition of the aortic valve characterized, in itsfinal stage, by dystrophic calcification of the valve leaflets(Freeman and Otto, 2005; Goldbarg et al., 2007; Beckmannet al., 2010). It is the most frequent valvular disease, with aprevalence of 3–9%, and themain cause for valve replacement inthe adult population (Bach et al., 2007). The aortic valvedegeneration starts with a normal trileaflet aortic valve; initialphases of the disease include mild thickening of the leafletswhereas more advanced stages are associated with impairedleaflet motion and increased resistance to forward blood flow(Freeman and Otto, 2005). These conditions are known asaortic valve sclerosis (AVSc) and aortic valve stenosis (AVS),respectively.
Despite the high prevalence and mortality associated withaortic valve calcification, little is known about its pathogeneticmechanisms. As a result, the current treatment of choice forsymptomatic AVS is surgical valve replacement (Cowell et al.,2004). Other treatment options, such as percutaneous valvereplacement or aortic valvuloplasty, offer some benefits interms of lower invasiveness and hospitalization time, but arenot applicable to all patients (Balmer et al., 2004; Perin et al.,2009). Notably, surgical valve replacement in any of its formsleaves the underlying mechanism that caused the originalvalvular degeneration, untreated.
Degenerative lesions in human valves are characterizedby endothelial cell (EC) disruption, increased cellularity andextracellular matrix deposition, accumulation of oxidizedlipoproteins, non-foam cell and foam cell macrophages, and
occasional T cells within the valve interstitium (O’Brien et al.,1996; Olsson et al., 1999). These histological findings resembleearly sclerotic lesions of the vasculature, and together with theshared risk factors, suggest that, as in atherosclerosis, theinitiation of aortic valvular sclerosis involves chronicinflammatory and neo-angiogenesis processes potentiated bysystemic factors. In advanced calcific valvular lesions, prominentfeatures include mineralized deposits composed ofhydroxyapatite, several bone matrix proteins, and matureosteoblasts and osteoclasts (O’Brien et al., 1995; Mohler et al.,2001; Rajamannan et al., 2003) (Fig. S1). Therefore, valvular
Additional Supporting Information may be found in the onlineversion of this article.
Contract grant sponsor: National Heart, Lung and Blood Institute;Contract grant number: RC1HL100035.Contract grant sponsor: NIH.Contract grant sponsor: University of Pennsylvania School ofMedicine (Harrison Memorial Fund).
*Correspondence to: Giovanni Ferrari, Research AssistantProfessor of Surgery, University of Pennsylvania School ofMedicine, 500 S. Ridgeway Ave, Room 257, Glenolden, PA 19036.E-mail: [email protected]
Received 25 August 2010; Accepted 9 November 2010
Published online in Wiley Online Library(wileyonlinelibrary.com), 6 December 2010.DOI: 10.1002/jcp.22549
ORIGINAL RESEARCH ARTICLE 2139J o u r n a l o fJ o u r n a l o f
CellularPhysiologyCellularPhysiology
� 2 0 1 0 W I L E Y - L I S S , I N C .

calcification, rather than being attributable to passive,unregulated precipitation of calcium phosphate, appears to bean active, highly regulated biomineralization process (Simmonset al., 2005).
Multiple biologic pathways are responsible for aortic valvedegeneration. EC activation and neo-vascularization of thecusps characterize the valvular degeneration process since theaortic sclerosis phase. Pro-angiogenic factors are stilldetectable in the valve tissue when leafletmotion is impaired bycalcium deposition (Collett andCanfield, 2005). The angiogenicprocess starts concomitantly with EC disruption, inflammationand lipid deposition (Pastor-Perez andMarin, 2009). Therefore,in the pathogenesis of CAS, injury to the endothelium is animportant initial step causing thickening of the valve as aconsequence of abnormal deposition of extracellular matrix,and thus preventing oxygen supply by diffusion. Decreasedoxygen tension is a strong initiator of the angiogenic responsethrough the induction of vascular endothelial growth factor(VEGF) gene expression (Ferrara and Davis-Smyth, 1997;Ferrara, 1999). The over-expression of VEGF receptors, Flk-1and Flt-1, also increase with tissue hypoxia further supportingthe importance of angiogenesis in the initiation and progressionof CAS. The endothelium lining the surface of valve leaflets ispresumed to be involved in valve homeostasis and pathology.Calcified aortic valve (CAV) cusps show a stronger angiogenicresponse than non-calcified valve (NCAV) cusps (Soini et al.,2003; Chalajour et al., 2004, 2007; Collett and Canfield, 2005).Our results, and other related studies, show the formation oftubular-like sprouts from human tissue explants of CAV leafletsusing a three-dimensional (3D) culture system. Sproutformation points toward the angiogenic potential of thesetissues during the process of valve degeneration. 3D collagenand immunohistochemical studies reveal that tissues fromCAVleaflets have a high angiogenic response when compared toNCAV. The formation of angiogenic sprouts from stenoticvalves occurs significantly faster than from non-stenotic valves(Soini et al., 2003; Chalajour et al., 2007).
Here, we first characterize the role of osteopontin (OPN) inthe regulation of EC activation in vitro, and then analyzed itsrole on tissue excised fromCAS patients and controls.OPN is amultifunctional glycol-phosphoprotein that is known for itsregulatory function in bone remodeling. OPN is also involved inthe inhibition of biomineralization of dystrophic and ectopicsites, including aortic valve tissue (Yu et al., 2009).Characterization of the biological process leading to valvulardegeneration has been mostly focused on the formation ofcalcium nodules and the role of valvular interstitial celltransdifferentiation (Taylor et al., 2003). With this study, weintend to characterize the effect of OPNon cultured ECs and inexcised tissue from CAS patients and controls. Studying theprocess of EC activation could bring to light important insightsin the molecular mechanisms that drive the process of valvedegeneration and help in the development of a therapeutictarget before the calcification process occurs and surgicalintervention becomes inevitable.
MethodsCells, antibodies, and reagents
Human coronary artery smooth muscle cells, MEMAlpha medium,MEM medium, 10� DMEM, M200 medium, L-glutamine, fetalbovine serum, low serum growth supplement (LSGS) andpenicillin/streptomycin (P/S) solutions were purchased fromInvitrogen (Carlsbad, CA). Human recombinant OPN, anti-OPNneutralizing Ab and anti-VEGF neutralizing Ab were purchasedfrom R&D systems (Minneapolis, MN), Ab anti-phospho-ERK1/2and Ab anti-CD31 (PECAM-1) from Cell Signal (Beverly, MA), Abanti-ERK 2, and secondary Ab goat anti-rabbit horseradishperoxidase-conjugated from Santa Cruz Biotechnology (Santa
Cruz, CA), anti-aVb3 neutralizing Ab fromMillipore (Billerica, MA)and anti-CD44 neutralizing Ab from Ancell (Bayport, MN).Collagen I was purchased from BD Biosciences (San Jose, CA),VEGF from Peprotech (Rocky Hill, NJ) and UO126 fromCalbiochem (San Diego, CA). For the phosphorylation study,casein kinase II was purchased from New England Biolabs(Ipswich, MA).
Wound healing assay
Mechanical endothelial damage was created in confluent humanumbilical vein endothelial cells (HUVECs) growing in 12-well platesby using a sterile 200ml tip. Cultures were reefed with low serummedium in the presence or absence of OPN (50 ng/ml), (p)OPN(50 ng/ml),NAbavb3 (10mg/ml), NAbCD44 (10mg/ml) orUO126(10 nM). Images were taken at 0, 4, and 24 h time points. Afterwounding, cell migration in the recovery space was estimated byimage analysis using an invertedOlympus CK2microscope (HitechInstruments, Inc., Broomall, CA). Wound closure was determinedas the difference between wound width at 0 and 24 h.
In vitro calcification assay
In vitro calcification assay was performed using human coronaryartery smooth muscle cells. Cells were cultured in Alpha MEMcontaining P/S, L-glutamine and10% Fetal Bovine Serum (FBS), until70–80% confluent. Growth medium was replaced by calcificationmedia (a-MEM containing 3mM phosphate buffer) in all wellsexcept the control. Fifty nanograms of recombinantOPNwas useddirectly and casein kinase II phosphorylated recombinant OPN inseparate wells in presence or absence of UO126, NAb avb3, orNAb CD44. Medium was replaced every 2 days. At day 8, thesmooth muscle cells (SMCs) were incubated overnight in 0.6NHCL at 48C.Calciumwas estimated using the calcium assay reagent(Biovision, Mountain View, CA). After washing the cells twice with1� PBS, cells were harvested using 1NNaOHþ 3% SDS, and totalprotein was estimated. Amount of total calcium was expressed asmg of calcium/ml of medium. Pictures were taken at days 0 and 8using an inverted Olympus CK2 microscope.
MAP kinase profile
Analysis of phospho-MAP kinases was carried out using the HumanPhospho-MAPK Array Kit (R&D Systems, Minneapolis, MN)according to the manufacturer’s instructions. Briefly, the HUVECsand human coronary artery SMCs were grown to confluence andstarved overnight with the growth medium supplemented with0.5% FBS. Cells were then treated for 15min with 50 ng/ml ofnon-phosphorylated and phosphorylated recombinant OPN. Cellswere washed twice with PBS and lysates were prepared using thelysis buffer. Array included, 4 nitrocellulose membranes containing21 different anti-kinase antibodies and 7 different controls printedin duplicate. Thesemembranes were blocked for 2 h. One hundredmicrograms of cell lysate was incubated with the membraneovernight at 48C. Blots were washed twice and incubated withdetection antibody for 2 h at room temperature. Finally, the blotswere incubated with the Streptavidin-HRP for 30min at roomtemperature on a rocking platform shaker. Blots were developedusing the ECL substrate (Thermo Scientific). Phospho-MAPKArray data were quantitated by scanning the image and analyzingthe array image file using Image J software.
Western blotting
After SDS–PAGE, proteins were transferred to PVDF membranes(Invitrogen). After transfer, membranes were blocked using 5%nonfat dried milk in TBS, pH 7.4 and incubated overnight at 48Cwith the indicated antibodies. The membranes were washed threetimeswith TBS–0.1%Tween 20 and then incubatedwith secondaryantibodies (1:5,000) for 45min at room temperature. Afterwashing three-times with TBS–0.1% Tween 20, theimmunoreactive bands were visualized using enhanced
JOURNAL OF CELLULAR PHYSIOLOGY
2140 P O G G I O E T A L .

chemilumiescence (ECL) detection reagents. The membraneswere scanned using the LAS-3000 Fujifilm Systems (Stamford, CT),and the labeled bands were quantified using the software Image J.
Tissue collection
Tissue samples were collected, through an IRB approved protocolfrom The Hospital of the University of Pennsylvania and PennPresbyterian Medical Center. Patients were consented prior tointervention. The valve leaflets were collected at the time ofsurgical excision. The presence (or lack) of calcific AVS wasdetermined through the analysis of cardiac catheterization,2D-Echocardiography and clinical impression in situ. The sampleswere collected inmedia and kept at 48Cuntil embedded in collagen.Some control patient valves were collected through ourcollaboration with the Heart Failure Research Department at theUniversity of Pennsylvania.
Baseline patients characteristics
On the total of 7 subjects, 43% were male. Their age was61.3� 21.7 years. For the patients with CAS, the initial AVA was0.8� 0.2, two had systemic hypertension, three had significantcoronary artery disease and onewas a current smoker. All patientshad a comprehensive echocardiographic assessment including,M-mode, two-dimensional and color Doppler echocardiography.Supplemental Table I further characterizes the study groups. Aorticvalve calcification score was 3.5� 0.59 in CAS patients and1.3� 0.49 in controls (P< 0.001).
In vitro angiogenesis assay
Collagen I was prepared according to the manufacturer’sinstruction: 80% Collagen I, 10% 10� DMEM, and 10% 0.01MNaOH. The solutionwas then titratedwith 0.1MNaOH to pH 7.4.In a 24-well plate, the tissue sample (1mm2) was embedded in 1mlof collagen solution and allowed to polymerize for 30min at 378C(5% CO2) prior to media addition. M200 medium wassupplemented with 10% FBS, 1% L-Glu, 10% LSGS, and 1% P/S. Themedia were changed every other day. The following treatmentswere added: M200 Medium; VEGF 30 ng/ml; OPN 50 ng/ml;(p)OPN 50 ng/ml; UO126 10mM;NAbCD44 10mg/ml; NAbaVb3
10mg/ml; NAb OPN; and NAb VEGF. Pictures were taken every3 days until day 14.
Immunohistochemistry
Paraffin embedded sections for both CAV and NCAV tissues werestained using the detection kit from DAKO (Glostrup, Denmark).A standard protocol was followed. Primary CD31 antibodyconcentration was optimized by titration and conjugated with aHRP linked secondary (DAKO). After color development usingDABþ (DAKO), a hematoxylin stain (Vector LaboratoriesBurlingame, CA) was applied. Slides were mounted with tolueneand imaged. Alizarin Red and H&E staining were provided by theHospital of the University of Pennsylvania’s Pathology Laboratory.
ResultsOsteopontin promotes endothelial cell migrationthrough ERK1/2 activation
Since the endothelium is one of the primary targets duringvalvular degenerationwe first characterized the endothelial cell(EC) response to OPN in vitro (Fig. 1) and its intracellularsignaling pathway (Figs. 2 and 4).
To test the effect that OPN has on ECs, we performed awound healing assay on cultured HUVECs and measure the ECresponse. As shown in Figure 1, OPN promotes EC migrationin vitro.Differences in ECmigration can be observed after 4 h oftreatment (not shown) and are evident after 24 h of treatment(Fig. 1A,B). These experiments confirm that OPN has an activerole in promoting EC migration in vitro. Similar experimentswere performed with human aortic ECs, human microvascularECs, and bovine ECs with similar results.
It has been reported that the positive effect of OPN onneo-vascularization is mediated by PI3K–ERK1/2 intracellularsignaling pathway in vivo (Dai et al., 2009). To test theintracellular signaling pathway that mediates EC migration, wefirst analyzed the MAPK activation of HUVECs in thepresence or absence of recombinant OPN. As reported inFigure 2A (and Supplement Fig. S2B) the MAPK profile arrayshows significant activation of ERK1/2 intracellular signalingpathway. To confirm these results, HUVECs were treated for
Fig. 1. OPN induces in vitroendothelial cellmigration.A:Woundhealingassayof confluentHUVECs in thepresenceorabsenceof recombinantOPN (50 ng/ml) analyzed after 24 h.Magnification 4T. B: Bar graph representation of endothelial cellmigration. The results of countedmigratedcells are representative of three independent experiments each done in triplicate; P<0.01
JOURNAL OF CELLULAR PHYSIOLOGY
O S T E O P O N T I N A N D C A L C I F I C A O R T I C S T E N O S I S 2141

the indicated time with recombinant OPN and then analyzedin Western blotting with phospho-specific ERK1/2 antibody(Fig. 2B). This experiment confirms ERK1/2 intracellularsignaling activation under OPN treatment. We thereforeutilized a widely used ERK1/2 chemical inhibitor (UO126) totest if ECmigration in vitrowasmediated by ERK1/2. In Figure 2,we demonstrated using the wound healing assay, that blockingERK1/2 activation results in the abolishment of EC migration invitro (Fig. 2C,D).
OPN biological activity is controlled by itsphosphorylation status in both endothelial cells andsmooth muscle cells
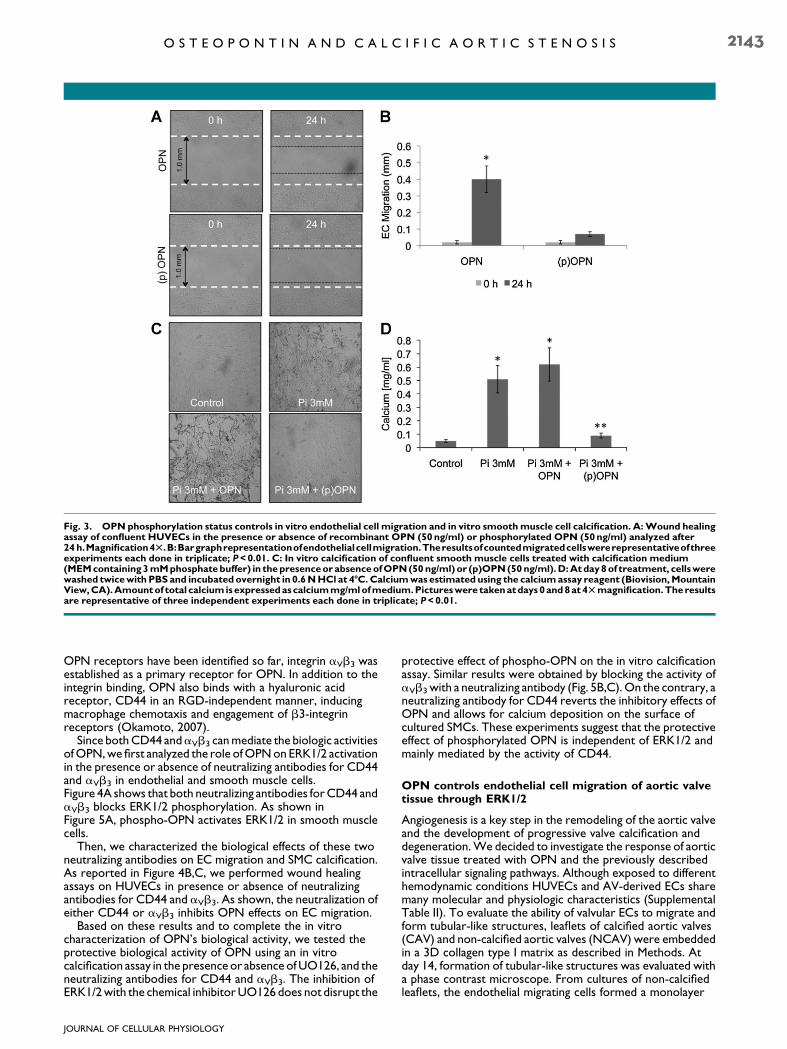
To further characterize the role of OPN on the regulation ofEC migration, we considered the effects of OPN’spost-translational modifications on EC activation. RecombinantOPN was phosphorylated in vitro using casein kinase II(Fig. S2A). The extent of the phosphorylation of OPN, as wellas, the phosphorylation occurring on specific sites, may play animportant role in its physiological functions (Jono et al., 2000).It has been described that the ability of OPN to inhibitcalcification depends on its phosphorylation status (Saad et al.,2008) (Fig. 3C,D). Since cultured smooth muscle cells (SMCs)and valvular interstitial cells (VICs) share many molecular andphysiological characteristics (Supplemental Table II), an in vitrocalcification assay has been extensively used as a model to testthe biological activity of proteins involved in the calciumdeposition related to aortic valve degeneration (Gericke et al.,2005; Speer et al., 2005).
We phosphorylated the recombinant OPN in vitro andtested its ability to control calcium deposition on a SMC basedcalcification assay. As shown in Figure 3C,D, dephosphorylatedrecombinant OPN did not inhibit human SMCbiomineralization, while phosphorylated recombinant OPNinhibits calcium deposition in vitro. These experiments confirmthat the phosphorylation status controls the ability of OPN toinhibit biomineralization.
We then performed wound healing experiments in thepresence or absence of in vitro phosphorylated OPN toinvestigate the role of this post-translationalmodification on ECactivation. As shown in Figure 3A,B, phospho-OPN does notactivate EC migration in vitro, whereas, as shown in Figure 1,dephosphorylated OPN promotes EC migration. Theseresults suggest a different mechanism of action of OPN ondifferent cellular populations. The phosphorylation statuscontrols OPN’s biological activity on both ECs and SMCs: inSMC in vitro calcification assay OPN requires phosphorylationto inhibit calcium deposition; on the contrary,dephosphorylated OPN has no effects in preventing calciumdeposition on the cellular surface. On ECs, dephosphorylatedOPNpromotes ECmigrationwhereas phospho-OPNpreventsEC migration.
Role of Osteopontin on CD44 and aVb3 signaling inendothelial cell migration compared to smooth musclecell calcification
The primary receptors for OPN are those integrins that bindthe central integrins attachment motif RGD. Since no specific
Fig. 2. OPN promotes in vitro endothelial cell migration through ERK1/2 phosphorylation. A: Phospho-MAP kinase profile array of HUVECstreatedwith recombinantOPN(50 ng/ml) showing (p)ERK1/2activation.B:Westernblotting for (p)ERK1/2ofOPN-treatedHUVECs.TotERK2wasusedtonormalizetheresultsandasa loadingcontrol.C:WoundhealingassayofconfluentHUVECstreatedwithFBS10%asapositivecontrol,andOPN(50ng/ml) in thepresenceor absenceofERK1/2’s chemical inhibitorUO126 (10mM).Endothelial cellmigrationwasphotographedafter24 h at 4Tmagnification. D: Bar graph representation of endothelial cell migration. The results of counted migrated cells are representative ofthree experiments each done in triplicate; P<0.01.
JOURNAL OF CELLULAR PHYSIOLOGY
2142 P O G G I O E T A L .
OPN receptors have been identified so far, integrin aVb3 wasestablished as a primary receptor for OPN. In addition to theintegrin binding, OPN also binds with a hyaluronic acidreceptor, CD44 in an RGD-independent manner, inducingmacrophage chemotaxis and engagement of b3-integrinreceptors (Okamoto, 2007).
Since bothCD44 andaVb3 canmediate the biologic activitiesofOPN,we first analyzed the role ofOPNonERK1/2 activationin the presence or absence of neutralizing antibodies for CD44and aVb3 in endothelial and smooth muscle cells.Figure 4A shows that both neutralizing antibodies forCD44 andaVb3 blocks ERK1/2 phosphorylation. As shown inFigure 5A, phospho-OPN activates ERK1/2 in smooth musclecells.
Then, we characterized the biological effects of these twoneutralizing antibodies on EC migration and SMC calcification.As reported in Figure 4B,C, we performed wound healingassays on HUVECs in presence or absence of neutralizingantibodies for CD44 and aVb3. As shown, the neutralization ofeither CD44 or aVb3 inhibits OPN effects on EC migration.
Based on these results and to complete the in vitrocharacterization of OPN’s biological activity, we tested theprotective biological activity of OPN using an in vitrocalcification assay in the presence or absence ofUO126, and theneutralizing antibodies for CD44 and aVb3. The inhibition ofERK1/2with the chemical inhibitorUO126 does not disrupt the
protective effect of phospho-OPN on the in vitro calcificationassay. Similar results were obtained by blocking the activity ofaVb3 with a neutralizing antibody (Fig. 5B,C).On the contrary, aneutralizing antibody for CD44 reverts the inhibitory effects ofOPN and allows for calcium deposition on the surface ofcultured SMCs. These experiments suggest that the protectiveeffect of phosphorylated OPN is independent of ERK1/2 andmainly mediated by the activity of CD44.
OPN controls endothelial cell migration of aortic valvetissue through ERK1/2
Angiogenesis is a key step in the remodeling of the aortic valveand the development of progressive valve calcification anddegeneration.We decided to investigate the response of aorticvalve tissue treated with OPN and the previously describedintracellular signaling pathways. Although exposed to differenthemodynamic conditions HUVECs and AV-derived ECs sharemany molecular and physiologic characteristics (SupplementalTable II). To evaluate the ability of valvular ECs to migrate andform tubular-like structures, leaflets of calcified aortic valves(CAV) and non-calcified aortic valves (NCAV) were embeddedin a 3D collagen type I matrix as described in Methods. Atday 14, formation of tubular-like structures was evaluated witha phase contrast microscope. From cultures of non-calcifiedleaflets, the endothelial migrating cells formed a monolayer
Fig. 3. OPN phosphorylation status controls in vitro endothelial cell migration and in vitro smoothmuscle cell calcification. A:Wound healingassay of confluent HUVECs in the presence or absence of recombinant OPN (50ng/ml) or phosphorylated OPN (50ng/ml) analyzed after24 h.Magnification4T.B:Bargraphrepresentationofendothelialcellmigration.Theresultsofcountedmigratedcellswererepresentativeofthreeexperiments each done in triplicate; P<0.01. C: In vitro calcification of confluent smooth muscle cells treated with calcification medium(MEMcontaining3mMphosphatebuffer) in thepresenceor absenceofOPN(50 ng/ml)or (p)OPN(50ng/ml).D:Atday8of treatment, cellswerewashed twicewith PBS and incubated overnight in 0.6NHCl at 4-C.Calciumwas estimated using the calciumassay reagent (Biovision,MountainView,CA).Amountof total calcium isexpressedascalciummg/mlofmedium.Pictureswere takenatdays0and8at4Tmagnification.Theresultsare representative of three independent experiments each done in triplicate; P<0.01.
JOURNAL OF CELLULAR PHYSIOLOGY
O S T E O P O N T I N A N D C A L C I F I C A O R T I C S T E N O S I S 2143

around the valve tissue with few tubular-like structures.Calcified leaflets showed extensive tubular-like structures(Fig. 6A,B).
Among the angiogenic factors, VEGF has been shown to playa major role in both physiologic and pathologic conditions, dueto its unique biologic capacity to induce migration andproliferation of ECs, to enhance vascular permeability and tomodulate thrombogenicity (Ferrara and Davis-Smyth, 1997).Accordingly, in Figure 6A,B we show that VEGF treatment ofboth CAV and NCAV enhances the angiogenic response ofcultured valve tissues. These results confirm the angiogenicpotential of ECs originating from aortic valve leaflets. CAVtissue shows more angiogenic activity than NCAV tissue.
To test if OPN has a similar effect on valvular ECs, werepeated the cell sprouting assay in the presence or absence ofOPN and we measured the cell number, migration distanceand the formation of tubular-like structures of ECs derivedfrom both NCAV and CAV tissue. As reported in Figure 6C,OPN promotes migration and tubular-like formation in our 3Dangiogenesis assay, suggesting that both HUVEC and valvularEC migration are controlled by OPN. Figure 6D is a bar
graph representation of the number of branches per fieldanalyzed.
We then tested the effect of OPN on valvular tissue toconfirm the ERK1/2mediation of the effects ofOPNon valvularEC migration. As reported in Figure 6C,D, OPN enhances ECmigration into the 3D collagen matrix and shows that UO126(a chemical inhibitor of ERK1/2) blocks such migration.Together, these results suggest that both HUVECs and aorticvalve-derived ECs are activated by OPN and that the activationis mediated by an ERK1/2 intracellular signaling pathway.
Finally, we tested the role of both CD44 and aVb3 on theformation of tubular-like structures on aortic valve derivedtissue (Fig. 7). In accord with our in vitro experiments, bothCD44 and aVb3 are necessary for EC migration and collagensprouting. The neutralization of either CD44 or aVb3 blocksthe cellular reorganization into the collagen matrix. Theseresults confirm that cultured ECs and aortic valve-derivedtissue share the same cellular responsewhen treatedwithOPNor phospho-OPN. The intracellular signaling pathway activatedby OPN is mediated by CD44 and aVb3; this results in ERK1/2activation (Fig. 7A,B). VEGF, the major EC derived pro-
Fig. 4. aVb3andCD44arenecessary forOPN-inducedendothelial cellmigration.A:Westernblottingof total proteinextractofHUVECs to testERK1/2phosphorylationunderdifferenttreatments.Fromleft toright:FBS10%,VEGF(30 ng/ml),untreated,OPN(50ng/ml), (p)OPN(50 ng/ml),UO126 (10mM) in the presence of OPN or (p)OPN, neutralizing antibody for aVb3 (10mg/ml) in the presence or absence of OPN or (p)OPNand neutralizing antibody for CD44 (10mg/ml) in the presence or absence of OPN or (p)OPN. B: Wound healing assay of confluent HUVECstreated with OPN (50 ng/ml) in the presence of neutralizing antibody for CD44 or aVb3. Endothelial cell migration was photographed after24 h at 4Tmagnification. C: Bar graph representation of endothelial cell migration. The results of counted migrated cells are representative ofthree experiments each done in triplicate; P<0.01.
JOURNAL OF CELLULAR PHYSIOLOGY
2144 P O G G I O E T A L .

angiogenic factor, increasesOPN andaVb3-integrin expressionin HUVECs and stimulates integrin dependent EC motility(Senger et al., 1996). Recently, it has been reported thatOPN increased VEGF expression in tumor cells stimulatingneo-vascularization. In turn, VEGF also induced theoverexpression of OPN in tumor cells (Chakraborty et al.,2008). Thus, experimental evidence suggests that OPN mayaffect angiogenesis by acting directly on ECs.
Since VEGF increases OPN and aVb3-integrin expression inHUVECs and stimulates integrin dependent ECmotility (Sengeret al., 1996), we tested the effect of neutralizing anti-VEGF andneutralizing anti-OPN on cellular migration of AV-derivedtissue. As expected, both treatments resulted in the inhibitionof cellular motility (Fig. S3). Analyzing the cellular andmolecularbiology responses of EC activation could bring to light veryimportant insights on the mechanisms that drive the process ofvalve degeneration and help in development of a therapeutictarget before the calcification process occurs and surgicalintervention becomes inevitable.
Discussion
With an estimated prevalence of 5.2 million affected people inthe United States, CAS represents the most common type ofvalvular disease (Bach et al., 2007). The prevalence increaseswith age, so that approximately 30% of all individuals aged 65þyears have AVSc and 4% have AVS (Cowell et al., 2004). As a
result of the steady population growth and rising lifeexpectancy, the demand for aortic valve operations willsignificantly increase in the future (Northrup et al., 2002;Freeman and Otto, 2005; Thom et al., 2006; Beckmann et al.,2010). The current treatment of choice for AVS is surgical valvereplacement (Cowell et al., 2004), either with mechanical orbiological prostheses. Currently, there is no FDA-approvedtreatment to halt the progression of calcific aortic valvedegeneration.
Although calcification is the most common problemassociated with aortic valve disease, the cellular physiology andmolecular events that underlie this process are not fullyelucidated.
The assumption that CAS is only a passive, age-relateddisease has been strongly questioned by numerous studiesshowing that the pathogenesis of this disease actively requiresthe activation and transdifferentiation of several cellularcomponents. The process of calcification is believed to be ahighly programmed sequence of different intercellularsignaling events, involving different cellular populations, whichinfluence many fundamental cellular functions, such asproliferation, migration, gene transcription, cell–cell adhesionand cell–matrix adhesion (Gu and Masters, 2010).
Cardiac VICs, the primary cellular component involved in thebiomineralization process, are a heterogeneous and dynamicpopulation. They expressmolecularmarkers similar to those ofSMCs performing a complex and highly sophisticated series of
Fig. 5. CD44 but not aVb3 regulated (p)OPN-induced smooth muscle cell bio-mineralization. A: Phospho-MAP kinase profile array ofhuman smooth muscle cells treated with recombinant OPN (50ng/ml) showing (p)ERK 1/2 activation. B: In vitro calcification of confluentsmooth muscle cells treated with calcification medium (MEM containing 3mM phosphate buffer) in the presence of the indicated treatments.C: At day 8 of treatment, calcium was estimated using the calcium assay reagent. Amount of total calcium is expressed as calcium mg/ml ofmedium. Pictures were taken at 4Tmagnification. The results are representative of three independent experiments each done in triplicate;P<0.01.
JOURNAL OF CELLULAR PHYSIOLOGY
O S T E O P O N T I N A N D C A L C I F I C A O R T I C S T E N O S I S 2145

functions over awide range of hemodynamic conditions (Tayloret al., 2003).Within the aortic valve leaflets, a number of cellularphenotypes can be distinguished; VICs represent the mostabundant and more studied cellular population. However, agrowing number of studies have been recently focused on theendothelium (El-Hamamsy et al., 2009; Simmons, 2009). Thesecells, actively interacting with the non-cellular component ofthe leaflets, are responsible for many valvular functions. Injuryto the EC covering the valve leaflets is an important initial step inthe pathogenesis of calcific aortic valve degeneration. As aresult, EC activation and neo-vascularization within the leaflettissue is a hallmark of aortic valve sclerosis and stenosis. Theoverexpression of pro-angiogenic factors such as VEGF, Flk-1and Flt-1 further supports the assumption that angiogenesis isclosely related to the progression of aortic valve degeneration.
OPN is a phosphorylated, acidic, RGD-containingglycoprotein that binds certain CD44 variants and integrinreceptors, including aVb3 (Denhardt et al., 2001). The OPN–aVb3 interaction is important for osteoclast migration andresorption, as well as, SMC migration and adhesion. OPN acts
as a cytokine, playing important roles in the migration andinvasion of several tumor cells (Rittling and Chambers, 2004;Tuck et al., 2007). In addition, OPN is regarded to be animportant angiogenic factor in several pathologies. High levelsof OPN have been demonstrated in both tissue and plasma ofpatients with aortic valve sclerosis and stenosis. Here wedescribed the biological effects of OPN in vitro and in excisedaortic valve tissue. Our results, combined with previouslyreported studies, confirm that the biological function ofOPN on aortic valve degeneration is not exclusively related tothe biomineralization process of VICs. OPN also plays animportant role on different cellular targets, such as ECs.Calcified rheumatic valves have also been shown to expressneo-angiogenesis factors (Rajamannan, 2005). Althoughangiogenesis is considered to be part of the early stages ofcalcification, pro-angiogenic factors are expressed even whenthe valve is completely calcified, suggesting that this mechanismis still present and active during the entire degenerationprocess. Immunohistochemistry for the EC markers CD31,CD34, von-Willebrand factor and Tie-2 and for the cell
Fig. 6. OPN induces in vitro angiogenesis of excised human aortic valve tissues through ERK1/2. A: In vitro angiogenesis assay of excised tissuefrom calcified and non-calcified aortic valve. Tissue was embedded in the collagen as described in the Methods Section and treated with VEGF(30ng/ml). B: Bar graph representation of the in vitro angiogenesis assay. The number of tubular-like structures was counted as a number ofbranches per field. Pictureswere taken at 10Tmagnification. C:Non-calcified and calcified tissue embedded in collagen and treatedwithOPN inthe presence or absence ofUO126 (10mM). B: Bar graph representation of the in vitro angiogenesis assay. The number of tubular-like structureswas counted as a number of branches per field. Pictures were taken at 10Tmagnification. The results are representative of three independentexperiments each done in triplicate; P<0.01.
JOURNAL OF CELLULAR PHYSIOLOGY
2146 P O G G I O E T A L .

adhesion molecules, reveals an intense neovascularization ofstenotic cusps in contrast to non-stenotic cusps. In Figure S1,we show immunohistochemistry staining of CD31 in NCAVandCAV tissue. Correspondingly, it has been shown that ECs ofstenotic but not those of non-stenotic valves exhibitCEACAM1, a cell adhesion molecule expressed in angiogenic,but not in quiescent ECs (Ergun et al., 2000; Chalajour et al.,2004; Yetkin and Waltenberger, 2009). Chalajour et al. (2004)have demonstrated that the formation of angiogenic sproutsfrom calcified valves occurs significantly faster than fromnon-calcified valves. The capillary-like tubes sprouting fromexplanted tissue shows a positive staining for CD31.Immunoreactivity of CD44 and vWF has also beendemonstrated in the tubular-like structures (Chalajour et al.,2004). We report our analysis of the role of OPN on culturedECs and SMCs, two cellular phenotypes that, due to theirphysiological and molecular characteristics, resemble the twomajor cellular populations of the aortic valve leaflets. We thencompared these results with the effects of OPN on aortic valvederived tissue form CAS patients and controls. Our analysisshows that OPN activated the same intracellular signalingpathways in HUVECs and excised aortic valve tissue. Inaddition, we reported a differential OPN activated intracellularsignaling pathway in EC and SMC (Fig. 8).
Based on the presented results, EC migration of bothHUVEC and aortic valve derived cells is dependent on CD44and aVb3 and results in ERK1/2 phosphorylation. Since nospecific OPN receptors have been identified, the receptors forOPN are those integrins that bind the central integrinsattachment motif RGD, with the integrin aVb3 considered oneof the primary mediators of OPN’s activity on different celltypes. In addition to integrin binding, OPN also binds CD44 inan RGD-independent manner, inducing macrophagechemotaxis and engagement of b3-integrin receptors(Okamoto, 2007). Interestingly, OPN’s biological effect onSMCs is dependent on the phosphorylation status ofOPNand itis mediated exclusively by CD44. It is possible that differentOPN post-translational modifications enable the protein to
bind different receptors with variable affinity and thereforeregulate a differential biological response.
In the aortic valve, lipid deposits and calcific lesions occurprimarily in the fibrosa, the layer of the valve immediatelybeneath the endotheliumon the aortic side of the valve (Otto etal., 1994; O’Brien et al., 1996; Simmons et al., 2005). Thepreferential susceptibility to lesion formation on the aorticrather than ventricular surface of the aortic valve may resultfrom coordinated regulation of gene expression by therespective endothelia, resulting in side-specific endothelialphenotypes that favor or inhibit calcification. Furthermore,during the cardiac cycle, the aortic valve endothelium issubjected to complex fluid dynamics that are distinctly differenton either side of the valve (Nicosia et al., 2003; Simmons et al.,2005). Thus, there is a spatial correlation between the nature ofcalcific lesions and the local hemodynamic environment, similarto that observed in the regions of large arteries moresusceptible to atherogenesis. Local environmental factors andbiomechanical forces may also contribute to the developmentof differential endothelial phenotypes with differentsusceptibility to focal calcification. This observation couldgenerate the question of whether AV-derived cells exhibitdifferential properties when compared to AV tissue due todifferent mechanical stresses in response to directional flow.However, our preliminary histological and cytological analysisshows equivalence in the EC surface and intracellular signalingpathways activation on both sides of the aortic valve leaflets,suggesting that, although the hemodynamic conditions aredifferent, the cellular response is similar. Knowledge of thecomplex mechanisms of cell–cell and cell–matrix interaction isessential in order to understand how different cell populationscontribute to the progressive degeneration of the valve. Thesedata provide new insights into the cellular and molecularmechanisms involving stenotic aortic valvular tissue and ECs.These results help us better understand the interactionbetween different cell populations within the valve tissue andbring new potential perspectives to the development of futurepreventive treatments for this condition.
Fig. 7. aVb3 andCD44 are necessary forOPN-induced in vitro angiogenesis of excised human aortic valve tissues. A: Bar graph representationof the in vitro angiogenesis assay. Tissues were embedded in the collagen as described in the Methods Section and treated as indicated. Thenumber of tubular-like structures was counted as the number of branches per field. Pictures were taken at 10T magnification. The results arerepresentative of three independent experiments each done in triplicate; P<0.01.
JOURNAL OF CELLULAR PHYSIOLOGY
O S T E O P O N T I N A N D C A L C I F I C A O R T I C S T E N O S I S 2147

Acknowledgments
This project is currently supported by award numberRC1HL100035 from the National Heart, Lung, And BloodInstitute, NIH (GF, JG, PP, and BF) and by the ‘‘HarrisonMemorial Fund’’ of the University of Pennsylvania School ofMedicine (RS and WS). We thank Kenneth B. Margulies, fromthe Heart Failure Research Department at the University ofPennsylvania for his precious collaboration in the collection ofheart valve specimens.
Literature Cited
Bach DS, Radeva JI, Birnbaum HG, Fournier AA, Tuttle EG. 2007. Prevalence, referralpatterns, testing, and surgery in aortic valve disease: Leaving women and elderly patientsbehind? J Heart Valve Dis 16:362–369.
Balmer C, Beghetti M, Fasnacht M, Friedli B, Arbenz U. 2004. Balloon aortic valvoplasty inpaediatric patients: Progressive aortic regurgitation is common. Heart 90:77–81.
Beckmann E, Grau JB, Sainger R, Poggio P, Ferrari G. 2010. Insights into the use of biomarkersin calcific aortic valve disease. J Heart Valve Dis 19:441–452.
Chakraborty G, Jain S, KunduGC. 2008. Osteopontin promotes vascular endothelial growthfactor-dependent breast tumor growth and angiogenesis via autocrine and paracrinemechanisms. Cancer Res 68:152–161.
Chalajour F, Treede H, Ebrahimnejad A, Lauke H, Reichenspurner H, Ergun S. 2004.Angiogenic activation of valvular endothelial cells in aortic valve stenosis. Exp Cell Res298:455–464.
Chalajour F, Treede H, Gehling UM, Ebrahimnejad A, Boehm DH, Riemer RK, Ergun S,Reichenspurner H. 2007. Identification and characterization of cells with high angiogenicpotential and transitional phenotype in calcific aortic valve. Exp Cell Res 313:2326–2335.
Collett GD, Canfield AE. 2005. Angiogenesis and pericytes in the initiation of ectopiccalcification. Circ Res 96:930–938.
Cowell SJ, Newby DE, Boon NA, Elder AT. 2004. Calcific aortic stenosis: Same old story?Age Ageing 33:538–544.
Dai J, Peng L, Fan K,Wang H,Wei R, Ji G, Cai J, Lu B, Li B, Zhang D, Kang Y, Tan M, QianW,Guo Y. 2009. Osteopontin induces angiogenesis through activation of PI3K/AKT andERK1/2 in endothelial cells. Oncogene 28:3412–3422.
Denhardt DT, Noda M, O’Regan AW, Pavlin D, Berman JS. 2001. Osteopontin as a means tocope with environmental insults: Regulation of inflammation, tissue remodeling, and cellsurvival. J Clin Invest 107:1055–1061.
El-Hamamsy I, Balachandran K, Yacoub MH, Stevens LM, Sarathchandra P, Taylor PM,Yoganathan AP, Chester AH. 2009. Endothelium-dependent regulation of the mechanicalproperties of aortic valve cusps. J Am Coll Cardiol 53:1448–1455.
Ergun S, Kilik N, Ziegeler G, Hansen A, Nollau P, Gotze J, Wurmbach JH, Horst A, Weil J,FernandoM,WagenerC. 2000. CEA-related cell adhesionmolecule 1: A potent angiogenicfactor and a major effector of vascular endothelial growth factor. Mol Cell 5:311–320.
Ferrara N. 1999. Molecular and biological properties of vascular endothelial growth factor. JMol Med 77:527–543.
Ferrara N, Davis-Smyth T. 1997. The biology of vascular endothelial growth factor. EndocrRev 18:4–25.
Freeman RV, Otto CM. 2005. Spectrum of calcific aortic valve disease: Pathogenesis, diseaseprogression, and treatment strategies. Circulation 111:3316–3326.
Gericke A, Qin C, Spevak L, Fujimoto Y, Butler WT, Sorensen ES, Boskey AL. 2005.Importance of phosphorylation for osteopontin regulation of biomineralization. CalcifTissue Int 77:45–54.
Goldbarg SH, Elmariah S, Miller MA, Fuster V. 2007. Insights into degenerative aortic valvedisease. J Am Coll Cardiol 50:1205–1213.
GuX,MastersKS. 2010. Regulationof valvular interstitial cell calcification by adhesive peptidesequences. J Biomed Mater Res A 93:1620–1630.
Jono S, Peinado C, Giachelli CM. 2000. Phosphorylation of osteopontin is required forinhibition of vascular smooth muscle cell calcification. J Biol Chem 275:20197–20203.
Mohler ER III, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. 2001. Boneformation and inflammation in cardiac valves. Circulation 103:1522–1528.
Nicosia MA, Cochran RP, Einstein DR, Rutland CJ, Kunzelman KS. 2003. A coupledfluid-structure finite element model of the aortic valve and root. J Heart Valve Dis 12:781–789.
Northrup WF III, Dubois KA, Kshettry VR, Teskey JM, Nicoloff DM. 2002. Trends in aorticvalve surgery in a largemulti-surgeon,multi-hospital practice, 1979–1999. JHeart ValveDis11:768–778; discussion 778–779.
Fig. 8. Schematic representationofOPNand (p)OPNeffect on endothelial cells, smoothmuscle cells and excised aortic valve tissues fromCASpatients and controls.
JOURNAL OF CELLULAR PHYSIOLOGY
2148 P O G G I O E T A L .

O’Brien KD, Kuusisto J, Reichenbach DD, Ferguson M, Giachelli C, Alpers CE, Otto CM.1995. Osteopontin is expressed in human aortic valvular lesions. Circulation 92:2163–2168.
O’Brien KD, Reichenbach DD, Marcovina SM, Kuusisto J, Alpers CE, Otto CM. 1996.Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of‘degenerative’ valvular aortic stenosis. Arterioscler Thromb Vasc Biol 16:523–532.
Okamoto H. 2007. Osteopontin and cardiovascular system. Mol Cell Biochem 300:1–7.Olsson M, Thyberg J, Nilsson J. 1999. Presence of oxidized low density lipoprotein innonrheumatic stenotic aortic valves. Arterioscler Thromb Vasc Biol 19:1218–1222.
Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD. 1994. Characterization ofthe early lesion of ‘degenerative’ valvular aortic stenosis. Histological andimmunohistochemical studies. Circulation 90:844–853.
Pastor-Perez F, Marin F. 2009. Hypertension, aortic sclerosis and the prothrombotic state:Understanding the complex interaction. J Hum Hypertens 23:287–288.
Perin MA, Brito FS, Jr., Almeida BO, Pereira MA, Abizaid A, Tarasoutchi F, Grube E. 2009.Percutaneous aortic valve replacement for the treatment of aortic stenosis: Earlyexperience in Brazil. Arq Bras Cardiol 93:299–306.
Rajamannan NM. 2005. Calcific aortic stenosis: Medical and surgical management in theelderly. Curr Treat Options Cardiovasc Med 7:437–442.
Rajamannan NM, SubramaniamM, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T,Fullerton DA, Tajik AJ, Bonow RO, Spelsberg T. 2003. Human aortic valve calcification isassociated with an osteoblast phenotype. Circulation 107:2181–2184.
Rittling SR, Chambers AF. 2004. Role of osteopontin in tumour progression. Br J Cancer90:1877–1881.
Saad FA, Salih E, Glimcher MJ. 2008. Identification of osteopontin phosphorylation sitesinvolved in bone remodeling and inhibition of pathological calcification. J Cell Biochem103:852–856.
SengerDR, Ledbetter SR, Claffey KP, Papadopoulos-SergiouA, Peruzzi CA,DetmarM. 1996.Stimulation of endothelial cell migration by vascular permeability factor/vascular
endothelial growth factor through cooperative mechanisms involving the alphavbeta3integrin, osteopontin, and thrombin. Am J Pathol 149:293–305.
SimmonsCA. 2009. Aortic valvemechanics: An emerging role for the endothelium. J AmCollCardiol 53:1456–1458.
Simmons CA, Grant GR, Manduchi E, Davies PF. 2005. Spatial heterogeneity of endothelialphenotypes correlates with side-specific vulnerability to calcification in normal porcineaortic valves. Circ Res 96:792–799.
Soini Y, Salo T, Satta J. 2003. Angiogenesis is involved in the pathogenesis of nonrheumaticaortic valve stenosis. Hum Pathol 34:756–763.
Speer MY, Chien YC, Quan M, Yang HY, Vali H, McKee MD, Giachelli CM. 2005. Smoothmuscle cells deficient in osteopontin have enhanced susceptibility to calcification in vitro.Cardiovasc Res 66:324–333.
Taylor PM, Batten P, Brand NJ, Thomas PS, Yacoub MH. 2003. The cardiac valve interstitialcell. Int J Biochem Cell Biol 35:113–118.
Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, Zheng ZJ, Flegal K,O’Donnell C, Kittner S, Lloyd-Jones D, Goff DC, Jr., Hong Y, Adams R, Friday G, Furie K,Gorelick P, Kissela B, Marler J, Meigs J, Roger V, Sidney S, Sorlie P, Steinberger J,Wasserthiel-Smoller S,WilsonM,Wolf P. 2006.Heart disease and stroke statistics—2006update: A report from the American Heart Association Statistics Committee and StrokeStatistics Subcommittee. Circulation 113:e85–e151.
Tuck AB, Chambers AF, Allan AL. 2007. Osteopontin overexpression in breast cancer:Knowledge gained and possible implications for clinical management. J Cell Biochem102:859–868.
Yetkin E, Waltenberger J. 2009. Molecular and cellular mechanisms of aortic stenosis. Int JCardiol 135:4–13.
Yu PJ, Skolnick A, Ferrari G, Heretis K, Mignatti P, Pintucci G, Rosenzweig B, Diaz-Cartelle J,Kronzon I, Perk G, Pass HI, Galloway AC, Grossi EA, Grau JB. 2009. Correlation betweenplasma osteopontin levels and aortic valve calcification: Potential insights into thepathogenesis of aortic valve calcification and stenosis. J ThoracCardiovasc Surg 138:196–199.
JOURNAL OF CELLULAR PHYSIOLOGY
O S T E O P O N T I N A N D C A L C I F I C A O R T I C S T E N O S I S 2149


















