
Noemi · Web viewFor all experiments, cow bone powder was manually drilled with sterile...
44
Minimising laboratory-induced decay in bone proteomics Noemi Procopio 1 , Michael Buckley 1 *. 1 Manchester Institute of Biotechnology, The University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK 1
Transcript of Noemi · Web viewFor all experiments, cow bone powder was manually drilled with sterile...

Minimising laboratory-induced decay in bone proteomics
Noemi Procopio1, Michael Buckley1*.
1Manchester Institute of Biotechnology, The University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK
*Correspondence to [email protected]; +44(0)161 306 5175
Abstract
Proteomics methods are being increasingly used to study archaeological and
palaeontological bone, assisting in species identification and phylogenetic studies as well
as improving our understanding of bone diagenesis. More recently, there are developing
interests in the study of post-translation modifications (PTMs), some of which are
potentially diagnostic of decay, but none of the previous extraction methods have been
developed in light of this. To be able to record close to natural deamidation levels of
samples an extraction procedure should minimise laboratory-induced decay, such as
asparagine and glutamine deamidations, which are considered most strongly related with
decay and known to occur frequently with standard laboratory procedures. Here we tested
numerous methods to identify an optimal approach of extracting proteins from bone whilst
minimising artificial decay. Using a weak acid to partially demineralize the bone sample,
then subsequent incubation of the acid insoluble fraction with guanidine hydrochloride and
enzymatic digestion in ammonium acetate, we observed a ~50% reduction in deamidation
whilst also substantially decreasing the protocol length. We propose this optimised method
as appropriate for studies of archaeological, palaeontological as well as potentially forensic
investigations using proteomics where decay measurements could act as ‘molecular
timers’.
Keywords
Bone proteome – deamidation – forensics – post-mortem interval – shotgun proteomics
1

Introduction
In recent years proteomics methods have been increasingly applied to the study
and characterization of bone, both modern1–3 and ancient4–6. Although the majority of
publications have investigated the dominant protein collagen for phylogenetic inferences7–9
and species identification10–13 to address archaeological and paleontological questions, the
suite of non-collagenous proteins (NCPs) like serum proteins and collagen-interacting
proteins present in sub-fossil bones are proving an exciting potential source for further
taxonomic information14. Unsurprisingly, most bone proteomic techniques have been
optimised for extracting the greatest number of different proteins (i.e., proteome
complexity) from the sample. However, the study of post-translational modifications
(PTMs) of proteins, such as oxidation15,16, deamidation17–19 and racemization20,21 have
become of greater interest because they can be used as degradative markers potentially
useful for post-mortem decay related studies17,22. The recording of some of these particular
PTMs is not only of interest to studies investigating the decay state, and therefore relative
age of ancient remains under similar burial histories, but of potential use in investigating
the timing of more recent decay in the field of forensics.
Apart from the differences in their chemistry, each of these PTMs is also
characterised by different kinetic rates, which can be influenced by several different factors
like the protein structure, the temperature, the pH and the ionic strength23. For example,
racemization of some amino acid residues requires more time to naturally occur than other
modifications such as deamidation and oxidation24. Where protein oxidation is thought to
be more strongly related with ageing phenomena in vivo15 with post-mortem oxidation
observed in muscle tissue25, protein deamidation can be more readily influenced by
several different factors, such as protein structure, temperature, pH and ionic strength26,
and has been shown to act physiologically as a molecular clock for protein turnover,
development and ageing27–31. Furthermore, deamidation can also increase post-mortem18,22
and so has been suggested potentially useful in palaeontological studies as a means for
authenticating the protein as being endogenous rather than contaminant32.
However, inappropriate handling and manipulation of bone samples to extract
proteomic information, including PTM data, could lead to incorrect interpretations of the
biological phenomena. In particular, protein deamidation levels have already been shown
to be strongly affected by the laboratory procedure used to extract proteins from the
specimen33–35, which will be evaluated in this study.
2

Bone structure and its proteomeBone is the most likely tissue to survive long periods of burial, characterised by
different components described generally as an inorganic phase (50-70%), organic phase
(20-40%) and a water component (5-10%). The mineral phase is composed of calcium
phosphates (hydroxyapatite) and provides mechanical rigidity and strength, whereas the
organic phase is composed fundamentally of proteins and a small fraction of lipids (<3%),
and provides elasticity and flexibility36,37. About 90% of this organic component is
represented by long fibrils of Type I collagen, a structural molecule in the extracellular
matrix (ECM) characterised by a triple-helix domain composed of three α chains known as
tropocollagen36,37; in mammals, each tropocollagen molecule is made up of two identical
α1(I) chain and one α2(I) chain36. The remaining 10% organic matter is primarily
composed of NCPs but includes minor amounts of other biomolecules such as lipids and
DNA. Of the NCPs there are four main groups: glycosaminoglycan (GAG)-bearing proteins
(e.g., aggrecan, versican, decorin, biglycan and fibromodulin), glycoproteins (e.g., alkaline
phosphatase, osteonectin, tetranectin, thrombospondin, fibronectin, vitronectin,
osteopontin and bone sialoprotein), -carboxy-glutamic acid (Gla)-containing proteins
(osteocalcin and matrix-Gla-protein) and numerous other proteins such as proteolipids,
collagen-degrading metalloproteinases, bone morphogenetic proteins, growth factors,
serum-derived proteins and cell-binding proteins38,39. Many of these have functions relating
to the promotion of mineralization and other aspects of bone formation and remodelling.
Existing approaches to post-mortem interval estimationOne of the most common questions that forensic experts face is the estimation of
post-mortem interval (PMI), which can be informative on the historical truth behind a
forensic case. There are currently several standard strategies that are used to investigate
PMI, such as thanatochemistry40,41 and forensic entomology42,43, which typically investigate
the soft tissue phase of decomposition44. In doing so they suffer from poor precision and
reliability where different experts often arrive at different conclusions from analyses of the
same samples40,45,46. To improve reliability, biomolecular methods have been previously
investigated, ranging from post-mortem DNA degradation47,48 and RNA degradation
studies49 to the measurement of the levels of some proteins accumulated in the tissues
after death50–52. They have proven promising but have so far only been investigated for
short PMIs and are not ideal for longer post-mortem periods. Other strategies have been
developed to get information from bodies heavily decomposed and skeletonized which
3

vary substantially from approaches based on morphological studies44,53 and histological
changes44 to analytical methods. Specific to bone analysis, these include the evaluation of
their loss of nitrogen44, loss of fluorescence pattern44,53 and loss of immunologic activity44.
More obscure methods involve the evaluation of radioisotopes present in the tissue54, the
citrate content of bones55, and the use of luminol on old skeletal remains for the detection
of blood56. Some protein-based methods have been reported, such as the measurement of
the reduction of particular amino acid levels in skeletal remains57, but with notable limits
regarding the accuracy of results44. Hence, given the recent advancements of proteomics
techniques, the development of alternative protein-based approaches to decay
measurements could be useful to determine the correct PMI from bone samples; this study
aims to set the scene for the use of proteomics as one such approach to decay estimation
– in this case the measurement of protein deamidation.
Post-translational modificationsIn addition to those already introduced (e.g., deamidation and oxidation), proteins
undergo a wide range of other modifications during the ageing process, such as
carbonylation58,59, glycation60,61, cross-linking62,63, racemization64,65, nitration65,66. However,
the deamidations of asparagine (Asn) and glutamine (Gln) are particularly ideal for use as
a ‘molecular clock’ because they occur physiologically in vivo in a time-dependent way67.
Protein deamidation is a non-enzymatic process that is characterised by the hydrolysis of
the amide linkage in Asn and Gln residues leading to the formation of a succinimide
intermediate and followed by the generation of the carboxylic acids aspartic acid and
glutamic acid respectively68. Moreover, deamidation can also occur by direct hydrolysis in
an acidic environment, without the formation of the succinimide intermediate which instead
is more commonly created under neutral or alkaline conditions68. With some previous
analyses linking deamidation levels of in situ archaeological collagen with ageing18,22, and
others demonstrating a correlation between in vivo protein deterioration and deamidation
rates67,69, we speculate that the study of shorter-term decay markers could have
applications in forensic investigations with further research. Specific studies have shown
that deamidation in vivo is influenced by protein primary28, secondary70 and tertiary
structure71, but also by several other factors such as temperature72,73, pH conditions34,73 and
ionic strength73. As such, deamidations are also known to occur in vitro67 during sample
processing steps and can arise during several phases of the protein extraction and
digestion methods most commonly used, particularly under strong pH and long exposures
4

to high temperatures34,35 resulting in an overestimation of deamidation levels. The aims of
this research are to address this issue.
Previous approaches to reducing deamidationIn order to investigate the study of deamidation levels in bone proteins it is
fundamental to minimise those introduced during laboratory protocols. Two main
improvements to the in-solution digestion process with trypsin have been proposed; one
approach is the reduction of digestion times down to less than one hour rather than
overnight or day-long processes35 whereas another approach is the use of different
digestion buffers33. In the case of the former it can be inferred that the reduced digestion
length will not always be applicable to complex mixtures of proteins (such as a complete
proteomes from a biological sample) because such a short incubation with the enzyme
reduces digestion efficiency34. In the case of the latter it has been shown that laboratory
processing-induced Asn deamidation is less pronounced in ammonium acetate than in
other buffers more commonly used in proteomics methods, such as ammonium
bicarbonate. There are likely several other parameters that are also influential in reducing
processing-induced deamidations, such as incubation length within the protein extraction
buffers and the pooling of different extractions. Therefore, the purpose of our study was to
optimise the existing protocols for protein bone extraction14,74,75 taking into consideration
the suggested digestion improvements previously reported33,35 as well as other factors
likely to influence levels of protein decay (decalcification agent and extraction length), in
order to identify the optimum compromise between obtaining a diverse proteome whilst
reducing processing-induced deamidations.
Experimental SectionMaterials
A single modern cow tibia was acquired from a local butcher. Dentist’s Protaper
Universal shaping files used for the drilling procedures were purchased from Henry Schein
Minerva Dental (UK). Hydrochloric acid (HCl), acetonitrile (ACN) and
ethylenediaminetetraacetic acid (EDTA) were purchased from Fisher Scientific (UK); nitric
acid (HNO3) was purchased from VWR International; formic acid (FA) and ammonium
bicarbonate (ABC) were purchased from Fluka (UK); Tris, dithiothreitol (DTT),
iodoacetamide (IAM), trifluoroacetic acid (TFA), guanidine hydrochloride (GuHCl),
ammonium acetate (AMAC) and acetic acid (AcOH) were purchased from Sigma-Aldrich
5

(UK), 10K Molecular Weight Cut Off (MWCO) ultrafiltration units were purchased from
Vivaspin (UK); sequencing grade trypsin was purchased from Promega (UK), and C18
reversed-phase Zip-Tips were purchased from Agilent Technologies (UK).
MethodsAs part of method development, several different variable parameters were tested in
order of assumed importance (Fig. 1). The starting protocol in which different modifications
have been introduced was taken from Wadsworth and Buckley14 and the variants have
been chosen from previously published methods33,74,76,77. For all experiments, cow bone
powder was manually drilled with sterile dentist’s Protaper Universal shaping files in order
to minimise the decay induced by electric drilling procedures. Approximately 25 mg of
powder was used for each analysis, carried out in triplicate starting from three different
biological samples taken from the same anatomical position of the bone to minimise the
variability within different samples.
Figure 1 - Flow charts of the overall experimental design followed in this study: A) initial tests of different acids, demineralization lengths (0.5 M EDTA only) and digestion buffer and lengths (0.5 M EDTA only) and B) further evaluation of fraction pooling.
Demineralization processFive different acids (0.6 M HCl, 0.5 M EDTA, 6% AcOH, 5% HNO3 and 10% FA) for
the demineralization of the bone sample were tested with 1 mL of acid added to each
sample in triplicate, mixed thoroughly and placed at 4°C for 66 h. After demineralization,
samples were centrifuged at 14,000 × g for 1 min and the acid soluble supernatant
removed from the acid insoluble pellet and frozen. The insoluble pellet was then incubated
with a buffer containing 6 M GuHCl and 100 mM Tris at pH 7.4 at 4°C for 66 h. Then both
6

soluble and insoluble fractions were centrifuged as above and the supernatants
exchanged into 50 mM ABC using 10 kDa MWCO ultrafiltration units, collecting the
retentate with 200 µL ABC. The proteins in this solution were then reduced using 8.4 µL
5 mM DTT for 40 min at room temperature and alkylated using 33.6 µL 15 mM IAM in the
dark for 45 min at room temperature. The alkylation step was then quenched by adding a
further volume of 5 mM DTT and samples were digested using 1 μg trypsin at 37°C for
18 h.
Demineralization lengthDifferent time intervals were tested for the demineralization of the bone sample in
order to obtain the best compromise between proteome complexity (i.e., the diversity of
the proteins observed measured as a protein count) and minimum observed levels of
protein decay (measure of peptide deamidations). To do so, EDTA was added to
demineralize the samples over 6, 24 or 48 h and the subsequent steps for the proteome
extraction were carried out as described above.
Digestion buffer and digestion lengthTo compare the use of 50 mM ABC (at pH 8.7) with 50 mM AMAC33 (at pH 6.7) as
an alternative buffer for tryptic digestion, following demineralization with EDTA (~66 h at
4°C) and incubation of the acid insoluble pellets (at 4°C for ~66 h) the pooled acid soluble
and acid insoluble fractions were ultrafiltered into either buffer and then reduced and
alkylated as described above but subsequently digested for either 1, 5 or 18 h.
GuHCl incubation length and protocol refinementTo investigate extraction incubation length, following demineralization with EDTA
(4°C for 6 h) the samples were centrifuged at 14,000 × g for 1 min and the acid insoluble
fraction incubated in a 6 M GuHCl buffer containing 100 mM Tris at pH 7.4 at 4°C for either
18, 42 or 66 h. Then both the acid soluble fraction and acid insoluble fractions were pooled
and ultrafiltrated together into 50 mM AMAC and the retentate collected with 200 μL of
AMAC. For practical reasons, the samples were then frozen overnight and subsequently
reduced, alkylated, quenched and digested for 5 h as described above.
The same experiment was then repeated to refine the methodology with two
modifications; firstly, the overnight freezing of the samples before the digestion was
avoided to exclude bias in the results, secondly, the soluble fraction of the samples was
separated and excluded from the analyses, and only the acid insoluble fraction was
7

ultrafiltrated in order to reduce the relative concentrations of collagen within the samples.
All successive steps were carried out as described above.
LC-Orbitrap Mass SpectrometryFor all methods tested, 1% TFA was added to the samples to stop the digest
(making 0.1% TFA), which were then desalted, purified and concentrated using OMIX C18
reversed-phase Zip-Tips following manufacturer’s protocols. Tips were prepared with two
volumes of 100 μL 0.1% TFA/50% ACN and then washed twice with 100 μL of 0.1% TFA.
The sample was then introduced to the C18 tips 5-10 times to allow peptides to bind,
followed by two wash steps with 100 μL of 0.1% TFA and a final elution in 100 μL of 50%
ACN and 0.1% TFA. Samples were dried under a fume cupboard for one day (at room
temperature) and then re-suspended in 20 μL of 0.1% TFA/5% ACN for subsequent
LC/MS/MS analysis. Re-suspended samples were analysed by LC/MS/MS using an
UltiMate® 3000 Rapid Separation LC (RSLC, Dionex Corporation, Sunnyvale, CA, USA)
coupled to an Orbitrap Elite (Thermo Fisher Scientific, Waltham, MA, USA) mass
spectrometer (120 k resolution, full scan, positive mode, normal mass range 350–1500).
Peptides were separated on an Ethylene Bridged Hybrid (BEH) C18 analytical column (75
mm × 250 µm i.d., 1.7 μM; Waters) using a gradient from 92% A (0.1% FA in water) and
8% B (0.1% FA in ACN) to 33% B in 44 min at a flow rate of 300 nL min–1. Peptides were
then automatically selected for fragmentation by data-dependent analysis; six MS/MS
scans (Velos ion trap, product ion scans, rapid scan rate, Centroid data; scan event: 500
count minimum signal threshold, top 6) were acquired per cycle, dynamic exclusion was
employed, and one repeat scan (i.e. two MS/MS scans total) was acquired in a 30 second
repeat duration with that precursor being excluded for the subsequent 30 seconds
(activation: collision-induced dissociation (CID), 2+ default charge state, 2 m/z isolation
width, 35 eV normalized collision energy, 0.25 Activation Q, 10.0 ms activation time).
Data analysisSpectra obtained via LC/MS/MS were searched as .mgf files (created assuming 0
#C13s with peak list generation using ExtractMSN) against the Swiss-Prot database
(540,052 entries) using the Mascot search engine (version 2.4.1; Matrix Science, London,
UK). Each search included the fixed carbamidomethyl modification of cysteine (+57.02 Da)
and the variable modifications for deamidation (+0.98 Da) of asparagine/glutamine and
oxidation (+15.99 Da) of lysine, proline and methionine residues, to account for PTMs and
diagenetic alterations; the oxidation of lysine and proline is equivalent to hydroxylation.
8

Enzyme specificity was set up to trypsin with up to 2 missed cleavages allowed, mass
tolerances were set at 5 parts per million for the precursor ions and 0.5 Daltons for the
fragment ions, with all spectra considered as having either 2+ or 3+ precursors. A search
with semiTrypsin specificity was also performed maintaining all other parameters; results
can be found in the Supporting Information.
It should be noted that the data were searched without any allowance for incorrect
monoisotopic peak detection to avoid the inclusion of false positive results at the cost of an
increased number of false negatives; as the occurrence of incorrect peak detection is
random this does not bias the results in these analyses. The identification rates could
potentially be improved by using a process that recalculates the monoisotopic peak from
the raw data such as Matrix Science’s Mascot Distiller, but this would not significantly
affect the experimental outcome.
To compare samples counting the number of proteins as well as the number of
deamidated peptides, Scaffold software version 4.4.1 (Proteome Software Inc., Portland,
OR) was used. Peptide identifications were accepted if they could be established at
greater than 95% probability by the Peptide Prophet algorithm78 with Scaffold delta-mass
correction. Protein identifications were accepted if they could be established at greater
than 99.0% probability and contained at least two identified peptides; protein probabilities
were assigned by the Protein Prophet algorithm79. Proteins that contained similar peptides
and could not be differentiated based on MS/MS analysis alone were grouped to satisfy
the principles of parsimony. The display option chosen for this work was “total spectrum
count” which shows the total number of spectra acquired for each protein during the
analysis and allows semi-quantitative measurement of the proteins present in the sample.
In Scaffold, the three replicates were grouped together and each experimental condition
considered as a distinct group. Subsequently, only proteins containing the keyword
“bovine” were filtered and selected in order to avoid the presence of proteins unrelated
with the bone sample. In addition, the filter related to the presence of deamidation was
also used to extrapolate data for the deamidation analysis. Then data were exported into
an Excel spreadsheet followed by the calculations of averages and standard deviations
(SD) for each group of replicates for all proteins matched.
STRING software (version 10.0) was used to create networks of the proteins
obtained with the optimised protocol. These protein interactions include direct (physical)
and indirect (functional) associations derived from “genomic context”, “high-throughput
experiments”, “co-expression” and “previous knowledge”. The clustering option chosen for
9

the graphs was KMEANS = 4, and the view utilised was the “confidence view” (in which
the line thickness between proteins was proportionally related to the strength of the
associations).
Results and Discussion
Ideally an optimised protocol for protein extraction should recover a complex
proteome (i.e., relatively high number of different proteins detected) without generating
processing-induced deamidations during the procedure. To improve upon some of the
current methods used for protein extraction from bone samples, we tested different
parameters considered to be crucial for the production of processing-induced
deamidations and explored alternatives for each. We initially evaluated the effects that two
parameters could have on the demineralization process: 1) the acid used for
demineralization (0.6 M HCl, 0.5 M EDTA, 6% AcOH, 5% HNO3 and 10% FA), and 2) the
length of this demineralization step (6, 24 and 48 h). We then investigated the digestion
step, testing different buffers (50 mM ABC and 50 mM AMAC) and different time lengths
(1, 5 and 18 h). Finally, we not only evaluated the effects of different incubation times (18,
42 and 66 h) in GuHCl for the acid insoluble fraction of the protein extract, but also
compared results between analyses performed pooling both fractions of the extract (acid
soluble and extracted insoluble fractions), and analyses using only the acid insoluble
fraction. We found this latter experiment appropriate given the likelihood that the much
more dominant collagen peptides present in the acid soluble fraction would interfere with,
and ultimately reduce, the remaining NCPs to the extent that previous studies have
attempted to specifically remove as much collagen as possible14.
Optimum demineralizing agentWhen extracting proteins from bone, demineralization/decalcification is a
fundamental step to remove calcium salts from the protein-rich organic matrix, which
substantially improves the efficiency of the protein extraction compared with the protein
extraction method with undemineralized bone74. Although several studies have evaluated
various reagents to improve the extraction efficiency whilst also limiting the laboratory-
induced decay on samples76,77,80,81, none have been done that consider the impact of the
use of different acids on the processing-induced deamidation of proteins. Our hypothesis
is that strong acids, like NA and HCl in this study, could yield a better quality proteome but
at the same time they could increase processing-induced deamidation levels compared
10

with weak acids, such as AA, FA and EDTA. To test this, we evaluated both the proteome
recovery and the ratio of deamidated peptides in relation to the total amount of peptides in
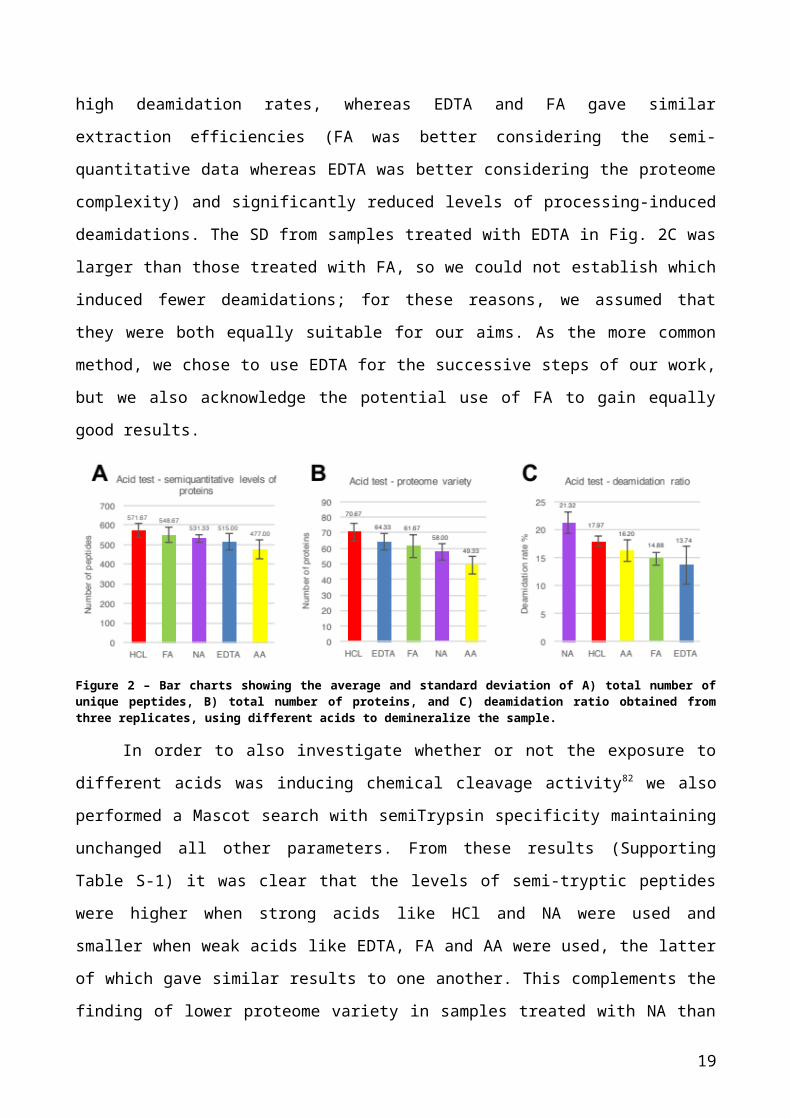
the sample (Fig. 2). As shown in Fig. 2A and 2B, the strongest acid (HCl) resulted in the
largest number of unique peptides matched, as well as the greatest proteome variability
compared with the other acids used in this test (572 unique peptides and 71 different
proteins). Acetic acid on the contrary gave the lowest numbers (477 unique peptides and
49 different proteins). Nitric acid yielded a poor proteome variety (58 different proteins)
although the semi-quantitative levels of proteins in this case were better than the levels
obtained using EDTA and AA (531 unique peptides versus 515 matched with EDTA and
477 matched with AA). Formic acid, with 549 unique peptides and 62 different proteins,
was respectively the second best in terms of semi-quantitative levels just after HCl and the
third best in terms of proteome variety, after HCl and EDTA (Fig. 2).
Deamidation rates, calculated by dividing the number of deamidated unique
peptides by the total number of unique peptides obtained from each sample, indicated that
the most deamidated peptides were obtained with NA (21%), followed by HCl (18%), AA
(16%), FA (15%) and EDTA (14%). Overall, as could be expected, weak acids were the
most suitable to minimise the processing-induced deamidation of peptides and, although
HCl yielded a greater proteome variety, it also introduced relatively high levels of
deamidation. Overall, NA performed most poorly, giving similar results to weak acids in
terms of protein recovery but high deamidation rates, whereas EDTA and FA gave similar
extraction efficiencies (FA was better considering the semi-quantitative data whereas
EDTA was better considering the proteome complexity) and significantly reduced levels of
processing-induced deamidations. The SD from samples treated with EDTA in Fig. 2C was
larger than those treated with FA, so we could not establish which induced fewer
deamidations; for these reasons, we assumed that they were both equally suitable for our
aims. As the more common method, we chose to use EDTA for the successive steps of
our work, but we also acknowledge the potential use of FA to gain equally good results.
11

Figure 2 – Bar charts showing the average and standard deviation of A) total number of unique peptides, B) total number of proteins, and C) deamidation ratio obtained from three replicates, using different acids to demineralize the sample.
In order to also investigate whether or not the exposure to different acids was
inducing chemical cleavage activity82 we also performed a Mascot search with semiTrypsin
specificity maintaining unchanged all other parameters. From these results (Supporting
Table S-1) it was clear that the levels of semi-tryptic peptides were higher when strong
acids like HCl and NA were used and smaller when weak acids like EDTA, FA and AA
were used, the latter of which gave similar results to one another. This complements the
finding of lower proteome variety in samples treated with NA than with the other weaker
acids when results were searched with only the normal trypsin specificity.
Demineralization lengthConsidering the pH conditions in which samples are typically incubated during the
demineralization process, it was important to evaluate the impact that different
demineralization lengths could have on the final proteome and deamidation rates
observed. Given that shorter periods of incubation in acid should decrease the processing-
induced decay of proteins we performed three different tests incubating the bone powder
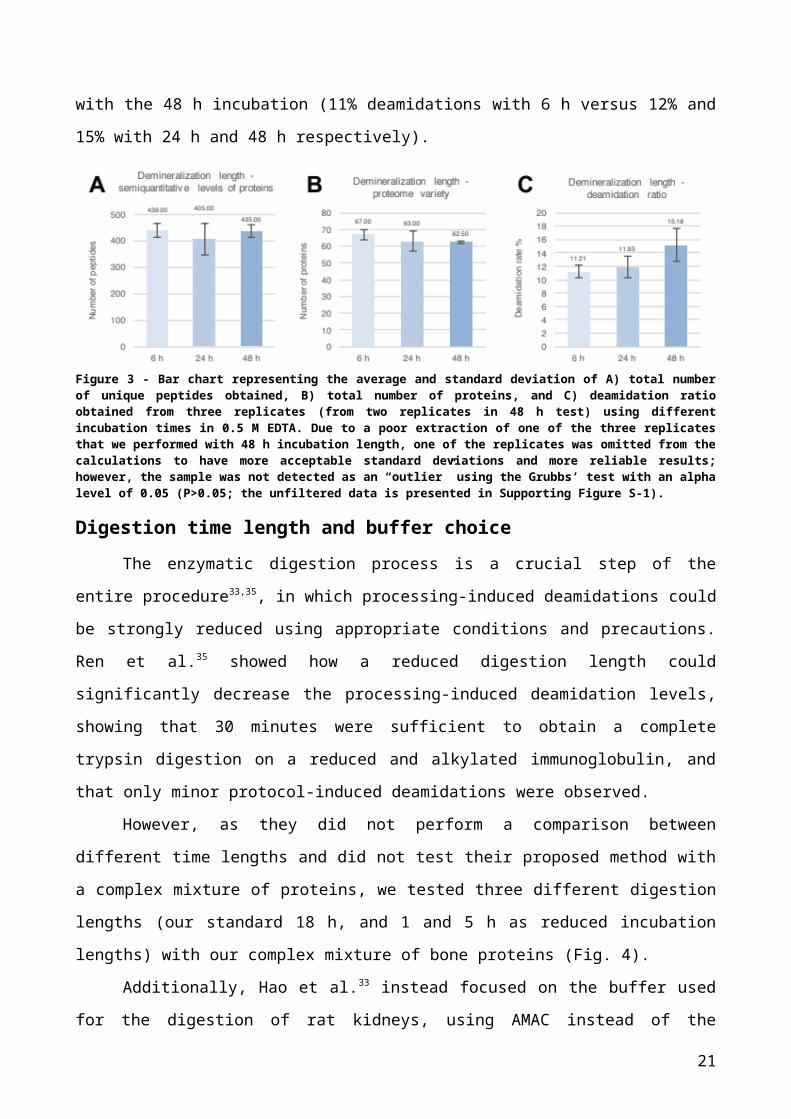
with 0.5 M EDTA for 6, 24 and 48 h, spanning most commonly used approaches.
Interestingly the shortest incubation proved to be sufficient at extracting a good quantity
and variety of proteins (Fig. 3A and 3B) and even yielded slightly better results than longer
demineralization periods. It is likely that longer incubation periods were damaging (i.e.,
chemically cleaving) proteins, preventing their identification during the successive steps of
the analysis. This hypothesis was confirmed through Mascot searches with semiTrypsin
specificity (Supporting Table S-1), where prolonged demineralization time periods in
EDTA, despite being a weak acid, caused an increase in proteolysis reducing the
capability to detect full tryptic peptides under normal search conditions. Generally, shorter
demineralization periods are more appropriate than longer ones to avoid protein disruption
12

and consequent loss. It was clear that the demineralization length was affecting the
processing-induced deamidation of samples as deamidation rates were lower using
shorter incubation times (Fig. 3C); interestingly, 6 h of incubation reduced the overall
deamidation rates by 26.7% compared with the 48 h incubation (11% deamidations with 6
h versus 12% and 15% with 24 h and 48 h respectively).
Figure 3 - Bar chart representing the average and standard deviation of A) total number of unique peptides obtained, B) total number of proteins, and C) deamidation ratio obtained from three replicates (from two replicates in 48 h test) using different incubation times in 0.5 M EDTA. Due to a poor extraction of one of the three replicates that we performed with 48 h incubation length, one of the replicates was omitted from the calculations to have more acceptable standard deviations and more reliable results; however, the sample was not detected as an “outlier” using the Grubbs’ test with an alpha level of 0.05 (P>0.05; the unfiltered data is presented in Supporting Figure S-1).
Digestion time length and buffer choiceThe enzymatic digestion process is a crucial step of the entire procedure33,35, in
which processing-induced deamidations could be strongly reduced using appropriate
conditions and precautions. Ren et al.35 showed how a reduced digestion length could
significantly decrease the processing-induced deamidation levels, showing that 30 minutes
were sufficient to obtain a complete trypsin digestion on a reduced and alkylated
immunoglobulin, and that only minor protocol-induced deamidations were observed.
However, as they did not perform a comparison between different time lengths and
did not test their proposed method with a complex mixture of proteins, we tested three
different digestion lengths (our standard 18 h, and 1 and 5 h as reduced incubation
lengths) with our complex mixture of bone proteins (Fig. 4).
Additionally, Hao et al.33 instead focused on the buffer used for the digestion of rat
kidneys, using AMAC instead of the standard ABC to reduce levels of artificial Asn
deamidations for complex proteomic samples by reducing the pH of the digest34,83; we also
explored this observation but for the bone tissue.
The results indicate that the digestion length appears to be directly proportional to
the number of unique peptides obtained from each sample (Fig. 4A) as well as to the
13

variety of the recovered proteome (Fig. 4B). We found that 1 h digestion was not sufficient
to get an adequate proteome coverage starting from a complex mixture of proteins,
whereby we observed 35% fewer unique peptides than the 18 h digestion, with 29.8% less
proteome variety. With 5 h digestion, we observed intermediate results both considering
the semi-quantitative levels of matched proteins and the proteome variety, losing only
16.6% of the unique number of peptides and the 15.9% of the proteome variety compared
with the longest incubation. These results were also supported through searches with
semiTrypsin specificity (Supporting Table S-1) where prolonged digestion length increased
the number of fully tryptic peptides matched, consequently reducing the levels of
semitryptic peptides observed in the sample. On the other hand, deamidation rates were
generally reduced using shorter incubation lengths, and this was observed for both buffers
tested.
The buffer choice also yielded interesting results where, in accordance with Hao et
al.33, the lowest levels of deamidations were observed in the five hour digests in AMAC
(Fig. 4C; 6% with 5 h digestion in AMAC versus 11% with same digestion length in ABC
and 14% with 18 h digestion in ABC). The more commonly used ABC on the other hand
usually resulted in a more diverse proteome than AMAC (around six more proteins were
frequently obtained with ABC than with AMAC) and greater semi-quantitative results (72
more unique peptides using ABC for 1h compared with AMAC, 70 more peptides with the
5 h and 32 more peptides with the 18 h digestion). Overall, AMAC appeared to greatly
reduce the decay compared with ABC as we observed as much as 43.3% more
deamidated peptides in the 5 h samples digested in ABC, 25.5% in the 1 h samples, and
37.3% more in the 18 h samples. We therefore encourage the use of AMAC as digestion
buffer in future studies relating to this aspect of studying endogenous protein decay.
Additionally, the digestion length of 5 h seemed most appropriate as a compromise
between reducing processing-induced deamidation levels and the complexity of proteome
observed. Further improvements not within the scope of this study, such as more rapid
digestion methods with greater enzyme concentrations, could also be investigated in the
future to reduce deamidation levels further while increasing the digestion efficiency.
14

Figure 4 - Bar chart representing the average and the standard deviation of A) total number of unique peptides obtained, B) total number of proteins, and C) deamidation ratio obtained from three replicates using different digestion buffers (ABC, orange and AMAC, green) and using different digestion length.
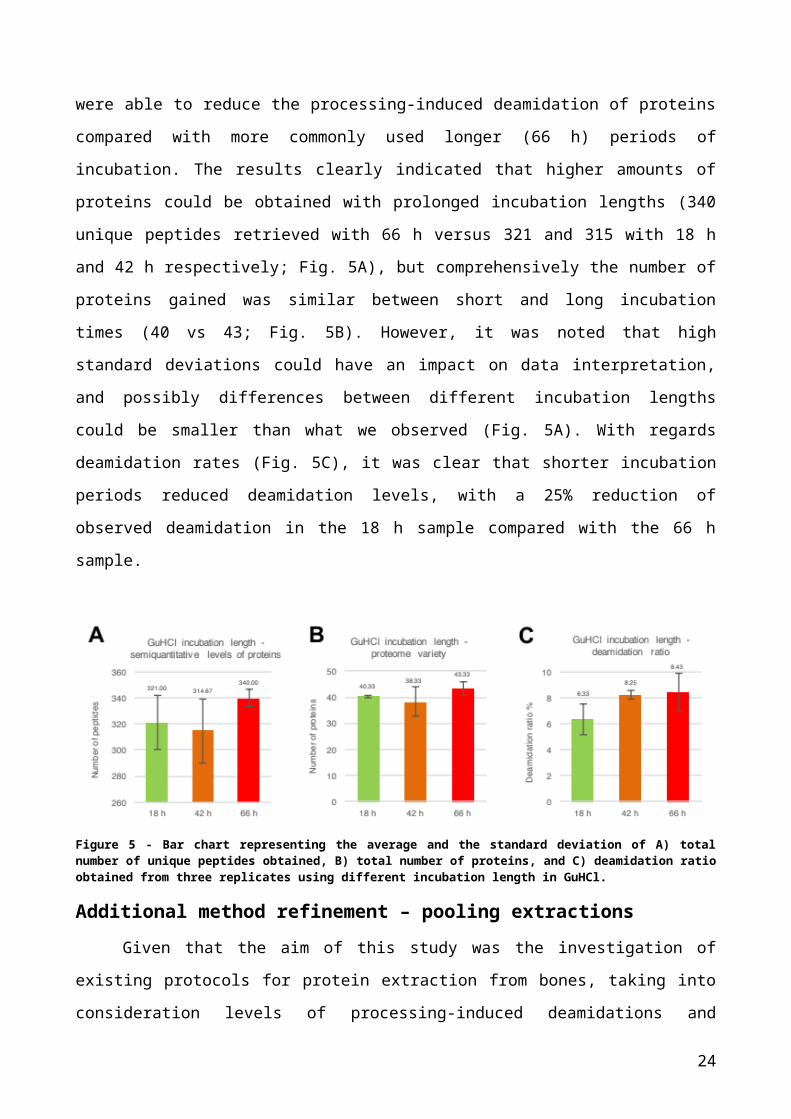
GuHCl incubation lengthThe final parameter evaluated was the variation in incubation length of the acid
insoluble pellet in extraction buffer. Even though the pH of the GuHCl protein denaturing
solution is near neutral (7.4), we evaluated whether shorter incubation periods were able
to reduce the processing-induced deamidation of proteins compared with more commonly
used longer (66 h) periods of incubation. The results clearly indicated that higher amounts
of proteins could be obtained with prolonged incubation lengths (340 unique peptides
retrieved with 66 h versus 321 and 315 with 18 h and 42 h respectively; Fig. 5A), but
comprehensively the number of proteins gained was similar between short and long
incubation times (40 vs 43; Fig. 5B). However, it was noted that high standard deviations
could have an impact on data interpretation, and possibly differences between different
incubation lengths could be smaller than what we observed (Fig. 5A). With regards
deamidation rates (Fig. 5C), it was clear that shorter incubation periods reduced
deamidation levels, with a 25% reduction of observed deamidation in the 18 h sample
compared with the 66 h sample.
15

Figure 5 - Bar chart representing the average and the standard deviation of A) total number of unique peptides obtained, B) total number of proteins, and C) deamidation ratio obtained from three replicates using different incubation length in GuHCl.
Additional method refinement – pooling extractionsGiven that the aim of this study was the investigation of existing protocols for protein
extraction from bones, taking into consideration levels of processing-induced deamidations
and limiting them as much as possible, we evaluated various steps of standard procedures
that can induce some deamidations due to their temperature, pH and ionic strength.
Comparing the results obtained using the newly defined protocol versus the initial results
obtained during the first acid test, we noticed a substantial reduction of both the proteome
variety and the quantity (40 different proteins and 321 unique peptides versus the starting
71 different proteins and 572 unique peptides).
Previous analyses14,74 have shown that the reduction of collagen levels in the
samples allowed for the observations of greater varieties of NCPs; the relative abundance
of the collagenous proteins are thought to impair the detection of less abundant proteins
during LC/MS/MS analyses, ending with a limited number of other proteins observed. Two
strategies have previously been explored to decrease levels of collagen and/or to increase
levels of NCPs; one used bacterial collagenase to digest the dominant collagen in the
extracted sample14, whereas another74 proposed the use of multiple extraction steps,
keeping separate extractions such as GuHCl incubation to extract relatively more NCPs
than collagen in other fractions.
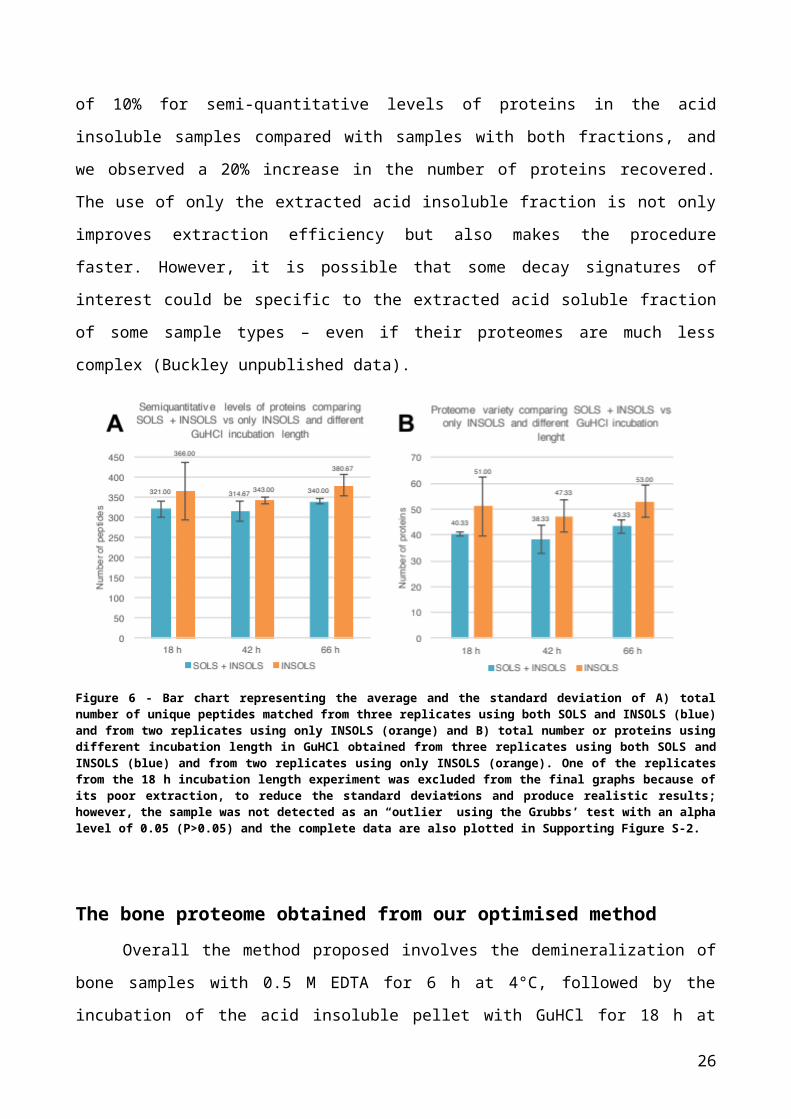
In order to improve results in terms of proteome variety, we tested a method
involving the incubation of the acid insoluble pellet with GuHCl followed by the extraction
of only this fraction for the successive steps of the procedure, without pooling fractions as
done previously. As expected, a substantial improvement was observed in keeping the two
fractions separate and analysing only the acid insoluble fraction, as the proteome variety
was greater as well as the number of unique peptides matched (Fig. 6A and 6B). In
particular, we observed an increase of 10% for semi-quantitative levels of proteins in the
acid insoluble samples compared with samples with both fractions, and we observed a
20% increase in the number of proteins recovered. The use of only the extracted acid
insoluble fraction is not only improves extraction efficiency but also makes the procedure
faster. However, it is possible that some decay signatures of interest could be specific to
the extracted acid soluble fraction of some sample types – even if their proteomes are
much less complex (Buckley unpublished data).
16

Figure 6 - Bar chart representing the average and the standard deviation of A) total number of unique peptides matched from three replicates using both SOLS and INSOLS (blue) and from two replicates using only INSOLS (orange) and B) total number or proteins using different incubation length in GuHCl obtained from three replicates using both SOLS and INSOLS (blue) and from two replicates using only INSOLS (orange). One of the replicates from the 18 h incubation length experiment was excluded from the final graphs because of its poor extraction, to reduce the standard deviations and produce realistic results; however, the sample was not detected as an “outlier” using the Grubbs’ test with an alpha level of 0.05 (P>0.05) and the complete data are also plotted in Supporting Figure S-2.
The bone proteome obtained from our optimised methodOverall the method proposed involves the demineralization of bone samples with
0.5 M EDTA for 6 h at 4°C, followed by the incubation of the acid insoluble pellet with
GuHCl for 18 h at 4°C, ultrafiltration into 50 mM AMAC and, following reduction and
alkylation at room temperature, digestion with trypsin for 5 h at 37°C. Despite this method
being satisfactory for the intended purposes of minimising deamidations whilst obtaining a
complex proteome, we consider it important to point out that the use of FA instead of
EDTA may be more appropriate for several practical and technical reasons. The EDTA
extractions proved to be substantially more time-consuming than all other acids during the
ultrafiltration step. The presence of sodium ions present in EDTA may also cause some
problems with mass spectrometry analysis if not sufficiently removed, creating adducts
with the protein in the gas phase84. Moreover, FA is becoming increasingly common in
many other proteomics applications in various fields85,86, and normally used to resuspend
proteins before LC/MS/MS analysis; therefore the eventual presence of small amounts of
this acid in the sample after the ultrafiltration will not as greatly affect subsequent
analyses. For these reasons, and because EDTA and FA appear to yield comparable
17

results, future investigations could consider FA for analysis for optimal deamidation
minimisation.
Figure 7 - Scheme of the optimised protocol for the reduction of the artificial decay.
Table 1 - Comparisons between the average number of unique peptides, unique proteins, NCPs, the number of NCPs obtained in the best of the three replicates and the average of the deamidation ratios obtained using the standard protocol (first column) and the proposed optimised one (second column).
18
Overall we have shown that our optimised protocol (Fig. 7) reduces processing-
induced deamidation levels in the samples up to ~50% of other protocols (Table 1), but
can also be carried out much more quickly: in fact, most published methods for bone
proteome extraction require the best part of one week (i.e., at least 3-4 days), whereas our
optimised one only requires two working days to the point of mass spectrometric analysis.
The extracted bone proteomeTo identify associations between the extracted proteins and to visualise the
proteome variety we used STRING software to build a map of protein associations within
the matched pooled proteome obtained using the optimised method (Fig. 8). Our method
allowed the recovery of bone-specific proteins (Fig. 8 yellow nodes), plasma proteins with
functions in bone development and/or turnover (Fig. 8 green nodes), plasma proteins with
coagulation functions (Fig. 8 blue nodes) and other proteins with less well-known
associations (Fig. 8 red nodes). The bone-specific proteins are characterised in particular
by different types of collagen, to which some other bone matrix NCPs are strongly linked,
like osteonectin (SPARC), lumican (LUM) and bone sialoprotein (IBSP). The plasma
proteins formed two main clusters, one of which centred around albumin (ALB), a protein
also present in bone that can be only partially exchanged with the plasma albumin, since
the majority of this protein is permanently fixed in the mineralized matrix of the bone 87. In
addition to apolipoproteins (APOA1, APOA2) involved in the plasma transport of
lipoproteins, transferrin (TF) and fetuin (AHSG) were also part of this cluster. TF is a
plasma protein able to deliver iron to the bone marrow to load newly formed erythrocytes 88,
whereas AHSG is a plasma protein bound to the bone matrix with important functions in
regulating the bone turnover39. The cluster of plasma proteins with coagulation-related
functions includes prothrombin (F2), coagulation factors (F9, F10), fibrinogen alpha and
beta chains (FGA and FGB), vitamin K-dependent protein C (PROC), vitamin K-dependent
protein S (PROS1), and antithrombin-III (SERPIN C1). Finally, the various proteins that do
not cluster strongly with others observed have a multitude of different functions, including
many proteins that are specific to bone such as thrombospondin (THBS1), which bind to
osteonectin, calcium and osteoblasts39, and decorin (DCN), a NCP that is expressed both
in cartilage and in bone39.
19

Figure 8 – Association map of the pooled proteome extracted using our optimised extraction method. Yellow nodes indicate bone specific proteins, green nodes indicate plasma proteins linked with the mineralized phase in bones, blue nodes indicate plasma specific proteins with coagulation functions, and red nodes indicate various remaining proteins with different functions. The black stars indicate proteins observed in each of the three replicates.
20
It is noteworthy that although a substantial number of bone-related proteins were
observed (both collagenous and non-collagenous) even with our minimally damaging
methods, there was a relatively large number of serum proteins matched, along with
numerous more ubiquitously expressed proteins. The successful extraction of a good
variety of NCPs such as albumin, fetuin and decorin, makes this extraction method
potentially suitable for phylogenetic studies and a means of species identification, as it has
been shown that these and others NCPs obtained in this study are characterised by a
higher amino acid variability between different species compared with collagen4.
Furthermore, some of these NCPs can also survive longer in archaeological bones5,89 than
DNA, making them useful for species identification purposes even when ancient DNA is
too degraded4. However, of more specific interest to potential forensic applications is that
the different types of proteins observed may offer an appropriate range of different
sequence environments within which the Asn and Gln residues deamidate at distinctly
different rates.
Conclusions
In this study we have developed an improved method for protein extraction from
bone samples with minimum laboratory-induced decay which provides a good range of
proteins. We observed a ~50% reduction in deamidation levels using this new protocol
compared with results obtained using the protocol used by Wadsworth and Buckley14. In
addition to the strong decrease of deamidation levels, our improved protocol also
dramatically decreased the length of the procedure that originally was almost one week
long down to within two days. We suggest that this method could be considered
appropriate for future studies of archaeological, palaeontological as well as forensic
investigations using proteomics where decay measurements are of interest.
AcknowledgmentsThe authors are grateful to the Royal Society for funding both a PhD studentship
(NP) and university research fellowship (MB) under grants RG130453 and UF120473
respectively. We also acknowledge the technical support of The University of
Manchester’s Faculty of Life Sciences Biomolecular Analysis core facility.

Supporting InformationFigure S-1 – Semi-quantitative levels of proteins obtained, proteome variety and
deamidation ratios from demineralization length test including the first replicate from the 48
h test.
Figure S-2 – Semi-quantitative levels of proteins obtained, proteome variety and
deamidation ratios from demineralization length test including the first replicate from the 18
h test.
Table S-1 – Results of Mascot searches with semiTrypsin specificity performed for acid
test, demineralization length test and digestion test.
Table S-2 – Scaffold peptide results for the analyses of different acids. Table S-3 – Scaffold peptide results for the analyses of different buffer protocols.
Table S-4 – Scaffold peptide results for the analyses of different demineralization lengths.
Table S-5 – Scaffold peptide results for the analyses of different GuHCl incubation
lengths.
Table S-6 – Scaffold peptide results for the analyses of different GuHCl incubation lengths
using only the acid-insoluble fraction.
Bibliography (1) Byrum, S.; Montgomery, C. O.; Nicholas, R. W.; Suva, L. J. The Promise of Bone
Cancer Proteomics. Ann. N. Y. Acad. Sci. 2010, 1192 (1), 222–229.
(2) Ruiz-Romero, C.; Blanco, F. J. The Role of Proteomics in Osteoarthritis
Pathogenesis Research. Curr. Drug Targets 2009, 10 (6), 543–556.
(3) Zhang, H.; Recker, R.; Lee, W.-N. P.; Xiao, G. G. Proteomics in Bone Research.
Expert Rev. Proteomics 2010, 7 (1), 103–111.
(4) Buckley, M.; Wadsworth, C. Proteome Degradation in Ancient Bone: Diagenesis and
Phylogenetic Potential. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2014, 416, 69–79.
(5) Cappellini, E.; Jensen, L. J.; Szklarczyk, D.; Ginolhac, A.; da Fonseca, R. A. R.;
Stafford Jr, T. W.; Holen, S. R.; Collins, M. J.; Orlando, L.; Willerslev, E. Proteomic
Analysis of a Pleistocene Mammoth Femur Reveals More than One Hundred
Ancient Bone Proteins. J. Proteome Res. 2011, 11 (2), 917–926.
(6) Hill, R. C.; Wither, M. J.; Nemkov, T.; Barrett, A.; D’Alessandro, A.; Dzieciatkowska,
M.; Hansen, K. C. Preserved Proteins from Extinct Bison Latifrons Identified by
Tandem Mass Spectrometry; Hydroxylysine Glycosides Are a Common Feature of

Ancient Collagen. Mol. Cell. Proteomics 2015, 14 (7), 1946–1958.
(7) Buckley, M.; Fraser, S.; Herman, J.; Melton, N. D.; Mulville, J.; Pálsdóttir, A. H.
Species Identification of Archaeological Marine Mammals Using Collagen
Fingerprinting. J. Archaeol. Sci. 2014, 41, 631–641.
(8) Buckley, M. Ancient Collagen Reveals Evolutionary History of the Endemic South
American “ungulates.” In Proc. R. Soc. B; The Royal Society, 2015; Vol. 282, p
20142671.
(9) Brown, S.; Higham, T.; Slon, V.; Pääbo, S.; Meyer, M.; Douka, K.; Brock, F.;
Comeskey, D.; Procopio, N.; Shunkov, M. Identification of a New Hominin Bone from
Denisova Cave, Siberia Using Collagen Fingerprinting and Mitochondrial DNA
Analysis. Sci. Rep. 2016, 6.
(10) Collins, M.; Buckley, M.; Grundy, H. H.; Thomas-Oates, J.; Wilson, J.; van Doorn, N.
ZooMS: The Collagen Barcode and Fingerprints. SpectroscopyEurope 2010, 22 (2),
6.
(11) Buckley, M.; Kansa, S. W.; Howard, S.; Campbell, S.; Thomas-Oates, J.; Collins, M.
Distinguishing between Archaeological Sheep and Goat Bones Using a Single
Collagen Peptide. J. Archaeol. Sci. 2010, 37 (1), 13–20.
(12) Buckley, M.; Kansa, S. W. Collagen Fingerprinting of Archaeological Bone and
Teeth Remains from Domuztepe, South Eastern Turkey. Archaeol. Anthropol. Sci.
2011, 3 (3), 271–280.
(13) Buckley, M.; Collins, M.; Thomas‐Oates, J.; Wilson, J. C. Species Identification by
Analysis of Bone Collagen Using Matrix‐assisted Laser Desorption/ionisation Time-
of-flight Mass Spectrometry. Rapid Commun. mass Spectrom. 2009, 23 (23), 3843–
3854.
(14) Wadsworth, C.; Buckley, M. Proteome Degradation in Fossils: Investigating the
Longevity of Protein Survival in Ancient Bone. Rapid Commun. Mass Spectrom.
2014, 28 (6), 605–615.
(15) Stadtman, E. R. Protein Oxidation in Aging and Age‐related Diseases. Ann. N. Y.
Acad. Sci. 2001, 928 (1), 22–38.
(16) Finkel, T.; Holbrook, N. J. Oxidants, Oxidative Stress and the Biology of Ageing.
Nature 2000, 408 (6809), 239–247.
(17) Wilson, J.; van Doorn, N. L.; Collins, M. J. Assessing the Extent of Bone
Degradation Using Glutamine Deamidation in Collagen. Anal. Chem. 2012, 84 (21),
9041–9048.
(18) Perez Hurtado, P.; O’Connor, P. B. Deamidation of Collagen. Anal. Chem. 2012, 84
(6), 3017–3025.
(19) Schroeter, E. R.; Cleland, T. P. Glutamine Deamidation: An Indicator of Antiquity, or
Preservational Quality? Rapid Commun. Mass Spectrom. 2016, 30 (2), 251–255.
(20) Muhammed, A. Amino Acid Racemization from Tooth for Age Estimation-An
Overview. Malaysian J. Forensic Sci. 2012, 3 (1), 41–45.
(21) Poinar, H. N.; Höss, M.; Bada, J. L.; Pääbo, S. Amino Acid Racemization and the
Preservation of Ancient DNA. Science (80-. ). 1996, 272 (5263), 864–866.
(22) Van Doorn, N. L.; Wilson, J.; Hollund, H.; Soressi, M.; Collins, M. J. Site-Specific
Deamidation of Glutamine: A New Marker of Bone Collagen Deterioration. Rapid
Commun. Mass Spectrom. 2012, 26 (19), 2319–2327.
(23) Cloos, P. A. C.; Christgau, S. Non-Enzymatic Covalent Modifications of Proteins:
Mechanisms, Physiological Consequences and Clinical Applications. Matrix Biol.
2002, 21 (1), 39–52.
(24) Collins, M. J.; Waite, E. R.; Van Duin, A. C. T. Predicting Protein Decomposition:
The Case of Aspartic–acid Racemization Kinetics. Philos. Trans. R. Soc. London B
Biol. Sci. 1999, 354 (1379), 51–64.
(25) Bernevic, B.; Petre, B. A.; Galetskiy, D.; Werner, C.; Wicke, M.; Schellander, K.;
Przybylski, M. Degradation and Oxidation Postmortem of Myofibrillar Proteins in
Porcine Skeleton Muscle Revealed by High Resolution Mass Spectrometric
Proteome Analysis. Int. J. Mass Spectrom. 2011, 305 (2), 217–227.
(26) Robinson, A. B.; Scotchler, J. W. Sequence Dependent Deamidation Rates for
Model Peptides of Histone IV. Int. J. Pept. Protein Res. 1974, 6 (5), 279–282.
(27) Robinson, N. E.; Robinson, A. B. Molecular Clocks. Proc. Natl. Acad. Sci. 2001, 98
(3), 944–949.
(28) Robinson, A. B.; McKerrow, J. H.; Cary, P. Controlled Deamidation of Peptides and
Proteins: An Experimental Hazard and a Possible Biological Timer. Proc. Natl. Acad.
Sci. U. S. A. 1970, 66 (3), 753–757.
(29) Robinson, A. B.; Rudd, C. J. Deamidation of Glutaminyl and Asparaginyl Residues in
Peptides and Proteins. Curr. Top. Cell. Regul. 1974, 8, 247–295.
(30) McKerrow, J. H.; Robinson, A. B. Primary Sequence Dependence of the
Deamidation of Rabbit Muscle Aldolase. Science 1974, 183 (4120), 85.
(31) Takemoto, L.; Fujii, N.; Boyle, D. Mechanism of Asparagine Deamidation during
Human Senile Cataractogenesis. Exp. Eye Res. 2001, 72 (5), 559–563.

(32) Buckley, M.; Walker, A.; Ho, S. Y. W.; Yang, Y.; Smith, C.; Ashton, P.; Oates, J. T.;
Cappellini, E.; Koon, H.; Penkman, K. et al. Comment on “Protein Sequences from
Mastodon and Tyrannosaurus Rex Revealed by Mass Spectrometry.” Science 2008,
319 (5859), 33c–33c.
(33) Hao, P.; Ren, Y.; Datta, A.; Tam, J. P.; Sze, S. K. Evaluation of the Effect of Trypsin
Digestion Buffers on Artificial Deamidation. J. Proteome Res. 2015, 14 (2), 1308–
1314.
(34) Hao, P.; Ren, Y.; Alpert, A. J.; Sze, S. K. Detection, Evaluation and Minimization of
Nonenzymatic Deamidation in Proteomic Sample Preparation. Mol. Cell. Proteomics
2011, 10 (10), O111.009381.
(35) Ren, D.; Pipes, G. D.; Liu, D.; Shih, L.-Y.; Nichols, A. C.; Treuheit, M. J.; Brems, D.
N.; Bondarenko, P. V. An Improved Trypsin Digestion Method Minimizes Digestion-
Induced Modifications on Proteins. Anal. Biochem. 2009, 392 (1), 12–21.
(36) Clarke, B. Normal Bone Anatomy and Physiology. Clin. J. Am. Soc. Nephrol. 2008, 3
Suppl 3, S131-9.
(37) Turner-Walker, G. The Chemical and Microbial Degradation of Bones and Teeth. In
Advances in Human Palaeopathology; Pinhasi, R., Mays, S., Eds.; John Wiley &
Sons, Ltd: Chichester, UK, 2007; pp 3–29.
(38) Zhu, W.; Robey, P. G.; Boskey, A. L. The Regulatory Role of Matrix Proteins in
Mineralization of Bone. Fundam. osteoporosis. Elsevier Acad. Press. San Diego
2009, 153–202.
(39) Heinegård, D.; Oldberg, A. Structure and Biology of Cartilage and Bone Matrix
Noncollagenous Macromolecules. FASEB J. 1989, 3 (9), 2042–2051.
(40) Madea, B. Is There Recent Progress in the Estimation of the Postmortem Interval by
Means of Thanatochemistry? Forensic Sci. Int. 2005, 151 (2–3), 139–149.
(41) Salam, H. A.; Shaat, E. A.; Aziz, M. H. A.; MoneimSheta, A. A.; Hussein, H. A. S. M.
Estimation of Postmortem Interval Using Thanatochemistry and Postmortem
Changes. Alexandria J. Med. 2012, 48 (4), 335–344.
(42) Catts, E. P.; Goff, M. L. Forensic Entomology in Criminal Investigations. Annu. Rev.
Entomol. 1992, 37 (1), 253–272.
(43) Villet, M. H.; Richards, C. S.; Midgley, J. M. Contemporary Precision, Bias and
Accuracy of Minimum Post-Mortem Intervals Estimated Using Development of
Carrion-Feeding Insects. In Current concepts in forensic entomology; Springer,
2009; pp 109–137.
(44) Swift, B. Essentials of Autopsy Practice. In Essentials of Autopsy Practice; Rutty, G.
N., Ed.; Springer London: London, 2006; pp 189–214.
(45) Catts, E. P. Problems in Estimating the Postmortem Interval in Death Investigations.
J. Agric. Entomol. 1992, 9 (4), 245–255.
(46) Amendt, J.; Richards, C. S.; Campobasso, C. P.; Zehner, R.; Hall, M. J. R. Forensic
Entomology: Applications and Limitations. Forensic Sci. Med. Pathol. 2011, 7 (4),
379–392.
(47) Di Nunno, N. R.; Costantinides, F.; Bernasconi, P.; Bottin, C.; Melato, M. Is Flow
Cytometric Evaluation of DNA Degradation a Reliable Method to Investigate the
Early Postmortem Period? Am. J. Forensic Med. Pathol. 1998, 19 (1), 50–53.
(48) Johnson, L. A.; Ferris, J. A. J. Analysis of Postmortem DNA Degradation by Single-
Cell Gel Electrophoresis. Forensic Sci. Int. 2002, 126 (1), 43–47.
(49) Bauer, M.; Gramlich, I.; Polzin, S.; Patzelt, D. Quantification of mRNA Degradation
as Possible Indicator of Postmortem Interval—a Pilot Study. Leg. Med. 2003, 5 (4),
220–227.
(50) Kang, S.; Kassam, N.; Gauthier, M. L.; O’Day, D. H. Post-Mortem Changes in
Calmodulin Binding Proteins in Muscle and Lung. Forensic Sci. Int. 2003, 131 (2–3),
140–147.
(51) Sabucedo, A. J.; Furton, K. G. Estimation of Postmortem Interval Using the Protein
Marker Cardiac Troponin I. Forensic Sci. Int. 2003, 134 (1), 11–16.
(52) Wehner, F.; Wehner, H.-D.; Schieffer, M. C.; Subke, J. Delimitation of the Time of
Death by Immunohistochemical Detection of Thyroglobulin. Forensic Sci. Int. 2000,
110 (3), 199–206.
(53) Saukko, P.; Knight, B. The Establishment of Identity of Human Remains. In Knight’s
Forensic Pathology Fourth Edition; CRC Press: London, 2015; pp 95–132.
(54) Suchey, J. M.; Payen, L. A.; Slota, P. J.; Taylor, R. E. The Use of Radiocarbon (14
C) to Identify Human Skeletal Materials of Forensic Science Interest. J. Forensic Sci.
1989, 34 (5), 1196–1205.
(55) Schwarcz, H. P.; Agur, K.; Jantz, L. M. A New Method for Determination of
Postmortem Interval: Citrate Content of Bone. J. Forensic Sci. 2010, 55 (6), 1516–
1522.
(56) Introna, F. J.; Di Vella, G.; Campobasso, C. P. Determination of Postmortem Interval
from Old Skeletal Remains by Image Analysis of Luminol Test Results. J. Forensic
Sci. 1999, 44 (3), 535–538.

(57) Knight, B.; Lauder, I. Methods of Dating Skeletal Remains. Hum. Biol. 1969, 41 (3),
322–341.
(58) Cabiscol, E.; Tamarit, J.; Ros, J. Protein Carbonylation: Proteomics, Specificity and
Relevance to Aging. Mass Spectrom. Rev. 2014, 33 (1), 21–48.
(59) Nyström, T. Role of Oxidative Carbonylation in Protein Quality Control and
Senescence. EMBO J. 2005, 24 (7), 1311–1317.
(60) Vanhooren, V.; Navarrete Santos, A.; Voutetakis, K.; Petropoulos, I.; Libert, C.;
Simm, A.; Gonos, E. S.; Friguet, B. Protein Modification and Maintenance Systems
as Biomarkers of Ageing. Mech. Ageing Dev. 2015, 151, 71–84.
(61) Ulrich, P.; Cerami, A. Protein Glycation, Diabetes, and Aging. Recent Prog. Horm.
Res. 2001, 56, 1–21.
(62) Wang, Z.; Lyons, B.; Truscott, R. J. W.; Schey, K. L. Human Protein Aging:
Modification and Crosslinking through Dehydroalanine and Dehydrobutyrine
Intermediates. Aging Cell 2014, 13 (2), 226–234.
(63) Chellan, P.; Nagaraj, R. H. Protein Crosslinking by the Maillard Reaction:
Dicarbonyl-Derived Imidazolium Crosslinks in Aging and Diabetes. Arch. Biochem.
Biophys. 1999, 368 (1), 98–104.
(64) Ritz-Timme, S.; Collins, M. J. Racemization of Aspartic Acid in Human Proteins.
Ageing Res. Rev. 2002, 1 (1), 43–59.
(65) Šoškić, V.; Groebe, K.; Schrattenholz, A. Nonenzymatic Posttranslational Protein
Modifications in Ageing. Exp. Gerontol. 2008, 43 (4), 247–257.
(66) Beal, M. F. Oxidatively Modified Proteins in Aging and Disease 1, 2. Free Radic.
Biol. Med. 2002, 32 (9), 797–803.
(67) Robinson, N. E.; Robinson, A. B. Deamidation of Human Proteins. Proc. Natl. Acad.
Sci. 2001, 98 (22), 12409–12413.
(68) Li, X.; Lin, C.; O’Connor, P. B. Glutamine Deamidation: Differentiation of Glutamic
Acid and Gamma-Glutamic Acid in Peptides by Electron Capture Dissociation. Anal.
Chem. 2010, 82 (9), 3606–3615.
(69) Hains, P. G.; Truscott, R. J. W. Age-Dependent Deamidation of Lifelong Proteins in
the Human Lens. Invest. Ophthalmol. Vis. Sci. 2010, 51 (6), 3107–3114.
(70) Xie, M.; Schowen, R. L. Secondary Structure and Protein Deamidation. J. Pharm.
Sci. 1999, 88 (1), 8–13.
(71) Kossiakoff, A. A. Tertiary Structure Is a Principal Determinant to Protein
Deamidation. Science 1988, 240 (4849), 191–194.

(72) Stratton, L. P.; Kelly, R. M.; Rowe, J.; Shively, J. E.; Smith, D. D.; Carpenter, J. F.;
Manning, M. C. Controlling Deamidation Rates in a Model Peptide: Effects of
Temperature, Peptide Concentration, and Additives. J. Pharm. Sci. 2001, 90 (12),
2141–2148.
(73) Scotchler, J. W.; Robinson, A. B. Deamidation of Glutaminyl Residues: Dependence
on pH, Temperature, and Ionic Strength. Anal. Biochem. 1974, 59 (1), 319–322.
(74) Jiang, X.; Ye, M.; Jiang, X.; Liu, G.; Feng, S.; Cui, L.; Zou, H. Method Development
of Efficient Protein Extraction in Bone Tissue for Proteome Analysis. J. Proteome
Res. 2007, 6 (6), 2287–2294.
(75) Cleland, T. P.; Voegele, K.; Schweitzer, M. H. Empirical Evaluation of Bone
Extraction Protocols. PLoS One 2012, 7 (2), e31443.
(76) Sanjai, K.; Kumarswamy, J.; Patil, A.; Papaiah, L.; Jayaram, S.; Krishnan, L.
Evaluation and Comparison of Decalcification Agents on the Human Teeth. J. Oral
Maxillofac. Pathol. 2012, 16 (2), 222–227.
(77) Liu, L.; Yue, S.; Jiang, H.; Lu, T. Comparison of Demineralization of Different
Organic Acid to Enamel. West China J. Stomatol. 1998, 16 (2), 103–104, 113.
(78) Keller, A.; Nesvizhskii, A. I.; Kolker, E.; Aebersold, R. Empirical Statistical Model to
Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database
Search. Anal. Chem. 2002, 74 (20), 5383–5392.
(79) Nesvizhskii, A. I.; Keller, A.; Kolker, E.; Aebersold, R. A Statistical Model for
Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem. 2003, 75 (17),
4646–4658.
(80) Cho, A.; Suzuki, S.; Hatakeyama, J.; Haruyama, N.; Kulkarni, A. B. A Method for
Rapid Demineralization of Teeth and Bones. Open Dent. J. 2010, 4 (1).
(81) González-Chávez, S. A.; Pacheco-Tena, C.; Macías-Vázquez, C. E.; Luévano-
Flores, E. Assessment of Different Decalcifying Protocols on Osteopontin and
Osteocalcin Immunostaining in Whole Bone Specimens of Arthritis Rat Model by
Confocal Immunofluorescence. Int. J. Clin. Exp. Pathol. 2013, 6 (10), 1972–1983.
(82) Han, K.-K.; Richard, C.; Biserte, G. Current Developments in Chemical Cleavage of
Proteins. Int. J. Biochem. 1983, 15 (7), 875–884.
(83) Kasserra, H. P.; Laidler, K. J. pH Effects in Trypsin Catalysis. Can. J. Chem. 1969,
47 (21), 4021–4029.
(84) Banerjee, S.; Mazumdar, S. Electrospray Ionization Mass Spectrometry: A
Technique to Access the Information beyond the Molecular Weight of the Analyte.

International Journal of Analytical Chemistry. 2012, pp 1–40.
(85) Gorton, R. L.; Seaton, S.; Ramnarain, P.; McHugh, T. D.; Kibbler, C. C. Evaluation of
a Short, on-Plate Formic Acid Extraction Method for Matrix-Assisted Laser
Desorption Ionization–time of Flight Mass Spectrometry-Based Identification of
Clinically Relevant Yeast Isolates. J. Clin. Microbiol. 2014, 52 (4), 1253–1255.
(86) Matsuda, N.; Matsuda, M.; Notake, S.; Yokokawa, H.; Kawamura, Y.; Hiramatsu, K.;
Kikuchi, K. Evaluation of a Simple Protein Extraction Method for Species
Identification of Clinically Relevant Staphylococci by Matrix-Assisted Laser
Desorption Ionization–time of Flight Mass Spectrometry. J. Clin. Microbiol. 2012, 50
(12), 3862–3866.
(87) Owen, M.; Triffitt, J. T. Extravascular Albumin in Bone Tissue. J. Physiol. 1976, 257
(2), 293–307.
(88) Rama, R.; Sanchez, J. Transferrin Uptake by Bone Marrow Macrophages Is
Independent of the Degree of Iron Saturation. Br. J. Haematol. 1992, 82 (2), 455–
459.
(89) Schmidt-Schultz, T. H.; Schultz, M. Well Preserved Non‐collagenous Extracellular
Matrix Proteins in Ancient Human Bone and Teeth. Int. J. Osteoarchaeol. 2007, 17
(1), 91–99.
TOC graphics
For TOC only


















