
Optic Nerve Hypoplasia and the Syndrome of Nevus Sebaceous of Jadassohn
7
Optic Nerve Hypoplasia and the Syndrome of Nevus Sebaceous of jadassohn A New Association BARRETI KATZ, MD,*t CLAYTON A. WILEY, MD, PhD,* VINCENT W. LEE, MD* Abstract: The nevus sebaceous of Jadassohn (NSJ) syndrome is a not un- common pediatric dermatosis, with malignant potential. It is the cutaneous manifestation of another phakomatosis, characterized by neurologic, ophthal- mic, cardiovascular, skeletal, and urogenital involvement. The features of this syndrome overlap those of the oculo-auriculo-vertebral dysplasia of Goldenhar and tuberous sclerosis. The extent of system involvement suggests a develop- mental insult during the first few weeks of gestation. A clearly genetic basis has not been established. An infant with NSJ syndrome is described who had associated optic nerve hypoplasia. His clinical, pathologic, and radiologic find- ings, including computed tomography (CT) and magnetic resonance imaging, are presented. [Key words: linear nevus sebaceous syndrome of Jadassohn, linear nevus syndrome, optic nerve hypoplasia, phakomatosis.] Ophthalmology 94:1570-1576, 1987 The nevus sebaceous of Jadassohn (NSJ) syndrome is a dermatosis consisting of congenital nevi characterized by hyperplasia of the sebaceous gland and papillary acanthosis. 1 The nevus itself is the cardinal feature of a more generalized abiotrophy, now recognized as a syn- drome. This syndrome consists of the triad of a linear nevus sebaceous, seizures, and mental retardation. 2 It has been expanded to encompass a widening spectrum of neurologic, ophthalmologic, cardiovascular, urogeni- tal, and skeletal anomalies. We present an infant with the NSJ syndrome and associated optic nerve hypoplasia. We believe the associ- ation is new. We include the first reported magnetic resonance imaging of a patient with this syndrome. From the Departments of Ophthalmology,* Neurosciences,t and Pathol- ogy,:j: University of California, San Diego, La Jolla. Presented in part at the 18th Frank Walsh Society Meeting, Seattle, Wash- ington, February 21, 1986. Reprint requests to Barrett Katz, MD, University of California, San Diego Eye Center, M-018, La Jolla, CA 92093. 1570 CASE REPORT A male infant was the product of a nonconsanguineous marriage to a 20-year-old primiparous woman. The pregnancy was full term and prenatal course unremarkable, without his:- tory of exposure to toxins. At birth, the infant weighed 3.41 kg (25th percentile) and measured 52 em (25th percentile) in length, with a head circumference of38.5 (98th percentile). He was noted to have a pair of colinear, waxy, pale pink plaques from the tip of his nose to the mid-forehead, on both sides of the mouth and across the upper lip (Fig 1 ). A darkly pigmented verruca! lesion was visible on the right fronto-parietal scalp and was associated with an underlying bony defect which transilluminated (Fig 2). Another series of three darkly pig- mented hairy nevi with raised irregular borders was noted on the scalp (Fig 3). Heart, lungs, abdomen, and genitalia were normal. There was no evidence of skeletal anomaly other than the bony defect of the calvarium. No organomegaly was iden- tified. Results of neurologic examination showed right facial weakness, facial asymmetry, and decreased tone symmetri- cally. Results of ophthalmologic examination showed fleshy epidermoid tumors about the upper and lower quadrants of the right globe (Fig 4). Pupils were normal and ductions full.
Transcript of Optic Nerve Hypoplasia and the Syndrome of Nevus Sebaceous of Jadassohn

Optic Nerve Hypoplasia and the Syndrome of Nevus Sebaceous of jadassohn
A New Association
BARRETI KATZ, MD,*t CLAYTON A. WILEY, MD, PhD,* VINCENT W. LEE, MD*
Abstract: The nevus sebaceous of Jadassohn (NSJ) syndrome is a not uncommon pediatric dermatosis, with malignant potential. It is the cutaneous manifestation of another phakomatosis, characterized by neurologic, ophthalmic, cardiovascular, skeletal, and urogenital involvement. The features of this syndrome overlap those of the oculo-auriculo-vertebral dysplasia of Goldenhar and tuberous sclerosis. The extent of system involvement suggests a developmental insult during the first few weeks of gestation. A clearly genetic basis has not been established. An infant with NSJ syndrome is described who had associated optic nerve hypoplasia. His clinical, pathologic, and radiologic findings, including computed tomography (CT) and magnetic resonance imaging, are presented. [Key words: linear nevus sebaceous syndrome of Jadassohn, linear nevus syndrome, optic nerve hypoplasia, phakomatosis.] Ophthalmology 94:1570-1576, 1987
The nevus sebaceous of Jadassohn (NSJ) syndrome is a dermatosis consisting of congenital nevi characterized by hyperplasia of the sebaceous gland and papillary acanthosis. 1 The nevus itself is the cardinal feature of a more generalized abiotrophy, now recognized as a syndrome. This syndrome consists of the triad of a linear nevus sebaceous, seizures, and mental retardation.2 It has been expanded to encompass a widening spectrum of neurologic, ophthalmologic, cardiovascular, urogenital, and skeletal anomalies.
We present an infant with the NSJ syndrome and associated optic nerve hypoplasia. We believe the association is new. We include the first reported magnetic resonance imaging of a patient with this syndrome.
From the Departments of Ophthalmology,* Neurosciences,t and Pathology,:j: University of California, San Diego, La Jolla.
Presented in part at the 18th Frank Walsh Society Meeting, Seattle, Washington, February 21, 1986.
Reprint requests to Barrett Katz, MD, University of California, San Diego Eye Center, M-018, La Jolla, CA 92093.
1570
CASE REPORT
A male infant was the product of a nonconsanguineous marriage to a 20-year-old primiparous woman. The pregnancy was full term and prenatal course unremarkable, without his:tory of exposure to toxins. At birth, the infant weighed 3.41 kg (25th percentile) and measured 52 em (25th percentile) in length, with a head circumference of38.5 (98th percentile). He was noted to have a pair of colinear, waxy, pale pink plaques from the tip of his nose to the mid-forehead, on both sides of the mouth and across the upper lip (Fig 1 ). A darkly pigmented verruca! lesion was visible on the right fronto-parietal scalp and was associated with an underlying bony defect which transilluminated (Fig 2). Another series of three darkly pigmented hairy nevi with raised irregular borders was noted on the scalp (Fig 3). Heart, lungs, abdomen, and genitalia were normal. There was no evidence of skeletal anomaly other than the bony defect of the calvarium. No organomegaly was identified. Results of neurologic examination showed right facial weakness, facial asymmetry, and decreased tone symmetrically. Results of ophthalmologic examination showed fleshy epidermoid tumors about the upper and lower quadrants of the right globe (Fig 4). Pupils were normal and ductions full.
KATZ et al • NEVUS SEBACEOUS OF JADASSOHN
Fig 1. Colinear, waxy, pale pink plaque-like lesions extending from the forehead to the tip of the nose, in the mid-line.
Fig 2. Darkly pigmented, verruca! lesion on the right fronto-parietal scalp, associated with underlying bony defect which transilluminated.
Fig 3. Series of three highly pigmented raised hairy nevi, with irregular borders, present on the left scalp.
Fig 4. F1eshy epidermoid tumors present at the superior and inferior temporal quadrants of the right globe.
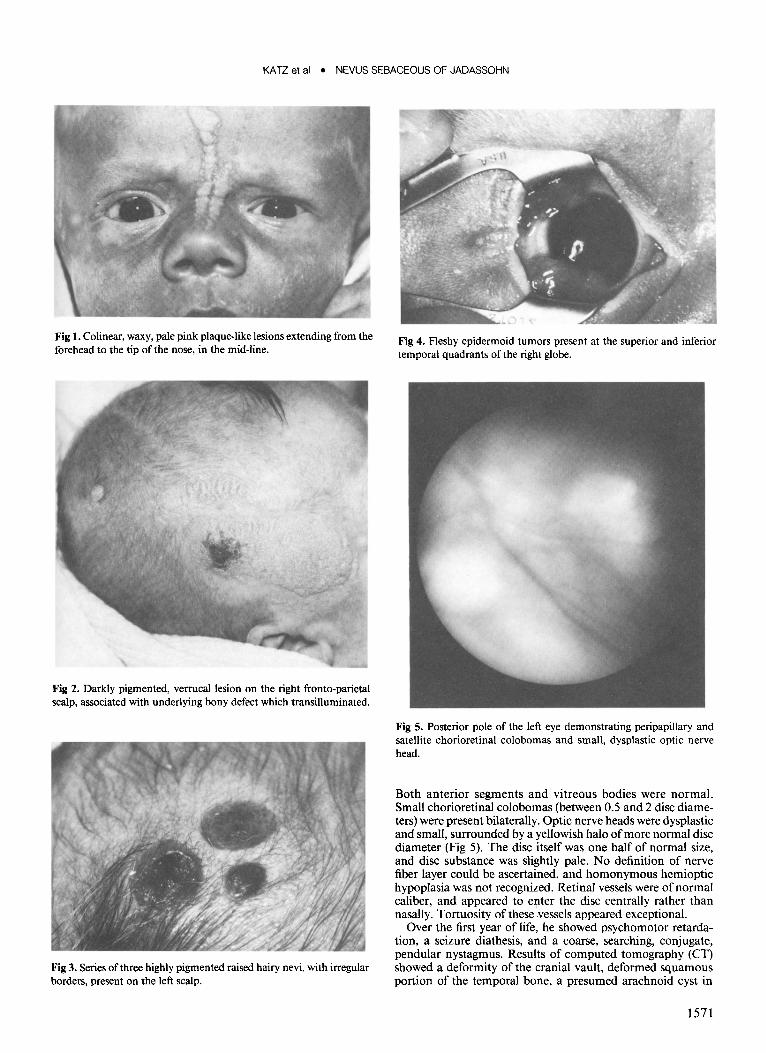
Fig 5. Posterior pole of the left eye demonstrating peripapillary and satellite chorioretinal colobomas and small, dysplastic optic nerve head.
Both anterior segments and vitreous bodies were normal. Small chorioretinal colobomas (between 0.5 and 2 disc diameters) were present bilaterally. Optic nerve heads were dysplastic and small, surrounded by a yellowish halo of more normal disc diameter (Fig 5). The disc itself was one half of normal size, and disc substance was slightly pale. No definition of nerve fiber layer could be ascertained, and homonymous hemioptic hypoplasia was not recognized. Retinal vessels were of normal caliber, and appeared to enter the disc centrally rather than nasally. Tortuosity of these vessels appeared exceptional.
Over the first year of life, he showed psychomotor retardation, a seizure diathesis, and a coarse, searching, conjugate, pendular nystagmus. Results of computed tomography (CT) showed a deformity of the cranial vault, deformed squamous portion of the temporal bone, a presumed arachnoid cyst in
1571

OPHTHALMOLOGY • DECEMBER 1987 • VOLUME 94 • NUMBER 12
Fig 6. Contrast enhanced axial CT through the middle cranial fossa demonstrating a cerebrospinal fluid density cystic structure at the temporal tip, a small and malformed temporal lobe with calcification, and bulging of the temporal bone.
the right middle fossa, calcification of the right temporal lobe, asymmetric ventricles, and a dysplastic small right cerebral hemisphere (Fig 6, 7). Magnetic resonance imaging showed a small anomalous right hemisphere with ipsilateral ventricular enlargement, prominent sulcal pattern, temporal fossa cyst, and increased signal characteristics of the right hemisphere interpreted as evidence of dysgenesis (Fig 8,9).
A skin biopsy showed immature sebaceous gland lobules, with abortive hair follicles, underlying apocrine glands, and papillomatous epidermal hyperplasia diagnostic of the NSJ (Fig I 0). The child underwent craniotomy for excision of a presumed arachnoid cyst and correction of skull deformity. The wall of the cyst showed glia-like tissue, reactive astrocytosis, fibroblastic proliferation, and focal hyalinization consistent with a wall of an arachnoid cyst. The tissue stained negative for GFAP and positive with trichrome. The epibulbar dermoids were excised. Results of pathologic examination of that tissue demonstrated ectopic lacrimal gland associated with hyalinized cartilage within the deep dermis and portions of the subcutaneum (Fig 11 ).
DISCUSSION
Jadassohn 1 introduced the term organ naevus as a distinct clinical and histopathologic entity for discrete,
1572
Fig 7. A more rostral axial scan demonstrating a small right hemisphere associated with prominent sulcal pattern and enlarged ventricles.
congenital, cutaneous lesions consisting of excesses or deficiencies of normal skin constituents. He particularly differentiated this lesion from a hyperplasia or adenoma, and used the term to distinguish it from pigmented "nevus cell" nevi. Robinson3 introduced the term Nevus Sebaceous of Jadassohn (NSJ) into American literature, using it to describe four cases of organoid nevi which also showed papillomatous epidermal hyperplasia and excessive, mature sebaceous glands.
Mehregan and Pinkus4 reported 150 cases of NSJ syndrome, attesting to its prevalence. They suggested that the life history of NSJ syndrome consists of three separable stages, and so a biopsy of any single lesion would look differently, depending on when it was obtained. An early stage, during the patient's infancy and childhood, is characterized by underdevelopment of hair and sebaceous glands. A second stage begins at puberty and is characterized by massive development of sebaceous glands, papillomatous epidermoid hyperplasia, and maturation of apocrine glands. A third stage is characterized by the development of benign and malignant neoplasms in the original nevus. In their series, tumorous degeneration developed in 52 of 150 (35%) nevi sebaceous. The majority was basal cell epitheliomas (21/52). Adenomas of various types (syringadenoma, hydradenoma, cystadenoma) occasionally occurred. Squamous carcinoma and sebaceous carcinoma were

KATZ et al • NEVUS SEBACEOUS OF JADASSOHN
Fig 8. Partial saturation magnetic resonance image, coronal section, through middle fossa, demonstrating a small and anomalous hemi&phere with prominent sulcal pattern and ipsilateral ventricular enlargement.
rarely observed. In a separate series, Jones and Heyl5
reviewed 140 older patients and confirmed the above observation of malignant degeneration. Domingo and Helwig6 reviewed 997 patients with nevus sebaceous and syringcystadenoma, and found 103 cases with associated epithelial neoplasm, 9 of which were aggressive malignant carcinomas. Fergin et af attributed the difference in the incidences of malignancy reported by various authors to the variation in the study population and the lack of uniformity in the clinical definition of the sebaceous nevus. Nevertheless, the possibility of malignancy as well as the curative nature of surgical excision seems established.
The NSJ syndrome has been suggested to be the cutaneous manifestation of another phakomatosis defined by the triad of linear nevus sebaceous, seizure disorder, and mental retardation. A wider spectrum of involvement extending to multiple organ systems has been acknowledged to include neurologic cutaneous, ophthalmic, cardiovascular, skeletal, and urogenital findings. The cardinal feature of the syndrome, the nevus, is only part of a more generalized abiotrophy. The dermatosis consisting of the nevus without any other organ system involvement is known to occur and has been considered a "form fruste" of the syndrome. 8
Fig 9. Trweighted magnetic resonance image axial scan through middle fossa and cyst, demonstrating increased signal characteristics within the dysgenetic right hemisphere.
Neurologic abnormalities described previously include apneic spells,9-11 seizures,9·11
-17 athetotic move
ments,14 abnormal gait, 14 developmental delay,2·15 mental retardation,2·11 ·14·15·18·19 arachnoid cyst, 10 cortical blindness, 16 cranial neuropathy (II1,20 VII, 12·14 VIII12), cortical atrophy, 15·21 ·22 elevated CSF protein, 11 hydrocephalus,9·23 hemiparesis, 10·12 hypesthesia, 10 homonymous field defect, 10 nystagmus,9·14- 16·19 arteriovenous malformation/4 intracavernous aneurysm,24 and nonfunctioning dural sinuses.Z4 Central nervous system (CNS) complications are more likely to .be associated when epidermal nevi occur on the head, and when structural CNS change is present it is most often ipsilateral to the skin lesion.24
Ophthalmologic involvement is second in frequency only to cutaneous disease and CNS dysfunction. Sixtyeight percent of the patients reported to have the NSJ syndrome had an ophthalmologic anomaly. Periorbital involvement occurs commonly, and is characterized by colobomas of the upper eyelid. 13·14·25·26 These can be large, and bilateral, though without distinguishing features. A downward slanting of the palpebral fissures has been recorded,9 as has ptosis 12·13·20·27 and ectropion. 14
Occasionally, the eyelid may be the site of hamartomatous tissue.28
Conjunctiva involvement usually appears as a firm, flat, pink, epibulbar tumor, commonly vascularized.9·10·12-15·20-31 There is no predilection for quadrant, and the tumors may be bilateral. Such tumors have been
1573

OPHTHALMOLOGY • DECEMBER 1987 • VOLUME 94 • NUMBER 12
pathologically shown to be normal conjunctival tissue with underlying substantia propia containing glands similar to the lacrimal gland, and associated with ectopic cartilage, fat, and connective tissueY Pathologic description includes dermoid pannus,30 lipodermoids,9
•28
teratoma, 13 dermolipoma/ 1 and choristoma?9
The cornea may be vascularized, either partially19 or completely,28 appearing as a leukoma. The cornea itself may harbor a dermoid tumor15 or choristoma, 22 or show diffuse scarring. 27 Pathologic study of such corneal changes has shown replacement of the corneal epithelium by that of conjunctival epithelium, without demonstrable Bowman's membrane, and associated with nerve fibers and capillaries within stroma. 28
Scleral involvement has included tumorous infiltration by hemangioma,9 fibroma, 10 and ectopic bone and cartilage.22 The sclera has also been dysplastic with prominent posterior staphyloma. 23
Involvement of the iris and ciliary body has been limited to typically appearing colobomas, occurring both superiorly and inferiorly; these, too, may be unilateral or bilateral. 9
•14
•18
•19 Incomplete opening of the anterior
chamber angle has been recognized.2•31 Pathologic de
scription has shown excessive vacuolization of the epithelium of the iris31 with mononuclear cells2 and adherence of Descemet's membrane to iris.2 Histologic evidence for flattening of the ciliary body, anterior insertion of the ciliary processes, and lack of development of the circular fibers is reportedY
Lenticular changes are occasionally noted and described as nonspecific cortical cataract.22
•31
•32 Results of
histologic analysis of such a lens showed vacuolization in the anterior and posterior cortical regions. 31
Chorioretinal involvement also reflects colobomatous formation, 9
•10
•18
•22
•31 commonly extending from the disc
though not exclusively so. 19 Occasionally, these colobomas involve the macula; they can be unilateral or bilateral, and do not have pathognomonic features. Peripapillary choroidal atrophy has been noted22 as has exudative retinal detachment and fibrovascular membrane formation. 22 A generalized retinal degeneration has been clinically described,9 and histologic analysis of one case has documented normal inner and outer nuclear layers though with almost complete absence of ganglion cellsY In addition, this retina showed increased vascularity with many large vacuolated cells with small eccentric hyperchromatic nucleiY
Optic nerve anomalies have been recognized, though clinical evaluation of the disc is often obscured by concomitant corneal leukoma. An optic nerve with ablatio falciformis has been described, 13 as well as optic disc pallor15
•22 and marked cupping.26
•31 In the case reported
with histologic confirmation of loss of retinal ganglion cells, clinical evaluation of the nervehead was precluded by corneal anomalies; the size of the optic nerve was not addressed in pathologic description. 31
Other ophthalmologic anomalies reported in association with the NSJ syndrome include strabismus,9
•14
•18
·-
24•33-35 unilateral proptosis,20 and microphthalmos. 13•14•31
Anomalies involving other organ systems include pa-
1574
Fig 10. Paraffin section of skin biopsy. A mature sebaceous gland is seen below a hyperkeratotic and papillomatous epidermis. A distended apocrine gland is seen beneath the sebaceous gland, in the deep dermis (hematoxylin-eosin, X 10).
Fig 11. Paraffin section from the epibulbar dermoid. In the deep dermis, ectopic lacrimal gland is seen adjacent to hyalinized cartilage (hematoxylin-eosin, X50).
tent ductus arteriosus,28 patent foramen ovale,31 ventricular septal defect, 16 coarctation of the aorta,9
•19
•23
•31 hy
poplasia of the aorta,9 atrial flutter/fibrillation,23 atrial/ ventricular premature systoles, 16 asymmetry of sphe-

KATZ et al • NEVUS SEBACEOUS OF JADASSOHN
noid wings,20 slanting of auricles, 9 highly arched palate, 12 hypoplastic dentition,9·19 lytic defect of ribs, 15
clavicular malformation, 15 scoliosis,9·12 hypoplastic iliac wing, 12 pes valgus deformity, 12 osteomalacic changes, 15·18
horseshoe kidney,28 duplicate urinary collection system,10 undescended testicles, 12 enlarged clitoris,23 hepatosplenomegaly, 12 and failure to thrive.9·19 Tumorous associations have also been recognized. These include nephroblastoma, 16 hemangioma of the leptomeninges, 28
abdominal wall12 and penis/0 cystic adenoma of the liver,28 ameloblastoma of the mandible, 11 and lipomas. 14
The NSJ syndrome does not have a clearly defined genetic basis. The nevus sebaceous syndrome has never been described in any family members of a proband, although seizures2·11 ·34·36 and mental retardation 11 ·34
have been. Chromosomal studies are described as normal. 19
The features of this syndrome overlap those of the oculo-auriculo-vertebral dysplasia of Goldenhar, and tuberous sclerosis. In the dysplasia ofGoldenhar, anomalies of skull and orbit are seen, as are epibulbar dermoids and colobomas. Both tuberous sclerosis and the NSJ syndrome share a sebaceous skin lesion, seizures, and mental retardation. The pathology of the skin lesion clearly differentiates the two syndromes. 16 Periventricular calcifications, commonly seen in children with tuberous sclerosis and observable by CT37 are absent in children with the NSJ syndrome. The CT and magnetic resonance imaging scans of our patient support this distinguishing feature. 35
Optic nerve hypoplasia represents a spectrum38·39 of developmental anomaly of the retina and optic disc. There is no predilection for sex.40 The defect may be bilateral or unilateral40 and may accompany systemic malformations or occur in an otherwise normal person.40.41 Visual acuity varies from profound loss42 to normal43 and there can be associated field defects. 43 A cardinal feature is a subnormal diameter of the optic nerve head, often one third to one half of the normal diameter.40·42 The disc is usually but not always surrounded by a white-yellow halo or double ring.40·44 The disc substance itself is pale and often spotted with pigment, making it appear dirty.40 The retinal vessels are of normal caliber and appear to enter the disc centrally rather than nasally, as expected.41 An afferent pupillary defect is common38
•40 and patients may exhibit a pen
dular or rotary nystagmus in cases of bilateral involvement with poor visual acuity.41
The etiologic events and associations of optic nerve hypoplasia are poorly defined. In a study of 22 cases of optic nerve hypoplasia, Hotchkiss noted that two thirds were associated with an abnormal pregnancy, half of which included a history of hydramnios,39 Drug effects to the growing fetus and maternal systemic conditions including hypertension, diabetes, Rh-incompatibility, German measles, syphilis, arachnoiditis, phlebitis, and obesity have been implicated. 39.43,45,46 None of the above conditions existed in the maternal history of our patient.
Optic nerve hypoplasia has been associated with a wide range of systemic pathology.41 By far, the most
common association has been CNS abnormalities. These include anencephaly, hydranencephaly, hypopituitarism, agenesis of the septum pellucidum, cerebral palsy, psychomotor retardation, epilepsy, and porencephaly.46·47 The association of optic nerve hypoplasia and the NSJ syndrome is new, although not unexpected. Shocoe1 described the typical pathologic finding of optic nerve hypoplasia in the retina of a patient with NSJ syndrome: complete absence of ganglion cells in the presence of normal amacrine and bipolar cells. The pathogenesis of optic nerve hypoplasia is incompletely understood. Early investigators considered it a primary agenesis of some or all retinal ganglion cells in the sixth or seventh week of embryonic development.41 More recent work has suggested it to be a result of degeneration of ganglion cells and their axons caused by an insult to the developing visual pathway any time during prenatal development.48 Our patient's concomitant porencephalic cyst and dysplastic hemisphere likely disturbed the developing visual pathways either by interference with the normal trajectory of the growing fibers, by removal of an appropriate target organ, or by secondary destruction of established pathways or connections.47 We conclude that the optic nerve hypoplasia seen in our patient likely represents an example of excessive degeneration of optic nerve axons and retinal ganglion cells which occurred as a result of nonspecific insult to the visual pathways and as part of NSJ syndrome, presumably preventing these axons from forming or maintaining appropriate connections at their target sites.
REFERENCES
1. Jadassohn J. Bemerkungen zur Histologie der systematisierten Naevi und Ober "Talgdrusen-Naevi." Arch tor Derm Syph 1895; 33:355-72.
2. Wilkes SR, Campbell RJ, Waller RR. Ocular malformation in association with ipsilateral facial nevus of Jadassohn. Am J Opthalmol1981; 92:344-52.
3. Robinson SS. Neavus sebaceus (Jadassohn). Arch Dermatol Syph 1932; 26:663-70.
4. Mehregan AH, Pinkus H. Life history of organoid nevi. Special reference to nevus sebaceus of Jadassohn. Arch Dermatol 1965; 91:574-88.
5. Jones EW, Heyl T. Naevus sebaceus. A report of 140 cases with special regard to the development of secondary malignant tumours.
Br J Derm 1970; 82:99-117. 6. Domingo J, Helwig EB. Malignant neoplasms associated with naevus
sebaceus of Jadassohn. JAm Acad Derm 1979; 1:545-56. 7. Fergin PE, Chu AC, MacDonald OM. Basal cell carcionama compli
cating naevus sebaceus. Clin Exp Derm 1981; 6:111-5. 8. Lentz CL, Altman J, Mopper C. Nevus sebaceus of Jadassohn:
report of a case with multiple and extensive lesions and an unusual linear distribution. Arch Dermatol1968; 97:294-6.
9. Marden PM, Venters HD. A new neurocutaneous syndrome. Am J Dis Child 1966; 112:79-81.
10. Holden KR, Dekaban AS. Neurological involvement in nevus unius lateris and nevus linearis sebaceus. Neurology 1972; 22:879-87.
11. Lovejoy FH, Boyle WE. Linear nevus sebaceous syndrome. report of two cases and a review of the literature. Ped 1973; 52:382-7.
12. Solomon LM, Fretzin OF, Dewald RL. The epidermal nevus syndrome. Arch Dermatol1968; 97:273-85.
1575

OPHTHALMOLOGY • DECEMBER 1987 • VOLUME 94 • NUMBER 12
13. Meythaler H. Augenbefunde beim Syndrom nach Schimmelpenning, Feuerstein und Mims. Klin Monatsbl Augenheilk 1975; 166:244-5.
14. Zaremba J, Wislawski J, Bidzinski J, et al. Jadassohn's naevus phakomatosis: 1. A report of two cases. J Ment Defic Res. 1978; 22:91-102.
15. Sugarman GJ, Reed WB. Two unusual neurocutaneous disorders with facial cutaneous signs. Arch Neurol 1969; 21:242-7.
16. Lansky LL, Funderburk S, Cuppage FE, et al. Linear sebaceous nevus syndrome: a hamartoma variant. Am J Dis Child 1972; 123:587-90.
17. Herbst BA, Cohen ME. Linear nevus sebaceus. A neurocutaneous syndrome associated with infantile spasms. Arch Neurol 1971; 24:317-22.
18. Moorjani R, Shaw DG. Feuerstein and Mims syndrome with resistant rickets. Pediatr Radiol1976; 5:120-2.
19. Monahan RH, Hill CW, Venters HD. Multiple choristomas, convulsions and mental retardation as a new neurocutaneous syndrome. Am J Ophthalmol 1967; 64:529-32.
20. Haslam RHA,Wirtschafter JD. Unilateral external oculomotor nerve palsy and nevus sebaceous of Jadassohn. Arch Ophthalmol 1972; 87:293-300.
21. Moynahan EJ, Wolff OH. A new neuro-cutaneous syndrome (skin, eye and brain) consisting of linear naevus, bilateral lipo-dermoid of the conjunctivae, cranial thickening, cerebral cortical atrophy and mental retardation. Br J Derm 1967; 79:651-2.
22. Wilkes SR, Campbell RJ, Waller RR. Ocular malformation in association with ipsilateral facial nevus of Jadassohn. Am J Ophthalmol 1981; 92:344-52.
23. Tripp JH, Joseph MC, Reay HAJ. A new "neurocutaneous" syndrome (skin, eye, brain and heart syndrome). Proc Roy Soc Med 1971; 64:23-4.
24. Baker RS, Ross PA, Baumann RJ. Neurological complications of the epidermal nevus syndrome. Arch Neurol 1987; 227-32.
25. Gi:irduren S. Aberrant lacrimal gland associated with other congenital abnormalities. Br J Ophthal 1962; 46:277-80.
26. Lantis S, Leyden J, Thew M, Heaton C. Nevus sebaceus of Jadassohn: part of a new neurocutaneous syndrome? Arch Dermatol1968; 98:117-23.
27. Berg JM, Creme L. A possible case of atypical tuberous sclerosis. J Men! Defic Res 1960; 4:24-31.
28. Mollica F, Pavone L, Nuciforo G. Linear sebaceous nevus syndrome in a newborn. Am J Dis Child 1974; 128:868-71.
29. Lall K. Teratoma of conjunctiva (associated with nevus systematicus and epilepsia symptomatica). Acta Ophthalmol1962; 40:555-8.
30. Kalis B, Degos R, Harmel L, Reset-Charles J. Naevus sebace verru-
1576
queux avec manifestations neurologique-oculaire-osseuse (Naevus de Solomon). Ann Dermatol Syph 1973; 100:380-1.
31. Shochot Y, Romano A, Barishak YR, et al. Eye findings in the linear sebaceous nevus syndrome: a possible clue to the pathogenesis. J Craniofac Genet Dev Bioi 1982; 2:289-94.
32. Leyh F, Loewel R. Ein Fall von Schimmelpenning-Feuerstein-Mims Syndrom mit allmahlicher Kataraktentwicklung. Z Hautkr 1973; 48:695-8.
33. Besser FS. Linear sebaceous naevi with convulsions and mental retardation (Feuerstein-Mims Syndrome), vitamin-D-resistant rickets. Proc Roy Soc Med 1976; 69:518-20.
34. Bianchine JW. The nevus sebaceous of Jadassohn: a neurocutaneous syndrome and a potentially premalignant lesion. Am J Dis Child 1970; 120:223-8.
35. Kurokawa T, Sasaki K, Hanai T, et al. Linear nevus sebaceus syndrome: report of a case with Lennox-Gastaut syndrome following infantile spasms. Arch Neurol1981; 38:375-7.
36. Zaremba J. Jadassohn's naevus phakomatosis: 2. A study based on a review of thirty-seven cases. J Men! Defic Res 1978; 22:103-23.
37. Monaghan H, Krafchik BR, MacGregor DL, Fitz CR. Tuberous sclerosis complex in children. Am J Dis Child 1981; 135:912-7.
38. Frisen L, Holmegaard L. Spectrum of optic nerve hypoplasia. Br J Ophthalmol1978; 62:7-15.
39. Hotchkiss ML, Green WR. Optic nerve aplasia and hypoplasia. J Pediatr Ophthalmol Strabismus 1979; 16:225-40.
40. Walton DS, Robb RM. Optic nerve hypoplasia: a report of 20 cases. Arch Ophthal 1970; 84:572-8.
41. Brown GC, Tasman WS. Congenital Anomalies of the Optic Disc. New York: Grune & Stratton, 1983; 195-206.
42. Edwards WC, Layden WE. Optic nerve hypoplasia. Am J Ophthalmol 1970; 70:950-9.
43. Peterson RA, Walton DS. Optic nerve hypoplasia with good visual acuity and visual field defects: a study of children with diabetic mothers. Arch Ophthalmol1977; 95:254-8.
44. Mosier MA, Lieberman MF, Green WR, Knox DL. Hypoplasia of the optic nerve. Arch Ophthalmol1978; 96:1437-42.
45. McKinna AJ. Quinine induced hypoplasia of the optic nerve. Can J Ophthalmol 1966; 1 :261-6.
46. Hoyt CS, Billson FA Maternal anticonvulsants and optic nerve hypoplasia. Br J Ophthalmol 1978; 62:3-6.
47. Greenfield PS, Wilcox LM Jr, Weiler JJ, Adelman L. Hypoplasia of the optic nerve in association with porencephaly. J Pediatr Ophthalmol Strabismus 1980; 17:75-80.
48. Skarf B, Hoyt CS. Optic nerve hypoplasia in children, association with anomalies of the endocrine and CNS. Arch Ophthalmol 1984; 102:62-7.


















