
Nonoxidative Activation of Methane - Texas A&M … files/crse-45-151-03...temperature and...
53
Nonoxidative Activation of Methane Tushar V. Choudhary, # Erhan Aksoylu, and D. Wayne Goodman * Department of Chemistry, Texas A&M University, College Station, Texas, USA ABSTRACT ...................................................... 152 I. INTRODUCTION ................................................. 152 II. FUNDAMENTAL SURFACE SCIENCE STUDIES ................... 154 A. Tungsten Surfaces ............................................. 155 B. Ni Surfaces ................................................... 155 C. Platinum and Palladium Surfaces ................................ 158 D. Electron and Photo-Induced Dissociation of Methane on Pt(111) . . . 159 E. Rh and Ir Surfaces ............................................. 160 F. Ru Surfaces ................................................... 160 III. LOW TEMPERATURE COUPLING OF METHANE ................. 161 IV. PYROLYSIS AND HIGH-TEMPERATURE COUPLING OF METHANE ....................................................... 166 A. Methane Pyrolysis and Dehydrogenative Coupling of Methane in Homogeneous Systems ...................................... 167 B. Catalytic Pyrolysis and Nonoxidative Coupling of Methane at High-Temperatures ............................................ 170 1. Carbonaceous, Oxide, and Metal Catalysts ................... 170 2. Zeolite-Supported Catalysts ................................. 171 V. PRODUCTION OF HYDROGEN ................................... 175 151 DOI: 10.1081/CR-120017010 0161-4940 (Print); 1520-5703 (Online) Copyright q 2003 by Marcel Dekker, Inc. www.dekker.com # Current address: Phillips Petroleum Company, Bartlesville, OK 74004, USA. * Correspondence: D. Wayne Goodman, Department of Chemistry, Texas A&M University, College Station, Texas, USA; Fax: (979) 845-6822; E-mail: [email protected]. CATALYSIS REVIEWS Vol. 45, No. 1, pp. 151–203, 2003 MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016 ©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
Transcript of Nonoxidative Activation of Methane - Texas A&M … files/crse-45-151-03...temperature and...

Nonoxidative Activation of Methane
Tushar V. Choudhary,# Erhan Aksoylu, and D. Wayne Goodman*
Department of Chemistry, Texas A&M University, College Station, Texas, USA
ABSTRACT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
I. INTRODUCTION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
II. FUNDAMENTAL SURFACE SCIENCE STUDIES . . . . . . . . . . . . . . . . . . . 154
A. Tungsten Surfaces . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 155
B. Ni Surfaces . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 155
C. Platinum and Palladium Surfaces . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 158
D. Electron and Photo-Induced Dissociation of Methane on Pt(111) . . . 159
E. Rh and Ir Surfaces . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 160
F. Ru Surfaces . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 160
III. LOW TEMPERATURE COUPLING OF METHANE . . . . . . . . . . . . . . . . . 161
IV. PYROLYSIS AND HIGH-TEMPERATURE COUPLING OF
METHANE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
A. Methane Pyrolysis and Dehydrogenative Coupling of Methane
in Homogeneous Systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 167
B. Catalytic Pyrolysis and Nonoxidative Coupling of Methane at
High-Temperatures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 170
1. Carbonaceous, Oxide, and Metal Catalysts . . . . . . . . . . . . . . . . . . . 170
2. Zeolite-Supported Catalysts . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
V. PRODUCTION OF HYDROGEN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 175
151
DOI: 10.1081/CR-120017010 0161-4940 (Print); 1520-5703 (Online)
Copyright q 2003 by Marcel Dekker, Inc. www.dekker.com
#Current address: Phillips Petroleum Company, Bartlesville, OK 74004, USA.*Correspondence: D. Wayne Goodman, Department of Chemistry, Texas A&M University, College
Station, Texas, USA; Fax: (979) 845-6822; E-mail: [email protected].
CATALYSIS REVIEWSVol. 45, No. 1, pp. 151–203, 2003
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

VI. FORMATION OF CARBON FILAMENTS . . . . . . . . . . . . . . . . . . . . . . . . . . 179
VII. SYNTHESIS OF DIAMOND FILMS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 183
VIII. CONCLUDING REMARKS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 184
ACKNOWLEDGMENTS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 185
REFERENCES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 185
ABSTRACT
Effective utilization of methane remains one of the long-standing problems in catalysis.
Over the past several years, various routes, both direct and indirect, have been
considered for the conversion of methane to value-added products such as higher
hydrocarbons and oxygenates. This review will focus on the range of issues dealing with
thermal and catalytic decomposition of methane that have been addressed in the last few
years. Surface science studies (molecular beam methods and elevated-pressure reaction
studies) involving methane activation on model catalyst systems are extensively
reviewed. These studies have contributed significantly to our understanding of the
fundamental dynamics of methane decomposition. Various aspects of the nonoxidative
methane to higher hydrocarbon conversion processes such as high-temperature coupling
and two-step low-temperature methane homologation have been discussed.
Decomposition of methane results in the production of COx-free hydrogen (which is
of great interest to state-of-the-art low-temperature fuel cells) and various types of
carbon (filamentous carbon, carbon black, diamond films, etc.) depending on the
reaction conditions employed; these issues will be briefly addressed in this review.
Key Words: Surface science; Model catalysts; Carbon filaments; Methane coupling;
Aromatization; Diamond films; Natural gas; Hydrogen production.
I. INTRODUCTION
Over the past several years, considerable time and effort has been invested in the study
of methane activation.[1 – 16] The main goal of these efforts has been to find effective
processes to develop the technology for the efficient and environmentally benign utilization
of natural gas. Activation of methane represents an intensely challenging problem due to its
refractory nature. A plethora of investigations have addressed the issue of methane
conversion over the past three decades and a number of excellent reviews can be found in
the literature[17 – 24] Conversion of methane to hydrocarbons can be effected via an indirect
or a direct route as observed in Fig. 1. The indirect route involves the production of
hydrocarbons via intermediates formed from the reaction of methane with steam, oxygen,
HCl, etc.,[17] whereas the direct route involves coupling of methane in the presence of
oxygen (oxidative coupling of methane) or nonoxidative coupling (high-temperature
coupling and low-temperature two-step methane homologation). Methane can also be
converted to carbon and hydrogen via catalytic/homogeneous decomposition and the COx-
free hydrogen so produced is further utilized in current low-temperature fuel cells.
Choudhary, Aksoylu, and Goodman152
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

Steam reforming of methane, currently the major route to methane conversion, can be
represented as:
CH4 þ H2O ! CO þ 3H2
This highly endothermic process results in the formation of synthesis gas that can be
further processed into methanol and ammonia. The methanol to gasoline (MTG) process
can then be utilized for the production of gasoline.[25] Alternatively, synthesis gas can be
directly processed into hydrocarbons via the Fischer–Tropsch process.[26] Steam
reforming of methane has been extensively reviewed[27] and will not be a focus of this
review.
Hydrocarbon production via synthesis gas is expensive and rather circuitous,
therefore there was considerable excitement in the scientific world when Keller and
Bhasin[28] first reported the direct conversion of methane into ethylene in 1982. Following
this pioneering work, oxidative coupling of methane (OCM) became one of the most
pursued topics of research in methane activation. Detailed reviews of the process can be
found in the literature.[29 – 31] Usually OCM is carried out between 900 and 1200K under
atmospheric pressures. Basic oxides promoted with alkali metal salts and/or alkaline earth
metal salts are important OCM catalysts.[30,32] The reaction results in C2 hydrocarbon
yields of less than 25%–30% because the selectivity of the desired C2 products is
hampered by the undesired COx species formed due to the presence of oxygen in the feed.
Enhancement of the C2 yield can be obtained by separating the desired products from the
reactants by avoiding further oxidation of these products. Aris and co-workers utilized a
simulated counter-current moving-bed chromatographic reactor to obtain significantly
higher yields of C2 hydrocarbons,[33] whereas Vayenas and co-workers employed a gas
recycled electro-catalytic reactor-separator to obtain a 85% yield of ethylene.[34]
However, economic considerations prohibit the large-scale implementation of such
Figure 1. Simplistic schematic representation of different methods of activating methane.
Nonoxidative Activation of Methane 153
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

reactors. Choudhary et al.,[35] have suggested distribution of oxygen feed for increasing
both the conversion and selectivity and also reducing the hazardous nature of the OCM
process. The large input of work (catalyst synthesis and reactor design) into OCM has
significantly improved the process over the years[36 – 41] but inspite of the improvements, it
is currently far from being commercially viable. Catalytic CO2 reforming of methane to
syngas in the presence of steam and/or oxygen has also been extensively investigated in
the last few years.[42 – 44]
The thermal and catalytic decomposition of methane has been extensively studied for
fundamental and applied reasons. In this review we will focus on the range of issues that
have been addressed in the last few years related to methane activation. Numerous articles
have dealt with methane decomposition on single crystal surfaces, studies that have
provided important, fundamental insights into the dynamics of methane decomposition.
These studies will be comprehensively covered in this review. Issues related to low-
temperature and high-temperature nonoxidative coupling of methane will be addressed.
Decomposition of methane to produce hydrogen has been proposed as an economical
process to produce hydrogen.[45,46] Various studies related to the process will be reviewed.
We would also like to dwell on the various applications such as formation of pyrolytic
carbon, diamond films, and carbon-nanofibers/filaments arising from decomposition of
methane. This review will not address issues dealing with the reaction of methane with
oxygen, chlorine, steam, ammonia, etc.
II. FUNDAMENTAL SURFACE SCIENCE STUDIES
The last three decades have seen the development of an array of surface analytical
techniques that allows surfaces to be defined at the atomic level. Ultra-high vacuum
(UHV) surface science studies have played a key role in relating the atomic structure,
composition, and electronic properties of surfaces to catalytic activity and selectivity.[47 –
49] The limitation of the “pressure gap” has been overcome by coupling an apparatus for
measurement of reaction kinetics at elevated pressures with a contiguous UHV chamber
for surface analysis.[50 – 53] This approach has facilitated a direct comparison of reaction
rates measured on single crystal surfaces with those measured on supported high-surface
area catalysts. Moreover, it has allowed detailed studies addressing reaction mechanisms,
activity relationships, the effect of promoters and inhibitors on catalytic activity, and in
certain cases, the identification of reaction intermediates by post-reaction surface
analysis.[54,55]
Methane dissociation has attracted attention in recent years due to its industrial
relevance. It is considered to be the rate-determining step for the steam reforming of
natural gas and this has led to considerable interest in investigating the reactive sticking
process of methane on metal single crystals.[56] Methane chemisorption has been studied
on a variety of transition metal surfaces by employing both “molecular beams” as well as
“bulb techniques.” These studies have shown that dissociation of methane on transition
metals proceeds via either a direct dissociative mechanism (DDM) or a precursor/trapping
mediated mechanism (PMM). In DDM, dissociation takes place on impact with the
surface. Initial adsorption probabilities for DDM do not strongly depend upon surface
temperature and the initial reaction probability is expected to increase with the increase in
Choudhary, Aksoylu, and Goodman154
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

incident kinetic energy. For PMM, a molecular precursor/trapped state forms that is
accommodated to the surface temperature. This trapped precursor can then either desorb
from the surface or undergo dissociative chemisorption. This leads to a strong dependence
of the initial adsorption probabilities on the surface temperature. Contrary to DDM, for
PMM the initial reaction probability is expected to decrease with an increase in the
incident kinetic energy. There has been considerable debate in the literature over the
controversial issue of the mechanism of methane dissociation over transition metal
surfaces. The studies undertaken by various groups on different metal surfaces are briefly
summarized below.
A. Tungsten Surfaces
Winters investigated the dissociative chemisorption of CH4 and its various deuterated
forms (CD3H, CD4, CDH3, and CD2H2) on tungsten wires. Methane dissociation results
showed two temperature regimes of activated adsorption behavior exhibiting activation
energies of 11.3 kJ/mol ð600K , T , 1000KÞ and 42.6 kJ/mol
ð1200K , T , 2600KÞ:[57] Interestingly, he observed an absence of temperature
dependence on the ratio of sticking probability (S0) of one methane isotope to another.[58]
Based on experimental results and model calculations, Winters proposed a vibrationally
activated tunneling mechanism for the dissociation of CH4 on tungsten.
Rettner et al. were the first to employ molecular beam techniques for investigating the
dissociative chemisorption of methane.[59] Molecular beam studies permit considerable
control with respect to reactant parameters such as translational energy (Etrans) and
incidence angle. Their studies showed a dramatic increase (,105) in sticking probability
of CH4 on W(110) for a 100 kJ/mol increase in translational energy. A translationally
activated tunneling model was proposed to explain their results. In contrast to these
studies, investigations undertaken by Lo and Ehrlich[60] on the CH4(CD4)/W(211) surface
led them to believe that a tunneling mechanism was not consistent for the dissociative
chemisorption of methane, instead they advocated a mechanism involving vibrational
excitation without the influence of tunneling.
B. Ni Surfaces
Since steam reforming catalysts are Ni-based, considerable attention has been given
to the interaction of methane with Ni surfaces. Bootsma and coworkers[61] showed that
there was no methane dissociation on Ni(110) at room temperature without the aid of
electron sources. The authors observed a sticking coefficient in the order of 1028 at
temperatures between 473 and 579K. Two possible kinetic decomposition processes
involving activation energies of 88 and 130 kJ mol21 were proposed; however, the authors
did not distinguish between the two due to lack of sufficient accuracy in their kinetic
results. Subsequently, they also investigated the sticking probabilities of methane with the
other low-index Ni crystal phases at temperatures between 523 and 618K for methane
pressures of ca. 1022 Torr.[62] While a sticking probability of 5 £ 1029 was observed for
Nonoxidative Activation of Methane 155
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

Ni(100), there was no reaction on the Ni(111) surface (within detection limits) indicating
an initial sticking coefficient of # 1 £ 10210:Ceyer and coworkers employed molecular beam experiments concomitant with high-
resolution electron-energy spectroscopy (HREELS) to investigate the interaction of
methane with Ni(111) surface.[63] The molecular beam of methane was produced by high-
pressure, adiabatic expansion of a CH4 diluted in He mixture. The dissociation probability
showed an exponential increase by two orders of magnitude on increasing the translational
energy (Etrans) from 50 to 71 kJ mol21 at a constant surface temperature of 475K. Studies
using HREELS indicated the presence of methyl (CH3) species on the surface after the
methane chemisorption experiments. Interestingly, the authors observed that the surface
temperature (Ts) had no influence on the dependence of the sticking probability with
translational energy. Experiments with CD4 showed a strong kinetic isotopic effect. Based
on their results the following was concluded:
(i) Methane dissociation proceeded via DDM.
(ii) Vibrational energy (Evib) and (Etrans) were both effective in promoting methane
dissociation.
(iii) Increase in Evib/Etrans promoted the deformation of the molecule such that the C
atom was effectively brought closer to the Ni surface. Moreover, it was not
essential to completely deform the molecule as the light H-atom could tunnel
through the barrier once it was sufficiently narrow.
Beebe, et al.[64] employed bulb experiments to thoroughly investigate methane
interaction kinetics with the low-index planes of Ni surfaces. These experiments were
carried out under high-incident methane flux (1 Torr) conditions as compared to molecular
beam studies. Time-dependent carbon buildup experiments at 450K showed that the
methane reactivity increased in the order Nið111Þ , Nið100Þ , Nið110Þ: Table 1
summarizes the activation energy and sticking probability results for the systems
investigated. The difference in sticking probabilities for the different low-index planes
clearly illustrates the structure sensitivity of the methane dissociation process on Ni. The
activation energies for CH4/Ni(111) and CH4/Ni(110) systems were quite similar, but the
activation energy for CH4/Ni(100) was smaller by a factor of 2. A large kinetic effect for
CH4 vs. CD4 was observed for Ni(100), whereas no such effect was observed for Ni(110)
surface. The rates measured in this work were about a magnitude higher than the adjusted
rates for catalytic methane steam reforming, rationalized by the fact that in these studies,
initial rates were measured on a clean surface. Based on this, the authors claimed that
Table 1. Summary of activation energies and initial
methane sticking co-efficients at 500K.[64]
Surface
Activation energy
CH4 (CD4), kJ mol21Initial sticking
co-efficient (CH4)
Ni(111) 52.7 (—) 1 £ 1028
Ni(100) 26.8 (62.3) 6 £ 1028
Ni(110) 55.6 (52.3) 1 £ 1027
Choudhary, Aksoylu, and Goodman156
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

the rates measured in their work effectively represented the upper limit for the unpromoted
steam reforming catalyst. As a part of this work the authors also provided a comparison of
this study with other studies performed on similar systems. Bootsma and coworkers
reported much lower sticking coefficients and an absence of dependence of initial sticking
coefficient on surface temperature.[61,62] This was in contrast to work by Beebe et al. who
reported a temperature dependence for both Ni(111) surface and Ni(100) surface and
higher sticking coefficients. This deviation was attributed to the presence of an
equilibrated gas above the Ni single crystals (at high flux such as 1 Torr) in the case of
Beebe et al. as opposed to the nonequilibrated behavior expected at pressures of 1023 to
1022 Torr used by Bootsma et al.[62] Though there was an excellent agreement for the
CH4/Ni(111) system studied by Beebe et al. (bulb method: high flux experiments) and
Ceyer and coworkers [molecular beam technique[63]], the results involving bulb
experiments for the CH4/Ni(100)[64] system differed appreciably from the molecular beam
experiments performed by Hamza and Madix on the same system.[65] It is also interesting
to note that Hamza and Madix reported a linear dependence of the sticking coefficient with
Etrans and much larger sticking coefficients (2 to 3 order magnitude) at a given incident
energy for the CH4/Ni(100) system as compared to the exponential dependence seen by
Ceyer and coworkers for the CH4/Ni(111) system.[63] Chorkendorff et al. employed x-ray
photo-electron spectroscopy (XPS) to study the CH4/Ni(100) system[66] and reported an
activation energy which was twice that reported by Beebe et al. on the same system.[64]
Bulb experiments performed by Yates and coworkers[67] showed that the dissociative
chemisorption of CH4 on Ni(111) proceeded via DDM at pressures relevant to catalytic
processes. At 1 Torr CH4 pressure and surface temperature of 600K, each methane
molecule collision was found to result in the deposition of 4 £ 1028 C atoms.
Beckerle et al.,[68,69] showed that methane dissociation could also be affected by
collision of inert gas atoms (Ne, Ar, and Kr) with CH4 physisorbed on Ni(111) surface
(surface temperature ðTsÞ ¼ 46KÞ: They proposed a hammer mechanism, which involved
impulsive transfer of some kinetic energy of the inert gas atom (Ar or Ne) to the
physisorbed CH4, followed by dissociative chemisorption of methane upon its collision
with the Ni surface to yield an adsorbed methyl group. Subsequently, the same group
reported the transformation of methane to benzene under UHV conditions.[70] The methyl
radicals formed from by the hammer method (as described previously) dissociated into CH
species at higher surface temperature and finally formed benzene via an ethylene
intermediate.
Jiang and Goodman[71] investigated the effect of sulfur poisoning on the
chemisorption of methane on Ni(100) surface. The influence of sulfur on the initial
methane decomposition activity is summarized in Table 2. The authors proposed a “simple
site-blocking mechanism” to explain their experimental results and the fit to a simple first-
order Langmurian form suggested that each sulfur atom was responsible for blocking three
“dissociation sites.” Campbell et al.,[72] reported that methane dissociation on Ni(100)
proceeded via PMM. The reactivity of methane was found to be lower on NiO thin film
[compared to Ni(100)] and exhibited an apparent activation energy of ca. 37 kJ/mol.
Employing both molecular beam experiments and bulb experiments, Chorkendorff and
coworkers[73,74] observed that the CH4 chemisorption on Ni(100) proceeded via DDM,
which was in contradiction to the results by Campbell et al.[72] In one of their previous
studies on the CH4/Ni(100) system, they had observed an apparent activation energy of
Nonoxidative Activation of Methane 157
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

52 kJ/mol[66] which was less than the value (ca. 59 kJ/mol) they reported in their later
work.[74] They attributed this to the use of a thermal finger in their later work, which
ensured that the methane was completely equilibrated to the crystal.
Recently, Juurlink et al.,[75] combined the use of laser excitation and molecular beam
experiments to probe the CH4/Ni(100) system. Enhancement in dissociative chemisorp-
tion of methane by singular excitation of v3 (antisymmetric stretching vibration) was
observed for all the translational energies employed in the study. Translational energy was
found to be more effective in enhancing the sticking probability than the vibrational
excitation of v3.
C. Platinum and Palladium Surfaces
Employing molecular beam methods on the CH4/Pt(111) system, Luntz and Bethune
found that the sticking probability (S0) did not show a perfect exponential dependence on
the translational energy (Etrans).[76] This behavior was contrary to that observed on other
systems[59,63] where S0 was found to increase exponentially with Etrans. Moreover, a
modest dependence of S0 with surface temperature (Ts) was observed. To explain the
dependence of S0 on all three parameters viz. Etrans, Evib, and Ts a model invoking
thermally assisted tunneling was proposed.[77] The coupling of a tunneling barrier to the
lattice could successfully explain the Ts dependence of S0, which was important since a
simple DDM involving tunneling could not account for the Ts dependence of S0. A second
study related to the same system by Valden et al.,[78] was in good agreement with the work
by Luntz and Bethune.[76] The presence of pre-adsorbed oxygen was found to be
detrimental to CZH activation on Pt(111). It was believed that the mechanism for
dissociation of methane on the clean and oxygen-precovered Pt(111) surface was similar.
Chesters and coworkers studied methane dissociation on both Pt(111) and Pt0.25Rh0.75
alloy and found that the methane sticking behavior was similar for both substrates.[79]
Klier and coworkers investigated methane dissociation on Pd(679) single crystal
employing bulb techniques.[80] The apparent activation energy for the process was
estimated to be ca. 44.7 kJ/mole. No CH4 dissociation occurred below a threshold pressure
Table 2. Methane decomposition rate on Ni(100) shown at different
sulfur coverages and temperatures.[71]
Sulfur coverage
(ML)
Temperature
(K)
Initial methane decomposition rate
(molecules site21 sec21)
0.008 550 0.013
600 0.019
0.04 550 0.011
600 0.017
0.1 600 0.014
0.2 550 0.003
0.28 550 0.002
600 0.003
Choudhary, Aksoylu, and Goodman158
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

of 0.2 Torr at 500K, based on which the authors suggested DDM as the route for
dissociative chemisorption of methane. The dissociation of methane resulted in formation
of carbon in form of disordered fractional monolayers at 400–500K and disordered
multilayer clusters at temperatures greater than 500K.
Based on molecular beam experiments, Valden et al. concluded that dissociative
chemisorption of methane on Pd(110) proceeded via DDM.[81] The linear decrease in S0
with increasing preadsorbed oxygen coverage was similar to that observed in case of Pt
(111).[78] The first-order Langmuir kinetics provided a good fit for their experimental data
and indicated that each oxygen atom was responsible for poisoning ca. 2 dissociation sites
for methane.[82]
Recently Walker and King carried out a comprehensive molecular beam study on the
CH4/Pt(110) system.[83] A striking feature of this work was the decrease in S0 until
Etrans ¼ 100 meV: Thereafter, a sharp increase in S0 was observed with increasing Etrans.
The authors felt that the anomalous behavior of S0 at low Etrans did not involve a precursor
state (as suggested in [84]) but was in fact related to dynamic steering in a direct
dissociation process. Thus, supposedly there were two different potential energy pathways
operating at low and high Etrans. The surface temperature was found to promote the direct
dissociation processes in good agreement with previous work.[73,76,85]
D. Electron and Photo-Induced Dissociation of Methane on Pt(111)
Vacuum-ultraviolet (VUV) photons are required for dissociation of gas-phase
methane as it is transparent in the visible to ultraviolet region. But use of VUV photons for
this purpose is not pragmatic, as it is expensive and arduous to obtain from conventional
sources of light. Gruzdov et al. overcame this problem by photo-dissociating methane on
Pt(111).[86] Methane physisorbed at 40K on Pt(111) was shown to undergo dissociation
when irradiated with 193 nm photons. Adsorbate photo-dissociation on metal surfaces is
postulated to proceed[87] either via direct excitation of adsorbed molecules by photons or
the transfer of an electron to the low-lying negative state of the adsorbate initiated by
substrate excitation. The authors favored the latter mechanism to explain the photo-
induced dissociation of methane on Pt(111). To acquire further rinformation, Yoshinobu
and coworkers studied the same system using infrared reflection absorption spectroscopy
(IRAS).[88] Employing elegantly designed experiments, they showed that distortion of
methane symmetry from tetrahedral (Td) to C3v was essential for its photo-dissociation.
Only a part of the first layer [on Pt(111)] was deformed to C3v symmetry and, hence,
photo-dissociated. The reaction became self-limiting as some of the reaction products
rendered the other methane molecules inactive by modifying the surface.
White and coworkers investigated the electro-induced dissociative chemisorption of
CH4 on Pt(111).[89] Irradiation of physisorbed CH4 on Pt(111), a ðTs ¼ 55KÞ with
electrons resulted in the selective production of CH3 and H species. The authors felt that
the selective formation of CH3 species was due to the fact that dissociation of CH4 on
surfaces was restricted to fewer number of pathways as quenching of the excited CH4 state
by the substrate played an important role (unlike that for gas-phase dissociation). It was
believed that the dissociation chemistry was initiated by impact excitation of the
physisorbed methane.
Nonoxidative Activation of Methane 159
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

E. Rh and Ir Surfaces
Brass and Ehrlich[90] proposed that activation energy measurements could help in
providing an understanding about the dissociation mechanism. Domination by
translational activation would result in single-activation energy for measurements on an
isothermal gas and surface as well as for a hot gas incident on a cold surface. But
involvement of internal degrees of freedom in the activation process would result in two
different activation energies for the two cases. For the CH4/Rh film system, they observed
an activation energy of 20.9 kJ/mol for isothermal gas and surface conditions.[91,92] The
activation energy was found to be much higher ca. 46 kJ/mol when the measurements were
carried out under nonisothermal conditions (i.e., hot gas striking the Rh film held at 273K).
The difference between the two activation energies was found to equal the estimated
desorption energy of molecular methane on the same surface. Based on these results the
authors claimed that the activation of methane on Rh films was dominated by vibrational
excitation of the gas.[93]
For the CH4/Ir(110) system, Seets et al.[84] observed an initial decrease (in the low-
energy region) in methane dissociation probability with increasing Etrans (up to a certain
value of Etrans). This was then followed by an increase in methane reactivity with
increasing Etrans. These results were interpreted in terms of PMM and DDM domination at
low and high Etrans, respectively. The authors found a good correspondence between their
molecular beam results and bulb experiments. Based on their results it was proposed that,
while DDM dominated at high gas temperatures (industrial relevant conditions), PMM
dominated at low gas temperatures. A similar set of studies were also carried out on the
Ir(111) surface.[94] The two surfaces were found to have a similar qualitative behavior.
Methane was found to be more reactive on the Ir(110) surface as compared to Ir(111)
surface. The experimental results indicated that PMM was more important for Ir(111)
surface than for the Ir(110) surface. In a separate study, Weinberg and coworkers
investigated the CH4(CD4)/Ir(111) system employing bulb methods.[95] PMM was
observed to dominate at low pressures (below 1023 Torr), whereas both pathways (PMM
and DDM) were believed to be active when the translational energy was increased by
diluting methane in argon at a total pressure of 1 Torr.
F. Ru Surfaces
Interference of the substrate signal precludes the effective use of Auger electron
spectroscopy to detect fractional monolayers of carbon on the Ru surface.[96] To
circumvent this, Wu and Goodman employed the d (CH) intensity obtained from HREELS
to estimate the sticking probability of methane on Ru(0001).[97] The estimated sticking
coefficients varied from 1026 to 1027 in the temperature range 500–650K. The apparent
activation energy for the methane dissociation on Ru(0001) was estimated to be
35.5 kJ/mol. Molecular beam studies on the same system provided excellent agreement for
the activation energy measurements (37 kJ/mol) and good agreement for the sticking
probabilities.[98]
An exhaustive study was undertaken in our laboratory to investigate the different
surface intermediates formed during methane chemisorption on Ru surfaces. Temperature
Choudhary, Aksoylu, and Goodman160
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

programmed desorption (TPD) and HREELS[99] were employed for identifying the
hydrocarbonaceous species formed on Ru(0001) and Ru(1120) surfaces. Methane
dissociation on Ru(1120) resulted in the formation of three different hydrocarbonaceous
species, namely methylidyne (CH), vinylidene (CCH2), and ethylidyne (CCH3) species. In
contrast, methane decomposition on Ru(0001) led to the formation of only CH and CCH2
species. For both substrates, only the graphitic phase was observed above 700K.
Additional information on the graphitic carbon (inactive form) was obtained by utilizing
scanning tunneling microscopy (STM).[100] On the Ru(0001) surface ðTs ¼ 800KÞ; the
carbon was present as small clusters (attached to the step edges of the surface) having a
diameter of 1.0–1.5 nm and apparent heights of 0.2–0.3 nm. The hexagonal super-
structure of graphite monolayers was observed at 1300K on the Ru(0001) surface, showing
a sharp corresponding LEED ð11 £ 11Þ pattern. In contrast, methane decomposition on
Ru(1120) surface produced three-dimensional particles of graphite. This difference in
growth patterns of carbon atoms was attributed to the structural differences between the
two substrates.
In order to confirm that similar surface intermediates are formed during methane
decomposition on supported real-world Ru catalysts, neutron vibrational spectroscopy
(NVS) studies were undertaken.[101] Neutron vibrational spectroscopy has several
advantages over conventional vibrational techniques in that it allows accurate qualitative,
as well as quantitative, analysis of surface hydrocarbonaceous species on supported metal
catalysts at ambient to high pressures. Furthermore, NVS is not limited by selection rules
and therefore in principle allows detection of all vibrational modes of the adsorbed
species. It is noteworthy that these studies showed the presence of similar surface species
(methylidyne, vinylidene, and ethylidyne) on the Ru/Al2O3 catalyst as those observed on
the model Ru catalysts after methane chemisorption. Moreover a similar trend was also
observed for the stability of the various surface intermediates on the Ru surfaces.
Since most of the surface science studies have focussed primarily on understanding
the fundamental methane chemisorption process, an extensive program was undertaken in
our laboratory to take these studies a step further, i.e., to investigate the transformation of
the surface species formed during methane chemisorption and relate them to the
transformation kinetics. These studies were undertaken to provide mechanistic
information about the low-temperature coupling of methane on Ru surfaces and are
described in the next section.
III. LOW TEMPERATURE COUPLING OF METHANE
The nonoxidative low-temperature homologation of methane has been proposed as an
alternative to OCM in recent years. Two research groups independently introduced the
process,[102,103] which involves methane decomposition in the first step followed by
hydrogenation of the surface carbonaceous species in the second step to obtain C2þ
hydrocarbons. Thermodynamics prohibits the direct formation of ethane from methane.
To circumvent this thermodynamic limitation, Van Santen and coworkers operated the
process at two different temperatures.[104] Dissociative adsorption of methane was carried
out on silica-supported transition metal catalysts (Ru, Rh, and Co) at 700K, followed by a
hydrogenation step at a lower temperature (ca. 373K). Temperature-programmed surface
Nonoxidative Activation of Methane 161
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

reaction of the carbonaceous species (after the chemisorption step) revealed three different
carbonaceous species on the surface (similar to the work by McCarty and Wise[105]). The
highly active carbidic phase Ca was held primarily responsible for the formation of higher
hydrocarbons. Cg, which gas graphitic in nature, was highly unreactive and had a
proclivity for methane formation at higher temperatures, whereas the Cb (amorphous
carbon) had intermediate reactivity-producing predominantly methane with very small
amounts of C2þ hydrocarbons. The hydrogenation products obeyed the Flory–Schulz–
Anderson distribution and a maximum yield of 13% for C2þ hydrocarbons was obtained at
an optimum surface coverage of 0.18 on the Ru/SiO2 catalyst.
Amariglio and coworkers carried out the two-step process under isothermal
conditions.[103] In the first step, pure methane (flow-rate: 400 ml min21) was passed over
100 mg of the EUROPT-1 catalystat 523K. Ethane was produced along with hydrogen but
was approximately an order of magnitude less than the latter. Hydrogenation (hydrogen
flow: 50 ml min21) of the adsorbed species at 523K in the second step resulted in the
formation of saturated hydrocarbons ranging from C1–C7; 19.3% of the adsorbed methane
was converted to C2þ hydrocarbons. Though the C2þ yield increased to ca. 29% on
carrying out the process at 473K, the relative amount of higher hydrocarbons produced
was lower due to the decrease in the amount of methane adsorbed in the chemisorption
step. Comparison studies of Co, Ru, and Pt catalysts for the homologation of methane
indicated that Ru was the most active catalyst.[106] The temperature at which maximum
hydrocarbon production took place for Ru, Pt, and Co was 433, 523, and 548K with
corresponding C2þ yields of 36.9%, 19.3%, and 7.5%, respectively. For Co and Ru the
product distribution shifted towards higher hydrocarbons with decrease in temperature,
whereas the reverse trend was observed for Pt.[107,108] This temperature effect was
attributed to differences in the hydrogenolysis activities; Pt being far less effective than
Ru. The C2þ yield was significantly improved for the EUROPT-1 catalyst by operating the
process in a batch reactor and employing Pd catalyst as hydrogen trap.[109,110] A 16-min
exposure to methane at 523K followed by hydrogenation at the same temperature gave
C2þ yield of ,42.5% with respect to the consumed methane. The total amount of methane
homologated for a 16-min exposure was 24.6m-moles, yet 9.34m-moles for a 1-min
exposure, indicating a weak influence of the exposure time on the amount of homologated
methane. The thermodynamic limitation was overcome in the isothermal process by
removing hydrogen from the surface in the first step at low pressures (high flow-rates of
methane were used) and then supplying it in the hydrogenation step at 1 bar.[111]
Though the basic idea behind both the dual temperature (DT) and isothermal
temperature (IT) homologation is the same, striking differences are apparent between the
two. A greater variety of C2þ hydrocarbons was observed in the isothermal process.[111]
The maximum yield for hydrocarbons in a DT process on Ru-based catalyst passed
through a maximum with respect to carbon surface coverage.[104] In contrast, the IT
process showed an increase in the amount of homologated products with increasing
surface coverage (methane exposure time).[109] Higher temperatures employed in the first
step of the DT process resulted in higher yields of hydrocarbons compared to the IT
process. The Cg form of carbon, which Van Santen and coworkers referred to, was not
observed in the IT process.[107] Another major difference was that, while van Santen and
coworkers insinuated that CZC bond formation was primarily involved in the
hydrogenation step, Amariglio and coworkers proposed CZC bond formation in
Choudhary, Aksoylu, and Goodman162
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

the chemisorption step.[112,113] This controversy involving CZC bond formation was
further brought to light in form of two letters.[114,115] Experiments carried out in vacuum
prompted Hlavathy et al. to propose that CZC bond formation could take place in the
chemisorption step as well as the hydrogenation step. In response, Amariglio et al. referred
to their previous work, which involved flushing of the surface with CO after the
chemisorption step.[112] This had resulted in production of a number of hydrocarbons (up
to C8), including olefins, providing evidence for formation of CZC bond in the first step.
In good agreement surface science studies in our laboratory (as described in the previous
section) have shown direct spectroscopic evidence for the CZC bond formation during the
methane chemisorption step on both model[99] as well as supported real-world Ru
catalysts.[101]
Solymosi and coworkers investigated the two-step process on different metal-support
systems.[116 – 118] Ruthenium was found to be the most active catalyst for methane
decomposition (per metal site) followed by Rh, Ir, Pd, and Pt, whereas the efficacy of C2
hydrocarbon production followed the order Pt . Ru . Rh . Ir . Pd: On Pd supported
catalysts, other than methane, the only hydrocarbons observed were ethane and
propane.[117] Dissolution of hydrogen (formed in the decomposition step) in the Pd
crystallites represented an interesting feature of the supported Pd/CH4 system. The
hydrogen desorption temperature on Pd\SiO2 catalyst was in good agreement to that
observed on Pd (679).[80]
Goodman and coworkers observed an ethane yield of ,15% for an optimized carbon
coverage of 30–35%[119] on Ru/SiO2 for the DT methane homologation process. Studies
related to effect of multiple cycles indicated a large drop in catalyst activity after three
cycles, after which the catalyst had to be regenerated to regain its initial activity.
Experiments undertaken at higher pressures did not result in an appreciable decrease in the
ethane yield. Carstens and Bell investigated the different forms of carbon formed after
methane dissociation on Ru/silica catalyst.[120] The distribution of the carbonaceous
species (Ca, Cb, and Cg) was found to be dependent on the aging time as well as carbon
coverage. Additionally, their studies indicated a higher yield of C2þ hydrocarbons on a
carburized Ru catalyst than on a noncarburized catalyst.
Smith and coworkers investigated the DT methane homologation on supported Co
catalysts.[121 – 123] The amount of methane decomposed per Co site was found to be higher
on Co/Al2O3 than on Co/SiO2, whereas the latter showed higher activity in the
hydrogenation step and greater C2þ selectivity. This was attributed to the more facile
metal to support migration of the carbonaceous species in case of Co/Al2O3. It was
emphasized that high C2þ yields in the second step required the formation of CH species
on the surface in the first step. Interestingly, higher selectivity for C2þ was obtained in the
second step when argon was used instead of hydrogen.
Boskovic et al. investigated the effect of potassium as a promoter for the methane
homologation reaction on cobalt catalysts.[124] Their results indicated that the effect
caused by potassium was strongly dependent on the supports utilized (silica and alumina).
The methane-decomposition activity per site showed a larger increase on alumina-
supported catalysts (as compared to Co/SiO2). For the hydrogenation step, a greater
increase in selectivity for C2þ hydrocarbons was observed on the silica-supported Co
catalyst and only a marginal increase in C2þ selectivity was observed for Co/Al2O3.
However, the addition of potassium was found to decrease the hydrogenation activity,
Nonoxidative Activation of Methane 163
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

resulting in a decrease in C2þ yield. Solymosi and Cserenyi observed an increase in ethane
selectivity in the methane chemisorption step on introduction of Cu in the Rh/SiO2
catalyst.[125] Additionally, the selectivity of C4–C6 hydrocarbons was also enhanced in the
hydrogenation step. As opposed to Cu addition, which altered the product distribution,
addition of molybdenum to Rh/Al2O3 induced the removal of coke by hydrogen at lower
temperatures.[126] The presence of Mo was believed to restrict the mobility of the residues,
and thereby the aging.
Methane chemisorption and the subsequent hydrogenation of the carbonaceous
species was investigated on bimetallic systems by Guczi and co-workers.[127 – 129] The
amount of methane chemisorbed was found to be much larger on the Co–Pt/NaY system
as compared to Co/NaY or Pt/NaY systems. This was attributed to the increase in
reducibility of Co ions in NaY due to the presence of the added Pt.[130] Moreover, the Co–
Pt/NaY system exhibited 100% conversion of adsorbed methane (present as adsorbed CHx
species) and a selectivity of 83.6% for C2þ hydrocarbons for the hydrogenation step. The
authors proposed that the enhancement was a result of a synergistic effect of Co–Pt on the
CZC bond formation in the hydrogenation step.
Garnier et al. utilized a Pd–Ag membrane reactor for the two-step methane
homologation on Ru catalyst.[131] As compared to a standard fixed bed reactor, the
temperature required for the same methane conversion (first step) was drastically reduced
for the Pd–Ag reactor. For a 60% methane conversion, the temperature required for the
Pd–Ag reactor was only 553K, whereas it was 673K for a fixed-bed reactor. The use of
low temperature in the first step had a positive effect on the C2þ selectivity in the
hydrogenation step. No deactivation was observed for the six cycles studied
(decomposition at 573K and hydrogenation at 373–393K). Effectively, the use of Pd–
Ag membrane resulted in the decrease of the “temperature gap” referred to by Van Santen
and coworkers for the DT process.
Marceau et al. investigated the hydrogenation step of the methane oligomerization
process on EUROPT-1 catalyst.[132] Based on their results, the authors proposed the
following:
(i) The first few minutes of the hydrogenation reaction resulted mainly in the
production of ethane and pentane.
(ii) Methane chemisorption in the first step led to the formation of cyclic precursors
of pentane.
(iii) CZC bond formation also took place in the hydrogenation step via a “statistical
and dynamical homologation,” which in turn led to the formation of C2–C5
alkanes.
(iv) Hydrogenolysis of the heavier carbonaceous compounds led to the formation
of higher hydrocarbons.
Recently, Bradford investigated the effect of pressure and metal-support interactions
on the methane homologation reaction on Ru- and Pt-based catalysts.[133] Increase in
pressure favored higher hydrocarbon formation in good agreement with previous studies
on Ni–Cu/SiO2 catalysts.[134] The results further indicated that employment of a support
exhibiting mild metal-support interactions could influence the product distribution.
Choudhary, Aksoylu, and Goodman164
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

As mentioned earlier, extensive efforts were undertaken in our laboratory to
understand the mechanism for the two-step low-temperature methane-coupling process on
Ru surfaces. To address this objective, the surface intermediates formed on the model
catalyst surfaces were identified by an array of surface science techniques.
Correspondingly, kinetic studies of the process were also undertaken on model and
the real-world catalyst surfaces. Since the studies involving the detection of surface
intermediates have been described in detail in the previous section, here emphasis will be
placed on describing the kinetic studies and relating the rates of the reactions with the
concentration of the surface reaction intermediates.
Kinetics studies were undertaken on Ru(0001) and Ru(1120) as a function of methane
chemisorption temperature and hydrogenation temperature.[135,136] Under the experimen-
tal conditions employed, the maximum yield in ethane/propane was obtained at 500K
(first-step temperature) on both Ru (0001) and Ru (1120) single-crystal catalysts. The
ethane yield obtained as a function of methane chemisorption temperature is shown in
Fig. 2; ethane formation rates passed through a maximum for both surfaces. The
hydrocarbon products, which were similar for the two surfaces, consisted of 16% ethane
and 2% propane under optimum conditions.
The mechanistic information was obtained by relating the stability of the surface
intermediates with the kinetic studies. The important points related to stability of the
surface species after methane chemisorption on the Ru model surfaces are summarized
below[99,100]:
1. Only graphitic species were observed on the surface after methane
chemisorption at $800K.
2. The ethylidyne species were stable only at low temperatures, and transformed to
vinylidene species upon heating.
3. The methylidyne and vinylidene species were stable from ,400–700K.
Since the ethane yield was significant only between ,400–700, the graphitic species
and the ethylidyne species were deemed unimportant for the methane coupling reaction.
Although the methylidyne and vinylidene species were both stable under the relevant
temperature region (corresponding to maximum ethane yield), the latter species were
considered to be the direct precursors for ethane, justified by the fact that the vinylidene
species can be directly hydrogenated to ethane, while methylidyne species require both
polymerization as well as hydrogenation for ethane formation.
Figure 2. Mechanism for the two-step methane homologation on Ru-based catalysts.[136]
Nonoxidative Activation of Methane 165
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

The following mechanism can then be proposed (illustrated in Fig. 3). The methane-
chemisorption step involves the formation of surface methylidyne and vinylidene species
along with hydrogen. In the hydrogenation step, the vinylidene species are directly
hydrogenated to ethane while methylidyne intermediates are involved in two competitive
reactions: (i) hydrogenation to methane, and (ii) transformation to vinylidene species. It is
noteworthy that similar ethane yields were obtained for the model Ru catalysts and real-
world Ru/SiO2 catalysts.[119] This, coupled with the fact that same surface intermediates
(along with similar trend in stability of surface intermediates) were observed for both
surfaces, implies that the proposed mechanism for the two-step low-temperature methane-
coupling is applicable to all Ru surfaces.[137] This combined study exemplifies the
effectiveness of surface science studies to probe catalytic reaction mechanisms.
IV. PYROLYSIS AND HIGH-TEMPERATURE COUPLING OF METHANE
The methane molecule is different from other paraffins in that it cannot be further
differentiated into lighter hydrocarbons. Coupling and formation of higher hydrocarbons
requires splitting of a CZH bond[138 – 141] in methane, but its high-dissociation energy
(435 kJ/mol) makes this highly endothermic. For this reason, high temperatures are
required for the direct (one-step) nonoxidative coupling of methane. This field has been
Figure 3. Ethane yield over model (B) Ru(001) and (X) Ru(1120) catalysts as a function of
temperature for the low-temperature methane-coupling reaction.[119]
Choudhary, Aksoylu, and Goodman166
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

a focus of large number of studies in the past few years. Here, these studies have been
classified into homogeneous (noncatalytic) and catalytic high-temperature coupling of
methane. The investigations involving catalytic coupling of methane have been further
split into two subsections based on the catalyst system employed, (i) carbonaceous and
oxide catalysts and (ii) zeolite-based catalysts.
A. Methane Pyrolysis and Dehydrogenative Coupling ofMethane in Homogeneous Systems
Thermodynamically, methane is unstable in terms of its elements (carbon and
hydrogen) from 803K, but is more stable than the other hydrocarbons up to 1303K[142]
(Fig. 4). Gueret et al. carried out an exhaustive investigation of the thermodynamics of
methane pyrolysis.[142] Based on these thermodynamic calculations, the authors predicted
the following:
(i) Low methane conversions below 1473K, with benzene as the principal
product, followed by ethylene and traces of acetylene.
(ii) High propensity of the CZC and CZH bonds to rupture above 1473K
(iii) Factors leading to stabilization of the radicals formed during the coupling
reaction would be favorable for the endothermic dissociation reaction.
Figure 4. Gibbs free energy of formation for various hydrocarbons as a function of
temperature.[142]
Nonoxidative Activation of Methane 167
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

(iv) The exothermic formation of the double bonds would enhance the dissociation
process.
(v) High pressures were detrimental to the splitting of the CZH bond and resulted
in low methane conversions.
Experimental studies by Holmen et al. revealed that high temperature and short
residence times were favorable for acetylene formation in the thermal coupling of
methane.[143] Acetylene yields greater than 85% could be obtained at temperatures higher
than 2000K and reaction times of ca. 1022 seconds. The maximum attainable yield of
acetylene declined sharply with decreasing temperatures. Auto-acceleration in the
methane decomposition process at low conversion was attributed to heat transfer from the
reactor wall to the gas. The rate of carbon formation escalated with increase in the partial
pressure of acetylene above a critical limit.[156] The inlet limiting pressure was found to
correspond to a maximum outlet partial pressure of acetylene of ca. 20 Torr. A recent study
by Sun et al. involving methane pyrolysis in a hot filament reactor yielded methane
conversion of 19.7% and a hydrocarbon selectivity of 68% at 1548K and residence time of
0.1 seconds.[144]
The overall reaction in the thermal coupling of methane is generally believed to
involve a stepwise dehydrogenation at high temperatures.[145 – 147]
2CH4 ! 2C2H6 þ H2 ! C2H4 þ H2 ! C2H2 þ H2 ! 2C þ H2
Arutyonov et al.[148] observed that at 1200K (static reactor), the concentration of all the
three C2 hydrocarbons (C2H6,C2H4, and C2H2) maximized simultaneously with time and,
hence, argued that although the above sequence accurately described the kinetics in the
low temperature region, it became less apparent at higher temperatures.
To date different experimental techniques such as tubular flow reactors, shock tube
reactors, and plasma reactors have been employed to investigate methane pyrolysis at the
temperatures high enough for the production of acetylene.[149 – 154] Studies undertaken in
shock-tube[155] reactors showed higher overall activation energies as compared to studies
in flow-reactors. This was attributed to the existence of a higher temperature difference
between the walls of the reactor and the bulk gas at high temperatures in case of flow-
reactors.[156] The overall methane decomposition was assigned a first-order reaction[157 –
159] by the majority of research groups in conflict with the observation by Eisenberg and
Bliss.[160]
Kinetic studies of the methane decomposition process (under static conditions) by
Arutyonov et al.[148] pointed to the possibility of obtaining C2 hydrocarbons in amounts
greater then the thermodynamic equilibrium quantities as the concentration of C2
hydrocarbons were found to pass through a maximum in pyrolysis. The concentration of
ethylene in the product mixture was ca. 10 times that of acetylene and ethane at 1100K,
whereas the concentration of acetylene and ethylene was comparable at 1300K. On
diluting methane with hydrogen ðCH4 : H2 ¼ 1 : 3Þ; there was a threefold decrease in the
pyrolysis rate constant.
Billaud et al.[161] investigated the influence of hydrogen dilution, temperature, and
residence time on the conversion and product yields during methane pyrolysis. Hydrogen
dilution had a detrimental effect on methane conversion, but decreased coke formation.
Choudhary, Aksoylu, and Goodman168
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

Ethylene and acetylene were obtained as the major products and ethane, propene,
propadiene, propane, butadiene, butenes, and cyclopentadiene as minor gaseous products.
Liquid products such as benzene, toluene, xylenes, and naphthalene were also observed.
The C2H4, C2H2, and C6H6 yields maximized between 7%–8% with residence time,
whereas the coke yield continued to increase. The studies suggested the existence of a
competition between C2 (ethylene and acetylene) formation and carbon deposition. The
formation of heavy products proceeded via acetylene as an intermediate rather than
ethylene at high conversions. The work concluded that reduction in coke formation and an
increase in C2 yields were possible by carrying out the pyrolysis of methane in presence of
hydrogen at high temperatures and low residence times.
The effects of hydrogen[161] (enhanced C2 yields, decreased benzene selectivity, and
suppressed coke formation) can be explained based upon a mechanism proposed by Olsvik
and Billaud[140]:
CH4 þ Hz , CH3zþ H2 ðIÞ
C2H4 þ CH3z , CH4 þ C2H3 ðIIÞ
C2H2 þ C2H3z , C4H5 ðIIIÞ
C2H2 þ C4H5z , C6H6 þ H ðIVÞ
In the elementary step (I), the addition of hydrogen leads to a smaller methane conversion
and to a lower CH3z concentration. As a result, the steps which included CH3z, specifically
(II), become less important, leading to a smaller C2H4 consumption and an increase in its
concentration. Since the concentration of C2H3 decreased, reaction (III) occurs at a slower
rate leading to a decrease in the production of C4H5. This effectively results in lower
production of C6H6 via (IV), thus, eventually leading to an increase in the concentration of
C2H2.
Coke (carbonaceous material containing hydrogen) is one of the undesired by-
products of the pyrolysis reaction.[162] According to one of the mechanisms, coke
formation proceeds via further reactions of aromatics (which are formed by the
trimerization of acetylene).[163] Further details regarding the mechanisms for carbon
formation during the methane decomposition reaction can be found in references.[163,164]
The stringent requirement of high temperatures for methane-coupling reactions places
very strong limitations on the reactor design. Heat recovery and transfer of heat pose a
severe challenge in the thermal-coupling process. Based on the method of heat supply and
removal from the reactor, the processes were grouped into four as follows[147]:
(i) The processes in which the energy source was an electric arc (Huels
process).[165]
(ii) The processes in which part of the methane feed was burnt in the reactor with
oxygen (BASF).[166]
(iii) Regenerative pyrolysis process, which employed a cyclic heating and pyrolysis
(Wulff process).[167]
(iv) Isothermal pyrolysis, which involved the introduction of heat through the
reactor wall by resistive heating.
Nonoxidative Activation of Methane 169
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

Detailed information on the complex mechanisms involved in the noncatalytic high-
temperature coupling of methane can be obtained in.[147]
B. Catalytic Pyrolysis and Nonoxidative Coupling of Methane
at High-Temperatures
1. Carbonaceous, Oxide, and Metal Catalysts
Catalyst systems have been investigated to increase the kinetics of the methane
coupling process. Fang and Yeh[168] investigated the catalytic effect of ThO2/SiO2
surfaces on the methane-coupling process. Operating at low conversions ðT ¼
1073K; Pmethane ¼ 15 TorrÞ; the authors observed a ca. 300 times faster product generation
rate than that in homogeneous methane decomposition process. Ethane and ethylene were
the major products formed, whereas propylene and acetylene were obtained in smaller
concentrations. Van Santen and coworkers investigated methane pyrolysis on a range of
catalysts with varied specific surface areas.[169] Their results indicated that the conversion
and product selectivities in the methane pyrolysis process were not catalyst-specific, but
instead were controlled by the specific surface area. Low specific areas resulted in highest
yields for gaseous (ethylene and acetylene) and liquid products (light aromatics), whereas
high surface areas resulted in the production of graphitic coke and hydrogen.
Methane pyrolysis on pitch-based carbon fibers at 1273K by Mochida and
coworkers[170] resulted in steady-state conversions of 20% and high selectivity (upto 67%)
for C2 hydrocarbons with low levels of carbon deposition. Although they admitted that the
reaction mechanism and role of the fiber surface was not very clear, they speculated that
the pyrolysis was initiated by the radical fission of methane either on the surface or in the
gas phase. The resulting methyl radicals first supposedly coupled on the surface into C2
species, leading to formation of higher hydrocarbons and, finally, formed pyrolitic carbon.
Kharatyan et al.[171] investigated the high-temperature kinetics of methane (2–
100 mm Hg, 1673–2473K) on Mo wire. Their results indicated that, with a pressure less
than or equal to 3 mm Hg, pyrolysis of methane obeyed a linear law at all temperatures. At
T , 1973K and P . 3 mm Hg; the process obeyed a linear law in the initial stages,
whereas the process was described by parabolic law after a certain time period. At
pressures equal or greater than 25 mm Hg, the parabolic law applied from the very
beginning. The pyrolysis rate of methane was found to be much higher on molybdenum
carbides than on tungsten carbides.[172] The other notable difference on the tungsten
surface was that, in this case, the methane pyrolysis complied with a linear law over the
whole range of temperatures (1673–2923K) and pressures (2–100 mm Hg) were
investigated.
Potapova et al.[173] carried out the decomposition reaction on indium oxide ðT ¼
973–1073KÞ with methane–ethane mixtures having 50–95 vol% methane. Results
showed that the ethane decomposition rate decreased in the presence of methane whereas
increasing ethane concentration in the mixture enhanced methane conversions. This effect
was attributed to the participation of atomic hydrogen in competitive reactions. The
detrimental effect of methane on ethane decomposition was limited in the presence of the
indium oxide catalyst (as compared to the homogeneous thermal-cracking process).
Choudhary, Aksoylu, and Goodman170
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

The authors claimed that the concentration of atomic hydrogen was far greater (3–8 times)
in the catalytic decomposition of ethane–methane mixtures than during the thermal
process, resulting in enhancement of the methane conversion rate and decrease in the
methane inhibition effect upon ethane conversion.
Investigation of methane pyrolysis by Kushch et al.[174] on fullerene black showed
that the methane conversion decreased with time due to pyrolitic carbon formation. Use of
methane diluted in argon instead of pure methane did not result in the inhibition of
pyrolytic carbon formation. The influence of H2 as a diluent (at low hydrogen contents)
was found to be identical to that of argon in the initial part of the reaction, but upon
accumulation of pyrocarbon, a higher selectivity to ethylene and lower selectivity for
propylene was obtained. The dehydrodimerization of methane became the dominant
process with the increase of the H2 content in the initial mixture (above 45%). The
methane conversion was found to be lower than that with a smaller H2 content (or a similar
argon dilution) and the only reaction products were ethylene and propylene. Since there
was no pyrocarbon accumulation, the activity and selectivity remained constant with time.
Kurosaka et al.[175] observed that 3 wt% Pt-added sulfated zirconia gave a steady
conversion of 0.23% at 773K for a period of 5 hr in a flow reactor (0.5 g cat, 10 ml/min
methane). Ethane and ethylene were obtained as products in a ratio of 9:1. Platinum
supported on zirconia without the sulfate was found to sinter, resulting in deactivation. The
authors emphasized that deployment of sulfur acid (as opposed to using ammonium
sulfate) as a sulfating reagent led to the synthesis of superior catalysts.
2. Zeolite-Supported Catalysts
The studies involving zeolite-based catalysts are different from the investigations
discussed previously in that the objective in this case is to convert methane selectively to
aromatic compounds. Wang et al.[176] pioneered the nonoxidative dehydrogenative
methane aromatization reaction on ZSM-5-based catalysts. HZSM-5 catalysts showed a
1.4% conversion of methane with 100% selectivity to benzene at 973K. The conversion of
methane was greatly enhanced (without any detrimental effect to benzene selectivity) by
loading the zeolite with Mo or Zn cations. Mo/HZSM-5 exhibited the best activity: 7.2%
and 4.4% for the SiO2/Al2O3 ratios of 50 and 25, respectively. The catalyst was found to be
stable without any loss in activity and selectivity under the reaction conditions employed
for the study. The authors claimed that the activation of methane proceeded via a
carbenium-ion mechanism with proton sites Zn2þ and Mo6þ acting as the hybrid acceptors
in HZSM-5, Zn/HZSM-5, and Mo/HZSM-5, respectively. Subsequent studies[177]
revealed that the Bronstead acidity, channel structure, and the state and location of Mo
species in the zeolite played a vital role in the performance of the catalyst. Increase in
temperature increased methane conversion and aromatics selectivity but decreased C2
selectivity. Molybdenum-loading studies showed that the methane conversion passed
through a maximum at 2–3% Mo loading. High calcination temperatures and High Mo
loadings (.6%) resulted in the blockage of channels, leading to loss in catalytic activity.
A mechanism involving heterolytic splitting of methane in the presence of Bronsted acid
sites of HZSM-5 zeolite (leading to the formation of a molybdenum-carbene-like complex
CH2 ¼ MoO3 intermediate) was proposed.
Nonoxidative Activation of Methane 171
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

Solymosi and coworkers investigated various aspects related to the methane
aromatization process.[178 – 183] Studies pertaining to MoO3 on various support oxides[178]
showed a common trend: initial induction period (varied with the support) followed by a
maximum in conversion, which finally decreased with time. The highest methane
conversion was observed on MoO3/SiO2 (but was less than that on MoO3/ZSM-5[176]) and
the least on MoO3/MgO. The dominant products were H2, CO, H2O, and CO2 in the initial
part of the reaction on MoO3/SiO2. Aromatic formation (benzene and trace amounts of
toluene) started late in the reaction (ca. 45 min) and its selectivity increased with reaction
time. Studies related to the induction period led the authors to believe that the partial
reduction of Mo6þ was required for the formation of hydrocarbons (as opposed to Ref.
[176] which assumed the active site to be Mo6þ species). Mo2C and Mo2C mixed with
HZSM-5 were found to be relatively inactive for the process, whereas Mo2C deposited in a
finely divided state on ZSM-5 (synthesized as in [184]) was found to be a highly active
catalyst for the dehydrogenation and aromatization of methane.[179,181] Molybdenum
carbide was proposed to be the active species responsible for the methane to ethylene (the
precursor for benzene formation) conversion.[181] Subsequently, the same group employed
surface science techniques (AES, XPS, TPD, HREELS) to study the generation and
reaction of CxHy species on the Mo2C/Mo(111) surface[182,183] in order to obtain
mechanistic information.
Lunsford and coworkers[185 – 189] carried out a series of investigations aimed at
determining the nature of the active catalytic sites. It is noteworthy to mention that in these
studies, nitrogen was used an internal standard so as to obtain accurate methane
conversions and to evaluate quantitatively coke formation. Catalyst results obtained at
973K ðGHSV ¼ 800 h21Þ showed an initial activation period, after which a sustained
selectivity of ca. 70% for benzene was obtained for several hours. Unlike benzene, the
selectivity for naphthalene passed though a maximum (ca. 20%) and then declined rapidly
with reaction time. The catalyst was stable (with methane conversions of 8–10%) and
showed a small decrease in catalytic activity over a period of 16 hr. The original activity of
the catalyst could be completely restored by oxidation treatment of the spent catalyst at
973K. Characterization of the catalyst with XPS indicated the gradual reduction of the
Mo6þ species with reaction time, finally leading to formation of Mo2C. Based on their
study of the induction period, the authors proposed that the coke-modified Mo2C surface
was the active species responsible for the formation of ethylene (reaction intermediate)
rather than clean Mo2C.[186] The XPS results indicated the presence of three different
types of surface carbon[188] on the active Mo/HZSM-5 catalyst:
(i) Graphitic carbon that was dominantly present in the zeolite channel system.
(ii) Carbidic carbon (from Mo2C) that was mainly located at the outer surface of
the catalyst.
(iii) Hydrogen-poor, sp-type carbon that was predominantly present on the outer
surface of the zeolite.
The last type of carbon was held responsible for the deactivation of the catalyst.
Subsequent studies on different transition metal ions (TMI)/H ZSM-5[187] indicated the
following trend in activities for methane conversion: Mo . W . Fe . V . Cr: The CO
pre-reduction treatments were found to enhance the catalytic activity. Catalysts obtained
Choudhary, Aksoylu, and Goodman172
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

by impregnation method showed a superior performance compared with those synthesized
by a solid-state ion exchange with a TMI salt. This was attributed to the presence of fewer
Bronsted acid sites in case of solid-state ion-exchanged catalysts. XPS studies revealed the
presence of transition metal suboxides[189] on all impregnated catalysts, with the exception
of Mo/HZSM-5; Mo2C was observed in this case. The authors concluded that the active
phase responsible for the reaction was different for different transition metals.
Characterization studes on the Mo/HZSM-5 catalyst related to Mo2C and the induction
period by Howe and coworkers[190] was in good correspondence with the work of
Lunsford and coworkers.[186]
Meriaudeau et al. carried out methane and ethylene aromatization over Mo/HZSM-5
and HZSM-5.[191] Surprisingly, the results indicated that ethylene aromatizion was more
facile on Mo/HZSM-5 (which has lower acid sites) than on HZSM-5. This led the authors
to express concern about ethylene being the key intermediate in methane aromatization
(which is the commonly accepted mechanism).[192] The authors suggested acetylene as
being the intermediate in methane aromatization. It was speculated that if ethylene was
indeed the key intermediate, then its transformation to benzene would not be a simple acid
(zeolitic protons) catalyzed reaction as proposed in the literature.[178,186]
Zhang et al.[193] compared the effect of different molecular sieve supports on the
catalytic performance of Mo-based catalysts and found that the zeolite structure exerted a
a significant influence on the catalytic activity. Zeolites possesing two-dimensional pore
diameters (such as ZSM-5, ZSM-8, and ZSM-11) in the vicinity of the dynamic diameter
of a benzene molecule (0.6 nm) were suggested to be excellent supports for methane
aromatization.
Jiang et al.[194] investigated the induction period of the methane aromatization
reaction on Mo/HZSM-5 employing pulse technique. In parallel to previous studies[178,186]
they observed that the induction period involved the gradual reduction of the Mo6þ
species. The carbonaceous residue resulting from further methane decomposition then
interacted with the partially reduced Mo species to form the active centers for the reaction.
In a second study involving the induction period, Ma et al.[195] concluded that the initial
methane activation was the most crucial step in the methane aromatization reaction.
Liu and Xu[196] utilized pyridine as a proble molecule to characterize the acid sites of
Mo/HZSM-5 and gain insights into the interaction between Mo species and HZSM-5.
Results indicated the presence of Mo species predominantly on OH-groups associated
with silanol groups and bridged hydroxyl group [Si(OH)Al]. Based on the catalytic
evaluation results and FT-IR results, the authors reiterated that Mo/HZSM-5 was a bi-
functional catalyst and claimed that ca. 60% of the original Bronsted acid sites in HZSM-5
were required for optimum performance in the methane dehydro-aromatization reaction.
Catalyst characterization studies by Xu and coworkers[197] indicated the presence of
two types of Mo species in the Mo/HZSM-5 catalyst: (i) polynuclear Mo species viz.
MoO3 species (with pressed octahedral coordination) and MoOx species (with square-
pyramidal coordination), present on the external surface; and (ii) mononuclear Mo species
located in the viscinity of framework aluminum. Elegantly designed NMR spectroscopic
experiments[198] were employed to follow the change in Bronstead protons during the
aromatization reaction. These results clearly illustrated the importance of the Bronsted
acid sites in the methane aromatization reaction. The same group further studied methane
dehydo-armatization on Mo supported on phosphoric and rare-earth containing
Nonoxidative Activation of Methane 173
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

penta-sil-type high-silica catalyst (HZRP-1).[199] The Mo/HZRP-1 was found to have
greater activity for the reaction than Mo/HZSM-5 at high Mo-loadings. This was attributed
to the better stability of HZRP1 zeolite framework at high-Mo loadings. NMR studies
were subsequently employed to characterize the Mo/HZRP-1 catalysts.[200] The
phosphorous atoms had a greater propensity to replace framework silicon atoms at
high-Mo loadings and calcination temperatures, which consequently resulted in the
variation of the acidic property of the catalyst. Under similar experimental conditions,
Mo/MCM-22[201] was found to give a greater yield of benzene, but a lower yield of
napthalene as compared to Mo/HZSM-5. Interestingly, the authors found a similarity in
the behavior of the aromatization reaction on both the catalysts.
Ichikawa and coworkers used Ar as an internal standard in order to obtain accurate
quantitative results on conversions and selectivities of the various products obtained
during the dehydro-aromatization of methane.[202] Along with benzene and toluene, higher
aromatics such as naphthalene, methyl-napthalene, phenanthrene, and anthracene and
their derivatives were also detected in trace amounts. The Mo/HZSM-5 catalyst having a
SiO2/Al2O3 ratio of 40 was found to give maximum benzene formation and minimum coke
formation.[202]
Addition of Fe and Co to the Mo/HZSM-5 catalyst resulted in a significant
enhancement in the conversion of methane to benzene and naphthalene.[203] Similarly,
introduction of Cu (by ion-exchange method) into Mo/HZSM-5 improved both the activity
as well benzene selectivity for the methane dehydro-aromatization process.[204] In
contrast, addition of phosphorous and lithium to Mo/HZSM-5 reduced the catalytic acidity
and activity.[205]
Choudhary et al. achieved high methane aromatization activity on H-galloaluminate
ZSM-5 type zeolite by co-feeding methane with higher alkenes and alkanes.[206] A
mechanism involving hydrogen transfer was invoked for describing the methane
activation mechanism. Ichikawa and coworkers included CO and CO2 in the methane feed
and observed an enhanced stability of Mo/HZSM-5 due to decrease in coke
formation.[207,208] Re/HZSM-5 catalyst was found to give comparable methane
conversions and product selectivities (to Mo/HZSM-5) in the methane dehydrogenation
and aromatization reaction. As in case of Mo/HZSM-5, addition of a few percent of
CO/CO2 in the methane feed was found favorable for the stability of Re/H-ZSM-5.[208]
Yuan et al. observed that addition of optimum amounts of oxygen was beneficial for the
stability of the Mo/HZSM-5 for aromatic formation.[209] But the presence of oxygen in
greater than a certain critical value resulted in total oxidation and OCM processes being
dominant.
Recently, Dantsin and Suslick[210] have employed a sonochemical preparation
method for preparation of a Mo2C/HZSM-5 catalyst. The synthesis procedure consisted of
irradiation of molybdenum hexacarbonyl and HZSM-5 with ultrasound at 20 kHz for 3 hr
at 358K in inert atmosphere. The resulting catalysts consituted of ca. 2 nm sized Mo2C
particles, which embellished the external surface of the HZSM-5 support. Methane
aromatization studies on the catalyst gave comparable conversion and selectivity to
benzene as compared to conventionally synthesized catalysts.
It is noteworthy that the studies described in the high-temperature coupling of
methane section provide valuable information for hydrogen production via methane
decomposition (described in the next section). The objective of the coupling reations is to
Choudhary, Aksoylu, and Goodman174
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
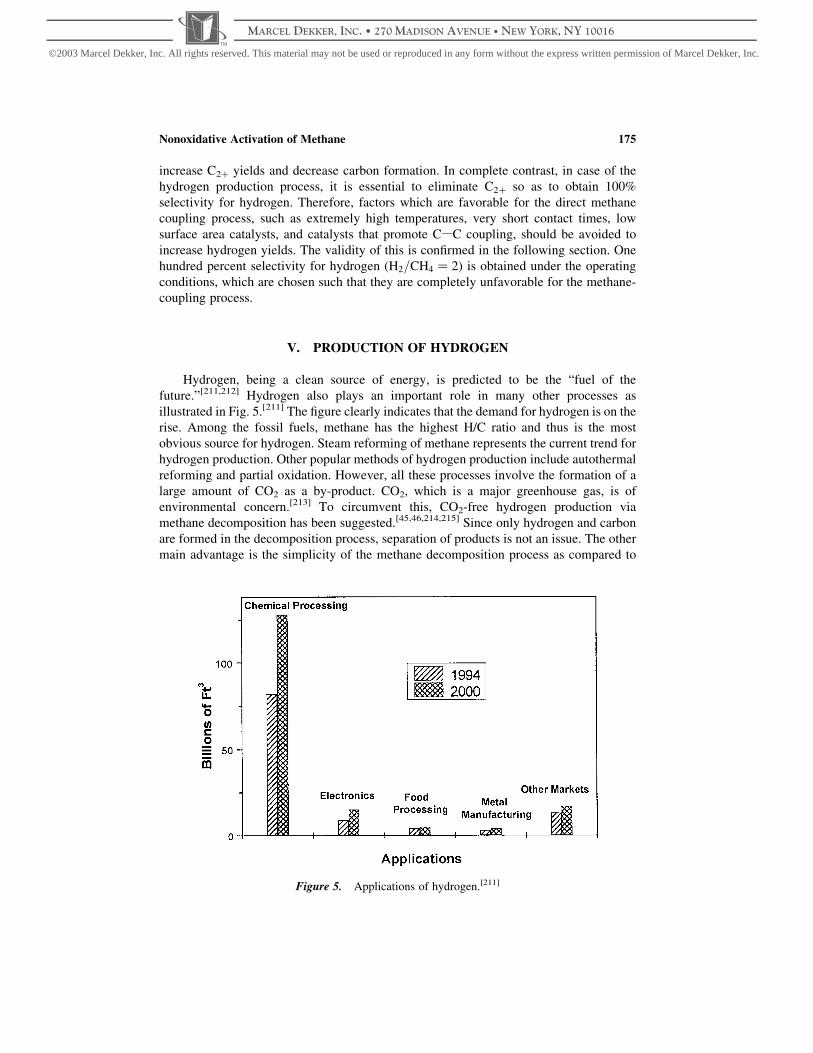
increase C2þ yields and decrease carbon formation. In complete contrast, in case of the
hydrogen production process, it is essential to eliminate C2þ so as to obtain 100%
selectivity for hydrogen. Therefore, factors which are favorable for the direct methane
coupling process, such as extremely high temperatures, very short contact times, low
surface area catalysts, and catalysts that promote CZC coupling, should be avoided to
increase hydrogen yields. The validity of this is confirmed in the following section. One
hundred percent selectivity for hydrogen ðH2=CH4 ¼ 2Þ is obtained under the operating
conditions, which are chosen such that they are completely unfavorable for the methane-
coupling process.
V. PRODUCTION OF HYDROGEN
Hydrogen, being a clean source of energy, is predicted to be the “fuel of the
future.”[211,212] Hydrogen also plays an important role in many other processes as
illustrated in Fig. 5.[211] The figure clearly indicates that the demand for hydrogen is on the
rise. Among the fossil fuels, methane has the highest H/C ratio and thus is the most
obvious source for hydrogen. Steam reforming of methane represents the current trend for
hydrogen production. Other popular methods of hydrogen production include autothermal
reforming and partial oxidation. However, all these processes involve the formation of a
large amount of CO2 as a by-product. CO2, which is a major greenhouse gas, is of
environmental concern.[213] To circumvent this, CO2-free hydrogen production via
methane decomposition has been suggested.[45,46,214,215] Since only hydrogen and carbon
are formed in the decomposition process, separation of products is not an issue. The other
main advantage is the simplicity of the methane decomposition process as compared to
Figure 5. Applications of hydrogen.[211]
Nonoxidative Activation of Methane 175
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

conventional methods. For example, the high- and low-temperature water-gas shift
reactions and CO2 removal step (involved in the conventional methods) are completely
eliminated.
Universal Oil Products developed the Hypro-process for hydrogen production based
on methane decomposition.[216] The process utilized a fluidized-bed reactor-regenerator
with 7% nickel on alumina as the catalyst. The thermal decomposition of methane was
carried out at ,1150K, whereas the regeneration took place at ,1475K. The reactor
effluent consisted of ca. 94% hydrogen ðH2=CH4 ¼ 2Þ; with the rest being mainly
unreacted methane. Noyes at the United Technologies Corporation proposed a process
which involved methane decomposition on nickel deposited on glass fibers.[217] The
process carried out at 1123K resulted in a production of high-density carbon and, thus,
required less frequent removal of carbon.
Steinberg proposed an interesting concept to reduce CO2 emission.[214] According to
the process, hydrogen produced via methane decomposition could be further used to
convert CO2 to methanol, which could then be used as fuel in automotive engines. The
process as he envisaged could decrease the CO2 emission by two-thirds from the
transportation and power generation sectors in use currently. Steinfeld et al. employed
solar power to crack methane for hydrogen production.[218,219] This approach further
decreases CO2 emissions by eliminating the fuel consumption required for the
endothermic methane-decomposition reaction. However, it should be noted that there
are severe technical difficulties involved in the large-scale use of solar reactors for
methane decomposition.
Muradov investigated methane decomposition on supported Ni and Fe catalysts over a
wide range of temperatures.[215] Results indicated that these catalysts required oxidative
regeneration to restore catalytic activity, resulting in the production of carbon-oxides in
the regeneration step. Carbon catalysts were used to circumvent the problem of carbon
removal. Among the different carbon materials studied, activated carbon produced from
coconut shells was found to have the highest initial activity, whereas graphite had the least.
This activity difference was attributed to the structure and size of the carbon crystallites.
The studies involving binary mixtures of methane with other hydrocarbons over inert
supports indicated that acetylene addition enhanced the steady-state methane
decomposition rate. No such effect was observed when propane was used. Carbon
resulting from acetylene decomposition was found to be more active towards methane
decomposition than that produced from methane.
Ishihara et al. achieved a significant increase in yield of hydrogen by using a
membrane reactor.[220] Ni supported on silica was the catalyst of choice and the hydrogen-
permeable membrane was made of a palladium–silver alloy. The permeated hydrogen was
swept by argon gas and increasing the argon flow-rate was found to have a positive effect
on methane conversion. A significant increase in methane decomposition was observed
above 723K, with the overall conversion exceeding equilibrium conversions. Results
indicated that contact time longer than 50 g cat h mol21 and sweep argon gas flow-rates
higher than 200 ml min21 were favorable for the process.
Fuel cells are gaining rapid importance, as they are environmentally benign and have
high efficiency. The stringent requirement of low levels of CO in the hydrogen stream
[ppm levels for the current proton-exchange membrane (PEM) fuel cells] adds substantial
cost to the operation of fuel cells using hydrogen from conventional sources such as steam
Choudhary, Aksoylu, and Goodman176
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

reforming, partial oxidation, and auto-thermal reforming of methane.[221] Recently, Zhang
and Amiridis investigated methane cracking over silica-supported catalysts[222] to produce
CO-free hydrogen. A 20% CH4 in He stream (T ¼ 823K and GHSV ¼ 30000 h21) gave
an initial methane conversion of 35%, which decreased with time until the catalyst was
completely deactivated after ca. 3 hr. Hydrogen (stoichiometric: H2=CH4 ¼ 2), along with
filamentous carbon were observed as the products. Regeneration with steam resulted in
complete restoration of the catalytic activity. A total of 3.4 moles of hydrogen were
produced per mole of methane consumed (decomposition þ regeneration). The TEM
results indicated that the external graphitic skin of the filamentous carbon was resistant
towards steam regeneration, whereas the inner less-graphitic carbon was easily removed in
the regeneration step. The external graphitic surface was roughly estimated to about 30%
of the total carbon deposited.
It has been recently noted that catalytic decomposition of methane may lead to CO
formation via reaction of the carbonaceous residue with the oxygen of the oxidic
support.[223] It is extremely important to monitor CO levels in the product stream as the
state-of-the-art PEM cells have very stringent CO-tolerance levels (ppm level). To address
this issue, the low levels of CO (in the hydrogen stream from methane decomposition)
were quantitatively estimated in our laboratory by methanation of CO and subsequent
analysis by flame ionization detection.[224 – 226] The step-wise methane steam-reforming
process for the production of CO-free hydrogen[224,227] involves the catalytic
decomposition of methane in step I to produce CO-free hydrogen and surface carbon
and/or hydrocarbonaceous species, followed by a separate step in which the surface
hydrocarbonaceous species is removed via reaction with water (step II), regenerating the
catalyst with respect to step II. Figure 6 shows the amount of methane reacted in step I and
the amount of carbon removed in step II as a function of cycle number (for a catalyst
loading of 200 mg at 648K). The amount of methane reacted is denoted by data points
below the abscissa, whereas the gas-phase carbon containing products obtained in step II
are shown above the abscissa. On an average, 93% of the carbon deposited in step I was
removed in steam gasification step and the catalytic activity was sustained throughout the
16 cycles studied. The average molar amount of hydrogen produced per mole of methane
consumed in step I was 1.1 (remaining hydrogen was present on the surface as
hydrocarbonaceous species) and contained less than 20 ppm CO. The total molar amount
of hydrogen produced per mole of methane consumed in Steps I and II was ca. 3.0. The
overall process was found to run optimally between 648–673K and a surface coverage of
0.1–0.2 monolayer equivalents.
While the above experiments were carried in a pulse mode at low temperatures,
Amiridis and coworkers investigated the same process at higher temperature in a
continuous mode.[228] Methane was cracked on 15% Ni/SiO2 catalyst at 973K for 3 hr, and
the catalyst subsequently regenerated with steam. This process was successfully repeated
for 10 cycles. X-ray diffraction analysis, after successive cracking–regeneration cycles
showed no change in the structure of nickel particles. Choudhary and coworkers[229]
carried out the methane decomposition step and steam gasification in two parallel reactors
by switching between the feed gas (methane containing stream) and steam at known time
intervals. Operating in a cyclic manner the authors observed no increase in pressure drop
(which may occur due to excess carbon deposition on the catalyst surface). The feed
switch-time was found to play an important role in determining the performance of a given
Nonoxidative Activation of Methane 177
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.
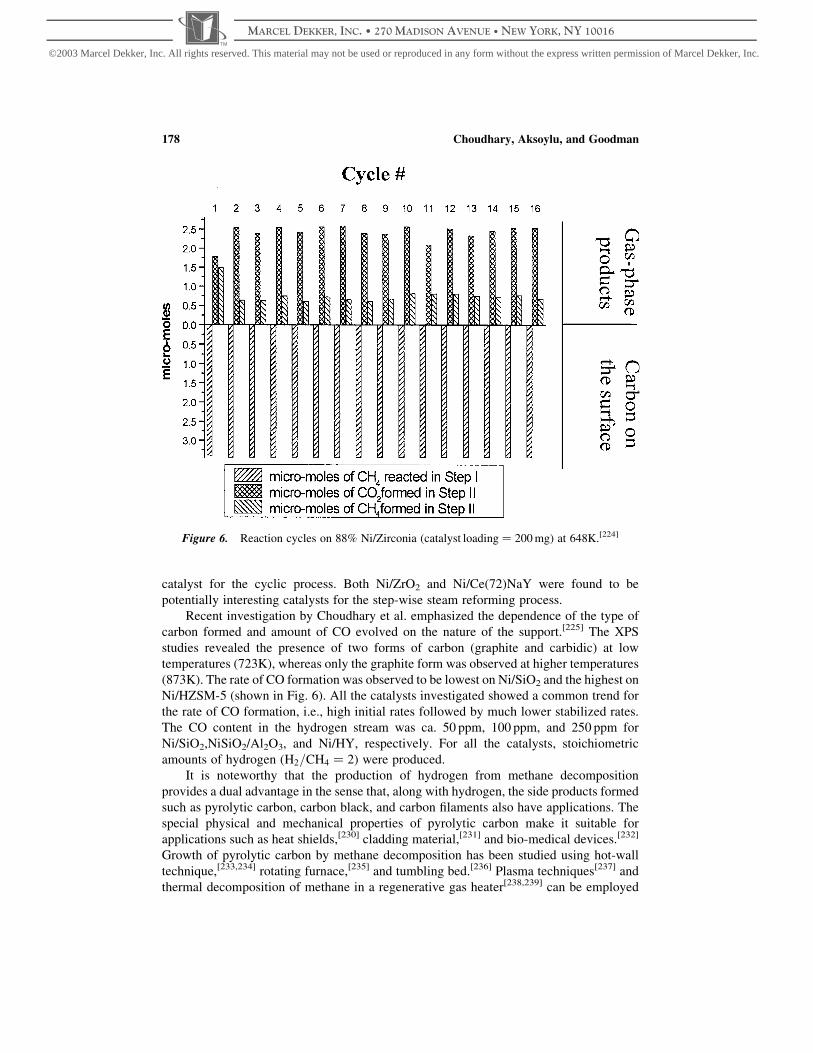
catalyst for the cyclic process. Both Ni/ZrO2 and Ni/Ce(72)NaY were found to be
potentially interesting catalysts for the step-wise steam reforming process.
Recent investigation by Choudhary et al. emphasized the dependence of the type of
carbon formed and amount of CO evolved on the nature of the support.[225] The XPS
studies revealed the presence of two forms of carbon (graphite and carbidic) at low
temperatures (723K), whereas only the graphite form was observed at higher temperatures
(873K). The rate of CO formation was observed to be lowest on Ni/SiO2 and the highest on
Ni/HZSM-5 (shown in Fig. 6). All the catalysts investigated showed a common trend for
the rate of CO formation, i.e., high initial rates followed by much lower stabilized rates.
The CO content in the hydrogen stream was ca. 50 ppm, 100 ppm, and 250 ppm for
Ni/SiO2,NiSiO2/Al2O3, and Ni/HY, respectively. For all the catalysts, stoichiometric
amounts of hydrogen ðH2=CH4 ¼ 2Þ were produced.
It is noteworthy that the production of hydrogen from methane decomposition
provides a dual advantage in the sense that, along with hydrogen, the side products formed
such as pyrolytic carbon, carbon black, and carbon filaments also have applications. The
special physical and mechanical properties of pyrolytic carbon make it suitable for
applications such as heat shields,[230] cladding material,[231] and bio-medical devices.[232]
Growth of pyrolytic carbon by methane decomposition has been studied using hot-wall
technique,[233,234] rotating furnace,[235] and tumbling bed.[236] Plasma techniques[237] and
thermal decomposition of methane in a regenerative gas heater[238,239] can be employed
Figure 6. Reaction cycles on 88% Ni/Zirconia ðcatalyst loading ¼ 200 mgÞ at 648K.[224]
Choudhary, Aksoylu, and Goodman178
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

for formation of carbon black and hydrogen. Carbon black finds applications in the rubber
industry. Amongst the various side-products formed during hydrogen generation from
methane cracking, carbon filaments are considered to be the most interesting. The issues
related to carbon filaments are addressed in the next section.
VI. FORMATION OF CARBON FILAMENTS
Carbon formed from the interaction of hydrocarbons with metals has been extensively
investigated due to its significance in various catalysis reactions.[240 – 244] Particularly,
interesting is the formation of carbon filaments on supported metal catalysts. Figure 7
shows a single filament of carbon. It is noteworthy that the formation of these carbon
filaments (CF) are studied for two entirely different reasons. In steam-reforming of
methane, formation of such filamentous carbon can eventually cause destruction of the
catalyst.[245] It is therefore of interest to study the mechanism and various other aspects of
CF formation to restrict the formation of this type of carbon. On the other hand, these
materials (CF) possess a variety of unique properties with prospective applications such as
catalyst supports, reinforcement material, selective adsorption agents, and in energy
storage devices.[246 – 252] Therefore, in the last few years a great deal of effort have been
directed towards optimization of the process condition for CF formation and to tune the
properties of the CF for desired applications.
Baker et al. proposed an influential model for the growth of CF.[253] According to this
basic model, the first step involves the adsorption and decomposition of hydrocarbons on
certain faces of the metal particle. The second step involves dissolution of some of the
carbon species into the bulk and diffusion through the metal particle from the hotter
Figure 7. TEM image of single filament of carbon.[225]
Nonoxidative Activation of Methane 179
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

leading faces (as a result of exothermic hydrocarbon decomposition) to the cooler rear
faces, where carbon is precipitated from the solution to form CF. Further growth is
inhibited when the hydrocarbon decomposition is prevented due to encapsulation of the
leading face of the metal particle by a carbon layer. The above model and others as well
advocate the presence of a temperature gradient as the driving force for the bulk diffusion
of carbon through the metal particle.[253,254]
However, the temperature gradient-driven diffusion model encounters difficulty in
giving a completely satisfactory explanation for endothermic surface reactions, such as
methane decomposition, which have led to the postulate that a concentration gradient is
the driving force for the diffusion.[255 – 260] Rostrup-Nielsen and Trimm proposed the
presence of a concentration gradient[255] due to the higher solubility of carbon in metal in a
metal-gas system as compared to that of a metal carbon system. Studies by Sacco,
et al.,[256] and Kock et al.[257,261] suggested that metal carbides were necessary
intermediates for CF growth. Based on the magnetization measurements and temperature-
programmed reduction experiments, Kock et.al.[257] proposed that the driving force for
filament growth was a gradient in carbon content of “nonstoichiometric carbides.”
Drawing from the work by Schouten et al.[61,62] involving methane decomposition on
single crystals, Alstrup modified the “carbide intermediates” model, suggesting the
presence of a surface carbide (and not bulk carbides) during the steady-state growth of
carbon filaments.[258] Lund and coworkers, on the other hand, disputed the necessity for
the assumption of the surface carbide species.[262] Recently, Snoeck et al.[260] have
presented a thermodynamic basis for the difference in solubility at the metal–gas and
metal–carbon interface. The gas-phase carbon solubility was found to equal the solubility
of CF in nickel at the coking threshold.
As mentioned earlier, CF formation in processes such as steam reforming can lead to
catalyst destruction and clogging of the reactor. Based on controlled-atmosphere electron-
microscopy experiments, Baker and coworkers proposed a mechanism for coke formation
in steam-cracker tubes.[246] According to this mechanism, CF formed on Ni or Fe–Ni
particles, on account of having large surface area, provide excellent collection centers for
amorphous carbon. Inhibition of these CF would thus result in diminishing the problem of
accumulation of amorphous carbon and, hence, the coking problem. With this objective,
Baker and coworkers embarked on a series of investigations dealing with various aspects
of CF formation.[263] The effect of a number of oxide additives on the yield of CF formed
from the interaction of acetylene with Ni–Fe surfaces was investigated. The studies
indicated that while MoO3, WO3, and Ta2O5 hindered CF formation by only reducing the
carbon solubility in the catalyst particles, SiO2 reduced both carbon solubility as well as
diffusion through the catalyst particle.
Rostrup-Nielsen investigated the effect of added sulfur on carbon formation from
methane decomposition as well as methane steam-reforming. It was found that the added
sulfur had a larger retarding effect on the rate of carbon formation than on the steam-
reforming reaction.[264] The explanation provided for the above was that the nucleation of
carbon required a larger number of active catalyst sites compared to the steam-reforming
reaction. Above a sulfur surface coverage of 0.7, filamentous carbon was no longer
observed and instead amorphous carbon having a plate-like shape or octopus carbon
(having several amorphous-like carbon threads attached to a single metal particle) was
observed. Bernardo et al. extended this study to the silica-supported Ni–Cu alloy
Choudhary, Aksoylu, and Goodman180
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

systems.[265,266] The investigation indicated that low loadings of Cu resulted in the
formation of primarily filamentous carbon, whereas octopus carbon was predominantly
formed at 80 at% Cu content.
Carbon filaments have various features such as high surface area, unique adsorption
properties, and interesting mechanical/electrical properties, all of which manifest their
potential in various applications. The abundance of methane makes it a prime feedstock
for growth of filamentous carbon. Several investigations have been undertaken with
regard to CF growth and properties optimization. Tibbets and coworkers studied
methane decomposition on nanometer-sized Fe particles for CF formation.[259] It was
found that deposition of ca. 2.8 nm layer of carbon resulted in the inhibition of CF
growth. Bennissad et al.[267,268] investigated CF formation on Fe-based catalysts using
CH4–H2 mixtures at temperatures to 1423K. Under these condition, thicker fibers (ca
1m) were obtained, but when heating was stopped at 1323K, the normal structure of CF
was observed.
Baker and coworkers investigated CF formation from acetylene, ethylene, and
methane decomposition on Ni and Ni–Cu catalysts.[269,270] The investigation revealed
that the nature of the catalyst particles strongly affected the structural characteristics of
CF. While particles rich in Ni resulted in the formation of smooth filaments, Cu-rich alloy
particles gave rise to filaments having a spiral conformation. The filament size (25–
100 nm) was found to be strongly dependent on particle size of the catalyst. Rodriguez,[247]
in a review on CF, stressed that a “supported-metal particle arrangement” was favorable
for tuning the CF size within a desired range.
Fenelonov et al. investigated CF formation on various Ni-based catalysts.[271] The
texture of the filamentous carbon resulting from methane decomposition was found to
weakly depend on the texture and composition of the catalysts employed. The adsorption
properties of CF as measured by the authors were a clear indication of the potential use of
these materials as adsorbents. Shaikhutdinov and coworkers carried out an extensive study
on CF formation from methane on coprecipitated Ni–alumina and Ni–Cu–alumina
catalysts.[272 – 274] The XRD analysis indicated that, unlike the Ni–alumina catalyst, the
Ni–Cu–alumina catalyst did not exist in a spinel form.[272] Surface characterization using
AES indicated that introduction of Cu resulted in greater dispersion of alumina over the
surface. The amount of CF formed per gram of the catalyst (Gc) increased with increasing
Ni content in the Ni–alumina catalyst, but was dramatically small for pure Ni powder.[273]
The maximum Gc ¼ 240 gCF=gcat; was obtained for a 3% Cu–87% Ni catalyst. Addition
of Cu decreased the rate of CF formation but greatly increased the life-time of the catalyst.
Also, for samples with more than 9 wt% Cu, some octopus carbon was observed in good
agreement with studies by Rostrup-Nielsen.[266] Based on their study, the authors divided
the CF formation process into three stages. The first step, which was the induction period,
involved the dissolution of carbon into the Ni particles leading to the formation of pear-
shaped particles in the Ni–alumina catalysts and quasi-octahedral shaped ones in case of
the Ni–Cu–alumina catalysts. The second stage involved the lengthening of the
filamentous carbon. Finally, the last stage was catalyst deactivation, which was attributed
to fragmentation or encapsulation of the catalyst particle by carbon. The morphology and
surface structure of CF (F1), produced on Ni–alumina catalysts, and carbon (F2),
produced in case of Ni–Cu–alumina catalysts, were studied by STM and high-resolution
transmission electron microscopy (HRTEM).[274] The carbon surface of the filaments was
Nonoxidative Activation of Methane 181
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

found to be rough and was proposed to be formed by misoriented edge planes of graphite
crystallites. In case of the F1, the basal graphite planes lay inclined to the fiber axis,
whereas the basal planes were perpendicular to the filament axis for F2. The HRTEM
micrographs indicated a closed-layer structure on the edges for F2, which was contrary to
the open structure observed for graphite crystallites.
Chen et al.[275] found that the decomposition of CH4/CO on Ni–MgO catalysts
resulted in the formation of small- and uniform-diameter carbon nano-tubes (outer
diameter: 15–20). Experimental conditions such as feed gas-employed, space velocity,
and reaction temperature were found to influence the rate-determining step of the process.
In a subsequent study, Holmen and coworkers[276] observed that the rate determining step
of the filamentous carbon growth on Ni/a-alumina was dependent on the carbon
deposition. Also, the induction and autoacceleration periods were influenced by the
presence of hydrogen and promoters such as Mg and Ca.
Kuvshinov and coworkers observed that the variation of the CH4:H2 feed ratio
could result in modification of the CF texture.[277] Their work also suggested that the
surface area of carbon growth centers was an important parameter for determining the
maximum CF yield on Ni catalysts using pure methane. Li et al employed a Ni–
alumina catalyst prepared from Feitknecht compound for producing CF from
methane.[278] The total amount of CF formed was found to increase with increasing Ni
content of the catalyst. Though the rate of CF formation was higher in this work
(compared to Shaikhutdinov and coworkers[274]) the total amount of CF formed in the
latter case was greater.
Contrary to previous studies,[255,257,270,274] no induction period was observed in this
work. The total amount of CF formed was dependent on the reduction temperature as well
as the reaction temperature. Although the rate of CF formation increased at higher
temperature, there was a decrease in the total yield of CF due to rapid deactivation of the
catalyst. The study was extended to study the doping effects of Cu on the CF
formation.[279] In this work, addition of small amounts of Cu not only increased the total
amount of CF formed, but also increased the growth rate at 873K. This was unlike the
work by Shaikhutdinov et al.,[274] where addition of small amount of Cu had decreased the
growth rate but increased the overall CF yield by greatly increasing the lifetime of the
catalyst. As previously observed,[274] addition of larger amounts of Cu had a detrimental
effect on the performance of the catalyst. Interestingly, it was found that the high-Cu-
content catalyst was the most effective catalyst for CF formation at higher temperature
when methane was co-fed with hydrogen.
Recent studies by Shaikhutdinov et al.[280] indicated that CF produced on the Cu–
Ni–alumina catalyst was an excellent support material for Ni in the methane
decomposition reaction. Comparison of this work with their previous studies[274] led
the authors to believe that the initial growth mechanism for CF formation was
independent of the support. However, the texture and composition of the support was
found to have a profound effect on the stability of the catalyst for accumulation of
carbon. X-ray diffraction studies[281] on high-loaded Ni–silica showed that the nickel
particles seemed to “self-organize” to the optimum size (30–40 nm) during the course
of the methane decomposition reaction, i.e., smaller particles underwent sintering to
form larger particles, whereas larger particles were found to undergo dispersion. The
authors proposed that deactivation occurred when the distance separating the metal
Choudhary, Aksoylu, and Goodman182
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

particles (present on filament ends) increased to an extent such that reversible merging
and dispersion of the Ni particles was prevented.
Carbon filaments formation on Co–alumina catalysts was found to be maximized at
60–75% content of Co.[282] No induction period was observed for the Co–
alumina catalysts, which was contrary to their previous experience involving Ni–alumina
catalysts.[273] Also in this case, a different variety of filaments (not observed on Ni-based
catalysts) with hallow-like core morphology were observed. Recently, Ermakova and
coworkers[283,284] achieved a very large yield of CF from methane decomposition at 823K.
The Ni-based catalyst employed for the process was synthesized by impregnation of nickel
oxide with a solution of the precursor of a textural promoter (silica, alumina, titanium
dioxide, zirconium oxide, and magnesia). The 90% Ni–10% silica catalyst was found to
be the most effective catalyst with a total CF yield of 375 gCF/gcat. The maximum yield of
CF was found to strongly depend on the Ni particle size. The optimum particle size
(10–40 nm) was obtained by varying the calcination temperature of NiO.
Recent studies by our group addressing methane decomposition on Ni-based catalysts
revealed a dependence of the type of carbon formed on the support employed.[225]
Filamentous carbon was observed on Ni/SiO2, Ni/HY, and Ni/SiO2/Al2O3 over the entire
temperature range studied (723–973K) whereas filamentous carbon was observed on
Ni/HZSM-5 at lower temperatures (723K), but not at high temperatures (.923K). At
higher temperatures an encapsulating type of carbon was observed which resulted in rapid
deactivation of the catalyst.
VII. SYNTHESIS OF DIAMOND FILMS
In this section, the formation of diamond-like (DCL) films from methane mixtures
will be briefly addressed. The interest in these films stem from their unique properties;
good thermal conductivity, extreme hardness, chemical intertness, low friction coefficient,
and optical transparency. These properties can be exploited in applications such as optical
coatings, wear-resistant coatings, heat sinks for high-power devices and microelectronic
devices.
Diamonds occurring naturally or those prepared at high pressures are in the form of
powder or grains, but the material in this form precludes the exploitation of its properties
in practical applications. Working at low pressures, Derjaguin et al.[285] Aisenberg and
Chabot,[286] Weissmantel et al.,[287] and Angus et al.[288] pioneered the synthesis of
diamond in the form of thin films. The DLC films are generally deposited by the chemical-
vapor deposition (CVD) methods, including hot-filament CVD[289 – 291] electron-assisted
CVD,[292,293] direct current,[294,295] radio frequency[296 – 299] microwave-plasma
CVD,[300 – 302] laser-induced CVD,[303] and combustion methods.[304]
Methane is one of the most commonly used carbon sources for growth of diamond-
like films. Work in this area has been primarily directed towards obtaining good-quality
DLC films at fast deposition rates and understanding the various mechanisms involved in
the film synthesis.[305 – 310] The general (simplistic form) mechanism involves the
dissociation of molecular hydrogen into H atoms, which then assists the reactions between
diluted gaseous hydrocarbons. The hydrocarbonaceous radicals (transient active species)
formed then subsequently transform to diamond. The concentration of methane in
Nonoxidative Activation of Methane 183
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

the starting mixture is found to strongly affect the quality of the synthesized
film.[306,311,312] Since large initial-methane concentration results in increasing the rate
of DLC film synthesis, efforts have been directed towards obtaining good-quality DLC
films from high-concentration mixtures of methane.[306,313] Based on isotopic labeling and
high gas flow-rate studies, methane was found to be distinctly more efficient than
acetylene for DLC film growth.[314] A large number of techniques have been used to
evaluate the quality of the DLC films viz. Raman spectroscopy,[315 – 317] x-ray
diffraction,[316,318,319] scanning electron microscopy,[305,320,321] EELS,[322] XPS,[320] and
TEM.[322] Detailed information pertaining to diamond-like films may be obtained from a
number of excellent reviews found in the literature.[323 – 326]
VIII. CONCLUDING REMARKS
The scientific community has put enormous efforts into the study of methane
activation. Fundamental surface science studies related to methane on transition metal
surfaces have been effectively employed to address the problem. These studies have
provided invaluable information relevant to promoter/poison effects, reaction
mechanisms, structure sensitivity/insensitivity for reactions, etc., in heterogeneous
catalysis. High-temperature coupling of methane to produce C2þ hydrocarbons has
been studied extensively from the early part of the twentieth century and the process is
now well understood. Unfortunately, the stringent conditions of extremely high
temperatures and low yields has resulted in limiting the widespread use of this
technology. The low-temperature two-step methane coupling process represents an
interesting approach, but suffers from disadvantages such as low yields and catalyst
deactivation. The dehydroaromatization is scientifically very interesting; however, the
process is severely limited by thermodynamics. Further work is required before any of
the above techniques become commercially viable. Methane decomposition to produce
hydrogen for fuel-cell devices seems to be an attractive method for natural gas
utilization. The by-products of the decomposition process such as carbon nanotubes,
pyrolytic carbon, and carbon black also have applications. Currently considerable
efforts are being directed toward exploring this possibility in our laboratory and
elsewhere. Filamentous carbon formation has been extensively studied to meet two
different objectives: first to eliminate or drastically reduce the formation of carbon
filaments on the catalysts in the steam reforming of methane, and second, to enhance
the rate of filamentous carbon formation. The unique properties exhibited by these
nano-structures can be exploited in a wide variety of applications. Continued research
efforts have been also made towards producing better-quality diamond films at
acceptable deposition rates.
Along with the aforementioned processes, various other routes of methane conversion
(steam, oxygen, etc.) are being explored (these have not been reviewed in this work).
Though no technology currently represents a panacea for the methane conversion
problem, the relentless efforts of the scientific community should hopefully soon lead to
the successful utilization of methane more economically and energy efficiently, while
minimizing environmental pollution.
Choudhary, Aksoylu, and Goodman184
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

ACKNOWLEDGMENTS
We would like to thank all the researchers whose contribution has been used in this
review. We would like to acknowledge the support of Department of Energy, Office of
Basic Sciences, and the Robert Welch Foundation. TVC acknowledges V.R. Choudhary
for helpful discussions and the Link Foundation for the Link Energy Fellowship.
REFERENCES
1. Hickman, D.A.; Schmidt, L.D. Production of syngas by direct catalytic-oxidation
of methane. Science 1993, 259, 343–346.
2. Ito, T.; Lunsford, J.H. Synthesis of ethylene and ethane by partial oxidation of
methane over lithium-doped magnesium-oxide. Nature 1985, 314, 721–722.
3. Ashcroft, A.T.; Cheetham, A.K.; Foord, J.S.; Green, M.L.H.; Grey, C.P.; Murrell,
A.J.; Vernon, P.D.F. Selective oxidation of methane to synthesis gas-using
transition-metal catalysts. Nature 1990, 344, 319–321.
4. Periana, R.A.; Taube, D.J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Platinum
catalysts for the high-yield oxidation of methane to a methanol derivative. Science
1998, 280, 560–564.
5. Solymosi, F.; Kutsan, G.; Erdohelyi, A. Catalytic reaction of CH4 with CO2 over
alumina-supported Pt metals. Catal. Lett. 1991, 11, 149–156.
6. Wu, M.C.; Truong, C.M.; Coulter, K.; Goodman, D.W. Investigations of active-
sites for methane activation in the oxidative coupling reaction over pure and Li-
promoted MgO catalysts. J. Catal. 1993, 140, 344–352.
7. Periana, R.A.; Taube, D.J.; Evitt, E.R.; Loffler, D.G.; Wentrcek, P.R.; Voss, G.;
Masuda, T. A mercury-catalyzed, high-yield system for the oxidation of methane to
methanol. Science 1993, 259, 340–343.
8. Choudhary, V.R.; Rajput, A.M.; Prabhakar, B. Low-temperature oxidative
conversion of methane to syngas over NiO–CaO catalyst. Catal. Lett. 1992, 15,
363–370.
9. Poirier, M.G.; Sanger, A.R.; Smith, K.J. Direct catalytic conversion of methane.
Can. J. Chem. Eng. 1991, 69, 1027–1035.
10. Yamagata, N.; Tanaka, K.; Sasaki, S.; Okazaki, S. Oxidative coupling of methane
over BaO mixed with CaO and MgO. Chem. Lett. 1987, 81–82.
11. Bharadwaj, S.S.; Schmidt, L.D. Synthesis gas-formation by catalytic-oxidation of
methane in fluidized-bed reactors. J. Catal. 1994, 146, 11–21.
12. Kasztelan, S.; Moffat, J.B. Partial oxidation of methane by oxygen over silica.
J. Chem. Soc.—Chem. Commun. 1987, 1663–1664.
13. Sigl, M.; Bradford, M.C.J.; Knozinger, H.; Vannice, M.A. CO2 reforming of
methane over vanadia-promoted Rh/SiO2 catalysts. Top. Catal. 1999, 8, 211–222.
14. Rezgui, S.; Liang, A.; Cheung, T.K.; Gates, B.C. Methane conversion to ethane in
the presence of iron- and manganese-promoted sulfated zirconia. Catal. Lett. 1998,
53, 1–2.
15. Campbell, K.D.; Zhang, H.; Lunsford, J.H. Methane activation by the lanthanide
oxides. J. Phys. Chem. 1998, 92, 750–753.
Nonoxidative Activation of Methane 185
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

16. Pereira, P.; Lee, S.H.; Somorjai, G.A.; Heinemann, H. The conversion of methane
to ethylene and ethane with near total selectivity by low-temperature (less-than
6108C) oxydehydrogenation over a calcium–nickel–potassium oxide catalyst.
Catal. Lett. 1990, 6, 255–262.
17. Anderson, J.R. Methane to higher hydrocarbons—review. Appl. Catal. 1989, 47,
177–196.
18. Lunsford, J.H. Catalytic conversion of methane to more useful chemicals and fuels:
a challenge for the 21st century. Catal. Today 2000, 63, 165–174.
19. Renesme, G.; Saint-Just, J.; Muller, Y. Transportation fuels and chemicals directly
from natural-gas—how expensive. Catal. Today 1992, 13, 371–380.
20. Parkyns, N.D.; Warburton, C.I.; Wilson, J.D. Natural-gas conversion to liquid fuels
and chemicals—where does it stand. Catal. Today 1993, 18, 385–442.
21. RostrupNielsen, J.R. Catalysis and large-scale conversion of natural-gas. Catal.
Today 1994, 21, 257–267.
22. Crabtree, R.H. Current ideas and future-prospects in metal-catalyzed methane
conversion. Nat. Gas Conversion II 1994, 81, 85–92.
23. Guczi, L.; Vansanten, R.A.; Sarma, K.V. Low-temperature coupling of methane.
Catal. Rev.—Sci. Eng. 1996, 38, 249–296.
24. Gesser, H.D.; Hunter, N.R. A review of C-1 conversion chemistry. Catal. Today
1998, 42, 183–189.
25. Chang, C.D. Mobils methanol-to-gasoline process—reaction-mechanism. Abstr.
Pap. Am. Chem. Soc. 1998, 195, 240-Iec.
26. Vannice, M.A. Catalytic synthesis of hydrocarbons from carbon-monoxide and
hydrogen. Catal. Rev.—Sci. Eng. 1976, 14, 153–191.
27. Rostrup-Nielsen, J.R. Catalytic steam reforming. In Catal. Sci and Eng.; Anderson,
J.R., Boudart, M., Eds.; Springer: Berlin, 1984; Vol. 5, 1; RostrupNielsen, J.R.
Production of synthesis gas. Catal. Today, 1993; 18, 305–324.
28. Keller, G.E.; Bhasin, M.M. Synthesis of ethylene via oxidative coupling of
methane. 1. Determination of active catalysis. J. Catal. 1982, 73, 9–19.
29. Lee, J.S.; Oyama, S.T. Oxidative coupling of methane to higher hydrocarbons.
Catal. Rev.—Sci. Eng. 1988, 30, 249–280.
30. Lunsford, J.H. The catalytic conversion of methane to higher hydrocarbons. Catal.
Today 1990, 6, 235–259.
31. Qiu, X.Q.; Wong, N.B.; Tin, K.C.; Zhu, Q.M. Low-temperature catalysts for
oxidative coupling of methane. J. Chem. Technol. Biotechnol. 1996, 65, 303–308.
32. Choudhary, V.R.; Uphade, B.S.; Mulla, S.A.R. Oxidative coupling of methane over
a Sr-promoted La2O3 catalyst supported on a low surface-area porous catalyst
carrier. Ind. Eng. Chem. Res. 1997, 36, 3594–3601.
33. Tonkovich, A.L.; Carr, R.W.; Aris, R. Enchanced C2 yields from methane
oxidative coupling by means of a separative chemical reactor. Science 1993, 262,
221–223.
34. Jiang, Y.; Yentekakis, I.V.; Vayenas, C.G. Methane to ethylene with 85-percent
yield in a gas recycle electrocatalytic reactor-separator. Science 1994, 264,
1563–1566.
35. Choudhary, V.R.; Chaudhari, S.T.; Rajput, A.M.; Rane, V.H. Beneficial effect of
oxygen distribution on methane conversion and C-2-selectivity in oxidative
Choudhary, Aksoylu, and Goodman186
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

coupling of methane to C-2-hydrocarbons over lanthanum-promoted magnesium-
oxide. J. Chem. Soc.—Chem. Commun. 1989, 1526–1527.
36. Bjorklund, M.C.; Carr, R.W. The simulated countercurrent moving-bed
chromatographic reactor—a catalytic and separative reactor. Catal. Today 1995,
25, 159–168.
37. Choudhary, V.R.; Sanasare, S.D.; Rajput, A.M. Two-Step Process for Production of
Liquid Hydrocarbons from Natural Gas. U. S. Patent 5,306,854, April 26 1994.
38. Choudhary, V.R.; Rane, V.H.; Gadre, R.V. Influence of precursors used in
preparation of MgO on its surface-properties and catalytic activity in oxidative
coupling of methane. J. Catal. 1994, 145, 300–311.
39. Cordi, E.M.; Pak, S.; Rosynek, M.P.; Lunsford, J.H. Steady-state production of
olefins in high yields during the oxidative coupling of methane: utilization of a
membrane contractor. Appl. Catal. A—Gen. 1997, 155, L1–L7.
40. Levan, T.; Che, M.; Kermarec, M.; Louis, C.; Tatibouet, J.M. Structure sensitivity
of the catalytic oxidative coupling of methane on lanthanum oxide. Catal. Lett.
1990, 6, 395–400.
41. Reyes, S.C.; Kelkar, C.P.; Iglesia, E. Kinetic transport models and the design of
catalysts and reactors for the oxidative coupling of methane. Catal. Lett. 1993, 19,
167–180.
42. Ashcroft, A.T.; Cheetham, A.K.; Green, M.L.H.; Vernon, P.D.F. Partial oxidation
of methane to synthesis gas-using carbon-dioxide. Nature 1991, 352, 225–226.
43. Choudhary, V.R.; Rajput, A.M.; Prabhakar, B. NiO/CaO-catalyzed formation of
syngas by coupled exothermic oxidative conversion and endothermic CO2 and
steam reforming of methane. Angew. Chem.—Int. Ed. Engl. 1994, 33, 2104–2106.
44. Bradford, M.C.J.; Vannice, M.A. CO2 reforming of CH4. Catal. Rev.—Sci. Eng.
1999, 41, 1–42.
45. Muradov, N.Z. How to produce hydrogen from fossil-fuels without CO2 emission.
Int. J. Hydrogen Energy 1993, 18, 211–215.
46. Poirier, M.G.; Sapundzhiev, C. Catalytic decomposition of natural gas to hydrogen
for fuel cell applications. Int. J. Hydrogen Energy 1997, 22, 429–433.
47. Goodman, D.W. Model catalysts—from extended single-crystals to supported
particles. Surf. Rev. Lett. 1995, 2, 9–24.
48. Goodman, D.W. Correlations between surface science models and real-world
catalysts. J. Phys. Chem. 1996, 100, 13090–13102.
49. Goodman, D.W. Catalysis by metals: from extended single crystals to small
clusters. Surf. Rev. Lett. 1994, 1, 449–455.
50. Goodman, D.W. Model studies in catalysis using surface science probes. Chem.
Rev. 1995, 95, 523–536.
51. Szanyi, J.; Goodman, D.W. Combined elevated pressure reactor and ultrahigh-
vacuum surface-analysis system. Rev. Sci. Instrum. 1993, 64, 2350–2352.
52. Sexton, B.A.; Somorjai, G.A. Hydrogenation of CO and CO2 over polycrystalline
rhodium—correlation of surface composition, kinetics and product distributions.
J. Catal. 1997, 46, 167–189.
53. Campbell, C.T. Studies of model catalysts with well-defined surfaces combining
ultrahigh-vacuum surface characterization with medium-pressure and high-
pressure kinetics. Adv. Catal. 1989, 36, 1–54.
Nonoxidative Activation of Methane 187
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

54. Rodriguez, J.A.; Goodman, D.W. High-pressure catalytic reactions over single-
crystal metal-surfaces. Surf. Sci. Rep. 1991, 14, 1–107.
55. Choudhary, T.V.; Goodman, D.W. Methane activation on Ni and Ru model
catalysts. J. Mol. Catal. A—Chem. 2000, 163, 9–18.
56. Larsen, J.H.; Chorkendorff, I. From fundamental studies of reactivity on single
crystals to the design of catalysts. Surf. Sci. Rep. 1999, 35, 165–222.
57. Winters, H.F. Activated, dissociative chemisorption of methane on tungsten.
J. Chem. Phys. 1975, 62, 2454–2460.
58. Winters, H.F. Kinetic isotope-effect in dissociative chemisorption of methane.
J. Chem. Phys. 1976, 64, 3495–3500.
59. Rettner, C.T.; Pfnur, H.E.; Auerbach, D.J. Dissociative chemisorption of CH4 on
W(110)—dramatic activation by initial kinetic-energy. Phys. Rev. Lett. 1985, 54,
2716–2719.
60. Lo, T.C.; Ehrlich, G. Activated chemisorption of methane and tunneling. Surf. Sci.
1987, 179, L19–L25.
61. Schouten, F.C.; Kaleveld, E.W.; Bootsma, G.A. Aes-Leed-ellipsometry study of
kinetics of interaction of methane with Ni(110). Surf. Sci. 1977, 63, 460–474.
62. Schouten, F.C.; Gijzeman, O.L.J.; Bootsma, G.A. Interaction of methane with
Ni(111) and Ni(100)—diffusion of carbon into nickel through the (100) surface-
Aes-Leed study. Surf. Sci. 1979, 87, 1–12.
63. Lee, M.B.; Yang, Q.Y.; Ceyer, S.T. Dynamics of the activated dissociative
chemisorption of CH4 and implication for the pressure gap in catalysis—a
molecular-beam high-resolution electron-energy loss study. J. Chem. Phys. 1987,
87, 2724–2741.
64. Beebe, T.P.; Goodman, D.W.; Kay, B.D.; Yates, J.T. Kinetics of the activated
dissociative adsorption of methane on the low index planes of nickel single-crystal
surfaces. J. Chem. Phys. 1987, 87, 2305–2315.
65. Hamza, A.V.; Madix, R.J. The activation of alkanes on Ni(100). Surf. Sci. 1987,
179, 25–46.
66. Chorkendorff, I.; Alstrup, I.; Ullmann, S. XPS study of chemisorption of CH4 on
Ni(100). Surf. Sci. 1990, 227, 291–296.
67. Hanley, L.; Xu, Z.; Yates, J.T. Methane activation on Ni(111) at high-pressures.
Surf. Sci. 1991, 248, L265–L273.
68. Beckerle, J.D.; Yang, Q.Y.; Johnson, A.D.; Ceyer, S.T. Collision-induced
dissociative chemisorption of adsorbates—chemistry with a hammer. J. Chem.
Phys. 1987, 86, 7236–7237.
69. Beckerle, J.D.; Johnson, A.D.; Yang, Q.Y.; Ceyer, S.T. Collision-induced
dissociative chemisorption of CH4 on Ni(111) by inert-gas atoms—the mechanism
for chemistry with a hammer. J. Chem. Phys. 1989, 91, 5756–5777.
70. Yang, Q.Y.; Johnson, A.D.; Maynard, K.J.; Ceyer, S.T. Synthesis of benzene
from methane over a Ni(111) catalyst. J. Am. Chem. Soc. 1989, 111,
8748–8749.
71. Jiang, X.; Goodman, D.W. The effect of sulfur on the dissociative adsorption of
methane on nickel. Catal. Lett. 1990, 4, 173–180.
72. Campbell, R.A.; Szanyi, J.; Lenz, P.; Goodman, D.W. Methane activation on clean
and oxidized Ni(100). Catal. Lett. 1993, 17, 39–46.
Choudhary, Aksoylu, and Goodman188
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

73. Holmblad, P.M.; Wambach, J.; Chorkendorff, I. Molecular-beam study of
dissociative sticking of methane on Ni(100). J. Chem. Phys. 1995, 102,
8255–8263.
74. Nielsen, B.O.; Luntz, A.C.; Holmblad, P.M.; Chorkendorff, I. Activated
dissociative chemisorption of methane on Ni(100)—a direct mechanism under
thermal conditions. Catal. Lett. 1995, 32, 15–30.
75. Juurlink, L.B.F.; Mccabe, P.R.; Smith, R.R.; Dicologero, C.L.; Utz, A.L.
Eignestate-resolved studies of gas-surface reactivity: CH4 v3 dissociation on
Ni(100). Phys. Rev. Lett. 1999, 83, 868–871.
76. Luntz, A.C.; Bethune, D.S. Activation of methane dissociation on a Pt(111)
surface. J. Chem. Phys. 1989, 90, 1274–1280.
77. Harris, J.; Simon, J.; Luntz, A.C.; Mullins, C.B.; Rettner, C.T. Thermally
assisted tunneling—CH4 dissociation on Pt(111). Phys. Rev. Lett. 1991, 67,
652–655.
78. Valden, M.; Xiang, N.; Pere, J.; Pessa, M. Dissociative chemisorption of methane
on clean and oxygen precovered Pt(111). Appl. Surf. Sci. 1996, 99, 83–89.
79. Newell, H.E.; Oakes, D.J.; Rutten, F.J.M.; Mccoustra, M.R.S.; Chesters, M.A.
Impact-induced dissociation of methane and ethane on Pt(111) and
Pt0.25Rh0.75(111). Faraday Discussions 1996, 193–207.
80. Wang, Y.N.; Herman, R.G.; Klier, K. Dissociative adsorption of methane on
Pd(679) surface. Surf. Sci. 1992, 279, 33–48.
81. Valden, M.; Pere, J.; Xiang, N.; Pessa, M. Influence of preadsorbed oxygen on
activated chemisorption of methane on Pd(110). Chem. Phys. Lett. 1996, 257,
289–296.
82. Valden, M.; Pere, J.; Hirsimaki, M.; Suhonen, S.; Pessa, M. Activated adsorption of
methane on clean and oxygen-modified Pt{111} and Pd{110}. Surf. Sci. 1997, 377,
605–609.
83. Walker, A.V.; King, D.A. Dynamics of the dissociative adsorption of methane on
Pt{110} (1 £ 2). Phys. Rev. Lett. 1999, 82, 5156–5159.
84. Seets, D.C.; Wheeler, M.C.; Mullins, C.B. Mechanism of the dissociative
chemisorption of methane over Ir(110): trapping-mediated or direct? Chem. Phys.
Lett. 1997, 266, 431–436.
85. Luntz, A.C.; Winters, H.F. Dissociation of methane and ethane on Pt(110)—
evidence for a direct mechanism under thermal conditions. J. Chem. Phys. 1994,
101, 10980–10989.
86. Gruzdkov, Y.A.; Watanabe, K.; Sawabe, K.; Matsumoto, Y. Photochemical CZH
bond activation of methane on a Pt(111) surface. Chem. Phys. Lett. 1994, 227,
243–247.
87. Zhou, X.L.; Zhu, X.Y.; White, J.M. Photochemistry at adsorbate metal interfaces.
Surf. Sci. Rep. 1991, 13, 73–220.
88. Yoshinobu, J.; Ogaswara, H.; Kawai, M. Symmetry-controlled surface photo-
chemistry of methane on Pt(111). Phys. Rev. Lett. 1995, 75, 2176–2179.
89. Alberas-Sloan, D.J.; White, J.M. Low-energy electron irradiation of methane on
Pt(111). Surf. Sci. 1996, 365, 212–228.
90. Brass, S.G.; Ehrlich, G. Activated chemisorption—internal degrees of freedom and
measured activation-energies. Phys. Rev. Lett. 1983, 57, 2532–2535.
Nonoxidative Activation of Methane 189
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

91. Brass, S.G.; Ehrlich, G. Internal molecular motions and activated chemisorption—
CH4 on rhodium. J. Chem. Phys. 1987, 87, 4285–4293.
92. Brass, S.G.; Ehrlich, G. Dissociative and molecular adsorption of methane on
rhodium. Surf. Sci. 1987, 187, 21–35.
93. Brass, S.G.; Ehrlich, G. Activated chemisorption of CH4 and internal molecular
motions. Surf. Sci. 1987, 191, L819–L824.
94. Seets, D.C.; Reeves, C.T.; Ferguson, B.A.; Wheeler, M.C.; Mullins, C.B.
Dissociative chemisorption of methane on Ir(111): evidence for direct and
trapping-mediated mechanisms. J. Chem. Phys. 1997, 107, 10229–10241.
95. Jachimowski, T.Q.; Hagedorn, C.J.; Weinberg, W.H. Direct and trapping-mediated
dissociative chemisorption of methane on Ir(111). Surf. Sci. 1997, 393, 126–134.
96. Goodman, D.W.; White, J.M. Measurement of active-carbon on ruthenium(110)—
relevance to catalytic methanation. Surf. Sci. 1979, 90, 201–203.
97. Wu, M.C.; Goodman, D.W. High-resolution electron-energy-loss measurements of
sticking coefficients of methane decomposition on Ru(0001). Surf. Sci. 1994, 306,
L529–L533.
98. Larsen, J.H.; Holmblad, P.M.; Chorkendorff, I. Dissociative sticking of CH4 on
Ru(0001). J. Chem. Phys. 1999, 110, 2637–2642.
99. Wu, M.C.; Goodman, D.W. High-resolution electron energy-loss studies of
hydrocarbon formation from methane decomposition on Ru(0001) and Ru(1120)
catalysts. J. Am. Chem. Soc. 1994, 116, 1364–1371.
100. Wu, M.C.; Xu, Q.; Goodman, D.W. Investigations of graphitic overlayers formed
from methane decompostion on Ru(0001) and Ru(1120) catalysts with scanning-
tunneling-microscopy and high-resolution electron-energy-loss spectroscopy.
J. Phys. Chem. 1994, 98, 5104–5110.
101. Sivadinarayana, C.; Choudhary, T.V.; Daemon, L.; Eckert, J.; Goodman, D.W.
Characterization of C2(CxHy) intermediates from adsorption and decomposition of
methane on supported-metal catalysts by in-situ inelastic neutron scattering
vibrational spectroscopy. Angew. Chem. Int. Ed. 2002, 41, 144–164.
102. Koerts, T.; Vansanten, R.A. A low-temperature reaction sequence for methane
conversion. J. Chem. Soc.—Chem. Commun. 1991, 1281–1283.
103. Belgued, M.; Pareja, P.; Amariglio, A.; Amariglio, H. Conversion of methane into
higher hydrocarbons on platinum. Nature 1991, 352, 789–790.
104. Koerts, T.; Deelen, M.J.A.G.; Vansanten, R.A. Hydrocarbon formation from
methane by a low-temperature 2-step reaction sequence. J. Catal. 1992, 138,
101–114.
105. McCarty, J.G.; Wise, H. Hydrogenation of surface carbon on alumina-supported
nickel. J. Catal. 1979, 57, 406–416.
106. Belgued, M.; Amariglio, H.; Pareja, P.; Amariglio, A.; Saintjust, J. Low-
temperature catalytic homologation of methane on platinum, ruthenium, and
cobalt. Catal. Today 1992, 13, 437–445.
107. Belgued, M.; Amariglio, A.; Lefort, L.; Pareja, P.; Amariglio, H. Oxygen-free
conversion of methane to higher alkanes through an isothermal two-step reaction on
ruthenium. J. Catal. 1996, 161, 282–291.
108. Amariglio, A.; Belgued, M.; Pareja, P.; Amariglio, H. Oxygen-free conversion of
methane to higher hydrocarbons through a dual-temperature two-step reaction
Choudhary, Aksoylu, and Goodman190
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

sequence on platinum and ruthenium—1. Chemisorption of CH4 at a fixed
temperature. J. Catal. 1998, 177, 113–120.
109. Amariglio, A.; Pareja, P.; Beglued, M.; Amariglio, H. Possibility of obtaining
appreciable yields in methane homologation through 2-step reaction at 2508C on a
platinum catalyst. J. Chem. Soc.—Chem. Commun. 1994, 561–562.
110. Pareja, P.; Amariglio, A.; Belgued, M.; Amariglio, H. Increasing the yield in
methane homologation through an isothermal 2-reaction sequence at 2508C on
platinum. Catal. Today 1994, 21, 423–430.
111. Amariglio, H.; Pareja, P.; Amariglio, A. Periodic operation of a catalyst as a means
of overcoming a thermodynamic constraint—the case of methane homologation on
metals. Catal. Today 1995, 25, 113–125.
112. Amariglio, H.; Belgued, M.; Pareja, P.; Amariglio, A. Carbon-monoxide induced
desorption of alkanes and alkenes up to C-8 after chemisorption of methane on
platinum. Catal. Lett. 1995, 31, 19–26.
113. Belgued, M.; Amariglio, A.; Pareja, P.; Amariglio, H. Oxygen-free conversion of
methane to higher alkanes through an isothermal two-step reaction on platinum
(Europt-1). 1. Chemisorption of methane. J. Catal. 1996, 159, 441–448.
114. Hlavathy, Z.; Paal, Z.; Tetenyi, P. Oxygen-free conversion of methane to higher
alkanes through an isothermal two-step reaction on platinum (Europt-1)—
comment. J. Catal. 1997, 166, 118–120.
115. Amariglio, A.; Pareja, P.; Amariglio, H. Does CZC bonding proceed during
exposure of adequate metal surfaces to CH4? Reply. J. Catal. 1997, 166, 121–123.
116. Solymosi, F.; Erdohelyi, A.; Cserenyi, J. A comparative-study on the activation and
reactions of CH4 on supported metals. Catal. Lett. 1992, 16, 399–405.
117. Solymosi, F.; Erdohelyi, A.; Cserenyi, J.; Felvegi, A. Decomposition of CH4 over
supported Pd catalysts. J. Catal. 1994, 147, 272–278.
118. Solymosi, F.; Cserenyi, J. Decomposition of CH4 over supported Ir catalysts. Catal.
Today 1994, 21, 561–569.
119. Koranne, M.M.; Goodman, D.W.; Zajac, G.W. Direct conversion of methane to
higher hydrocarbons via an oxygen-free, low-temperature route. Catal. Lett. 1995,
30, 219–234.
120. Carstens, J.N.; Bell, A.T. Methane activation and conversion to higher
hydrocarbons on supported ruthenium. J. Catal. 1996, 161, 423–429.
121. Zadeh, J.S.M.; Smith, K.J. Kinetics of CH4 decomposition on supported cobalt
catalysts. J. Catal. 1998, 176, 115–124.
122. Boskovic, G.; Smith, K.J. Methane homologation and reactivity of carbon species
on supported Co catalysts. Catal. Today 1997, 37, 25–32.
123. Zadeh, J.S.M.; Smith, K.J. Two-step homologation of methane on supported Co
catalysts. J. Catal. 1999, 183, 232–239.
124. Boskovic, G.; Zadeh, J.S.M.; Smith, K.J.K. Promotion of Co catalysts for the two-
step methane homologation reaction. Catal. Lett. 1996, 39, 163–168.
125. Solymosi, F.; Cserenyi, J. Enhanced formation of ethane in the conversion of
methane over Cu–Rh/SiO2. Catal. Lett. 1995, 34, 343–350.
126. Wang, Z.J.; Rochester, C.H.; Anderson, J.A. Decomposition of methane and
subsequent reaction of carbonaceous residues over Rh/Mo/Al2O3 catalysts. J. Catal.
1999, 184, 213–223.
Nonoxidative Activation of Methane 191
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

127. Guczi, L.; Sarma, K.V.; Borko, L. Non-oxidative methane coupling over Co–
Pt/NaY bimetallic catalysts. Catal. Lett. 1996, 39, 43–47.
128. Guczi, L.; Koppany, Z.; Sarma, K.V.; Borko, L.; Kiricsi, I. Structure and catalytic
activity of Co-based bimetallic systems in NaY zeolite: low temperature methane
activation. Prog. Zeolite Microporous Mater., Pts A–C 1997, 105, 861–868.
129. Guczi, L.; Sarma, K.V.; Borko, L. Low-temperature methane activation under
nonoxidative conditions over supported ruthenium–cobalt bimetallic catalysts.
J. Catal. 1997, 167, 495–502.
130. Lu, G.M.; Hoffer, T.; Guczi, L. Reducibility and co-hydrogenation over Pt and Pt–
Co bimetallic catalysts encaged in NaY-zeolite. Catal. Lett. 1992, 14, 207–220.
131. Garnier, O.; Shu, J.; Grandjean, B.P.A. Membrane-assisted two-step process for
methane conversion into hydrogen and higher hydrocarbons. Ind. Eng. Chem. Res.
1997, 36, 553–558.
132. Marceau, E.; Tatibouet, J.M.; Che, M.; Saint-Just, J. Influence of the hydrogenation
step on selectivity during the nonoxidative oligomerization of methane to alkanes
on Pt/SiO2 catalysts (Europt-1). J. Catal. 1999, 183, 384–395.
133. Bradford, M.C.J. Isothermal, non-oxidative, two-step CH4 conversion over
unsupported and supported Ru and Pt catalysts. Catal. Lett. 2000, 66, 113–120.
134. Simon, F.; Molina, S.; Amariglio, A.; Pareja, P.; Amariglio, H.; Szabo, G.
Cyclohexane, methyl- and 1,2-dimethyl-cyclohexane as the major C2 þ products
of an oxygen-free CH4 conversion. Catal. Today 1998, 46, 217–222.
135. Lenz-Solomun, P.; Wu, M.C.; Goodman, W. Methane coupling at low-
temperatures on Ru(0001) and Ru(1120) catalysts. Catal. Lett. 1994, 25, 75–86.
136. Wu, M.C.; Lenzsolomun, P.; Goodman, D.W. 2-step, oxygen-free route to higher
hydrocarbons from methane over ruthenium catalysts. J. Vacuum Sci. Technol.
A—Vacuum Surf. Films 1994, 12, 2205–2209.
137. Choudhary, T.V.; Goodman, D.W. Methane activation on ruthenium: the nature of
surface intermediates. Top. Catal. 2002, 20, 35–42.
138. Billaud, F.; Gueret, C.; Weill, J. Thermal-decomposition of pure methane at 1263-
K—experiments and mechanistic modeling. Thermochim. Acta 1992, 211,
303–322.
139. Kuguchin, A.E.; Vnukov, S.P. Effect of inert-gases on the rate of pyrocarbon
formation in the thermal-decomposition of methane. Kinet. Catal. 1990, 31,
641–643.
140. Olsvik, O.; Billaud, F. Modeling of the decomposition of methane at 1273K in a
plug-flow reactor at low conversion. J. Anal. Appl. Pyrolysis 1993, 25, 395–405.
141. Zaslonko, I.S.; Smirnov, V.N.; Tereza, A.M. A kinetic-model of high-temperature
decomposition of methane and ethane. Kinet. Catal. 1994, 35, 1–11.
142. Gueret, C.; Daroux, M.; Billaud, F. Methane pyrolysis: thermodynamics. Chem.
Eng. Sci. 1997, 52, 815–827.
143. Holmen, A.; Rokstad, O.A.; Solbakken, A. High-temperature pyrolysis of
hydrocarbons. 1. Methane to acetylene. Ind. Eng. Chem. Process Design Dev.
1976, 15, 439–444.
144. Sun, Q.; Tang, Y.C.; Gavalas, G.R. Methane pyrolysis in a hot filament reactor.
Energy Fuels 2000, 14, 490–494.
145. Kassel, L.S. J. Am. Chem. Soc. 1932, 54, 3949.
Choudhary, Aksoylu, and Goodman192
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

146. Olsvik, O.; Billaud, F. Thermal coupling of methane—a comparison between
kinetic-model data and experimental-data. Thermochim. Acta 1994, 232, 155–169.
147. Holmen, A.; Olsvik, O.; Rokstad, O.A. Pyrolysis of natural-gas-chemistry and
process concepts. Fuel Process. Technol. 1995, 42, 249–267.
148. Arutyunov, V.S.; Vedeneev, V.I.; Moshkina, R.I.; Ushakov, V.A. Pyrolysis of
methane under static conditions at 1100–1400K. Kinet. Catal. 1991, 32,
234–240.
149. Egloff, G. Decomposition of the paraffins hydrocarbons. J. Phys. Chem. 1930, 34,
1617.
150. Storch, H.H. Acetylene formation in thermal decomposition of hydrocarbons. Ind.
Eng. Chem. 1934, 26, 56.
151. Kramer, L.; Happel, J. Acetylene by the Pyrolysis of Light Hydrocarbons, the
Chemistry of Petroleum Hydrocarbons; Brooks, B.T., Kurtz, S.S., Bond, C.E.,
Schmerling, L., Eds.; Reinhold: New York, 1955; Vol. 2, 71.
152. Khan, M.S.; Crynes, B.L. Survey of recent methane pyrolysis literature—a survey
of methane pyrolysis data is presented and discussed. Ind. Eng. Chem. 1970, 62, 54.
153. Omar, K.I. The pyrolysis of methane. Chem. Eng. Res. Bull. 1982, 6, 29.
154. Billaud, F.; Baronnet, F.; Freund, E.; Busson, C.; Weill, J. Thermal-decomposition
of methane—biliographic study and proposal of a mechanism. Revue De L Institut
Francais Du Petrole 1989, 44, 813–823.
155. Skinner, G.B.; Ruehrwein, R.A. Shock tube studies of the pyrolysis and oxidation
of methane. J. Phys. Chem. 1959, 63, 1736–1742.
156. Holmen, A.; Rokstad, O.A.; Solbakken, A. High-temperature pyrolysis of
hydrocarbons. 2. Naphtha to acetylene. Ind. Eng. Chem. Process Design Dev.
1979, 18, 653–657.
157. Khan, M.S.; Crynes, B.L. Survey of recent methane pyrolysis literature—a survey
of methane pyrolysis data is presented and discussed. Ind. Eng. Chem. 1970, 62,
54–59.
158. Kunugi, T.; Tamura, T.; Naito, T. New acetylene process uses hydrogen dilution.
Chem. Eng. Prog. 1961, 57, 43–49.
159. Kevorkian, V.; Health, C.; Boudart, M. The decomposition of methane in shock
waves. J. Phys. Chem. 1960, 64, 964–968.
160. Eisenberg, B.; Bliss, H. Kinetics of methane pyrolysis. Chem. Eng. Prog., Symp.
Ser. 1967, 63 (72), 3–17.
161. Billaud, F.G.; Baronnet, F.; Gueret, C.P. Thermal coupling of methane in a tubular
flow reactor—parametric study. Ind. Eng. Chem. Res. 1993, 32, 1549–1554.
162. Blekkan, E.A.; Myrstad, R.; Olsvik, O.; Rokstad, O.A. Characterization of tars and
coke formed during the pyrolysis of methane in a tubular reactor. Carbon 1992, 30,
665–673.
163. Lahaye, J.; Prado, G. Mechanisms and Nucleation and Growth of Carbon Black;
ACS Symp. Ser. 21; 1976; 335.
164. Palmer, H.B.; Lahaye, J.; Hou, K.C. On kinetics mechanism of thermal
decomposition of methane in a flow system. J. Phys. Chem. 1968, 72, 348.
165. Gladdish, H. How huels makes acetylene by DC arc. Pet. Refinery 1962, 41, 159.
166. Forbath, T.P.; Gaffney, B.J. Acetylene by the BASF process. Pet. Refinery 1954,
33, 160–165.
Nonoxidative Activation of Methane 193
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

167. Bogart, M.J.P.; Long, R.H. Pyrolysis of liquid hydrocarbons via the Wulff process.
Chem. Eng. Prog. 1962, 58, 91.
168. Fang, T.; Yeh, C. Interactions of methane with ThO2–SiO2 surface at 1073-K.
J. Catal. 1981, 69, 227–229.
169. Van der Zwet, G.P.; Hendriks, P.A.J.M.; Van Santen, R.A. Pyrolysis of methane
and the role of surface area. Catal. Today 1989, 4, 365–369.
170. Mochida, I.; Aoyagi, Y.; Fujitsu, H. Pyrolysis of methane into higher hydrocarbons
on carbon-fibers. Chem. Lett. 1990, 1525–1526.
171. Kharatyan, S.L.; Chatilyan, A.A.; Merzhanov, A.G. Laws governing the high-
temperature pyrolysis of methane on a molybdenum carbide surface. Kinet. Catal.
1990, 31, 772–777.
172. Kharatyan, S.L.; Chatilyan, A.A.; Merzhanov, A.G. Kinetics of tungsten interaction
with methane. Khim. Fiz. 1987, 6, 225–233.
173. Potapova, L.L.; Cherches, B.K.; Egiazarov, Y.G. The kinetics of the pyrolysis of
methane–ethane mixtures in the presence of indium-oxide catalyst. Kinet. Catal.
1993, 34, 339–343.
174. Kushch, S.D.; Moravskii, A.P.; Muradyan, V.Y.; Fursikov, P.V. Activation of
methane on fullerene black. Pet. Chem. 1997, 37, 112–118.
175. Kurosaka, T.; Matsuhashi, H.; Arata, K. Dehydrogenative coupling of methane
catalyzed by platinum-added sulfated zirconia and characterization of the catalyst
surface. J. Catal. 1998, 179, 28–35.
176. Wang, L.S.; Tao, L.X.; Xie, M.S.; Xu, G.F.; Huang, J.S.; Xu, Y.D.
Dehydrogenation and aromatization of methane under nonoxidizing conditions.
Catal. Lett. 1993, 21, 35–41.
177. Xu, Y.D.; Liu, S.T.; Wang, L.S.; Xie, M.S.; Guo, X.X. Methane activation without
using oxidants over Mo/HZSM-5 zeolite catalysts. Catal. Lett. 1995, 30, 135–149.
178. Solymosi, F.; Erdohelyi, A.; Szoke, A. Dehydrogenation of methane on supported
molybdenum oxides—formation of benzene from methane. Catal. Lett. 1995, 32,
4353.
179. Solymosi, F.; Szoke, A.; Cserenyi, J. Conversion of methane to benzene over Mo2C
and Mo2C/ZSM-5 catalysts. Catal. Lett. 1996, 39, 157–161.
180. Szoke, A.; Solymosi, F. Selective oxidation of methane to benzene over
K2MoO4/ZSM-5 catalysts. Appl. Catal. A—Gen. 1996, 142, 361–374.
181. Solymosi, F.; Cserenyi, J.; Szoke, A.; Bansagi, T.; Oszko, A. Aromatization of
methane over supported and unsupported Mo-based catalysts. J. Catal. 1997, 165,
150–161.
182. Solymosi, F.; Bugyi, L.; Oszko, A.; Horvath, I. Generation and reactions of CH2
and C2H5 species on Mo2C/Mo(111) surface. J. Catal. 1999, 185, 160–169.
183. Solymosi, F.; Bugyi, L.; Oszko, A. Formation and reactions of CH3 species over
Mo2C/Mo(111) surface. J. Catal. Lett. 1999, 57, 103–107.
184. Lee, J.S.; Oyama, S.T.; Boudart, M. Molybdenum carbide catalysts. 1. Synthesis of
unsupported powders. J. Catal. 1987, 106, 125–133.
185. Wang, D.J.; Lunsford, J.H.; Rosynek, M.P. Catalytic conversion of methane to
benzene over Mo/ZSM-5. Top. Catal. 1996, 3, 289–297.
186. Wang, D.J.; Lunsford, J.H.; Rosynek, M.P. Characterization of a Mo/ZSM-5
catalyst for the conversion of methane to benzene. J. Catal. 1997, 169, 347–358.
Choudhary, Aksoylu, and Goodman194
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

187. Weckhuysen, B.M.; Wang, D.J.; Rosynek, M.P.; Lunsford, J.H. Conversion of
methane to benzene over transition metal ion ZSM-5 zeolites—I. Catalytic
Characterization. J. Catal. 1998, 175, 338–346.
188. Weckhuysen, B.M.; Rosynek, M.P.; Lunsford, J.H. Characterization of surface
carbon formed during the conversion of methane to benzene over Mo/H-ZSM-5
catalysts. Catal. Lett. 1998, 52, 31–36.
189. Weckhuysen, B.M.; Wang, D.J.; Rosynek, M.P.; Lunsford, J.H. Conversion of
methane to benzene over transition metal ion ZSM-5 zeolites—II. Catalyst
characterization by x-ray photoelectron spectroscopy. J. Catal. 1998, 175,
347–351.
190. Zhang, J.Z.; Long, M.A.; Howe, R.F. Molybdenum ZSM-5 zeolite catalysts for the
conversion of methane to benzene. Catal. Today 1998, 44, 293–300.
191. Meriaudeau, P.; Ha, V.T.T.; Tiep, L.V. Methane aromatization over Mo/H-ZSM-5:
on the reaction pathway. Catal. Lett. 2000, 64, 49–51.
192. Xu, Y.D.; Lin, L.W. Recent advances in methane dehydro-aromatization over
transition metal ion-modified zeolite catalysts under non-oxidative conditions.
Appl. Catal. A—Gen. 1999, 188, 53–67.
193. Zhang, C.L.; Li, S.A.; Yuan, Y.; Zhang, W.X.; Wu, T.H.; Lin, L.W. Aromatization
of methane in the absence of oxygen over Mo-based catalysts supported on
different types of zeolites. Catal. Lett. 1998, 56, 207–213.
194. Jiang, H.; Wang, L.S.; Cui, W.; Xu, Y.D. Study on the induction period of methane
aromatization over Mo/HZSM-5: partial reduction of Mo species and formation of
carbonaceous deposit. Catal. Lett. 1999, 57, 95–102.
195. Ma, D.; Shu, Y.Y.; Cheng, M.J.; Xu, Y.D.; Bao, X.H. On the induction period of
methane aromatization over Mo-based catalysts. J. Catal. 2000, 194, 105–114.
196. Liu, W.; Xu, Y.D. Methane dehydrogenation and aromatization over Mo/HZSM-5:
in situ FT-IR characterization of its acidity and the interaction between Mo species
and HZSM-5. J. Catal. 1999, 185, 386–392.
197. Ma, D.; Shu, Y.Y.; Bao, X.H.; Xu, Y.D. Methane dehydro-aromatization under
nonoxidative conditions over Mo/HZSM-5 catalysts: EPR study of the Mo species
on/in the HZSM-5 zeolite. J. Catal. 2000, 189, 314–325.
198. Ma, D.; Shu, Y.Y.; Zhang, W.P.; Han, X.W.; Xu, Y.D.; Bao, X.H. In situ H-1 MAS
NMR spectroscopic observation of proton species on a Mo-modified HZSM-5
zeolite catalyst for the dehydroaromatization of methane. Angew. Chem.—Int. Ed.
2000, 39, 2928–2931.
199. Shu, Y.Y.; Ma, D.; Bao, X.H.; Xu, Y.D. Methane dehydro-aromatization over a
Mo/phosphoric rare earth-containing penta-sil type zeolite in the absence of
oxygen. Catal. Lett. 2000, 66, 161–167.
200. Shu, Y.Y.; Ma, D.; Liu, X.C.; Han, X.W.; Xu, Y.D.; Bao, X.H. An MAS NMR
study on the Mo-modified phosphoric rare earth (HZRP-1) penta-sil zeolite
catalyst. J. Phys. Chem. B 2000, 104, 8245–8249.
201. Shu, Y.Y.; Ma, D.; Xu, L.Y.; Xu, Y.D.; Bao, X.H. Methane dehydro-aromatization
over Mo/MCM-22 catalysts: a highly selective catalyst for the formation of
benzene. Catal. Lett. 2000, 70, 67–73.
202. Liu, S.T.; Wang, L.; Ohnishi, R.; Ichikawa, M. Bifunctional catalysis of
Mo/HZSM-5 in the dehydroaromatization of methane to benzene and naphthalene
Nonoxidative Activation of Methane 195
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

XAFS/TG/DTA/Mass/FTIR characterization and supporting effects. J. Catal. 1999,
181, 175–188.
203. Liu, S.T.; Dong, Q.; Ohnishi, R.; Ichikawa, M. Remarkable non-oxidative
conversion of methane to naphthalene and benzene on Co and Fe modified
Mo/HZSM-5 catalysts. Chem. Commun. 1997, 1455–1456.
204. Li, S.; Zhang, C.L.; Kan, Q.B.; Wang, D.Y.; Wu, T.H.; Lin, L.W. The
function of Cu(II) ions in the Mo/Cuh-ZSM-5 catalyst for methane
conversion under non-oxidative condition. Appl. Catal. A—Gen. 1999, 187,
199–206.
205. Chen, L.Y.; Lin, L.W.; Xu, Z.S.; Li, X.S.; Zhang, T. Dehydro-oligomerization of
methane to ethylene and aromatics over molybdenum/HZSM-5 catalyst. J. Catal.
1995, 157, 190–200.
206. Choudhary, V.R.; Kinage, A.K.; Choudhary, T.V. Low-temperature nonoxidative
activation of methane over H-galloaluminosilicate (MFI) zeolite. Science 1997,
275, 1286–1288.
207. Liu, S.T.; Dong, Q.; Ohnishi, R.; Ichikawa, M. Unique promotion effect of Co and
Co2 on the catalytic stability for benzene and naphthalene production from methane
on Mo/HZSM-5 catalysts. Chem. Commun. 1998, 1217–1218.
208. Wang, L.S.; Ohnishi, R.; Ichikawa, M. Selective dehydroaromatization of methane
toward benzene on Re/HZSM-5 catalysts and effects of CO/CO2 addition. J. Catal.
2000, 190, 276–283.
209. Yuan, S.D.; Li, J.; Hao, Z.X.; Feng, Z.C.; Xin, Q.; Ying, P.L.; Li, C. The effect of
oxygen on the aromatization of methane over the Mo/HZSM-5 catalyst. Catal. Lett.
1999, 63, 73–77.
210. Dantsin, G.; Suslick, K.S. Sonochemical preparation of a nanostructured
bifunctional catalyst. J. Am. Chem. Soc. 2000, 122, 5214–5215.
211. Armor, J.N. The multiple roles for catalysis in the production of H2. Appl. Catal.
A—Gen. 1999, 176, 159–176.
212. Dicks, A.L. Hydrogen generation from natural gas for the fuel cell systems of
tomorrow. J. Power Sources 1996, 61, 113–124.
213. Hileman, B. Climate change—UN report says global warming’s impact is already
being felt. Chem. Eng. News 2001, 79, 10.
214. Steinberg, M. Production of hydrogen and methanol from natural gas with reduced
CO2 emission. Int. J. Hydrogen Energy 1998, 23, 419–425.
215. Muradov, N.Z. CO2-Free production of hydrogen by catalytic pyrolysis of
hydrocarbon fuel. Energy Fuels 1998, 12, 41–48.
216. Cox, K., Williamson, K., Eds.; Hydrogen: Its Technology and Implications; CRC
Press: Boca Raton, FL, 1977; Vol. 1.
217. Noyes, G.P. Methane Conversion Reactor. US Patent No. 4,836,898, June 6,
1989.
218. Steinfeld, A.; Kirillov, V.; Kuvshinov, G.; Mogilnykh, Y.; Reller, A. Production of
filamentous carbon and hydrogen by solarthermal catalytic cracking of methane.
Chem. Eng. Sci. 1997, 52, 3599–3603.
219. Meier, A.; Kirillov, V.A.; Kuvshinov, G.G.; Mogilnykh, Y.I.; Weidenkaff, A.;
Steinfeld, A. Production of catalytic filamentous carbon by solar thermal
decomposition of hydrocarbons. J. Phys. IV 1999, 9, 393–398.
Choudhary, Aksoylu, and Goodman196
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

220. Ishihara, T.; Miyashita, Y.; Iseda, H.; Takita, Y. Decomposition of methane over
Ni/SiO2 catalysts with membrane reactor for the production of hydrogen. Chem.
Lett. 1955, 93–94.
221. Choudhary, T.V.; Sivadinrayana, C.; Goodman, D.W. Catalytic ammonia
decomposition: COx-free hydrogen production for fuel cell applications. Catal.
Lett., 2001, 72, 197–201.
222. Zhang, T.J.; Amiridis, M.D. Hydrogen production via the direct cracking of
methane over silica-supported nickel catalysts. Appl. Catal. A—Gen. 1998, 167,
161–172.
223. Ferreira-Aparicio, P.; Rodriguez-Ramos, I.; Guerrero-Ruiz, A. Methane interaction
with silica and alumina supported metal catalysts. Appl. Catal. A—Gen. 1997, 148,
343–356.
224. Choudhary, T.V.; Goodman, D.W. CO-free production of hydrogen via stepwise
steam reforming of methane. J. Catal. 2000, 192, 316–321.
225. Choudhary, T.V.; Sivadinarayana, C.; Chusuei, C.; Klinghoffer, A.; Goodman,
D.W. Hydrogen production via catalytic decomposition of methane. J. Catal. 2001,
199, 9–19.
226. Choudhary, T.V.; Sivadinarayana, C.; Klinghoffer, A.; Goodman, D.W. Stud. Surf.
Sci. Catal. 2001, 136, 197.
227. Choudhary, T.V.; Goodman, D.W. Stepwise methane steam reforming: a route to
CO-free hydrogen. Catal. Lett. 1999, 59, 93–94.
228. Aiello, R.; Fiscus, J.E.; Zur Loye, H.C.; Amiridis, M.D. Hydrogen production via
the direct cracking of methane over Ni/SiO2: catalyst deactivation and
regeneration. Appl. Catal. A—Gen. 2000, 192, 227–234.
229. Choudhary, V.R.; Banerjee, S.; Rajput, A.M. Continuous production of H2 at low
temperature from methane decomposition over Ni-containing catalyst followed by
gasification by steam of the carbon on the catalyst in two parallel reactors operated
in cyclic manner. J. Catal. 2001, 198, 136–141.
230. Hughes, M.C.; Copelend, J.P.; Singleton, R.H.; Undercoffer, K.E. Interim Report
for Period Dec 1972–Jan 1974, Vol. 1, AFRPL_TR-74-15.
231. Lefevre, R.L.R.; Price, M.S.T. Coated nuclear-fuel particles—coating process and
its model. Nucl. Technol. 1977, 35, 263–278.
232. Bokros, J.C. Carbon biomedical devices. Carbon 1977, 15, 355–371.
233. Tesner, P.A. Symposium on Combustion 7th, London (1958); 5. 546, Academic
Press: New York, 1959.
234. Bammidipati, S.; Stewart, G.D.; Elliott, J.R.; Gokoglu, S.A.; Purdy, M.J. Chemical
vapor deposition of carbon on graphite by methane pyrolysis. AICHE J. 1996, 42,
3123–3132.
235. Grisdale, R.O. J. Appl. Phys. 1953, 34, 1082.
236. Je, J.H.; Lee, J.Y. A study of the deposition of pyrolytic carbons from
hydrocarbons. Carbon 1984, 22, 563–570.
237. Fulcheri, L.; Schwob, Y. From methane to hydrogen, carbon-black, and water. Int.
J. Hydrogen Energy 1995, 20, 197–202.
238. Popov, R.G.; Shprilrain, E.E.; Zaytchenko, V.M. Natural gas pyrolysis in the
regenerative gas heater—part I: natural gas thermal decomposition at a hot matrix
in a regenerative gas heater. Int. J. Hydrogen Energy 1999, 24, 327–334.
Nonoxidative Activation of Methane 197
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

239. Popov, R.G.; Shpilrain, E.E.; Zaytchenko, V.M. Natural gas pyrolysis in the
regenerative gas heater—part II: natural gas pyrolysis in the free volume of the
regenerative gas heater. Int. J. Hydrogen Energy 1999, 24, 335–339.
240. Audier, M.; Oberlin, A.; Oberlin, M.; Coulon, M.; Bonnetain, L. Morphology and
crystalline order in catalytic carbons. Carbon 1981, 19, 217–224.
241. Koestner, R.J.; Vanhove, M.A.; Somorjai, G.A. Origins of the behavior of
hydrocarbons on metal-surfaces. ChemTech 1983, 13, 376–384.
242. Rodriguez, J.A.; Goodman, D.W. Dissociative adsorption and hydrogenolysis of
ethane over clean and Ni-covered Pt(111). J. Phys. Chem. 1990, 94, 5342–5347.
243. Reyniers, M.F.S.G.; Froment, G.F. Influence of metal-surface and sulfur addition
on coke deposition in the thermal-cracking of hydrocarbons. Ind. Eng. Chem. Res.
1995, 34, 773–785.
244. Duprez, D.; Fadili, K.; Barbier, J. Regeneration of nickel catalysts deactivated by
filamentous carbon. Ind. Eng. Chem. Res. 1997, 36, 3180–3187.
245. Trimm, D.L. Formation and removal of coke from nickel-catalyst. Catal. Rev.—
Sci. Eng. 1977, 16, 155–189.
246. Baker, R.T.K. Catalytic growth of carbon filaments. Carbon 1989, 27, 315–323.
247. Gadd, G.E.; Blackford, M.; Moricca, S.; Webb, N.; Evans, P.J.; Smith, A.N.;
Jacobsen, G.; Leung, S.; Day, A.; Hua, Q. The world’s smallest gas cylinders?
Science 1997, 277, 933–936.
248. Chen, P.; Wu, X.; Lin, J.; Tan, K.L. High H-2 uptake by alkali-doped carbon
nanotubes under ambient pressure and moderate temperatures. Science 1999, 285,
91–93.
249. Rodriguez, N.M.; Kim, M.S.; Baker, R.T.K. Carbon nanofibers—a unique catalyst
support medium. J. Phys. Chem. 1994, 98, 13108–13111.
250. Rodriguez, N.M. A review of catalytically grown carbon nanofibers. J. Mater. Res.
1993, 8, 3233–3250.
251. Likholobov, V.A.; Fenelonov, V.B.; Okkel, L.G.; Goncharova, O.V.; Avdeeva,
L.B.; Zaikovskii, V.I.; Kuvshinov, G.G.; Semikolenov, V.A.; Duplyakin, V.K.;
Baklanova, O.N.; Plaksin, G.V. New carbon-carbonaceous composites for catalysis
and adsorption. React. Kinet. Catal. Lett. 1995, 54, 381–411.
252. De Jong, K.P.; Geus, J.W. Carbon nanofibers: catalytic synthesis and applications.
Catal. Rev.—Sci. Eng. 2000, 42, 481–510.
253. Baker, R.T.K.; Barber, M.A.; Waite, R.J.; Harris, P.S.; Feates, F.S. Nucleation and
growth of carbon deposits from nickel catalyzed decomposition of acetylene.
J. Catal. 1972, 26, 51.
254. Yang, R.T.; Chen, J.P. Mechanism of carbon-filament growth on metal-catalysts.
J. Catal. 1989, 115, 52–64.
255. Nielsen, J.R.; Trimm, D.L. Mechanisms of carbon formation on nickel-containing
catalysts. J. Catal. 1977, 48, 155–165.
256. Sacco, A.; Thacker, P.; Chang, T.N.; Chiang, A.T.S. The initiation and growth of
filamentous carbon from a-iron in H2, CH4, H2O, and CO gas-mixtures. J. Catal.
1984, 85, 224–236.
257. Kock, A.J.H.M.; Debokx, P.K.; Boellaard, E.; Klop, W.; Geus, J.W. The formation
of filamentous carbon on iron and nickel-catalysts. 2. Mechanism. J. Catal. 1985,
96, 468–480.
Choudhary, Aksoylu, and Goodman198
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

258. Alstrup, I. A new model explaining carbon-filament growth on nickel, iron, and
Ni–Cu alloy catalysts. J. Catal. 1988, 109, 241–251.
259. Tibbetts, G.G.; Devour, M.G.; Rodda, E.J. An adsorption–diffusion isotherm and
its application to the growth of carbon filaments on iron catalyst particles. Carbon
1987, 25, 367–375.
260. Snoeck, J.W.; Froment, G.F.; Fowles, M. Filamentous carbon formation and
gasification: thermodynamics, driving force, nucleation, and steady-state growth.
J. Catal. 1997, 169, 240–249.
261. Debokx, P.K.; Kock, A.J.H.M.; Boellaard, E.; Klop, W.; Geus, J.W. The formation
of filamentous carbon on iron and nickel-catalysts. 1. Thermodynamics. J. Catal.
1985, 96, 454–467.
262. Safvi, S.A.; Bianchini, E.C.; Lund, C.R.F. The dependence of catalytic carbon-
filament growth-kinetics upon gas-phase carbon activity. Carbon 1991, 29,
1245–1250.
263. Baker, R.T.K.; Chludzinski, J.J. Filamentous carbon growth on nickel-iron
surfaces—the effect of various oxide additives. J. Catal. 1980, 64, 464–478.
264. RostrupNielsen, J.R. Sulfur-passivated nickel-catalysts for carbon-free steam
reforming of methane. J. Catal. 1984, 85, 31–43.
265. Bernardo, C.A.; Alstrup, I.; Rostrupnielsen, J.R. Carbon deposition and methane
steam reforming on silica-supported Ni–Cu catalysts. J. Catal. 1985, 96, 517–534.
266. Tavares, M.T.; Bernardo, C.A.; Alstrup, I.; Rostrupnielsen, J.R. Reactivity of
carbon deposited on nickel copper alloy catalysts from the decomposition of
methane. J. Catal. 1986, 100, 545–548.
267. Benissad, F.; Gadelle, P.; Coulon, M.; Bonnetain, L. Formation of carbon-fibers
from methane. 1. Catalytic growth and pyrolytic thickening. Carbon 1988, 26,
61–69.
268. Benissad, F.; Gadelle, P.; Coulon, M.; Bonnetain, L. Carbon-fibers from methane.
3. Impact of catalyst precursors. Carbon 1989, 27, 585–592.
269. Kim, M.S.; Rodriguez, N.M.; Baker, R.T.K. The interaction of hydrocarbons with
copper nickel and nickel in the formation of carbon filaments. J. Catal. 1991, 131,
60–73.
270. Kim, M.S.; Rodriguez, N.M.; Baker, R.T.K. The role of intergacial phenomena in
the structure of carbon deposits. J. Catal. 1992, 134, 253–268.
271. Fenelonov, V.B.; Avdeeva, L.B.; Zheivot, V.I.; Okkel, L.G.; Goncharova, O.V.;
Pimneva, L.G. Texture and adsorption properties of fibrous carbon prepared by
decomposition of carbon-containing gases on metal-catalysts. Kinet. Catal. 1993,
34, 483–487.
272. Shaikhutdinov, S.K.; Avdeeva, L.B.; Goncharova, O.V.; Kochubey, D.I.;
Novgorodov, B.N. Coprecipitated Ni–Al and Ni–Cu–Al catalysts for methane
decomposition and carbon deposition. 1. Genesis of calcined and reduced catalysts.
Appl. Catal. A—Gen. 1995, 126, 125–139.
273. Avdeeva, L.B.; Goncharova, O.V.; Kochubey, D.I.; Zaikovskii, V.I.; Plyasova,
L.M.; Novgorodov, B.N.; Shaikhutdinov, S.K. Coprecipitated Ni–alumina and
Ni–Cu–alumina catalysts of methane decomposition and carbon deposition. 2.
Evolution of the catalysts in reaction. Appl. Catal. A—Gen. 1996, 141,
117–129.
Nonoxidative Activation of Methane 199
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

274. Shaikhutdinov, S.K.; Zaikovskii, V.I.; Avdeeva, L.B. Coprecipitated Ni–alumina
and Ni–Cu–alumina catalysts of methane decomposition and carbon deposition. 3.
Morphology and surface structure of the carbon filaments. Appl. Catal. A—Gen.
1996, 148, 123–133.
275. Chen, P.; Zhang, H.B.; Lin, G.D.; Hong, Q.; Tsai, K.R. Growth of carbon nanotubes
by catalytic decomposition of CH4 or CO on a Ni–MgO catalyst. Carbon 1997, 35,
1495–1501.
276. Chen, D.; Lodeng, R.; Holmen, A. Kinetic study of initiation and growth of
filamentous carbon during methane cracking over Ni/a-Al2O3. Catal. Deactivation
1999, 126, 473–476.
277. Kuvshinov, G.G.; Mogilnykh, Y.I.; Kuvshinov, D.G.; Zaikovskii, V.I.; Avdeeva,
L.B. Peculiarities of filamentous carbon formation in methane decomposition on
Ni-containing catalysts. Carbon 1998, 36, 87–97.
278. Li, Y.D.; Chen, J.L.; Chang, L. Catalytic growth of carbon fibers from methane on a
nickel–alumina composite catalyst prepared from Feitknecht compound precursor.
Appl. Catal. A—Gen. 1997, 163, 45–57.
279. Li, Y.D.; Chen, J.L.; Chang, L.; Qin, Y.N. The doping effect of copper on the
catalytic growth of carbon fibers from methane over a Ni/Al2O3 catalyst prepared
from Feitknecht compound precursor. J. Catal. 1998, 178, 76–83.
280. Shaikhutdinov, S.K.; Avdeeva, L.B.; Novgorodov, B.N.; Zaikovskii, V.I.;
Kochubey, D.I. Nickel catalysts supported on carbon nanofibers: structure and
activity in methane decomposition. Catal. Lett. 1997, 47, 35–42.
281. Ermakova, M.A.; Ermakov, D.Y.; Plyasova, L.M.; Kuvshinov, G.G. XRD studies
of evolution of catalytic nickel nanoparticles during synthesis of filamentous
carbon from methane. Catal. Lett. 1999, 62, 93–97.
282. Avdeeva, L.B.; Kochubey, D.I.; Shaikhutdinov, S.K. Cobalt catalysts of methane
decomposition: accumulation of the filamentous carbon. Appl. Catal. A—Gen.
1999, 177, 43–51.
283. Ermakova, M.A.; Ermakov, D.Y.; Kuvshinov, G.G.; Plyasova, L.M. New nickel
catalysts for the formation of filamentous carbon in the reaction of methane
decomposition. J. Catal. 1999, 187, 77–84.
284. Ermakova, M.A.; Ermakov, D.Y.; Kuvshinov, G.G. Effective catalysts for direct
cracking of methane to produce hydrogen and filamentous carbon part I. Nickel
catalysts. Appl. Catal. A—Gen. 2000, 201, 61–70.
285. Derjaguin, B.V.; Fedoseev, D.V.; Lykuanovich, V.M.; Sptisyn, B.V.; Ryanov,
V.A.; Lavrentyev, A.V. Filamentary diamond crystals. J. Cryst. Growth 1968, 2,
380.
286. Aisenberg, S.; Chabot, R. Ion-beam deposition of thin films of diamondlike carbon.
J. Appl. Phys. 1971, 42, 2953–2958.
287. Weissmantel, C.; Bewilogua, K.; Dietrich, D.; Erler, H.J.; Hinneberg, H.J.; Klose,
S.; Nowick, W.; Reisse, G. Structure and properties of quasi-amorphous films
prepared by ion-beam techniques. Thin Solid Films 1980, 72, 19–31.
288. Angus, J.C.; Will, H.A.; Stanko, W.S. Growth of diamond seed crystals by vapor
deposition. J. Appl. Phys. 1968, 39, 2915–2922.
289. Matsumoto, S.; Sato, Y.; Tsutsumi, M.; Setaka, N. Growth of diamond particles
from methane–hydrogen gas. J. Mater. Sci. 1982, 17, 3106–3112.
Choudhary, Aksoylu, and Goodman200
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

290. Sommer; Smith, F.W. In New Diamond Science and Technology; Messier, R.F.,
Glass, J.T., Butler, M.J.E., Roy, R., Eds.; Material Research Society: Pittsburgh,
1991; 443.
291. Hirose, Y.; Teraswa, Y. Synthesis of diamond thin-films by thermal cvd using
organic-compounds. Jpn. J. Appl. Phys. Part 2—Lett. 1986, 25, L519–L521.
292. Sawabe, A.; Inuzuku, T. Growth of diamond thin-films by electron-assisted
chemical vapor-deposition. Appl. Phys. Lett. 1985, 46, 146–147.
293. Sawabe, A.; Inuzuka, T. Growth of diamond thin-films by electron-assisted
chemical vapor-deposition and their characterization. Thin Solid Films 1986, 137,
89–99.
294. Suzuki, K.; Sawabe, A.; Yasuda, H.; Inuzuka, T. Growth of diamond thin-films by
Dc plasma chemical vapor-deposition. Appl. Phys. Lett. 1987, 50, 728–729.
295. Nakao, S.; Watatani, H.; Maruno, S.; Noda, M. Effects of discharge current and
substrate-temperature on the microstructure of diamond films deposited by Dc-
plasma CVD. J. Cryst. Growth 1991, 115, 313–317.
296. Lamb, J.D.; Woollam, J.A. Dielectric-properties of diamondlike carbon prepared
by Rf plasma deposition. J. Appl. Phys. 1985, 57, 5420–5423.
297. Matsumoto, S. Chemical vapor-deposition of diamond in Rf glow-discharge.
J. Mater. Sci. Lett. 1985, 4, 600–602.
298. Meyer, D.E.; Dillon, R.O.; Woollam, J.A. Radio-frequency plasma chemical
vapor-deposition growth of diamond. J. Vacuum Sci. Technol. A—Vacuum Surf.
Films 1989, 7, 2325–2327.
299. Rudder, R.A.; Posthill, J.B.; Hudson, G.C.; Malta, D.; Thomas, R.E.; Markunas,
R.J.; Humphrey, T.P.; Nemanich, R.J. In New Diamond Science and Technology;
Messier, R.F., Glass, J.T., Bulter, M.J.E., Roy, R., Eds.; Material Research Soceity:
Pittsburgh, 1991; 425.
300. Saito, Y.; Matsuda, S.; Nogita, S. Synthesis of diamond by decomposition of
methane in microwave plasma. J. Mater. Sci. Lett. 1986, 5, 565–568.
301. Kamo, M.; Sato, Y.; Matsumoto, S.; Setaka, N. Diamond synthesis from gas-phase
in microwave plasma. J. Cryst. Growth 1983, 62, 642–644.
302. Lee, J.C.; Hong, B.Y.; Messier, R.; Collins, R.W. Real time spectroellipsometry
for optimization of diamond film growth by microwave plasma-enhanced
chemical vapor deposition from Co/H-2 mixtures. J. Appl. Phys. 1996, 80,
6489–6495.
303. Kitahama, K.; Hirata, K.; Nakamatsu, H.; Kawai, S.; Fujimori, N.; Imai, T.;
Yoshino, H.; Doi, A. Synthesis of diamond by laser-induced chemical vapor-
deposition. Appl. Phys. Lett. 1986, 49, 634–635.
304. Matsui, Y.; Yuuki, A.; Sahara, M.; Hirose, Y. Flame structure and diamond growth-
mechanism of acetylene torch. Jpn. J. Appl. Phys. Part 1—Regular Pap. Short Notes
Rev. Pap. 1989, 28, 1718–1724.
305. Kaneko, H.; Kamada, M.; Kuwae, R.; Sawabe, A.; Inuzuka, T. Influence of
substrate-temperatures and atmosphereic-temperatures on diamond deposition.
Appl. Surf. Sci. 1988, 33-4, 546–552.
306. Celii, F.G.; Butler, J.E. Diamond chemical vapor-deposition. Annu. Rev. Phys.
Chem. 1991, 42, 643–684.
Nonoxidative Activation of Methane 201
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

307. Saito, Y.; Sato, K.; Tanaka, H.; Miyadera, H. Diamond-like carbon-films prepared
from CH4–H2–H2O mixed gas-using a microwave plasma. J. Mater. Sci. 1989, 24,
293–297.
308. Hsu, W.L. Gas-phase kinetics during microwave plasma-assisted diamond
deposition—is the hydrocarbon product distribution dictated by neutral–neutral
interactions. J. Appl. Phys. 1992, 72, 3102–3109.
309. Barshilia, H.C.; Mehta, B.R.; Vankar, V.D. Growth of diamond thin films by
microwave plasma chemical vapor deposition process. J. Mater. Res. 1996, 11,
1019–1024.
310. Fan, Q.H.; Pereira, E.; Gracio, J. Diamond deposition on copper: studies on
nucleation, growth, and adhesion behaviors. J. Mater. Sci. 1999, 34, 1353–1365.
311. Ganesan, D.; Sharma, S.C. A study of texture in diamond films as functions of
methane concentration during chemical-vapor-deposition and postgrowth hydrogen
treatment. J. Mater. Res. 1995, 10, 1764–1771.
312. Liang, G.T.; Hong, F.C.N. Diamond growth by hollow cathode arc plasma
chemical vapor deposition. J. Mater. Res. 1998, 13, 3114–3121.
313. Matsubara, H.; Sakuma, T. Diamond deposition on cemented carbide by chemical
vapor-deposition using a tantalum filament. J. Mater. Sci. 1990, 25, 4472–4476.
314. Johnson, C.E.; Weimer, W.A.; Cerio, F.M. Efficiency of methane and acetylene in
forming diamond by microwave plasma assisted chemical vapor-deposition.
J. Mater. Res. 1992, 7, 1427–1431.
315. Clark, C.D.; Dickerson, C.B. Raman and photoluminescence spectra of as-grown
CVD diamond films. J. Phys.—Condensed Matter 1992, 4, 869–878.
316. Park, D.W.; Yun, J.S. Structural characterization of diamond thin films prepared by
plasma jet. Thin Solid Films 1999, 345, 60–66.
317. Bonnot, A.M. Raman microspectroscopy of diamond crystals and thin-films
prepared by hot-filament-assisted chemical vapor-deposition. Phys. Rev. B 1990,
41, 6040–6049.
318. Catledge, S.A.; Vohra, Y.K. Effect of nitrogen addition on the microstructure and
mechanical properties of diamond films grown using high-methane concentrations.
J. Appl. Phys. 1999, 86, 698–700.
319. Asakura, Y.; Chattopadhyay, K.K.; Matsumoto, S.; Hirakuri, K. Diamond synthesis
in capacitively coupled 13.56 MHz radio frequency plasma using parallel plate
electrodes with the addition of direct current power. J. Vacuum Sci. Technol. A—
Vacuum Surf. Films 1998, 16, 3185–3189.
320. Reinke, P.; Kania, P.; Oelhafen, P. Investigation of the nucleation mechanism in
bias-enhanced diamond deposition on silicon and molybdenum. Thin Solid Films
1995, 270, 124–129.
321. Ramesham, R.; Ellis, C.D.; Olivas, J.D.; Bolin, S. Fabrication of diamond
membranes for MEMS using reactive ion etching of silicon. Thin Solid Films 1998,
330, 62–66.
322. Zhou, D.; Krauss, A.R.; Qin, L.C.; Mccauley, T.G.; Gruen, D.M.; Corrigan, T.D.;
Chang, R.P.H.; Gnaser, H. Synthesis and electron field emission of nanocrystalline
diamond thin films grown from N-2/CH4 microwave plasmas. J. Appl. Phys. 1997,
82, 4546–4550.
Choudhary, Aksoylu, and Goodman202
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.

323. Lettington, A.; Steeds, J.W. Thin Film Diamond; Chapman and Hall: London,
1994.
324. Devries, R.C. Synthesis of diamond under metastable conditions. Annu. Rev.
Mater. Sci. 1987, 17, 161–187.
325. Grill, A.; Meyerson, B.; Spear, K.E.; Dismukes, J.P. Synthetic Diamond: Emerging
CVD Science and Technology; Wiley: New York, 1994; 91.
326. Grilll, A. Diamond-like carbon: state-of-the-art. Diamond Relat. Mater. 1999, 8,
428–434.
Nonoxidative Activation of Methane 203
MARCEL DEKKER, INC. • 270 MADISON AVENUE • NEW YORK, NY 10016
©2003 Marcel Dekker, Inc. All rights reserved. This material may not be used or reproduced in any form without the express written permission of Marcel Dekker, Inc.














![Phase Equilibrium Measurements of Methane + Benzene and ... · the methane + methylbenzene system at 198.15 K, a higher temperature than has been reported or was expected [1]. The](https://static.fdocuments.in/doc/165x107/601228e197b50977543f89e2/phase-equilibrium-measurements-of-methane-benzene-and-the-methane-methylbenzene.jpg)



