
Novel derivatives of ansa-titanocenes procured from 6-phenylfulvene: a combined experimental and...
10
Novel derivatives of ansa-titanocenes procured from 6-phenylfulvene: a combined experimental and theoretical study Shona Fox a , John P. Dunne a,1 , Matthias Tacke a, * , John F. Gallagher b a Chemistry Department, Centre for Synthesis and Chemical Biology (CSCB), Conway Institute of Biomolecular and Biomedical Research, University College Dublin, Belfield, Dublin 4, Ireland b School of Chemical Sciences, Dublin City University, Glasnevin, Dublin 9, Ireland Received 25 July 2003; accepted 2 August 2003 Abstract The previously prepared trans-[(1,2-diphenyl-1,2-dicyclopentadienyl)ethanediyl] titanium(IV) dichloride, [1,2-(Ph) 2 C 2 H 2 {g 5 - C 5 H 4 } 2 ]Ti(Cl) 2 , was synthesised using an alternative procedure, from which its crystal structure was determined. Using this compound, a variety of other ansa-titanocene derivatives were synthesised by replacement of the chloride ligands with selected substituents. Thus RTi(X)(Y) systems where R ¼ 1; 2-(Ph) 2 C 2 H 2 g 5 -C 5 H 42 ; X ¼ Y ¼ CH 3 ; X ¼ CH 3 , Y ¼ Cl; X ¼ Y ¼ NCS; X ¼ Y ¼ NCO; X ¼ Y ¼ OPh and (X/Y) ¼ O have been synthesised and characterised. DFT calculations were performed on the complexes trans-[(1,2-diphenyl-1,2-dicyclopentadienyl)-ethanediyl] titanium(IV) dichloride, bis-(6,6-diphenylfulvene)titanium and bis-(6,6-diphenylfulvene)iron. This demonstrated the role that the metal centre plays in their formation, generating either an ansa-metallocene, a Ôtucked inÕ fulvene complex or a metallocene coordinating fulvene anions. Ó 2003 Elsevier B.V. All rights reserved. Keywords: Titanium; ansa-Metallocenes; X-ray crystal structure; Density functional theory 1. Introduction Since their discovery, ansa-metallocenes have played an increasingly important role in organometallic chem- istry, as it soon became evident that the presence of the bridging moiety introduces interesting modifications into the properties of the molecules [2,3]. An example of such a change can be seen in the tetramethylethylene chromocene carbonyl complex. In this case the cyclo- pentadienyl rings are tethered by a two-carbon bridge, which produces a stable complex, unlike its unbridged counterpart [4]. This rationale has also been applied to the titanocene derivatives (Cp 2 TiX 2 ) to improve upon existing and develop (these include their potential to act as anti-cancer drugs) new applications. But by far the most important application is their utilisation as cata- lysts in olefin polymerisations [5]. The placement of a bridging moiety on the sandwich structure, thereby generating ansa-titanocenes, enhances the activity of the catalyst and can also change the chemical and physical properties of the polymer [6,7]. The widespread success of this application has centred on the ansa-titanocenes ability to promote stereospecific polymerisation [8] when activated by a co-catalyst, e.g. MAO (methylaluminox- ane) [9]. Numerous methods exist to bring about the reductive dimerisation of fulvenes, which are versatile precursors for the synthesis of ansa-metallocenes. This can be achieved through the use of metal vapour synthesis Inorganica Chimica Acta 357 (2004) 225–234 www.elsevier.com/locate/ica * Corresponding author. Tel.: +35317168428; fax: +35317162127. E-mail address: [email protected] (M. Tacke). 1 Present address: Department of Chemistry, University of York, Heslington, York YO10 5DD, UK. 0020-1693/$ - see front matter Ó 2003 Elsevier B.V. All rights reserved. doi:10.1016/S0020-1693(03)00496-1
Transcript of Novel derivatives of ansa-titanocenes procured from 6-phenylfulvene: a combined experimental and...

Inorganica Chimica Acta 357 (2004) 225–234
www.elsevier.com/locate/ica
Novel derivatives of ansa-titanocenes procured from6-phenylfulvene: a combined experimental and theoretical study
Shona Fox a, John P. Dunne a,1, Matthias Tacke a,*, John F. Gallagher b
a Chemistry Department, Centre for Synthesis and Chemical Biology (CSCB),
Conway Institute of Biomolecular and Biomedical Research, University College Dublin, Belfield, Dublin 4, Irelandb School of Chemical Sciences, Dublin City University, Glasnevin, Dublin 9, Ireland
Received 25 July 2003; accepted 2 August 2003
Abstract
The previously prepared trans-[(1,2-diphenyl-1,2-dicyclopentadienyl)ethanediyl] titanium(IV) dichloride, [1,2-(Ph)2C2H2{g5-
C5H4}2]Ti(Cl)2, was synthesised using an alternative procedure, from which its crystal structure was determined. Using this
compound, a variety of other ansa-titanocene derivatives were synthesised by replacement of the chloride ligands with selected
substituents. Thus RTi(X)(Y) systems where R ¼ 1; 2-(Ph)2C2H2g5-C5H42; X¼Y¼CH3; X¼CH3, Y¼Cl; X¼Y¼NCS;
X¼Y¼NCO; X¼Y¼OPh and (X/Y)¼O have been synthesised and characterised. DFT calculations were performed on the
complexes trans-[(1,2-diphenyl-1,2-dicyclopentadienyl)-ethanediyl] titanium(IV) dichloride, bis-(6,6-diphenylfulvene)titanium and
bis-(6,6-diphenylfulvene)iron. This demonstrated the role that the metal centre plays in their formation, generating either an
ansa-metallocene, a �tucked in� fulvene complex or a metallocene coordinating fulvene anions.
� 2003 Elsevier B.V. All rights reserved.
Keywords: Titanium; ansa-Metallocenes; X-ray crystal structure; Density functional theory
1. Introduction
Since their discovery, ansa-metallocenes have played
an increasingly important role in organometallic chem-
istry, as it soon became evident that the presence of the
bridging moiety introduces interesting modificationsinto the properties of the molecules [2,3]. An example of
such a change can be seen in the tetramethylethylene
chromocene carbonyl complex. In this case the cyclo-
pentadienyl rings are tethered by a two-carbon bridge,
which produces a stable complex, unlike its unbridged
counterpart [4]. This rationale has also been applied to
the titanocene derivatives (Cp2TiX2) to improve upon
existing and develop (these include their potential to actas anti-cancer drugs) new applications. But by far the
* Corresponding author. Tel.: +35317168428; fax: +35317162127.
E-mail address: [email protected] (M. Tacke).1 Present address: Department of Chemistry, University of York,
Heslington, York YO10 5DD, UK.
0020-1693/$ - see front matter � 2003 Elsevier B.V. All rights reserved.
doi:10.1016/S0020-1693(03)00496-1
most important application is their utilisation as cata-
lysts in olefin polymerisations [5]. The placement of a
bridging moiety on the sandwich structure, thereby
generating ansa-titanocenes, enhances the activity of the
catalyst and can also change the chemical and physical
properties of the polymer [6,7]. The widespread successof this application has centred on the ansa-titanocenes
ability to promote stereospecific polymerisation [8] when
activated by a co-catalyst, e.g. MAO (methylaluminox-
ane) [9].
Numerous methods exist to bring about the reductive
dimerisation of fulvenes, which are versatile precursors
for the synthesis of ansa-metallocenes. This can be
achieved through the use of metal vapour synthesis

226 S. Fox et al. / Inorganica Chimica Acta 357 (2004) 225–234
techniques [10,11] or more conventionally by solution
methods such as; activated metal powders [12–14], low
valent transition metal complexes [15] and one-electron
reducing agents [4].
For the purposes of this study, we were interested in
preparing ansa-titanocene derivatives with a two-carbon
bridging unit, which distorts the cyclopentadienyl rings
out of the normal arrangement they adopt in the un-bridged analogue. The synthesis involved the reductive
dimerisation of our ligand of choice, 6-phenylfulvene
[16], with activated calcium producing an intermediate
compound, [1,2-(Ph)2C2H2{g5-C5H4}2]Ca(THF)2 (1)
[17] on which a transmetallation with 3TiCl3 �AlCl3yielded the desired complex, 2. This compound was then
used as the point of origin for the synthesis and spec-
troscopic characterisation of a variety of derivatives.Our choice of 6-phenylfulvene in these procedures was a
result of a theoretical study carried out on a series of
fulvenes using DFT methods [16]. This ascertained the
effect of varying the fulvene exocyclic substituents, on
the radical anion generated as an intermediate in these
reactions. This was further expanded here to include a
comparative study of the titanocene structures, 2 and 3,
prepared from two different fulvenes. A third calculationwas then carried out using complex 4 and this was to
study the effect of varying the metal atom on the
metallocene structure.
2. Experimental
2.1. Synthetic work – general
The preparation of 6-phenylfulvene [18] and 1 [17]
were carried out according to the literature procedures.
Solvents were purified and degassed by three-freezepump thaw cycle prior to use. Benzene-d6 was dried over
sodium and collected by vacuum transfer. Air- and
moisture-sensitive compounds were stored and handled
in an argon glove box and manipulations of these
substances were carried out using standard Schlenk
techniques.
NMR spectra were recorded on a VARIAN 300
MHz spectrometer and a JEOL GX 270 MHz spec-trometer. Chemical shifts (d) are reported in ppm and
are referenced to TMS. The mass spectra were measured
on a Finnigan MAT Incos 50 B mass spectrometer in EI
mode. X-ray data were collected on a Bruker P4
diffractometer.
2.2. Computational details
DFT calculations were performed using the GAUSS-GAUSS-
IANIAN 98 [19] programme implemented on an Origin 200
eight-processor cluster (SGI 180 MHz CPU/2 GB
RAM). These were carried out at the B3LYP level, using
the 6-31G** basis set for all atoms. A correction value,
namely the zero-point energy (ZPE), must be added on
to these values to give the overall energies (E0) of the
molecules.
2.3. Synthesis of [1,2-(Ph)2C2H2g5-C5H42]Ti(Cl)2 (2)
3TiCl3 �AlCl3 (0.65 g, 3.25 mmol) was added under
argon to a freshly prepared solution of ansa-calcocene
[17] (1.13 g, 3.25 mmol) in dry THF, which was cooled
to )50 �C. This was allowed to slowly warm to room
temperature and was stirred for ca. 10 h. The solutionwas filtered and the filtrate was cooled to )40 �C, whilemaintaining stirring. To this cooled solution, 6 M HCl
(4 ml) was added dropwise and stirring was continued
for an hour, during which it was gradually allowed to
warm to room temperature. Dichloromethane (50 ml)
was added and the solution was dried over MgSO4. This
was removed by filtration and the deep red solution was
held at )30 �C to afford 0.902 g of a red crystallineproduct (yield of 65%). 1H NMR (CDCl3, d): 4.83 (s,
2H, 2x PhCHCp), 6.17, 6.35, 6.86 (m, 6H, C5H4), 7.17–
7.26 (m, 2H, C5H4, 10H, C6H5).13C NMR (CDCl3, d):
54.1 (PhCHCp), 109.9, 117.2, 127.5, 133.9, 137.3
(C5H4), 126.5, 127.6, 128.8, 140.1 (C6H5). Principle IR
absorptions (KBr cm�1): 3076 (m), 3030 (m), 2925 (m),
2853 (m), 1586 (m), 1492 (m), 1454 (m), 1384 (m), 1261
(w), 1106 (s), 1042 (w), 823 (s), 736 (s), 698 (s). MS (EI):m/z 428 (C24H
4820 Ti37Cl35Cl, 9.5%), 426 (Mþ, 13.6%),
391 (Mþ–Cl, 5.2%), 355 (Mþ–2Cl, 1.0%), 272 (Mþ–fulv,100%), 237 (Mþ–fulv, –Cl, 16.9%), 200 (Mþ–fulv, –2Cl,36.9%), 154.1 (fulv, 26.3%), 153.1 (fulv–Hþ, 84.0%).
UV–Vis (CH2Cl2 nm) kmax 294, 382, 520.
2.4. Synthesis of [1,2-(Ph)2C2H2g5-C5H42]Ti(CH3)2(5)
CH3Li dissolved in diethylether (2.93 ml, 4.1 mmol)
was added to a solution of 2 (0.365 g, 0.85 mmol) in dry
toluene (30 ml). This was refluxed for 1 h to give a
musty-yellow solution. The solution was then filtered
and the toluene removed under reduced pressure to give
0.323 g (98% yield) of a bright yellow powder. 1H NMR
(CDCl3, d): 0.25 (s, 6H, 2x 2CH3), 3.64 (s, 2H, 2xPhCHCp), 5.21, 5.32, 6.85 (m, 6H, C5H4), 6.89–7.02 (m,
2H, C5H4, 10H, C6H5).13C NMR (CDCl3, d): 42.6 (2x
CH3), 52.5 (PhCHCp), 105.5, 111.0, 117.1, 121.3, 126.1

S. Fox et al. / Inorganica Chimica Acta 357 (2004) 225–234 227
(C5H4), 125.4, 126.8, 127.0, 140.9 (C6H5). Principle IR
absorptions (KBr cm�1): 3078 (m), 3062 (m), 3030 (m),
2879 (m), 2793 (w), 1598 (m), 1490 (m), 1425 (m), 1260
(s), 1073 (s), 1042 (s), 819 (s), 749 (m), 719 (s), 694 (s).
MS (EI): m/z 371 (Mþ–Me, 23.8%), 355 (Mþ–2Me,51.4%), 217 (Mþ–fulv, –Me, 1.1%), 200 (Mþ–fulv, –
2Me, 15.1%), 154 (fulv, 10.2%), 153 (fulv–Hþ, 15.3%).
UV–Vis (CH2Cl2 nm) kmax 297, 362, 514.
2.5. Synthesis of [1,2-(Ph)2C2H2g5-C5H42]Ti(CH3)Cl
(6)
A solution of 5 (0.323 g, 0.83 mmol) in toluene (30ml) was treated with Me3SiCl (0.14 ml, 1.12 mmol)
followed by H2O (7� 10�3 ml, 0.41 mmol). This was
stirred for 1 h at room temperature to produce an amber
coloured solution. The solvent was removed under re-
duced pressure and the residue was washed with pentane
to afford an amber-red powder (0.321 g, 95% yield). 1H
NMR (CDCl3, d): 0.29 (s, 3H, CH3), 4.23 (s, 2H, 2xPhCHCp), 5.67, 6.69 (m, 6H, C5H4), 6.96–7.03 (m, 2H,C5H4, 10H, C6H5).
13C NMR (CDCl3, d): 44.2 (CH3),
53.6 (PhCHCp), 107.9, 115.2, 124.8, 132.5, 135.2
(C5H4), 126.0, 127.0, 127.4, 139.5 (C6H5). Principle IR
absorptions (KBr cm�1): 3073 (m), 3028 (w), 2922 (m),
2850 (m), 1641 (m), 1490 (s), 1453 (s), 1383 (w), 1424
(m), 1261 (s), 1094 (s), 1042 (s), 820 (s), 710 (m). MS
(EI): m/z 391 (Mþ–Me, 4.5%), 355 (Mþ–Me, –Cl, 0.8%),
252 (Mþ–fulv, 0.7%), 237 (Mþ–fulv, –Me, 22.7%), 217(Mþ–fulv, –Cl, 1.7%), 200 (Mþ–fulv, –Me, –Cl, 76.2%),
154 (fulv, 29.5%), 153 (fulv–Hþ, 100.0%). UV–Vis
(CH2Cl2 nm) kmax 295, 376, 527.
2.6. Reactions of [1,2-(Ph)2C2H2g5-C5H42]Ti(CH3)Cl
(6)
(a) A solution of 6 (0.3 g, 0.7 mmol) in dry toluene (30ml) was freshly prepared. CH3Li dissolved in dieth-
ylether (1.2 ml, 1.68 mmol) was slowly added and
the solution was refluxed for 30 min to give a
musty-yellow solution. Filtration and removal of
the solvent yielded a bright yellow powder (0.26 g,
96%), which analysis proved to be 5.
(b) To a solution of 6 (0.3 g, 0.7 mmol) in toluene (30
ml) was added Me3SiCl (0.11 ml, 0.95 mmol) fol-lowed by H2O (6� 10�3 ml, 0.34 mmol). This was
stirred at room temperature for 1 h to give a bright
red solution. The work-up is the same as for 6,
which yielded a pale red powder (0.28 g, 97%). Anal-
ysis of this compound revealed it to be 2.
2.7. Synthesis of [1,2-(Ph)2C2H2g5-C5H42]Ti(NCS)2(7)
Complex 2 (0.124 g, 0.29 mmol) was dissolved in
THF (50 ml) and to this was added KSCN (0.062 g, 0.64
mmol). The mixture was refluxed for 2 h, filtered while
still hot and the solvent was removed in vacuo. This
gave 0.1311 g of a brick-red powder (96% yield). 1H
NMR (CDCl3, d): 4.96 (s, 2H, 2x PhCHCp), 6.17, 6.44,
6.62, 6.93 (m, 8H, C5H4), 7.17–7.25 (m, 10H, C6H5).13C
NMR (CDCl3, d): 54.5 (PhCHCp), 110.9, 117.2, 122.5,
129.4, 138.2 (C5H4), 127.6, 127.9, 128.9, 139.7 (C6H5),
122.3 (NCS). Principle IR absorptions (KBr cm�1) 3076
(w), 3028 (w), 2963 (m), 2059 (vs), 2009 (vs), 1637 (s),
1618 (s), 1490 (m), 1452 (m), 1261 (s), 1082 (s), 1042 (s),
823 (s), 801 (s), 711 (m), 694 (m). MS (EI): m/z 472 (Mþ,28.9%), 414 (Mþ–NCS, 41.2%), 355 (Mþ–(NCS)2,
6.2%), 318 (Mþ–fulv, 74.8%), 260 (Mþ–fulv, –NCS,42.8%), 200 (Mþ–fulv, –(NCS)2, 37.4%), 164 (Ti(NCS)2,
7%), 154 (fulv, 39.7%), 153 (fulv–Hþ, 100.0%). UV–Vis
(CH2Cl2 nm) kmax 283, 345, 430, 571.
2.8. Synthesis of [1,2-(Ph)2C2H2g5-C5H42]Ti(NCO)2(8)
A solution of 2 (0.16 g, 0.38 mmol) in THF (50 ml)was prepared and to it was added freshly ground KOCN
(0.067 g, 0.83 mmol). This was refluxed for 4 h and fil-
tered while hot. Removal of the solvent under reduced
pressure gave a red powder (0.138 g, 84). 1H NMR
(C6D6, d): 4.25 (s, 2H, 2x PhCHCp), 5.38, 5.67, 6.44,
6.70 (m, 8H, C5H4), 6.91–7.10 (m, 10H, C6H5).13C
NMR (C6D6, d): 53.8 (PhCHCp), 109.1, 116.5, 126.1,
133.8, 136.4 (C5H4), 127.3, 127.8, 128.6, 140.6 (C6H5),122.4 (NCO). Principle IR absorptions (KBr cm�1) 3080
(m), 3020 (m), 2920 (w), 2222 (vs), 2204 (vs), 1602 (m),
1489 (s), 1454 (m), 1234 (w), 1086 (s), 1044 (s), 956 (w),
814 (s), 766 (m). MS (EI): m/z 440 (Mþ, 1.4%), 398
(Mþ–NCO, 0.5%), 355 (Mþ–(NCO)2, 0.3%), 286 (Mþ–fulv, 16.9%), 244 (Mþ–fulv, –NCO, 2.5%), 200 (Mþ–fulv, –(NCO)2, 89.6%), 154 (fulv, 27.9%), 153 (fulv–Hþ,100.0%). UV–Vis (CH2Cl2 nm) kmax 297, 378, 511.
2.9. Synthesis of [1,2-(Ph)2C2H2g5-C5H42]Ti(OPh)2(9)
The dropwise addition of LiOPh to a solution of 2
(0.16 g, 0.38 mmol) in toluene (50 ml) was carried out at
)30 �C. (LiOPh was prepared by dissolving PhOH
(0.071 g, 0.75 mmol) in benzene (15 ml) and reacting itwith nBuLi 2.0 M in Pentane (0.375 ml, 0.75 mmol) at
)10 �C). The solution was gradually warmed to room
temperature and was stirred for 18 h. After filtration, the
solvent was removed in vacuo and the beige coloured
powder was washed twice with pentane (2� 20 ml). This
produced a pale yellow powder (0.148 g, 73% yield). 1H
NMR (C6D6, d): 4.55 (s, 2H, 2x PhCHCp), 5.77, 5.91,
6.14, 6.43 (m, 8H, C5H4), 6.90–7.12 (m, 16H, 10x C6H5;6x OC6H5), 7.31–7.38 (m, 4H, 4x OC6H5).
13C NMR
(C6D6, d): 52.9 (PhCHCp), 104.7, 111.8, 118.9, 128.2,
134.1 (C5H4), 125.8, 126.9, 127.3, 140.5 (C6H5), 116.9,

228 S. Fox et al. / Inorganica Chimica Acta 357 (2004) 225–234
118.1, 128.3, 169.4 (OC6H5). Principle IR absorptions
(KBr cm�1) 2950 (m), 2920 (w), 1586 (m), 1495 (m),
1480 (s), 1233 (w), 1213 (w), 1184 (w), 1095 (s), 1050 (w),
1014 (s), 944 (m), 868 (w), 809 (w). MS (EI): m/z 449
(Mþ–OPh, 2.9%), 388 (Mþ–fulv, 2.1%), 371 (Mþ–OPh,–Ph, 0.5%), 355 (Mþ–2OPh, 0.5%), 310 (Mþ–fulv, –Ph,1.2%), 293 (Mþ–fulv, –OPh, 3.5%), 234 [Ti(OPh)2,
2.7%], 215 (Mþ–fulv, –OPh, –Ph, 4.3%), 200 (Mþ–2OPh, 2.5%), 155 (fulv–Hþ, 100.0%), 154 (fulv, 30.4%),
153 (fulv–Hþ, 33.9%). UV–Vis (CH2Cl2 nm) kmax 286,
322, 500.
2.10. Synthesis of [1,2-(Ph)2C2H2g5-C5H42]Ti@O (10)
To a solution of 2 (0.32 g, 0.749 mmol) in THF (50
ml) was added KOH (0.11 g, 1.89 mmol). This was
treated with H2O (2 ml) and stirred for 1 h at room
temperature, to give a pale yellow solution. Following
filtration, the solvent was removed in vacuo and the
residue extracted into pentane. Removal of the pentane
gave a yellow powder (0.184 g, 66%). 1H NMR (CDCl3,d): 4.43 (s, 2H, 2x PhCHCp), 6.11, 6.19, 6.33, 6.44 (m,
8H, C5H4), 6.97–7.15 (m, 10H, C6H5). Principle IR ab-
sorptions (KBr cm�1) 3103 (m), 3062 (m), 2958 (m),
1602 (w), 1448 (m), 1442 (w), 1093 (s), 1051 (m), 1013
(s), 866 (w), 806 (s). MS (EI): m/z 372 (Mþ, 0.6%), 355
(Mþ–O, 3.1%), 310 (Mþ–Ti@O, 4.8%), 218 (Mþ–fulv,8.9%), 200 (Mþ–fulv, –O, 8.9%), 155 (fulv–Hþ, 100.0%),
154 (fulv, 33.5%), 153 (fulv–Hþ, 25.7%). UV–Vis(CH2Cl2 nm) kmax 352, 373, 505.
3. Results and discussion
3.1. Synthetic procedure for 2
The ansa-titanocene, 2, has previously preparedthrough the reduction of 6-phenylfulvene with
TiCl2 � 2THF in refluxing toluene [1]. This yielded a
mixture of cis and trans isomers in a 45:55 ratio. In an
attempt to improve the stereochemical control, we
sought an alternative route, namely the reductive di-
merisation of 6-phenylfulvene, with activated calcium at
0 �C to produce ansa-calcocene [17]. This is the most
important step in the whole process as it is here that thebridge formation occurs. There are a number of factors
Scheme 1. Synthetic p
that determine whether or not this happens, an impor-
tant factor being the type of fulvene used. This dictates
the degree to which the sole production of the bridge
occurs. Choosing a fulvene, that is easily reduced and
stable enough as a radical anion to undergo selectivedimerisation, gives the desired results. Much of the early
work in these ansa-metallocenes was carried out using
6,6-dimethylfulvene and regardless of whether the re-
action was carried out in solution [20] or using metal
vapour synthesis [10], an unbridged isopropyl impurity
was always present. Later it was revealed that, not only
was this side product present, but so too was an isop-
ropenyl metallocene [13]. From our own studies on thereducibility and selectivity of a series of fulvenes [16], we
also see the inability of this fulvene to give sole forma-
tion of the bridge and find that 6-phenylfulvene is our
ligand of choice.
Following the preparation of the ansa-calcocene, a
transmetallation was carried out at )50 �C with 3TiCl3 �AlCl3. Gradual warming to room temperature and
stirring for ca. 10 h gave a red–brown mixture, whichwas filtered to give a clear solution. An oxidation was
then carried out by first cooling the solution and then
adding the required volume of HCl. Stirring in air after
warming back to room temperature afforded the desired
product (Scheme 1). (The transmetallation can also be
carried out using TiCl4, giving the same quality of
product, but slightly lower yields, 59%.) This resulted in
an overall yield of 65% and a cis:trans ratio of 30:70.Due to the particular work-up of 2, only the trans iso-
mer is crystallised out of solution and this is evident
from both the 1H NMR and X-ray structure itself. The1H NMR exhibits three distinct multiplets, d 6.17, 6.35,
and 6.86, corresponding to three of the four-cyclopen-
tadienyl ring protons, the fourth overlaps with the
multiplet found between d 7.17 and 7.26 due to the
phenyl rings. If the cis isomer were present, additionalmultiplets would occur slightly downfield from these
values. It was upon this trans conformer that the deri-
vation reactions were carried out. Upon closer inspec-
tion of the results, we see that 2 crystallised in the trans
form with a R;R:S; S ratio of 30:70. However, the syn-
thetic route carried out by Eisch and co-workers [1],
where the MCl2 was reacted directly with 6-phenylful-
vene, produced a cis:trans ratio of 45:55 and was quotedas having high yields. Separation of these isomers was
possible using flash column chromatography.
rocedure for 2.
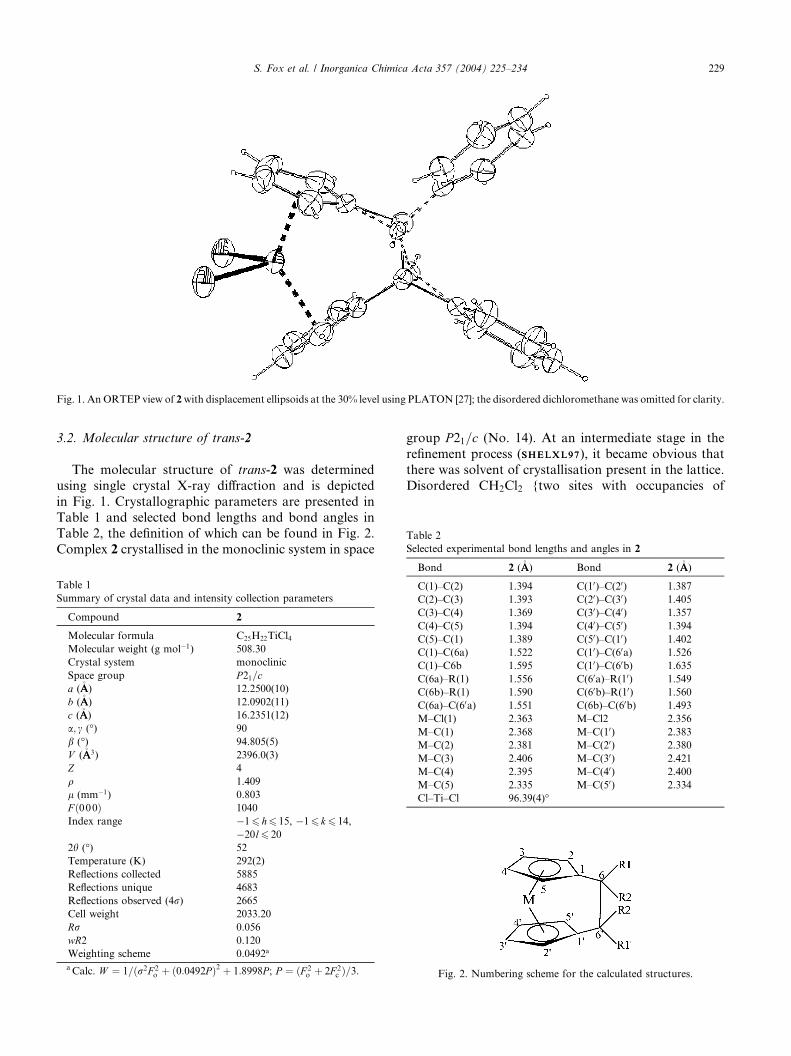
Fig. 1. An ORTEP view of 2with displacement ellipsoids at the 30% level using PLATON [27]; the disordered dichloromethane was omitted for clarity.
Table 2
Selected experimental bond lengths and angles in 2
S. Fox et al. / Inorganica Chimica Acta 357 (2004) 225–234 229
3.2. Molecular structure of trans-2
The molecular structure of trans-2 was determined
using single crystal X-ray diffraction and is depicted
in Fig. 1. Crystallographic parameters are presented in
Table 1 and selected bond lengths and bond angles inTable 2, the definition of which can be found in Fig. 2.
Complex 2 crystallised in the monoclinic system in space
Table 1
Summary of crystal data and intensity collection parameters
Compound 2
Molecular formula C25H22TiCl4Molecular weight (g mol�1) 508.30
Crystal system monoclinic
Space group P21=ca (�AA) 12.2500(10)
b (�AA) 12.0902(11)
c (�AA) 16.2351(12)
a; c (�) 90
b (�) 94.805(5)
V (�AA3) 2396.0(3)
Z 4
q 1.409
l (mm�1) 0.803
F ð000Þ 1040
Index range �16 h6 15, �16 k6 14,
�20l6 20
2h (�) 52
Temperature (K) 292(2)
Reflections collected 5885
Reflections unique 4683
Reflections observed (4r) 2665
Cell weight 2033.20
Rr 0.056
wR2 0.120
Weighting scheme 0.0492a
aCalc. W ¼ 1=ðr2F 2o þ ð0:0492P Þ2 þ 1:8998P ; P ¼ ðF 2
o þ 2F 2c Þ=3.
group P21=c (No. 14). At an intermediate stage in the
refinement process (SHELXL97SHELXL97), it became obvious that
there was solvent of crystallisation present in the lattice.
Disordered CH2Cl2 {two sites with occupancies of
Bond 2 (�AA) Bond 2 (�AA)
C(1)–C(2) 1.394 C(10)–C(20) 1.387
C(2)–C(3) 1.393 C(20)–C(30) 1.405
C(3)–C(4) 1.369 C(30)–C(40) 1.357
C(4)–C(5) 1.394 C(40)–C(50) 1.394
C(5)–C(1) 1.389 C(50)–C(10) 1.402
C(1)–C(6a) 1.522 C(10)–C(60a) 1.526
C(1)–C6b 1.595 C(10)–C(60b) 1.635
C(6a)–R(1) 1.556 C(60a)–R(10) 1.549
C(6b)–R(1) 1.590 C(60b)–R(10) 1.560
C(6a)–C(60a) 1.551 C(6b)–C(60b) 1.493
M–Cl(1) 2.363 M–Cl2 2.356
M–C(1) 2.368 M–C(10) 2.383
M–C(2) 2.381 M–C(20) 2.380
M–C(3) 2.406 M–C(30) 2.421
M–C(4) 2.395 M–C(40) 2.400
M–C(5) 2.335 M–C(50) 2.334
Cl–Ti–Cl 96.39(4)�
Fig. 2. Numbering scheme for the calculated structures.

230 S. Fox et al. / Inorganica Chimica Acta 357 (2004) 225–234
0.721(7) and 0.235(7)} was treated using soft DELU/
ISOR restraints for the anisotropic displacement pa-
rameters. Further disorder was deduced from the
bridging carbon atoms C(6)/C(60) over two sites with
occupancies of ca. 0.70:0.30 and refining to 0.708(15):0.292(15). This is not uncommon [21] in ethylene
bridged systems and in this structure is consistent with
SS (major) and RR (minor) configurations at the C(1)/
C(10) atom sites. The disorder is present due to the
fortuitous and close fit of the RR=SS systems into a
loosely packed crystal lattice.
The bridge bond lengths for the major/minor sites of
1.553(14)/1.49(3) �AA, are not significantly different, thehigh esd�s are due to the disorder and lower site occu-
pancy so that a discussion of the differences is not really
viable. The only distances that can be discussed com-
prehensively are those involving the Ti–Cl, Ti � � �Cgwhere Cg are the ring centroids and these will be dis-
cussed in more detail later on. No disorder model was
attempted for the cyclopentadienyl ring or phenyl ring
systems and the fit between the R;R and S; S is very goodfor these groups on analysis of Fourier maps through
the g5-C5 planes.
The phenyl groups adopt a trans arrangement, with
respect to one another and the metal is g5 coordinated
to the cyclopentadienyl rings (Fig. 1). However, the
structure does not adopt perfect C2 symmetry about the
Ti atom and the following torsion angles highlight this
deviation: Cl(1) � � �Ti � � �Cg(1) � � �C(11) is )123.5(3)�,Cl(1) � � �Ti � � �Cg(2) � � �C(21) is 130.9(3)�, Cl(2) � � �Ti� � �Cg(1) � � �C(11) is 134.3(3)� and finally Cl(2) � � �Ti� � �Cg(2) � � �C(21) is )126.6(3)� (Cg is a g5-C5 ring
centroid). The two Cg � � �Ti distances are 2.071(2) �AA for
Cg(1) and 2.064(2) �AA for Cg(2) and they intersect at an
Scheme 2. Synthesis of ansa-tit
angle (d) of 128.33(9)� at the Ti centre. The cyclopen-
tadienyl rings are oriented at an angle (a) of 54.75(15)�and the angle formed between the Cl(1)–Ti–C(l2) atoms
is 96.39(4)�. These results can be compared with an
unsubstituted ansa-titanocene, (CH2)2(C5H4)2TiCl2 [13].The Cg � � �Ti distances, 2.06 �AA, and resulting angle,
128.2�, are similar to 2 and the bridge length is 1.49 �AA. A
slight decrease is observed for the two angles, the ring
tilt angle (a), 51.8�, and the Cl(1)–Ti–Cl(2) angle, 94.8�.Crystallographic data have been deposited with the
Cambridge Crystallographic Data Centre as CCDC-
206414. Copies of this information may be obtained free
of charge from the Director, CCDC, 12 Union Road,Cambridge, CD21E2, UK (www.ccdc.cam.ac.uk, email:
3.3. Derivatives obtained from 2
The synthesis of 2 allows formation of a new and
broad range of derivatives through ligand exchange.
Compounds 5 to 10 are a direct result (Scheme 2).The dimethyl species, 5, was obtained in a high yield
of 98% by reacting MeLi with a solution of 2 in refluxing
toluene for an hour. A variety of colour changes oc-
curred during this period, from the original red to amber
to pale yellow and finally to a musty yellow, the product
was isolated as a bright yellow solid. The effect of the
ligand exchange can be seen in the 1H NMR for 5. Here
an upfield shift occurs with the bridging hydrogens oc-curring at d 3.64 (d 4.83 for 2) and the cyclopentadienyl
hydrogens at d 5.21, 5.32, and 6.85, with the fourth
being present within the phenyl hydrogens range of d6.89–7.02. This is a result of the increased shielding ex-
perienced by the protons due to the replacement of an
anocene derivatives 5–10.

S. Fox et al. / Inorganica Chimica Acta 357 (2004) 225–234 231
electron-withdrawing group with an electron releasing
one.
Derivatisation of compound 5 yielded the interme-
diary compound 6, whereby one of the methyl groups
was replaced with a chloride. This fact was demon-strated effectively in the 1H NMR, where its multiplets
lie midway between those of 2 and 5. Synthesis was
achieved by treating a solution of 5 with Me3SiCl, fol-
lowed by H2O and stirring for an hour, an amber so-
lution being the end result. In order to purify the
compound, the solvent was removed and the residue
washed twice with pentane to give an amber-red powder
(95% yield). This ligand arrangement has also beenproposed to occur during the course of olefin poly-
merisations, where a titanocene dichloride is used as a
catalyst in conjunction with methylaluminoxane
(MAO). The MAO is thought to displace one of the
chloride groups with a methyl group, which is then
followed by the removal of the final chloride to produce
an active site, from where chain propagation can occur.
From 6 it is possible to synthesise 5 or 2, simply bychoosing the required reagents. Treating 6 with MeLi,
removes the chloride and replaces it with another methyl
group. Alternatively, reacting this titanocene with
Me3SiCl, followed by an addition of H2O, displaces the
methyl group for a chloride to yield 2. A source of error
can also occur in the synthesis of 6. Adding an excess of
Me3SiCl to the original reaction with 5 yields 2 as the
final product, 6 only occurs as an intermediate, which isthen consumed to yield the dichloride derivative.
A further derivative, 7, was synthesised by refluxing a
solution of 2 in THF with KSCN for 2 h and filtering the
hot solution removed any unreactedKSCN. The addition
ofNCS groups causes slightlymore proton deshielding to
occur with respect to 2. Cyclopentadienyl hydrogens are
found in the following positions: d 6.17, 6.44, 6.62, and
6.93, in this case each ring proton is represented by adistinct multiplet. The assertion that these groups are in
fact isothiocyanate and not thiocyanates arises from
spectroscopic analysis. The carbon of the NCS group
occurs at d 122.3 in the 13C NMR, which is within the
expected isothiocyanate range and downfield from where
thiocyanates occur (d 95–115). Furthermore, two broad
and very intense absorptions occur in the IR spectrum of
7, at 2059 and 2009 cm�1 typical of isothiocyanates (range2140–1990 cm�1). Titanium bound sulphur would give
absorption bands between 2175 and 2160 cm�1.
Synthesis of 8 is similar to that already discussed for
7, with KNCO being utilised and resulting in a red
powder (84% yield). For an isocyanate bound ligand the
carbon signal was located at d 122.4, within the expected
isocyanate range. However, from this, it cannot be de-
finitively stated that bonding occurs through the nitro-gen, as this value is borderline with the cyanate range of
d 105–120. From IR data, the N-bonding is obvious, as
two intense absorptions occur at 2222 and 2204 cm�1.
Formation of the phenolate derivative, 9, was carried
out by reacting a freshly prepared solution of LiOPh
with 2 at )30 �C. This yielded a yellow/brown solution,
which after 18 h was filtered to give a beige powder.
Spectroscopic analysis confirmed this to be 9. The in-fluence of the phenoxy groups can be seen to contribute
to the deshielding of the complex, although not to the
same extent as for 2. Also peaks are visible for all the
cyclopentadienyl H atoms at d 5.77, 5.91, 6.14, and 6.43,
respectively. It is also evident in the 13C NMR that some
degree of deshielding is occurring causing a more upfield
position from those of 2, but not to the same degree as
seen in 5.The final derivative prepared was that of 10. Treat-
ment of a solution of 2 with KOH, followed by H2O
yielded a pale yellow solution, which upon workup gave
the desired compound. Unfortunately spectroscopic
analysis was more limited in this case, as due to poor
intensity levels, no 13C NMR could be determined.
However in the 1H NMR, it is apparent that the protons
are being deshielded to a greater extent than in 9 due tothe oxy group, with peaks at d 6.11, 6.19, 6.33, and 6.44.
3.4. Theoretical study on 2, 3, and 4
Density functional theory calculations were carried
out at the B3LYP level and using 6-31G�� as the basis set,results from which can be found in Table 3. The first of
these calculations was performed on compound 2 itselfand a direct comparison can be made between these re-
sults and those from X-ray diffraction. Both the experi-
mental and calculated structures show a typical bent
ansa-metallocene geometry. However, the C–C andM–C
bond length ranges are comparable, with the estimated
carbon ring bond lengths from 1.404 to 1.427�AA and Ti–C
distances of 2.369–2.454�AA. The ipso carbons are closer to
Ti, 2.369 and 2.416 �AA, whereas the other carbons havelonger Ti–C, e.g. 2.454 and 2.438 �AA. The length of the
two-carbon bridge spanning the cyclopentadienyl rings
was found to be 1.563 �AA. It has been shown that the size
of a two-carbon bridge [22] is such that its presence on a
metallocene causes a distortion away from its �natural�arrangement. In the case of titanocenes, which have a
bent structure, the bridging unit amplifies the distortion
causing the Cg � � �Ti � � �Cg angle to decrease from 131.0�[23] in the unbridged metallocene to 128.3� in 2. However
despite the fact that the complex is being pulled from its
preferred configuration, no additional strain is imposed
on the bridge. Additional strain is on the other hand
generated due to the presence of the two-phenyl groups,
with the result that the carbon–carbon single bond
lengthens, to relieve this steric strain. The calculated
bridge length of 1.563�AAdemonstrates this effect. This toohas also been seen with a variety of transition metal met-
allocenes. An analogous coboticenium species, trans-[(1,
2-diphenyl-1,2-dicyclopentadienyl) ethanediyl]–cobalt(III)

Table 3
Selected calculated bond lengths for compounds 2, 3 and 4 using the B3LYP/6-31G** level of theory
Bond Bond length (�AA) (2) Bond (�AA) (3) Bond length (�AA) (4)
M–C(1) 2.369 2.149 2.213
M–C(2) 2.438 2.290 2.061
M–C(3) 2.454 2.450 2.039
M–C(4) 2.416 2.419 2.043
M–C(5) 2.419 2.253 2.055
M–C(6) n/a 2.385 3.348
M–C(10) 2.416 2.149 2.213
M–C(20) 2.454 2.290 2.055
M–C(30) 2.438 2.450 2.043
M–C(40) 2.369 2.419 2.039
M–C(50) 2.419 2.253 2.061
M–C(60) n/a 2.385 3.348
C1–C(2) 1.427 1.448 1.458
C2–C(3) 1.422 1.410 1.418
C3–C(4) 1.404 1.414 1.427
C4–C(5) 1.424 1.417 1.418
C5–C(1) 1.416 1.449 1.461
C1–C(6) 1.514 1.454 1.411
C6–C(60) 1.563 n/a n/a
C6–C(Ph(1)) 1.525 1.512 1.478
C6–C(Ph(2)) n/a 1.503 1.470
C(10)–C(20) 1.416 1.448 1.461
C(20)–C(30) 1.424 1.410 1.418
C(30)–C(40) 1.404 1.414 1.427
C(40)–C(50) 1.422 1.417 1.418
C(50)–C(10) 1.427 1.449 1.458
C(10)–C(60) 1.514 1.454 1.411
C(6)–C(Ph(1))0 n/a 1.512 1.470
C(6)–C(Ph(2))0 1.525 1.503 1.478
232 S. Fox et al. / Inorganica Chimica Acta 357 (2004) 225–234
hexafluorophosphate has a slightly shorter C–C bond
length than 2, of 1.540 �AA [12] while the C–C bond length
in 1,10-tetramethylethyleneferrocene has a longer bridg-ing C–C bond of 1.584 �AA [24]. This is attributed to the
increased steric crowding caused by replacing the phenyl
and hydrogen groups with methyl groups.
Varying the metal component and fulvenyl exocyclic
substituents can have an even more dramatic effect on
structure, i.e., the ansa-metallocene structure is not the
only possible outcome, two further bonding modes
could also form instead. Formation of complex 2(bonding mode a) involves the reduction of 6-phenyl-
fulvene to generate its corresponding radical anion. This
intermediate is stable enough, due to the presence of the
phenyl group, to undergo dimerisation and exclusively
yield 2, the structure of which is depicted in Scheme 5.
Increasing the steric load on the exocyclic carbon to the
extent that bridge formation is suppressed and using an
electron deficient metal, results in �tucked in� or fulvenecomplexes (bonding mode b) shown in Scheme 4. An
example of this is the 16VE titanocene derivative 3 [25].
Replacing the Ti in this complex with Fe gives yet an-
other type of structure (Scheme 3). In this case, the
presence of iron eliminates the possibility for a �tuckedin� configuration due to its higher electron count. In-
stead the fulvene ligands are reduced and complexed by
a M2þ metal centre, forming an intermediate species,
which can either dimerise or undergo hydride extraction
from the solvent. Support for this postulate was ob-
tained following the elucidation of the structure of 4, anintermediate formed during the reaction of iron atoms
with 6,6-diphenylfulvene [26]. Initial formation of this
species is thought to involve the complexation of neutral
fulvenes with an iron centre whose oxidation state is
zero (bonding mode c). Reduction of each of these li-
gands takes place as a result of electron transfer from
the metal centre. This species, then undergoes hydrogen
abstraction giving the unbridged metallocene, a routethat minimises the steric clashes between the bulky
phenyl groups (Scheme 3).
From the crystal structure of 4, the question arises as
to whether this complex exists in a singlet or triplet state.
DFT calculations obtained using B3LYP/6-31g**
showed a minimum in the singlet state. Using the X-ray
crystal structure of 4 as a starting geometry, the triplet
species failed to optimise, thereby showing that it is notan electronic state on the potential energy surface for
this system. This indicates that in 4 the radicals at the
C(6) and C(60) positions are paired, but are unable to
form a C–C bond for steric reasons.
The effect of these structural details can also be seen
in the calculated reaction enthalpies for the formation of
2, 3, and 4. The reaction between titanium atoms and
6,6-diphenylfulvene results in the most exothermic

Scheme 4. Reaction of titanium atoms with 6,6-diphenylfulvene to yield 3, whose structure depicts bonding mode b.
Scheme 5. Reaction of TiCl2 with 6-phenylfulvene to yield 2.
Scheme 6. Reaction of iron atoms with 6,6-diphenylfulvene to yield 4.
Scheme 3. Reaction mechanism for the synthesis of metallocenes, showing the role of the bis-fulvene iron complex 4.
S. Fox et al. / Inorganica Chimica Acta 357 (2004) 225–234 233
value, )136.8 kcal/mol, out of the three complexes
(Scheme 4). The reaction enthalpy for 2 was also cal-
culated, this time from the reaction of unsolvated TiCl2with 6-phenylfulvene (Scheme 5). A lower value of
)114.4 kcal/mol was obtained. By replacing the Ti
centre with an Fe one, the reaction enthalpy for the
formation of 4 becomes )79.5 kcal/mol (Scheme 6). This
is significantly lower than the previous enthalpy of for-mation, a difference attributable to their structural
variations. Experimental and theoretical results support
the postulate that 3 adopts the �tucked in� structure.
Both of these data sources are comparable, with the
theoretical range for the cyclopentadienyl carbon–
carbon bonds being 1.410–1.449 �AA and the experimental
range being 1.377–1.455 �AA. The metal to ring carbon
range was calculated to be 2.149–2.450 �AA, while struc-
turally this is 2.121–2.400 �AA. But the most significant
bond length in this complex is that between the titanium
centre and the exocyclic carbon, 2.385 �AA theoretically,2.347 �AA experimentally. It is the presence and strength
of this bond in 3 that results in the greater exothermic
output during its formation. However, formation of an

234 S. Fox et al. / Inorganica Chimica Acta 357 (2004) 225–234
equivalent bond in 4 is not possible, due to the increased
distance between the metal centre and the exocyclic
carbon. Theoretically, this was found to be 3.484 �AA, 0.06�AA greater than the crystallographically determined va-
lue. As for the rest of the structural arrangement in 4,again theoretical and experimental results are in good
agreement. The calculated ring carbon–carbon bonds
occupy a range of 1.418–1.458 �AA with the measured
range being 1.406–1.452 �AA. From these values, it can be
noted that the cyclopentadienyl rings have increased
aromatic character. The metal–carbon distances, theo-
retically and experimentally are found to be 2.039–2.213�AA and 2.035–2.209 �AA, respectively. Elongation has alsooccurred in the exocyclic bond, causing increased single
bond character, theoretically this was found to be 1.411�AA and experimentally to be 1.408 �AA. It is clear from
these results that the true structural nature of 4 is that of
a ferrocene derivative, in which the Fe(II) centre coor-
dinates to two fulvene anions. The complex is also found
to be in a singlet state, which shows that the spins of the
two-fulvene radical anions are paired.
4. Conclusion
A new synthetic route to the previously isolated
ansa-metallocene, trans-[(1,2-diphenyl-1,2-dicyclopenta-
dienyl)ethanediyl] titanium(IV) dichloride (2) was carried
out, in good yield. This involved the reductive dimerisa-tion of 6-phenylfulvene with activated calcium powder,
followed by transmetallation using 3TiCl3 � � �AlCl3. This
compound was then used to synthesise a variety of other
ansa-titanocene compounds, 5–10, in high yield through
ligand replacement. As the ansa-metallocene framework
remained untouched, the influence of these ligands on the
titanocene itself, could be ascertained spectroscopically.
The reaction of 6,6-diphenylfulvene with Ti atoms wascalculated using B3LYP/6-31G** and the reaction en-
thalpy for the formation of this �tucked in� titanocenedichloride, 3, was )136.8 kcal/mol. However the same
reaction involving Fe atoms had a lower reaction en-
thalpy of )79.5 kcal/mol and resulted in the formation of
a singlet ferrocene, 4, in which coordination to fulvene
anions occurred. The formation of a C2 bridged ferrocene
was not possible due to the steric load of two phenylsubstituents on each of the fulvene systems. The calcu-
lated reaction enthalpy for the reaction of TiCl2 with 6-
phenylfulvene to yield 2, an example of the third type of
structural geometry available, was found to be )114.4kcal/mol. Structural comparisons were also carried out
between each of these calculated complexes, giving fur-
ther verification of their differing geometric arrangement.
Acknowledgements
This work was supported by the Centre for High-
Performance Computing Applications at University
College Dublin, Ireland. M.T. and S.F. also wish tothank Enterprise Ireland for financial support (Grant
SC/1997/570).
References
[1] J.J. Eisch, X. Shi, F.A. Owuor, Organometallics 17 (1998) 5219.
[2] H.L. Lentzner, W.E. Watts, Tetrahedron 27 (1971) 4343.
[3] R.W. Heo, T.R. Lee, J. Organomet. Chem. 578 (1999) 31.
[4] H.H. Brintzinger, D. Fischer, R. Mulhaupt, B. Rieger, R.M.
Waymouth, Angew. Chem., Int. Ed. Engl. 34 (1995) 1143.
[5] S.R. Hitchcock, J.J. Situ, J.A. Covel, M.M. Olmstead, M.H.
Nantz, Organometallics 14 (1995) 3732.
[6] F.R.W.P. Wild, L. Zsolnai, G. Huttner, H.H. Brintzinger, J.
Organomet. Chem. 232 (1982) 233.
[7] H.G. Alt, J. Chem. Soc., Dalton Trans. (1999) 1703.
[8] J.A. Ewen, R.L. Jones, A. Razavi, J.D. Ferrara, J. Am. Chem.
Soc. 110 (1988) 6255.
[9] J.A. Ewen, J. Am. Chem. Soc. 106 (1984) 6355.
[10] T.S. Tan, J.L. Fletcher, M.J. McGlinchey, J. Chem. Soc., Chem.
Commun. (1975) 771.
[11] M. Tacke, Organometallics 13 (1994) 4124.
[12] S. Fox, J.P. Dunne, M. Tacke, D. Schmitz, R. Dronskowski, Eur.
J. Inorg. Chem. (2002) 3039.
[13] P.J. Sinnema, P.J. Shapiro, B. Hohn, T.E. Bitterwolf, B. Twamley,
Organometallics 20 (2001) 2883.
[14] (a) M. Rieckhoff, U. Pieper, D. Stalke, F.T. Edelmann, Angew.
Chem., Int. Ed. Engl. 32 (1993) 1079;
(b) A. Recknagel, F.T. Edelmann, Angew. Chem., Int. Ed. Engl.
30 (1991) 693;
(c) I.L. Fedushkin, S. Dechert, H. Schumann, Angew. Chem., Int.
Ed. Engl. 40 (2001) 561.
[15] J.J. Eisch, X. Shi, F.A. Owuor, Organometallics 17 (1998) 5219.
[16] M. Tacke, S. Fox, L. Cuffe, J.P. Dunne, F. Hartl, T. Mahabier-
sing, J. Mol. Struct. 559 (2001) 331.
[17] K.M. Kane, P.J. Shapiro, A. Vij, R. Cubbon, A.L. Rheingold,
Organometallics 16 (1997) 4571.
[18] K.J. Stone, R.D. Little, J. Org. Chem. 49 (1984) 1849.
[19] M.J. Frisch, et al., GAUSSIANGAUSSIAN 98, Revision A.7, Gaussian Inc.,
Pittsburgh, PA, 1998.
[20] K.L. Rinehart, A.K. Frerichs, P.A. Kittle, L.F. Westman, D.H.
Gustafson, R.L. Pruett, J.E. McMahon, J. Am. Chem. Soc. 82
(1960) 1111.
[21] M. Horacek, P. Stepnicka, R. Gyepes, I. Cisarova, I. Tislerova, J.
Zemanek, J. Kubista, K. Mach, Chem. Eur. J. 13 (2000) 2397.
[22] P.J. Shapiro, Eur. J. Inorg. Chem. (2001) 321.
[23] J.C. Green, Chem. Soc. Rev. 27 (1998) 263.
[24] M.B. Laing, K.N. Trueblood, Acta Cryst. 19 (1965) 373.
[25] J.A. Bandy, V.S.B. Mtetwa, K. Prout, J.C. Green, C.E. Davies,
M.L.H. Green, N.J. Hazel, A. Izquierdo, J.J. Martin-Polo, J.
Chem. Soc., Dalton. Trans. (1985) 2037.
[26] R. Teuber, G. Linti, M. Tacke, J. Organomet. Chem. (1997) 545,
105.
[27] A.L. Spek, PLATON, University of Utrecht, The Netherlands.


















