
Oral Presentations: Novel Synthetic Strategies · 2016-06-03 · Oral Presentations: Novel...
31
Oral Presentations: Novel Synthetic Strategies 12 th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron Novel Triptycene-containing Multiblock Copolymers of Sulfonated Poly(arylene ether sulfone) for Proton Exchange Membrane Fuel Cells (PEMFCs). Joseph Aboki, Ruilan Guo Department of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, IN 46556-5637 Sulfonated poly(arylene ether sulfone) multiblock copolymers with alternating hydrophilic and hydrophobic blocks have been shown to hold great potential as alternatives to Nafion ® for polyelectrolyte membrane fuel cells (PEMFCs). Previous research has shown that long hydrophilic sequences (>10,000 g/mol) are typically needed to effect nanophase separation and form continuous proton conducting nanochannels enabling high proton conductivity at low RH. However, this usually comes at the expense of excessive water uptake and swelling, resulting in severe deterioration of the dimensional stability of PEM membranes. To address this problem, triptycene units were introduced into the hydrophobic sequences of multiblock copolymers. Specifically, a series of triptycene-containing poly(arylene ether sulfone) multiblock copolymers were synthesized via coupling reaction between biphenol-based hydrophilic oligomers (BPS100) and triptycene-containing hydrophobic oligomers (TRP0). The molecular weight (5,000 – 15,000 g/mol) and end-group functionality of oligomers were precisely controlled by adjusting the stoichiometric feeding ratio of monomers. The resulting multiblock copolymers yielded tough and ductile membranes via solution casting and fundamental membrane properties were characterized. In general, the multiblock copolymer membranes showed increasing water uptakes and proton conductivities with increasing block length. Anisotropic swelling behavior was observed for all membranes suggesting nanophase-separated lamellae-like morphology. The copolymers with long hydrophilic sequences (>10,000 g/mol) showed greatly reduced swelling ratios as compared to Nafion ® 212 due to the ability of the triptycene units to induce supramolecular chain threading and interlocking interactions that suppress membrane swelling and improve dimensional stability. For example, the copolymer with hydrophilic block length of 15,000 g/mol had a water uptake of 107%, proton conductivity of 0.155 S/cm in R.T. liquid water (almost 3 times that of Nafion ® 212) and a volume swelling ratio of just 27% (> 65% reduction compared to Nafion ® 212). These multiblock copolymer membranes hold tremendous potential for application in proton exchange membrane fuel cells. 1
Transcript of Oral Presentations: Novel Synthetic Strategies · 2016-06-03 · Oral Presentations: Novel...

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Novel Triptycene-containing Multiblock Copolymers of Sulfonated Poly(arylene ether
sulfone) for Proton Exchange Membrane Fuel Cells (PEMFCs).
Joseph Aboki, Ruilan Guo
Department of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, IN
46556-5637
Sulfonated poly(arylene ether sulfone) multiblock copolymers with alternating hydrophilic and
hydrophobic blocks have been shown to hold great potential as alternatives to Nafion® for polyelectrolyte
membrane fuel cells (PEMFCs). Previous research has shown that long hydrophilic sequences (>10,000
g/mol) are typically needed to effect nanophase separation and form continuous proton conducting
nanochannels enabling high proton conductivity at low RH. However, this usually comes at the expense
of excessive water uptake and swelling, resulting in severe deterioration of the dimensional stability of
PEM membranes. To address this problem, triptycene units were introduced into the hydrophobic
sequences of multiblock copolymers. Specifically, a series of triptycene-containing poly(arylene ether
sulfone) multiblock copolymers were synthesized via coupling reaction between biphenol-based
hydrophilic oligomers (BPS100) and triptycene-containing hydrophobic oligomers (TRP0). The
molecular weight (5,000 – 15,000 g/mol) and end-group functionality of oligomers were precisely
controlled by adjusting the stoichiometric feeding ratio of monomers. The resulting multiblock
copolymers yielded tough and ductile membranes via solution casting and fundamental membrane
properties were characterized. In general, the multiblock copolymer membranes showed increasing water
uptakes and proton conductivities with increasing block length. Anisotropic swelling behavior was
observed for all membranes suggesting nanophase-separated lamellae-like morphology. The copolymers
with long hydrophilic sequences (>10,000 g/mol) showed greatly reduced swelling ratios as compared to
Nafion®212 due to the ability of the triptycene units to induce supramolecular chain threading and
interlocking interactions that suppress membrane swelling and improve dimensional stability. For
example, the copolymer with hydrophilic block length of 15,000 g/mol had a water uptake of 107%,
proton conductivity of 0.155 S/cm in R.T. liquid water (almost 3 times that of Nafion®212) and a volume
swelling ratio of just 27% (> 65% reduction compared to Nafion®212). These multiblock copolymer
membranes hold tremendous potential for application in proton exchange membrane fuel cells.
1

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
ABSTRACT
Synthesis of Star and Network Architectures from Reactive Molecular
Bottlebrushes
Esra Altay, Javid Rzayev [email protected]. Department of Chemistry, University at
Buffalo, 359 Natural Sciences Complex, Buffalo, New York 14260-3000.
Well-defined star-brush polymer architectures were prepared by cross-linking of
bottlebrush copolymers containing reactive end blocks. Linear-brush poly(glycidyl
methacrylate-b-(2-(bromoisobutyryl)ethyl methacrylate-g-(methyl methacrylate-r-
coumarin methacrylate)) (poly(GMA-b-(BIEM-g-(MMA-r-CMA))) copolymers were
prepared by a grafting-from method. First, poly(GMA-b-BIEM) diblock copolymer
backbones were synthesized using reversible addition-fragmentation chain-transfer
(RAFT) polymerization. Subsequent atom transfer radical polymerization (ATRP) of
methyl methacrylate and coumarin methacrylate using poly(BIEM) block as the
polyinitiator resulted in densely grafted bottlebrush copolymers with narrow molecular
weight distributions. The pendant epoxide groups in the poly(GMA) block were further
modified using 5-pentenoic acid to provide an olefin-functionalized cross-linkable end-
block. Intermolecular cross-linking of pentenyl groups in the block copolymers via olefin
metathesis at low concentrations and mild conditions led to core-cross-linked stars with
high yields. Resulting aggregates displayed globular conformation with mostly uniform
distributions. Network architectures were prepared by a similar route from triblock
bottlebrush copolymers with two reactive end blocks. Intramolecular photo-cross-linking
of coumarin functionalities led to the formation of rigidified linkers. The obtained
nanomaterials were characterized by electron microscopy and x-ray scattering methods.
2

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Active Networks Prepared via Degradable Thiol-Ene Acetal Photopolymerization
Douglas V. Amato Dahlia N. Amato, Olga V. Mavrodi, Dwaine A. Braasch, William B. Martin, Dmitri V. Mavrodi, and Derek L. Patton*
School of Polymers and High Performance Materials University of Southern Mississippi Hattiesburg, MS 39406, USA
Dr. O. V. Mavrodi, Prof. D. V. Mavrodi Department of Biological Sciences University of Southern Mississippi Hattiesburg, MS 39406, USA
Antibiotics that are dispersed throughout a polymeric network traditionally suffer from unwanted leaching. In this presentation, we describe the synthesis of new acetal containing monomers derived from intrinsically antimicrobial compounds. Upon polymerization, these active agents are fully incorporated within a stable thiol-ene network. However, exposure to aqueous environments results in rapid degradation and release of the antibiotic in its active form. Herein, we demonstrate the synthesis of a library of monomers, photopolymerization kinetics, degradation kinetics, and antimicrobial evaluation of both the monomers and their films. The antimicrobial activity is shown to increase with concentration of monomer within the film for gram-positive (B. subtilis and S. aureus) and gram-negative (E. coli, P. aeruginosa and B. cenocepacia) bacteria. This thiol-ene system represents a new approach for tailoring the release of antimicrobial agents from crosslinked polymeric networks.
3

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Non-Random Sulfonation of Poly(ether ether ketone) via Post Polymerization Functionalization of Thermoreversible Gels Lindsey J. Anderson, Xijing Yuan, Robert B. Moore* Department of Chemistry, Macromolecules Innovation Institute, Virginia Tech, Blacksburg, Virginia 24061, United States A novel post-polymerization method for the non-random sulfonation of poly(ether ether ketone) (PEEK) is introduced. Thermoreversible gels of PEEK were prepared and functionalized to yield sulfonated PEEK (SPEEK) with controllable ion content and a high degree of maintained crystallinity. By sulfonating PEEK in the gel state, the sulfonation reagent is sterically excluded from the crystalline domains within the gel and penetrates only the solvent-swollen amorphous fraction, resulting in selective sulfonation of the amorphous domains. Thus a non-random, blocky architecture is obtained with significantly different crystallization behavior than conventionally prepared SPEEK and SPEEK prepared in the solution state. In this work, SPEEK with variable degrees of sulfonation was prepared by both solution-state and gel-state sulfonation. The resulting thermal transitions and crystallization behavior of the random and non-random architectures generated were compared using differential scanning calorimetry. At similar ion contents, non-random ionomers displayed greater crystallizability and faster crystallization rates than their random ionomer analogues, thereby confirming a blocky architecture in the non-random ionomer. By utilizing this gel-state reaction, it should be possible to obtain SPEEK membranes with both high ion content and high crystallinity.
4

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
High Purity Benzoxazine Monomer Synthesis via Continuous High-Shear Reactor
Andrew S. Frazee and Dr. Jeffrey S. Wiggins*
The School of Polymers and High Performance Materials, the University of Southern
Mississippi, 118 College Dr. #5050, Hattiesburg, MS 39406, USA
This research develops a continuous high-shear reactor adept to synthesize benzoxazine
monomers, and alloys thereof, under solvent-free conditions. Validated by 1H NMR, the
continuous high-shear reactor demonstrates throughputs that are 6-40x faster with increased
efficacy in reaction kinetics, such as targeting specific monomeric conformations, as compared to
current batch and continuous reactor technology. Furthermore, comparison of purified 1H NMR
spectra of monomer synthesized in a batch reactor to the unpurified 1H NMR of monomer
synthesized in the aforementioned reactor design, demonstrates that utilization of the continuous
high-shear reactor for benzoxazine monomer synthesis yields a high purity product eliminating the
need for post-processing purification. Synergistically, these attributes significantly increase the
throughput, synthetic control, and reduce the cost of melt synthesized benzoxazine monomers.
5

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Catalyst migration in conjugated polymer synthesis
Peter K. Goldberg, Anne. J. McNeil
Conjugated polymers combine mechanical flexibility with light absorption/emission and charge conductivity, stimulating their use in transistors, light emitting diodes, and solar cells. Living, chain-growth methods enable polymeric properties (e.g. miscibility, charge transport) to be tailored by controlling molecular weight, end-groups, and copolymer sequence. However, synthetic challenges have limited the scope of conjugated copolymers produced in this fashion. The distinguishing feature of chain-growth polymerizations of conjugated monomers is that the catalyst remains associated with a single polymer chain throughout the polymerization. This association is due to a metal-polymer pi-complex, which may be able to migrate along the polymer to react at both ends - preventing sequence and end-group control. We have recently demonstrated that Pd-based PEPPSI-IPr is a promising catalyst for thiophene and phenylene copolymers, which were previously inaccessible. However, control over polymer sequence can only be exerted if the catalyst reacts from a single polymer end. We developed a model system to investigate catalyst migration by terminating polymerizations with a capping agent. Reaction (with the capping agent) at both polymer ends indicates catalyst migration. While PEPPSI-IPr yielded similar results to the commonly used Ni(dppe)Cl2 and Ni(dppp)Cl2 catalysts in phenylene polymerizations, only PEPPSI-IPr produced migration products in the model system. Therefore, PEPPSI-IPr cannot produce sequence controlled copolymers containing phenylene. Although pre-functionalized IPr-Pd(R)Cl catalysts could, in principle, minimize the impact of catalyst migration, we found these catalysts were either too unstable or too slow to initiate. While PEPPSI-IPr migration limits our ability to define sequence in phenylene copolymerizations, we have exploited this migration to expand the limited monomer scope of Ni and access a broader range of solid-state and electronic properties.
6

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
PRESENTATION TYPE: Oral
TITLE: Regulating the Synthesis of Nanostructured Polymers by Atom Transfer Radical Polymerization in Microemulsion
AUTHORS (FIRST NAME, LAST NAME): Robert Graff1, Xiaofeng Wang1, Yi Shi1, Haifeng Gao1
INSTITUTIONS (ALL): 1. 365 Stepan Hall, UND Chemistry Biochemistry, Notre Dame, IN, United States.
ABSTRACT BODY:
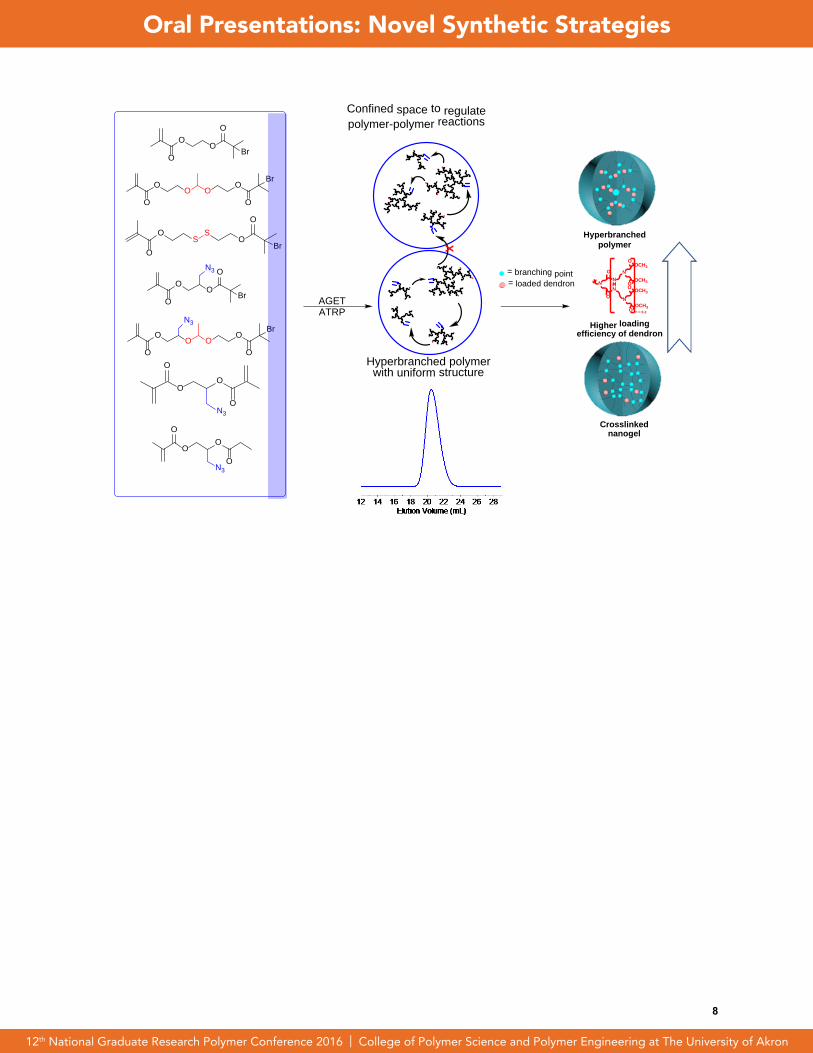
Abstract: New strategies to synthesize hyperbranched polymers with controlled molecular weights (MW = 200-1300 kg/mol), low polydispersities (PDI < 1.6), and tunable degree of branching ( DB = 0.26 - 0.41) over a wide variety of polymer compositions are presented. This was achieved by the polymerization of AB* ini-mers in a microemulsion dispersed system which was utilized as a confined nano-space which regulated the polymerization process. Using this process, the structure-property relationship of azido-containing hyperbranched polymers and cross-linked nanogels on their loading efficiency of alkynyl-containing dendron molecules was studied. The order of dendron loading efficiencies (i.e., final conversion of alkynyl-containing dendron) was as hyperbranched polymers > nanogels synthesized by ATRP > nanogels synthesized by RP. We look to further expand this method as a new synthetic strategy to help control the structure of polymers over various different polymer topologies and to be utilized as a tool for comparison between their structures and properties.
TOC:
7
Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
X
Confined space to regulate polymer-polymer reactions
O
OO
Br
O
OO O
O
O
Br
O
O
OO
Br
ON3
SO
O
BrS
O
O
O
OO O
O
O
BrN3
AGETATRP
Hyperbranched polymer with uniform structure
= branching point= loaded dendron
Hyperbranchedpolymer
Crosslinkednanogel
NNH
O
HN
O
N
N
OCH3
OCH3O
O
OCH3
OCH3
O
O
n = 0-2
Higher loading efficiency of dendron
O
O
O
N3O
O
OO
N3O
8

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
3D printing all-aromatic high-performance polyimides using μSLA; processing the non-processable.
Maruti Hegde1, Donald C. Aduba Jr.2,Nicholas A. Chartrain2, Christopher B. Williams2, and Timothy E.
Long1
Email: [email protected]
1Virginia Tech, Department of Chemistry and Macromolecules and Innovations Institute (MII), Blacksburg, Virginia, US, 24061. 2 Virginia Tech, Department of Mechanical Engineering and Macromolecules and Innovations Institute (MII), Blacksburg, Virginia, US, 24061.
The advent of 3D printing has revolutionized manufacturing by enabling the production of complex
shapes. In so far as micro-stereolithography (µSLA) is concerned, the polymeric resins commonly utilized
generally have an aliphatic backbone with glass transition temperatures (Tg) < 100 oC.1 As far as we
know, there have been no reports on using µSLA for 3D printing all-aromatic high-performance polymers.
All-aromatic polyimides represent a class of polymers with exceptional thermal and thermo-
mechanical properties. The semi-crystalline rigid-rod all-aromatic high-performance polyimide based on
pyromellitic dianhydride (PMDA) and 4,4’-oxydianiline (ODA) (commonly known as KaptonTM) is
thermally stable up to 500 oC and has a Tg at ~400 oC. This high Tg makes processing using traditional
methods impossible.
We will present how 3D printed parts of PMDA-ODA can be produced using µSLA. Polyimide films
are traditionally prepared by a 2-step process involving the synthesis of a soluble polyamic acid precursor
followed by film casting and thermal imidization up to 300 oC to produce the final polyimide. By
synthesizing a soluble poly (amic diethyl acrylate esters),2 we prepared a soluble precursor polymer that
can be photo-crosslinked in solution, an essential pre-requisite for µSLA. Thermal imidization after photo-
crosslinking of the precursor yields PMDA-ODA polyimide. We will discuss the photo-crosslinking
process and reaction kinetics using results from UV-Vis spectroscopy, photo-DSC and photo-Rheology.
The application of these results for 3D printing will also be shown. Detailed discussions on the
morphology and mechanical properties of the crosslinked and final polyimide structures along with
discussion on structure-property relationships will be presented. 1 Schultz et al, ACS Macro Lett., 2014, 3, 1205-1209. 2 Rubner R., J. Photopoly. Sci. Tech., 2004, 17, 685-691.
9

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Towards Truly Sustainable Polymers: Metal-Free Recyclable Polyester from
Bio-renewable Non-Strained γ-Butyrolactone
Miao Hong and Eugene Y.-X. Chen*
Department of Chemistry, Colorado State University, Fort Collins, CO 80523-1872
Cyclic esters (lactones and lactides) with relatively high strain energy are the desired building
blocks for the construction of aliphatic polyesters through ring-opening polymerization
(ROP). The five-membered γ-butyrolactone (γ-BL) would also be a desirable bio-derived
monomer for ROP. However, it has been a challenge to convert the non-strained γ-BL,
commonly referred as “non-polymerizable” in textbooks and literatures. Recently, we
discovered the first successful ROP of γ-BL into poly(γ-butyrolactone) (PγBL) by metal (La,
Y)-based catalysts which were found to be the most effective method to achieve high
molecular weight (MW) (Mn up to 30 kg/mol) and high monomer conversion (up to 90%).1
However, in contrast, commonly used organic catalysts highly effective for the ROP of
typical cyclic esters, produce PγBL with low MW and low conversion (Mn ≤ 6.2 kg/mol, yield
≤ 33%). Considering the ROP of γ-BL to PγBL as the chemical route to the biomaterial
poly(4-hydroxybutyrate) (P4HB), it is highly desired to develop an effective metal-free
organopolymerization of γ-BL that produces high MW polyester with good activity.
Presented here is the first effective organocatalytic ROP of γ-BL to high-MW polyester.
This ROP can achieve high monomer conversion up to 90% and high MW up to 26.7 kg/mol.
The resulting metal-free PγBL can be completely recycled back to its monomer γ-BL upon
heating the bulk material at high temperature. The proposed mechanistic pathways will also
be presented. Overall, the PγBL reported herein represents a truly sustainable polymer, which
is biorenewable, organically synthesized, and completely recyclable.
Reference:
1. Hong, M.; Chen, E. Y.-X. Nat. Chem. 2016, 8, 42−49.
10

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Synthesis and Characterization of Tetraamino Diphenyl Sulfone
(TADPS) Based Polybenzimidazoles and Post-modification for Gas
Separation Membranes
R. Liu, R. M. Joseph, S. R. Choudhury, H. Borjigin and J. S. Riffle
*
The Macromolecules and Interfaces Institute
Department of Chemistry – (0212)
Virginia Tech
Blacksburg, VA 24061
Tel: (540) 231-8214; Fax: (540) 231-8517 *Email: [email protected]
Josh Moon, Kevin Stevens, Benny D. Freeman
Department of Chemical Engineering
University of Texas at Austin, Austin, TX 78712
A series of tetraamine diphenyl sulfone (TADPS) based polybenzimidazoles were
synthesized. The monomer, TADPS, was synthesized via a novel and economical
route. The monomer has a sulfonyl linking moiety, which results in a kinked
morphology in the polymer backbone (Figure 1). Compared to the commercial
polybenzimidazoles, Celazole®, these novel polybenzimidazoles exhibit better
solubilities in common solvents without sacrificing thermal or chemical integrity.
Additionally, these polybenzimidazoles have excellent gas transportation properties at
elevated temperatures. 1
Our approaches to improve the gas transport properties are: 1. blending the
polybenzimidazole with volatile component such as polyethylene oxide (PEO) or
polypropylene oxide (PPO). 2. grafting propylene oxide onto the polybenzimidazole
backbone by grafting from the imidazole amines by anionic polymerization. For both
methods, by degrading volatile components, blended PEO, PPO or grafted PPO blocks,
through controlled heat treatment, nanoscale voids are expected to be introduced into
the film, enhancing gas permeability.
References:
1. H. Borjigin, K. A. Stevens, R. Liu, J. D. Moon, A. Shaver, S. Swinnea, B. D. Freeman, J. S. Riffle, J. E. Mcgrath.
Polymer., 2015, 71,135-142
Figure 1. Difference in chemical structure between Celazole® monomer (left)
and TADPS (right) illustrating introduction of kinked morphology in the
polymer backbone.
11

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Enhanced supramolecular self-assembly of P3HT by copolymerization with pyrene functionalized methacrylate
Taniya M.S.K. Pathiranage†, Xiangcheng Pan‡, Michael C. Biewer†, Krzysztof Matyjaszewski‡ and Mihaela C. Stefan†* †Department of Chemistry, University of Texas at Dallas, 800 West Campbell Road, Richardson, Texas 75080, United States ‡Center for Macromolecular Engineering, Department of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, Pennsylvania 15213, United States A diblock copolymers of semiconducting poly(3-hexylthiophene) (P3HT) and methacrylates with pyrene side chains was synthesized with the combination of Grignard metathesis polymerization (GRIM) and Initiators for Continuous Activator Regeneration Atom Transfer Radical Polymerization (ICAR-ATRP). Since we were targeting to use these polymers in semiconductor applications, it was always necessary to subject the polymers to rigorous cleaning to ensure removal of copper residues after conventional ATRP. With ICAR-ATRP we could reduce the amount of copper used up to 90% while assuring good incorporation of the second block maintaining narrow PDI. Pyrene, a discotic liquid crystalline mesogen with thermotropic properties was incorporated into P3HT thus making a block copolymer as potential material for thermal sensors. The composition of synthesized copolymer was determined by 1H-NMR analysis. The effect of the supramolecular assembly on the opto-electronic properties of the resulting block copolymers was investigated. Improved long range ordering of polymer with unique morphological features was observed in TMAFM images. Characterization of the thermotropic liquid crystalline behavior was performed in a combined study with polarizing optical microscopy with heating stage and differential scanning calorimetry. The liquid crystalline mesophases of the copolymer appeared above the glass transition of P3HT which helps to trigger long range ordering of P3HT by co-crystallization followed by annealing above thermotropic mesophase transition. The softer nature of the P3HT segment above glass transition supports enhanced self-assembly. The effect of annealing on self-assembly and morphological changes of the block copolymer was studied with tapping mode atomic force microscopy (TMAFM), thin film X-ray diffraction (XRD), and UV-VIS analysis. The field effect mobilities of the P3HT diblock copolymer was determined in organic field effect transistors (OFET). Mobilities as high as 10-2 cm2/Vs were measured.
12

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Organo-soluble polybenzoxazoles (PBOs) for high performance gas separation membranes:
a novel synthesis route and gas transport properties
Ashish Kushwaha ([email protected]), Shuangjiang Luo ([email protected]), Ruilan Guo ([email protected])
Membrane gas separation holds great promise as alternative to conventional separation technologies
because of its higher energy efficiency and lower carbon footprints. Polybenzoxazoles (PBOs) are highly
attractive membrane materials because of their superior gas separation parameters (high permeability and
high selectivity) and excellent mechanical and thermal stability. However, the practical application of PBO-
based membranes is frequently frustrated by their poor processability due to insolubility problem.
In this work, we present a novel synthetic approach of one-pot, silylation-assisted solution conversion of
ortho-functional polyamide precursors that produces organo-soluble PBOs. Since it is an in-situ thermal
conversion in solution, the process is highly efficient, and also prevents undesired thermogenic degradation
occurred during high temperature solid-state conversion process. Both molecular dynamics (MD) studies
and model experiments suggest that silylation and catalytic effects of pyridinium chloride (Py-HCl) salt
formed in situ significantly lower the activation energy of conversion reaction by forming a silylated
intermediate. The feasibility of this new synthetic approach was confirmed by using a variety of monomers
to form a series of organo-soluble PBOs, which were found equally robust as the conventional insoluble
PBOs. The gas transport properties for all the PBO membranes were investigated with a range of operating
pressures (2 – 17 atm) for H2, N2, CH4, Ar and CO2. All the PBO membranes synthesized in this work
showed excellent transport properties, comparable with their corresponding high-temperature-converted
insoluble PBOs, some of which demonstrated exceptional separation performances for CO2/CH4 and
H2/CH4 gas pairs exceeding the upper bounds. Thus, provided their organo-solubility and cost-
effectiveness, the novel synthetic route to soluble PBOs reported in this work represents a vital step towards
the practical application of PBOs.
13

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Synthesis of mannose- and glucose-based hyperbranched glycopolymers
Cesar Lopez, Hao Chen, William Horn, Carolyn Scherger and Coleen Pugh*
Carbohydrate-based polymers play a central role in biological processes such as cell
adhesion or pathogen recognition.1 Glycopolymers are synthesized to mimic natural
polysaccharides, with the aim to use them as models to understand their key features in
biological systems.2 Glycoinimers were homopolymerized using activators regenerated
by electron transfer (ARGET) and simultaneous normal and reverse initiated atom
transfer radical polymerization (SN&RI ATRP) to synthesize hyperbranched
glycopolymers. We established polymerization conditions to control the molecular
weight of the polymers, and synthesized linear and hyperbranched polymers in the
same molecular weight range to study the role of the polymer architecture. The relative
hydrodynamic volumes of the linear and hyperbranched glycopolymers were studied
using gel permeation chromatography (GPC) as a function of absolute molecular weight
obtained by light scattering.
1. Bertozzi, C. R.; Kiessling, L. L. Science 2001, 291, 2357–2364.
2. a) Yilmaz, G.; Becer, C.R. Front. Bioeng. Biotechnol. 2014, 2, 39. b) Ting, S. R.
S; Chen, G; Stenzel, M. H. Polym. Chem. 2010, 1, 1392–1412.
14

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Synthesis and Self-Assembly of Ultrahigh Molecular Weight Linear
Block Copolymers
Jose Kenneth Mapas1 and Javid Rzayev1
1Department of Chemistry, University at Buffalo, The State University of New York, Buffalo, New York 14260-3000
ABSTRACT
Ultrahigh molecular weight block copolymers (Mn ≥ 1 × 106 g/mol), which grants access
to periodic nanostructures with large domain spacings, afford the fabrication of materials
with interesting properties and enables a variety of applications. However, the phase
behavior and other physical properties of high molecular weight linear block copolymers
remain largely unexplored due to their highly entangled conformations. Furthermore, the
production of these copolymers require challenging synthetic protocols, which limits the
availability of these materials for non-synthetic experts and diminishes their potential
impact. In this work, we report a “user-friendly” RDRP method for the rapid synthesis of
ultrahigh molecular weight linear diblock copolymer using a combination of ATRP and
RAFT polymerization to generate block copolymers that can self-assemble into ordered
nanostructures with domain spacings exceeding 100 nm.
15

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Organocatalyzed Atom Transfer Radical Polymerization for the Synthesis of Precise
Polymer Architectures
Controlled radical polymerizations (CRPs) have changed the paradigm of polymeric
functional materials by providing the precise control needed to access complex architectures on
the macromolecular scale. Atom Transfer Radical Polymerization (ATRP) is one of the most
commonly used types of CRP and has been used to successfully synthesize star-shaped polymers,
brush copolymers, polymer networks, and dendritic polymers. Since molecular weight directly
affects the macromolecular behavior of polymers, ATRP can be used to tune the macroscopic
properties of materials comprised of these complex polymer architectures. Thus, ATRP has opened
the door to synthesizing polymers with well-defined shapes and sizes used for a variety of
applications including photonics, water purification, stimuli-responsive materials, and drug
delivery. However, synthesizing these useful architectures with traditional copper-catalyzed
ATRP limits the use of the resulting materials in biomedical or electronic applications due to
difficulty removing remaining metal from the polymer matrix. Inspired by the recent development
of organocatalyzed ATRP (O-ATRP) using organic photoredox catalysts, efforts have been put
forth to broaden the monomer scope of this system and access complex architectures. The synthesis
of linear, telechelic, and star-shaped copolymers using O-ATRP will be discussed. Several
monomers have been incorporated into these structures, which could inspire structural
modification of the photoredox catalysts in order to optimize each system. Additionally, electronic
modification of the photoredox catalysts provides more insight into the mechanism of the system.
Successfully synthesizing these macromolecular structures demonstrates the utility of our O-
ATRP system and highlights the potential for O-ATRP to impact the growing fields of electronics
and biomaterials while addressing a major limitation of metal-catalyzed ATRP.
16

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Poly(β-thioesters) Derived from the Thiol Michael ‘Click’ Reaction for Advanced Materials
Nicholas G. Moon1, Fiorella Mazzini1, Timothy E. Long1
Email: [email protected]
1Virginia Tech, Department of Chemistry, Macromolecules and Interfaces Institute (MII), Blacksburg, Virginia, US
The thiol Michael ‘click’ reaction attracts significant attention in the field of polymer chemistry due to its
rapidity, lack of byproducts, chemoselectivity, and impressive functional group tolerance. While many of
the current applications of the thiol Michael reaction focus either on network materials or the modification
of existing polymers and biopolymers, linear polymers obtained via the thiol Michael reaction remain
relatively understudied. The mild nature of the reaction and wide variety of synthetically accessible
monomers presents a significant opportunity for discovery. Herein we report our efforts to expand the
application of linear poly(β-thioesters) derived from the thiol Michael reaction with a goal towards
developing advanced materials. We seek to incorporate hydrogen-bonding motifs such as oxamides and
ureas into thiol Michael-derived polymers to enhance their properties. The effect of hydrogen-bonding
motif incorporation on the thermomechanical properties of the resulting poly(β-thioesters) will be reported.
The thiol Michael reaction is also capable of yielding telechelic, acrylate-terminated poly(β-thioesters)
through simple use of stoichiometric control. We will explore structure-property relationships of
photopolymers made from these oligomers, as well as their potential as precursors for 3D printing
applications.
17

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Photoredox-Mediated Methods for Ring-Opening Metathesis Polymerization
Laura M. M. Pascual, Adam E. Goetz, Damian G. Dunford, Kelli A. Ogawa, Yosuke Ashikari, Andrew J. Boydston*
Department of Chemistry, University of Washington, Seattle, WA 98195 [email protected]
Metal-free ring-opening metathesis polymerization (MF-ROMP) is an emerging technique for ring-opening
metathesis polymerization (ROMP) that utilizes a pyrylium photoredox mediator and vinyl ether initiator, in
contrast to the metal-based initiators traditionally used in ROMP. In this study, a variety of modularly substituted
pyrylium and thiopyrylium tetrafluoroborate salts were examined as photoredox mediators for MF-ROMP. Photo-
oxidants with more electron-withdrawing functional groups, or weaker oxidants, achieved higher conversion in the
polymerization. Thiopyrylium photo-oxidants consistently led to higher conversion than their similarly
functionalized pyrylium counterparts. Additionally, polymerizations with thiopyrylium photo-oxidants displayed
higher rates of conversion faster than the corresponding pyryliums. While our previous studies used 2,4,6-
tris(methoxyphenyl)pyrylium tetrafluoroborate, the corresponding thiopyrylium has emerged as a promising
photo-oxidant for rapidly achieving high conversion. Ongoing work includes evaluating alternative photo-oxidants
for photoredox-mediated ring-opening metathesis polymerization.
18
Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Random L-lactide/ bioaromatics copolymerizations for increasing the glass transition
temperature of PLA
Ha Thi Hoang Nguyen, Gabriel Short, Stephen A. Miller*
Butler Polymer Laboratory, Chemistry Department, University of Florida
Polylactic acid (PLA) is one of the most successful bioplastics. Nevertheless it has some
limitations due to a low glass transition temperature and poor mechanical properties. Many
copolymerizations have been studied in order to improve those properties and widen the
application of the PLA. It has been noticed that only block copolymers of lactide and an AB type
oligomer were reported, and that a prepolymer made from an AB monomer was used as an initiator
for the ring opening polymerization with lactide. Moreover, thermal and mechanical properties
and degradability are highly dependent on crystallinity, type of copolymers (random or block), as
well as ratios of aromatic/aliphatic in the main chain. Herein, we reported a generalized and
efficient ring opening polymerization (ROP) polycondensation methodology in melt to form
random copolymers of L-lactide and a bioaromatic as the comonomer. The randomness of
copolymers were well achieved and confirmed by 1H NMR. The copolymer strategy improved
significantly the thermal stability of the PLA from 235 to 420 °C, and the glass transition
temperature (Tg) of the homopolymer PLA from 50 to 107 °C. In addition, the polymerization
methodology was further applied to other ROP systems. For example, random copolymers of -caprolactone and bioaromatics were successfully synthesized with the Tg of the homopolymer PCL
drastically increased from -60 to 105 °C. These thermal properties open up the gate for PCL to the
high temperature applications. The ability of tuneable glass transition temperature permitted
productions of many copolymers having thermal properties match those of PET and PS. These
copolymers potentially can be served as PET and PS replacements.
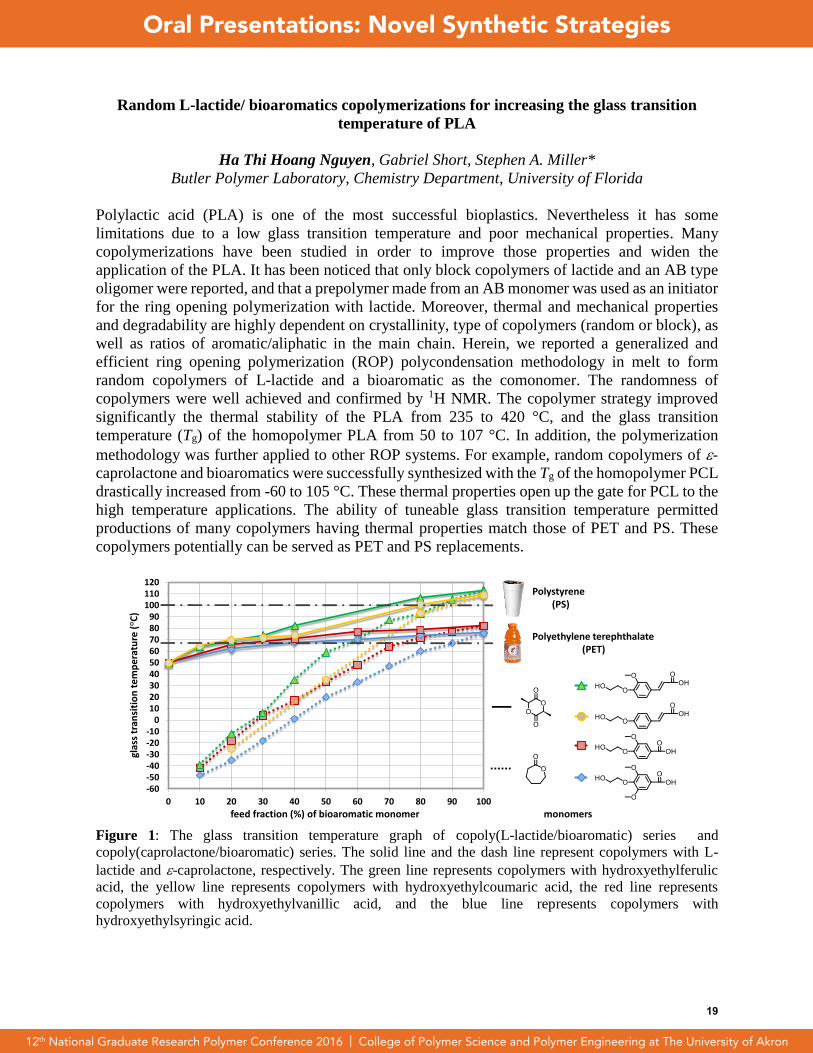
Figure 1: The glass transition temperature graph of copoly(L-lactide/bioaromatic) series and
copoly(caprolactone/bioaromatic) series. The solid line and the dash line represent copolymers with L-
lactide and -caprolactone, respectively. The green line represents copolymers with hydroxyethylferulic
acid, the yellow line represents copolymers with hydroxyethylcoumaric acid, the red line represents
copolymers with hydroxyethylvanillic acid, and the blue line represents copolymers with
hydroxyethylsyringic acid.
-60-50-40-30-20-10
0102030405060708090
100110120
0 10 20 30 40 50 60 70 80 90 100
feed fraction (%) of bioaromatic monomer
glas
s tr
ansi
tio
n t
em
per
atu
re ( C
)
monomers
5060708090
100110120130140150160170
0102030405060708090100
Poly(ferulic acid/dihydroferulic acid)
Poly(ferulic acid/dihydrocoumaric acid)
Poly(dihydroferulic acid/coumaric acid)
Poly(dihydroferulic acid/dihydrocoumaric acid)
Poly(coumaric acid/dihydrocoumaric acid)
5060708090
100110120130140150160170
0102030405060708090100
Poly(ferulic acid/dihydroferulic acid)
Poly(ferulic acid/dihydrocoumaric acid)
Poly(dihydroferulic acid/coumaric acid)
Poly(dihydroferulic acid/dihydrocoumaric acid)
Poly(coumaric acid/dihydrocoumaric acid)
5060708090
100110120130140150160170
0102030405060708090100
Poly(ferulic acid/dihydroferulic acid)
Poly(ferulic acid/dihydrocoumaric acid)
Poly(dihydroferulic acid/coumaric acid)
Poly(dihydroferulic acid/dihydrocoumaric acid)
Poly(coumaric acid/dihydrocoumaric acid)
5060708090
100110120130140150160170
0102030405060708090100
Poly(ferulic acid/dihydroferulic acid)
Poly(ferulic acid/dihydrocoumaric acid)
Poly(dihydroferulic acid/coumaric acid)
Poly(dihydroferulic acid/dihydrocoumaric acid)
Poly(coumaric acid/dihydrocoumaric acid)
Polyethylene terephthalate(PET)
Polystyrene (PS)
19

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Novel discovery of biorenewable and water degradable polylactam esters from itaconic acid
Pengxu Qi, Ha Thi Nguyen and Stephen A. Miller
As the commercial plastics industry was born and thrived in the last century, the overutilization of these petroleum-based
commodity plastics has revealed dire environmental issues. The depletion of fossil fuel resources, along with persistence in the
environment have stimulated academic and industrial research programs that seek degradable polymers derived from biorenewable
resources. Among these research, the most successful, fully biorenewable polyester is polyactic acid (PLA), was made from corn
starch or other carbohydrates. While PLA is 100% biobased, its useful temperature range is limited by its low glass transition
temperature (Tg) of 50°C and it composts very poorly in the environment, thus requiring industrial composting conditions. In
continued pursuit for better replacement, incorporating ring system possessing decent rigidity and degradability into the polymer
could be one of the plausible pathway to improve the thermal properties and degradability, respectively. Recently, pyrrolidone
functional group was caught on our eyes because 1) bifunctional monomers containing this group can be easily prepared from
biorenewable materials, 2) the rigidity of this ring system is comparable with that of aromatic, and 3) this highly polar ring
containing amide functional group may significantly assist the degradability of the polymer. Itaconic acid, a naturally occurring
compound mass-produced via fermentation of glucose, was reacted with ethanolamine or ethylene diamine to afford hydroxy-acid
or di-acid monomers containing this 2-pyrrolidone lactam. Homopolymerization or copolymerization with diols, respectively,
yielded polylactam esters with higher glass transition temperatures and faster hydrolytic degradation compared to the commercial
polyester polylactic acid (PLA).
20

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Sustainable polyvinyl acetals from vanillin and other biobased aromatic aldehydes. Biodegradable, non-toxic and potentially renewable polyvinyl alcohol was condensed with vanillin and 11 other aromatic aldehydes (most of them naturally occurring, biorenewable and/or edible) to obtain polyvinyl acetals. Polyvinyl alcohol (PVA) is a commercial polymer with combined production of several hundred thousand tons annualy, including its derivatives polyvinyl formal and polyvinyl butyral. PVA is water soluble and biodegradable, making it and excellent candidate as a part of advanced sustainable and degradable commodity plastic replacements. Vanillin is a renewable aromatic hydroxyaldehyde which can be obtained from the pyrolysis of lignin. There are many other aromatic aldehydes available that are either lignin derived, naturally occurring, and/or utilized in the flavors and fragrances industry. We have selected 12 of those molecules to be incorporated into PVA to improve its thermal properties: 4-hydroxybenzaldehyde, vanillin, syringaldehyde, ethylvanillin, ortho-vanillin, isovanillin, salicylaldehyde, ortho-anisaldehyde, para-anisaldehyde, benzaldehyde, cinnamaldehyde, and cuminaldehyde. Acetalization was achieved under acidic catalysis in two hours. The degree of acetalization was determined by 1H NMR and found to be in the range of 37.5–79.2 %, giving calculated number average molecular weights of 31,500 to 48,300. The thermal properties of PVA were substantially improved, with the resulting polyvynil aromatic acetals being amorphous and having glass transition temperatures (Tg) ranging from 114 to 157 °C. An heterogeneous degradation study performed on polyvinyl vanillin acetal showed the products become soluble in water after exposure to acidic aqueous media.
Acetal formation from the reaction of polyvinyl alcohol and vanillin to yield aromatic polyvinyl vanillin acetal with improved thermal properties.
21

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Matthew D. Ryan, Chern-Hooi Lim, Jordan C. Theriot, Charles B. Musgrave, and Garret M. Miyake* Title Organocatalyzed Atom Transfer Radical Polymerization Abstract Atom transfer radical polymerization (ATRP) is one of the most studied and utilized controlled radical polymerization methodologies. ATRP can produce polymers with complex higher order structure and diverse monomer functionality, making it a powerful tool for the production of synthetic materials with highly engineered structure and function. However, contamination of the polymer product by metal catalysts remains a limitation. To address this problem, our research efforts have been focused on developing organic catalysts capable of mediating the complex catalytic cycle defining an ATRP process. We have employed computationally-directed catalyst discovery to identify a number of catalyst features that have proven key for control over the polymerization process. With these design principles in hand we have developed a class of organic photoredox catalysts for mediating ATRP that operate with visible light sources, including sunlight, as a low energy driving force of reaction. These catalysts consistently produce polymers of low dispersity, predictable molecular weight, and near quantitative initiator efficiency. Strategies for small molecule regulation of the proposed catalytic cycle and their mechanistic implications will also be presented.
22

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Utilizing thiol-ene click chemistry to fabricate highly emissive QD films/patterns for polymer based optical systems
Marcus J. Smith†, Sidney T. Malak†, Jaehan Jung†, Chun Hao Lin†, Sunghan Kim†
Timothy J. White‡, Timothy J. Bunning‡, Zhiqun Lin†, & Vladimir V. Tsukruk†
†School of Materials Science and Engineering, Georgia Institute of Technology,
Atlanta, Georgia 30332, United States
‡Air Force Research Labs-Materials & Manufacturing Directorate WPAFB, OH 45433
Keywords: Quantum dot, QD, soft lithography, pattern, nano, quantum yield, thiol-ene
This work demonstrates a facile and versatile method for generating QD-‐polymer films and patterned photonic structures with ultra-‐high QD loading and minimal phase-‐separation. QD-‐polymer films are fabricated using thiol-‐ene click chemistry, in which cross-‐linked polymer networks are rapidly produced in ambient conditions via UV polymerization. Thiol-‐ene click chemistry is an ideal approach for minimizing phase separation through rapid photopolymerization in the presence of QDs. For example, we demonstrate the fabrication of QD-‐polymer films with ultra-‐high QD loading (>30-‐40%) and minimal phase-‐separation. Furthermore, we demonstrate that the cross-‐linked thiol-‐ene composites are robust with interesting mechanical properties and chemically inert. Using soft-‐imprint lithography it possible to pattern highly loaded QD structures while preserving the optical properties making them suitable candidates for high gain and low loss devices. The versatility of thiol-‐ene click chemistry to produce ultra-‐dense QD films potentially makes it an important technique for polymer based PT-‐symmetric optical metamaterials, where efficient light propagation is critical, like waveguides, sensors, and optical gain films.
23

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Polymerization-induced Shrinkage Stress in Thiol–Ene Photopolymerizations
Harry L. van der Laan, Jae Hwan Jung, Junting Li, Brian H. Clarkson, Timothy F. Scott
With an estimated 90% of adults in the US suffering from a form of dental caries, resulting in over 100 million dental fillings being placed each year, dental caries is a significant public health concern. Although conventional methacrylate-based composite dental restoratives have excellent thermal and mechanical properties as well as good aesthetics, the frustrated shrinkage and concomitant shrinkage stress that accompanies the polymerization of these materials can lead to several deleterious consequences, including marginal gap formation or microcracking of the substrate tooth, leading to secondary cavities. Moreover, as methacrylate polymerizations proceed via chain growth, a significant amount of extractable material remains after cure. In order to address these and other problems, we employ a thiol–ene-based resin formulation as the continuous, polymeric phase of dental restorative materials. Here, we discuss the development of novel dental restorative materials that employ a pre-oligomerization method and incorporate addition-fragmentation functional groups in the polymer backbone. By using Fourier transform infrared spectroscopy (FT-IR), tensometry, and dynamic mechanical analysis (DMA), we demonstrated how our approach leads to improved cross-linked polymeric networks with significantly reduced polymerization shrinkage stresses.
24

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Chemo- and Stereoselective Polymerization of Polar Divinyl Monomers by Zirconocenium Catalysts to Functional Materials
Fernando Vidal-Peña† and Eugene Y.-X. Chen†
†Department of Chemistry, Colorado State University, 1301 Center Av. Fort Collins,
Colorado 80523-1801, USA
email: [email protected]
Linear polymers that carry accessible vinyl groups in every repeating unit are of great
interest since they can be photo-, thermal-, and chemically modified or crosslinked to obtain
new functional materials with specific properties. A leading strategy to produce such
materials is to develop chemoselesctive polymerization of bifunctional divinyl monomers,
such as vinyl methacrylate (VMA), in which the methacrylic double bond is selectively
polymerized while the unconjugated double bond remains intact. We recently reported the
use of chiral C2 symmetric single-site ansa-metallocenium catalysts to achieve the
simultaneous control over the chemoselectivity and stereoselectivity of the polymerization
reaction. The conjugate-addition coordination, living, and stereospecific polymerization by
such chiral metallocenium catalysts was responsible for the observed high degree of control
over the polymerization characteristics and stereochemistry. In this presentation, we will
describe the chemo- and syndiospecific polymerization of methacrylate-based divinyl
monomers by appropriate chiral, single-site ansa-zirconocenium catalysts. Highly
syndiotactic polymers bearing the reactive side-chain double bonds can be functionalized and
photocrosslinked through thiol-ene “click” reactions. Moreover, the helical conformation of
iso- and syndiotactic polymers allows the formation of new polymer stereocomplexes.
25

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
ABSTRACT
Block copolymer templating is a versatile approach for the generation of well-defined
porosity in a wide variety of framework chemistries. Here, we systematically investigate
how the composition of a poly(methoxy poly[ethylene glycol] methacrylate)-block-
poly(butyl acrylate) (PMPEG-PBA) template impacts the pore characteristics of
mesoporous cobalt oxide films. Three templates with a constant PMPEG segment length
and three different hydrophilic block volume fractions of 17%, 51% and, 68% for the
PMPEG-PBA are cooperatively assembled with cobalt nitrate hexahydrate and citric acid.
Irrespective of template composition, a spherical nanostructure is templated and elliptical
mesostructures are obtained on calcination due to uniaxial contraction of the film. The
average pore size increases from 11.4 ± 2.8 nm to 48.5 ± 4.3 nm as the length of the PBA
segment increases as determined from AFM. For all three templates examined, a maximum
in porosity (~35 % in all cases) and surface area is obtained when the precursor solids
contain 35-45 wt % PMPEG-PBA. This invariance suggests that the total polymer content
drives the structure through interfacial assembly. The composition for maximizing porosity
and surface area with the micelle templating approach results from a general decrease in
porosity with increasing cobalt nitrate hexahydrate content and the increasing mechanical
integrity of the framework to resist collapse during template removal / crystallization as
the cobalt nitrate hexahydrate content increases. Unlike typical evaporation induced self-
assembly with sol gel chemistry, the hydrophilic/hydrophobic composition of the block
iii
Structure Control of Functional Mesoporous Materials and Synthesis of Polydimethylsiloxane-Containing Block Copolymer
Author Siyang Wang
26

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
copolymer template is not a critical component to the mesostructure developed with
micelle-templating using metal nitrate-citric acid as the precursor.
A series of PS-PDMS with narrow molecular weight distributions with aimed molecular
weight was synthesized with PDMS-RAFT as a macro-chain transfer agent via reversible
addition fragmentation chain transfer (RAFT) polymerization. The PDMS-RAFT agent is
utilized to control the free radical polymerization of styrene monomer. Hydroxyl
terminated polydimethylsiloxane (PDMS-OH) is end functionalized through esterification
with a carboxylic acid functional RAFT agent. The thin films of the synthesized PS-PDMS
block copolymers are fabricated with spin coating and subsequently annealed with solvent
vapor annealing with soft shear (SVA-SS). Varied surface morphology can be obtained
with different volume fraction of polystyrene block in PS-PDMS, however, long range
ordered structures have not been obtained as expected. Moreover, tri-block copolymer
polydimethylsiloxane-block-poly ethyl acrylate-block-poly styrene (PDMS-PS-PEA) with
low polydispersity has been successful synthesized with low polydispersity (PDI) in order
to investigate more interesting morphologies.
iv
27

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
One-pot Synthesis of Hyperstar Polymers in Aqueous Dispersed Media
Xiaofeng Wang, Robert W. Graff, Yi Shi, and Haifeng Gao
Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame,
Indiana 46556-5670, United States
Abstract: A one-pot synthesis of hyperstar polymers with high molecular weight, low
polydispersity using a “grafting from” strategy was accomplished. The hyperbranched
polymer core with well-controlled was prepared by a microemulsion system via atom
transfer radical polymerization of inimers. A second batch of monovinyl monomers was
added in situ after the complete inimer conversion to subsequently grow hundreds of
radiating arms from these hyperbranched polymer macroinitiators (MIs). Two protecting
environments were used to avoid star-star coupling during the arm growth. One is the
polymerization of hydrophobic nBA from the core in a seeded emulsion and the second is
the electrostatic repulsion between polyelectrolyte arms. When inimers containing a
disulfide linker group were used in the synthesis of hyperbranched MIs, the produced
hyperstar polymers exhibited rapid core degradation in a reducing environment and
produced linear polymers as the degradation product.
Hydrophobic nBA swollen latex
Polymerization
Water-soluble CysMA
Polymerization
Electrostatic repulsion
Seeded emulsion Hyperstars protected in latex
Charged arms stretched out No star-star coupling
Steric repulsion
28

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
“Nanoatoms”-Based Macromolecular Engineering Strategy towardsGiant Molecules with Precisely-Controlled Heterogeneity
Wei Zhanga, Xinlin Lua, Gaoyan Mua, Wen-bin Zhangb, Yiwen Lia, Stephen Z.D. Chenga
aDepartment of Polymer Science, College of Polymer Science and Polymer Engineering, The University ofAkron, Akron, Ohio, 44325-3909, United StatesbKey Laboratory of Polymer Chemistry & Physics of Ministry of Education, College of Chemistry andMolecular Engineering, Center for Soft Matter Science and Engineering, Peking University, Beijing 100871,China.
In nature, “precision” biomacromolecules are prepared via replicating complex
hierarchical structures. Achieving the defined heterogeneities (such as size, composition,
functionality, topology, sequence and other primary structures) in synthetic
macromolecules at length scales ranging from sub-nanometers to micrometers remains to
be a grand challenge. In this report, we introduce a “nanoatom”-based strategy with a
unique and versatile synthetic methodology to prepare a library of polymer-nanoparticle
conjugates (giant molecules). These giant molecules with controlled heterogeneities are
obtained by accurate arrangement of precisely functionalized molecular nanoparticles
(here T8 polyhedral oligomeric silsesquioxanecages are used) via sequentially performed
orthogonal “click” reactions. The chemical structure in terms of composition, surface
functionality, sequence, and topology can be precisely controlled by these method. These
giant molecules are able to self-assemble into different supramolecular structures in the
solid state of which their phase transitions depend on composition, functionality and
topology. This strategy offers a promising opportunity to manipulate and control the
hierarchical structures of giant molecules via precise and modular assemblies of various
nano-building blocks.
References(1) Zhang, W.-B.; Yu, X.; Wang, C.-L.; Sun, H.-J.; Hsieh, I. F.; Li, Y.; Dong, X.-H.; Yue, K.; Van Horn, R.;
Cheng, S. Z. D. Macromolecules 2014, 47, 1221-1239.
(2) Zhang, W.; Huang, M.; Su, H.; Zhang, S.; Yue, K.; Dong, X.-H.; Li, X.; Liu, H.; Zhang, S.; Wesdemiotis, C.;Lotz, B.; Zhang, W.-B.; Li, Y.; Cheng, S. Z. D. ACS Central Science 2016, 2, 48-54.
29

Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
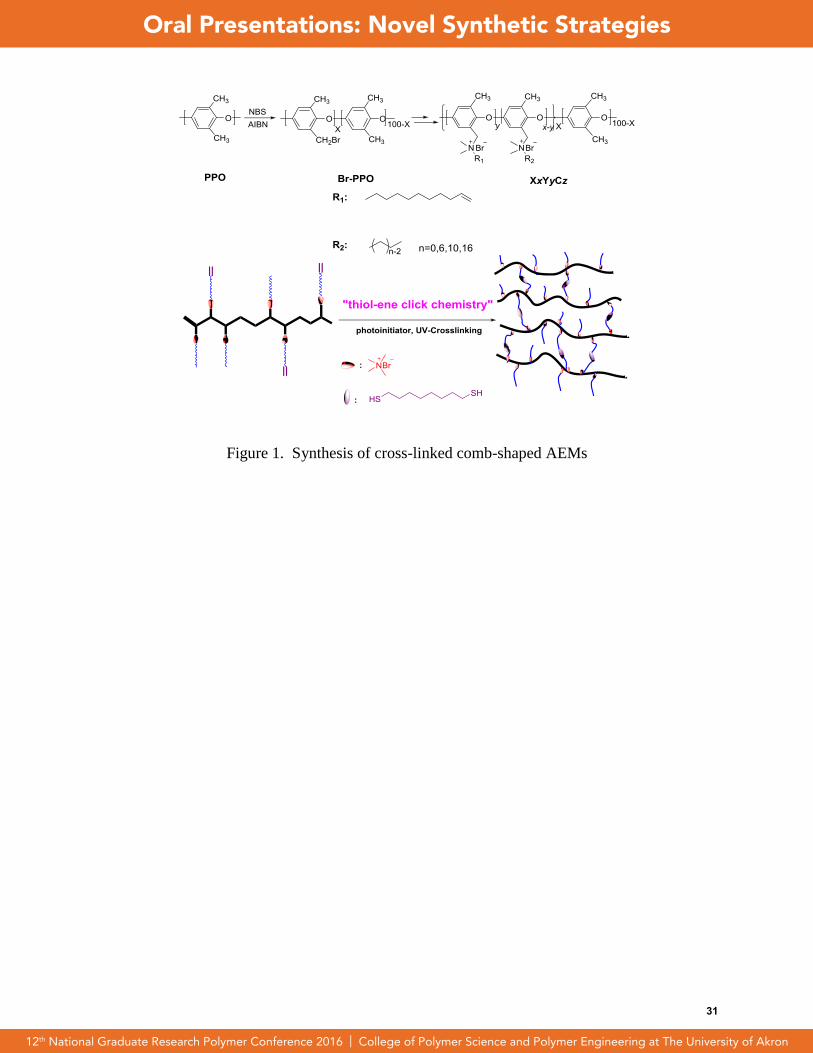
Cross-linking of Comb-Shaped Polymer Anion Exchange Membranes Via Thiol-ene
Click Chemistry
L. Zhu, Tawanda J. Zimudzi, Nanwen Li, Jing Pan, Bencai Lin, M. A. Hickner
Department of Materials Science and Engineering, The Pennsylvania State University,
Pennsylvania 16802, USA
Abstract
Anion exchange membrane fuel cells (AEMFCs) have been regarded as promising
energy conversion devices for stationary and mobile applications due to their potentially
low cost. To realize high-performance AEMFCs, new polymeric membranes are needed
that are highly conductive and chemically stable. In this work, to produce anion
conductive and durable polymer electrolytes for alkaline fuel cell applications, a series of
cross-linked quaternary ammonium functionalized poly(2,6-dimethyl-1,4-phenylene
oxide)s were synthesized via thiol-ene click chemistry. Firstly, 1H nuclear magnetic
resonance (NMR) spectroscopy and Fourier transform infrared spectroscopy (FTIR) were
used to confirm the chemical structure of the samples. In addition, from small angle X-
ray scattering (SAXS), it was found that the cross-linked membranes developed micro-
phase separation between the hydrophilic PPO backbone and the hydrophobic alkyl side
chains. The hydroxide ion conductivity of the cross-linked samples reached 60 mS/cm in
liquid water at room temperature. Finally, the chemical stabilities of the membranes were
evaluated under severe, accelerated aging conditions and degradation was quantified by
measuring ionic conductivity changes during aging. The cross-linked membranes retained
their relatively high ion conductivity and good mechanical properties in both 1 M and 4
M NaOH at 80 °C after 500 h. Attenuated total reflection (ATR) spectra were used to
study the degradation pathways of the membranes, and it was discovered that β-hydrogen
(Hofmann) elimination was likely to be the major pathway for degradation in these
membranes.
This talk will discuss details of how the cross-linking strategy enhanced both the
chemical and dimensional stability of the membranes.
30
Oral Presentations: Novel Synthetic Strategies
12th National Graduate Research Polymer Conference 2016 | College of Polymer Science and Polymer Engineering at The University of Akron
Figure 1. Synthesis of cross-linked comb-shaped AEMs
31


















