
Ocular Clinicopathologic Correlation of Hallervorden-Spatz Syndrome with Acanthocytosis and...
14
OCULAR CLINICOPATHOLOGIC CORRELATION OF HALLERVORDEN-SPATZ SYNDROME WITH ACANTHOCYTOSIS AND PIGMENTARY RETINOPATHY MARTHA W. LUCKENBACH, M.D., W. RICHARD GREEN, M.D., NEIL R. MILLER, M.D., HUGO W. MOSER, M.D., ARTHUR W. CLARK, M.D., AND GlHAN TENNEKOON, M.D. Baltimore, MaryL·nd We studied the eyes of a 10-year-old girl with retinal degeneration, acanthocytosis, and normal betalipoprotein levels. The ophthalmoscop- ic pattern was characterized initially by a flecked retina and later by bone-spicule formation and "bull's-eye" annular maculopathy. On ultrastructural study, the retinal pigment epithelium varied in size and contained large, round single-membrane-bound aggregates composed of complex melanolipofuscin granules. Cells that had migrated into the outer retinal layers contained similar melanolipofuscin aggregates; these cells were identified as macrophages and correlated with the flecks and macular annulus seen on ophthalmoscopy. The cells around the retinal blood vessels contained normal melanin pigment, were identified as retinal pigment epithelial cells, and correlated with the bone spicule pigmentation found on ophthalmoscopic examination. In 1922, Hallervorden and Spatz 1 de- scribed five sisters in a sibship of 12 who had a progressive neurodegenerative dis- order characterized by extrapyramidal motor signs, dysarthria, dementia, and death. In 1974, Dooling, Schoene, and Richardson 2 reviewed the 64 reported autopsy-proven cases, established strict clinical and histopathologic criteria, and found that 42 cases indicated a distinct clinicopathologic entity. In two cases, 3,4 Accepted for publication Oct. 29, 1982. From the Eye Pathology Laboratory, Wilmer Oph- thalmological Institute (Drs. Luckenbach, Green, and Miller), the John F. Kennedy Institute (Dr. Moser), and the Departments of Pathology (Drs. Green and Clark) and Neurology (Dr. Tennekoon), Johns Hopkins Medical Institutions, Baltimore, Maryland. This study was supported in part by the Pearl S. Stetter Research Fund (Dr. Luckenbach) and Research Grant 01684 from the National Eye Insti- tute (Dr. Green). Reprint requests to W. Richard Green, M. D., Eye Pathology Laboratory, Johns Hopkins Hospital, 600 N. Wolfe St., Baltimore, MD 21205. there was an associated acanthocytosis with normal lipid findings and normal betalipoprotein levels. Newell, Johnson, and Huttenlocher 5 found that almost one fourth of the 42 patients had retinal degeneration. They believed that such patients formed a dis- tinct group that had a much earlier onset and a much more rapid course, with death occurring in late childhood. Both of the earlier patients who had an associated acanthocytosis 3 ' 4 were classed in this group. In clinical studies, Newell, John- son, and Huttenlocher 5 observed retinal degeneration with retinal flecks and a "bull's-eye" maculopathy in dizygotic twins with the Hallervorden-Spatz syn- drome. We describe the ocular clinicopatho- logic features of a patient who died of Hallervorden-Spatz syndrome with an as- sociated acanthocytosis and normal beta- lipoprotein level. ©AMERICAN JOURNAL OF OPHTHALMOLOGY 95:369-382, 1983 369
Transcript of Ocular Clinicopathologic Correlation of Hallervorden-Spatz Syndrome with Acanthocytosis and...

OCULAR CLINICOPATHOLOGIC CORRELATION O F HALLERVORDEN-SPATZ SYNDROME W I T H ACANTHOCYTOSIS A N D
PIGMENTARY RETINOPATHY
M A R T H A W. LUCKENBACH, M.D. , W. R I C H A R D G R E E N , M.D. , N E I L R. M I L L E R , M.D. , H U G O W. M O S E R , M.D. ,
A R T H U R W. CLARK, M.D. , AND GlHAN T E N N E K O O N , M . D .
Baltimore, MaryL·nd
We studied the eyes of a 10-year-old girl with retinal degeneration, acanthocytosis, and normal betalipoprotein levels. The ophthalmoscop-ic pattern was characterized initially by a flecked retina and later by bone-spicule formation and "bull's-eye" annular maculopathy. On ultrastructural study, the retinal pigment epithelium varied in size and contained large, round single-membrane-bound aggregates composed of complex melanolipofuscin granules. Cells that had migrated into the outer retinal layers contained similar melanolipofuscin aggregates; these cells were identified as macrophages and correlated with the flecks and macular annulus seen on ophthalmoscopy. The cells around the retinal blood vessels contained normal melanin pigment, were identified as retinal pigment epithelial cells, and correlated with the bone spicule pigmentation found on ophthalmoscopic examination.
In 1922, Hallervorden and Spatz1 described five sisters in a sibship of 12 who had a progressive neurodegenerative disorder characterized by extrapyramidal motor signs, dysarthria, dementia, and death. In 1974, Dooling, Schoene, and Richardson2 reviewed the 64 reported autopsy-proven cases, established strict clinical and histopathologic criteria, and found that 42 cases indicated a distinct clinicopathologic entity. In two cases,3,4
Accepted for publication Oct. 29, 1982. From the Eye Pathology Laboratory, Wilmer Oph-
thalmological Institute (Drs. Luckenbach, Green, and Miller), the John F. Kennedy Institute (Dr. Moser), and the Departments of Pathology (Drs. Green and Clark) and Neurology (Dr. Tennekoon), Johns Hopkins Medical Institutions, Baltimore, Maryland. This study was supported in part by the Pearl S. Stetter Research Fund (Dr. Luckenbach) and Research Grant 01684 from the National Eye Institute (Dr. Green).
Reprint requests to W. Richard Green, M. D., Eye Pathology Laboratory, Johns Hopkins Hospital, 600 N. Wolfe St., Baltimore, MD 21205.
there was an associated acanthocytosis with normal lipid findings and normal betalipoprotein levels.
Newell, Johnson, and Huttenlocher5
found that almost one fourth of the 42 patients had retinal degeneration. They believed that such patients formed a distinct group that had a much earlier onset and a much more rapid course, with death occurring in late childhood. Both of the earlier patients who had an associated acanthocytosis3'4 were classed in this group. In clinical studies, Newell, Johnson, and Huttenlocher5 observed retinal degeneration with retinal flecks and a "bull's-eye" maculopathy in dizygotic twins with the Hallervorden-Spatz syndrome.
We describe the ocular clinicopathologic features of a patient who died of Hallervorden-Spatz syndrome with an associated acanthocytosis and normal betalipoprotein level.
©AMERICAN JOURNAL OF OPHTHALMOLOGY 95:369-382, 1983 369

370 AMERICAN JOURNAL OF OPHTHALMOLOGY MARCH, 1983
C A S E REPORT
The patient was the full-term product of a consanguineous mating between an 11-year-old girl and her 16-year-old half-brother. The infant's parents had the same mother. During the pregnancy, the young mother had taken dextroamphetamine diet pills and had received a rubella immunization one month before delivery.
The infant was placed in permanent foster care, and when examined during the newborn period was thought to be normal. She held her head up at 5 to 6 months of age, rolled over at 5 months, was rolling well at 9 months, sat at 5 to 6 months, pulled to a standing position at 14 months, crawled at 15 months, and walked unsteadily at 16 months.
When the patient was 19 days old, her foster mother noted crossed eyes. The child was examined at the Wilmer Institute when she was 4 months old. She was found to have a 45-diopter alternating esotropia with a symmetric hyperopic, astigmatic refractive error. Ophthalmoscopic findings were normal. Spectacles were prescribed, and the esotropia was followed up at three-month intervals.
Shortly after the first ophthalmologic evaluation, a peripheral blood smear disclosed acanthocytosis. The diagnosis of abetalipoproteinemia (Bassen-Kornzweig syndrome) was considered, but lipid levels were normal (cholesterol was 171 mg/100 ml and triglycérides were 8 mg/100 ml) and alpha-, beta-, and pre-beta-lipoproteins were present on paper electro-phoresis.
The child grew well but had some difficulty in chewing and swallowing and had to be fed strained foods. She never developed the ability to speak. At the age of two years seven months, she was unstable on her feet, frequently reeling backwards and falling. She was examined at the National Institutes of Health when she was about 3 years of age. Neurologic examination disclosed mental retardation, ataxia, and pyramidal tract signs, compatible with a spino-cerebellar degeneration. Lipid and lipoprotein levels were normal; carotene was 176 μg/100 ml (normal, 50 to 300 μg/100 ml); vitamin A was 122 IU/100 ml (normal, 65 to 275 IU/100 ml); sickle-cell preparation was negative, and protein electrophoresis was normal, as were the SMA-6, SMA-12, and X-rays for bone age. It was thought, however, that the patient had functionally defective betalipoproteins because normal red blood cells formed acanthocytes when incubated in her serum.
When she was examined at the National Eye Institute, she was found to have red hair and a yellow and lightly pigmented skin. Ophthalmoscopic examination disclosed that the macula had a granular appearance and there was a poor foveal reflex in each eye. There was a diffuse, mild granularity of pigmentation extending to the equatorial retina, but bone-spicule pigmentation was absent. The electroretino-gram of each eye was flat.
During the following years, the child underwent frequent ocular examinations at the Wilmer Institute and general tests at the John F. Kennedy Institute. The ophthalmoscopic appearance evolved and led to
a clinical diagnosis of "retinitis punctata albescens" when she was 5 years old. When she was 8 years old, her macula had developed a bull's-eye appearance; white flecks, without bone spicules, were noted in the posterior pole and equatorial retina.
At the age of 8 years 2 months, she developed involuntary athetoid movements in her head, neck, and arms. She developed increasing spasticity, with dystonic posturing (primarily in her arms), hyperex-tension of her head and neck, and the appearance of tonic labyrinthine reflex. Results of a computed axial tomographic scan of the head were normal. She gradually lost the ability to sit or walk without support, was unable to chew and eat, and had difficulty in swallowing because of traumatic deterioration in her coordination.
She was again admitted for testing at the age of WÎ years. A neurologic examination disclosed severe retardation, with no expressive language but receptive language appropriate for a 21/2-year-old. At rest she had choreiform movement, unusual posturing of her limbs, and occasional ballistic-type movement. Her reflexes were then about 1 + , with no response to plantar stimulation. She also had evidence of a peripheral neuropathy, Laboratory findings remained unchanged. There was no evidence of liver disease or vitamin E deficiency as a cause for the acanthocytosis. However, a one-hour postprandial triglycéride test showed that there was only a slight increase in the total triglycérides. The patient was thought to have some metabolic difficulty in forming chylomicrons, and thus reduced ability to absorb fat and fat-soluble vitamins. An electroencephalogram was slightly abnormal for her age, with a mildly disorganized low-voltage tracing and sporadic anterior minor spikes. No seizure discharges or localizing signs were present. Results of tests of nerve conduction velocity were normal. Results of a skin biopsy for fibroblast culture were noncontributory. A computed tomographic scan then showed dilated ventricles, cortical sulci, and vermian folia. Dilation of the sylvian fissures and basal cisterns was also present. There was no abnormal enhancement or increased density in the basal ganglia that would indicate iron-pigment deposits.
Using the technique of Barr and associates,6 we determined the bicaudate index. The ratio of the bicaudate diameter to the distance between the outer tables of the skull (bicaudate index, CC:OTCC) was 0.127. The ratio of the maximum distance of the frontal horns tó the distance between the heads of the caudate nuclei (FH:CC) was 2.09. These ratios were similar to those seen in cerebral atrophy.
Laboratory tests showed a hemoglobin level of 12.3 g/100 ml and a platelet count of 338,000/cu mm. Serum copper was 149 μg/100 ml (normal, 27 to 153 μg/100 ml) of blood, and the ceruloplasmin level was normal. Red blood cell enzyme evaluation disclosed normal hexokinase and pyruvate kinase levels. Lipid analysis of ghost red blood cells showed normal levels of cholesterol, sphingomyelin, and phospho-lipid. Spin-label studies by electron-spin resonance7
of the red blood cells showed no difference between the spread or distribution of order factors

VOL. 95, NO. 3 HALLERVORDEN-SPATZ SYNDROME 371
in her test compared with that of a normal control subject.8
Ophthalmologic examination disclosed a slightly inconsistent right nystagmus in primary gaze, with mild symmetric ophthalmoplegia on attempted upward and rightward gaze. There was an exotropia on downward gaze. Ophthalmoscopy showed a bull's-eye appearance of the macula, with a central zone of mild granular hyperpigmentation surrounded by a ring of yellow material located deep in the retinal vessels (Fig. 1). There was minimal peripheral bone-spicule pigmentation. The optic disk appeared to be normal. The arterioles were moderately attenuated. Many yellow-white flecks of various sizes and shapes gave the fundus a mottled appearance (Fig. 2), These flecks were most evident in the equatorial retina but were also scattered throughout the posterior pole. A conjunctive biopsy performed for metabolic screening disclosed an abnormal accumulation of lipofuscin in the fibroeytes.
The patient died at the age of 10 years of broncho-interstitial pneumonia and probable sepsis.
Pathologic findings—At autopsy the body showed severe cachexia, with thy-mic and liver atrophy consistent with malnutrition. Signs of spasticity included flexion contracture of the left arm and bilateral equinovarus deformities of the feet, with tight heel cords. There was bilateral purulent otitis media, with gross evidence of osteomyelitis, and Escher-
Fig. 1 (Luckenbach and .ASOUcttuv Macular area appearance with granular hyperpigmentation of the fo deep in the retinal vessels.
ichia colt organisms were cultured from the middle-ear canals after death. There was extensive acute and organizing bron-chointerstitial pneumonia, with much pulmonary congestion and edema.
BRAIN FINDINGS—The brain was grossly normal in size and weighed 1,120 g. A section showed a red-brown discoloration in the regions of the globus pallidus and internal capsule, bilaterally. Otherwise, the gross findings were unremarkable.
Microscopic examination disclosed innumerable eosinophilic spheroids in the medial globus pallidus corresponding to the zone of discoloration. These spheroids were also apparent in the adjacent internal capsule. They were not found in other areas of the brain. Specifically, the substantia nigra was not affected. The spheroids were not stained with standard silver techniques for axons. Some stained positively with the PAS stain. Stains for iron disclosed iron-containing granules located primarily extracellularly, but sometimes intracellularly in the same region. The iron-positive granules were never found within the spheroids.
of right (left) and left (right) eyes showing bull's-eye yea. An annular deposition of yellow material is located

372 AMERICAN JOURNAL· OF OPHTHALMOLOGY MARCH, 1983
Fig. 2 (Luckenbach and associates). Midperipheral areas of retina with many yellow-white flecks of various sizes and shapes located deep in the retinal blood vessels.
ULTRASTRUCTURAL FINDINGS—Transmission electron microscopy showed that the spheroid bodies observed by light microscopy corresponded to nuclei of degenerating astrocytes. Oligodendroglial cells were swollen, with pale ballooned cytoplasm. By electron microscopy, the cytoplasm contained homogeneous membrane-bound material consistent with lipid in appearance. The myelin did not seem to be abnormal, and axonal swellings were not observed. There were no noteworthy lesions in the substantia nigra, brain stem, or cerebellum.
M A T E R I A L AND METHODS
The eyes were preserved in 4% buffered formaldehyde, then washed overnight and placed in 60% alcohol for gross examination. The right eye was prepared for light microscopic examination, and serial sections through the plane of the pupil, optic nerve head, and macula were prepared. The left globe was placed in a mixture of 1% glutaraldehyde and 4% formaldehyde, postfixed in 2% osmium
tetroxide, dehydrated in graded acetones, and embedded in Araldite and epoxy resin. For phase-contrast microscopy, sections 1 μιη thick were stained with paraphenylenediamine. For electron microscopy, ultrathin sections were doubly stained with uranyl acetate and lead citrate. Specimens for scanning electron microscopy were prepared by peeling the retina from the surface of the retinal pigment epithelium, dehydrating in graded acetones, critical-point drying with liquid carbon dioxide, and sputter-coating with gold palladium.
Deparaffinized unstained sections were examined by ultraviolet fluorescent microscopy, using high-intensity halogen light sources, and a dark-field condenser and exciter (BG-12), and type 47, 50, GG-14, and OG-53 barrier filters.
Gross examination—The eyes were of normal size and shape. Abnormalities were limited to the retina, where a moderate amount of bone-spicule pigmentation was present from anterior to the equator to the ora serrata.

VOL. 95, NO. 3 HALLERVORDEN-SPATZ SYNDROME 373
Light microscopy—Moderate autolysis was present. Abnormalities were limited to the retina. There was total loss of the outer segments of the photoreceptor cells and near-total loss of the inner segments, with only a few stubby inner segments remaining throughout the retina. The outer nuclear layer was much thinned or absent throughout (Figs. 3 and 4). Pig-mented cells were located in the outer aspect of the retina in many areas (Figs. 3 and 4). The retinal pigment epithelium showed variation in the size and shape of individual cells (Fig. 5). Intermixed among cells of normal size and shape were greatly enlarged retinal pigment epithelial cells. These enlarged cells were either broad and flattened or of normal diameter but much increased in height. Contained mostly within the enlarged retinal pigment epithelial cells, but also occasionally found in normal-sized cells, were large round pigment aggregates composed of small irregularly shaped granules. The retinal pigment epithelial cells were moderately well pig-mented throughout and contained many individual pigment granules in addition to the large, round aggregates. A 60-minute bleached section disclosed that after the melanin pigment in these CIUS-
"•ÌfÉSf·»
Fig. 3 (Luckenbach and associates). Temporal mid-peripheral retina with total loss of the photoreceptor cell layer. Pigmented cells are present in the outer aspect of the retina (hematoxylin and eosin, x 220).
* *
Ά* \\ '*. V. ^
> -là*
Fig. 4 (Luckenbach and associates). Macular area with loss of photoreceptor cell layer and presence of pigmented cells in outer retina (PAS, x 525).
ters was removed, small golden-brown granules remained. The pigment in the retinal pigment epithelium did not stain with the PAS stain nor did it show auto-fluorescence.
In the perimacular retina there was migration of pigment-laden cells into the outer retinal layers (Fig. 4), with some of
4%* · ·
Fig. 5 (Luckenbach and associates). Posterior area showing much distension of the retinal pigment epithelial cells, which vary in size and shape and contain large, round pigment aggregates (hematoxylin and eosin, x 900).

374 AMERICAN JOURNAL OF OPHTHALMOLOGY MARCH, 1983
them clinging to the outer limiting membrane on either side of the center of the macula. Similar pigment-laden cells were present focally in the outer retinal layers peripheral to the perimacular area; they extended to the ora serrata. There was minimal-to-moderate migration of the retinal pigment epithelium into the inner retinal layers in a perivascular location, from the postequatorial retina to the ora serrata (Fig. 6). The ganglion-cell, nerve-fiber, and inner-plexiform layers were normal, as were cross sections of the optic nerve. The choriocapillaris and choroid were normal throughout.
Electron microscopy—Transmission electron microscopic examination disclosed similar findings in the macular, equatorial, and peripheral areas of the retinal pigment epithelium. The pigment-laden cells varied in size and shape, with basally located mitochondria and nuclei. The cells contained several different types of pigment granules, in addition to a few normal-appearing melano-somes. The most common pigment was in the form of giant single-membrane-bound melanolipofuscin aggregates;
Fig. 6 (Luckenbach and associates). Equatorial area showing retinal blood vessel surrounded by pigmented cells (PAS, x 280).
these were round in shape, measured 3 to 4 μηι in diameter, and were similar in size to the cell nucleus (Fig. 7). These large round aggregates were composed primarily of many cigar-shaped or round melanin granules ranging from 0.2 to 0.5 μπι in diameter and embedded in a moderately dense lipofuscin matrix composed of regular, membranous stacks or whorls of lamellae, with individual membrane thickness of 14 nm and a periodicity of 14 nm. Rare pigment aggregates measuring 1.4 μπι in diameter contained lipofuscin granules, with little or no melanin (Fig. 8). Other common pigment granules were pleomorphic lipofuscin granules and single-membrane-bound melanolipofuscin granules, which were found both individually and in aggregates that measured up to 1.4 μιη in diameter (Fig. 9).
Macrophages were present in the outer nuclear layer and contained giant, round single-membrane-bound melanolipofuscin aggregates, measuring 3 to 4 μιη in diameter (Fig. 10). These were similar to the aggregates present in the retinal pigment epithelium. We also found smaller melanolipofuscin granules and clusters.
The perivascular pigment-laden cells were identified as hyperplastic retinal pigment epithelium because of polarity and proliferation in an acinar pattern, with apical microvilli facing the lumen and basement membrane at the base (Fig. 11). Complex intercellular junctions were present near the apexes of the cells. These cells contained round mel-anosomes measuring 0.05 to 0.12 nm in diameter and, rarely, lipofuscin granules.
Occasional vascular pericytes contained dense lipofuscin granules (Fig. 11, top inset). Neither ganglion cells nor the other retinal neural cells contained inclusions or lipofuscin. The choriocapillaris and Bruch's membrane were normal. There was no evidence of demyelination in the optic nerve.
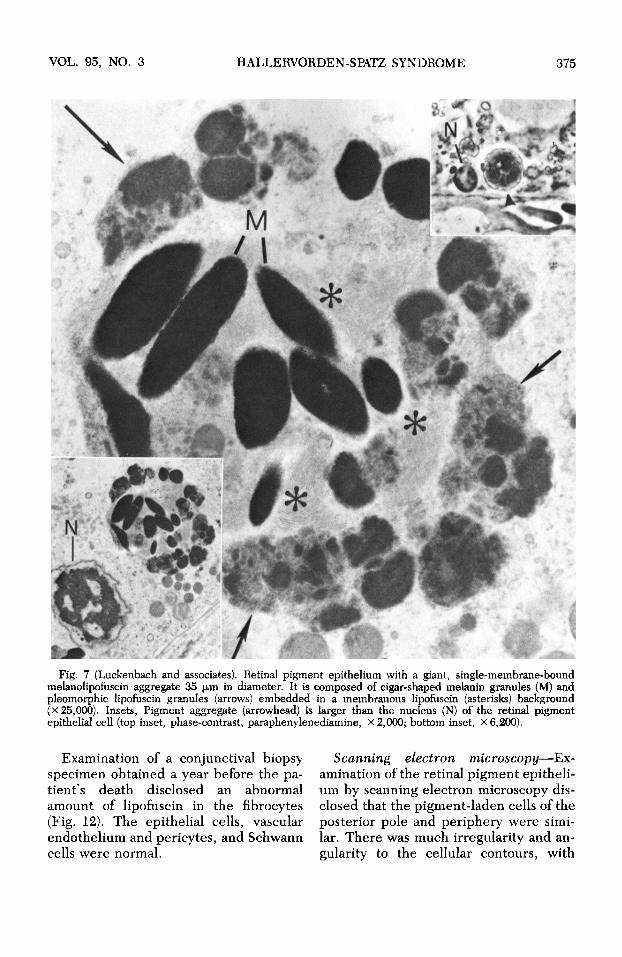
VOL. 95, NO. 3 HALLERVORDEN-SPATZ SYNDROME 375
Fig. 7 (Luckenbach and associates). Retinal pigment epithelium with a giant, single-membrane-bound melanolipofuscin aggregate 35 μπι in diameter. It is composed of cigar-shaped melanin granules (M) and pleomorphic lipofuscin granules (arrows) embedded in a membranous lipofuscin (asterisks) background (x 25,000). Insets, Pigment aggregate (arrowhead) is larger than the nucleus (N) of the retinal pigment epithelial cell (top inset, phase-contrast, paraphenylenediamine, x 2,000; bottom inset, X 6,200).
Examination of a conjunctival biopsy Scanning electron microscopy—Ex-specimen obtained a year before the pa- amination of the retinal pigment epitheli-t i en t s death disclosed an abnormal urn by scanning electron microscopy dis-amount of lipofuscin in the fibrocytes closed that the pigment-laden cells of the (Fig. 12). The epithelial cells, vascular posterior pole and periphery were simi-endothelium and pericytes, and Schwann lar. There was much irregularity and an-cells were normal. gularity to the cellular contours, with

Fig. 8 (Luckenbach and associates). Retinal pigment epithelium with pigment aggregate that measures 1.4 μιη in diameter and is composed of lipofuscin in the form of membranous (circled areas), pleomorphic lipofuscin granules (arrowheads), and partially degraded melanin granules (arrows) (x 40,000).
Fig. 9 (Luckenbach and associates). Retinal pigment epithelium with single-membrane-bound (arrowhead) melanolipofuscin aggregate that measures 1.4 u.m in diameter and contains partially degraded melanin (arrows) in a background of finely granular lipofuscin and dense pleomorphic lipofuscin granules (X 40,000).

VOL. 95, NO. 3 HALLERVORDEN-SPATZ SYNDROME 377
Fig. 10 (Luckenbach and associates). Macrophage in outer retina contains giant melanolipofuscin aggregate that measures 3.8 μηι in diameter. The aggregate is composed of cigar-shaped melanin granules (arrowheads) embedded in a granular lipofuscin matrix (asterisks) (x 15,300). Inset, The aggregate is bounded by a single membrane (arrow) (x 60,000).
large cells mixed equally among smaller cells. The cells varied in size from 3 to 20 μπι in diameter.
D I S C U S S I O N
Dooling, Schoene, and Richardson2
listed four clinical characteristics of Hallervorden-Spatz syndrome: (1) onset at a young age, generally after earliest childhood; (2) extrapyramidal motor disorder with dystonic posturing, muscular rigidity, choreo-athetoid movements, ataxia, hyperreflexia, and spasticity; (3) dementia; and (4) a relentlessly progressive course leading to death in early adulthood.
The neuropathologic characteristics are (1) symmetric, partially destructive le
sions of the globus pallidus, especially its internal segment and the pars reticulata of the substantia nigra, characterized by some loss of myelinated fibers and neurons, with gliosis; (2) widely disseminated, rounded or oval nonnucleated structures ("spheroids") identifiable as swollen axons, especially numerous in the globus pallidus and pars reticulata; and (3) accumulations of pigment, much of it iron-containing, in the affected regions.
Our patient had all of the above criteria, except that the pars reticulata of the substantia nigra was normal. Our ultra-structural studies were unable to confirm the presence of axonal swellings; however, Vakili and associates,9 who first described the ultrastructural morphology of
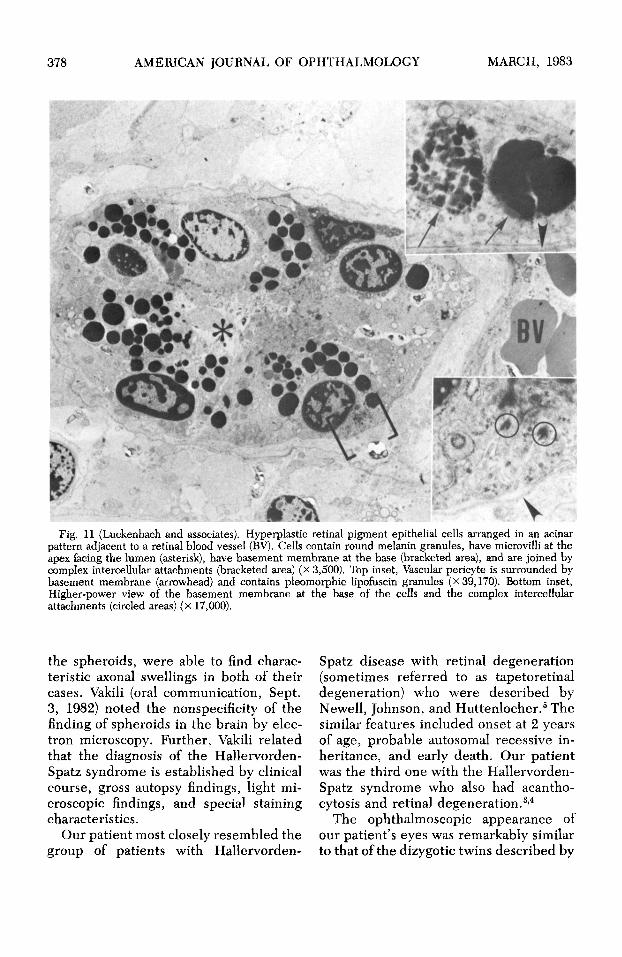
378 AMERICAN JOURNAL OF OPHTHALMOLOGY MARCH, 1983
Fig. 11 (Luckenbach and associates). Hyperplastic retinal pigment epithelial cells arranged in an acinar pattern adjacent to a retinal blood vessel (BY). Cells contain round melanin granules, have microvilli at the apex facing the lumen (asterisk), have basement membrane at the base (bracketed area), and are joined by complex intercellular attachments (bracketed area) (x 3,500). Top inset, Vascular pericyte is surrounded by basement membrane (arrowhead) and contains pleomorphic lipofuscin granules (x 39,170). Bottom inset, Higher-power view of the basement membrane at the base of the cells and the complex intercellular attachments (circled areas) (x 17,000).
the spheroids, were able to find characteristic axonal swellings in both of their cases. Vakili (oral communication, Sept. 3, 1982) noted the nonspecificity of the finding of spheroids in the brain by electron microscopy. Further, Vakili related that the diagnosis of the Hallervorden-Spatz syndrome is established by clinical course, gross autopsy findings, light microscopic findings, and special staining characteristics.
Our patient most closely resembled the group of patients with Hallervorden-
Spatz disease with retinal degeneration (sometimes referred to as tapetoretinal degeneration) who were described by Newell, Johnson, and Huttenlocher.5 The similar features included onset at 2 years of age, probable autosomal recessive inheritance, and early death. Our patient was the third one with the Hallervorden-Spatz syndrome who also had acantho-cytosis and retinal degeneration.3'4
The ophthalmoscopic appearance of our patient's eyes was remarkably similar to that of the dizygotic twins described by

VOL. 95, NO. 3 HALLERVORDEN-SPATZ SYNDROME 379
Fig. 12 (Luckenbach and associates). Conjunctival fibrocyte contains membrane-bound (arrows) lipofuscin granules. These granules are composed of a fine granular material, with interspersed dense homogeneous areas often located in the periphery (x 24,000). Insets, Higher-power views of the lipofuscin granules (x 50,000).
Newell, Johnson, and Huttenlocher.6 In our patient, the electroretinogram was already extinguished at the age of 2 years, but ophthalmoscopic examination at that time disclosed only pigmentary granularity and a decreased foveal reflex. When she was 5 years old, the retina was flecked, and when she was 8 years old, bull's-eye maculopathy was present. A mild to moderate amount of equatorial bone-spicule pigmentation did not become evident until she was 9Vs years old.
There has been one previous report3 of the light microscopic findings in the eyes of a patient with Hallervorden-Spatz syndrome with acanthocytosis and normal levels of betalipoproteins. Clinically, in that patient, there was thinning of the macula and a diffuse gray clouding of the paramacular areas. Bone-spicule pigmentation was noted in the equatorial retina,
but the optic disks and vessels were normal. On light microscopy, the photore-ceptor cells were absent and the outer nuclear and outer plexiform layers were quite attenuated or absent in most areas. The inner retinal layers were normal, as was the optic nerve. There were accumulations of pigment, both intracellu-larly and extracellularly, predominantly around equatorial blood vessels. The retinal pigment epithelium was described as irregular.
In our case, the light microscopic findings were similar in that only a few photoreceptor cell inner segments remained, the outer nuclear layer was markedly thinned, and there were peri-vascular pigment-laden cells along the retinal vessels. However, we noted much variation in the size of the retinal pigment epithelial cells. The enlarged cells had

380 AMERICAN JOURNAL OF OPHTHALMOLOGY MARCH, 1983
many large round pigment aggregates, but the smaller cells rarely had these aggregates. The retinal pigment epithelial cells were moderately well pigmented throughout. In addition to perivascular pigment-laden cells, we found clusters of such cells in the outer retinal layers—■ both around the macula and focally throughout the retina.
By electron microscopy, the macula, peripheral retinal pigment epithelium, and the pigment-laden macrophages found in the outer retinal layers contained similar, round single-membrane-bound aggregates, 3 to 4 μπι thick, composed of cigar-shaped melanin granules enmeshed in a membranous or granular lipofuscin background. Although many lipofuscin and melanin pigment granules were present, most were in melanolipo-fuscin complexes arranged singly or, more commonly, in aggregates. Similarly sized single-membrane-bound pigment aggregates have been described in two previous cases of retinitis pigmentosa.10,11
Kroll and Kuwabara10 found these aggregates in a patient with Leber's congenital amaurosis; however, the aggregates were found only in the foveal pigment epithelium, and were clusters of lipofuscin, not melanolipofuscin. In that patient, the pigment epithelial cells were more nearly uniform in size. Katz, Stone, and Dratz12 also noted lipofuscin-laden cells in the outer retinal layers by fluorescence. In a case of retinitis pigmentosa, Szamier and Benson11 found spherical aggregates of melanolipofuscin complexes that were similar to, but smaller than, the ones observed in our case. They observed large round granules in macular and peripheral retinal pigment epithelial cells, and pigment-laden macrophages in the outer retina. The melanolipofuscin content, however, greatly diminished outside of the macula, and the pigment epithelium was relatively uniform in shape.
In contrast to the pigment-laden cells
in the outer retinal layers, those cells arranged around vessels were not macrophages, but were identified as retinal pigment epithelial cells containing large spherical melanosomes, similar to those described in normal retinal pigment epithelium.13 Others11,14 have noted a similar tendency for perivascular migrations of retinal pigment epithelial cells to contain round melanin granules.
The ophthalmoscopic appearance of bone-spicule pigmentation clearly correlated with the perivascular proliferation of retinal pigment epithelial cells containing normal spherical melanin granules. The cause of the flecked-retina appearance and the bull's-eye maculopathy was not as obvious. There were no changes in the choriocapillaris, and, although the retinal pigment epithelial cells varied markedly in size, scanning electron microscopy did not disclose the distinctly shaped clusters of greatly enlarged cells found by Eagle and associates15 to explain the flecks observed in fundus flavi-maculatus.
The most likely explanation for the flecks and the bull's-eye maculopathy was the presence of clusters of pigment-laden macrophages containing round melanolipofuscin aggregates located in the outer retinal layers, both around the macula and throughout the retina focally. The melanolipofuscin pigment probably appears ophthalmoscopically as golden-yellow to white, resulting in the yellow-orange annulus around the macula, and also the yellow-white flecks. The enlarged retinal pigment epithelium containing many large melanolipofuscin aggregates may also play a part in the ophthalmoscopic appearance.
The pathogenesis of Hallervorden-Spatz syndrome is unknown, but a common feature appears to be an abnormal accumulation of lipofuscin in certain susceptible tissues. Several investigators9,16,17
have conducted histochemical and ultra-

VOL. 95, NO. 3 HALLERVORDEN-SPATZ SYNDROME 381
structural studies and have concluded that the pigment found in the basal ganglia is primarily lipofuscin and neuro-melanin. We found an abnormal accumulation of lipofuscin in conjunctival fibro-cytes and retinal pigment epithelial cells, and also in the retinal vessel pericytes and macrophages.
Lipofuscin or "age pigment" accumulates in the retinal pigment epithelium with age.18 Young19 and Young and Bok,20
using radioactive markers, established that rod outer-segment disks in frogs were shed continually, and that the retinal pigment epithelium phagocytosed them and later eliminated the material. In the process of metabolic degradation, these membranous disks become lipofuscin material. Feeney18 found that, with an increase in age, there is not only an increase in lipofuscin content in the retinal pigment epithelium, but also a tendency for melanin to combine with lipofuscin to become melanolipofuscin granules. She theorized that the phago-somes fuse with other lysosomes to become compound phagosomes, in an effort to hydrolyze the membranous material. The relative preservation of most of the melanin granules in the giant aggregates in our case may indicate (in these patients) a relative inability to hydrolyze the melanin and lipofuscin, despite many attempts by lysosomal fusion.
Although carotene and betalipoprotein levels were normal in our patient, test results suggested that she might have had some metabolic difficulty in handling fat-soluble vitamins. Vitamin E and vitamin A are both fat-soluble vitamins. Vitamin E has been shown to act as an important antioxidant and to protect against membrane damage. Experimental studies have shown that deficiencies of vitamin E or vitamin A, or both, in rats12,21,22 and monkeys23 result in loss of photoreceptor cells, damage to the outer nuclear layer, and accumulation of lipofuscin in the reti
nal pigment epithelium. A neurologic pattern, similar to that of infantile neuro-axonal dystrophy, with axonal spheroids occurs in vitamin E deficient rats.24
Low levels of vitamins A and E in the serum25 are a consistent feature of abeta-lipoproteinemia (Bassen-Kornzweig disease), as also are pigmentary retinopathy, acanthocytosis, fat malabsorption, and a progressive neurologic syndrome. The associated retinal degeneration has histo-pathologic features, observed by light microscopy, that are consistent with those found in advanced retinitis pigmentosa.26
Therapy with high dosages of vitamins A and E slows the progression of the associated neurologic diseases.25 If such therapy proves to be effective in abetalipo-proteinemia, it might also prove to be beneficial in the Hallervorden-Spatz syndrome.
There is no specific test that establishes the diagnosis of Hallervorden-Spatz disease before death. By electron microscopy, we found that peripheral nerves were normal, but a premortem conjunctival biopsy specimen showed only nonspecific changes characterized by an abnormal accumulation of lipofuscin in the fibro-cytes. Vakili and associates,9 however, demonstrated increased uptake of B9Fe in the region of the basal ganglia in two patients with the clinical diagnosis of Hallervorden-Spatz disease. Dooling, Richardson, and Davis27 noted the usefulness of measuring the ratio of the span of the frontal horns to the distance between the heads of the caudate nuclei (FH:CC ratio), and the ratio between the inter-caudate distance and the distance between the outer tables of the skull at the same level (CC:OTCC ratio). In a case of Hallervorden-Spatz syndrome, they found ratios of 1.2 and 0.29 respectively, which were most consistent with Hunt ingtons chorea, whereas we found ratios of 0.127 and 2.09, which were most consistent with cerebral atrophy. As the

382 AMERICAN JOURNAL OF OPHTHALMOLOGY MARCH, 1983
disease worsened, repeated computed to-mographic scans showed progressive changes in both patients.
REFERENCES 1. Hallervorden, J., and Spatz, H.: Eigenartige
Erkrankung im extrapyramidalen System mit besonderer Beteiligung des Globus pallidus unter der Substantia nigra. Z. Neurol. Psychiatr. 79:254, 1922.
2. Dooling, E. C , Schoene, W. C , and Richardson, E. P., Jr.: Hallervorden-Spatz syndrome. Arch. Neurol. 30:70, 1974.
3. Roth, A. M., Hepler, R. S., Mukoyama, M., Cancilla, P. A., and Foos, R. Y.: Pigmentary retinal dystrophy in Hallervorden-Spatz disease. Clini-copathological report of a case. Surv. Ophthalmol. 16:24, 1971.
4. Swisher, C. N., Menkes, J. H., Cancilla, P. A., and Dodge, P. R.: Coexistence of Hallervorden-Spatz disease with acanthocytosis. Trans. Am. Neurol. Assoc. 97:212, 1972.
5. Newell, F. W., Johnson, R. O., II, and Hut-tenlocher, P. R. : Pigmentary degeneration of the retina in the Hallervorden-Spatz syndrome. Am. J. Ophthalmol. 88:467, 1979.
6. Barr, A. N., Heinze, W. J., Dobbin, G. D., Valvassori, G. E., and Sugar, O.: Bicaudate index in computerized tomography of Huntington's disease and cerebral atrophy. Neurology 28:1196, 1978.
7. Berlinger, L. J. (ed.): Spin Labeling. Theory and Applications. New York, Academic Press, 1976, pp. 1-592.
8. Gaflhey, B. J., Drachman, D. B., Lin, D. C., and Tennekoon, G. : Spin-label studies of erythro-cytes in myotonic dystrophy. No increase in membrane fluidity. J. Neurol. 30:272, 1980.
9. Vakili, S., Drew, A. L., Von Schuching, S., Becker, D., and Zeman, W.: Hallervorden-Spatz syndrome. Arch. Neurol. 34:729, 1977.
10. Kroll, A. J., and Kuwabara, T.: Electron microscopy of a retinal abiotrophy. Arch. Ophthalmol. 71:683, 1964.
11. Szamier, R. B., and Benson, E. L.: Retinal ultrastructure in advanced retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 16:947, 1977.
12. Katz, M. L., Stone, W. L., and Dratz, E. A.: Fluorescent pigment accumulation in retinal pigment epithelium of antioxidant-deficient rats. Invest. Ophthalmol. Vis. Sci. 17:1049, 1978.
13. Kaczurowski, M. I.: The pigment epithelium of the human eye. Am. J. Ophthalmol. 53:79, 1962.
14. Kolb, H., and Gouras, P.: Electron microscopic observations of human retinitis pigmentosa, dominantly inherited. Invest. Ophthalmol. Vis. Sci. 13:487, 1974.
15. Eagle, R. C., Jr., Lucier, A. C., Bernardino, V. B., Jr., and YanofF, M.: Retinal pigment epithelial abnormalities in fundus flavimaculatus. Ophthalmology 87:1189, 1980.
16. Yanagisawa, N., Shiraki, H., Minakawa, M., and Narabayshi, H.: Clinico-pathological and histo-chemical studies of Hallervorden-Spatz disease with torsion dystonia with special reference to diagnostic criteria of the disease from the clinico-pathological viewpoint. Prog. Brain Res. 21:373, 1966.
17. Park, B. E., Netsky, M. G., and Betsill, W. L., Jr. : Pathogenesis of pigment and spheroid formation in Hallervorden-Spatz syndrome and related disorders. Neurology 25:1172, 1975.
18. Feeney, L,: Lipofuscin and melanin of human retinal pigment epithelium. Fluorescence, enzyme cytochemical, and ultrastructural studies. Invest. Ophthalmol. Vis. Sci. 17:583, 1978.
19. Young, R. W. : The renewal of photoreceptor cell outer segments. J. Cell Biol. 33:61, 1967.
20. Young, R. W., and Bok, D.: Participation of the retinal pigment epithelium in the rod outer segment renewal process. J. Cell Biol. 42:392, 1969.
21. Robison, W. G., Jr., Kuwabara, T., and Bieri, J. G.: Déficiences of vitamins E and A in the rat. Retinal damage and lipofuscin accumulation. Invest. Ophthalmol. Vis. Sci. 19:1030, 1980.
22. : Vitamin E deficiency and the retina. Photoreceptor and pigment epithelial changes. Invest. Ophthalmol. Vis. Sci. 18:683, 1979.
23. Hayes, K. C : Retinal degeneration in monkeys induced by deficiencies of vitamin E or A. Invest. Ophthalmol. 13:499, 1974.
24. Pentschew, A., and Schwartz, K.: Systemic axonal dystrophy in vitamin E deficient rats. Acta Neuropathol. 1:313, 1962.
25. Illingworth, D. R., Connor, W. E., and Miller, R. G.: Abetalipoproteinemia. Report of two cases and review of therapy. Arch. Neurol. 37:659, 1980.
26. von Sallmann, L. K., Gelderman, A. H., and Laster, L. : Ocular histopathologic changes in a case of a-beta-lipoproteinemia. Doc. Ophthalmol. 26:451, 1969.
27. Dooling, E. C , Richardson, E. P., Jr., and Davis, K. R. : Computed tomography in Hallervorden-Spatz disease. Neurology 30:1128, 1980.


















